

Abstract
Mitochondria of all tissues convert various metabolic substrates into two forms of energy – ATP and heat. Historically, the primary focus of research in mitochondrial bioenergetics was on the mechanisms of ATP production, while mitochondrial thermogenesis received significantly less attention. Nevertheless, mitochondrial heat production is crucial for the maintenance of body temperature, regulation of the pace of metabolism, and prevention of oxidative damage to mitochondria and the cell. In addition, mitochondrial thermogenesis has gained significance as a pharmacological target for treating metabolic disorders. Mitochondria produce heat as the result of H+ leak across their inner membrane. This review provides a critical assessment of the current field of mitochondrial H+ leak and thermogenesis, with a focus on the molecular mechanisms involved in the function and regulation of uncoupling protein 1 (UCP1) and the ADP/ATP carrier (AAC), the two proteins that mediate mitochondrial H+ leak.
Keywords: mitochondria, thermogenesis, uncoupling, H+ leak, ADP/ATP carrier, uncoupling proteins
INTRODUCTION
Mitochondria are intracellular organelles, bound by two membranes, that supply the eukaryotic cell with energy. Together with the upstream energy metabolism pathways, mitochondria convert diverse sources of chemical energy (nutrients) into adenosine triphosphate (ATP) and heat. Despite being often overlooked and understudied as compared to mitochondrial ATP production, mitochondrial heat production (thermogenesis) has crucial physiological functions. It supports core body temperature, regulates body weight by setting a balance between energy consumption and expenditure, and controls mitochondrial production of free radicals to preserve mitochondrial and cell integrity. Mitochondrial thermogenesis is sometimes represented as a byproduct of mitochondrial ATP production, resulting from the inherent inefficiency of mitochondrial ATP production pathways. Although the mitochondrial ATP production machinery is not 100% efficient and thus dissipates some heat, this inefficiency is not the topic of this review. Here we consider a specialized thermogenic pathway associated with a regulated H+ leak across the inner mitochondrial membrane (IMM), which draws energy from the mitochondrial electrochemical H+ gradient in parallel to the ATP production pathway.
The subject of the mitochondrial H+ leak and thermogenesis holds significant promise in terms of addressing metabolic disorders and mitochondrial dysfunction. Unfortunately, the same field has also been an area of intense controversy. Traditionally, the mitochondrial H+ leak was addressed with two very different approaches. One approach was based on measurements of mitochondrial uncoupled respiration (“uncoupled” means not associated with ATP production). This method studied the mitochondrial H+ leak in intact mitochondria or cells but was indirect (not measuring H+ leak but rather an associated oxygen consumption by the electron transport chain [ETC]) and offered very limited control over the experimental conditions. The second method employed reconstitution of the proteins mediating the mitochondrial H+ leak in artificial lipid bilayers to measure their activity more directly and under more controlled conditions; however, these assays were criticized for lacking the native membrane environment. These two assays helped establish the phenomenon of the mitochondrial H+ leak and thermogenesis, but their limitations resulted in controversies and often misunderstanding of the mitochondrial H+ leak. Therefore, we introduced a different method to study the mitochondrial H+ leak – its direct patch-clamp measurement across the whole IMM. This method combines the best qualities of the two classical methods while avoiding most of their pitfalls. It studies the proteins mediating the mitochondrial H+ leak in the native membrane environment, directly and under perfectly controlled experimental conditions. The patch-clamp technique is universally considered a method of choice for understanding membrane transport proteins.
This review provides a critical assessment of the field of the mitochondrial H+ leak and thermogenesis in light of recent findings enabled by the mitochondrial patch-clamp. This review is not intended to be comprehensive but aims rather to address and reevaluate some of the most essential aspects and implications of the mitochondrial H+ leak.
MITOCHONDRIA AS ORGANELLES THAT GENERATE ATP AND HEAT
Mitochondria: beyond ATP production
Mitochondria are intracellular organelles primarily known for producing ATP, the universal energy source of the cell. However, all mitochondria also produce heat. Arguably, it would even be difficult to claim that ATP is the dominant form of energy produced by mitochondria. This is because mitochondria of the specialized thermogenic tissue brown fat function specifically to produce heat (1; 2). However, even mitochondria of tissues other than brown fat, such as skeletal muscle, heart, and liver, can convert as much as 20% of nutrient energy into heat (3).
A simplified scheme of mitochondrial energy conversion is shown in Fig. 1. Mitochondria convert the chemical energy of nutrients into electrical voltage, which is then converted into ATP and heat. At a more mechanistic level, in the first step in mitochondrial energy conversion, nutrients enter the Krebs cycle and are oxidized to generate high-energy electron donors NADH and FADH2. In the second step, NADH and FADH2 donate their high-energy electrons to the mitochondrial ETC that eventually passes them onto oxygen where they have significantly lower energy. The ETC uses the energy released in such electron transfer to pump H+ outside the mitochondrial matrix and to generate an electrochemical H+ gradient across the IMM. This gradient primarily consists of voltage across the IMM (ΔΨ ~ −160 mV in the mitochondrial matrix as compared to cytosol), while its chemical component is relatively small (ΔpH ~ 0.5) (4). In the last step, ΔΨ is converted into ATP and heat. ATP is generated by ATP synthase that passes H+ down ΔΨ and uses the released energy to generate ATP from ADP and inorganic phosphate. In contrast, heat is generated by so-called uncoupling proteins (UCPs) that also pass H+ down ΔΨ but do not generate ATP, instead letting energy dissipate as heat (Fig. 1).
Figure 1. Mitochondrial ATP and heat production.
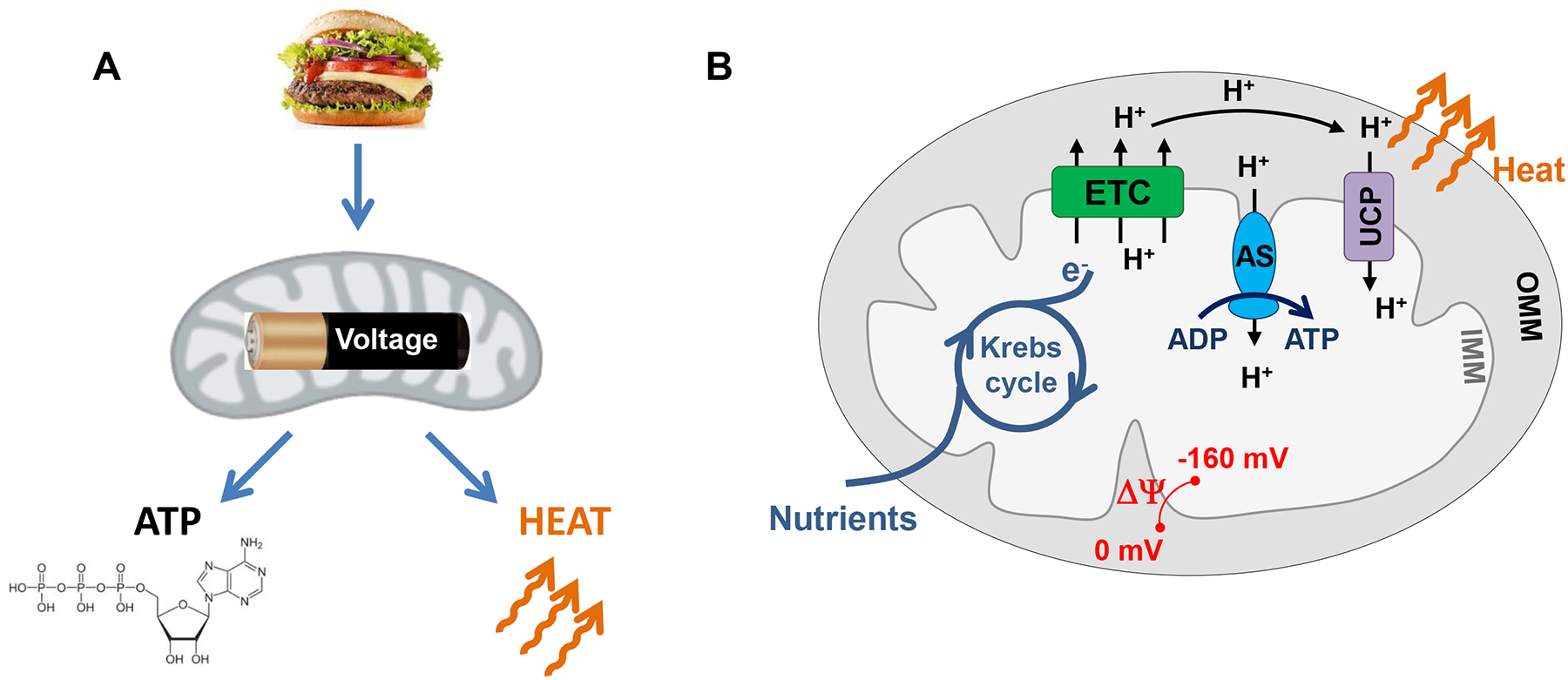
(A) General diagram of energy conversion within mitochondria. The chemical energy of nutrients is converted into a mitochondrial voltage, which is then converted into ATP and heat. (B) Mechanism of mitochondrial ATP and heat production. Mitochondria have two membranes, the inner mitochondrial membrane (IMM) and outer mitochondrial membrane (OMM). The OMM is freely permeable for ions and small metabolites with a molecular weight less than 2000 Da but presents a barrier for transport of proteins. In contrast, the IMM permeability is tightly controlled for all ions, metabolites and proteins. The IMM contains the machinery for ATP synthesis and confines the enzymes of the Krebs cycle within the mitochondrial matrix. The electron transport chain (ETC) uses the energy of electrons supplied by the Krebs cycle (in the form of electron donors NADH and FADH2) to pump H+ outside the IMM and generates membrane potential (ΔΨ) across the IMM. ΔΨ is then used by the ATP synthase (AS) to generate ATP and by uncoupling proteins (UCPs) to generate heat.
UCPs are called “uncoupling proteins” because they break a perfect coupling between the H+ flows via the ETC and ATP synthase. The uncoupling H+ current across the IMM mediated by UCPs is called H+ leak. Free fatty acids (FA) are the physiological activators of H+ leak via UCPs (5).
The mechanisms of mitochondrial H+ leak are poorly understood. Although UCP1 has been established as the UCP responsible for mitochondrial H+ leak in the specialized thermogenic tissues brown and beige fat (1; 6–10), the molecular identity of the UCP(s) of all other tissues is actively debated. Even for UCP1, the mechanism by which FA activate H+ leak is elusive, and several conflicting models have been proposed (1).
Because the molecular mechanisms of the native mitochondrial H+ leak remained largely unknown and its pharmacological control impossible, H+ conductance of the IMM was induced artificially using chemical protonophores such 2,4-dinitrophenol (DNP) and cyanide-4-(trifluoromethoxy) phenylhydrazone (FCCP). This helped to reveal the physiological significance of the mitochondrial H+ leak. FCCP and DNP are hydrophobic, membrane-soluble weak acids that can induce H+ leak by carrying H+ across the IMM without the help of membrane transport proteins (11; 12). While FCCP was primarily used in laboratory research, DNP initially found broad application in the clinic. DNP was used as an explosive during World War I, when its thermogenic effect was serendipitously discovered at munition factories (13). DNP was then widely used in humans in the 1930s and 1960s and became known as one of the most efficient weight loss medications. Indeed, DNP increases the metabolic rate, shifting the energy balance toward expenditure and reducing fat deposits (13). Although no significant safety concerns were identified originally (14), side effects, especially cataracts and even overdose-associated deaths, were reported later, and the drug was then banned by the US Food and Drug Administration (FDA) (15). Recently, there has been a significant resurgence of interest in DNP with the discovery that it is extraordinarily efficient against type II diabetes, fatty liver, and metabolic syndrome in animal models (16). However, the use of DNP in humans remains prohibited.
Controlling the mitochondrial H+ leak for therapeutic purposes holds significant promise, but its realization depends on the identification of UCPs and understanding the molecular mechanism of their operation.
Mitochondrial H+ leak vs mitochondrial uncoupling
Although the transport proteins of the IMM responsible for H+ leak are called uncoupling proteins, it should be mentioned that mitochondrial uncoupling can be caused not only by H+ leak. Increasing conductance of the IMM for any type of ion will dissipate ΔΨ and uncouple the activities of the ETC and ATP synthase. Among notable examples, the IMM channel known as mitochondrial Ca2+ uniporter (MCU) can dissipate ΔΨ and induce mitochondrial uncoupling (4). Other ion channels have been reported in the IMM including several types of K+ channels and the permeability transition pore (PTP) (17; 18). Any of them, even if they have a low open state probability, can allow abundant cytosolic K+ to enter mitochondria and uncouple them. Finally, certain electrogenic transporters of the IMM may also dissipate ΔΨ. All these modes of electrogenic transport will reduce the efficiency of mitochondrial ATP synthesis and make mitochondria generate more heat.
However, among all electrogenic conductances of the IMM involved in mitochondrial uncoupling, only H+ leak is a specialized thermogenic mechanism. The rest have other primary physiological functions, such as Ca2+-dependent stimulation of the Krebs cycle (MCU) or regulation of mitochondrial cell death (PTP), with uncoupling and the associated thermogenesis serving as byproducts of these primary functions. The central role of H+ leak in thermogenesis is illustrated by the specialized thermogenic tissue brown fat where mitochondrial heat production is due to H+ leak via uncoupling protein 1 (UCP1). H+ leak is the simplest way to connect the H+ transport activity of the ETC with thermogenesis, resulting in a futile cycling of H+ across the IMM that causes mitochondria to generate heat.
We mention the alternative mitochondrial uncoupling pathways in this review, because they introduce significant complexity into mitochondrial bioenergetics and could be confused with H+ leak when indirect methods are used.
Methods to study mitochondrial H+ leak
There have been two main approaches to studying mitochondrial H+ leak. The first measures the rate of mitochondrial uncoupled respiration either in isolated mitochondria or intact cells. In this assay, oligomycin, a highly specific inhibitor of ATP synthase, is used to eliminate H+ flow through the ATP synthase. Under these conditions, the outward H+ flux generated by the ETC should return back to the matrix only via H+ leak, and thus, the oxygen consumption by the ETC reflects the magnitude of H+ leak (Figure 1B)(2; 19). The benefit of this method is that it studies H+ leak in the intact native system (mitochondria or cells). However, H+ leak is not measured directly, and a change in the respiration rate of the ETC, an enormous molecular complex, can be caused by other factors. The assumption that the uncoupled respiration of the ETC is always associated with H+ leak is questionable, as other IMM conductances also are capable of uncoupling, such as K+, PTP, or even compromised IMM integrity. Finally, the experimental conditions such as the compositions of matrix and cytosolic solutions, concentration of membrane FA, pH gradient across the IMM, and ΔΨ are poorly controlled in respiration studies, and their variation during the experiment can further complicate the interpretation of the results.
