

Abstract
Introduction
Drawing on the amyloid/tau/neurodegeneration (AT[N]) model, the study examined whether the tau positron emission tomography (PET) biomarker [18F]AV‐1451 was associated with episodic memory problems beyond what was predicted by the amyloid beta (Aβ) PET in Down syndrome (DS).
Methods
Data from 123 non‐demented adults with DS (M = 47 years, standard deviation = 6.34) were analyzed. The Cued Recall Test assessed episodic memory. Tau PET standardized update value ratio (SUVR) was assessed across Braak regions as continuous and binary (high tau [TH] vs. low tau [TL]) variable. Global PET Aβ SUVR was assessed as binary variable (Aβ– vs. Aβ+).
Results
In models adjusting for controls, tau SUVR was negatively associated with episodic memory performance in the Aβ+ but not Aβ– group. The Aβ+/TH group evidenced significantly worse episodic memory than the Aβ+/TL group.
Discussion
Similar to late‐onset and autosomal dominant Alzheimer's disease (AD), high tau was an indicator of early prodromal AD in DS.
Keywords: Alzheimer's disease, amyloid, Down syndrome, memory, positron emission tomography, tau
1. BACKGROUND
There is a critical need to identify biomarkers of Alzheimer's disease (AD) pathogenesis that are associated with early cognitive decline in the Down syndrome (DS) population. Due to trisomy 21, virtually all adults with DS exhibit AD pathology by age 40 years, 1 , 2 with 70% to 90% developing clinical dementia by their 60s or 70s. 3 , 4 , 5 The extra copy of chromosome 21, which includes the amyloid precursor protein gene, leads to an overproduction of amyloid beta (Aβ), which is an early hallmark feature of AD. 6 A growing number of imaging studies have examined Aβ accumulation using positron emission tomography (PET) biomarkers and its relation to early cognitive decline in the DS population. 3 , 7 , 8 , 9 Building on these efforts, there is now a need to examine imaging biomarkers of other aspects of AD pathogenesis, including pathology that may signal further progression toward the onset of clinical dementia in the DS population. The goal of the current study was to determine whether an imaging biomarker for tau PET was associated with cognitive impairment beyond the presence of Aβ accumulation in non‐demented adults with DS. Information on sensitivity of tau PET for capturing early cognitive decline prior to the onset of dementia in the DS population can inform therapeutic targets, guide the selection of clinical trial participants, and aid in the monitoring of intervention efficacy.
The AT(N) framework has been proposed as a biomarker‐descriptive classification scheme for AD pathology that is used in non‐DS populations. 6 In this framework, “A” refers to biomarkers of Aβ (including PET Aβ or cerebrospinal fluid [CSF] Aβ42), “T” refers to biomarkers of tau (tau PET or CSF phosphorylated tau), and “(N)” refers to biomarkers of neurodegeneration or neuronal injury ([18F] fluorodeoxyglucose [FDG]–PET, structural magnetic resonance imaging [MRI], or CSF total tau). In the AT(N) framework, A and T biomarkers have been analyzed as both continuous variables (e.g., standardized uptake value ratio [SUVR] of PET Aβ) and in a binary fashion as high versus low or positive (+) versus negative (–), resulting in multiple classifications of AD pathology (e.g., A–/T–, A+/T–, A+/T+), with A–/T+ posited to indicate non‐AD pathology. The AT(N) framework is independent of clinical diagnostic classifications (e.g., dementia status) and performance on cognitive measures. Thus, a focus of research on non‐DS populations has been to examine whether the AT(N) scheme maps onto the emergence of early cognitive declines in AD. 10 , 11 , 12
In the DS population, cross‐sectional and longitudinal imaging studies have found associations between PET‐measured Aβ, assessed as both a continuous (SUVR) and binary variable (Aβ+ vs. Aβ–), and decline in cognitive functioning prior to dementia. 7 , 13 , 14 These associations were often strongest in measures of episodic memory such as list learning tasks, 13 , 15 suggesting that this domain of cognitive functioning may be affected earliest in the progression to the prodromal stage of AD, as has been found in non‐DS populations. 16 , 17 Measures of episodic memory have also been found to distinguish adults with DS who were clinically deemed to have mild cognitive impairment (MCI) or dementia from those who are cognitively stable. 18 , 19 , 20 , 21 , 22
RESEARCH IN CONTEXT
Systematic review: A review of published literature and meeting abstracts and presentations on biomarkers of tau in Alzheimer's disease (AD) was conducted. Drawing on the amyloid/tau/neurodegeneration (AT[N]) framework, imaging biomarkers of positron emission tomography (PET) tau are a meaningful biomarker for early AD pathology in late‐onset AD and autosomal dominant AD. It is not clear if tau PET is similarly a meaningful biomarker of the early prodromal stage in the Down syndrome (DS) type of AD.
Interpretation: Findings indicate that the AT(N) framework has relevance in DS. Specifically, tau PET was found to be a more sensitive biomarker than amyloid beta (Aβ) PET alone for predicting early problems in episodic memory in adults with DS.
Future directions: The article proposes that the AT(N) framework is relevant to the DS type of ADand proposes using the AT(N) framework to classify AD pathology in DS. The field should consider using biomarkers of PET tau, in addition to Aβ in selection of participants for AD clinical trials in DS, and to monitor disease progression.
