

Summary
High-dimensional mass cytometry provides unparalleled insight into the cellular composition of the immune system. Here, we describe a mass-cytometry-based protocol to examine memory CD4+ T cell and memory B cell (MBC) responses in human peripheral blood. This approach allows for the identification of >50 distinct memory CD4+ T cell and MBC populations from a single clinical sample. This highly reproducible protocol has been successfully applied to multiple infectious disease settings to identify correlates of susceptibility or protection from infection.
For complete details on the use and execution of this protocol, please refer to Ioannidis et al. (2021).
Subject areas: Cell Biology, Flow Cytometry/Mass Cytometry, Health Sciences, Immunology, Systems biology
Graphical abstract
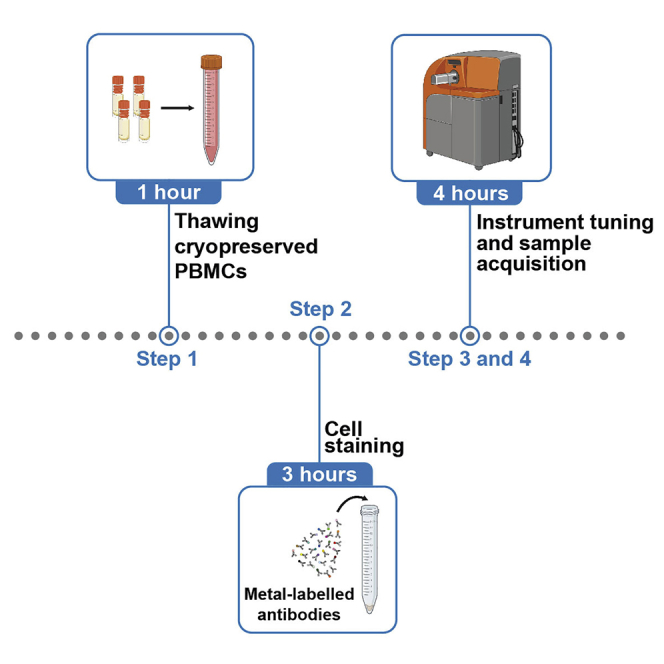
Highlights
-
•
CyTOF mass-cytometry-based protocol to analyze human peripheral blood mononuclear cells
-
•
Optimized protocol for clinical samples
-
•
Allows in-depth analysis of human memory CD4+ T cell and memory B cell responses
-
•
Applicable to multiple disease settings
High-dimensional mass cytometry provides unparalleled insight into the cellular composition of the immune system. Here, we describe a mass-cytometry-based protocol to examine memory CD4+ T cell and memory B cell (MBC) responses in human peripheral blood. This approach allows for the identification of >50 distinct memory CD4+ T cell and MBC populations from a single clinical sample. This highly reproducible protocol has been successfully applied to multiple infectious disease settings to identify correlates of susceptibility or protection from infection.
Before you begin
This protocol describes a CyTOF mass cytometry-based method to characterize memory CD4+ T cell and MBC populations among human peripheral blood mononuclear cells (PBMCs). Here, we provide a detailed description of the basic workflow of a CyTOF experiment from sample preparation to data acquisition, as well as an overview of the different strategies that can be used to analyze CyTOF data. One of the major strengths of our protocol is that it can be used to analyze clinical samples collected from field studies where cell number in samples is a limiting factor. Although our protocol recommends staining 2 × 106 PBMCs, we have obtained robust results with as few as 5 × 105 cells.
The example provided in this study used PBMCs from healthy donors. However, we have also used this protocol to examine memory CD4+ T cell and MBC responses to infectious diseases including malaria, dengue fever, Chagas disease and COVID-19. This protocol could also be adapted to examine other cell populations of interest in human peripheral blood, including CD8+ T cells, NK cells and monocytes by adding antibodies against additional surface markers (e.g., CD8, CD14, CD16 and CD56) to our CyTOF panel.
Obtain human PBMCs
The cryopreserved PBMCs used in this study were isolated from peripheral blood collected from healthy volunteers at the Australian Red Cross Blood Service or the Walter and Eliza Hall Institute of Medical Research Volunteer Blood Donor Registry. This study was approved by the Human Research Ethics Committee of the Walter and Eliza Hall Institute of Medical Research.
Preparation of PBMC media and staining buffer
Timing: 30 min
-
1.Prepare PBMC media.
-
a.Add heat-inactivated (56°C, 1 h) fetal bovine serum (HI-FBS), penicillin-streptomycin and L-glutamine to RPMI-1640.
-
b.Filter sterilize and store at 4°C.
-
a.
-
2.Prepare staining buffer.
-
a.Add bovine serum albumin (BSA) and sodium azide to Dulbecco’s phosphate buffered saline (DPBS).
-
b.Incubate for 20–30 min at room temperature (20°C–25°C) to allow bovine serum albumin to completely dissolve.
-
c.Filter sterilize and store at 4°C.
-
a.
CRITICAL: Many common laboratory detergents contain heavy metals such as Barium. To avoid heavy metal contamination, do not use any material (e.g., reusable glass beakers, pipettes, etc.) that has been in contact with detergent when preparing the staining buffer. We recommend running a small aliquot of staining buffer on a Helios mass cytometer before use to ensure that there is no heavy metal contamination.