Another classical approach attempts to recapitulate the phenomenon of mitochondrial H+ leak in artificial membranes. In this approach, candidate UCPs are reconstituted in lipid bilayers or liposomes and the properties of the resultant H+ leak are studied (20; 21). This method helped to reduce the complexity of the system, achieve more direct measurements of H+ leak, and improve control over the experimental conditions. However, this method was criticized because the UCPs are not studied in their native membrane environment, potentially altering their functional properties and possible even assigning the uncoupling role to proteins that do not mediate H+ leak in vivo. Although the mitochondrial respiration studies and the reconstitution studies laid important foundations for our understanding of mitochondrial H+ leak, the data collected with these two methods often supported very different conclusions, resulting in controversies.
Very recently another method for studying mitochondrial H+ leak has emerged. It was demonstrated that patch-clamp electrophysiology can be used to directly measure H+ leak across the whole IMM of isolated mitochondria devoid of the outer membrane (mitoplasts) (8; 22–24). The patch-clamp technique is a gold standard in studies of membrane transport proteins. The vast experience with application of this method in neuroscience, cardiac, and muscle physiology demonstrates that it generates reproducible and physiologically relevant results. In mitochondria, it measures H+ leak in the native IMM, with exceptional amplitude and time resolution as well as reliable control of all critical experimental conditions such as transmembrane voltage and solution compositions on both sides of the IMM. This method can record mitochondrial H+ leak in isolation from other IMM conductances, thus distinguishing it from K+ channels, PTP, or compromised membrane integrity.
Therefore, the patch-clamp electrophysiology approach has eliminated all major disadvantages associated with the respiration and reconstitution approaches, but it must be considered that the IMM cristae are likely to unfold during mitoplast preparation, making the IMM less intact compared to its state in the respiration studies. In this review, we often compare the results obtained with these three methods to provide better insight into the molecular mechanisms of mitochondrial H+ leak and address the decades-long controversies in this field.
MITOCHONDRIAL UNCOUPLING PROTEINS
Historical perspective on mitochondrial UCPs
UCP1 was the first UCP identified (25; 26). It has been unambiguously demonstrated to mediate mitochondrial H+ leak in the specialized thermogenic tissues brown fat (6; 7; 9; 25; 27) and beige fat (8; 10) by studies using the UCP1 inhibitor guanosine 5-diphospahte (GDP) and a UCP1-deficient mouse model. However, because UCP1 expression is limited to brown and beige fat, the UCPs of other tissues remained unknown.
Soon after the identification of UCP1, it was discovered the FA-induced uncoupled respiration in isolated skeletal muscle and liver mitochondria is reduced by carboxyatractyloside (CATR), a highly specific inhibitor of the mitochondrial ADP/ATP carrier (AAC) (28–30). These results indicated that AAC, which is responsible for the exchange of matrix ATP for cytosolic ADP (31), may also mediate H+ leak in the presence of FA, the physiological activators of mitochondrial H+ leak and thermogenesis. However, around the same time, AAC was also implicated as the PTP (32–34), a large-conductance non-selective pore activated under certain conditions in the IMM. Similar to H+ leak, the PTP is induced by FA and uncouples mitochondria (35; 36). Thus, it was difficult to unambiguously determine whether AAC induces mitochondrial uncoupling via a selective H+ leak or PTP, or both.
Genetic evidence for the involvement of AAC in mitochondrial H+ leak was scarce. Humans have four AAC isoforms, encoded by four different genes, AAC1-AAC4 (31; 37; 38). The AAC3 gene is absent in mice (39; 40). The existence of several AAC isoforms with likely redundant functions has complicated analysis of knockout phenotypes. In addition, the primary focus was on the roles of AAC in ADP/ATP exchange and the PTP. As a result, a loss of ADP/ATP exchange and disruption of mitochondrial ATP production was confirmed along with an important but perhaps non-essential role of AAC in the PTP (40–43). However, the involvement of AAC in mitochondrial H+ leak was only partially addressed. In an AAC1 knockout mouse, only basal mitochondrial uncoupled respiration (without FA added) was measured, and contrasting results were reported, from no effect to a decrease (40–42). Double AAC1/AAC2 knockout in mouse liver unexpectedly led to increased basal mitochondrial uncoupled respiration (43). Such discrepancy is not surprising given the fundamental role of AAC in mitochondrial physiology. The broad effects of AAC knockouts on mitochondrial function (42) can give rise to multiple phenotypes depending of the experimental conditions used. Thus, the involvement of AAC in mitochondrial H+ leak, especially FA-induced H+ leak, remained unclear.
The implication of UCP1 and AAC in H+ leak was followed by suggestions that other mitochondrial transporters, such as the phosphate carrier (44; 45), aspartate-glutamate carrier (46), and dicarboxylate carrier (46–48), could also facilitate FA-induced mitochondrial H+ leak. All these transporters belong to the solute carrier 25 (SLC25) family of mitochondrial anion carriers and are related to AAC and UCP1. However, the pharmacology used to demonstrate the involvement of these three additional carriers in H+ leak was not specific. In addition, similar to AAC, the phosphate carrier was also implicated in the PTP (49), complicating its functional analysis. Mouse models deficient for phosphate and aspartate-glutamate carriers have been generated, but similar to the AAC knockouts, the focus was not on H+ leak but other transport modes of these proteins (50–55). Some of these studies measured basal uncoupled respiration, but no significant abnormalities in carrier-deficient models were observed to implicate the phosphate or aspartate-glutamate carrier in H+ leak or mitochondrial uncoupling (51; 54; 55). In addition to its role in transporting inorganic phosphate, knockout of the phosphate carrier revealed its possible involvement in the PTP (50). To the best of our knowledge, no dicarboxylate carrier knockout mice have been reported.
With the onset of the genomic era, mitochondrial carriers UCP2 (56; 57) and UCP3 (58–60) were identified as close UCP1 homologs. In contrast to UCP1, UCP2 and UCP3 are expressed ubiquitously, and thus, could mediate mitochondrial uncoupling in tissues other than brown fat. By manipulating the levels of UCP2/3 expression (genetic ablation or overexpression) or using the UCP inhibitor GDP, numerous studies implicated UCP2 and UCP3 in mitochondrial H+ leak and uncoupling. UCP2 and UCP3 were also shown to facilitate H+ leak after reconstitution in artificial phospholipid membranes (61; 62). However, several other studies did not support that UCP2 and UCP3 are bona fide UCPs (reviewed in (63–66)). Despite the controversy, UCP2 and UCP3 are still generally believed to mediate mitochondrial H+ leak, at least under certain conditions, and the literature elaborating on their involvement in mitochondrial uncoupling is immense. Apart from their putative roles in mediating mitochondrial H+ leak, UCP2 and UCP3 have also been implicated in metabolite transport and regulation of glucose and glutamine oxidation (67–69).
Because the UCP candidates discussed above are all members of the SLC25 superfamily of mitochondrial solute carriers, it has even been suggested that all ~50 members of this family can contribute to H+ leak (3; 70).
To summarize, while UCP1 was confirmed to be the bona-fide mitochondrial UCP responsible for mitochondrial H+ leak and thermogenesis in brown/beige fat, the identity and number of UCPs in other tissues remained elusive. An important barrier to the identification of the UCP(s) in tissues other than brown fat was that all candidate proteins had other major transport functions in addition to the putative H+-transport activity. Thus, their knockout could have very broad effects on mitochondrial function including mitochondrial respiration, and thus, the respiration assays were not well-suited to study H+ leak specifically. The situation was further complicated by the fact that H+ leak in tissues other than brown fat is not robust enough to be easily detected on the background of other possible changes. Given the complexity of the mitochondrial transport of ions and metabolites, addressing the molecular mechanisms of the mitochondrial H+ leak required the development of a method for its direct measurement.
UCP1 – mitochondrial uncoupling protein of specialized thermogenic tissues
Brown adipose tissue is the main location for non-shivering adaptive thermogenesis in response to stimuli such as cold or diet, and it is essential for body temperature maintenance (1; 71). Research to understand the mechanisms of non-shivering adaptive thermogenesis has been energized by the enormous interest in human metabolic diseases linked to obesity, including type 2 diabetes and fatty liver disease (72). Indeed, brown fat possesses unique mitochondria that are extremely efficient in heat production and use fat as a preferred source of energy. These mitochondria express the uncoupling protein UCP1, a sophisticated H+ leak pathway responsible for brown fat thermogenesis.
The unusual behavior of isolated brown fat mitochondria was first detected in the 1960s (73). In contrast to mitochondria of other tissues, brown fat mitochondria were profoundly uncoupled when isolated using standard procedures (73). It was then determined that these mitochondria can be recoupled by adding purine nucleotides such as GDP or by removing free FA with albumin (74). Eventually, FA and purine nucleotides were determined to be the principal physiological regulators of a putative protein (referred to as thermogenin or UCP) responsible for H+ leak across the IMM of brown fat (1; 74; 75). This set the stage for attempts to identify this protein, which eventually was accomplished by the Klingenberg and Ricquier groups (6; 9). Brown fat UCP was a protein with six predicted transmembrane helixes, which could be grouped into three homologous repeats of two helixes each. Surprisingly, it was homologous to the previously identified AAC (6), establishing the SLC25 family of mitochondria solute carriers. The protein was later renamed UCP1 upon the identification of UCP2 and 3, two other SLC25 members. The molecular identification of UCP1 helped to clarify the mechanistic basis of its purine nucleotide inhibition (31), but the mechanism by which UCP1 facilitates H+ transport in the presence of free long-chain FA has been one of the most controversial topics in the field of bioenergetics (70; 76; 77). In fact, the actual transport function of UCP1 is still debated, as in some models UCP1 transports FA anions rather than H+ (70). The mechanism by which UCP1 facilitates H+ leak in the presence of FA will be addressed in subsequent sections of this review.
Direct patch-clamp recording from the IMM of brown fat demonstrated that the UCP1-medited H+ current is very large (23). In fact, it is one of the largest H+ conductances measured across any biological membrane, and in this respect, it can be compared only to the currents mediated by voltage-gated H+ channel Hv1 (78). However, UCP1 is a transporter that should have a significantly slower H+ turnover rate as compared to a H+ channel. The large H+ current mediated by UCP1 is due to the extremely high density of this protein in the IMM of brown fat (25). The FA-dependent H+ current across the IMM of brown fat is eliminated in UCP1 knockout mice (23).
Originally, brown fat was considered the only site for adaptive thermogenesis. However, it was later discovered that under certain conditions, such as prolonged cold exposure, increased sympathetic tone, or physical exercise, white fat develops islands of cells with the morphology of brown adipocytes. Instead of a single large lipid droplet and scarce mitochondria (monolocular adipocytes characteristic of white fat), these cells contain numerous small lipid droplets and abundant mitochondria that give them a brownish appearance (so-called multilocular adipocytes, characteristic of brown fat). Thus, these multilocular cells were named beige (or brite) adipocytes (79–82). Early on, it was suggested that all beige adipocytes express UCP1, and UCP1 was universally used as a marker to distinguish beige adipocytes from white adipocytes within white fat depots. Also, it was generally accepted that all beige adipocytes were capable of UCP1-dependent mitochondrial thermogenesis. However, direct patch-clamp characterization of H+ leak across the IMM of subcutaneous and abdominal beige fat demonstrated the existence of both UCP1-positive and UCP1-negative beige adipocytes (8). All mitochondria from subcutaneous (inguinal) beige fat, when analyzed with patch-clamp electrophysiology, had UCP1-dependent H+ leak similar to that found in brown fat. In contrast, only about 15% of the mitochondria in abdominal (epididymal) beige fat had the UCP1-dependent H+ leak, with UCP1 currents being undetectable in the remaining 85% (8). Thus, mitochondrial patch-clamp analysis revealed a new population of beige adipocytes that do not use UCP1 for thermogenesis (8). In mice, the UCP1-negative beige adipocytes are abundant in abdominal fat, whereas UCP1-positive beige adipocytes are the predominant type in subcutaneous fat. The UCP1-negative and UCP1-positive beige adipocytes share significant similarities such as being multilocular and having a large mitochondrial network, a robust thermogenic gene program and a distinctive OXPHOS profile different from that of brown fat (8). Moreover, both UCP1-negative and UCP1-positive beige adipocytes are capable of heat production via a futile cycle of creatine phosphorylation/dephosphorylation (8; 71; 83).
Thus, UCP1 was unambiguously identified as a UCP of specialized thermogenic tissues brown and beige fat. However, UCP1-dependent thermogenesis is not necessarily the only thermogenic mechanism present in beige fat. Indeed, not all beige adipocytes express UCP1, and beige fat possesses a futile cycle of creatine phosphorylation/dephosphorylation as an alternative thermogenic mechanism. However, UCP1 is by far the dominant mechanism of adaptive thermogenesis in beige and brown fat, and its deficiency leads to profound disruption of thermoregulation and diet-induced thermogenesis in mice.
AAC – ubiquitous mitochondrial uncoupling protein
H+ leak is not a distinctive property of the mitochondria of the specialized thermogenic tissues. Even though thermogenesis is not the primary function of other somatic tissues such as skeletal muscle and liver, their mitochondria still have H+ leak. It has been estimated that H+ leak could be responsible for up to 52% of the total mitochondrial oxygen consumption in the resting rat skeletal muscle (84; 85) and for up to 26% in resting (not engaged in gluconeogenesis and ureagenesis) rat hepatocytes (86). These numbers are reduced to 34% and 22%, respectively, as the activity of these tissues increases (85), but this still demonstrates that mitochondrial H+ leak and thermogenesis can have a profound effect on the bioenergetics of these tissues and of organisms in general. It was estimated that the mitochondrial H+ leak of all tissues combined could represent as much as 20% of the resting metabolic rate of the entire body (85).
Although H+ leak is an essential aspect of mitochondrial physiology, the molecular identity of the transport protein(s) mediating the thermogenic H+ leak across the IMM in tissues other than brown/beige fat remained elusive for decades. Recently, the patch-clamp technique was applied to directly measure H+ leak across the whole IMM of tissues other than brown fat including skeletal muscle, heart, liver, and kidney. The results showed that the mitochondria of all these tissues possess a robust FA-induced H+ leak (22), supporting the previous data demonstrating that mitochondria of all tissues are capable of thermogenesis and heat is an essential component of mitochondrial energy output.
