

Abstract
Osteonecrosis of the femoral head (ONFH) involves necrosis of bone and bone marrow of the femoral head caused by ischemia with unknown etiology. Previous genetic studies on ONFH failed to produce consistent results, presumably because ONFH has various causes with different genetic backgrounds and the underlying diseases confounded the associations. Steroid-associated ONFH (S-ONFH) accounts for one-half of all ONFH, and systemic lupus erythematosus (SLE) is a representative disease underlying S-ONFH. We performed a genome-wide association study (GWAS) to identify genetic risk factors for S-ONFH in patients with SLE. We conducted a two-staged GWAS on 636 SLE patients with S-ONFH and 95 588 non-SLE controls. Among the novel loci identified, we determined S-ONFH-specific loci by comparing allele frequencies between SLE patients without S-ONFH and non-SLE controls. We also used Korean datasets comprising 148 S-ONFH cases and 37 015 controls to assess overall significance. We evaluated the functional annotations of significant variants by in silico analyses. The Japanese GWAS identified 4 significant loci together with 12 known SLE susceptibility loci. The four significant variants showed comparable effect sizes on S-ONFH compared with SLE controls and non-SLE controls. Three of the four loci, MIR4293/MIR1265 [odds ratio (OR) = 1.99, P-value = 1.1 × 10−9)], TRIM49/NAALAD2 (OR = 1.65, P-value = 4.8 × 10−8) and MYO16 (OR = 3.91, P-value = 4.9 × 10−10), showed significant associations in the meta-analysis with Korean datasets. Bioinformatics analyses identified MIR4293, NAALAD2 and MYO16 as candidate causal genes. MIR4293 regulates a PPARG-related adipogenesis pathway relevant to S-ONFH. We identified three novel susceptibility loci for S-ONFH in SLE.
Introduction
Osteonecrosis of the femoral head (ONFH) involves necrosis of bone and bone marrow of the femoral head caused by ischemia (1). A Japanese nationwide study estimated that 11 400 patients developed ONFH in 2004, with an incidence of 2.51 per 100 000 person-years (2). The annual number of patients seeking medical care increased by 1.5 times in 2004 compared with 1994 (3). Hip pain from femoral head collapse in the progressive stage of ONFH is often the initial subjective symptom of the disease. Femoral head collapse usually produces gait disturbance and patients often require surgery, including total hip arthroplasty or joint-preserving procedures (4). ONFH often manifests in people aged from their 30s to 50s. Because these young-to-middle-aged patients frequently require surgical interventions, the disease has socioeconomic impacts. Consequently, there is a huge demand for determination of the elucidating disease mechanism and development of non-surgical treatments and predictive tools for its onset.
Steroid intake is the most prevalent risk factor for ONFH (one-half of patients have history of steroid administration), followed by excessive alcohol consumption (2). Systemic lupus erythematosus (SLE) is the major underlying disease (33.1%) for steroid-associated ONFH (S-ONFH) (2). Nevertheless, two-thirds of SLE patients with steroid administration did not develop ONFH (5,6), strongly suggesting that factors other than steroid administration also contribute to the disease onset.
Genetic factors are involved in ONFH, but only a few associated loci have been established. A Chinese nationwide epidemiological survey revealed that family history increased the risk of ONFH [odds ratio (OR) = 5.33, P < 0.0001)] (7). Many genetic analyses have focused on particular candidate genes or families (8–13). A genome-wide association study (GWAS) is a useful approach to identify genetic risk factors for complex diseases by comprehensively searching for association signals across genomes in a hypothesis-free manner. Previous GWASs for ONFH identified significant loci around GRIN3A (14), BMP7, LINC00251, PROX1-AS1 (15) and CACNA1E (16). However, the studies involved very small cohorts (~400 cases) and did not produce consistent associations. We previously performed a GWAS for ONFH and identified two significant loci: 12q24 and 20q12 (17). Rs3858704 on 12q24 was significantly associated with alcohol consumption and alcohol-associated ONFH (OR = 2.73, P-value = 1.23 × 10−21), while rs6038718 on 20q12 was significantly associated with ONFH regardless of risk factors and was suggestively associated with S-ONFH with a distinct effect size (OR = 1.12, P-value = 6.84 × 10−7). These findings indicate that ONFH has multiple phenotypes with distinct genetic backgrounds.
Here, we conducted a two-staged GWAS on 636 SLE patients with S-ONFH [SLE_ON(+)] and 95 588 controls in Japan and conducted a subsequent meta-analysis using 148 SLE_ON(+) and 37 015 controls in Korea (Supplementary Material, Table S1, Fig S1). We also analyzed SLE patients without S-ONFH [SLE_ON(−)] to confirm S-ONFH-specific associations rather than associations with SLE susceptibility.