A growing number of DS studies have examined biomarkers of tau using plasma or CSF. Within cross‐sectional studies, plasma concentrations of total tau had poor diagnostic performance in differentiating non‐demented adults with DS from those with early cognitive declines and those with dementia. 3 In contrast, CSF concentrations of total tau and phosphorylated‐tau‐181 (p‐tau181) strongly differentiated demented adults, those with early cognitive declines, and those who were non‐demented. 3 To date, scant research has examined the utility of PET imaging biomarkers of tau in DS. In a sample of 12 non‐demented adults with DS, PET tau measured via 18F‐AV‐1451 was evident in Braak regions II, V, and VI in three non‐demented adults with DS and tau PET was positively correlated with age and Aβ PET and negatively associated with cognitive performance. 23 In a sample of 92 adults with DS, tau PET measured with 18F‐AV‐1451 was also associated with trajectories of decline in episodic memory and motor control and planning and an increase in dementia symptoms. 24 It is not yet clear, however, if tau PET biomarkers contribute understanding about the transition to the prodromal stage of AD beyond Aβ PET biomarkers in the DS population. If so, then the AT(N) model may offer DS researchers a system for the selection of non‐demented adults with DS into clinical trials who are at different phases in the trajectory toward AD. Moreover, the model would offer a way to track AD progression independent of clinical status and cognitive performance.
The current study drew on the AT(N) framework to examine whether the tau PET biomarker [18F]AV‐1451 as assessed as both a continuous and binary (high tau [TH] vs. low tau [TL]) measure was associated with episodic memory problems beyond those predicted by the Aβ PET biomarker [11C]PiB status (Aβ– vs. Aβ+) in DS. In other words, the study sought to determine whether non‐demented adults with DS classified as Aβ+/TH exhibited worse episodic memory than those who were Aβ+/TL. Tau PET was evaluated as a continuous measure using the SUVR assessed across the six Braak stages, corresponding to different topographical regions. For the binary measure, our focus was on high versus low tau PET SUVR using a two standard deviation (SD) approach given the lack of established thresholds for tau positivity in DS. Global Aβ PET SUVR was assessed across the neocortex and in the striatum, due to the striatal‐first pattern of accumulation in DS 25 and negative (Aβ–) versus positive (Aβ+) status based on global Aβ were determined using previously developed thresholds. 26 Episodic memory was assessed through the Cued Recall Test, a list learning task. Tau PET SUVR was predicted to be negatively associated with episodic memory in non‐demented adults with DS classified as Aβ+ but not in those classified as Aβ–. Moreover, non‐demented adults with DS classified as Aβ+/TH were predicted to exhibit worse episodic memory than those who were Aβ–/TL or Aβ+/TL.
2. METHOD
2.1. Participants
Data from 123 adults with DS who were part of the Alzheimer's Biomarker Consortium–Down Syndrome (ABC‐DS) were included. Participants came from the University of Wisconsin–Madison (N = 48), University of Pittsburgh (N = 47), University of Cambridge (N = 18), Barrow Neurological Institute (N = 6), and Washington University–St. Louis (N = 4) sites of ABC‐DS. Inclusion criteria included being 25 years or older, to capture early AD pathology,1,2 and a mental age of ≥ 30 months, given cognitive requirements of the neuropsychological battery. Inclusion criteria also included genetic testing to confirm DS, no conditions contraindicative for brain imaging scans (e.g., pregnant and metal in the body), or psychiatric or physical health conditions that impacted cognition. In addition, for current analyses, the participant must have completed a PET Aβ and tau scan. The study was approved by the institutional review boards of each site and all participants consented and/or assented. Standardized operation manuals and monitoring ensured consistent procedures across sites. Participants were aged 25 to 58 years (M = 38.37 years, SD = 7.84), about half were female (N = 60, 49%), and the majority (N = 86, 70%) lived with family. Table 1 provides sample sociodemographics. Main analyses are on the 123 non‐demented adults with DS depicted in Table 1. However, five demented adults with DS were included along with the 123 non‐demented adults with DS to create the two‐SD threshold for high tau (TH). These five adults came from the University of Wisconsin–Madison (N = 2) and University of Pittsburgh (N = 3) sites and had an average age of 59 years (SD = 4.32).
TABLE 1.
Sample sociodemographics (N = 123)
| Site (N, [%]) | |
| University of Wisconsin–Madison | 48 (39.0%) |
| University of Pittsburgh | 47 (38.2) |
| Cambridge University | 18 (14.6%) |
| Barrow Institute | 6 (4.9%) |
| Washington University–St. Louis | 4 (3.3%) |
| Chronological age (M, [SD]) | 38.37 (7.84) |
| Premorbid intellectual disability level | |
| Mild | 38 (30.9%) |
| Moderate | 83 (67.5%) |
| Severe | 2 (1.6%) |
| Clinical status (N, [%]) | |
| Cognitively stable | 117 (95%) |
| Mild cognitive impairment | 6 (5%) |
| Residence (N, [%]) | |
| Family | 86 (69.9%) |
| Independent | 22 (17.9%) |
| Group home | 14 (11.4%) |
2.2. Measures
2.2.1. Clinical AD status
Clinical AD status was based on a case consensus process involving a psychologist and physician and staff experienced in AD in DS who were blind to imaging and biofluid data. This process involved review of caregiver‐reported and directly administered measures of dementia symptoms, cognitive functioning, and adaptive behavior. A detailed description has been previously provided. 27 Raters were blind to episodic memory scores, the cognitive measure of interest in this study. Clinical AD status categories were (1) cognitively stable; (2) mild cognitive impairment‐DS (MCI‐DS), indicating mild cognitive declines limited in scope; (3) dementia, indicating marked cognitive decline and decreases in daily functioning; and (4) unable to determine.
2.2.2. Control variables
Age was calculated in years using the date of birth and date of study visit. The Peabody Picture Vocabulary Test‐Fourth Edition 28 was used to assess verbal mental age. This measure of receptive language is reliable and valid in DS and highly correlated with IQ. 29 The following mental age equivalent scores were used to determine intellectual disability: mild: ≥ 9 years, moderate: 4 to 8 years, and severe: ≤ 3 years. Data collection site was coded: University of Pittsburgh = 1, University of Wisconsin–Madison = 2, Cambridge University = 3, Barrow Neurological Institute = 4, and Washington University–St. Louis = 5.