Most importantly, the patch-clamp technique helped determine the molecular identity of the mitochondrial UCP in non-adipose tissues. Being the closest UCP1 homologs, the ubiquitously expressed UCP2 and UCP3 have received much attention (64), but their contributions to mitochondrial uncoupling have remained controversial (63; 67). Electrophysiological analysis demonstrated that the amplitude of the FA-induced H+ leak in skeletal muscle mitoplasts isolated from wild-type (WT), UCP2−/− and UCP3−/− mice were comparable. Additionally, the UCP inhibitor GDP (61; 63; 64) had no effect on the H+ leak. Thus, UCP2 and UCP3 do not contribute significantly to H+ leak (22).
Strikingly, the electrophysiological analysis demonstrated that AAC is the protein responsible for the FA-induced mitochondrial H+ leak in skeletal muscle, heart, liver, and kidney. Indeed, an AAC-specific inhibitor CATR inhibited FA-induced H+ leak in all these tissues. Moreover, another specific AAC inhibitor, bongkrekic acid (BKA), abolished H+ leak in skeletal muscle and heart (22). This pharmacological identification was reinforced by the measurement of H+ leak in AAC1- and AAC2-deficient mice. AAC1 and AAC2 are the two somatic isoforms of AAC in mice. The third somatic isoform AAC3 is present is humans but absent in mice (39; 40), while AAC4 is expressed mainly in germ cells and pluripotent stem cells (38). AAC1 expression is highest in heart and skeletal muscle, and AAC2 is most abundant in kidney (39; 87). In AAC1−/− mice (40), H+ leak completely disappeared in heart mitoplasts and was reduced about 70% in skeletal muscle (22). Importantly, the remaining H+ leak in skeletal muscle was CATR-sensitive, suggesting the involvement of AAC2 in H+ leak. AAC1 knockout did not result in reduction of H+ leak in kidney, a tissue with significant expression of AAC2, while H+ leak was still inhibited by CATR (22). In AAC2 hypomorphic mice (87), in which AAC2 was considerably reduced but not completely eliminated, H+ leak was not significantly altered in heart and kidney (22), likely due to compensation by AAC1 as previously observed in these mice (87). These results indicate that AAC1 plays a crucial role in H+ leak, at least in heart and skeletal muscle. Although the reduction of H+ leak in AAC2 hypomorphic mice was not significant, the recording of robust CATR-sensitive H+ leak in skeletal muscle of AAC1−/− mice demonstrates that AAC2 is capable of mediating H+ leak. Thus, direct patch-clamp recording from the IMM demonstrated that AAC is responsible for the FA-induced mitochondrial H+ leak.
Respiration experiments in isolated mitochondria and intact cells further confirmed the crucial role of AAC in mitochondrial H+ leak. Uncoupled respiration induced by long-chain FA was eliminated in cardiac mitochondria isolated from AAC1−/− mice (22). The role of AAC in mitochondrial H+ leak and uncoupled respiration was also analyzed in a C2C12 mouse cell line deficient for AAC1 and AAC2 (AAC1/AAC2 double knockout). Despite being fully AAC-deficient, double knockout cells had unaltered mitochondrial biomass and their mitochondria have robust respiratory capacity. The basal and FA-dependent uncoupled respiration rates were dramatically reduced both in isolated double knockout mitochondria and intact cells (22). The dramatic effect of AAC deficiency on mitochondrial uncoupled respiration confirms the conclusion from the electrophysiological experiments that AAC is responsible for mitochondrial H+ leak in tissues other than brown/beige fat. Thus, there are two bona-fide thermogenic UCPs: ubiquitous AAC and fat-specific UCP1.
Previously, AAC was proposed as a candidate mitochondrial UCP in tissues other than brown/beige fat, but assigning this functional identity to AAC unambiguously was difficult based on the indirect H+ leak measurements using mitochondrial respiration. AAC was first implicated in mitochondrial uncoupling by Skulachev’s group based on the observation that CATR inhibits FA-induced mitochondrial uncoupled respiration in skeletal muscle and liver (29). Later, the Brand group proposed that the basal uncoupled respiration (a small uncoupled respiration observed in the presence of FA acceptor albumin and which is assumed to be FA-independent) is also caused by AAC (41). However, these experiments fell short at convincing the field that AAC operates as a bona-fide UCP. First, the inhibition of mitochondrial uncoupling by CATR was often partial (88; 89). Second, in addition to H+ leak, AAC was implicated in the PTP (4), which is also activated by FA and causes mitochondrial uncoupling. Furthermore, because AAC is one of the most abundant proteins in the IMM, changes in the AAC conformational state induced by AAC ligands (CATR, etc.) can vastly affect mitochondrial morphology (condensed vs orthodox) and potentially change respiration rates (31; 90). Finally, the degree of AAC contribution to the H+ leak as compared to other possible UCPs remained uncertain. Mitochondrial proteins such as the phosphate carrier (44; 45), aspartate-glutamate carrier (46; 91), dicarboxylate carrier (46–48), UCP2 and UCP3 (64; 66), and eventually all members of the SLC25 family were implicated in mitochondrial H+ leak (3). The uncertainties in the interpretation of respiration data and the numerous possible UCP candidates have confused the field and distracted attention from AAC.
The direct patch-clamp measurements of mitochondrial H+ leak across the native IMM were instrumental in reevaluating the identity of the protein(s) that control mitochondrial uncoupling in tissues other than brown and beige fat. In the patch-clamp measurements, the “basal H+ leak” was not observed in the absence of long-chain FA, while application of FA induced robust H+ current across the IMM that appeared to be solely mediated by AAC based on pharmacological and knockout experiments (22). Interestingly, the patch-clamp experiments demonstrated that although long-chain FA can activate H+ leak via AAC in both the “c” and “m” conformational states, CATR only inhibits H+ leak in the “c” state (CATR is a c-state specific inhibitor)(22). This could explain, at least partially, why in mitochondrial respiration studies CATR often showed incomplete inhibition of the FA-induced H+ leak (88; 89).
The fact that AAC is the most highly expressed of all SLC25 family members could explain why this transporter is the main UCP. However, it is also obvious that if the remaining ~50 members of the SLC25 family also can mediate H+ leak, there would be a significant AAC-independent H+ current mediated by the combination of the remaining SLC25 members. Moreover, some SLC25 family members (such as phosphate carrier or aspartate-glutamate carrier) are expressed at levels comparable to that of AAC (92; 93). However, as recorded by patch-clamp electrophysiology, AAC dominates the FA-induced H+ leak in skeletal muscle, heart, liver, and kidney (22).
Thus, AAC has two modes of transport, the classical ADP/ATP exchange and the FA-dependent H+ leak (Fig. 2). These two transport functions control the most basic aspects of mitochondrial bioenergetics, ATP production and thermogenesis.
Figure 2. The two transport functions of ADP/ATP carrier (AAC).
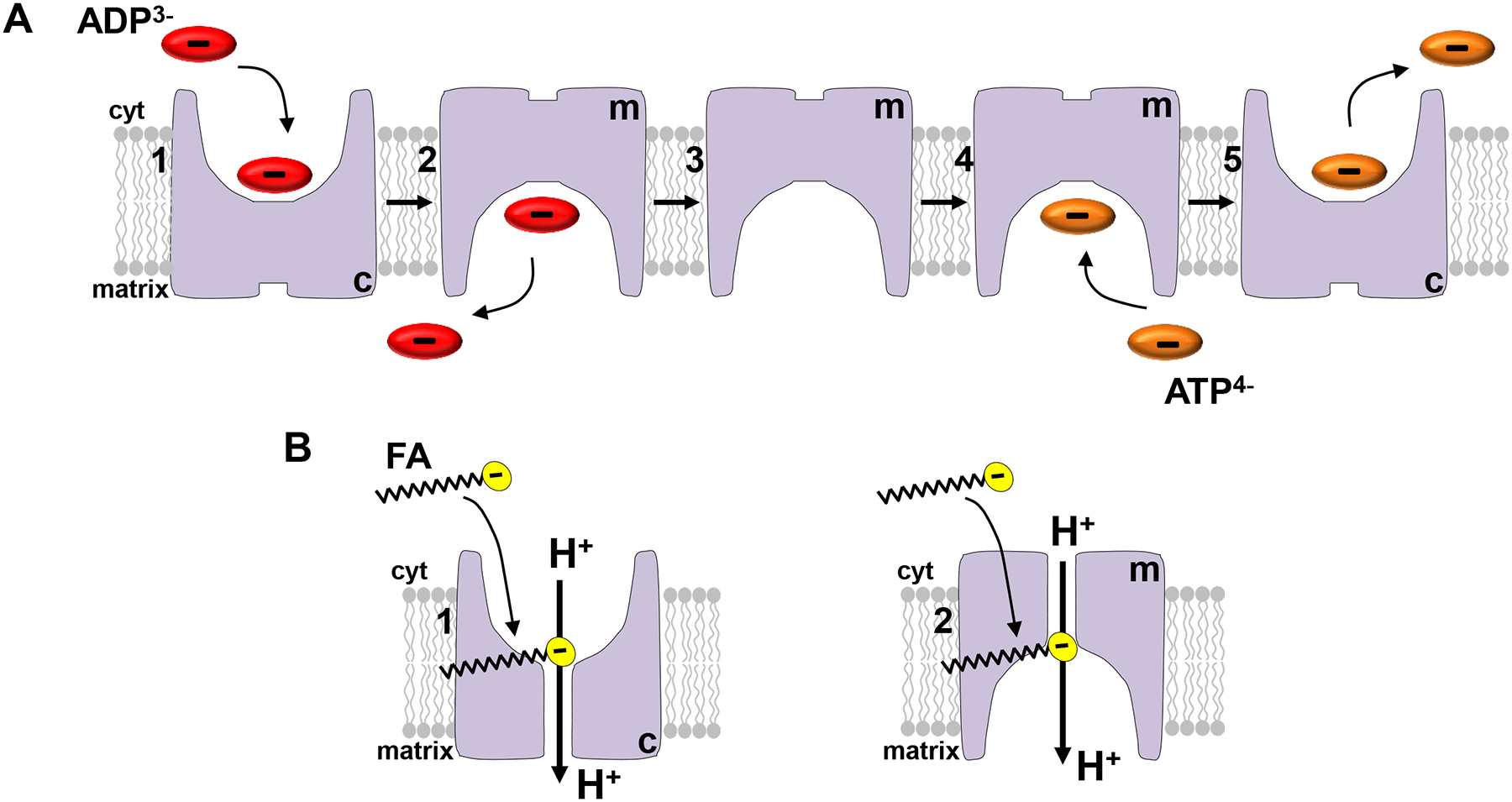
(A) ADP/ATP exchange. Cytosolic Mg2+-free ADP binds to the c-state of AAC (the substrate binding site exposed to the cytosol) [1], AAC changes its conformation to the m-state (the substrate binding site exposed to the matrix), and ADP dissociates into the matrix [2]. AAC dwells in the m-state [3]. When matrix Mg2+-free ATP binds to the m-state [4], AAC changes its conformation to the c-state, and ATP dissociates into cytosol [5] (31). (B) The fatty acid (FA)-dependent H+ leak. When ADP and ATP are not associated with the AAC substrate binding site, cytosolic free long-chain FA can bind either to the c-state [1] or m-state [2] of AAC and activate H+ leak. Matrix long-chain FA cannot activate H+ leak via AAC in either of the two states (22).
PHYSIOLOGICAL REGULATION OF MITOCHONDRIAL UNCOUPLING PROTEINS AND H+ LEAK
Does “basal mitochondrial H+ leak” exist?
Although long-chain FA increase mitochondrial H+ leak in all tissues (5; 89; 94), mitochondria remain partially uncoupled even in the presence of the FA acceptor albumin when free FA are believed to be completely eliminated. Thus, the existence of a constitutive mitochondrial H+ leak requiring no activators was postulated. Brand and coauthors called it “basal H+ leak” and suggested that it is largely mediated by AAC and UCP1 (3; 19; 41; 95). Although the precise mechanism of the “basal H+ leak” has not been explained, it was tentatively proposed to occur via the interface between AAC/UCP1 and the lipid bilayer and to depend only on the membrane density of these proteins and not their specific transport activities (41; 95). The long-chain FA would increase H+ leak via UCP1 and AAC further, but through a mechanism that could be different from that of the “basal H+ leak”. The existence of the “basal H+ leak” would imply that the IMM is constitutively leaky for H+, and there is a significant constitutive inefficiency in mitochondrial ATP production.
It is important to understand that the “basal H+ leak” has never been measured directly but rather assumed from mitochondrial uncoupled respiration in the presence of oligomycin and the FA acceptor albumin. There is an undeniable phenomenon of mitochondrial uncoupled respiration under these conditions (so-called basal mitochondrial respiration). However, explaining such respiration with “basal H+ leak” may not be accurate. Respiration studies alone are not capable of such definitive mechanistic insight, and there might be explanations for the basal mitochondrial respiration other than “basal H+ leak”.
First, complete FA extraction from a suspension of isolated mitochondria using albumin would be impossible. Thus, the “basal H+ leak” could simply be explained by residual FA-dependent H+ leak. The belief that albumin can completely extract long-chain FA from the IMM to achieve “the strict absence of long-chain FA” associated with UCP1 or AAC could be overstated. Indeed, addition of a finite amount of albumin to a large amount of long-chain FA contained within a mitochondrial suspension (as done in the respiration experiments) will never achieve a zero concentration of FA. The situation is further complicated by the fact that long-chain FA and other extractable lipids could be continuously generated within the mitochondrial membranes, and all these lipids will eventually saturate the albumin buffer (albumin is not specific for FA and can bind other lipids). A phospholipase activity associated with mitochondria (such as that of phospholipase A2 [PLA2]), which has been reported by several groups (8; 23; 96–98), can continuously generate free long-chain FA from membrane phospholipids. Thus, complete extraction of long-chain FA by albumin in respiration studies is not a certainty.
Second, the basal respiration can also be due to IMM conductances other than H+ leak. Indeed, mitochondria can also be uncoupled by several types of K+ channels, non-selective cationic channels, and the PTP that have been reported in the IMM (99). Possible contributions of these channels are often overlooked in the respiratory analysis. Finally, compromised IMM integrity resulting from the isolation procedure is another factor that can contribute to basal respiration in suspensions of isolated mitochondria. Unfortunately, respiration experiments cannot reliably distinguish between these various pathways to clearly elucidate the mechanism of the basal respiration.