Results
GWAS for SLE_ON(+) in Japan
We conducted first GWAS on 436 SLE_ON(+) and 63 726 BioBank Japan (BBJ) controls which is composed on 47 common diseases (18,19) and conducted the second GWAS on 200 SLE_ON(+) and 31 862 BBJ that passed quality control (QC) in Japanese population. Then, we conducted meta-analysis. For these studies, we used very stringent criteria for cases, namely, patients who were diagnosed as ONFH by X-rays or MRI and as SLE by the 1982 or 1997 American College of Rheumatology (ACR) criteria (20,21), to ensure that the cases had common pathology and thus genetic backgrounds. We took the approach of comparing cases with BBJ controls rather than with SLE_ON(−) because the limited number of definitive controls (MRI was essential to exclude ONFH) restricted the statistical power of the study. Thus, we took advantage of data for definitive controls to confirm the associations, as shown later. As a result, we identified 16 significant loci (Fig. 1, Table 1). The genomic control inflation factor showed very slight inflation of chi-square statistics (lambda: 1.047) and the linkage disequilibrium score regression (LDSC) analysis indicated minimal bias in the results (estimated mean χ2: 1.0575 and intercept: 1.012) (22). Therefore, we did not apply genomic control correction.
Figure 1.
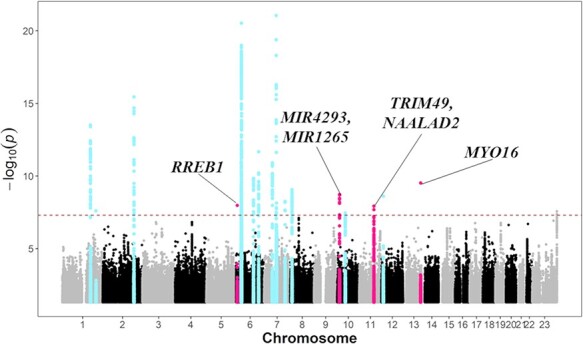
Manhattan plot of the GWAS comprising 636 SLE patients with S-ONFH and 95 588 BBJ controls. X- and Y-axes indicate genomic positions and minus log10-transformed association P-values, respectively. Dashed line shows the genome-wide significant threshold (P < 5 × 10−8). Red and blue plots indicate SLE_ON(+)-associated loci and known SLE susceptibility loci, respectively. The nearest genes are shown in SLE_ON(+)-associated loci.
Table 1.
Significant loci in two-staged GWAS in 636 SLE_ON(+) and 95 588 BBJ controls
| CHR POS |
EA NEA |
Gene | Function | Meta-analysis | First GWAS | Second GWAS | |||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| OR [95%CI] |
P-value | EAF case control |
OR [95%CI] |
P-value | EAF case control |
OR [95%CI] |
P-value | R2 | Novel or known | ||||
| SLE_ON(+)-associated loci | |||||||||||||
| 6 7 236 620 |
T C |
RREB1 | Intronic | 3.11 [2.11–4.59] |
1 × 10−8 | 0.032 0.016 |
3.34 [2.12–5.26] |
2 × 10−7 | 0.027 0.015 |
2.57 [1.22–5.45] |
0.014 | 0.46 | Novel |
| 10 14 436 836 |
A G |
MIR4293 MIR1265 |
Intergenic | 2.1 [1.65–2.67] |
2 × 10−9 | 0.056 0.028 |
2.12 [1.58–2.83] |
4.6 × 10−7 | 0.055 0.027 |
2.05 [1.33–3.16] |
0.0011 | 1 | Novel |
| 11 89 538 685 |
C T |
TRIM49 NAALAD2 |
Intronic | 1.78 [1.46–2.16] |
1.2 × 10−8 | 0.128 0.091 |
1.77 [1.39–2.24] |
3.1 × 10−6 | 0.13 0.092 |
1.8 [1.27–2.56] |
0.0011 | 0.66 | Novel |
| 13 109 491 871 |
A G |
MYO16 | Intronic | 4 [2.6–6.15] |
2.8 × 10−10 | 0.019 0.006 |
3.86 [2.29–6.49] |
3.5 × 10−7 | 0.019 0.006 |
4.32 [2.01–9.31] |
1.9 × 10−4 | 0.73 | Novel |
| SLE susceptibility loci | |||||||||||||
| 1 173 191 475 |
T G |
TNFSF4 LOC100506023 |
Intergenic | 1.62 [1.43–1.84] |
3.7 × 10−14 | 0.264 0.183 |
1.59 [1.37–1.85] |
2.1 × 10−9 | 0.277 0.182 |
1.69 [1.35–2.11] |
3.2 × 10−6 | 1 | Known |
| 1 206 735 148 |
A C |
RASSF5 | Intronic | 3.66 [2.35–5.71] |