2.2.3. Episodic memory
The Cued Recall Test 30 is a list learning task that assesses episodic memory. Participants were shown three cards, each with four pictures of objects. After the learning trials, participants are asked to freely recall the pictures (i.e., Free Recall). For pictures not freely recalled, a category cue is provided (e.g., “fruit” for picture of grapes) in a cued trial. The Free and Cued Recall scores are summed to create a Free and Cued Total score. 30 This measure was shown to be associated with Aβ PET SUVR in adults with DS 13 , 14 and differentiated adults with DS with versus without AD. 31
2.2.4. Image acquisition and processing
PET‐measured Aβ and tau was assessed using nominal injections of [11C]PiB and [18F]AV‐1451. The average injected [11C]PiB dose was 571.88 MBq (SD = 52.17) and 365.38 MBq (SD = 34.50) for [18F]AV‐1451, excluding Cambridge, which administered an average dose of 301.97 MBq (SD = 40.65) and 191.89 (SD = 13.16), respectively. The PET scanners included the Siemens HR+ (Wisconsin, Pittsburgh), GE Signa PET/MR (Cambridge), GE Discovery 710 (Barrow/Banner), and the Siemens Biograph mCT (Washington University, Pittsburgh). The PET data included 5‐minute time frames for 50 to 70 minutes (PiB) and 80 to 100 minutes (AV‐1451) post‐injection. Data were reconstructed using iterative methods and corrected for deadtime, normalization, attenuation, scatter, and radioactive decay. PET data images were examined, and if necessary, corrected on a frame‐by‐frame basis for motion. The multi‐frame images were then averaged to produce a single frame image representing [11C]PiB uptake over 50 to 70 minutes post‐injection and 18F]AV‐1451 over 80 to 100 minutes.
T1‐weighted MRI brain scans were conducted on 3.0T MRI scanners incuding GE SIGNA 750 (Wisconsin, Barrow/Banner), Siemens Prisma scanner (Pittsburgh), GE Signa PET/MR (Cambridge), and Siemens Prisma (Washington University). The single‐frame PET images were registered with the corresponding MRI images. MRI scans were processed via FreeSurfer 5.3 to segment images via the Desikan‐Killany atlas modified by the addition of the Clinical Imaging Center (CIC) atlas for segmentation of the striatum. 24 , 25 , 26 The FreeSurfer segmentations were used to sample the single‐frame PET image activity. For each FreeSurfer region, Aβ and tau were quantified as SUVR using the cerebellar gray matter as the reference. 25 These biomarkers did not greatly differ between the different scanners in previous work from our groups. 32
2.2.5. Aβ and tau processing
For Aβ index, a global PiB score was determined and represented the volume‐weighted average of the SUVR in nine signature brain regions, including the frontal cortex, anterior cingulate, posterior cingulate, parietal cortex, precuneus, lateral temporal cortex, insula, orbito‐frontal, and the striatum, each constructed from FreeSurfer segmentation components. A previously established global SUVR threshold of 1.36 for Aβ+ versus Aβ– in DS 24 was used.
For tau, six image‐based Braak regions corresponding to the six Braak stages 33 were used for the quantitation of [18F]AV‐1451 SUVR. The Braak stages show the progression of the neurofibrillary changes related to AD. Stage I is limited to lesions in the transentorhinal region. Stage II displays lesions extending into the entorhinal region. Stages III and IV show disease progression into the temporal neocortex. Stage V includes neocortical pathology extending to the frontal, superolateral, and occipital directions. Last, Stage VI shows the pathology reaching the primary neocortical areas and into the striatum. Braak region SUVR were determined from a volume‐weighted average of SUVR in sets of FreeSurfer regions as described in Schöll et al. 34 except that the striatum was not included in Braak region V. To examine high versus low tau, a binary variable was created by defining high tau (TH) as two SDs above the mean region of interest (ROI) SUVR for each Braak region based on the 123 adults with DS as well as five additional adults with DS with a clinical status of dementia. The resulting TH thresholds were: 1.63 for Braak I, 1.61 for Braak II, 1.62 for Braak III, 1.53 for Braak IV, 1.60 for Braak V, and 1.44 for Braak VI. Every numbered region was exclusive of others; in other words, Braak II did not include Braak I, and so on.
2.3. Data analysis plan
Histograms and boxplots were used to examine the distribution of variables and screen for outliers. Descriptive statistics were used to examine the number and prevalence of A/T classification groups by clinical status (cognitively stable vs. MCI). To assess differences between Aβ+ versus Aβ– participants in tau SUVR in each of the six Braak regions, two‐sample t tests were performed. To assess the association between Braak ROIs and episodic memory, linear regression models were performed for each Braak ROI region in models that adjusted for age, premorbid level of intelligence, and site. Models were conducted separately for the Aβ– versus Aβ+ groups, as higher tau SUVR was only predicted to be associated with worse episodic memory for the Aβ+ group. Finally, linear regression models were conducted to determine whether non‐demented adults in the Aβ+/TH group exhibited worse episodic memory than those in the Aβ+/TL group and in the Aβ–/TL group. These models also controlled for site, age, and premorbid level of intelligence. Model assumptions were checked by examining residual plots.
3. RESULTS
Aβ and tau PET SUVR and the episodic memory score had a normal distribution without skew and there were no outliers. Table 2 displays the frequency and percent of participants in each A/T classification group. No one was classified as Aβ–/TH and thus this group is not depicted in the table. Most participants (N = 81, 66%) were classified as Aβ–/TL across all Braak regions. In contrast, 29% (N = 36) to 33% (N = 41) of participants were classified as Aβ+/TL across Braak regions, and 1% (N = 1) to 5% (N = 6) were Aβ+/TH. Two‐sample t‐tests were performed to examine differences between the Aβ+ versus Aβ– participants in tau SUVR in each of the six Braak regions. As shown in Table 3, the Aβ+ group (N = 42) had significantly higher tau SUVR across Braak regions I to V than the Aβ– group (N = 81).
TABLE 2.
