

Abstract
Amyloid-β peptide (Aβ) aggregation is the hallmark of Alzheimer’s disease (AD). The imbalance between the production and clearance of Aβ results in the accumulation and aggregation of Aβ in the brain. Thus far, few drugs are available for AD treatment, but exercise has been recognized for its cognition-enhancing properties in AD patients. The underlying mechanisms remain unclear. Our recent study showed that long-term running exercise could activate the lysosomal function in the brains of mice. In this study, we investigated whether exercise could reduce Aβ accumulation by activating lysosomal function in APP/PSEN1 transgenic mice. Started at the age of 5 months, the mice were trained with a running wheel at the speed of 18 r/min, 40 min/d, 6 d/week for 5 months, and were killed at the end of the 10th month, then brain tissue was collected for biochemical analyses. The cognitive ability was assessed in the 9th month. We showed that long-term exercise significantly mitigated cognitive dysfunction in AD mice, accompanied by the enhanced lysosomal function and the clearance of Aβ in the brain. Exercise significantly promoted the nuclear translocation of transcription factor EB (TFEB), and increased the interaction between nuclear TFEB with AMPK-mediated acetyl-CoA synthetase 2, thus enhancing transcription of the genes associated with the biogenesis of lysosomes. Exercise also raised the levels of mature cathepsin D and cathepsin L, suggesting that more Aβ peptides could be degraded in the activated lysosomes. This study demonstrates that exercise may improve the cognitive dysfunction of AD by enhancing lysosomal function.
Keywords: Alzheimer’s disease, exercise, Aβ clearance, lysosomal function, TFEB, ACSS2, APP/PSEN1 transgenic mice
Introduction
Alzheimer’s disease (AD) is the most prevalent dementia among old people. Neuropathologically, AD is characterized by the accumulation of extracellular β-amyloid (Aβ) peptide, which is generated from the sequential cleavage of the amyloid precursor protein by β-secretases and γ-secretases [1], and intracellular neurofibrillary tangles [2, 3]. The pathogenesis of AD is complex, among which the amyloid plaques hypothesis postulates that Aβ (1–42 aa) has a pivotal role in this process. With the progress of AD, the accumulation of Aβ produces cytotoxicity, and leads to neuronal dysfunction and cell death, and eventually cognitive dysfunction [4]. Aβ plaques also accelerate tau spreading and cognitive decline in human AD [5]. As there are few effective treatments of AD in the clinic at the present, it is particularly important to find new therapeutic targets and prophylactic pathways for AD [6].
Lysosomes are the main digestive organelles in eukaryotic cells, first discovered in the 1950s by Christian de Duve [7]. These membrane-enclosed organelles are equipped with an acidic pH (pH 3.8–5.0) and >50 hydrolases in the lumen [8, 9]. Extracellular and intracellular substrates are delivered to lysosomes for degradation through endocytosis or autophagic pathways [10]. Lysosomal degradation of substrates is crucial to remove damaged organelles and misfolded proteins in cells. Recent research has revealed that lysosomal dysfunction damages the homeostasis of the central nervous system (CNS). Several studies have shown that lysosomal dysfunction is linked to Parkinson’s disease (PD), AD, and Huntington’s disease [11], which are all characterized by the accumulation of certain specific undegraded substrates [12–14]. Autophagic/lysosomal activity is robustly affected in AD, where a large group of autophagic vacuoles (AVs) is accumulated in neurons [15]. Lysosomal proteolysis and autophagy are disrupted by Alzheimer-related PS1 mutations [16]. Similarly, the lysosomal function is important to lower the levels of β-amyloid [17]. These observations suggest a crucial role of lysosomes in AD, and restoring the normal function of lysosomes may be a new therapeutic direction for the treatment of AD.
Undoubtedly, long-term regular exercise is beneficial to human health. Exercise enhances the function of the heart and lungs, improves energy metabolism, and mitigates the function of the circulatory system [18]. In addition to these widespread benefits, exercise is also involved in preventing aging of the nervous system, enhancing neuronal plasticity, and enhancing cognitive function. Thus, exercise has a positive impact on neurodegenerative diseases such as AD [19–21]. Several mechanisms have been suggested for the preventive effects of exercise. It has been reported that exercise delays the progression of AD by improving mitochondrial function and upregulating brain-derived neurotrophic factor levels [22, 23]. We and others have found that exercise can activate the autophagy-lysosomal pathway and plays an important role in the construction of skeletal muscle [24]. Recently, we reported that long-term running exercise could activate the lysosomal function in the brains of mice [25]. Here we aimed to investigate whether exercise could reduce the accumulation of Aβ peptides by activating lysosomal function. Mutations in APP and Presenilin1/2 (PSEN1/2) lead to an increase in the production of Aβ1–42 peptides and the risk for AD. Thus, the APP/PSEN1 transgenic mice were used in the current research to examine the relationship between AD, exercise, and lysosomes.
Materials and methods
Antibodies and reagents
The following antibodies were used for immunofluorescence (IF) and Western blot (WB) analysis: LAMP1 (Sigma; #SAB3500285), LC3 (Novus; #NB600-1384), p62 (Sigma; #P0067), Atg5 (Cell Signaling; #12994), β-Actin (Sigma; #A5441), APP (Novus;#nbp1-76910), BACE (Cell Signaling; #5606), cathepsin D (SANTA CRUZ; # SC-377299), cathepsin L (Abcam; #ab6314), GAPDH (Abcam; #ab9484), Aβ1–42 (Proteintech; #25524-1-ap), p-Tau(Ser396) (Immunoway;# YP0263), Tau (Proteintech; 66499-1), AMPK-mediated acetyl-CoA synthetase 2 (ACSS2) (Cell Signaling; #3658), translocation of transcription factor EB (TFEB) (Sigma; # 3110428), Histone H3 (Cell Signaling; #4499), DAPI (Sigma; #D9564), AMPK (Cell Signaling; #5832), p-AMPK (Cell Signaling; #2535), p-ULK1(Ser 555) (Cell Signaling; #5869), ULK1 (Cell Signaling; #8054), p-mTOR (Cell Signaling; #5536), mTOR (Cell Signaling; #2979), p-Raptor (Cell Signaling; #2280 T), Raptor (Cell Signaling; #2083 T), P70S6K (Abcam; #ab32529), p-P70S6K (Cell Signaling; #9206), RIP1 (BD Biosciences; #BD-610659), p-ERK1/2 (SantaCruz; #sc-81492), Calcineurin A (Cell Signaling; #2614), Calcineurin B (R&D Systems; #AF1348).
Animals
The AD transgenic mice and the background-matched WT mice (C57BL/6) used in this study were obtained from the Model Animal Research Center of Nanjing University (Animal strain: B6;C3-Tg (APPswe, PSEN1dE9)/Nju). All mice were housed individually in standard mice cages under laboratory conditions (22–24°C, 50%–60% relative humidity, air circulation), with ad libitum access to food and water, and maintained on a standard 12/12 h light/dark cycle with lights on at 8:00 a.m. All experimental procedures were approved by the Experimental Animal Care and Use Committee at Soochow University. At the age of 5 months, both male WT littermate mice and APP/PS1 transgenic mice were randomly divided into three groups: wild-type control group (WT), APP/PS1 control group (AD-con), APP/PS1 exercise group (AD-EXE) with 12 mice in each group. The mice of the long-term exercise group were trained with a running wheel at the speed of 18 r/min, 40 min/d, 6 d/week for 5 months.