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| 141Pr-conjugated anti-human CCR6 (clone 11A9) (1:50 dilution) | Fluidigm | Cat#3141014A |
| 143Nd-conjugated anti-human CD45RA (clone HI100) (1:50 dilution) | Fluidigm | Cat#3143006B |
| 146Nd-conjugated anti-human IgD (clone IA6-2) (1:50 dilution) | Fluidigm | Cat#3146005B |
| 147Sm-conjugated anti-human CD20 (clone 2H7) (1:50 dilution) | Fluidigm | Cat#3147001B |
| 151Eu-conjugated anti-ICOS (clone DX29) (1:50 dilution) | Fluidigm | Cat#3151020B |
| 152Sm-conjugated anti-human CD21 (clone BL13) (1:50 dilution) | Fluidigm | Cat#3152010B |
| 153Eu-conjugated anti-human CXCR5 (clone RF8B2) (1:50 dilution) | Fluidigm | Cat#3153020B |
| 156Gd-conjugated anti-human CXCR3 (clone G025H7) (1:50 dilution) | Fluidigm | Cat#3156004B |
| 158Gd-conjugated anti-human CD10 (clone HI10a) (1:50 dilution) | Fluidigm | Cat#3158011B |
| 165Ho-conjugated anti-human CD19 (clone HIB19) (1:50 dilution) | Fluidigm | Cat#3165025B |
| 167Er-conjugated anti-human CD27 (clone L128) (1:50 dilution) | Fluidigm | Cat#3167006B |
| 170Er-conjugated anti-human CD3 (clone UCHT1) (1:50 dilution) | Fluidigm | Cat#3170001B |
| 172Yb-conjugated anti-human IgM (clone MHM-88) (1:50 dilution) | Fluidigm | Cat#3172004B |
| 174Yb-conjugated anti-human CD4 (clone SK3) (1:50 dilution) | Fluidigm | Cat#3174004B |
| 175Lu-conjugated anti-human PD-1 (clone EH12.2H7) (1:50 dilution) | Fluidigm | Cat#3175008B |
| 176Yb-conjugated anti-human CD127 (clone A019D5) (1:50 dilution) | Fluidigm | Cat#3176004B |
| 161Dy-conjugated anti-T-bet (clone 4B10) (1:50 dilution) | Fluidigm | Cat#3161014B |
| Human TruStain FcX (1:10 dilution) | BioLegend | Cat#422302 |
| Biological samples | ||
| Human PBMCs from healthy male and female donors, 25–59 years of age. | The Australian Red Cross Blood Service or The Walter and Eliza Hall Institute of Medical Research Volunteer Blood Donor Registry | N/A |
| Chemicals, peptides, and recombinant proteins | ||
| RPMI-1640 | Life Technologies | Cat#11875101 |
| 200 mM L-glutamine | Gibco | Cat#25030081; CAS: 56-85-9 |
| Penicillin-streptomycin | Gibco | Cat#15140122 |
| Fetal bovine serum | Sigma-Aldrich | Cat#12007C; CAS: 1943609-65-1 |
| Dulbecco’s phosphate buffered saline (DPBS) | Gibco | Cat#14190144 |
| Bovine serum albumin | Sigma-Aldrich | Cat#A7906-100G; CAS: 9048-46-8 |
| Sodium azide | Sigma-Aldrich | Cat#S8032-25G; CAS: 26628-22-8 |
| Trypan blue | Sigma-Aldrich | Cat#T8154-100ML; CAS: 72-57-1 |
| Cell-ID Cisplatin - 5 mM | Fluidigm | Cat#201064 |
| Cell-ID Intercalator Ir - 125 μM | Fluidigm | Cat#201192A |
| Critical commercial assays | ||
| Maxpar nuclear antigen staining buffer set | Fluidigm | Cat#201063 |
| Maxpar fix and perm buffer | Fluidigm | Cat#201067 |
| Software and algorithms | ||
| CyTOF software V 7.0.8493 | Fluidigm | N/A |
| Premessa normalizer | Parker Institute for Cancer Immunotherapy | https://github.com/ParkerICI/premessa/ |
| tSNE algorithm | Cytobank | (Kotecha et al., 2010) |
| FlowSOM algorithm | Cytobank | (Kotecha et al., 2010) |
| FlowJo version 10 | BD | N/A |
| Other | ||
| 10 mL polypropylene tubes | Sarstedt | Cat#62.9924.284 |
| Cell strainers | Falcon | Cat#352340 |
| Hemacytometer | Merck | Cat#Z359629-1EA |
| 96-well u-bottom plates | Falcon | Cat#353077 |
| 1.7 mL microcentrifuge tubes | Axygen | Cat#72.690.001 |
| 500 mL 0.2 μm filter units | Nalgene | Cat#566-0020 |
| CyTOF wash solution | Fluidigm | Cat#201070 |
| CyTOF tuning solution | Fluidigm | Cat#201072 |
| Four element EQ beads | Fluidigm | Cat#201078 |
| Cell strainer tubes, 5 mL, 12 × 75 mm, 35 μm mesh size | Falcon | Cat#352235 |
| Round-Bottom Polystyrene tubes, 5 mL, 12 × 75 mm | Falcon | Cat#352058 |
| Helios Mass cytometer | Fluidigm | N/A |
Materials and equipment
PBMC media
| Reagent | Final concentration | Amount |
|---|---|---|
| RPMI-1640 | N/A | 440 mL |
| HI-FBS | 10% (v/v) | 50 mL |
| Penicillin-streptomycin | 100 U/mL penicillin and 100 μg/mL streptomycin | 5 mL |
| 200 mM L-glutamine | 2 mM | 5 mL |
| Total | N/A | 500 mL |
Store at 4°C for up to one month.