The existence of the FA-independent basal H+ leak via UCP1 has been challenged by the groups of Martin Klingenberg and Keith Garlid (20; 21). These two groups performed experiments under more controlled conditions using UCP1 reconstituted in proteoliposomes. In this system, UCP1 transport activity can be isolated from that of other transport proteins, and the concentration of long-chain FA can be controlled better due to the lack of mitochondrial phospholipases or other enzymes that can supply UCP1 activators. These studies demonstrated that without long-chain FA, there is no H+ leak via UCP1 (20; 21). However, their use of artificial membranes to study UCP1 has been criticized by supporters of the “basal H+ leak”.
Mitochondrial patch-clamp helped to address the problem of the “basal H+ leak” in a rather precise manner. The first direct recording of the H+ leak mediated by UCP1 (23) and AAC (22) across the native IMM confirmed the strict requirement of FA. In these experiments, conditions such as the solution compositions on both sides of the IMM and the transmembrane voltage are well-controlled. The H+ leak is measured in isolation from other mitochondrial conductances and can also be easily distinguished from an unselective leak caused by a compromised membrane. Importantly, extremely low concentrations of long-chain FA can be achieved during whole-IMM patch-clamp recordings. Specifically, the recording chamber contains a single IMM (mitoplast) attached to the patch pipette, and the bath solution containing albumin or cyclodextrin is continuously perfused through the recording chamber. Albumin and cyclodextrins can also be added into the pipette (matrix side of the IMM). Therefore, a very limited amount of FA contained within a single IMM is exposed to almost an infinite amount of FA acceptors, bringing the FA concentration to zero. Under these conditions, no H+ transport activity of UCP1 (8; 23) or AAC (22) is detected in the absence of FA.
It should also be mentioned that the “basal H+ leak” does not align well with the postulates of the chemiosmotic theory. An unregulated “basal H+ leak” would render mitochondria permanently uncoupled and permanently inefficient for ATP production, while the chemiosmotic theory postulates that the H+ permeability of the IMM should be tightly controlled. It would only make sense that such a fundamental physiological phenomenon as mitochondrial H+ leak is precisely regulated, because ATP production and the metabolic integrity of the whole organism are at stake.
To conclude, the concept of the “basal H+ leak” has been rightfully challenged based on evidence provided by direct mitochondrial patch-clamp measurements and reconstitution studies. Although the existence of the “basal H+ leak” cannot be completely excluded, it must be realized that the experimental evidence in its support is weak.
Free long-chain FA as activators of mitochondrial H+ leak
In the early 1950s, it was suggested that cells might contain a substance that uncouples oxidative phosphorylation, thus exerting control over cell metabolism. This theory was further extended to postulate that the uncoupling substance is not always present in an active form but is liberated from a precursor under certain conditions. This postulate was primarily based on the observation that freshly prepared liver mitochondria were not significantly uncoupled, but as they were incubated at room temperature, they developed many of the characteristics of freshly prepared mitochondria uncoupled with a protonophoric agent such as DNP (48; 100). The mysterious substance was later identified as a mixture of various nonesterified long-chain FA (48; 100–102).
Later, in the 1960s, it was discovered that mitochondria isolated from brown fat behaved differently from mitochondria isolated from other tissues and were completely uncoupled immediately upon isolation (73; 103). Shortly after, a link between the uncoupled state of brown fat mitochondria and long-chain FA was established (104; 105). The Nicholls group demonstrated that the action of FA in brown fat mitochondria was associated with an in increased H+ conductance of IMM (74). Eventually, this H+ conductance was shown to be mediated by a highly abundant protein of brown fat mitochondria, UCP1 (6; 9; 27).
In a parallel line of research, experiments with pigeons, seals and rodents demonstrated that acute exposure to cold markedly increased uncoupling of skeletal muscle mitochondria isolated from these animals (reviewed in (70)). This uncoupling was inhibited by the FA acceptor albumin and later demonstrated to be associated with an increased concentration of long-chain FA (70; 106). This research eventually led to the identification of AAC as the protein that could at least partially explain the FA-dependent increase in H+ leak across the IMM of skeletal muscle, heart, liver and kidney (28; 29; 88; 89).
In addition to long-chain FA, other UCP1 activators were proposed, most notably long chain acyl-CoA (107), coenzyme Q (108) and 4-hydroxy 2-nonenal (109), but their ability to activate UCP1 and AAC remained controversial (61; 110; 111). Patch-clamp recordings demonstrated that oleoyl-CoA strongly inhibited the UCP1-dependent H+ leak instead of inducing it, whereas coenzyme Q6 had no effect (23). 4-Hydroxy 2-nonenal also showed no effect on UCP1 current, it but potentiated FA-dependent H+ leak via AAC (22; 23). Importantly, 4-hydroxy 2-nonenal could not increase the AAC-dependent H+ leak without the presence of FA (22). Thus, free long-chain FA are the only established physiological activators of UCP1 and AAC.
In brown fat, it has been proposed that free FA for UCP1 activation are generated via the hydrolysis of cytoplasmic lipid droplets (1; 2; 110). This process is under the control of the sympathetic nervous system that innervates brown adipocytes and activates adaptive thermogenesis mediated by these cells. In response to cold, diet, and perhaps other physiological stimuli, the sympathetic nervous system releases norepinephrine, which activates β3-adrenergic receptors on the surface of brown adipocytes, eventually leading to the hydrolysis of cytoplasmic lipid droplets (1). Free cytosolic long-chain FA released via the hydrolysis of lipid droplets are postulated to serve both as UCP1 activators and the substrate for mitochondrial heat production (1; 2).
The assumption that H+ leak via UCP1 is activated by free long-chain FA derived from the hydrolysis of cytoplasmic lipid droplets was based, to a great extent, on in vitro assays in which exogenous FA were infused into a suspension of isolated brown fat mitochondria (2; 112). In this experimental setting, UCP1-dependent uncoupling is indeed activated by extramitochondrial FA. However, the role of the free long-chain FA generated by hydrolysis of cytosolic lipid droplets in UCP1 activation is less clear in intact brown adipocytes in vivo. The concentration of free long-chain FA is tightly controlled via FA binding to proteins, esterification, and other modifications (113), rendering them unable to activate H+ leak via UCP1/AAC. Mitochondria in particular have a sophisticated system for the modification and metabolism of free FA (114), and thus, delivery of free FA across the outer membrane to UCP1 located in the IMM could be challenging. Thus, free long-chain FA generated within the IMM can have a comparative advantage for UCP1 activation as compared to those coming from the cytosol. Interestingly, direct patch-clamp studies of the UCP1-dependent H+ leak in brown and beige fat mitochondria suggested that free FA are locally produced via the hydrolysis of phospholipids of the IMM (8; 23). This indicates the existence of a protein endowed with a phospholipase activity (likely PLA2) in the IMM of brown fat. By hydrolyzing IMM phospholipids, this protein can strongly contribute to H+ leak activation via UCP1 along with hydrolysis of cytosolic lipid droplets. Overall, the signaling pathway that connects the engagement of adrenergic receptors on the surface of brown adipocytes and UCP1 activation in the IMM could be more complex than is currently assumed.
The sources of free long-chain FA for activation of H+ leak via AAC in non-adipose tissues are even less explored. Because under normal conditions the density of cytosolic lipid droplets in such tissues is much less than that in brown fat, other mechanisms may play more significant roles. Thus, local membrane FA production could be an important mode of activation of H+ leak via AAC. In this regard, PLA2 activity has been reported not only in mitochondria of brown/beige fat (8; 23) but also in mitochondria of various non-adipose tissues. In fact, mitochondrial PLA2 activity was first identified as the reason why isolated liver mitochondria lose their capacity for coupled respiration as they age upon isolation (115; 116). Later, PLA2 activity was implicated in different physiological and pathophysiological aspects of mitochondrial function such as uncoupling and energy metabolism, remodeling of mitochondrial phospholipids to protect against oxidative damage, PTP activation, and cell death (96; 97; 117–126). It is suggested that Ca-independent PLA2 (iPLA2) is the predominant mitochondrial PLA2 in non-adipose tissues such skeletal muscle, liver, heart, kidney and brain (96–98; 117; 118; 121; 122; 124), but the actual identity of the enzyme responsible for the hydrolysis of mitochondrial phospholipids and generation of free long-chain FA remains elusive. Also, there is a possibility that the FA production is not mitochondrial but associated with remnants of other cellular organelles tethered to mitochondria after isolation, such as endoplasmic reticulum or lipid droplets.
The physiological concentrations of extramitochondrial long-chain FA needed to induce H+ leak via UCP1 and AAC are still under debate, ranging broadly from low nanomolar to high micromolar concentrations. Such discussion has only a limited applicability to those free FA that are produced within the IMM. However, the question is still relevant when the source of free long-chain FA is cytosolic. Early studies found that nanomolar FA concentrations (10–50nM) are sufficient to activate H+ leak via UCP1 in isolated brown fat mitochondria respiring in vitro (94). However, micromolar concentrations are required to activate H+ leak either via UCP1 (127; 128) or AAC (129) functionally reconstituted in lipid bilayers. Direct patch-clamp analysis of UCP1 and AAC in the native IMM also demonstrated that low micromolar FA concentrations are needed to activate H+ currents via these proteins (8; 22; 23). Finally, it was estimated that free cytosolic FA can reach millimolar concentrations during active lipolysis in brown adipocytes (130), demonstrating that achieving micromolar concentrations of FA in the immediate proximity of UCP1 is not impossible physiologically.
With several groups finding that low micromolar concentrations of FA are required for activation of H+ leak via UCP1, why did the early results with isolated brown fat mitochondria suggest that the required FA concentrations were two orders of magnitude lower? One possible explanation is the use of the albumin buffer for free long-chain FA in these studies (94). In these experiments, the ~ 60 μM bovine serum albumin buffer was mixed with tens to hundreds of micromoles of free palmitate to achieve nanomolar concentrations of free, unbound FA (94). The calibration of this buffer was performed in the absence of isolated mitochondria. However, when this albumin-based FA buffer was mixed with a mitochondrial suspension, bound FA were likely displaced from albumin by various lipids released from the mitochondrial membranes. Importantly, the FA-binding sites of albumin are not selective for FA (131), and FA can be displaced from albumin by many types of lipids. The situation is further exacerbated by continuous generation of free FA and other lipids within the mitochondrial membranes, because these could eventually overwhelm the albumin buffer. One example is the mitochondrial PLA2 activity reported by several groups (8; 23; 96–98). Thus, the actual concentration of free FA provided by the so-called FA/albumin buffer will be significantly higher in the presence of mitochondria, potentially approaching the total concentration of FA (hundreds of μM) added into the albumin buffer initially. Thus, use of the albumin buffer in respiration experiments could result in underestimation of the free FA concentration required to activate UCP1 by about two orders of magnitude (94). Such underestimation was also noted by others (21).
Direct patch-clamp analysis offers the best possible control of FA concentrations while studying UCPs in the native IMM. Only one microscopic vesicle of the whole IMM (mitoplast) is present in the 500-μl recording chamber. This single mitoplast does not affect the concentration of FA in the large, continuously perfused recording chamber. In this experimental setting, the free FA concentration can be controlled directly by simple addition of the required concentration. This electrophysiological analysis demonstrated that low micromolar concentrations of free long-chain FA are required to activate H+ currents via UCP1 (22; 23), supporting the data obtained with purified UCP1 and AAC incorporated in lipid bilayers (127–129).
To summarize, free long-chain FA are the only established physiological activators of H+ leak via UCP1 and AAC. Although we still cannot exclude a possibility that other activators of UCP1 and AAC exist within the cell, in order to activate H+ leak via UCP1 and AAC, they may need to share certain chemical properties with FA. These properties and the proposed mechanism by which FA activate H+ leak via UCP1 and AAC are discussed in the next section.
Mechanism by which FA activate H+ leak via UCP1 and AAC
UCP1 is the first and most studied mitochondrial UCP. Almost everything we know about the mechanism by which long-chain FA activate mitochondrial H+ leak comes from studies of this protein. Several different models of how UCP1 facilitates H+ translocation in the presence of FA have been proposed. These have been discussed in previous reviews (1; 77). These models vary significantly in two aspects: 1) the species transported by UCP1 (H+ vs. OH-, vs. FA anions) and 2) the requirement of FA for UCP1-dependent H+ leak (required vs. not required). These models were proposed based on indirect measurements of the UCP1-dependent H+ leak, such as mitochondrial respiration or flux studies in proteoliposomes reconstituted with UCP1. When the method to directly measure UCP1-dependent H+ leak using patch-clamp electrophysiology was introduced (23), it generated a trove of new data regarding the functional properties of UCP1. In particular, the high amplitude and time resolution of patch-clamp electrophysiology enabled the first measurement of transient currents generated by UCP1 in the presence of low-pKa analogs of long-chain FA. Because in the presence of such analogs UCP1 does not translocate H+, these transient currents revealed that UCP1 moves long-chain FA within the membrane and undergoes a conformational change, as transporters do in the presence of a bona-fide substrate. Another notable observation enabled by patch-clamp electrophysiology was a striking asymmetry of UCP1 in terms of its activation by cytosolic vs matrix long-chain FA. Long-chain FA failed to activate UCP1 currents when applied on the matrix side of the IMM, but activated robust currents when applied on the cytosolic side (23).
The combination of all UCP1 functional properties revealed by patch-clamp electrophysiology could not be fully explained by any previous model of UCP1 function (23). Therefore, the need to develop a new model that incorporates all previous and new observations arose. UCP1 is a member of the SLC25 family of mitochondrial anion carriers. All members of this family are postulated to share a common mechanism of action (132). They all have a single substrate binding site located in the center of the membrane, which is alternatingly exposed to the opposite IMM sides through the conformational change of the carrier. There are two principal conformation states of SLC25 family members, the “c-state” with the substrate binding site exposed to the cytosol and the “m-state” with the substrate binding site exposed to the mitochondrial matrix. Based on the symmetry analysis of the amino acid residues at its putative substrate binding site, UCP1 was predicted to transport small carboxylic or keto acids in symport with protons (133). Transport of FA by UCP1 was also postulated in the FA-cycling model of UCP1 function, though H+ ions were assumed to be transported outside of UCP1 through a FA flip-flop across the lipid bilayer (20). When UCP1 currents were analyzed by patch-clamp electrophysiology, it was determined that UCP1 likely operates as a FA anion/H+ symporter; however, this appeared to be true only for short-chain FA (23). Short-chain FA anions could bind to UCP1 on both sides of the IMM and be transported to the opposite side. In contrast, long-chain FA anions could only bind on the cytosolic face of the IMM. Also, they appeared to be “anchored” inside UCP1 and produced transient currents in response to changes in transmembrane voltage, as compared to steady currents generated by short-chain FA (23). This anchoring was due to the longer carbon tails of the long-chain FA, and thus, was mediated by a hydrophobic interaction (23).