Behavioral analysis
Novel object recognition test
The novel object recognition test is now among the most used behavioral tests for murine neuronal function, which is based on the innate tendency of rodents to explore novel objects over familiar ones. On the first day, mice were placed into an open-field (OF) box consisting of a quadrangular area (60 cm × 60 cm × 60 cm) and were habituated for 15 min. The next day, the training trial was performed by presenting the mice with two similar objects (A and A’, the two identical yellow dice) for a period of 5 min. The testing trial was performed for 4 h, one of the two objects (A or A’) was replaced near a new object (B, red triangle) and the animals were left in the OF box for 5 min. The exploration of the objects was considered when sniffing or deliberate contact occurred with the objects or when the animal’s snout was directed toward the object at a distance <1 cm. The amount of time taken to explore the new object provides an index of recognition memory. The exploration time for the familiar or the novel object during the test phase was recorded by an observer that was blinded to the group of the mice. The priority recognition index = time to explore the individual object/total exploration time to objects × 100%.
Rotarod test
The rotarod test is a common experimental method for testing the coordination ability of animals. The balance of the mouse on the rotating rod requires positional awareness and coordinated exercise ability. Each mouse was placed at the top around a rod in a separate lane. In the accelerating rotarod task, the speed of the rotating rod will accelerate from 10 rpm to 40 rpm in 5 min. The day before the test, the speed of the instrument was set to 4 r/min, allowing the mouse to balance on the rotating stick for 60 s. The next day, all mice were in place and then the rod began to move slowly at first, then faster and faster until the mouse fell into a trough of bedding material. Time until the fall was used as the index of motor capability.
Balance beam test
The balance beam test was used to score the ability of mice to traverse a stationary horizontal rod. Sensorimotor coordination was measured by the latency to cross the beam. Motor dysfunction affects balance beam performance. In the training period, the mice could pass the balance beam (91 cm × 35 mm × 57 cm) twice autonomously. During the test period, the time elapsed for the mice to crawl from the start of the balance beam to the end of the balance was recorded. During this period, only the crawl time was calculated, and the latency was not counted. Each mouse was tested once in the morning and once in the afternoon, and the average of the time was taken as the index.
Lashley III water maze test
The Lashley III maze is a complex task that can be solved by learning the correct sequence of right-left responses, or extra-maze cues, or both (Denenberg et al., 1991). The maze (80 cm × 50 cm × 20 cm) was constructed of double opaque plexiglass with a starting point, an endpoint, and four blind ends (S1–S4). The terminal platform was a safe area and animals could reach the end of the platform to avoid the danger of drowning. The warm water with a temperature of 20–25°C was injected into the box (water depth was 10 cm and the ambient temperature was above 20 °C). The elapsed time for the animal looking for the exit (latency period) was automatically recorded by the infrared sensor which was connected to the computer so that the spatial memory ability of the movement was valued, and the latent period was recorded.
The experiment was carried out during a 5-day period. The first 4 days were the training phase. In the beginning, the mice were free to explore for one minute in the water maze, and then the different blind spots (S1–S4) were used as starting points for the mouse to look for the end platform every day for 4 days. If the mouse could not find the end platform within 1 min, it would be guided ashore. Mice were trained twice a day in the morning and afternoon. On day 5, the mice were tested, and the latency of the mice was recorded.
Morris water maze test
The Morris water maze test was used to assess learning and memory ability. The pool was equally divided into four invisible quadrants, and a hidden platform was set in one quadrant, submerged 1 cm beneath the water surface. Several colorful signs (a triangle, a circular, a square, and a five-pointed star designed by construction paper) were placed in each quadrant around the pool which acted as clues for mice during the test. The inner wall of the pool was pasted with white wallpaper and a small amount of titanium dioxide (food grade) was used to make the water opaque. The pool temperature was maintained at 22 ± 1 °C by the temperature controller. A camera with a digital tracking system was fixed over the pool to automatically identify the swimming track of the mice (the swimming track from the starting point to the platform).
The experiment period was 7 days in all, contained a hidden platform test (Day 1–6) and probe test (Day 7). During the hidden platform test, the mice were randomly released into the water from each quadrant to explore and find the hidden platform by themselves for 60 s. Mice were tested in two different quadrants per day with an interval of 4 h and their latency was recorded respectively. To examine spatial reference memory, a probe test was performed 24 h after the last day of the hidden platform test. During the probe test, the platform was removed from the pool, and the mice were allowed to swim freely for 60 s. Likewise, the tracks were recorded through an overhead camera linked to a computer for a visualization purpose. The differences in latency and swimming speed were analyzed.
WB analysis
As described previously, the prefrontal cortex and hippocampus were added to the appropriate cell lysate, then sonicated thoroughly, then centrifuged for 15 min (4 °C, 12,500 r/min) and the supernatant was used as the sample for WB. The total protein level was measured with the BCA protein assay kit.
The protein was separated by electrophoresis on sodium dodecyl sulfate (SDS) polyacrylamide gel (30 μg), the separated target protein was then transferred to a nitrocellulose membrane and blocked for 1 h with 5% non-fat milk powder as a blocking solution. The membrane was incubated overnight at 4°C with the primary antibody. After washing, the membrane was incubated in horseradish peroxidase-labeled secondary antibody for 2 h at room temperature. The membranes were processed for protein detection using a two-color infrared laser imager.
Immunostaining and confocal microscopy
In brief, mice were anesthetized with 10% chloral hydrate and perfused with PBS. Brain was fixed with 4% paraformaldehyde (diluted in PBS) followed by gradient dehydration with 20% to 30% sucrose. The brain sections were cut with a cryotome (12 μm-thick).
The brain sections were permeabilized with 0.4% PBST (Triton-100 dissolved in PBS) and blocked in 5% goat serum in 0.4% PBST for 2 h. After that, the brain sections were incubated with primary antibodies (A-beta 1–42, 1:500; LAMP1, 1:500) overnight at 4°C and washed in PBST, and then incubated with Alexa-conjugated antibodies for 3 h at room temperature. The target protein was observed on an LSM510 Meta Laser Scanning Confocal Microscopy (CarlZeiss) using a 20-fold or 60-fold oil lens.
Transmission electron microscopy
The CA1 of the hippocampus was cut into 1 × 1 × 3 mm3 and fixed in 2.5% glutaraldehyde phosphate buffer, then dehydrated with ethanol and acetone. After embedded and dried, the tissue was cut into 70 nm-thick sections. Then the sections were stained with 3% uranyl acetate citrate. Photographs were taken from the H-7650 transmission electron microscopy (Hitachi High-Technologies Corporation) in the Analysis Center of Shanghai Jiao Tong University School of Medicine.
mRNA extraction and quantitative real-time PCR analysis
Total RNA from cortical tissue was extracted with Trizol (TaKaRa, #9109) reagent. The PrimeScript RT Master Mix and ddH2O were added to 500 ng RNA (200 ng/μL) for reverse transcription (37°C, 15 min; 85°C, 5 s). Real-time PCR was performed using SYBR Premix Ex Taq (TaKaRa, #RR420). For the quantitative analysis of PCR results, GAPDH was selected as the internal reference gene. According to the threshold of each gene Ct in the plate, the relative expression of the target gene was calculated according to the following formula:
△Ct = Ct (target gene)−Ct (GAPDH)
The primer sequence is as follows:
| Primer name | Sequence (5′−3′) |
|---|---|
| msGAPDH-R | CCTGTTGCTGTAGCCGTATTC |
| msGAPDH-F | TGGAGAAACCTGCCAAGTATG |
| msLAMP1-F | GGTCTGTGGAAGAGTGTGTTC |
| msLAMP1-R | GTTTGCCAGAAAGTGTGCCTC |
| msLAMP2-F | TCTGGAGCAACAGGGAACTG |
| msLAMP2-R | CAGCATAAGCCAGTGCCAC |
| msTFEB-F | GATGTTCTGTGACGCTGGCTG |
| msTFEB-R | GGCAGGCACTAAGTCCAAAC |
| msMOCLN-F | GCGCCTATGACACCATCAAG |
| msMOCLN-R | TATCCTGGCACTGCTCGATG |
| msV-ATPase-F | AAGCTTCTCAGAGTGGTGGG |
| msV-ATPase-R | CAGGCAAACAAACACGTGAC |
| mscathepsinL-F | TGTGAAGAACCAGGGCCAG |
| mscathepsinL-R | CGTCCTTCGCTTCATAGGG |
| mscathepsinD-F | GCTGTGAGGCTATTGTGGAC |
| mscathepsinD-R | TTCCACCCTGCGATACCTTG |
Co-immunoprecipitation
Co-immunoprecipitation is a popular technique to identify physiologically relevant protein–protein interactions. In our research, 2 μg TFEB antibody was diluted with PBS (100 μL) and incubated with 20 μL Protein G magnetic beads at 4 °C for 8–12 h. Then the unbound antibody was absorbed and extensively washed with lysis buffer. After lysed in Nonidet P40 (NP-40) lysis buffer (50 mM Tris (pH 7.4), 150 mM NaCl, 1% NP-40, 0.1% SDS), 600 μg lysed brain tissue was added to the magnetic beads, and incubation was continued for 6–8 h. then the magnetic beads were washed with lysis buffer to discard the unbound antibody. After that immunoprecipitants were added to 30 μL loading buffer and boiled for 5 min. The samples were resolved by SDS-polyacrylamide gel electrophoresis and analyzed the ACSS2 protein by WB. IgG was used as the negative control.