Number and percentage of participants by clinical status in each A/T group across Braak regions
| Tau Region | A/T group | Cognitively stable N (%) | MCI N (%) | Overall N (%) |
|---|---|---|---|---|
| Braak I | Aβ–/TL | 80 (68%) | 1 (17%) | 81 (66%) |
| Aβ+/TL | 35 (30%) | 1(17%) | 36 (29%) | |
| Aβ+/TH | 2 (2%) | 4 (67%) | 6 (5%) | |
| Braak II | Aβ–/TL | 80 (68%) | 1 (17%) | 81 (66%) |
| Aβ+/TL | 34 (29%) | 3 (50%) | 37 (30%) | |
| Aβ+/TH | 3 (3%) | 2 (17%) | 5 (4%) | |
| Braak III | Aβ–/TL | 80 (68%) | 1 (17%) | 81 (66%) |
| Aβ+/TL | 35 (30%) | 2 (33%) | 37 (30%) | |
| Aβ+/TH | 2 (2%) | 3 (50%) | 5 (4%) | |
| Braak IV | Aβ–/TL | 80 (68%) | 1 (17%) | 81 (66%) |
| Aβ+/TL | 36 (31%) | 1 (17%) | 37 (30%) | |
| Aβ+/TH | 1 (1%) | 4 (67%) | 5 (4%) | |
| Braak V | Aβ–/TL | 80 (68%) | 1 (17%) | 81 (66%) |
| Aβ+/TL | 37 (32%) | 2 (33%) | 39 (32%) | |
| Aβ +/ TH | 0 (0%) | 3 (50%) | 3 (2%) | |
| Braak VI | Aβ‐ / TL | 80 (68%) | 1 (17%) | 81 (66%) |
| Aβ +/ TL | 37 (32%) | 4 (67%) | 41 (33%) | |
| Aβ +/ TH | 0 (0%) | 1 (17%) | 1 (1%) |
Abbreviations: Aβ–, amyloid‐beta negative; Aβ+, amyloid‐beta positive; A/T, amyloid tau; TL, low tau; TH , high tau; MCI, mild cognitive impairment.
TABLE 3.
Mean, standard deviations, and differences in tau PET SUVR by Aβ status (+ vs. –)
| Tau region | Overall | Aβ– (n = 81) | Aβ+ (n = 42) | t statistics, P |
|---|---|---|---|---|
| Braak I | 1.21 (0.19) | 1.14 (0.10) | 1.34 (0.25) | −4.835, P = .000 |
| Braak II | 1.16 (0.19) | 1.09 (0.11) | 1.28 (0.24) | −4.808, P = .000 |
| Braak III | 1.15 (0.16) | 1.10 (0.06) | 1.23 (0.24) | −3.495, P = .001 |
| Braak IV | 1.12 (0.15) | 1.08 (0.06) | 1.19 (0.22) | −3.312, P = .002 |
| Braak V | 1.09 (0.16) | 1.04 (0.06) | 1.17 (0.25) | −3.238, P = .002 |
| Braak VI | 1.02 (0.14) | 1.005 (0.06) | 1.06 (0.22) | −1.634, P = .109 |
Abbreviations: Aβ–, amyloid‐beta negative; Aβ+, amyloid‐beta positive; PET, positron emission tomography; SUVR, standardized uptake value ratio.
Linear regression models shown in Table 4 indicated significant associations between tau SUVR and episodic memory across Braak regions for the Aβ+ group but not the Aβ– group in linear regression models controlling for site, age, and premorbid level of intelligence. As shown in Table 5, tau SUVR was significantly negatively associated with Free and Cued Recall Total score for the Aβ+ group across Braak regions I to V. In contrast, the limited range of tau SUVR observed in the Aβ– group was not significantly associated with Free and Cued Recall Total score. We then re‐ran these linear regression models but assessed both tau as a binary variable (TL versus TH; see Table 5). For Braak region I to VI, participants who were classified as Aβ+/TH evidenced significantly worse episodic memory than those who were Aβ+/TL. There was not a significant difference between the Aβ+/TL versus Aβ–/TL groups in any of the Braak regions (Figure 1).
TABLE 4.
Estimated effect of tau SUVR on episodic memory score for Aβ+ and Aβ– groups adjusting for site, premorbid intellectual level, and age
| Aβ– (N = 81) | Aβ+ (N = 42) | |||
|---|---|---|---|---|
| Estimated effect of tau | t statistics | Estimated effect of tau | t statistics | |
| Tau region | (95% CI) | (P) | (95% CI) | (P) |
| Braak I | 0.441 (–6.076, 6.959) | .133, P = .895 | −15.833 (–22.114, –9.553) | −4.941, P = .000 |
| Braak II | −1.517 (–8.308, 5.275) | −.438, P = .663 | −12.290 (–20.113, –4.467) | −3.079, P = .004 |
| Braak III | 1.282 (–10.537, 13.102) | .213, P = .832 | −15.039 (–21.877, –8.200) | −4.310, P = .000 |
| Braak IV | −1.975 (–13.053, 9.104) | −.349, P = .728 | −22.423 (‐28.175, –16.671) | −7.641, P = .000 |
| Braak V | 0.128 (–11.507, 11.763) | .022, P = .983 | −15.267 (–21.672, –8.862) | −4.672, P = .000 |
| Braak VI | −0.876 (–12.971, 11.219) | −.142, P = .888 | −10.364 (–18.686, –2.042) | −2.441, P = .021 |
Abbreviations: Aβ–, amyloid‐beta negative; Aβ+, amyloid‐beta positive; CI, confidence interval; SUVR, standardized uptake value ratio.
TABLE 5.