9.8 × 10−9 | 0.022 0.007 |
4.37 [2.64–7.22] |
8.8 × 10−9 | 0.013 0.007 |
1.96 [0.76–5.06] |
0.16 | 0.76 | Known |
| 2 191 943 742 |
T C |
STAT4 | Intronic | 1.6 [1.43–1.79] |
3 × 10−16 | 0.411 0.302 |
1.62 [1.41–1.86] |
4.3 × 10−12 | 0.405 0.306 |
1.57 [1.28–1.92] |
1.4 × 10−5 | 1 | Known |
| 6 32 614 963 |
A G |
HLA-DQA1 HLA-DQB1 |
Intergenic | 2.19 [1.86–2.58] |
4.3 × 10−21 | 0.209 0.144 |
2.1 [1.72–2.56] |
4.2 × 10−13 | 0.226 0.143 |
2.4 [1.81–3.18] |
1.3 × 10−9 | 0.58 | Known |
| 6 106 564 236 |
A G |
PRDM1 ATG5 |
Intergenic | 1.47 [1.3–1.65] |
1.7 × 10−10 | 0.372 0.291 |
1.48 [1.28–1.71] |
7 × 10−8 | 0.373 0.293 |
1.44 [1.17–1.78] |
6 × 10−4 | 0.94 | Known |
| 6 138 190 533 |
T C |
TNFAIP3 | Intronic | 1.93 [1.61–2.32] |
1.3 × 10−12 | 0.113 0.065 |
1.9 [1.52–2.37] |
1.1 × 10−8 | 0.113 0.064 |
2.01 [1.45–2.77] |
2.6 × 10−5 | 0.91 | Known |
| 7 50 307 334 |
A G |
C7orf72 IKZF1 |
Intergenic | 1.49 [1.33–1.67] |
1.1 × 10−11 | 0.612 0.517 |
1.48 [1.29–1.7] |
2.6 × 10−8 | 0.625 0.518 |
1.5 [1.22–1.84] |
9.3 × 10−5 | 0.99 | Known |
| 7 74 196 185 |
A C |
NCF1 | Intronic | 3.42 [2.67–4.37] |
1.2 × 10−22 | 0.099 0.065 |
2.7 [1.97–3.7] |
7.3 × 10−10 | 0.13 0.065 |
4.91 [3.32–7.26] |
1.6 × 10−15 | 0.36 | Known |
| 7 128 575 797 |
T TCTTAGCTATTGCTC |
KCP IRF5 |
Intergenic | 1.59 [1.36–1.85] |
3.9 × 10−9 | 0.158 0.105 |
1.6 [1.33–1.92] |
6.4 × 10−7 | 0.154 0.107 |
1.56 [1.18–2.05] |
0.0016 | 0.99 | Known |
| 8 11 354 097 |
T C |
BLK | Intronic | 1.55 [1.35–1.78] |
7.8 × 10−10 | 0.777 0.69 |
1.56 [1.32–1.85] |
2.2 × 10−7 | 0.767 0.691 |
1.51 [1.18–1.94] |
9.3 × 10−4 | 0.95 | Known |
| 10 50 076 325 |
T G |
WDFY4 | Intronic | 1.36 [1.22–1.52] |
3.8 × 10−8 | 0.484 0.406 |
1.4 [1.22–1.6] |
8.6 × 10−7 | 0.467 0.406 |
1.29 [1.06–1.57] |
0.011 | 1 | Known |
| 12 12 874 462 |
G A |
CDKN1B | 3’UTR | 2.64 [1.92–3.63] |
2.4 × 10−9 | 0.96 0.918 |
2.57 [1.75–3.76] |
1.4 × 10−6 | 0.961 0.917 |
2.81 [1.58–4.99] |
4.3 × 10−4 | 0.84 | Known |
SLE_ON(+), SLE with S-ONFH; CHR, chromosome; POS, position (hg19); EA, effect allele; NEF, non-effect allele; EAF, effect allele frequency.
R 2 shows imputation quality.
The association results apparently contained statistics for susceptibility to SLE and susceptibility to SLE_ON(+). We hypothesized that susceptibility to SLE_ON(+) is distinct from susceptibility to SLE and excluded 12 significant loci which were previously reported as SLE-susceptibility loci. As a result, we identified four remaining significant loci: RREB1 on 6p24.3 (OR = 3.11, P-value = 1 × 10−8), MIR4293/MIR1265 on 10p13 (OR = 2.1, P-value = 2 × 10−9), TRIM49/NAALAD2 on 11q14.3 (OR = 1.78, P-value = 1.2 × 10−8) and MYO16 on 13q33.3 (OR = 4, P-value = 2.8 × 10−10) (Table 1). Their ORs were quite comparable between first and second GWAS. While we did not find systemic inflation of statistics (extreme case–control imbalance would result in false-positives, especially for rare variants by violating assumptions of logistic regression model), we confirmed the association signals by firth logistic regression and down-sampling (case–control ratio of 1:19) analyses (Supplementary Material, Tables S2 and S3). Their effect sizes and P-values are consistent with logistic regression model.