Cell staining buffer
| Reagent | Final concentration | Amount |
|---|---|---|
| DPBS | N/A | 500 mL |
| BSA | 0.5% (w/v) | 2.5 g |
| Sodium azide | 0.02% (w/v) | 100 mg |
| Total | N/A | 500 mL |
Store at 4°C for up to two months.
Alternatives: Cell staining buffer can be purchased from Fluidigm (Cat#201068).
Step-by-step method details
Thawing cryopreserved PBMCs
This section describes the procedure for thawing cryopreserved PBMCs to use for cell staining.
-
1.
Collect vials of PBMCs from liquid nitrogen and place on dry ice.
-
2.Thaw cells.
-
a.Working with two vials of PBMCs at a time, rapidly thaw the cells by placing the vials in a 37°C water bath.
-
b.Allow cells to thaw in the water bath until only a pea sized piece of ice remains (1–2 min).
-
c.Transfer the contents of each vial to a pre-labeled 10 mL tube on ice.
-
d.Add 10 drops of cold PBMC media to each tube using a transfer pipette and then gently flick each tube to mix.
-
e.Repeat step 2d until the tube has been filled to 5 mL.
-
f.Use 1 mL of media from the 10 mL tube to rinse the cryovial and collect any remaining cells.
-
g.Place the cap on each tube and gently invert to mix.
-
h.Add 20 drops of cold PBMC media to each tube and then gently flick each tube to mix.
-
i.Repeat step 2h until the tubes have been filled to 10 mL.
-
j.Centrifuge the cells at 350 g for 5 min.
-
k.Aspirate the supernatant.
-
l.Flick the tubes to disrupt the cell pellets.
-
m.Thoroughly resuspend cells in 10 mL of cold PBMC media.
-
n.Place tubes on ice until the remaining vials of PBMCs have been thawed.
-
o.Repeat steps 2a–2n to thaw the remaining vials.Note: If cell clumps are observed, pass cell suspension through a 40 μm cell strainer to remove clumps before counting.
-
a.
-
3.Count cells using a hemocytometer.
-
a.Add 10 μL of trypan blue to the wells of a u-bottom 96-well plate (prepare one well for each sample to be counted).
-
b.Add 10 μL of cells to each well and pipette to mix.
-
c.Load 10 μL of this mixture into a hemacytometer.
-
d.Count cells.
-
e.Calculate number of cells per sample.
-
a.
-
4.
Centrifuge the cells at 350 g for 5 min.
-
5.
Aspirate the supernatant.
-
6.
Flick the tubes to disrupt the cell pellets.
-
7.
Resuspend cells to 2 × 106 cells/mL with PBMC media.
-
8.
Transfer 1 mL (2 × 106 cells) of cells to a fresh 10 mL tube and place on ice until you are ready to start the staining protocol.
Note: The timing for this step is based on thawing four vials of PBMCs.
We routinely freeze aliquots of 107 PBMCs. The cells from each vial of 107 PBMCs was resuspended in 10 mL of PBMC media prior to counting. This may need to be adjusted to obtain an accurate cell count depending on the number of PBMCs you have per vial. If cells are resuspended in a smaller volume for counting, top the tube up to 10 mL with PBMC media after the cell count has been performed (step 3).
Alternatives: An automated cell counter can be used instead of a hemocytometer. This may reduce the amount of time required for this step.
Cell staining
This section describes the procedure for staining PBMCs with metal-labeled antibodies. Cells are first stained with cisplatin, which is a viability stain used for dead cell discrimination, followed by a cocktail of metal-labeled antibodies against surface makers of interest. After surface staining, cells are fixed and permeabilized and then stained with metal-labeled antibodies against intracellular antigens. The cells are then fixed in the presence of an iridium-labeled DNA intercalator, which allows for the discrimination of nucleated cells from cellular debris. This protocol has been adapted from the Fluidigm MaxPar Nuclear Antigen Staining Protocol with Fresh Fix (https://fluidigm.my.salesforce.com/sfc/p/#700000009Daw/a/4u0000019gvy/k1.CbKFY8JN7swG2ZVHybPpEZldzMpDAZnJYQvxyrrc).
-
9.Prepare Cell-ID Cisplatin, Fc receptor block and surface marker antibody cocktail.
-
a.Cell-ID Cisplatin: Dilute 5 mM Cell-ID Cisplatin 1:1,000 in DPBS to make a 5 μM working stock (prepare 200 μL/sample).
-
b.Fc receptor block: Dilute Human TruStain FcX 1:10 in cell staining buffer (prepare 50 μL/sample).