Estimated effect of the PET Aβ and tau classification groups on episodic memory score in models adjusted for site, premorbid intellectual disability, and chronological age
| Age | |||
|---|---|---|---|
| Tau region | Variable | Estimate (SE) | F value |
| Braak I | Site | 2.082 | |
| WI vs. UP | 0.621 (0.672) | ||
| BN vs. UP | −0.385 (2.347) | ||
| UK vs. UP | −0.429 (0.944) | ||
| WU vs. UP | −4.030 (1.691) | ||
| Premorbid ID | 4.787* | ||
| Mild vs. moderate | −0.781 (0.649) | ||
| Mild vs. severe | −9.991 (3.337)* | 1.131 | |
| Chronological age | −0.054 (0.051) | ||
| Aβ/tau group status | 48.018* | ||
| Aβ+/TH vs. Aβ–/TL | −14.188 (1.595)* | ||
| Aβ+/TL vs. Aβ–/TL | 0.347 (0.808) | ||
| Aβ+/TH vs. Aβ+/TL | −14.535 (1.490)* | ||
| Braak II | Site | 1.401 | |
| WI vs. UP | 0.708 (0.882) | ||
| BN vs. UP | −0.488 (3.079) | ||
| UK vs. UP | −0.781 (1.239) | ||
| WU vs. UP | −4.036 (2.219) | ||
| Premorbid ID | −0.688 (0.854) | 2.018 | |
| Mild vs. moderate | |||
| Mild vs. severe | −8.468 (4.375) | ||
| Chronological age | −0.145 (0.065) | 4.947* | |
| Aβ/tau group status | |||
| Aβ+/TH vs. Aβ–/TL | −6.682 (2.156)* | 5.240* | |
| Aβ+/TL vs. Aβ–/TL | −0.167 (1.065) | ||
| Aβ+/TH vs. Aβ+/TL | −6.516 (2.061)* | ||
| Braak III | Site | 1.505 | |
| WI vs. UP | 0.891 (0.819) | ||
| BN vs. UP | −0.319 (2.857) | ||
| UK vs. UP | −0.166 (1.154) | ||
| WU vs. UP | −3.849 (2.058) | ||
| Premorbid ID | 2.776 | ||
| Mild vs. moderate | −0.903 (0.791)* | ||
| Mild vs. severe | −8.953 (4.059)* | ||
| Chronological age | −0.112 (0.061) | 3.356 | |
| Aβ/tau group status | 14.808* | ||
| Aβ+/TH vs. Aβ–/ TL | −10.472 (2.053)* | ||
| Aβ+/TL vs. Aβ–/ TL | −0.009 (0.983) | ||
| Aβ+/TH vs. Aβ +/ TL | −10.462 (1.941)* | ||
| Braak IV | Site | 2.366 | |
| WI vs. UP | 0.969 (0.654) | ||
| BN vs. UP | −0.202 (2.283) | ||
| UK vs. UP | 0.062 (0.921) | ||
| WU vs. UP | −3.789 (1.645) | ||
| Premorbid ID | 4.986* | ||
| Mild vs. moderate | −0.929 (0.632) | ||
| Mild vs. severe | −9.663 (3.244)* | ||
| Chronological age | −0.065 (0.049) | 1.773 | |
| Aβ/tau group status | 53.784* | ||
| Aβ+/TH vs. Aβ–/TL | −15.842 (1.657)* | ||
| Aβ+/TL vs. Aβ–/TL | 0.259 (0.784) | ||
| Aβ+/TH vs. Aβ+/TL | −16.102 (1.559)* | ||
| Braak V | Site | 1.738 | |
| WI vs. UP | 1.160 (0.845) | ||
| BN vs. UP | −0.334 (2.933) | ||
| UK vs. UP | −0.356 (1.182) | ||
| WU vs. UP | −3.746 (2.113) | ||
| Premorbid ID | 2.911 | ||
| Mild vs. moderate | −1.252 (0.816) | ||
| Mild vs. severe | −8.624 (4.165) | ||
| Chronological age | −0.145 (0.062) | 5.462* | |
| Aβ/tau group status | 11.291* | ||
| Aβ+/TH vs. Aβ–/TL | −11.768 (2.555)* | ||
| Aβ+/TL vs. Aβ–/TL | −0.146 (1.008) | ||
| Aβ+/TH vs. Aβ+/TL | −11.622 (2.475)* | ||
| Braak VI | Site | 1.522 | |
| WI vs. UP | 0.938 (0.899) | ||
| BN vs. UP | −0.506 (3.124) | ||
| UK vs. UP | −0.862 (1.259) | ||
| WU vs. UP | −3.831(2.251) | ||
| Premorbid ID | 2.266 | ||
| Mild vs. moderate | −1.173 (0.873)* | ||
| Mild vs. severe | −8.134 (4.435)* | ||
| Chronological age | −0.161 (0.066) | 5.985* | |
| Aβ/tau group status | 3.54* | ||
| Aβ+/TH vs. Aβ–/TL | −11.992 (4.507)* | ||
| Aβ+/TL vs. Aβ–/TL | −0.578 (1.067) | ||
| Aβ+/TH vs. Aβ+/TL | −11.414 (4.438)* |
Abbreviations: Aβ– , amyloid‐beta negative; Aβ+ , amyloid‐beta positive; BN, Barrow Neurological Institute; MCI , mild cognitive impairment; PET, positron emission tomography; TH , high tau; TL , low tau; UK, University of Cambridge: UP , University of Pittsburgh; WI , University of Wisconsin–Madison; WU , Washington University–St. Louis.
Notes: F value = null hypothesis is that the effect of the variable in each row on episodic memory is equal to zero.
P < .05; ** P ≤ 01.
FIGURE 1.