We evaluated the associations between these four variants and SLE by referring to our recent meta-analysis in Asian populations (23). As a result, regardless of the very large sample size of the latest meta-analysis, we did not find even trends for associations between the variants and susceptibility to SLE [ORs for nearby 1 being <1 in contrast to ORs ≥ 1.78 in SLE_ON(+)], indicating that these associations were not explained by SLE susceptibility (Supplementary Material, Table S4, Supplementary Material, Fig S2). To further confirm that these loci were associated with SLE_ON(+), we evaluated the associations of the four variants by comparing 636 SLE_ON(+) and 683 SLE_ON(−) or 95 588 BBJ controls, assuming similar allele frequencies between the BBJ controls and SLE_ON(−) patients. We conducted GWAS on the first study composing of 436 SLE_ON(+) and 377 SLE_ON(−) and conducted GWAS on the second study of composing 200 SLE_ON(+) and 306 SLE_ON(−) and conducted meta-analysis for these two GWASs (Supplementary Material, Table S5). All four variants had comparable effect size with association study between SLE_ON(+) and BBJ (or rather enhanced associations in terms of ORs) and P-values satisfying the nominal threshold (P-value < 0.0018) (Supplementary Material, Table S5, Fig. 2A–D). We conducted GWAS on the first study composing of 377 SLE_ON(−) and 64 103 BBJ controls and conducted the second study composing of 306 SLE_ON(−) and 31 862 BBJ controls and also meta-analysis. As a result, we confirmed the lack of associations between the four variants and SLE_ON(−) by comparing 683 SLE_ON(−) and 95 588 BBJ controls (Supplementary Material, Table S5). Conditional analyses showed no additional signals (data not shown).
Figure 2.
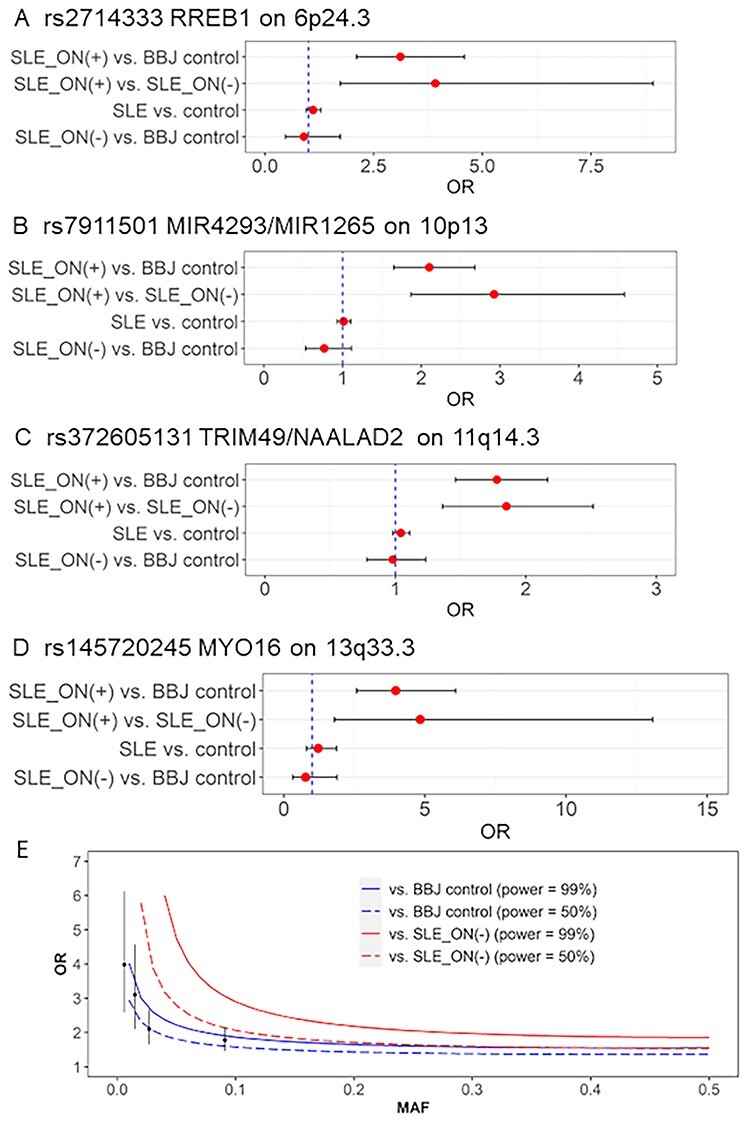
Associations of four S-ONFH associated loci in SLE. (A–D) The four association results in Japan, namely, SLE_ON(+) versus BBJ controls, SLE_ON(+) versus SLE_ON(−), SLE versus controls (23) and SLE_ON(−) versus BBJ controls, are shown for four SLE_ON(+)-associated variants. The numeric data are shown in Supplementary Material, Table S2. The statistical analysis for GWAS using SLE_ON(+) and SLE_ON(−) or BBJ controls is shown in (E). X- and Y-axes indicate MAFs and ORs, respectively. Alpha-error rate and statistical power are set to 5 × 10−8 and 0.99 or 0.5, respectively. Red and blue lines show statistical power of association studies of 636 SLE_ON(+) compared with 683 SLE_ON(−) and 95 588 BBJ controls, respectively. Solid and dashed lines show statistical power of 0.99 and 0.5, respectively. Plots and bars indicate ORs and 95% confidence intervals (CIs) of SLE_ON(+)-associated variants in current Japanese study, respectively.