-
c.Surface marker antibody cocktail: Dilute each surface marker antibody in cell staining buffer as indicated in Table 1 (prepare 50 μL/sample).
-
d.Place on ice until needed.
-
a.
-
10.Cisplatin staining.
-
a.Add 2 mL of DPBS to each tube of cells containing 1 mL (2 × 106) of cells from step 8.
-
b.Centrifuge the cells at 350 g for 5 min.
-
c.Aspirate the supernatant.
-
d.Flick the tubes to disrupt the cell pellets.
-
e.Add 200 μL of the Cell-ID cisplatin prepared in step 9a to each tube.
-
f.Pipette to mix and incubate for 5 min at room temperature (20°C–25°C).Note: Cell-ID cisplatin staining must be performed in serum-free medium.CRITICAL: Cells should be stained with Cell-ID cisplatin for a maximum of 5 min. Staining for longer than 5 min can result in significant cell death.
-
g.Add 2 mL of cell staining buffer to each tube.
-
h.Centrifuge the cells at 350 g for 5 min.
-
i.Aspirate the supernatant.
-
j.Flick the tubes to disrupt the cell pellets.
-
a.
-
11.Surface marker staining.
-
a.Add 50 μL of Fc receptor block prepared in step 9b to each tube.
-
b.Pipette to mix and incubate for 10 min at room temperature (20°C–25°C) on an orbital mixer at 100 rpm.
-
c.Add 50 μL of antibody cocktail prepared in step 9c to each tube.
-
d.Pipette to mix and incubate for 30 min at room temperature (20°C–25°C) on an orbital mixer at 100 rpm.
-
e.Add 2 mL of cell staining buffer to each tube.
-
f.Centrifuge the cells at 350 g for 5 min.
-
g.Aspirate the supernatant.
-
h.Flick the tubes to disrupt the cell pellets.
-
i.Repeat steps 11e–11h.
-
a.
-
12.Intranuclear staining.
-
a.Vortex samples to thoroughly disrupt the cell pellets.
-
b.Add 1 mL of nuclear antigen staining buffer to each tube.
-
c.Vortex to mix and incubate for 30 min at room temperature (20°C–25°C).
-
d.Add 2 mL of nuclear antigen staining perm to each tube.
-
e.Centrifuge the cells at 800 g for 5 min.Note: Cells are centrifuged at a higher speed after fixation to reduce cell loss.
-
f.Aspirate the supernatant.
-
g.Flick the tubes to disrupt the cell pellets.
-
h.Repeat steps 12d–12g.
-
i.Vortex samples to thoroughly disrupt the cell pellets.
-
j.Add 50 μL of a 1:50 dilution 161Dy-conjugated anti-T-bet (clone 4B10) antibody to each tube.
-
k.Pipette to mix and incubate for 30 min at room temperature (20°C–25°C).
-
l.Add 2 mL of nuclear antigen staining perm to each tube.
-
m.Centrifuge the cells at 800 g for 5 min.
-
n.Aspirate the supernatant.
-
o.Flick the tubes to disrupt the cell pellets.
-
p.Repeat steps 12l–12o.
-
q.Vortex samples to thoroughly disrupt the cell pellets.
-
r.Place cells on ice for 10 min.
-
a.
-
13.Iridium intercalator staining.
-
a.While cells are incubating on ice, prepare the DNA intercalator.
-
i.Dilute 125 μM Cell-ID Intercalator-Ir 1:1,000 in Maxpar Fix and Perm Buffer to make a 125 nM working stock (prepare 1 mL/sample).
-
i.
-
b.Add 1 mL of intercalator to each tube.
-
c.Vortex to mix and incubate at 4°C overnight (16–20 h).
-
a.
Note: Adequate fixation of cell suspensions is critical for maintaining cellular integrity during subsequent washes in ultra-pure water prior to acquisition. Failure to appropriately fix cell suspensions leads to loss of cells and clogging of the sample introduction system with debris during sample acquisition.
Alternatives: Cell suspensions can also be fixed in a minimum of 4% fresh formaldehyde with iridium intercalator for at least 4 h, however overnight (16–20 h) fixation at 4°C is recommended.
Pause point: We usually run the samples on a Helios mass cytometer the next day, however samples may be stored for 2–4 days at 4°C without apparent detrimental effects.
Table 1.
Surface marker antibody cocktail
| Antibody | Dilution factor | Volume per test |
|---|---|---|
| 141Pr-conjugated anti-human CCR6 (clone 11A9) | 50 | 1 μL |
| 143Nd-conjugated anti-human CD45RA (clone HI100) | 50 | 1 μL |
| 146Nd-conjugated anti-human IgD (clone IA6-2) | 50 | 1 μL |
| 147Sm-conjugted anti-human CD20 (clone 2H7) | 50 | 1 μL |
| 151Eu-conjugated anti-ICOS (clone DX29) | 50 | 1 μL |
| 152Sm-conjugated anti-human CD21 (clone BL13) | 50 | 1 μL |
| 153Eu-conjugated anti-human CXCR5 (clone RF8B2) | 50 | 1 μL |
| 156Gd-conjugated anti-human CXCR3 (clone G025H7) | 50 | 1 μL |
| 158Gd-conjugated anti-human CD10 (clone HI10a) | 50 | 1 μL |
| 165Ho-conjugated anti-human CD19 (clone HIB19) | 50 | 1 μL |
| 167Er-conjugated anti-human CD27 (clone L128) | 50 | 1 μL |
| 170Er-conjugated anti-human CD3 (clone UCHT1) | 50 | 1 μL |
| 172Yb-conjugated anti-human IgM (clone MHM-88) | 50 | 1 μL |
| 174Yb-conjugated anti-human CD4 (clone SK3) | 50 | 1 μL |
| 175Lu-conjugated anti-human PD-1 (clone EH12.2H7) | 50 | 1 μL |
| 176Yb-conjugated anti-human CD127 (clone A019D5) | 50 | 1 μL |
Instrument tuning
This step involves setting up the mass cytometer, followed by bead quality control (QC). Final sample preparation steps can be performed during instrument setup so that these can be acquired immediately on completing.