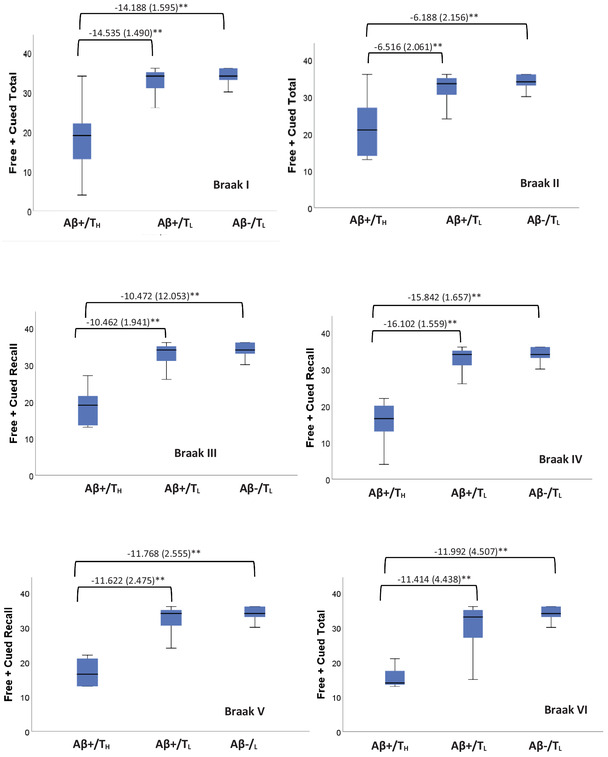
Boxplots depicting median and variability of Free + Cued Total score by positron emission tomography amyloid beta (Aβ– vs. Aβ+) and tau (TL vs. TH) status in Braak regions I to VI. Brackets highlight significant (**P < .010) group comparisons for episodic memory score in models adjusted for site, premorbid intellectual disability, and chronological age
4. DISCUSSION
There is a critical need for research that establishes biomarkers of AD pathogenesis in DS, including the usefulness of the AT(N) framework. 23 The current study built on previous research that has examined biomarkers of Aβ (e.g., Annus et al., 7 Lao et al., 8 and Zammit et al. 9 ) and plasma and CSF biomarkers of tau 3 in predicting early AD‐related cognitive declines in DS by examining a tau PET biomarker. Our goal was to determine whether the tau PET biomarker [18F]AV‐1451 was associated with early episodic memory impairment beyond PET‐measured Aβ accumulation in a sample of 123 non‐demented adults with DS.
We found that the majority (N = 81, 66%) of our sample of non‐demented adults with DS were classified as Aβ–/TL across all Braak regions. Of the remaining non‐demented adults with DS, 29% to 33% were classified as Aβ+/TL and, depending on the Braak region, 1% to 5% were classified as Aβ+/TH. Adults with DS who were Aβ+ had a higher tau SUVR across Braak I to V than did those who were Aβ–. Tau SUVR as a continuous variable in Braak I to V was negatively related to episodic memory performance for adults with DS who were Aβ+ but was not associated with episodic memory for those who were Aβ–, most likely due to the very limited range of tau SUVR in this latter group. These findings were in models controlling for age, site effects, and premorbid level of intellectual disability. Moreover, as a binary variable (TL vs. TH), tau PET predicted worse episodic memory performance beyond the presence of Aβ accumulation in models that were adjusted for the same control variables. In fact, Aβ deposition alone did not predict episodic memory performance. In the absence of elevated tau (i.e., TL), non‐demented adults with DS with versus without marked Aβ+ accumulation (Aβ+ vs. Aβ–) did not differ in episodic memory performance. This finding suggests that the presence of tau, but not Aβ alone, co‐occurs with subtle episodic memory declines early on in the trajectory to AD in DS.
Our findings of AD in DS are similar to reports for late‐onset AD (e.g., Gordon et al. 35 ) and autosomal dominant AD (e.g., Quiroz et al. 36 ) in the non‐DS population, which have also found that the presence of high tau beyond Aβ status is an indicator of the transition from preclinical to the early prodromal stage of AD. Also similar to our findings, episodic memory as opposed to other cognitive domains has been found to correspond to the presence of high tau in late‐onset AD and autosomal dominant AD. 37 , 38 These findings are consistent with those for blood (e.g., Aβ42/Aβ40 and p‐tau181) and cerebrospinal fluid (e.g., Aβ42, p‐tau, and total tau) biomarkers in supporting the AT(N) framework in DS. 39 , 40 Together, this work suggests that DS is a relevant model for AD pathogenesis in non‐DS populations, as both imaging and biofluid AT(N) biomarkers are informative in predicting AD in DS.
There are both strengths and limitations to the current study. In terms of strengths, analyses included a relatively large sample of adults with DS and used a validated directly administered measure of episodic memory in DS. To our knowledge, this is the first published study to examine whether tau PET predicts cognitive decline beyond Aβ alone in DS. Our focus on biomarkers that correlate with early cognitive change prior to the onset of dementia makes findings particularly relevant for AD clinical trials. In terms of limitations, the study is cross‐sectional and thus findings cannot speak to the time‐ordered effect of tau on episodic memory. Future longitudinal research is needed to track changes in Aβ and tau across time and should also include biomarkers of neurodegeneration (N in the AT[N] model). The field will also need to assess the clinical utility of tracking incremental increases in tau (continuous SUVR) prior to Aβ positivity, as our study found limited variability. In the current study, high versus low tau was determined using a two‐SD approach created from a sample that included the 123 non‐demented adults with DS reported on here, as well as five demented adults with DS. Future research will need to test this approach against visual reads and other quantification methods and in new samples. The current sample was largely White, non‐Hispanic adults with DS and there is a need for diverse samples to replicate findings. Although chronological age was controlled in models, our wide age range could mean that aging effects, outside of AD pathology, influenced episodic memory. Finally, adults with DS with a mental age below 30 months were not included; thus, it is not clear how the AT(N) system applies to this lower functioning group.
Findings have implications for advancing clinical AD trials in DS. Findings suggest that the AT(N) classification system has relevance in DS. Specifically, biomarkers of tau PET appear to be meaningful indicators of adults with DS who are further along in the progression toward AD. Moreover, tau PET may be a more sensitive biomarker than Aβ PET alone for predicting early AD‐related cognitive decline in the DS population similar to findings for late‐onset AD and autosomal dominant AD. 35 , 36 , 37 Biomarkers of tau PET may thus be useful in the selection of non‐demented adults with DS for AD clinical trials, and to aid in the monitoring of the early stages of disease progression.