Therefore, from the viewpoint of the classical mechanism of SLC25 function, short-chain FA could be considered bona-fide, although low-affinity, UCP1 substrates. In contrast, long-chain FA appear to “hijack” the UCP1 transport mechanism by lodging within the UCP1 substrate binding site with high affinity and becoming, de-facto, continuously associated substrates. While continuously associated with the UCP1 substrate binding site, a single long-chain FA can facilitate multiple c–m conformation changes and help to transport multiple H+ ions via UCP1 (Fig. 3A). Thus, in the presence of long-chain FA, UCP1 operates like a H+ uniporter (a transporter that has a single transport substrate). This mechanism of UCP1 function draws another analogy. The long-chain FA lodged within UCP1 can be considered a co-factor for H+ transport via UCP1. The Klingenberg group first proposed a FA-cofactor model for UCP1 function (21), in which FA bind within the UCP1 translocation pathway and its protonatable headgroup provides a missing “stepping stone” for H+ transport via UCP1. The model presented in Fig. 3A differs from their co-factor model in that FA are rather unusual co-factors capable of inducing a large conformational change within the apoprotein (UCP1). This conformational change is likely required for successful H+ translocation (23).
Figure 3. Mechanisms of fatty acid (FA)-dependent H+ transport by uncoupling protein 1 (UCP1) and ADP/ATP carrier (AAC).
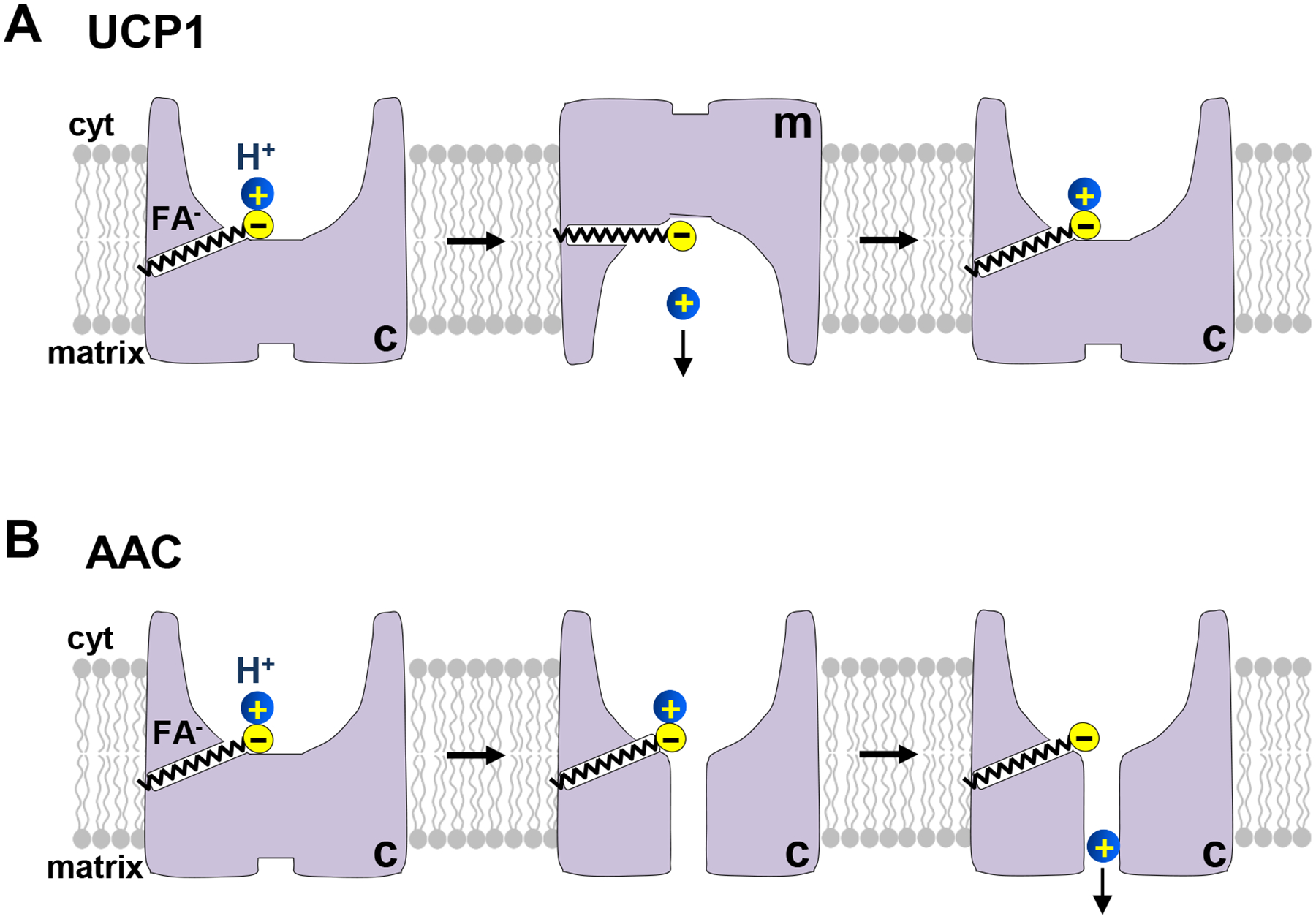
(A) The mechanism of H+ leak via UCP1. UCP1 functions as a FA anion/H+ symporter. FA are UCP1 transport substrates and induce UCP1 transition between the c- and m-state. When long-chain FA and H+ are bound, UCP1 undergoes conformation changes between the c- and m-state, enabling H+ translocation. However, long-chain FA do not dissociate from UCP1 because they are “caught” within UCP1 by hydrophobic interactions. Thus, each long-chain FA can facilitate transport of multiple H+. (B) The mechanism of H+ leak via AAC. In contrast to UCP1, AAC does not transport FA anions, and FA cannot induce AAC transition between the c- and m-state. Instead, binding of a long-chain FA anion to AAC might induce a minor conformational change to open a narrow translocation pathway for H+ across AAC. In addition, long-chain FA provide an essential binding site for H+ permeation through AAC.
What are the structural features of long-chain FA that help them to bind within UCP1 and activate H+ leak? As mentioned earlier, the length of the carbon tail is the crucial factor (21; 23), which is likely explained by the hydrophobic interactions established by FA when they bind to UCP1. There might be a hydrophobic pocket within UCP1 that accommodates the carbon tail of FA, as represented in Fig. 3A, or the carbon tail may protrude into the lipid bilayer, while the polar head of FA resides within UCP1. The degree of saturation of the carbon tail is not a major actor in H+ leak activation (21; 23). Finally, a protonatable polar head plays a crucial role in activation of UCP1 by FA. This is because it likely participates directly in the binding and translocation of H+. Non-protonatable low-pKa FA analogs (such as alkylsulfonates or halogenated FA) do not activate UCP1 and competitively inhibit H+ leak activated by regular FA (23; 77; 134). It must be kept in mind that although FA in solution have a pKa around 4.8, the majority of membrane-bound FA are protonated at physiological pH, as their pKa can increase to ~ 7 once they are incorporated into a phospholipid membrane (135; 136). In contrast, low-pKa FA analogs such as alkylsulfonates (solution pKa ≈−2) stay unprotonated at physiological pH even after incorporation into a membrane (137).
The mechanism of activation of H+ leak via AAC appears to be overall similar to that of UCP1 (22). First, AAC does not appear to conduct H+ in the absence of FA. Second, long-chain free FA activate H+ leak via AAC and their potency depends on the length of the carbon tail. Third, the capacity of FA to bind H+ at physiological pH is essential for activation of H+ leak via AAC, and low-pKa FA analogs induce no H+ current via AAC. Finally, FA activate H+ leak only when added on the cytosolic side of the IMM and not when added on the matrix side (22). Therefore, the presence of protonatable FA within the translocation pathway, enabled by hydrophobic interactions, appears to be the common criterion for H+ transport by UCP1 and AAC (22).
Interestingly, FA could not induce the c–m conformational change of AAC, as probed with the c-state specific AAC inhibitor CATR (22). This supports the concept that FA are not transport species of AAC, contrary to adenine nucleotides that induce the AAC c–m conformational change (22; 31; 138). Despite the inability of FA to induce the c–m conformational change in AAC, negatively charged low-pKa FA analogs induce low-amplitude AAC-dependent transient currents that are indicative of a small conformational change that FA anions can facilitate in AAC (22). Thus, a likely mechanism by which long-chain FA activate H+ leak via AAC is that of a cofactor for H+ translocation (Fig. 3B). Binding of a long-chain FA anion within AAC can enable H+ translocation by providing a missing binding site in the H+ translocation pathway and facilitating a small conformation change within AAC that may open a narrow H+ translocation pathway through the closed gate of AAC. To activate H+ leak via AAC, long-chain FA must bind from the cytosolic face of the IMM, but AAC can be in either the c- or m-state (Fig. 2B) (22).
Regulation of mitochondrial H+ leak by purine nucleotides
While FA are the principal physiological activators of H+ leak via UCP1 and AAC, Mg2+-free purine nucleotides are the main physiological inhibitors. Thus, the amount of H+ leak via UCP1 and AAC under physiological conditions is the result of a fine balance between activation by FA and inhibition by purine nucleotides. Despite some similarities, the mechanisms by which nucleotides inhibit H+ leak via UCP1 and AAC are not identical.
UCP1 and AAC possess binding sites that recognize only Mg2+-free purine nucleotides. However, these binding sites do not have the same nucleotide specificity. AAC binds ADP and ATP with an affinity about 10 times greater than that for GDP and GTP. In contrast, the affinities of UCP1 for adenine and guanyl nucleotides are about equal (31; 76). As GDP does not bind to AAC, it is often used in biochemical assays as a UCP1-specific ligand. Despite the ability of UCP1 to bind GDP and GTP, ADP and especially ATP are likely to be its principal physiological inhibitors in vivo because of their abundance.
The nucleotide-binding site for UCP1 is on the cytosolic face of the IMM, whereas that of AAC opens alternatively to both sides of the IMM (31; 76). This is because nucleotides are inhibitors of UCP1, whereas they are transport species of AAC. Moreover, nucleotides first bind to AAC with low affinity, but as AAC undergoes the c–m conformational change required for transport, the affinity of AAC for nucleotide binding increases in the transition state, in accordance with the induced transition fit of the transport catalysis (31; 76). In contrast, UCP1 binds nucleotides with higher affinity and does not undergo the nucleotide-induced transition fit, because it does not transport nucleotides (31; 76).
Nucleotide regulation is particularly well characterized for UCP1. In brown adipocytes, UCP1 is tonically inhibited by purine nucleotides, primarily ATP. Binding of purine nucleotides on the cytosolic side of UCP1 obstructs the UCP1 translocation pathway for H+ (76). To activate brown fat thermogenesis, FA must overcome this inhibition. However, whether FA remove purine nucleotide inhibition by direct competition or another mechanism has remained controversial (127; 139–142). Direct electrophysiological analysis of UCP1-dependent H+ current helped to demonstrate competition between FA and purine nucleotides for binding to UCP1 (23; 77). Structurally, FA anions and ATP are very different and unlikely to bind to the same site. However, the binding sites for FA anions and ATP may partially overlap or be located in immediate proximity, so that electrostatic repulsion between the two negatively charged molecules results in competition.
In contrast to the well-characterized purine nucleotide regulation of H+ leak via UCP1, little was known about the mechanism by which nucleotides regulate H+ leak via AAC. The recent application of the patch-clamp technique to directly record both the ADP/ATP exchange current and the FA-induced H+ leak mediated by AAC was instrumental in understanding the mechanism of their interaction (22). Remarkably, active nucleotide exchange significantly inhibits FA-dependent H+ leak but does not suppress it completely. This is not very surprising, because in contrast to their action in UCP1, nucleotides are not inhibitors of AAC but its transported species. Adenine nucleotide exchange and FA-induced H+ leak occur through the AAC translocation pathway and interfere with each other. However, the affinities with which FA and adenine nucleotides bind to AAC are comparable, and they cannot completely inhibit each other at their physiologically relevant concentrations (22).
The competition of nucleotide exchange and FA-dependent H+ leak in the AAC translocation pathway suggests intimate interconnection between mitochondrial ATP and heat production. By combining two transport modes, one controlling mitochondrial ATP production and another controlling mitochondrial thermogenesis, AAC can set the ratio between these two forms of mitochondrial energy output (Fig. 4).
Figure 4. ADP/ATP carrier (AAC) regulates the balance between mitochondrial ATP and heat production.
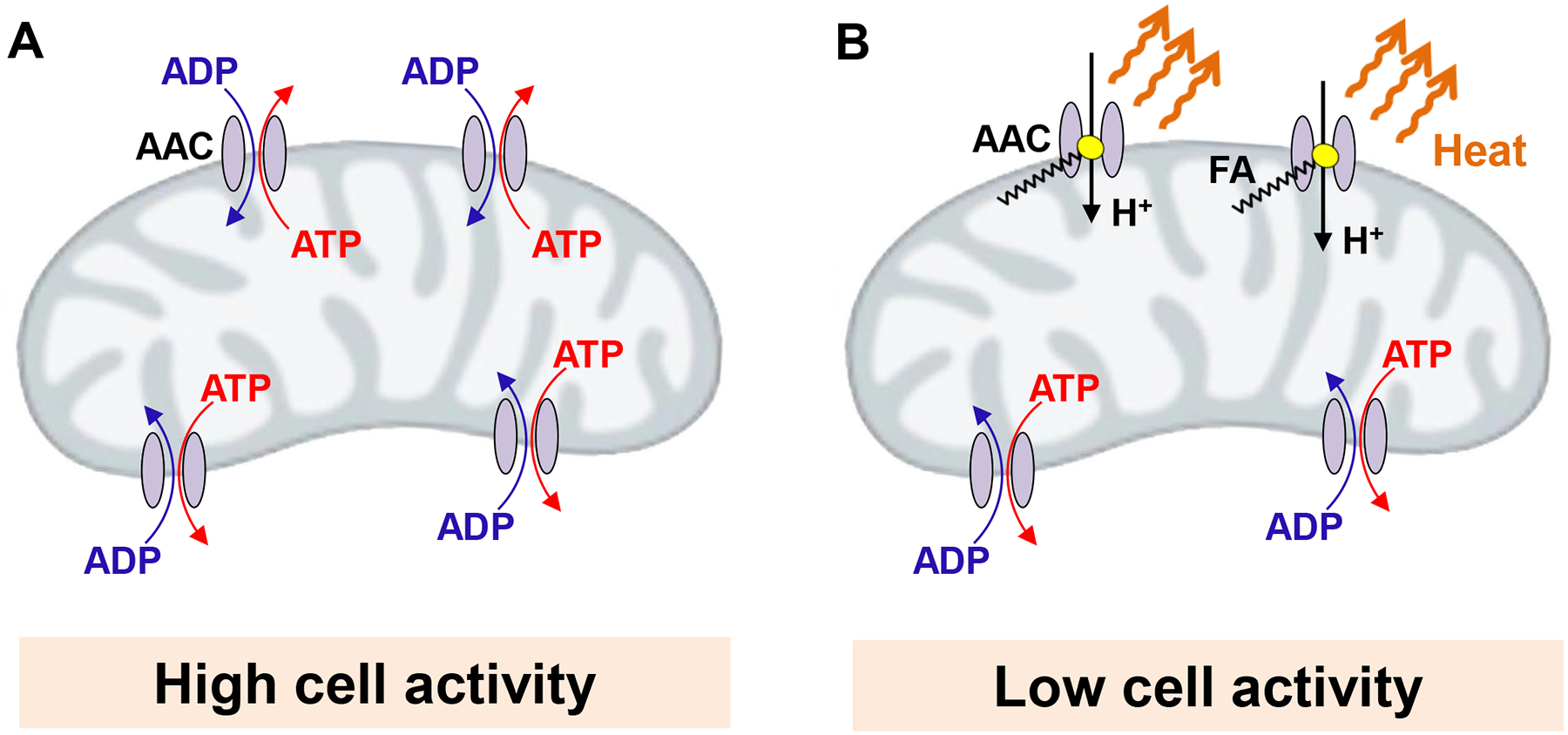
(A) During periods of high cell activity, such as in the working skeletal muscle, the requirement for ATP production is high, and the majority of AAC molecules are involved in ADP/ATP exchange. Under these conditions mitochondrial thermogenesis is low. (B) When cell activity is low, such as in the resting skeletal muscle, AAC molecules are less occupied by ADP/ATP exchange. Under these conditions, long-chain fatty acids (FA) can interact with more AAC molecules, generating H+ leak and activating thermogenesis.