Statistical power analysis for Japanese GWAS
We evaluated the statistical power for this study. The association study between SLE_ON(+) and BBJ controls had >99% power to identify variants with minor allele frequency (MAF) of 0.01 and OR of 4.02 and with MAF of 0.1 and OR of 1.87, with significance at P-value = 5 × 10−8 (Fig. 2E). This statistical power was not obtained when SLE_ON(−) patients were used as controls (Fig. 2E). Because there were no significant variants with MAF between 0.1 and 0.5 (after excluding SLE-associated variants), common variants (MAF > 0.1) with large effects (OR > 1.87) on SLE_ON(+) were unlikely to be present.
Association study in Korean subjects and meta-analysis with Japanese GWAS
We took advantage of Korean data for 148 SLE_ON(+) and 37 015 population controls to assess consistent associations of the four variants (24). Although rs7911501 on MIR4293/MIR1265 (OR = 1.52) and rs372605131 on TRIM49/NAALAD2 (OR = 1.14) are not significant (P > 0.05), they showed the same direction for their effects as the Japanese study (Supplementary Material, Table S6). Especially, rs7911501 had almost the same effect size (OR = 1.61) in association between SLE_ON(+) and SLE_ON(−), and opposite direction of effect size (OR = 0.9) in association between SLE_ON(−) and population control, which was the same trend as the Japanese study (Supplementary Material, Table S6). We noted that rs145720245 was too rare in Koreans (MAF = 0.005) to find even a single heterozygote in the limited number of case subjects assuming a similar effect size. Rs2714333 on RREB1 did not show even a trend of the same direction of association as the Japanese study (Supplementary Material, Table S6).
When we evaluated overall significance in a meta-analysis of the two datasets composing 784 SLE_ON(+) and 132 603 controls, rs7911501 on MIR4293/MIR1265 (OR = 1.99, P-value = 1.1 × 10−9), rs372605131 on TRIM49/NAALAD2 (OR = 1.65, P-value = 4.8 × 10−8) and rs145720245 on MYO16 (OR = 3.91, P-value = 4.9 × 10−10) showed genome-wide significance (Table 2). We did not observe heterogeneity among the three studies (P-heterogeneity > 0.05). However, rs2714333 on RREB1 did not satisfy genome-wide significance. Thus, we regarded MIR4293/MIR1265, TRIM49/NAALAD2 and MYO16 loci as statistically significant. To further evaluate potential shared genetic components among ONFH, in general, we evaluated associations of the three significant variants with idiopathic ONFH (I-ONFH) which have neither excessive alcohol consumption nor steroid intake (see Materials Methods). As a result, all three variants showed comparable directions of effects with SLE_ON(+) (Supplementary Material, Table S7).
Table 2.
Meta-analysis of two GWASs comprising 784 SLE_ON(+) and 132 603 controls in Japan and Korea
| CHR | POS | rsID | EA/NEA | Gene | Function | EAF | OR [95%CI] | P-value | Phet |
|---|---|---|---|---|---|---|---|---|---|
| Significant in the meta-analysis | |||||||||
| 10 | 14 436 836 | rs7911501 | A/G | MIR4293/MIR1265 | Intergenic | 0.028 | 1.99 [1.59–2.48] | 1.1 × 10−9 | 0.56 |
| 11 | 89 538 685 | rs372605131 | C/T | TRIM49/NAALAD2 | Intronic | 0.089 | 1.65 [1.38–1.98] | 4.8 × 10−8 | 0.21 |
| 13 | 109 491 871 | rs145720245 | A/G | MYO16 | Intronic | 0.006 | 3.91 [2.54–6] | 4.9 × 10−10 | 0.34 |
| Not significant in the meta-analysis | |||||||||
| 6 | 7 236 620 | rs2714333 | T/C | RREB1 | Intronic | 0.015 | 2.76 [1.9–4] | 8.4 × 10−8 | 0.1 |
SLE_ON(+), SLE with S-ONFH; rsID, reference single-nucleotide polymorphism ID; Phet, P heterogeneity among Japanese 2 studies and Korean study.
Rs372605131 locates in intron of TRIM49; however, it is an eQTL of NAALAD2.
Identification of potentially causal genes
We identified sets of 95% credible variants by fine-mapping in MIR4293/MIR1265 and TRIM49/NAALAD2 locus (Supplementary Material, Table S8) by using the FINEMAP software (25). As a result, TRIM49/NAALAD2 locus had a candidate causal variant [rs372605131; posterior probability (PP) > 0.5], on the other hand, MIR4293/MIR1265 did not (PP < 0.154), because of the LD structure. (MYO16 locus did not have significant variants <1 Mb from the lead variant (rs145720245).) We checked the functional annotations of rs145720245 on MYO16 locus, rs372605131 on TRIM49/NAALAD2 locus and a set of credible variants on MIR4293/MIR1265 and tried to investigate potentially causal genes in three significant loci (Fig. 3A–C). As a result, these variants did not include missense or loss-of-function mutations. In TRIM49/NAALAD2 locus, eQTL analysis using GTEx v8 (26) revealed that rs372605131 was significantly associated with increased NAALAD2 expression in the tibial artery (Fig. 4A and B, Supplementary Material, Fig S3, Supplementary Material, Table S9). Though most of GTEx v8 consist of European descent (26), Spearman’s rank correlation coefficient analyses showed correlated association signals (Spearman’s rank correlation = 0.438) and Locus compareR showed suggestive colocalization signal (PP.H4 = 38.3%) between GWAS and eQTL regardless of different LD structures between populations, suggesting that eQTL of NAALAD2 in the tibial artery might be colocalized with the SLE_ON(+) association signal (Fig. 4C). Because other novel loci had no variants with evidences of missense, loss-of-function or eQTL, we regarded MYO16 and MIR4293, the nearest genes from the lead or a set of 95% credible variants, as potentially causal genes (Fig. 3A and C).