CONFLICTS OF INTEREST
GE Healthcare holds a license agreement with the University of Pittsburgh for the [11C]PiB PET technology involved in this study. Dr. William Klunk is a co‐inventor of [11C]PiB and has financial interest in this license agreement. GE Healthcare did not provide financial support for this study nor did it have a role in designing the study, interpreting results, or manuscript writing. No other authors have relationships, activities, or interests to disclose related to this manuscript.
ACKNOWLEDGMENTS
The research was funded by the National Institute on Aging (R01AG031110, U01AG051406; U19 AG070043) and the National Institute on Child Health and Human Development (U54 HD090256, P50 HD105353).
We are grateful to the adults with Down syndrome and their caregivers who participated in this study for their time and efforts in supporting this research.
Hartley SL, Handen BL, Tudorascu D, et al. Role of tau deposition in early cognitive decline in Down syndrome. Alzheimer's Dement. 2022;14:e12256. 10.1002/dad2.12256
REFERENCES
- 1. Wiseman FK, Al‐Janabi T, Hardy J, et al. A genetic cause of Alzheimer disease: mechanistic insights from Down syndrome. Nat Rev Neurosci. 2015;16(9):564‐574. 10.1038/nrn3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2. Zigman WB, Devenny DA, Krinsky‐McHale SJ, et al. Alzheimer's disease in adults with Down syndrome. Int Rev Res Ment Retard. 2008;36:103‐145. 10.1016/S0074-7750(08)00004-9 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Fortea J, Vilaplana E, Carmona‐Iragui M, et al. Clinical and biomarker changes of Alzheimer's Disease in adults with Down Syndrome: a cross‐sectional study. Lancet. 2020;395:1988‐1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. McCarron M, McCallion P, Reilly E, Mulryan N. A prospective 14‐year longitudinal follow‐up of dementia in persons with Down syndrome. J Intellect Disabil Res. 2014;58:61‐70. 10.1111/jir.12074 [DOI] [PubMed] [Google Scholar]
- 5. Rubenstein E, Hartley SL, Bishop L. Epidemiology of dementia and Alzheimer disease in individuals with Down syndrome. JAMA Neurol. 2020;77(2):262‐264. 10.1001/jamaneurol.2019.3666366 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Jack CR Jr, Bennett DA, Blennow K, et al. A/T/N: an unbiased descriptive classification scheme for Alzheimer disease biomarkers. Neurology. 2016;87(5):539‐547. 10.1212/WNL.0000000000002923 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Annus T, Wilson LR, Hong YT, et al. The pattern of amyloid accumulation in the brains of adults with Down syndrome. Alzheimers Dement. 2016;12:538‐545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Lao PJ, Handen BL, Betthauser TJ, et al. Longitudinal changes in amyloid PET and volumetric MRI in the non‐demented Down syndrome population. Alzheimers Dement. 2017;9:2‐9. 10.1016/j.dadm.2017.05.001 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zammit MD, Laymon CM, Betthauser TJ, et al. Amyloid accumulation in Down syndrome measured with amyloid load. Alzheimers Dement (N Y). 2021;12:1‐9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Coutinho AM, Busatto GF, de Gobbi Porto FH, et al. Brain PET amyloid and neurodegeneration biomarkers in the context of the 2018 NIA‐AA research framework: an individual approach exploring clinical‐biomarker mismatches and sociodemographic parameters. Eur J Nucl Med Mol Imaging. 2020;47:2666‐2680. 10.1007/s00259-020-04714-0 [DOI] [PubMed] [Google Scholar]
- 11. Knopman DS, Haeberlein SB, Carrillo MC, et al. The National Institute on Aging and the Alzheimer's Association Research framework for Alzheimer's disease: perspectives from the research roundtable. Alzheimers Dement. 2018;14(4):563‐575. 10.1016/j.jalz.2018.03.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Yu JT, Li JQ, Suckling J, et al. Alzheimer's Disease Neuroimaging Initiative. Frequency and longitudinal clinical outcomes of Alzheimer's AT(N) biomarker profiles: a longitudinal study. Alzheimers Dement. 2019;15(9):1208‐1217. 10.1016/j.jalz.2019.05.006 [DOI] [PubMed] [Google Scholar]
- 13. Hartley SL, Handen BL, Devenny D, et al. Cognitive indicators of transition to preclinical and prodromal stages of Alzheimer's disease in Down syndrome. Alzheimers Dement (N Y). 2020;12:1‐10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Nelson LD, Siddarth P, Kepe V, et al. Positron emission tomography of brain amyloid and tau levels in adults with Down syndrome. Arch Neurol. 2011;68:768‐774. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Hartley SL, Handen BL, Devenny D, et al. Cognitive decline and brain amyloid‐β accumulation across 3 years in adults with Down syndrome. Neurobiol Aging. 2017;58:68‐76. 10.1016/j.neurobiolaging.2017.05.019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Gadliardi G, Epelbaum S, Houot M, et al. Which episodic memory performance is associated with Alzheimer's disease biomarkers in elderly cognitive complainers? Evidence from a longitudinal observational stud with four episodic memory tests (Insigh‐PREAD). Alzheimer's Dis. 2019;70:8110824. 10.3233/JAD-180966 [DOI] [PubMed] [Google Scholar]
- 17. Tromp D, Dufour A, Lithfous S, Pebayle T, Després O. Episodic memory in normal aging and Alzheimer disease: insights from imaging and behavioral studies. Ageing Res Rev. 2015;24(Pt B):232‐262. 10.1016/j.arr.2015.08.006 [DOI] [PubMed] [Google Scholar]