Statistical analysis
Data were presented as means ± SD and were calculated using Prism software. All statistical calculations were performed using GraphPad Prism 6 software (GraphPad Software, Inc., La Jolla, CA, USA). Statistical significance was considered when P < 0.05.
Results
Exercise improves the cognitive function and physical coordination of AD transgenic mice
To explore the importance of exercise in improving cognitive function, APP/PSEN1 transgenic mice were used for our study. First, the APP/PSEN1 transgenic mice confirmed by the genetic analysis were randomly divided into the non-exercise group (AD group) and the exercise group (AD + Exe group), and the wild-type littermate mice were used as the control (control group). Every mouse was marked by ear-ring with a number. Therefore, the running training situation, behavior test, and biochemical analysis of each mice could be recorded. The long-term exercise started at the age of 5 months old and ended at the age of 10 months old. Overall, the exercise is voluntary. Most of the mice were willing to run the wheel and complete the running time. If a mouse was not ready to take the run it would be gently carried on the wheel again to facilitate their voluntary running. In the case that a mouse eventually was unable to complete the running time for several days, it was removed from the exercise group. The cognitive ability test, including the Lashley water maze, the novel object recognition test, and the motor coordination test, including the balance beam and the rotarod experiment, were performed during this period. To avoid any potential effect of behavioral tests on lysosomal function in the sedentary group, we carried out the behavioral test at 9 months. After the behavioral test was completed, the sedentary group mice were kept in the cage for another three weeks, and the running mice were continued to be trained every day. At the end of 10 months, all the mice were killed, and the brain tissue was isolated to do a further biochemical test. The schedule was shown in Fig. 1a.
Fig. 1. Exercise improves the cognitive ability and physical coordination of AD transgenic mice.

a The schedule of this study. b The latency time of mice in Lashley water maze at age 5 months and 9 months (n = 8). The data were calculated by two-way ANOVA with Tukey’s multiple comparisons test. c Recognition index for new objects of mice at 9 months (n = 8). The data were calculated by one-way ANOVA test. d, e Time of mice on the balance beam and the rotarod experiments at 5 months and 9 months (n = 8). The data were calculated by two-way ANOVA with Tukey’s multiple comparisons test. All values are presented as the means ± SD from eight samples. *P < 0.05, **P < 0.01, ***P < 0.001, and ns P > 0.05 versus the control group.
The spatial learning and memory deficits are the commonly described alterations in the APP/PS1 mice. We found that there was no significant difference in the cognitive ability experiments and motor coordination tests among the groups when the mice were 5 months old. However, at 9 months, the latency time of AD mice in the Lashley water maze significantly increased, and the recognition ability of new objects significantly decreased compared with the control group. The data showed that the AD mice had the reduced cognitive function at 9 months old. In addition, compared with the control group, the time of the mice passing the balance beam was significantly extended in the AD group mice, whereas the keeping time on the rotatrod was shortened. The data suggest that the motor coordination ability of the AD group also decreased. However, compared with the AD group, long-term exercise could significantly shorten the latency of the Lashley water maze in AD mice (Fig. 1b) and improved the priority index of AD mice to identify new objects (Fig. 1c), which suggested that exercise could improve the cognitive ability of AD mice. In addition, AD mice subjected to long-term exercise could pass the balance beam in a shorter time (Fig. 1d) and stayed on the rotarod for a longer time (Fig. 1e), indicating that their motor coordination ability has also been improved. Together, these results suggest that long-term exercise could significantly delay the decline in cognitive function and motor coordination in AD.
As our previous data have shown that long-term exercise could enhance movement behavior and cognitive function in AD mice, then a question is whether the motor coordination deficit observed in AD mice affect their performance in maze and novel object recognition test? Thus, Morris water maze test was used to measure the mice’s spatial memory, meanwhile, the mice swimming track were also recorded by the computer. Similar to the results from Lashley water maze, the AD-EXE mice spent less time than AD mice to find the hidden platform on the last day of platform exploration (Fig. S1a-a’). When the platform was removed, the AD mice had an equal distribution of swimming track in each quadrant with no position preference, while the wild-type mice and AD-EXE mice still had position preference, which means that exercise improves the position memory of AD mice (Fig. S1a-b’). During the 6 days hidden platform test, we also found that the AD mice spent more time finding the hidden platform than the WT mice and the AD-EXE (Fig. S1b), which showed that long-term exercise enhanced the learning and spatial orientation of AD mice. There was also no significant difference in body weight in the feeding process of mice from each group (Fig. S1c). In addition, there is no obvious difference in swimming velocity among the three groups of mice (Fig. S1d). From the data of the swimming, distance shown in Fig. S1a and swimming speed shown in Fig. S1d gave us a clue that it is not the motor coordination deficit observed in APP/PS1 mice affected the performance in Morris maze test.
Exercise reduces Aβ accumulation in AD transgenic mice
We next explored the mechanism by which exercise improves cognitive dysfunction of AD. Owing to the potentially important role of the amyloid plaque in the pathogenesis of AD, we observed the amyloid plaque in the hippocampal regions and cortex with IF. In contrast to a large accumulation of Aβ peptides in AD mice, exercise significantly reduced the aggregation of Aβ peptides in the hippocampus and cortex of AD mice (Fig. 2a). In addition, exercise also significantly reduced phosphorylated Tau, which is a core component of neurofibrillary tangles in the brain of AD mice (Fig. 2b). Our results indicate that exercise could alleviate the molecular pathology of AD. Because some studies have shown that the accumulation of Aβ peptides could accelerate the phosphorylation of Tau, our research mainly focused on the effects of long-term exercise on Aβ peptides.
Fig. 2. Exercise reduces the accumulation of Aβ and the level of p-Tau (Ser396) in the brain of AD mice.