-
14.Tuning and QC is performed based on the standard Helios™ setup procedure as described in detail in the Helios User Guide. Note that other CyTOF™ systems such as the CyTOF2™ and CyTOF XT™ can also be used after following their specific setup procedures. Full tuning is recommended at the beginning of each day (described here), and with a full day analysis an additional quick tuning protocol is recommended to be run mid-way to ensure system responsiveness.
-
a.Connect the nebulizer, and in the CyTOF software switch on the heater, nebulizer gas and sample flow running ultra-pure water. Droplet formation is monitored by spraying onto the underside of a cell culture dish lid.
-
b.Stop the sample instruction before mounting the nebulizer and when the heater temperature is >195°C, ignite the plasma. Leave instrument to thermally stabilize for a minimum of 20 min. A tube of tuning solution is introduced using the sample loader and the 159Tb dual count signal monitored in masses per reading mode until it has stabilized.
-
c.In tuning manager, launch the full automated tuning protocol. Signal from 159Tb should be >6 × 105 counts per pg.
-
d.Remove tuning solution and wash for 3–5 min with wash solution, followed by 3–5 min with ultra-pure water.
-
e.Vigorously shake four element EQ bead bottle for 30 s and load a tube with 0.5 mL of beads into sample loader. Monitor bead rate using a rain plot with a protocol with bead channels open until event rate has stabilized. Collect 120 s of data.
-
f.After EQ bead sample has finished acquisition, in the Process mode run clustering on the resulting EQ file to identify the main bead populations: 151Eu, 153Eu median signals should be >1,000 counts, and total bead numbers should be >6000.
-
g.Load a tube of ultra-pure water into the sample loader and run until samples are ready for acquisition.
-
h.At the end of the day, following sample acquisition (described below), run wash solution for 5 min and ultra-pure water for a further 5 min prior to shutting the system down, and removing the nebulizer. Refill the nebulizer with ultra-pure water to avoid clogging in dry state.
-
a.
Sample acquisition
This section outlines the final sample preparation steps as well as acquisition of data from samples. Cell suspensions have previously been fixed and stained in a solution with iridium intercalator and have been stored at 4°C. They are retrieved and washed in batches throughout the course of the sample procedure to minimize degradation effects due to being resuspended in ultra-pure water.
The timing for this step is based on acquiring four samples, with approximately 400,000 events acquired per sample. However, the time needed for acquisition of each sample depends on the cells available and the number of processed events needed for downstream analysis. A rule of thumb calculation is that approximately 0.75–1 × 106 cells can be introduced into the system per hour of instrument run time, and with a typical transport efficiency of approximately 55%–60%, this equates to around 0.4–0.6 × 106 events collected per hour. The number of “useable” events for downstream analysis then depends on any loss during pre-processing steps such as doublet discrimination and viability gating – see expected outcomes. Ultimately there is no hard and fast rule to the number of events to acquire per sample, and this should be informed by experimental design and analysis strategies (Marsh-Wakefield et al., 2021).
-
15.Wash cell suspensions.
-
a.An EQ bead suspension is made fresh daily by vigorously shaking the storage bottle for 30 s prior to aliquoting of 1 mL beads to 9 mL water. This step can be performed during the subsequent centrifugation steps.
-
b.Samples that have been stained are retrieved from storage at 4°C. Each sample is in fixative and iridium intercalator in a volume of 1.0 mL.
-
c.Transfer suspension to a round-bottomed 5 mL snap-cap centrifuge tube, add 4 mL ultra-pure water, replace tube cap and invert to mix.
-
d.Centrifuge at 1,000 g for 8 min at ambient temperature in swinging bucket centrifuge, then decant supernatant.Note: The supernatant must be treated as toxic waste as it contains formaldehyde. Use appropriate PPE and follow institutional guidelines for waste disposal.
-
e.Gently flick cell pellet to resuspend, then add 4 mL of ultra-pure water, replace tube cap and invert to mix.
-
f.Centrifuge at 1,000 g for 8 min at ambient temperature (20°C–25°C) in swinging bucket centrifuge, then decant supernatant, leaving cell in pellet form and stored on ice or at 4°C until acquisition.