- 18. Firth NC, Startin CM, Hithersay R, et al. Aging related cognitive changes associated with Alzheimer's disease in Down syndrome. Ann Clin Transl Neurol. 2018;5:741‐751. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Holland AJ, Hon J, Huppert FA, Stevens F. Incidence and course of dementia in people with Down's syndrome: findings from a population‐based study. Intellect Disabil Res. 2000;44:138‐146. [DOI] [PubMed] [Google Scholar]
- 20. Krinsky‐McHale SJ, Devenny DA, Kittler P, Silverman W. Selective attention deficits associated with mild cognitive impairment and early stage Alzheimer's disease in adults with Down syndrome. Am Mental Retard. 2008;113:369‐386. [DOI] [PubMed] [Google Scholar]
- 21. Krinsky‐McHale SJ, Zigman WB, Lee JH, et al. Promising outcome measures of early Alzheimer's dementia in adults with Down syndrome. Alzheimers Dement (N Y). 2020;12:1‐11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Startin CM, Ashton NJ, Hamburg S, et al. Plasma biomarkers for amyloid, tau, and cytokines in Down syndrome and sporadic Alzheimer's disease. Alzheimers Res Ther. 2019;11:26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Rafii MS, Ances BM, Schupf N, et al. The AT(N) framework for Alzheimer's disease in adults with Down syndrome. Alzheimers Dement (Amst). 2020;12(1):e12062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Tudorascu DL, Laymon CM, Zammit M, et al. Relationship of amyloid beta and neurofibrillary tau deposition in Neurodegeneration in Aging Down Syndrome (NiAD) study at baseline. Alzheimers Dement (N Y). 2020;6(1):e12096. Published 2020 Oct 31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Lao PJ, Betthauser TJ, Hilmer AT, et al. The effects of normal aging on amyloid‐β deposition in a population of nondemented adults with Down syndrome as imaged by [11C] PIB. Alzheimer's Dem (Amst). 2016;12:380‐390. 10.1016/j.jalz.2015.05.013 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Tudorascu DL, Minhas DS, Lao PJ, et al. The use of centiloids for applying 11C PiB classification cutoffs across region‐of‐interest delineation methods. Alzheimer's Dem (Amst). 2018;10:332‐339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Handen BL, Lott IT, Christian BT, et al. The Alzheimer's Biomarker Consortium‐Down Syndrome: rationale and methodology. Alzheimer's Dement Diagn Assess Dis Monit. 2020;12. 10.1002/dad2.12065 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Dunn LM. PPVT‐III : Peabody Picture Vocabulary Test. Circle Pines, MN: American Guidance Service; 1997. [Google Scholar]
- 29. Phillips BA, Loveall SJ, Channell MM, Conners FA. Matching variables for research involving youth with Down syndrome: leiter‐R versus PPVT‐4. Res Develop Disabil. 2014;35:429‐438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Zimmerli E, Devenny DA, Paper presented at the Gatlinburg conference on research and theory in mental retardation and developmental disabilities. Cued recall as a screen for dementia in the MR population. Gatlinburg, TN; 1995.
- 31. Devenny DA, Zimmerli EJ, Kittler P, Krinsky‐McHale SJ. Cued recall in early‐stage dementia in adults with Down's syndrome. J Intellect Disabil Res. 2002;46(Pt 6):472‐483. 10.1046/j.1365-2788.2002.00417.x [DOI] [PubMed] [Google Scholar]
- 32. Minhas D, Yang Z, Muschelli J, et al. Satistical methods for processing neuroimaging data from two sites with a Down syndrome population application. Inform Process Manage Uncertainty Knowledge‐Based Syst. 2020;1239:367‐379. 10.1007/978-3-030-50153-L8. Published 2020 May 16. [DOI] [Google Scholar]
- 33. Braak H, Alafuzoff I, Arzberger T, Kretzschmar H, Del Tredici K. Staging of Alzheimer disease‐associated neurofibrillary pathology using paraffin sections and immunocytochemistry. Acta Neuropathol. 2006;112(4):389‐404. 10.1007/s00401-006-0127-z [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Schöll M, Lockhart SN, Schonhaut DR, et al. PET imaging of tau deposition in the aging human brain. Neuron. 2016;89(5):971‐982. 10.1016/j.neuron.2016.01.028 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Gordon BA, Blazey TM, Christensen J, et al. Tau PET in autosomal dominant Alzheimer's disease: relationship with cognition, dementia and other biomarkers. Brain. 2019;142(4):1063‐1076. 10.1093/brain/awz019 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Quiroz YT, Sperling RA, Norton DJ, et al. Association between amyloid and tau accumulation in young adults with autosomal dominant Alzheimer disease. JAMA Neurol. 2018;75(5):548‐556. 10.1001/jamaneurol.2017.4907 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hammond TC, Xing X, Wang C, et al. β‐amyloid and tau drive early Alzheimer's disease decline while glucose hypometabolism drives late decline. Commun Biol. 2020;3:352‐363. 10.1038/s42003-020-1079 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Tanner JA, Rabinovici GD. Relationship between tau and cognition in the evolution of Alzheimer's Disease: new insights from tau PET. J Nucl Med. 2021;62(5):612‐613. 10.2967/jnumed.120.257824 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Montoliu‐Gaya L, Strydom A, Blennow K, Zetterberg H, Ashton NJ. Blood biomarkers for Alzheimer's disease in Down syndrome. J Clin Med. 2021;10(16):3639. 10.3390/jcm10163639 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Petersen ME, Zhang F, Schupf N, et al. Proteomic profiles for Alzheimer's disease and mild cognitive impairment among adults with Down syndrome spanning serum and plasma: an Alzheimer's Biomarker Consortium‐Down Syndrome (ABC‐DS) study. Alzheimers Dement (Amst). 2020;12(1):e12039. 10.1002/dad2.12039 [DOI] [PMC free article] [PubMed] [Google Scholar]