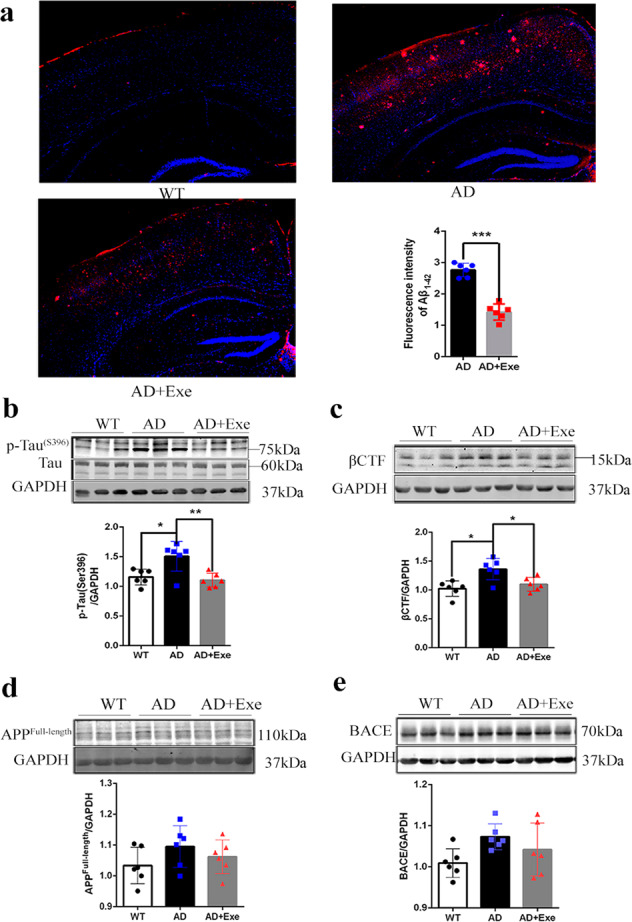
a Immunofluorescence staining for Aβ and DAPI (blue) of hippocampus and cortex in AD and AD + Exe mice at 10 months. The pictures were calculated with MJ software, the values were calculated with t test. b Western blot analysis of the level of p-Tau (Ser396) in the hippocampus. c–e Western blot analysis of βCTF, full-length APP, and BACE in the hippocampus. GAPDH was used as a loading control (n = 6). The data were calculated by one-way ANOVA with Tukey’s multiple comparisons test. All values are presented as the means ± SD. *P < 0.05, **P < 0.01, ***P < 0.001, and ns P > 0.05 versus the control group.
To determine the effects of exercise on APP processing, we compared the level of APP and its products in the brains of mice from the control group, AD group, and AD + Exe group. The 5 months running exercise caused a significant decrease in β-C-terminal fragment (βCTF) level in vivo (Fig. 2c), but did not affect the level of full-length APP and β-secretase BACE (Fig. 2d, e). This suggests that the decrease of the accumulation of Aβ peptide and βCTF level was not owing to the decrease in APP cleavage by BACE. Therefore, we considered that the decrease in the accumulation of Aβ by exercise may be caused by promoting the degradation of Aβ, rather than the decrease in the production of Aβ.
Exercise activates the lysosomal function and increases colocalization of lysosomes and Aβ in AD transgenic mice
Based on the result that exercise did not affect the generation of Aβ, the degradation of Aβ was further examined. As studies have shown that Aβ was mainly degraded through the autophagy-lysosome pathway, we detected the level of conjugation of Atg5-Atg12. The levels of Atg5-Atg12 in the brains of AD mice were significantly lower than those in the control mice. Exercise increased the Atg5-Atg12 level in AD mice, indicating that exercise activated autophagy in the brains of AD mice (Fig. 3a, b). However, compared with that in the control group, the increase in LC3II and p62 protein levels was found in the prefrontal cortex and hippocampus of AD mice, suggesting autophagic flux was also blocked in AD mice. Interestingly, exercise could reduce p62 and LC3II levels in AD mice (Fig. 3c, d). This suggested exercise could potentially rescue autophagic flux in the brains of AD mice.
Fig. 3. Exercise activates autophagy and rescues the impairment of autophagy flux both in the hippocampus and cortex of AD mice.

a, b Western blot analysis of Atg5 in hippocampus and cortex of mice at 10 months. Actin was used as a loading control (n = 3). c, d The levels of p62 and LC3II both in the hippocampus and cortex of mice at 10 months old were measured by Western blot analysis (n = 3). GAPDH was used as a loading control. Data represent means ± SD, *P < 0.05. **P < 0.01, ***P < 0.001. One-way ANOVA test.
To confirm the effects of exercise on lysosomes, we observed the morphology of lysosomes in the neurons of hippocampus CA1 region by transmission electron microscopy. We found that many lysosomes were enlarged and had some membrane-bound structure incompletely fused with lysosomes in AD mice, whereas the lysosome volume was normal and in deep staining in AD mice with long-term running exercise (Fig. 4a). We also detected lysosomal membrane proteins by immunoblotting and found that the level of LAMP1 protein was significantly elevated in the AD mice, which could be reversed with the treatment of exercise (Fig. 4b, c), consistent with the results of electron microscopy.
Fig. 4. Exercise activates lysosomal function and increases the colocalization of lysosomes with Aβ.

a Representative electron micrographs of lysosomes in pyramidal neurons from the CA1 area of the hippocampus from AD and AD + Exe mice. Arrows indicate lysosomes. (Scale bar, 1 μm). b, c Western blot of LAMP1 in hippocampus and cortex. d, e Maturation of cathepsin D in the hippocampus and cortex was assessed by Western blotting. f, g Maturation of cathepsin L in the cortex was assessed by Western blotting. GAPDH was used as a loading control (n = 4). h Representative colocalization of immunofluorescence image of Aβ and LAMP1. (Scale bar = 5 μm). Data represent means ± SD. *P < 0.05, **P < 0.01. One-way ANOVA test.
The maturation of lysosomal hydrolases was also analyzed. We found a decline in enzyme maturation in AD mice, which suggested that the lysosomal function in AD mice was compromised. However, exercise increased the level of the maturation of cathepsin D (Fig. 4d, e), and a similar phenomenon was found in cathepsin L (Fig. 4f, g), which indicated that exercise might enhance the lysosomal function of AD mice. The WB data including the increased LC3II, increased p62, increased Lamp1 and the decreased maturation of cathepsins in the brains of AD mice, consistent with the EM results, suggested that lysosomal function were abnormal in the brains of AD mice, and exercise could recover the function of lysosomes.
We next determined whether the improved lysosomal function by exercise was vital to the degradation of Aβ. The immunostaining analysis results showed that accumulated Aβ had little colocalization with LAMP1, a lysosome marker, in AD mice, but more Aβ colocalized with LAMP1 in AD mice after exercise (Fig. 4h). The data suggested exercise enhanced lysosomal function and accelerated the process of lysosomes to clear the harmful Aβ peptides.
Exercise likely promotes lysosomal biogenesis in AD mice
We next investigated the possible mechanisms associated with the improvement of lysosomal functions by exercise. TFEB is the transcriptional factor regulating the transcription of the genes associated with autophagy and lysosomes. We initially examined the nuclear localization of TFEB in the brains of AD mice and AD + Exe mice by the IF approach and found more TFEB was translocated in the nucleus after long-term exercise in AD mice (Fig. 5a). We further tested the TFEB level in the cortex by WB. We observed exercise increased the total level of TFEB (Fig. 5b). The TFEB levels in the nucleus in AD mice decreased compared with those in the control, and exercise increased the levels of TFEB in the nucleus (Fig. 5c). On the other hand, the levels of TFEB in the cytoplasm were not significantly different in the three groups (Fig. 5d). The data show that exercise may improve the nuclear translocation of TFEB.
Fig. 5. Exercise increases the level of TFEB in the nucleus and upregulates the expression of lysosomal genes.

a TFEB localization in mice cortex by immunofluorescent staining. b Total TFEB protein level was measured by Western blotting. c, d Western blot analysis of TFEB nuclear translocation by nuclear-cytoplasmic fractionation. GAPDH was used as a cytoplasmic loading control and histone H3 was used as a nuclear loading control (n = 3). e Analysis of the transcription level of lysosomal-associated genes regulated by TFEB by quantitative PCR (n = 7). Data represent means ± SD. *P < 0.05. **P < 0.01, ns P > 0.05 versus the control group. One-way ANOVA test.
Then, we tested the mRNA levels of several lysosomal-associated genes regulated by TFEB and found that the mRNA levels associated with lysosomes displayed a decreasing trend in AD mice. Compared with AD mice, the mRNA levels of lysosomal-associated proteins LAMP1, V-ATPase, MCLON1, cathepsin D, and cathepsin L significantly increased in AD mice with 5 months of running exercise (Fig. 5e). Our data suggest that exercise could enhance lysosomal function potentially by promoting TFEB nuclear translocation and improve the transcription of genes associated with lysosomal biogenesis in AD mice.