-
g.Resuspend cells at approximately 0.75–1 × 106 cells per mL in a 1 in 10 dilution of EQ beads in ultra-pure water by pipetting, and filter through the mesh of a cell strainer tube.Note: Cell suspensions must be filtered immediately prior to acquisition to avoid clogging.
-
a.
-
16.Acquire sample on Helios.
-
a.Set up a template that includes, at a minimum, all required parameters for the experiment. A full mass range template is recommended to monitor for unexpected metal contamination that could degrade instrument performance. The template can be set up during sample washing to save time.
-
b.Load sample and acquire in CyTOF software. Default sample collection settings are generally appropriate. Collection of .imd format files in addition to .fcs files is recommended as, in the unlikely event of poor event detection, data can be reprocessed to .fcs files.
-
c.Monitor the run using a rain plot to ensure stable sample delivery, absence of clogging, and absence of metal contamination. With the High Throughput (HT) injector, an appropriate event rate is approximately 300–400 events per second, though lower event rates will reduce co-incidences and decrease eventual doublet events. If the event rate is higher than recommended, sample acquisition can be paused, and additional water/EQ mix can be added to adjust event rate.
-
d.Following collection of data from each sample, load a tube of ultra-pure water on the sample introduction system and monitor the event rate in Preview mode until the event rate is < 1 per second. Note that processing of data into .fcs format may continue for some minutes after stopping data acquisition, and a tube of water can be run during this period to shorten washing time.
-
e.Between every 3–4 samples run wash solution for 3 min and then ultra-pure water for 3–5 min to remove lanthanides adhering to the sample tubing. Monitor the event rate in Preview mode until the event rate is < 1 per second.
-
a.
Note: In this example, a HT injector was used with cells suspended in a suspension of EQ beads in ultra-pure water. Use of a Wide Bore (WB) injector with cells resuspended in cell acquisition solution (not described here) is an alternative. Considerations for choice between use HT and WB injectors are instrument and sample dependent (Lee et al., 2019; Thrash et al., 2020).
EQ beads are included to normalize any signal drift during the run and is done post acquisition – see expected outcomes. Cell suspension samples are prepared throughout the day to avoid storage in pellet form for longer than necessary. A typical approach is to prepare two samples of approximately 106 cells per sample at one time, overlapping preparation times so that when one set is finished running, the first sample of the next set is ready to acquire.
Expected outcomes
The .fcs data files produced at the end of this procedure can be analyzed using standard single-cell cytometry packages such as FlowJo and FCS express, as well as high-dimensional data analysis algorithms. Before performing exploratory analysis, data normalization must be performed to correct for intra- and inter-sample signal drift (Finck et al., 2013). This step can be performed in CyTOF software or using R packages such as Premessa (https://github.com/ParkerICI/premessa/). Manual gating is then performed to exclude EQ beads and identify viable single cells (Figure 1). After these pre-processing steps have been completed, exploratory analysis can be performed using traditional biaxial manual gating.
Figure 1.

Pre-processing of CyTOF data
PBMCs from four healthy donors were stained with a panel of metal-labeled antibodies and analyzed by CyTOF. The resulting data was then normalized using Premessa. Manual gating was then used to remove EQ beads (140Ce high events) and exclude cell debris (193Ir-DNA2- cells) and dead cells (195Pt Cisplatin high cells) before exploratory data analysis was performed.
An example of a manual gating strategy to identify MBC subsets is shown in Figure 2A. This gating strategy allows for the identification of CD21-CD27- atypical MBCs, CD21-CD27+ activated MBCs and CD21+CD27+ classical MBCs. These subsets can be further divided into un-switched (IgM+IgD- and IgM+IgD+) and class-switched (IgM-IgD-) cells based on their surface expression of IgM and IgD. Although manual gating can be useful, it can be labor-intensive, relies on prior knowledge of the cell population of interest and may ignore novel and unexpected cell populations. To overcome these limitations, mass cytometry data can also be analyzed using unbiased high-dimensional data analysis algorithms such as tSNE and FlowSOM. This approach may reveal previously unappreciated heterogeneity within defined cell populations and uncover novel cell populations. Using this approach, we were able to identify 5 distinct MBC sub-populations, including un-switched and switched cells expressing variable levels of the chemokine receptors CXCR3, CXCR5 and CCR6 within the classical, atypical and activated MBCs subsets (Figures 2B–2D). The parameters and settings used for the tSNE analysis shown in Figure 2 are listed in Table 2. A similar strategy can also be used to examine the composition of the memory CD4+ T cell pool. Within the memory CD4+ T cell pool, expression of the chemokine receptors CCR6 and CXCR3 can be used to distinguish TH1 CD4+ T cells (CXCR3+CCR6-), TH2 CD4+ T cells (CXCR3-CCR6-) and TH17 CD4+ T cells (CXCR3- CCR6+), while CXCR5 can be used to identify circulating memory T follicular helper (TFH) cells (Figure 2E). Further analysis of these subsets using tSNE analysis and FlowSOM clustering, revealed a high degree of heterogeneity within these subsets and allowed for the identification of > 20 distinct memory CD4+ T cell sub-populations (Figures 2F–2I).
Figure 2.