Regulation of H+ leak by mitochondrial hyperpolarization and oxidative conditions
In addition to its role in thermogenesis, the mitochondrial H+ leak is an important regulator of mitochondrial production of reactive oxygen species (ROS). H+ leak reduces mitochondrial ROS production (19; 143) and has been implicated in preventing oxidative damage to mitochondria and cells, as well as in promoting longevity (144; 145).
Mitochondria are the primary source of ROS in the cell and contain several sites of ROS production, notably the ETC and particular enzymes for substrate catabolism in the mitochondrial matrix. and (146). ROS production in suspensions of isolated mitochondria is sharply increased when ΔΨ is hyperpolarized above a certain “threshold” (143). This is because high ΔΨ makes it more difficult for the ETC to pump H+, also slowing electron transport via the ETC. The slow electron transport leads to saturation of the electron carrier sites in the ETC and the upstream mitochondrial substrate catabolism pathways, causing the premature leak of the electrons to oxygen and excessive ROS production (146).
Mitochondria were proposed to possess a mechanism that prevents ROS generation caused by ΔΨ hyperpolarization. Specifically, the mitochondrial H+ leak was postulated to be “non-ohmic”; i.e., it increases sharply when ΔΨ becomes very negative (143). Such a sharp increase in H+ leak would depolarize ΔΨ, preventing excessive ROS production. This could be useful under conditions when a cell undergoes a fast transition from a high to low level of activity. As an example, in skeletal muscle transitioning from high activity to rest, ΔΨ could hyperpolarize because of the reduction in ATP consumption, leading to increased ROS production (143). However, the non-ohmic H+ leak prevents such ΔΨ hyperpolarization and associated ROS production. When the AAC-dependent H+ leak was recorded with patch-clamp electrophysiology, its sharp increase with ΔΨ hyperpolarization was confirmed (22). The H+ leak via UCP1 is also non-ohmic but to a lesser degree than that via AAC (23).
The early respiration studies also suggested that mitochondrial H+ leak is upregulated in the presence of superoxide, proposing that ROS potentiate mitochondrial H+ leak directly (147). This highlighted the second important negative feedback mechanism for the regulation of mitochondrial ROS production and indicated that UCPs should be potentiated by ROS. After UCP2 and UCP3 were identified, ROS activation of the mitochondrial H+ leak was first assigned to them (148). However, when the mitochondrial H+ leak was measured directly with the patch-clamp electrophysiology, it was determined that AAC, and not UCP2 or UCP3, is responsible for the potentiation of H+ leak under oxidative conditions (22). This oxidative potentiation of the AAC-dependent H+ leak is strictly dependent on prior activation by FA, which eliminates the possibility of any FA-independent, parallel mechanism of H+ leak activated by oxidation (22). Redox modifications of AAC cysteine residues have been suggested to regulate ADP/ATP exchange (31; 149). Similarly, potentiation of the FA-dependent H+ leak via AAC under oxidative conditions could be due to modification of cysteine residues, as was also recently reported for H+ leak mediated by UCP1 in brown fat (71; 150).
Acknowledgements:
This work was supported by NIH grant R35GM136415 and a grant from the UCSF Program for Breakthrough Biomedical Research (PBBR) to Y.K. A.M.B. was supported by an American Heart Association Career Development Award 19CDA34630062 and NIH grant 1R35GM143097.
ACRONYMS AND DEFINITIONS
- AAC
ADP/ATP carrier, also referred to as adenine nucleotide translocator (ANT)
- ADP
adenosine 5’-diphosphate
- ATP
adenosine 5’-triphosphate
- Beige fat
thermogenic brown-fat like adipocytes in white fat. Despite the morphological and functional similarities with brown fat they have different cellular origin
- Brown fat
specialized thermogenic tissue responsible for adaptive thermogenesis
- DNP
2,4-dinitrophenol, protonophore and mitochondrial uncoupler
- ETC
the electron transport chain
- FA
free fatty acids
- FCCP
cyanide-4-(trifluoromethoxy) phenylhydrazone, protonophore and mitochondrial uncoupler
- GDP
guanosine 5’- diphosphate
- H+ leak
passive H+ conductance of the inner mitochondrial membrane that uncouples H+ transport activities of the electron transport chain and ATP synthase
- IMM
the inner mitochondrial membrane
- PTP
permeability transition pore. A large-conductance pore activated in the inner mitochondrial membrane under pathological conditions to cause mitochondrial depolarization, release of some of the matrix contents and mitochondrial disfunction. It has also been proposed to operate in a partially activated flickering mode under certain physiological conditions
- SLC25
the superfamily of mitochondrial solute carriers
- Thermogenesis
heat production by the living organisms
- UCP
uncoupling protein. A protein of the inner mitochondrial membrane responsible for H+ leak and thermogenesis. This term does not necessarily mean UCP1-UCP4
- UCP1
uncoupling protein 1 responsible for H+ leak and thermogenesis in brown and beige fat
- UCP2
uncoupling protein 2. A protein of SLC25 family and a close homologue of UCP1
- UCP3
uncoupling protein 3. A protein of SLC25 family and a close homologue of UCP1
- Uncoupling
dissociation between the H+ transport activities of the electron transport chain and ATP synthase due to passive conductance of the inner mitochondrial membrane for certain ions
Footnotes
Disclosure: Y.K. is co-founder and advisor for Equator Therapeutics. A.M.B. declares no competing interests.
REFERENCES
- 1.Cannon B, Nedergaard J. 2004. Brown adipose tissue: function and physiological significance. Physiol Rev 84:277–359 [DOI] [PubMed] [Google Scholar]
- 2.Nicholls DG. 2017. The hunt for the molecular mechanism of brown fat thermogenesis. Biochimie 134:9–18 [DOI] [PubMed] [Google Scholar]
- 3.Jastroch M, Divakaruni AS, Mookerjee S, Treberg JR, Brand MD. 2010. Mitochondrial proton and electron leaks. Essays Biochem 47:53–67 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Bernardi P 1999. Mitochondrial transport of cations: channels, exchangers, and permeability transition. Physiol Rev 79:1127–55 [DOI] [PubMed] [Google Scholar]
- 5.Azzu V, Brand MD. 2010. The on-off switches of the mitochondrial uncoupling proteins. Trends Biochem Sci 35:298–307 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Aquila H, Link TA, Klingenberg M. 1985. The uncoupling protein from brown fat mitochondria is related to the mitochondrial ADP/ATP carrier. Analysis of sequence homologies and of folding of the protein in the membrane. EMBO J 4:2369–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Enerback S, Jacobsson A, Simpson EM, Guerra C, Yamashita H, et al. 1997. Mice lacking mitochondrial uncoupling protein are cold-sensitive but not obese. Nature 387:90–4 [DOI] [PubMed] [Google Scholar]
- 8.Bertholet AM, Kazak L, Chouchani ET, Bogaczynska MG, Paranjpe I, et al. 2017. Mitochondrial Patch Clamp of Beige Adipocytes Reveals UCP1-Positive and UCP1-Negative Cells Both Exhibiting Futile Creatine Cycling. Cell metabolism 25:811–22 e4 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Bouillaud F, Weissenbach J, Ricquier D. 1986. Complete cDNA-derived amino acid sequence of rat brown fat uncoupling protein. J Biol Chem 261:1487–90 [PubMed] [Google Scholar]
- 10.Shabalina IG, Petrovic N, de Jong JM, Kalinovich AV, Cannon B, Nedergaard J. 2013. UCP1 in brite/beige adipose tissue mitochondria is functionally thermogenic. Cell Rep 5:1196–203 [DOI] [PubMed] [Google Scholar]
- 11.McLaughlin SG, Dilger JP. 1980. Transport of protons across membranes by weak acids. Physiol Rev 60:825–63 [DOI] [PubMed] [Google Scholar]
- 12.Terada H 1990. Uncouplers of oxidative phosphorylation. Environ Health Perspect 87:213–8 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Harper JA, Dickinson K, Brand MD. 2001. Mitochondrial uncoupling as a target for drug development for the treatment of obesity. Obes Rev 2:255–65 [DOI] [PubMed] [Google Scholar]
- 14.Tainter ML, Cutting WC, Hines E. 1935. Effect Of Moderate Doses of dinitrophenol on the energy exchange and nitrogen metabolism of patients under conditions of restricted dietary. Journal of Pharmacology and Experimental Therapeutics 55:326–53 [Google Scholar]
- 15.Grundlingh J, Dargan PI, El-Zanfaly M, Wood DM. 2011. 2,4-dinitrophenol (DNP): a weight loss agent with significant acute toxicity and risk of death. J Med Toxicol 7:205–12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Perry RJ, Zhang D, Zhang XM, Boyer JL, Shulman GI. 2015. Controlled-release mitochondrial protonophore reverses diabetes and steatohepatitis in rats. Science 347:1253–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Szabo I, Leanza L, Gulbins E, Zoratti M. 2012. Physiology of potassium channels in the inner membrane of mitochondria. Pflugers Arch 463:231–46 [DOI] [PubMed] [Google Scholar]
- 18.Rasola A, Sciacovelli M, Pantic B, Bernardi P. 2010. Signal transduction to the permeability transition pore. FEBS letters 584:1989–96 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Divakaruni AS, Brand MD. 2011. The regulation and physiology of mitochondrial proton leak. Physiology (Bethesda) 26:192–205 [DOI] [PubMed] [Google Scholar]
- 20.Garlid KD, Jaburek M, Jezek P. 2001. Mechanism of uncoupling protein action. Biochemical Society transactions 29:803–6 [DOI] [PubMed] [Google Scholar]
- 21.Klingenberg M, Huang SG. 1999. Structure and function of the uncoupling protein from brown adipose tissue. Biochim Biophys Acta 1415:271–96 [DOI] [PubMed] [Google Scholar]
- 22.Bertholet AM, Chouchani ET, Kazak L, Angelin A, Fedorenko A, et al. 2019. H(+) transport is an integral function of the mitochondrial ADP/ATP carrier. Nature 571:515–20 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Fedorenko A, Lishko PV, Kirichok Y. 2012. Mechanism of fatty-acid-dependent UCP1 uncoupling in brown fat mitochondria. Cell 151:400–13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Bertholet AM, Kirichok Y. 2020. Patch-Clamp Analysis of the Mitochondrial H(+) Leak in Brown and Beige Fat. Front Physiol 11:326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ricquier D, Kader JC. 1976. Mitochondrial protein alteration in active brown fat: a soidum dodecyl sulfate-polyacrylamide gel electrophoretic study. Biochem Biophys Res Commun 73:577–83 [DOI] [PubMed] [Google Scholar]
- 26.Nicholls DG. 2001. A history of UCP1. Biochemical Society transactions 29:751–5 [DOI] [PubMed] [Google Scholar]
- 27.Heaton GM, Wagenvoord RJ, Kemp A Jr., Nicholls DG. 1978. Brown-adipose-tissue mitochondria: photoaffinity labelling of the regulatory site of energy dissipation. Eur J Biochem 82:515–21 [DOI] [PubMed] [Google Scholar]
- 28.Andreyev A, Bondareva TO, Dedukhova VI, Mokhova EN, Skulachev VP, Volkov NI. 1988. Carboxyatractylate inhibits the uncoupling effect of free fatty acids. FEBS letters 226:265–9 [DOI] [PubMed] [Google Scholar]
- 29.Andreyev A, Bondareva TO, Dedukhova VI, Mokhova EN, Skulachev VP, et al. 1989. The ATP/ADP-antiporter is involved in the uncoupling effect of fatty acids on mitochondria. Eur J Biochem 182:585–92 [DOI] [PubMed] [Google Scholar]
- 30.Skulachev VP. 1991. Fatty acid circuit as a physiological mechanism of uncoupling of oxidative phosphorylation. FEBS letters 294:158–62 [DOI] [PubMed] [Google Scholar]
- 31.Klingenberg M 2008. The ADP and ATP transport in mitochondria and its carrier. Biochim Biophys Acta 1778:1978–2021 [DOI] [PubMed] [Google Scholar]
- 32.Brustovetsky N, Klingenberg M. 1996. Mitochondrial ADP/ATP carrier can be reversibly converted into a large channel by Ca2+. Biochemistry 35:8483–8 [DOI] [PubMed] [Google Scholar]
- 33.Hunter DR, Haworth RA. 1979. The Ca2+-induced membrane transition in mitochondria. I. The protective mechanisms. Arch Biochem Biophys 195:453–9 [DOI] [PubMed] [Google Scholar]
- 34.Le Quoc K, Le Quoc D. 1988. Involvement of the ADP/ATP carrier in calcium-induced perturbations of the mitochondrial inner membrane permeability: importance of the orientation of the nucleotide binding site. Arch Biochem Biophys 265:249–57 [DOI] [PubMed] [Google Scholar]
- 35.Schonfeld P, Bohnensack R. 1997. Fatty acid-promoted mitochondrial permeability transition by membrane depolarization and binding to the ADP/ATP carrier. FEBS letters 420:167–70 [DOI] [PubMed] [Google Scholar]