We further studied the mechanisms underlying exercise-induced nuclear translocation of TFEB and our research suggests that TFEB translocation would be associated with calcineurin activation or the inhibition of the RIP1-ERK pathway, but appeared independent of the mTOR pathway, known to positively regulate TFEB nuclear translocation (Fig. S2).
Exercise potentially increases the interaction of TFEB with ACSS2 in the nucleus of AD mice
The nuclear activity of AMPK-mediated acetyl-CoA synthetase 2 (ACSS2) is required for glucose deprivation-induced lysosomal biogenesis. ACSS2 is the one of principal enzymes that generate acetyl-CoA for histone acetylation. ACSS2 translocates into the nucleus and binds to TFEB. The TFEB-ACSS2 complex binds and activates the promoter regions of a battery of lysosomal and autophagy genes to initiate transcription of these genes, where ACSS2 enhances the histone acetylation that is required for the transcriptional process [26]. We found that long-term exercise enhanced the level of phosphorylated AMPK (Fig. 6a) which stimulated the phosphorylation of ULK1, a substrate of AMPK (Fig. 6b). AMPK phosphorylates ACSS2, leading to ACSS2 nuclear translocation [26]. Thus, we speculated whether long-term exercise could promote the interaction of TFEB with ACSS2 in the nucleus of the brain of AD mice. We found that exercise enhanced the ACSS2 level of AD mice (Fig. 6c), and the immunoprecipitation showed that the long-term exercise increased the association of TFEB with ACSS2 in the nucleus (Fig. 6d). All these results indicate that long-term exercise could promote the interaction of TFEB and ACSS2, strengthening the gene transcription associated with lysosomal biogenesis.
Fig. 6. Exercise increases the interaction of TFEB with ACSS2 in the cortex.

a The p-AMPK and total AMPK levels in cortex. b The level of p=ULK1 (Ser 555), the substrate of AMPK. c ACSS2 protein level was measured by Western blot analysis. d Co-immunoprecipitation of ACSS2 with TFEB in the cortex of AD mice (n = 3). Data represent means ± SD. *P < 0.05. **P < 0.01, ns P > 0.05 versus the control group. One-way ANOVA test.
Exercise activates vesicle trafficking to promote the maturation of lysosomal hydrolases
Although our results indicated that long-term exercise enhanced lysosomal function, we found that the immature forms of cathepsin L and cathepsin D by WB (Fig. 4d–g) were significantly increased in the brain of AD mice while the levels of maturation of cathepsins were decreased (Fig. 7a). Owing to the importance of vesicle trafficking in the process of the maturation of lysosomal hydrolases, we observed the morphology of the Golgi complex, vital for immature lysosomal hydrolases processing. Long-term exercise maintained the normal form of the Golgi complex (Fig. 7b) and the normal level of GM130 in the brains of AD mice (Fig. 7c, f), suggesting that modification of lysosomal hydrolases would be compromised in the brain of AD mice owing to the dysfunction of the Golgi complex. In addition, the proteins related to the vesicle trafficking of lysosomal hydrolases, Rab7 and Rab9, were analyzed. The level of Rab7 and Rab9 in the hippocampus of AD mice decreased, which was restored after long-term exercise (Fig. 7d, e). Similar results were found in the prefrontal cortex of the three groups (Fig. 7g, h). In summary, our results indicated that long-term exercise promoted the maturation of lysosomal hydrolases via activating vesicle trafficking.
Fig. 7. Exercise improves the level of proteins related to vesicle trafficking.

a Comparison of the precursor and maturation of cathepsin L and cathepsin D in cortex and hippocampus. b Representative electron micrographs of Golgi complex in pyramidal neurons from the CA1 area of the hippocampus from AD and AD + Exe mice. c, f Western blot analysis of GM130 in the hippocampus and cortex. d, g Western blot analysis of Rab7 in the hippocampus and cortex. e, h Western blot analysis of Rab9 in the hippocampus and cortex. GAPDH was used as a loading control (n = 4). Data represent means ± SD. *P < 0.05. **P < 0.01, ns P > 0.05 versus the control group. One-way ANOVA test.
Discussion
Exercise would be a medicine for enhancing the learning ability of AD
With the prolongation of lifespan, neurodegenerative diseases are becoming a serious health care burden in aged individuals. AD is the most common cause of dementia, which accounts for 60% to 80% of dementia cases. Despite extensive research and several new medicines for the treatment of AD in the past few decades, they have resulted in a very limited therapeutic effect on AD patients. Basic research will be helpful to find a potential new strategy for AD treatment.
Studies have shown that exercise protects hippocampal neurons and improves cognitive abilities in AD mice [27, 28]. Here, we showed that long-term running exercise improved both learning and cognitive function and physical coordination of the APP/PS1 transgenic mice, consistent with previous research [11].
Although the motor coordination of APP/PS1 mice was worse than the wild-type mice in the rotarod test and balance beam test, the spatial learning and memory deficits shown in the water maze and new object recognition test would not be confounded by the motor coordination deficits because the AD mice just showed longer latencies and shorter percentages of time in the correct quadrant but not the shorter swimming length [29].
In addition, exercise significantly reduced the accumulation of Aβ and the level of p-Tau in the hippocampus. Reduced aggregation of Aβ also decreased the phosphorylation of Tau and would protect the neurons [30, 31].
Lysosomal function is impaired in AD
The autophagy-lysosomal pathway is particularly important in the degradation of mutant proteins in neurons because neurons are mitotically quiescent cells. It is reported that deficiency in the autophagy-lysosomal pathway exhibits some neurodegeneration, including AD [32, 33]. Pharmacologically induced autophagy to enhance the clearance of aggregated proteins has been considered as a treatment for improving cell and animal pathology [34, 35].
Lysosomes play a core role in the degradation of some harmful proteins like Aβ and in maintaining the homeostasis of intracellular protein [36]. Lysosomal dysfunction has been considered to be one of the major defects in neurons that leads to the progression of AD [37] and PD [38]. Recent work also found that β-amyloid deposits cause the blockade of lysosomal retrograde transport, leading to their accumulation in axons and affecting the maturation of lysosomal hydrolases [39].
Our results show that the lysosomes enlarged in the CA1 region of the hippocampus in AD mice and the fusion with various membrane structures were blocked. AD-related mutations in PSEN1 have been shown to lead to the failure of lysosomal acidification and hydrolase activity [40]. Lysosomal hydrolases are key molecules required to degrade cellular garbage. The fact that the lysosomes lacking multiple proteases accumulate near amyloid plaques also suggests that the dysfunctional lysosomes were unable to efficiently degrade the cellular garbage [39]. In our study, we also found that the levels of mature (activated) cathepsins L/D significantly reduced, suggesting there is lysosomal dysfunction in AD.
Exercise can enhance lysosomal function in AD mice
Studies have shown that aging causes a decrease in autophagosomes in skeletal muscle while is upregulated after exercise [41]. Zhao et al. [42] also found exercise could increase LC3 and decrease p62 in App/psn1 mice. This means exercise could increase autophagy levels. However, the effect of exercise on lysosomal function is still unknown. Surprisingly, we found that the morphology of lysosomes in the hippocampus of AD mice was restored to the normal structure with long-term exercise. We also found that long-term exercise increased the levels of mature cathepsin L/D in the cortex of AD mice, confirming that exercise restored the damaged lysosomal function in AD, and restored the autophagic flux. Studies have shown that long-term exercise can activate microglia in AD mice [43], which could phagocytose Aβ and transport it to lysosomes for degradation [44]. However, Aβ deposits might be dealt with by neurons instead. In fact, we found that more robust colocalization of Aβ1–42 and LAMP1 in AD mice after exercise, suggesting exercise accelerates the degradation of Aβ through lysosomes. However, whether it is the microglia, neurons, or glia that mediates this effect, we aim to verify this in our subsequent work.