CyTOF data analysis
(A) PBMCs from four healthy donors were stained with a panel of metal-labeled antibodies and analyzed by CyTOF. After data pre-processing, manual gating was used to identify classical MBCs (CD3-CD19+CD20+CD10-CD27+CD21+), atypical MBCs (CD3-CD19+CD20+CD10-CD27-CD21-) and activated MBCs (CD3-CD19+CD20+CD10-CD27+CD21-). tSNE analysis was then performed in Cytobank (Kotecha et al., 2010) before self-organizing maps (SOMs) were generated using hierarchical consensus clustering on the tSNE axes.
(B) tSNE and FlowSOM analysis of classical MBCs.
(C) tSNE and FlowSOM analysis of atypical MBCs.
(D) tSNE and FlowSOM analysis of activated MBCs.
(E) A similar approach was used to analyze the memory CD4+ T cell compartment. Manual gating was used to identify TH1 memory CD4+ T cells (CD19-CD3+CD4+CD45RA-CCR6-CXCR3+), TH2 memory CD4+ T cells (CD19-CD3+CD4+CD45RA-CCR6-CXCR3-), TH17 memory CD4+ T cells (CD19-CD3+CD4+CD45RA-CCR6+CXCR3-) and circulating memory TFH cells (CD19-CD3+CD4+CD45RA-CXCR5+) before tSNE analysis and FlowSOM clustering was performed for each memory CD4+ T cell sub-set.
(F) tSNE and FlowSOM analysis of TH1 memory CD4+ T cells.
(G) tSNE and FlowSOM analysis of TH2 memory CD4+ T cells.
(H) tSNE and FlowSOM analysis of TH17 memory CD4+ T cells.
(I) tSNE and FlowSOM analysis of circulating memory TFH cells. The tSNE plots in the upper left panel display cell density and represent the pooled data from four healthy donors, while the upper right panel shows a projection of the FlowSOM clusters on the tSNE plot. Heatmaps shows the mean marker expression for each FlowSOM cluster.
Table 2.
Parameters and setting used for tSNE analysis
| Cell population | Events per sample | Parameters | Settings |
|---|---|---|---|
| Classical, atypical and activated MBCs | 750 | IgM, IgD, CXCR5, CCR6, CXCR3, CD45RA and T-bet | iterations=1000, perplexity=30, theta= 0.5 |
| TH1 memory CD4+ T cells | 2,000 | CXCR5, ICOS, PD-1, CD27, CD25, CD127 and T-bet | iterations=1000, perplexity=30, theta= 0.5 |
| TH2, and TH17 memory CD4+ T cells | 2,000 | CXCR5, ICOS, PD-1, CD27, CD25 and CD127 | iterations=1000, perplexity=30, theta= 0.5 |
| Circulating memory TFH cells | 1,800 | CXCR3, CCR6, ICOS, PD-1, CD27, CD25, CD127 and T-bet | iterations=1000, perplexity=30, theta= 0.5 |
Quantification and statistical analysis
Manual gating or clustering algorithms such as FlowSOM can be used to determine the frequency of individual cell populations. Differentially abundant cell populations can then be identified using univariate tests (e.g., t-test, Mann-Whitney test, one-way ANOVA), while the association between cell frequencies and different outcomes can be explored using linear or logistic regression.
Limitations
This protocol uses commercially available metal-labeled antibodies purchased from Fluidigm. Although Fluidim has more than 800 metal-labeled antibodies against at least 400 human antigens, specific clones or antibodies against less common antigens may not be available. If a pre-conjugated antibody is not available, custom conjugations can be performed. A detailed protocol for custom conjugations is described elsewhere (Thrash et al., 2020).
The protocol here describes the acquisition of non-barcoded samples – however, sample barcoding is generally recommended as it has many advantages, including higher throughput (due to decreased between-sample washing), improved doublet discrimination during post-acquisition debarcoding, and decreased between-sample signal variation due to control of instrument signal drift. Barcoding approaches have been outlined elsewhere (Marsh-Wakefield et al., 2021; Rybakowska et al., 2020).
Troubleshooting
Problem 1
This protocol requires 2 × 106 PBMCs for each stain. However, this may not be possible when working with clinical samples (step 3).
Potential solution
We have found that approximately 70% of cells are lost during this staining protocol (Figure 3). Most of this cell loss occurs during the intranuclear staining step. If fewer cells are available, we recommend performing only surface staining to ensure that enough cells can be acquired for downstream analysis. Alternatively, other approaches may reduce cell loss during the staining procedure including, barcoding and pooling cells for staining, increasing the centrifugation time to 8 min, and optimizing the fixation and permeabilization conditions (e.g., tyring different fixation and permeabilization kits, testing different concentrations of PFA and saponin, increasing the length of incubations).
Figure 3.

Cell loss during cell staining
PBMCs from four healthy donors were stained with a panel of metal-labeled antibodies and analyzed by CyTOF. After each staining step, cells were counted using a hemacytometer and the percentage of remaining cells (% of starting number) was calculated. Data are represented as mean ± SD.
Problem 2
Low antibody signal intensity cell (steps 11 and 12).
Potential solution
We typically use pre-labeled commercial antibodies at a 1:50 dilution (1 μL per test). However, if low signal intensity is observed, antibody titrations should be performed to determine the optimal dilution factor for each antibody.