- 36.Wieckowski MR, Wojtczak L. 1998. Fatty acid-induced uncoupling of oxidative phosphorylation is partly due to opening of the mitochondrial permeability transition pore. FEBS letters 423:339–42 [DOI] [PubMed] [Google Scholar]
- 37.Stepien G, Torroni A, Chung AB, Hodge JA, Wallace DC. 1992. Differential expression of adenine nucleotide translocator isoforms in mammalian tissues and during muscle cell differentiation. J Biol Chem 267:14592–7 [PubMed] [Google Scholar]
- 38.Rodic N, Oka M, Hamazaki T, Murawski MR, Jorgensen M, et al. 2005. DNA methylation is required for silencing of ant4, an adenine nucleotide translocase selectively expressed in mouse embryonic stem cells and germ cells. Stem Cells 23:1314–23 [DOI] [PubMed] [Google Scholar]
- 39.Levy SE, Chen YS, Graham BH, Wallace DC. 2000. Expression and sequence analysis of the mouse adenine nucleotide translocase 1 and 2 genes. Gene 254:57–66 [DOI] [PubMed] [Google Scholar]
- 40.Graham BH, Waymire KG, Cottrell B, Trounce IA, MacGregor GR, Wallace DC. 1997. A mouse model for mitochondrial myopathy and cardiomyopathy resulting from a deficiency in the heart/muscle isoform of the adenine nucleotide translocator. Nat Genet 16:226–34 [DOI] [PubMed] [Google Scholar]
- 41.Brand MD, Pakay JL, Ocloo A, Kokoszka J, Wallace DC, et al. 2005. The basal proton conductance of mitochondria depends on adenine nucleotide translocase content. Biochem J 392:353–62 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Morrow RM, Picard M, Derbeneva O, Leipzig J, McManus MJ, et al. 2017. Mitochondrial energy deficiency leads to hyperproliferation of skeletal muscle mitochondria and enhanced insulin sensitivity. Proc Natl Acad Sci U S A 114:2705–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Kokoszka JE, Waymire KG, Levy SE, Sligh JE, Cai J, et al. 2004. The ADP/ATP translocator is not essential for the mitochondrial permeability transition pore. Nature 427:461–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zackova M, Kramer R, Jezek P. 2000. Interaction of mitochondrial phosphate carrier with fatty acids and hydrophobic phosphate analogs. Int J Biochem Cell Biol 32:499–508 [DOI] [PubMed] [Google Scholar]
- 45.Engstova H, Zackova M, Ruzicka M, Meinhardt A, Hanus J, et al. 2001. Natural and azido fatty acids inhibit phosphate transport and activate fatty acid anion uniport mediated by the mitochondrial phosphate carrier. J Biol Chem 276:4683–91 [DOI] [PubMed] [Google Scholar]
- 46.Skulachev VP. 1998. Uncoupling: new approaches to an old problem of bioenergetics. Biochim Biophys Acta 1363:100–24 [DOI] [PubMed] [Google Scholar]
- 47.Wieckowski MR, Wojtczak L. 1997. Involvement of the dicarboxylate carrier in the protonophoric action of long-chain fatty acids in mitochondria. Biochem Biophys Res Commun 232:414–7 [DOI] [PubMed] [Google Scholar]
- 48.Wojtczak L, Wieckowski MR. 1999. The mechanisms of fatty acid-induced proton permeability of the inner mitochondrial membrane. Journal of bioenergetics and biomembranes 31:447–55 [DOI] [PubMed] [Google Scholar]
- 49.Halestrap AP, Richardson AP. 2015. The mitochondrial permeability transition: a current perspective on its identity and role in ischaemia/reperfusion injury. J Mol Cell Cardiol 78:129–41 [DOI] [PubMed] [Google Scholar]
- 50.Kwong JQ, Davis J, Baines CP, Sargent MA, Karch J, et al. 2014. Genetic deletion of the mitochondrial phosphate carrier desensitizes the mitochondrial permeability transition pore and causes cardiomyopathy. Cell Death Differ 21:1209–17 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Seifert EL, Gal A, Acoba MG, Li Q, Anderson-Pullinger L, et al. 2016. Natural and Induced Mitochondrial Phosphate Carrier Loss: DIFFERENTIAL DEPENDENCE OF MITOCHONDRIAL METABOLISM AND DYNAMICS AND CELL SURVIVAL ON THE EXTENT OF DEPLETION. J Biol Chem 291:26126–37 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gutierrez-Aguilar M, Douglas DL, Gibson AK, Domeier TL, Molkentin JD, Baines CP. 2014. Genetic manipulation of the cardiac mitochondrial phosphate carrier does not affect permeability transition. J Mol Cell Cardiol 72:316–25 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Sinasac DS, Moriyama M, Jalil MA, Begum L, Li MX, et al. 2004. Slc25a13-knockout mice harbor metabolic deficits but fail to display hallmarks of adult-onset type II citrullinemia. Mol Cell Biol 24:527–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Jalil MA, Begum L, Contreras L, Pardo B, Iijima M, et al. 2005. Reduced N-acetylaspartate levels in mice lacking aralar, a brain- and muscle-type mitochondrial aspartate-glutamate carrier. J Biol Chem 280:31333–9 [DOI] [PubMed] [Google Scholar]
- 55.Llorente-Folch I, Rueda CB, Amigo I, del Arco A, Saheki T, et al. 2013. Calcium-regulation of mitochondrial respiration maintains ATP homeostasis and requires ARALAR/AGC1-malate aspartate shuttle in intact cortical neurons. J Neurosci 33:13957–71, 71a [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Fleury C, Neverova M, Collins S, Raimbault S, Champigny O, et al. 1997. Uncoupling protein-2: a novel gene linked to obesity and hyperinsulinemia. Nat Genet 15:269–72 [DOI] [PubMed] [Google Scholar]
- 57.Gimeno RE, Dembski M, Weng X, Deng N, Shyjan AW, et al. 1997. Cloning and characterization of an uncoupling protein homolog: a potential molecular mediator of human thermogenesis. Diabetes 46:900–6 [DOI] [PubMed] [Google Scholar]
- 58.Boss O, Samec S, Paoloni-Giacobino A, Rossier C, Dulloo A, et al. 1997. Uncoupling protein-3: a new member of the mitochondrial carrier family with tissue-specific expression. FEBS letters 408:39–42 [DOI] [PubMed] [Google Scholar]
- 59.Gong DW, He Y, Karas M, Reitman M. 1997. Uncoupling protein-3 is a mediator of thermogenesis regulated by thyroid hormone, beta3-adrenergic agonists, and leptin. J Biol Chem 272:24129–32 [DOI] [PubMed] [Google Scholar]
- 60.Vidal-Puig A, Solanes G, Grujic D, Flier JS, Lowell BB. 1997. UCP3: an uncoupling protein homologue expressed preferentially and abundantly in skeletal muscle and brown adipose tissue. Biochem Biophys Res Commun 235:79–82 [DOI] [PubMed] [Google Scholar]
- 61.Jaburek M, Garlid KD. 2003. Reconstitution of recombinant uncoupling proteins: UCP1, −2, and −3 have similar affinities for ATP and are unaffected by coenzyme Q10. J Biol Chem 278:25825–31 [DOI] [PubMed] [Google Scholar]
- 62.Jaburek M, Varecha M, Gimeno RE, Dembski M, Jezek P, et al. 1999. Transport function and regulation of mitochondrial uncoupling proteins 2 and 3. J Biol Chem 274:26003–7 [DOI] [PubMed] [Google Scholar]
- 63.Nedergaard J, Cannon B. 2003. The ‘novel’ ‘uncoupling’ proteins UCP2 and UCP3: what do they really do? Pros and cons for suggested functions. Exp Physiol 88:65–84 [DOI] [PubMed] [Google Scholar]
- 64.Krauss S, Zhang CY, Lowell BB. 2005. The mitochondrial uncoupling-protein homologues. Nature reviews 6:248–61 [DOI] [PubMed] [Google Scholar]
- 65.Bouillaud F, Alves-Guerra MC, Ricquier D. 2016. UCPs, at the interface between bioenergetics and metabolism. Biochim Biophys Acta 1863:2443–56 [DOI] [PubMed] [Google Scholar]
- 66.Brand MD, Esteves TC. 2005. Physiological functions of the mitochondrial uncoupling proteins UCP2 and UCP3. Cell metabolism 2:85–93 [DOI] [PubMed] [Google Scholar]
- 67.Criscuolo F, Mozo J, Hurtaud C, Nubel T, Bouillaud F. 2006. UCP2, UCP3, avUCP, what do they do when proton transport is not stimulated? Possible relevance to pyruvate and glutamine metabolism. Biochim Biophys Acta 1757:1284–91 [DOI] [PubMed] [Google Scholar]
- 68.Bouillaud F 2009. UCP2, not a physiologically relevant uncoupler but a glucose sparing switch impacting ROS production and glucose sensing. Biochim Biophys Acta 1787:377–83 [DOI] [PubMed] [Google Scholar]
- 69.Vozza A, Parisi G, De Leonardis F, Lasorsa FM, Castegna A, et al. 2014. UCP2 transports C4 metabolites out of mitochondria, regulating glucose and glutamine oxidation. Proc Natl Acad Sci U S A 111:960–5 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Skulachev VP. 1999. Anion carriers in fatty acid-mediated physiological uncoupling. Journal of bioenergetics and biomembranes 31:431–45 [DOI] [PubMed] [Google Scholar]
- 71.Chouchani ET, Kazak L, Spiegelman BM. 2019. New Advances in Adaptive Thermogenesis: UCP1 and Beyond. Cell metabolism 29:27–37 [DOI] [PubMed] [Google Scholar]
- 72.Pfeifer A, Hoffmann LS. 2015. Brown, beige, and white: the new color code of fat and its pharmacological implications. Annu Rev Pharmacol Toxicol 55:207–27 [DOI] [PubMed] [Google Scholar]
- 73.Smith RE, Roberts JC, Hittelman KJ. 1966. Nonphosphorylating respiration of mitochondria from brown adipose tissue of rats. Science 154:653–4 [DOI] [PubMed] [Google Scholar]
- 74.Nicholls DG, Lindberg O. 1973. Brown-adipose-tissue mitochondria. The influence of albumin and nucleotides on passive ion permeabilities. Eur J Biochem 37:523–30 [DOI] [PubMed] [Google Scholar]
- 75.Nicholls DG, Rial E. 1999. A history of the first uncoupling protein, UCP1. Journal of bioenergetics and biomembranes 31:399–406 [DOI] [PubMed] [Google Scholar]
- 76.Klingenberg M 2017. UCP1 - A sophisticated energy valve. Biochimie 134:19–27 [DOI] [PubMed] [Google Scholar]
- 77.Bertholet AM, Kirichok Y. 2017. UCP1: A transporter for H(+) and fatty acid anions. Biochimie 134:28–34 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Decoursey TE. 2003. Voltage-gated proton channels and other proton transfer pathways. Physiol Rev 83:475–579 [DOI] [PubMed] [Google Scholar]
- 79.Kozak LP, Koza RA, Anunciado-Koza R. 2010. Brown fat thermogenesis and body weight regulation in mice: relevance to humans. Int J Obes (Lond) 34 Suppl 1:S23–7 [DOI] [PubMed] [Google Scholar]
- 80.Walden TB, Hansen IR, Timmons JA, Cannon B, Nedergaard J. 2012. Recruited vs. nonrecruited molecular signatures of brown, “brite,” and white adipose tissues. Am J Physiol Endocrinol Metab 302:E19–31 [DOI] [PubMed] [Google Scholar]
- 81.Wu J, Cohen P, Spiegelman BM. 2013. Adaptive thermogenesis in adipocytes: is beige the new brown? Genes Dev 27:234–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Wu J, Bostrom P, Sparks LM, Ye L, Choi JH, et al. 2012. Beige adipocytes are a distinct type of thermogenic fat cell in mouse and human. Cell 150:366–76 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kazak L, Chouchani ET, Jedrychowski MP, Erickson BK, Shinoda K, et al. 2015. A creatine-driven substrate cycle enhances energy expenditure and thermogenesis in beige fat. Cell 163:643–55 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Rolfe DF, Brand MD. 1996. Contribution of mitochondrial proton leak to skeletal muscle respiration and to standard metabolic rate. The American journal of physiology 271:C1380–9 [DOI] [PubMed] [Google Scholar]
- 85.Rolfe DF, Newman JM, Buckingham JA, Clark MG, Brand MD. 1999. Contribution of mitochondrial proton leak to respiration rate in working skeletal muscle and liver and to SMR. The American journal of physiology 276:C692–9 [DOI] [PubMed] [Google Scholar]
- 86.Brand MD, Chien LF, Ainscow EK, Rolfe DF, Porter RK. 1994. The causes and functions of mitochondrial proton leak. Biochim Biophys Acta 1187:132–9 [DOI] [PubMed] [Google Scholar]
- 87.Cho J, Seo J, Lim CH, Yang L, Shiratsuchi T, et al. 2015. Mitochondrial ATP transporter Ant2 depletion impairs erythropoiesis and B lymphopoiesis. Cell Death Differ 22:1437–50 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Schonfeld P 1990. Does the function of adenine nucleotide translocase in fatty acid uncoupling depend on the type of mitochondria? FEBS letters 264:246–8 [DOI] [PubMed] [Google Scholar]
- 89.Wojtczak L, Schonfeld P. 1993. Effect of fatty acids on energy coupling processes in mitochondria. Biochim Biophys Acta 1183:41–57 [DOI] [PubMed] [Google Scholar]
- 90.Garlid KD, Halestrap AP. 2012. The mitochondrial K(ATP) channel--fact or fiction? J Mol Cell Cardiol 52:578–83 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Samartsev VN, Smirnov AV, Zeldi IP, Markova OV, Mokhova EN, Skulachev VP. 1997. Involvement of aspartate/glutamate antiporter in fatty acid-induced uncoupling of liver mitochondria. Biochim Biophys Acta 1319:251–7 [DOI] [PubMed] [Google Scholar]
- 92.Pagliarini DJ, Calvo SE, Chang B, Sheth SA, Vafai SB, et al. 2008. A mitochondrial protein compendium elucidates complex I disease biology. Cell 134:112–23 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Da Cruz S, Xenarios I, Langridge J, Vilbois F, Parone PA, Martinou JC. 2003. Proteomic analysis of the mouse liver mitochondrial inner membrane. J Biol Chem 278:41566–71 [DOI] [PubMed] [Google Scholar]