Figure 3.
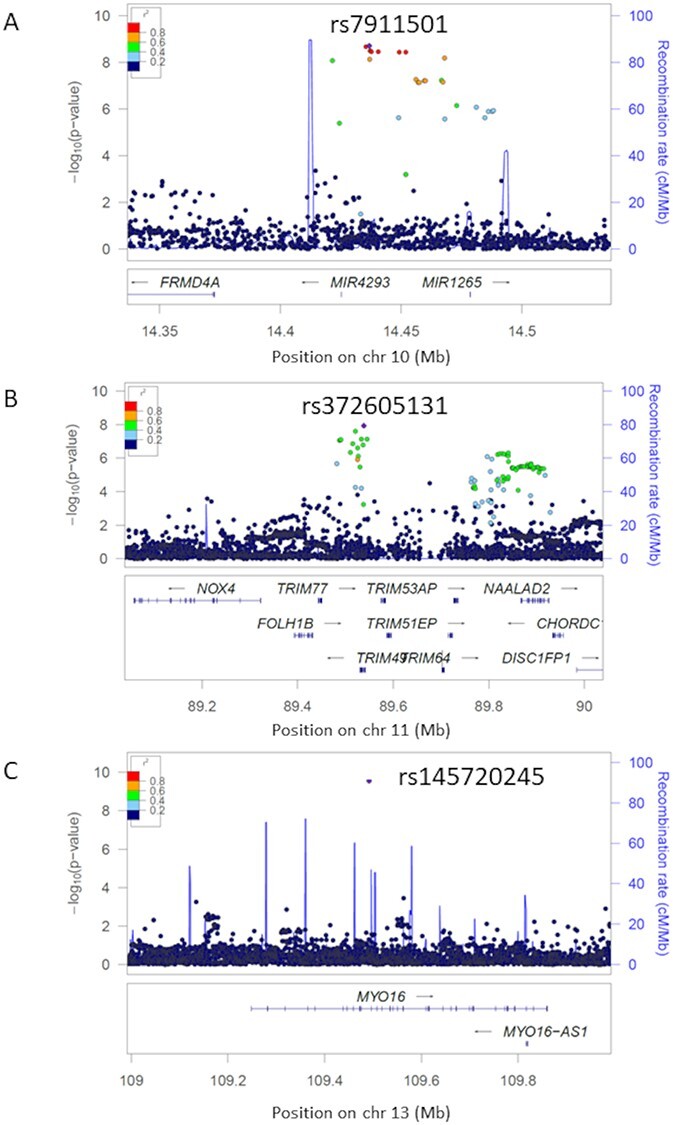
Regional plots of SLE_ON(+) susceptibility loci. Regional plots of SLE_ON(+) susceptibility loci. X- and Y-axes indicate genomic positions and minus log10-transformed association P-values, respectively. Colors of dots show strength of linkage disequilibrium (R-square) with the lead variant in each locus. (A) MIR4293/MIR1265 region on 10p13. (B) TRIM49/NAALAD2 region on 11q14.3. (C) MYO16 region on 13q33.3.
Figure 4.
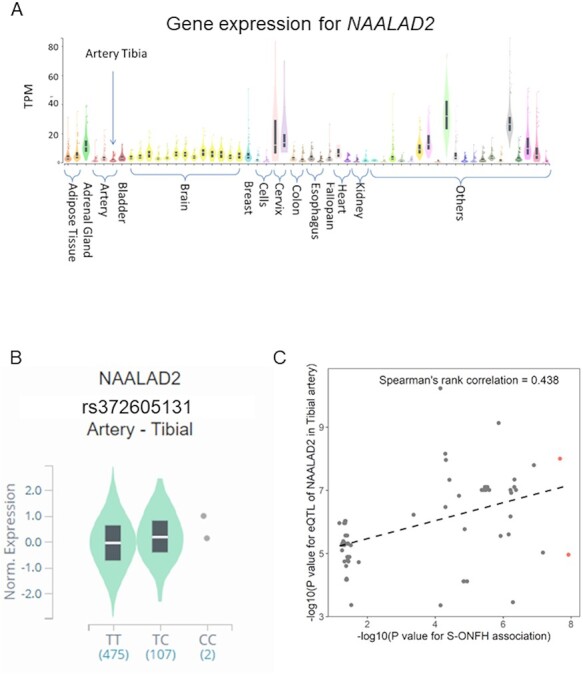
Functional annotation for SLE_ON(+) susceptibility locus on 11q14.3. (A) Expression levels of NAALAD2 in 54 tissues within GTEx v8. X- and Y-axes show groups of tissues and transcripts per million, respectively. Box plots show median and 25th and 75th percentiles. (B) Violin plot of NAALAD2 expression levels in the tibial artery for each genotype of rs372605131. X- and Y-axes show haplotype and normalized gene expression levels, respectively. Box plots show median and 25th and 75th percentiles. (C) Scatter plot of variants in the susceptibility locus. X-axis is minus log10-transformed association P-values for the association study between SLE_ON(+) and BBJ controls in Japan, and Y-axis is minus log10-transformed eQTL P-values for NAALAD2 in GTEx v8 in the tibial artery. Red dots indicate significant variants in the association study (P < 5 × 10−8). Dashed line indicates the regression line.