Our research is the first study examining the relationship between AD, exercise, and lysosomal function. We found that exercise improved cognitive dysfunction of AD by activating lysosomal functions, providing mechanistic insight into the benefits of exercise on AD intervention.
Exercise enhances lysosomal function by improving lysosomal biogenesis and the maturation of cathepsins
TFEB is a master gene transcription factor that regulates the transcription of genes associated with autophagy/lysosome biogenesis [45, 46]. Studies have shown that decreased TFEB is involved in AD, PD, and HD [47]. Overexpressing TFEB or promoting its nuclear translocation could alleviate these neurodegenerative diseases with the aggregation of mutant proteins [48, 49]. We found long-term exercise promoted the TEFB nuclear translocation and increased the expression of the downstream target genes [25] so that the lysosomal function can be activated which in turn accelerates the degradation of Aβ.
The activity of TFEB is related to its phosphorylation of specific amino acids [50, 51]. Our results have indicated that the increased nuclear level of TFEB after exercise might be mTOR pathway independent. Recent studies have shown that the inhibition of the RIP1-ERK1/2 pathway and the activation of calcineurin were also related to the TFEB nuclear translocation [46, 52]. We found an abnormal activation of the RIP1-ERK1/2 pathway and a significantly decreased level of calcineurin A/B in the brain of AD mice, which were reversed after long-term exercise. Therefore, our data suggested that the increase in the nuclear TFEB after exercise in AD mice would be owing to the cooperation of the two pathways.
Histone acetylation can dynamically regulate chromatin structure and gene expression [53, 54]. ACSS2 is highly expressed in the hippocampus of mice, and ACSS2 deficiency impairs the long-term spatial memory of adult mice [55]. After glucose deprivation, AMPK phosphorylates ACSS2 and enhances the nuclear translocation of ACSS2, then ACSS2 binds to TFEB in the nucleus and enhances TFEB-regulated gene transcription [26]. AMPK can be phosphorylated by the increased AMP/ATP ratio. Therefore, AMPK phosphorylation occurs in skeletal muscle both in humans and rodents during and after exercise [56, 57], also in the brain [25]. Our data shows that long-term exercise could activate AMPK and enhance the interaction of TFEB and ACSS2, which would be a reason why TFEB upregulated the transcription of genes associated with lysosomal biogenesis.
Mature cathepsin L and cathepsin D were decreased in AD, but the immature forms were abnormally increased, suggesting that the process of the maturation of lysosomal hydrolases in AD is impaired. Immature lysosomal hydrolases need to be transported to the lysosome by vesicle trafficking and be cleaved to produce the mature form. Studies have shown that vesicular trafficking dysfunction is associated with AD [58, 59]. Golgi is an important “transfer station” for protein sorting in vesicle trafficking. Rab7 and Rab9 are involved in the regulation of the transport of substances from endosomes to lysosomes, including cathepsins. Studies have shown that Golgi dysfunction is associated with AD [58]. In addition, overexpression of Rab7 in primary neurons and N2a cells accelerates the transport of Aβ to lysosomes for degradation [60, 61], and silencing of Rab7 increased the production of Aβ [62]. All these data suggest that the trafficking of proteins play an important role in neuronal health. Our results showed that exercise activated vesicle trafficking to promote the transport of hydrolases to lysosomes, thereby promoting the maturation of cathepsins.
In conclusion, as shown in the graphical abstract (Fig. 8), our present study suggests that long-term exercise could restore the damaged lysosomal function in AD mice to reduce Aβ toxicity potentially by enhancing the lysosomal gene transcription of TFEB and increasing the transportation of mature cathepsins. Yet, the exact mechanism of exercise-exerted benefit needs to be further clarified. As a full-body workout, the long-term running exercise would ameliorate AD from different aspects. There has not been any drug developed to control the disease progress well for AD patients as so far. Therefore, long-term exercise might be a good “medicine” for AD patients to improve their life quality. In addition, the effect of long-term exercise on lysosomal function may be a strategy to treat certain types of neurodegenerative diseases with mutant protein aggregations, including AD, PD, ALS, and HD.
Fig. 8. The proposed mechanisms by which exercise delays the progression of AD.
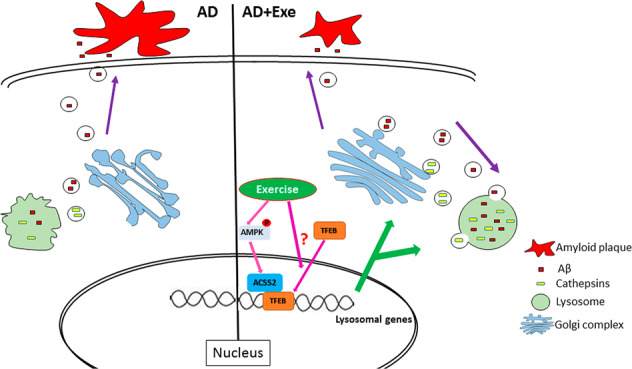
Exercise promotes lysosomal biosynthesis by promoting nuclear translocation of TFEB, and activates the maturation of lysosomal enzymes via proteins related to vesicle trafficking, thereby improving the degradation of Aβ, maintaining the protein homeostasis, and delaying the progress of AD. Lysosomes may be a new target for non-pharmacological treatment for AD.
Supplementary information
{kind=link}
{kind=link}
Acknowledgements
We are grateful to professor Shou-qing Luo at Plymouth University for his helpful suggestions for this paper. This work was supported by the National Natural Science Foundation of China (Grant no. 81571252, 2016) and the Priority Academic Program Development of the Jiangsu Higher Education Institutes (PAPD).
Author contributions
XW and YTZ completed most of the research work and wrote the manuscript; YLS, ZL, and JH performed some research work; YZ, YW, and JCW provided suggestions for the research and revised the manuscript. ZHQ and FL designed the research work and revised the manuscript.
Competing interests
The authors declare no competing interests.
Contributor Information
Zheng-hong Qin, Email: qinzhenhong@suda.edu.cn.
Fang Lin, Email: linfang-1@suda.edu.cn.
Supplementary information
The online version contains supplementary material available at 10.1038/s41401-021-00720-6.