Problem 3
Significant cell loss and clogging of the sample introduction system during acquisition (step 16).
Potential solution
We have found that inadequate fixation is one of the most common causes of significant cell loss and clogging of the sample introduction system during acquisition. A Helios mass cytometer has a transmission efficiency of approximately 55%–60%. If >50% cell loss observed or repeated clogging of the sample introduction system is experienced, prepare fresh fixative and ensure that samples are fixed for at least 4 h before acquisition.
Problem 4
Cisplatin staining reveals higher or lower than expected cell viability (step 10).
Potential solution
Cisplatin preferentially enters non-viable cells with compromised cell membranes and covalently binds to intracellular proteins. However, it is important to note that Cisplatin also binds to serum proteins in BSA and FBS. If higher than expected cell viability is observed, ensure that Cisplatin staining is performed in PBS and that cells are washed with PBS prior staining. Samples can also be spiked with dead cells prior to staining to confirm that that the Cisplatin labelling is working.
Incubating cells with Cisplatin for more than five minutes can result in significant cell death. If lower than expected cell viability is observed, ensure that cells are incubated with Cisplatin for no more than five minutes and then immediately washed with cell staining buffer to quench the Cisplatin. We also recommend dividing the Cisplatin stock solution into single-use aliquots, as using Cisplatin that has been freeze-thawed multiple times can result in increased non-specific binding.
Problem 5
Iodine contamination of samples (step 16).
Potential solution
Significant iodine contamination of samples can damage the 127I detector of the Helios. Ficoll, which is commonly used for PBMC enrichment, is known to contain high levels of iodine. It is therefore important to minimise the amount of Ficoll collected with PBMCs at the interface during PBMC enrichment. Additional washing of the collected cells with PBS may also help to reduce Ficoll contamination.
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Diana Hansen (hansen@wehi.edu.au).
Materials availability
This study did not generate new unique reagents.
Acknowledgments
In memory of our dear friend and colleague, Dr. Andrew Mitchell, who helped us find the invisible. This work was performed in part at the Materials Characterisation and Fabrication Platform (MCPF) at the University of Melbourne and the Victorian Node of the Australian National Fabrication Facility (ANFF). Supported by the Australian Government National Health and Medical Research Council (NHMRC) Independent Research Institutes Infrastructure Support Scheme and Project Grants 1028665 (D.S.H.) and 1137989 (D.S.H.), the Australian Academy of Science (D.S.H.), and the Victorian State Government Operational Infrastructure Support. The graphical abstract was created with BioRender.com.
Author contributions
L.J.I., A.J.M., and T.Z. developed the protocols. L.J.I., A.J.M., T.Z., and D.S.H. wrote the manuscript.
Declaration of interests
The authors declare no competing interests.
Contributor Information
Lisa J. Ioannidis, Email: ioannidis@wehi.edu.au.
Diana S. Hansen, Email: hansen@wehi.edu.au.
Data and code availability
This study did not generate any new code.
References
- Finck R., Simonds E.F., Jager A., Krishnaswamy S., Sachs K., Fantl W., Pe'er D., Nolan G.P., Bendall S.C. Normalization of mass cytometry data with bead standards. Cytometry A. 2013;83:483–494. doi: 10.1002/cyto.a.22271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ioannidis L.J., Pietrzak H.M., Ly A., Utami R.A., Eriksson E.M., Studniberg S.I., Abeysekera W., Li-Wai-Suen C.S., Sheerin D., Healer J., et al. High-dimensional mass cytometry identifies T cell and B cell signatures predicting reduced risk of Plasmodium vivax malaria. JCI Insight. 2021;6:e48086. doi: 10.1172/jci.insight.148086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kotecha N., Krutzik P.O., Irish J.M. Web-based analysis and publication of flow cytometry experiments. Curr. Protoc. Cytom. 2010;Chapter 10 doi: 10.1002/0471142956.cy1017s53. Unit10.17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee B.H., Kelly G., Bradford S., Davila M., Guo X.V., Amir E.-A.D., Thrash E.M., Solga M.D., Lannigan J., Sellers B., et al. A modified injector and sample acquisition protocol can improve data quality and reduce inter-instrument variability of the Helios mass cytometer. Cytometry A. 2019;95:1019–1030. doi: 10.1002/cyto.a.23866. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marsh-Wakefield F.M., Mitchell A.J., Norton S.E., Ashhurst T.M., Leman J.K., Roberts J.M., Harte J.E., McGuire H.M., Kemp R.A. Making the most of high-dimensional cytometry data. Immunol. Cell Biol. 2021;99:680–696. doi: 10.1111/imcb.12456. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rybakowska P., Alarcón-Riquelme M.E., Marañón C. Key steps and methods in the experimental design and data analysis of highly multi-parametric flow and mass cytometry. Comput. Struct. Biotechnol. J. 2020;18:874–886. doi: 10.1016/j.csbj.2020.03.024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Thrash E.M., Kleinsteuber K., Hathaway E.S., Nazzaro M., Haas E., Hodi F.S., Severgnini M. High-throughput mass cytometry staining for immunophenotyping clinical samples. STAR Protoc. 2020;1:100055. doi: 10.1016/j.xpro.2020.100055. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
This study did not generate any new code.