Pathway enrichment analysis for target genes of MIR4293
MIR4293 is a type of microRNA and is considered to regulate other genes (27). We identified 111 target genes with target score ≥ 80, which are likely regulated by MIR4293 by using miRDB (Supplementary Material, Table S10) (28). MIR4293 regulates SREBF2 in the sterol regulatory element-binding protein (SREBP) signaling pathway, which includes SREBP2 as a previously reported S-ONFH susceptibility gene by candidate gene analysis (9). Moreover, we conducted pathway enrichment analysis for 111 target genes by using FUMA (29). As a result, we found that a PPARG-related adipogenesis pathway, a relevant pathway for S-ONFH (30,31), was significantly enriched in the target genes regulated by MIR4293 (Supplementary Material, Table S11). Meanwhile, 47 genes regulated by MIR1265, another gene near the variant, did not show enrichment of relevant pathways.
LDSC-heritability enrichment analysis
We found some types of helper T cells and hematopoietic cell groups with strong enrichment of heritability in SLE_ON(+) [SLE_ON(+) vs. BBJ controls, Supplementary Material, Tables S12 and S13] by LDSC software with baseline model version2.2 (32). However, strong enrichment of SLE heritability in these cell types and cell groups could apparently explain the findings (33). Similarly, we did not find SLE_ON(+)-specific significant genetic correlations between SLE_ON(+) [SLE_ON(+) vs. BBJ controls] and 99 other traits (59 complex traits) (34) (40 diseases) (35)) (Supplementary Material, Table S14).
Discussion
The majority of ONFH patients have no evident cause of ischemia, such as decompression sickness (36), sickle cell disease (37) or trauma. How steroid administration leads to ischemia remains unknown. Many genetic studies have been performed on S-ONFH, but no definite susceptibility genes have been identified. One possible reason for the lack of established genetic loci for S-ONFH is that S-ONFH has various underlying diseases, including SLE, polymyositis/dermatomyositis, nephrotic syndrome and mixed connective tissue disease, and that the genetic backgrounds of these diseases can influence GWAS results. Another reason could be the low power of previous studies. Difficulty in recruiting S-ONFH cases and controls with harmonized backgrounds, especially from the viewpoint of underlying diseases, has made it difficult to identify consistent associations with S-ONFH.
In this study, we addressed two problems in GWAS for ONFH. First, we focused on S-ONFH cases stringently restricted to SLE patients. Second, we took the approach to use controls irrespective of their backgrounds (disease-mixed controls; a common strategy for GWAS (18,19)) in the initial stage to maximize statistical power and showed comparable effect sizes of significant variants using definitive controls with the same backgrounds as cases. As a result, three loci showed significant GWAS signals in the meta-analysis of the Japanese and Korean studies. Our study could serve as a model to identify susceptibility loci for intra-disease phenotypes. In the current study, the information of steroid dosage is not available. Since the steroid dosage tends to be related to SLE_ON(+) occurrence, whether the genetic risk of SLE_ON(+) is modulated by steroid dosage should be evaluated in detail in future studies.
Though the statistical power analysis showed that our association study should identify variants with MAF > 0.1 and OR > 1.87, our study did not identify such significant variants. This result indicates that common variants (MAF > 0.1) with large effect for SLE_ON(+), which may serve as biomarkers and predictive markers, are unlikely to be present as far as SLE_ON(+) associations are distinct from those for SLE susceptibility. It is therefore necessary to collect further samples and to identify common variants with a weak effect size or rare variants with modest to strong effect size.
We conducted association study for three significant variants of SLE_ON(+) on I-ONFH and BBJ controls to evaluate whether these variants have effect to other type of ONFH without SLE. As a result, all of variants shared effects between I-ONFH and SLE_ON(+). This result suggests that SLE_ON(+) and I-ONFH have partially common genetic background and pathology and may be compatible with the previous study reporting ONFH in some SLE patients without steroid administration and excessive alcohol consumption (38). We should increase sample sizes of SLE_ON(+) and I-ONFH to replicate the current findings in future studies. Moreover, because current data are composed on East Asian population, we attempted to replicate significant variants on UK BioBank (UKBB) data. However, UKBB did not have the data of ONFH. In the future, we should conduct a trans-ethnic meta-analysis among multiple populations.