References
- 1.Appelqvist H, Waster P, Kagedal K, Ollinger K. The lysosome: from waste bag to potential therapeutic target. J Mol Cell Biol. 2013;5:214–26. doi: 10.1093/jmcb/mjt022. [DOI] [PubMed] [Google Scholar]
- 2.Querfurth HW, LaFerla FM. Alzheimer’s disease. N Engl J Med. 2010;362:329–44. doi: 10.1056/NEJMra0909142. [DOI] [PubMed] [Google Scholar]
- 3.De Strooper B. Proteases and proteolysis in Alzheimer disease: a multifactorial view on the disease process. Physiol Rev. 2010;90:465–94. doi: 10.1152/physrev.00023.2009. [DOI] [PubMed] [Google Scholar]
- 4.De-Paula VJ, Radanovic M, Diniz BS, Forlenza OV. Alzheimer’s disease. Subcell Biochem. 2012;65:329–52. doi: 10.1007/978-94-007-5416-4_14. [DOI] [PubMed] [Google Scholar]
- 5.Bennett RE, DeVos SL, Dujardin S, Corjuc B, Gor R, Gonzalez J, et al. Enhanced tau aggregation in the presence of amyloid beta. Am J Pathol. 2017;187:1601–12. doi: 10.1016/j.ajpath.2017.03.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Solomon A, Mangialasche F, Richard E, Andrieu S, Bennett DA, Breteler M, et al. Advances in the prevention of Alzheimer’s disease and dementia. J Intern Med. 2014;275:229–50. doi: 10.1111/joim.12178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.De Duve C, Wattiaux R. Functions of lysosomes. Annu Rev Physiol. 1966;28:435–92. doi: 10.1146/annurev.ph.28.030166.002251. [DOI] [PubMed] [Google Scholar]
- 8.Mindell JA. Lysosomal acidification mechanisms. Annu Rev Physiol. 2012;74:69–86. doi: 10.1146/annurev-physiol-012110-142317. [DOI] [PubMed] [Google Scholar]
- 9.Serrano-Puebla A, Boya P. Lysosomal membrane permeabilization in cell death: new evidence and implications for health and disease. Ann N Y Acad Sci. 2016;1371:30–44. doi: 10.1111/nyas.12966. [DOI] [PubMed] [Google Scholar]
- 10.Fraldi A, Klein AD, Medina DL, Settembre C. Brain disorders due to lysosomal dysfunction. Annu Rev Neurosci. 2016;39:277–95. doi: 10.1146/annurev-neuro-070815-014031. [DOI] [PubMed] [Google Scholar]
- 11.Khodadadi D, Gharakhanlou R, Naghdi N, Salimi M, Azimi M, Shahed A, et al. Treadmill exercise ameliorates spatial learning and memory deficits through improving the clearance of peripheral and central amyloid-beta levels. Neurochem Res. 2018;43:1561–74. doi: 10.1007/s11064-018-2571-2. [DOI] [PubMed] [Google Scholar]
- 12.Boya P. Lysosomal function and dysfunction: mechanism and disease. Antioxid Redox Signal. 2012;17:766–74. doi: 10.1089/ars.2011.4405. [DOI] [PubMed] [Google Scholar]
- 13.Huang WJ, Zhang X, Chen WW. Gaucher disease: a lysosomal neurodegenerative disorder. Eur Rev Med Pharmacol Sci. 2015;19:1219–26. [PubMed] [Google Scholar]
- 14.Mazzulli JR, Zunke F, Isacson O, Studer L, Krainc D. alpha-Synuclein-induced lysosomal dysfunction occurs through disruptions in protein trafficking in human midbrain synucleinopathy models. Proc Natl Acad Sci USA. 2016;113:1931–6. doi: 10.1073/pnas.1520335113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Orr ME, Oddo S. Autophagic/lysosomal dysfunction in Alzheimer’s disease. Alzheimers Res Ther. 2013;5:53. doi: 10.1186/alzrt217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Lee JH, Yu WH, Kumar A, Lee S, Mohan PS, Peterhoff CM, et al. Lysosomal proteolysis and autophagy require presenilin 1 and are disrupted by Alzheimer-related PS1 mutations. Cell. 2010;141:1146–58. doi: 10.1016/j.cell.2010.05.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Bustos V, Pulina MV, Bispo A, Lam A, Flajolet M, Gorelick FS, et al. Phosphorylated presenilin 1 decreases beta-amyloid by facilitating autophagosome-lysosome fusion. Proc Natl Acad Sci USA. 2017;114:7148–53. doi: 10.1073/pnas.1705240114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hawley JA, Hargreaves M, Joyner MJ, Zierath JR. Integrative biology of exercise. Cell. 2014;159:738–49. doi: 10.1016/j.cell.2014.10.029. [DOI] [PubMed] [Google Scholar]
- 19.Aguiar AS, Jr., Stragier E, da Luz Scheffer D, Remor AP, Oliveira PA, Prediger RD, et al. Effects of exercise on mitochondrial function, neuroplasticity and anxio-depressive behavior of mice. Neuroscience. 2014;271:56–63. doi: 10.1016/j.neuroscience.2014.04.027. [DOI] [PubMed] [Google Scholar]
- 20.van Praag H. Exercise and the brain: something to chew on. Trends Neurosci. 2009;32:283–90. doi: 10.1016/j.tins.2008.12.007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Valenzuela PL, Castillo-Garcia A, Morales JS, de la Villa P, Hampel H, Emanuele E, et al. Exercise benefits on Alzheimer’s disease: State-of-the-science. Ageing Res Rev. 2020;62:101108. doi: 10.1016/j.arr.2020.101108. [DOI] [PubMed] [Google Scholar]
- 22.Bo H, Kang W, Jiang N, Wang X, Zhang Y, Ji LL. Exercise-induced neuroprotection of hippocampus in APP/PS1 transgenic mice via upregulation of mitochondrial 8-oxoguanine DNA glycosylase. Oxid Med Cell Longev. 2014;2014:834502. doi: 10.1155/2014/834502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Liu HL, Zhao G, Cai K, Zhao HH, Shi LD. Treadmill exercise prevents decline in spatial learning and memory in APP/PS1 transgenic mice through improvement of hippocampal long-term potentiation. Behav Brain Res. 2011;218:308–14. doi: 10.1016/j.bbr.2010.12.030. [DOI] [PubMed] [Google Scholar]
- 24.Jiang D, Chen K, Lu X, Gao HJ, Qin ZH, Lin F. Exercise ameliorates the detrimental effect of chloroquine on skeletal muscles in mice via restoring autophagy flux. Acta Pharmacol Sin. 2014;35:135–42. doi: 10.1038/aps.2013.144. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Huang J, Wang X, Zhu Y, Li Z, Zhu YT, Wu JC, et al. Exercise activates lysosomal function in the brain through AMPK-SIRT1-TFEB pathway. CNS Neurosci Ther. 2019;25:796–807. doi: 10.1111/cns.13114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li X, Yu W, Qian X, Xia Y, Zheng Y, Lee JH, et al. Nucleus-translocated ACSS2 promotes gene transcription for lysosomal biogenesis and autophagy. Mol Cell. 2017;66:684–97 e9. doi: 10.1016/j.molcel.2017.04.026. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Um HS, Kang EB, Koo JH, Kim HT, Jin-Lee, Kim EJ, et al. Treadmill exercise represses neuronal cell death in an aged transgenic mouse model of Alzheimer’s disease. Neurosci Res. 2011;69:161–73. doi: 10.1016/j.neures.2010.10.004. [DOI] [PubMed] [Google Scholar]
- 28.Lin TW, Shih YH, Chen SJ, Lien CH, Chang CY, Huang TY, et al. Running exercise delays neurodegeneration in amygdala and hippocampus of Alzheimer’s disease (APP/PS1) transgenic mice. Neurobiol Learn Mem. 2015;118:189–97. doi: 10.1016/j.nlm.2014.12.005. [DOI] [PubMed] [Google Scholar]
- 29.Ferguson SA, Sarkar S, Schmued LC. Longitudinal behavioral changes in the APP/PS1 transgenic Alzheimer’s disease model. Behav Brain Res. 2013;242:125–34. doi: 10.1016/j.bbr.2012.12.055. [DOI] [PubMed] [Google Scholar]
- 30.Jack CR, Jr., Knopman DS, Jagust WJ, Shaw LM, Aisen PS, Weiner MW, et al. Hypothetical model of dynamic biomarkers of the Alzheimer’s pathological cascade. Lancet Neurol. 2010;9:119–28. doi: 10.1016/S1474-4422(09)70299-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Jin M, Shepardson N, Yang T, Chen G, Walsh D, Selkoe DJ. Soluble amyloid beta-protein dimers isolated from Alzheimer cortex directly induce Tau hyperphosphorylation and neuritic degeneration. Proc Natl Acad Sci USA. 2011;108:5819–24. doi: 10.1073/pnas.1017033108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Komatsu M, Waguri S, Chiba T, Murata S, Iwata J, Tanida I, et al. Loss of autophagy in the central nervous system causes neurodegeneration in mice. Nature. 2006;441:880–4. doi: 10.1038/nature04723. [DOI] [PubMed] [Google Scholar]