- 94.Cunningham SA, Wiesinger H, Nicholls DG. 1986. Quantification of fatty acid activation of the uncoupling protein in brown adipocytes and mitochondria from the guinea-pig. Eur J Biochem 157:415–20 [DOI] [PubMed] [Google Scholar]
- 95.Parker N, Crichton PG, Vidal-Puig AJ, Brand MD. 2009. Uncoupling protein-1 (UCP1) contributes to the basal proton conductance of brown adipose tissue mitochondria. Journal of bioenergetics and biomembranes 41:335–42 [DOI] [PubMed] [Google Scholar]
- 96.Rauckhorst AJ, Broekemeier KM, Pfeiffer DR. 2014. Regulation of the Ca(2+)-independent phospholipase A2 in liver mitochondria by changes in the energetic state. J Lipid Res 55:826–36 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Seleznev K, Zhao C, Zhang XH, Song K, Ma ZA. 2006. Calcium-independent phospholipase A2 localizes in and protects mitochondria during apoptotic induction by staurosporine. J Biol Chem 281:22275–88 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gadd ME, Broekemeier KM, Crouser ED, Kumar J, Graff G, Pfeiffer DR. 2006. Mitochondrial iPLA2 activity modulates the release of cytochrome c from mitochondria and influences the permeability transition. J Biol Chem 281:6931–9 [DOI] [PubMed] [Google Scholar]
- 99.Szabo I, Zoratti M. 2014. Mitochondrial channels: ion fluxes and more. Physiol Rev 94:519–608 [DOI] [PubMed] [Google Scholar]
- 100.Hulsmann WC, Elliott WB, Slater EC. 1960. The nature and mechanism of action of uncoupling agents present in mitochrome preparations. Biochim Biophys Acta 39:267–76 [DOI] [PubMed] [Google Scholar]
- 101.Lardy HA, Pressman BC. 1956. Effect of surface active agents on the latent ATPase of mitochondria. Biochim Biophys Acta 21:458–66 [DOI] [PubMed] [Google Scholar]
- 102.Wojtczak L, Wojtczak AB. 1960. Uncoupling of oxidative phosphorylation and inhibition of ATP-Pi exchange by a substance from insect mitochondria. Biochim Biophys Acta 39:277–86 [DOI] [PubMed] [Google Scholar]
- 103.Lindberg O, de Pierre J, Rylander E, Afzelius BA. 1967. Studies of the mitochondrial energy-transfer system of brown adipose tissue. J Cell Biol 34:293–310 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Hittelman KJ, Lindberg O, Cannon B. 1969. Oxidative phosphorylation and compartmentation of fatty acid metabolism in brown fat mitochondria. Eur J Biochem 11:183–92 [DOI] [PubMed] [Google Scholar]
- 105.Rafael J, Ludolph HJ, Hohorst HJ. 1969. [Mitochondria from brown adipose tissue: uncoupling of respiratory chain phosphorylation by long fatty acids and recoupling by guanosine triphosphate]. Hoppe-Seyler’s Zeitschrift fur physiologische Chemie 350:1121–31 [PubMed] [Google Scholar]
- 106.Grav HI, Blix AS. 1979. A source of nonshivering thermogenesis in fur seal skeletal muscle. Science 204:87–9 [DOI] [PubMed] [Google Scholar]
- 107.Cannon B, Sundin U, Romert L. 1977. Palmitoyl coenzyme A: a possible physiological regulator of nucleotide binding to brown adipose tissue mitochondria. FEBS letters 74:43–6 [DOI] [PubMed] [Google Scholar]
- 108.Echtay KS, Winkler E, Klingenberg M. 2000. Coenzyme Q is an obligatory cofactor for uncoupling protein function. Nature 408:609–13 [DOI] [PubMed] [Google Scholar]
- 109.Echtay KS, Esteves TC, Pakay JL, Jekabsons MB, Lambert AJ, et al. 2003. A signalling role for 4-hydroxy-2-nonenal in regulation of mitochondrial uncoupling. EMBO J 22:4103–10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Nicholls DG, Locke RM. 1984. Thermogenic mechanisms in brown fat. Physiol Rev 64:1–64 [DOI] [PubMed] [Google Scholar]
- 111.Shabalina IG, Petrovic N, Kramarova TV, Hoeks J, Cannon B, Nedergaard J. 2006. UCP1 and defense against oxidative stress. 4-Hydroxy-2-nonenal effects on brown fat mitochondria are uncoupling protein 1-independent. J Biol Chem 281:13882–93 [DOI] [PubMed] [Google Scholar]
- 112.Locke RM, Rial E, Scott ID, Nicholls DG. 1982. Fatty acids as acute regulators of the proton conductance of hamster brown-fat mitochondria. Eur J Biochem 129:373–80 [DOI] [PubMed] [Google Scholar]
- 113.Kimura I, Ichimura A, Ohue-Kitano R, Igarashi M. 2020. Free Fatty Acid Receptors in Health and Disease. Physiol Rev 100:171–210 [DOI] [PubMed] [Google Scholar]
- 114.Houten SM, Violante S, Ventura FV, Wanders RJ. 2016. The Biochemistry and Physiology of Mitochondrial Fatty Acid beta-Oxidation and Its Genetic Disorders. Annu Rev Physiol 78:23–44 [DOI] [PubMed] [Google Scholar]
- 115.Scarpa A, Lindsay JG. 1972. Maintenance of energy-linked functions in rat-liver mitochondria aged in the presence of nupercaine. Eur J Biochem 27:401–7 [DOI] [PubMed] [Google Scholar]
- 116.Parce JW, Cunningham CC, Waite M. 1978. Mitochondrial phospholipase A2 activity and mitochondrial aging. Biochemistry 17:1634–9 [DOI] [PubMed] [Google Scholar]
- 117.Song H, Wohltmann M, Bao S, Ladenson JH, Semenkovich CF, Turk J. 2010. Mice deficient in group VIB phospholipase A2 (iPLA2gamma) exhibit relative resistance to obesity and metabolic abnormalities induced by a Western diet. Am J Physiol Endocrinol Metab 298:E1097–114 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Mancuso DJ, Sims HF, Yang K, Kiebish MA, Su X, et al. 2010. Genetic ablation of calcium-independent phospholipase A2gamma prevents obesity and insulin resistance during high fat feeding by mitochondrial uncoupling and increased adipocyte fatty acid oxidation. J Biol Chem 285:36495–510 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Levrat C, Louisot P. 1996. Increase of mitochondrial PLA2-released fatty acids is an early event in tumor necrosis factor alpha-treated WEHI-164 cells. Biochem Biophys Res Commun 221:531–8 [DOI] [PubMed] [Google Scholar]
- 120.Broekemeier KM, Iben JR, LeVan EG, Crouser ED, Pfeiffer DR. 2002. Pore formation and uncoupling initiate a Ca2+-independent degradation of mitochondrial phospholipids. Biochemistry 41:7771–80 [DOI] [PubMed] [Google Scholar]
- 121.Williams SD, Gottlieb RA. 2002. Inhibition of mitochondrial calcium-independent phospholipase A2 (iPLA2) attenuates mitochondrial phospholipid loss and is cardioprotective. Biochem J 362:23–32 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Brustovetsky T, Antonsson B, Jemmerson R, Dubinsky JM, Brustovetsky N. 2005. Activation of calcium-independent phospholipase A (iPLA) in brain mitochondria and release of apoptogenic factors by BAX and truncated BID. J Neurochem 94:980–94 [DOI] [PubMed] [Google Scholar]
- 123.Zhu D, Lai Y, Shelat PB, Hu C, Sun GY, Lee JC. 2006. Phospholipases A2 mediate amyloid-beta peptide-induced mitochondrial dysfunction. J Neurosci 26:11111–9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Kinsey GR, McHowat J, Patrick KS, Schnellmann RG. 2007. Role of Ca2+-independent phospholipase A2gamma in Ca2+-induced mitochondrial permeability transition. J Pharmacol Exp Ther 321:707–15 [DOI] [PubMed] [Google Scholar]
- 125.Jezek J, Jaburek M, Zelenka J, Jezek P. 2010. Mitochondrial phospholipase A2 activated by reactive oxygen species in heart mitochondria induces mild uncoupling. Physiol Res 59:737–47 [DOI] [PubMed] [Google Scholar]
- 126.Blum JL, Kinsey GR, Monian P, Sun B, Cummings BS, et al. 2011. Profiling of fatty acids released during calcium-induced mitochondrial permeability transition in isolated rabbit kidney cortex mitochondria. Toxicol In Vitro 25:1001–6 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Winkler E, Klingenberg M. 1994. Effect of fatty acids on H+ transport activity of the reconstituted uncoupling protein. J Biol Chem 269:2508–15 [PubMed] [Google Scholar]
- 128.Garlid KD, Orosz DE, Modriansky M, Vassanelli S, Jezek P. 1996. On the mechanism of fatty acid-induced proton transport by mitochondrial uncoupling protein. J Biol Chem 271:2615–20 [DOI] [PubMed] [Google Scholar]
- 129.Brustovetsky N, Klingenberg M. 1994. The reconstituted ADP/ATP carrier can mediate H+ transport by free fatty acids, which is further stimulated by mersalyl. J Biol Chem 269:27329–36 [PubMed] [Google Scholar]
- 130.Bieber LL, Pettersson B, Lindberg O. 1975. Studies on norepinephrine-induced efflux of free fatty acid from hamster brown-adipose-tissue cells. Eur J Biochem 58:375–81 [DOI] [PubMed] [Google Scholar]
- 131.Fasano M, Curry S, Terreno E, Galliano M, Fanali G, et al. 2005. The extraordinary ligand binding properties of human serum albumin. IUBMB Life 57:787–96 [DOI] [PubMed] [Google Scholar]
- 132.Ruprecht JJ, Kunji ERS. 2020. The SLC25 Mitochondrial Carrier Family: Structure and Mechanism. Trends Biochem Sci 45:244–58 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Robinson AJ, Overy C, Kunji ER. 2008. The mechanism of transport by mitochondrial carriers based on analysis of symmetry. Proc Natl Acad Sci U S A 105:17766–71 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Jezek P, Spacek T, Garlid K, Jaburek M. 2006. Undecanesulfonate does not allosterically activate H+ uniport mediated by uncoupling protein-1 in brown adipose tissue mitochondria. Int J Biochem Cell Biol 38:1965–74 [DOI] [PubMed] [Google Scholar]
- 135.Hamilton JA. 1998. Fatty acid transport: difficult or easy? J Lipid Res 39:467–81 [PubMed] [Google Scholar]
- 136.Ptak M, Egret-Charlier M, Sanson A, Bouloussa O. 1980. A NMR study of the ionization of fatty acids, fatty amines and N-acylamino acids incorporated in phosphatidylcholine vesicles. Biochim Biophys Acta 600:387–97 [DOI] [PubMed] [Google Scholar]
- 137.Guthrie JP. 1978. Hydrolysis of Esters of Oxy Acids - Pka Values for Strong Acids - Bronsted Relationship for Attack of Water at Methyl - Free-Energies of Hydrolysis of Esters of Oxy Acids - and a Linear Relationship between Free-Energy of Hydrolysis and Pka Holding over a Range of 20 Pk Units. Canadian Journal of Chemistry-Revue Canadienne De Chimie 56:2342–54 [Google Scholar]
- 138.Kunji ER, Aleksandrova A, King MS, Majd H, Ashton VL, et al. 2016. The transport mechanism of the mitochondrial ADP/ATP carrier. Biochim Biophys Acta 1863:2379–93 [DOI] [PubMed] [Google Scholar]
- 139.Huang SG. 2003. Binding of fatty acids to the uncoupling protein from brown adipose tissue mitochondria. Arch Biochem Biophys 412:142–6 [DOI] [PubMed] [Google Scholar]
- 140.Nicholls DG. 2006. The physiological regulation of uncoupling proteins. Biochim Biophys Acta 1757:459–66 [DOI] [PubMed] [Google Scholar]
- 141.Rial E, Poustie A, Nicholls DG. 1983. Brown-adipose-tissue mitochondria: the regulation of the 32000-Mr uncoupling protein by fatty acids and purine nucleotides. Eur J Biochem 137:197–203 [DOI] [PubMed] [Google Scholar]
- 142.Shabalina IG, Jacobsson A, Cannon B, Nedergaard J. 2004. Native UCP1 displays simple competitive kinetics between the regulators purine nucleotides and fatty acids. J Biol Chem 279:38236–48 [DOI] [PubMed] [Google Scholar]
- 143.Korshunov SS, Skulachev VP, Starkov AA. 1997. High protonic potential actuates a mechanism of production of reactive oxygen species in mitochondria. FEBS letters 416:15–8 [DOI] [PubMed] [Google Scholar]
- 144.Caldeira da Silva CC, Cerqueira FM, Barbosa LF, Medeiros MH, Kowaltowski AJ. 2008. Mild mitochondrial uncoupling in mice affects energy metabolism, redox balance and longevity. Aging Cell 7:552–60 [DOI] [PubMed] [Google Scholar]
- 145.Brand MD, Affourtit C, Esteves TC, Green K, Lambert AJ, et al. 2004. Mitochondrial superoxide: production, biological effects, and activation of uncoupling proteins. Free Radic Biol Med 37:755–67 [DOI] [PubMed] [Google Scholar]
- 146.Brand MD. 2016. Mitochondrial generation of superoxide and hydrogen peroxide as the source of mitochondrial redox signaling. Free Radic Biol Med 100:14–31 [DOI] [PubMed] [Google Scholar]
- 147.Liu SS. 1999. Cooperation of a “reactive oxygen cycle” with the Q cycle and the proton cycle in the respiratory chain--superoxide generating and cycling mechanisms in mitochondria. Journal of bioenergetics and biomembranes 31:367–76 [DOI] [PubMed] [Google Scholar]
- 148.Echtay KS, Roussel D, St-Pierre J, Jekabsons MB, Cadenas S, et al. 2002. Superoxide activates mitochondrial uncoupling proteins. Nature 415:96–9 [DOI] [PubMed] [Google Scholar]
- 149.Xiao H, Jedrychowski MP, Schweppe DK, Huttlin EL, Yu Q, et al. 2020. A Quantitative Tissue-Specific Landscape of Protein Redox Regulation during Aging. Cell 180:968–83 e24 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Chouchani ET, Kazak L, Jedrychowski MP, Lu GZ, Erickson BK, et al. 2016. Mitochondrial ROS regulate thermogenic energy expenditure and sulfenylation of UCP1. Nature 532:112–6 [DOI] [PMC free article] [PubMed] [Google Scholar]