- 33.Martini-Stoica H, Xu Y, Ballabio A, Zheng H. The autophagy-lysosomal pathway in neurodegeneration: a TFEB perspective. Trends Neurosci. 2016;39:221–34. doi: 10.1016/j.tins.2016.02.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang Q, Mao Z. Parkinson disease: a role for autophagy? Neuroscientist. 2010;16:335–41. doi: 10.1177/1073858409357118. [DOI] [PubMed] [Google Scholar]
- 35.Chen L, Huang Y, Yu X, Lu J, Jia W, Song J, et al. Corynoxine protects dopaminergic neurons through inducing autophagy and diminishing neuroinflammation in rotenone-induced animal models of Parkinson’s Disease. Front Pharmacol. 2021;12:642900. [DOI] [PMC free article] [PubMed]
- 36.de Duve C. The lysosome turns fifty. Nat Cell Biol. 2005;7:847–9. doi: 10.1038/ncb0905-847. [DOI] [PubMed] [Google Scholar]
- 37.Tammineni P, Jeong YY, Feng T, Aikal D, Cai Q. Impaired axonal retrograde trafficking of the retromer complex augments lysosomal deficits in Alzheimer’s disease neurons. Hum Mol Genet. 2017;26:4352–66. doi: 10.1093/hmg/ddx321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Burbulla LF, Song P, Mazzulli JR, Zampese E, Wong YC, Jeon S, et al. Dopamine oxidation mediates mitochondrial and lysosomal dysfunction in Parkinson’s disease. Science. 2017;357:1255–61. doi: 10.1126/science.aam9080. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Gowrishankar S, Yuan P, Wu Y, Schrag M, Paradise S, Grutzendler J, et al. Massive accumulation of luminal protease-deficient axonal lysosomes at Alzheimer’s disease amyloid plaques. Proc Natl Acad Sci USA. 2015;112:E3699–708. doi: 10.1073/pnas.1510329112. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Lee JH, McBrayer MK, Wolfe DM, Haslett LJ, Kumar A, Sato Y, et al. Presenilin 1 maintains lysosomal Ca2+ homeostasis via TRPML1 by regulating vATPase-mediated lysosome acidification. Cell Rep. 2015;12:1430–44. doi: 10.1016/j.celrep.2015.07.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Kim YA, Kim YS, Oh SL, Kim HJ, Song W. Autophagic response to exercise training in skeletal muscle with age. J Physiol Biochem. 2013;69:697–705. doi: 10.1007/s13105-013-0246-7. [DOI] [PubMed] [Google Scholar]
- 42.Zhao N, Zhang X, Song C, Yang Y, He B, Xu B. The effects of treadmill exercise on autophagy in hippocampus of APP/PS1 transgenic mice. Neuroreport. 2018;29:819–25. doi: 10.1097/WNR.0000000000001038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Ke HC, Huang HJ, Liang KC, Hsieh-Li HM. Selective improvement of cognitive function in adult and aged APP/PS1 transgenic mice by continuous non-shock treadmill exercise. Brain Res. 2011;1403:1–11. doi: 10.1016/j.brainres.2011.05.056. [DOI] [PubMed] [Google Scholar]
- 44.Bao J, Zheng L, Zhang Q, Li X, Zhang X, Li Z, et al. Deacetylation of TFEB promotes fibrillar Abeta degradation by upregulating lysosomal biogenesis in microglia. Protein Cell. 2016;7:417–33. doi: 10.1007/s13238-016-0269-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Settembre C, De Cegli R, Mansueto G, Saha PK, Vetrini F, Visvikis O, et al. TFEB controls cellular lipid metabolism through a starvation-induced autoregulatory loop (vol 15, pg 647, 2013) Nat Cell Biol. 2013;15:1016. doi: 10.1038/ncb2718. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Medina DL, Di Paola S, Peluso I, Armani A, De Stefani D, Venditti R, et al. Lysosomal calcium signalling regulates autophagy through calcineurin and TFEB. Nat Cell Biol. 2015;17:288. doi: 10.1038/ncb3114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Tsunemi T, Ashe TD, Morrison BE, Soriano KR, Au J, Roque RAV, et al. PGC-1 alpha rescues Huntington’s disease proteotoxicity by preventing oxidative stress and promoting TFEB function. Sci Transl Med. 2012;4:142ra97. [DOI] [PMC free article] [PubMed]
- 48.Kilpatrick K, Zeng YM, Hancock T, Segatori L. Genetic and chemical activation of TFEB mediates clearance of aggregated alpha-synuclein. PLoS ONE 2015;10:e0120819. [DOI] [PMC free article] [PubMed]
- 49.Xiao QL, Yan P, Ma XC, Liu HY, Perez R, Zhu A, et al. Enhancing astrocytic lysosome biogenesis facilitates A beta clearance and attenuates amyloid plaque pathogenesis. J Neurosci. 2014;34:9607–20. doi: 10.1523/JNEUROSCI.3788-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Martina JA, Chen Y, Gucek M, Puertollano R. MTORC1 functions as a transcriptional regulator of autophagy by preventing nuclear transport of TFEB. Autophagy. 2012;8:903–14. doi: 10.4161/auto.19653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Villarroel-Campos D, Gastaldi L, Conde C, Caceres A, Gonzalez-Billault C. Rab-mediated trafficking role in neurite formation. J Neurochem. 2014;129:240–8. doi: 10.1111/jnc.12676. [DOI] [PubMed] [Google Scholar]
- 52.Yonekawa T, Gamez G, Kim J, Tan AC, Thorburn J, Gump J, et al. RIP1 negatively regulates basal autophagic flux through TFEB to control sensitivity to apoptosis. Embo Rep. 2015;16:700–8. doi: 10.15252/embr.201439496. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Katada S, Imhof A, Sassone-Corsi P. Connecting threads: epigenetics and metabolism. Cell. 2012;148:24–8. doi: 10.1016/j.cell.2012.01.001. [DOI] [PubMed] [Google Scholar]
- 54.Kaelin WG, Jr., McKnight SL. Influence of metabolism on epigenetics and disease. Cell. 2013;153:56–69. doi: 10.1016/j.cell.2013.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mews P, Donahue G, Drake AM, Luczak V, Abel T, Berger SL. Acetyl-CoA synthetase regulates histone acetylation and hippocampal memory. Nature. 2017;546:381–6. doi: 10.1038/nature22405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Richter EA, Ruderman NB. AMPK and the biochemistry of exercise: implications for human health and disease. Biochem J. 2009;418:261–75. doi: 10.1042/BJ20082055. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.HM O’Neill. AMPK and exercise: glucose uptake and insulin sensitivity. Diabetes Metab J. 2013;37:1–21. doi: 10.4093/dmj.2013.37.1.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Bexiga MG, Simpson JC. Human diseases associated with form and function of the Golgi complex. Int J Mol Sci. 2013;14:18670–81. doi: 10.3390/ijms140918670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Musunuri S, Khoonsari PE, Mikus M, Wetterhall M, Haggmark-Manberg A, Lannfelt L, et al. Increased levels of extracellular microvesicle markers and decreased levels of endocytic/exocytic proteins in the Alzheimer’s disease brain. J Alzheimers Dis. 2016;54:1671–86. doi: 10.3233/JAD-160271. [DOI] [PubMed] [Google Scholar]
- 60.Eskelinen EL. Maturation of autophagic vacuoles in mammalian cells. Autophagy. 2005;1:1–10. doi: 10.4161/auto.1.1.1270. [DOI] [PubMed] [Google Scholar]
- 61.Li J, Kanekiyo T, Shinohara M, Zhang Y, LaDu MJ, Xu H, et al. Differential regulation of amyloid-beta endocytic trafficking and lysosomal degradation by apolipoprotein E isoforms. J Biol Chem. 2012;287:44593–601. doi: 10.1074/jbc.M112.420224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Udayar V, Buggia-Prevot V, Guerreiro RL, Siegel G, Rambabu N, Soohoo AL, et al. A paired RNAi and RabGAP overexpression screen identifies Rab11 as a regulator of beta-amyloid production. Cell Rep. 2013;5:1536–51. doi: 10.1016/j.celrep.2013.12.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.