We identified MIR4293, NAALAD2 and MYO16 as candidate causal genes for SLE_ON(+). MIR4293 regulates SREBF2 in the lipid metabolism pathway, including SREBP2, a previously reported S-ONFH susceptibility gene (9). Moreover, pathway enrichment analysis indicated that the 111 genes regulated by MIR4293 were significantly enriched in a PPARG-related adipogenesis pathway. PPARG was reported as a susceptibility gene for ONFH (16,30). A previous study indicated that glucocorticoid administration induced promotive differentiation of adipocytes and increased intraosseous pressure in steroid-treated rabbits (31). As a result, the femoral head of the rabbits exhibited osteonecrosis. From these results, we concluded that MIR4293 is a plausible candidate causal gene for SLE_ON(+). There were no available data for MIR4293 in GTEx v8, and we were unable to evaluate the eQTL effect of the lead variant on MIR4293. To clearly verify that MIR4293 is associated with SLE_ON(+), it is necessary to investigate gene expression and eQTL in ONFH-relevant tissues by in vitro or in vivo assays. Although most previous candidate gene analyses focused on protein-coding genes, recent transcriptome studies demonstrated that S-ONFH was correlated with non-coding RNAs, such as microRNAs (39,40) and long-non-coding RNAs (41). These results appear compatible with the present and previous findings. Actually, our previous GWAS (17) for ONFH identified, as a susceptibility gene, long non-coding RNA 01370 whose regulatory function to lipid-metabolism pathway was suggested in in silico analyses. NAALAD2 is a member of the N-acetylated alpha-linked acidic dipeptidase gene family and is ubiquitously expressed in tissues. A previous genetic study demonstrated that NAALAD2 was associated with basophil count (42). However, its detailed function was not revealed. We found an eQTL association of the lead variant with NAALAD2 in the tibial artery. The eQTL of NAALAD2 may share an effective variant with GWAS signals (Spearman’s correlation = 0.438, PP.H4 = 38.3%). High expression of NAALAD2 in arteries may increase the risk of SLE_ON(+). However, the colocalization signal is not very convincing. Since most of eQTL data are derived from European subjects, further colocalization analysis by using East Asian eQTL data and functional analyses in arterial tissues should be conducted to confirm the association. MYO16 is a member of the myosin superfamily and it may have an important role in neural development, although the detailed function has not yet been revealed (43). The allele frequency of rs145720245 is quite low, and the accuracy of the association of this locus should be evaluated in further samples. In conclusion, we identified three susceptibility loci for SLE_ON(+) and three candidate causal genes. Our approach will be useful for the detection of loci associated with intra-disease phenotypes. The present findings suggest that common variants with large effects on SLE_ON(+) are unlikely. We must collect further samples to identify susceptibility genes for SLE_ON(+) and elucidate the biological mechanisms underlying SLE_ON(+) and other types of ONFH.
Materials and Methods
Japanese subjects in the GWAS
We collected a total of 648 SLE_ON(+) and 697 SLE_ON(−) patients with SLE diagnosed by the 1982 (20) or 1997 ACR criteria (21). SLE_ON(+) was defined as ONFH findings on X-rays or MRI. SLE_ON(−) was defined as no ONFH findings on MRI at >6 months after starting steroid administration because almost all ONFH occurs within 3 months of steroid administration (5,6). Clinical information and peripheral blood for SLE_ON(+) and SLE_ON(−) patients were collected in 32 hospitals across Japan from 2012 to 2019. We used genome-wide screening data in BBJ controls for subjects with 47 common diseases as controls (18,19). After excluding SLE- and S-ONFH-related diseases, like autoimmune diseases and osteoporosis, data for 95 588 subjects were used as BBJ controls. Written informed consent was obtained from all patients and/or their guardians. The study was approved by the Institutional Review Board of Kyushu University, RIKEN and from all participating institutes for sample collection. All methods were carried out in accordance with relevant guidelines and regulations.
QC in Japanese GWAS
Genomic DNA was extracted from peripheral blood using a standard protocol. Genotyping was performed by Illumina HumanOmniExpressExome, HumanOmniExpress or HumanExome BeadChips (Supplementary Material, Table S1).
For QC of subjects, subjects with call rate < 0.98, high-degree relatedness with other subjects and outliers of East Asian ethnicity were excluded. For QC of variants, variants with call rate < 0.99, P-value for Hardy–Weinberg equilibrium < 1.0 × 10−6 and minor allele count < 10 were excluded (Supplementary Material, Table S1).
Imputation in Japanese GWAS
The reference panel for imputation was composed of 1000 Genomes Project phase 3 (version 5) (44) and Japanese whole-genome sequence data (Flaganan et al., manuscript in preparation) with 3256 high-depth subjects (≥30 read counts) and 4216 low-depth subjects (≤15 read counts). We performed pre-phasing using EAGLE2.4.1 (https://alkesgroup.broadinstitute.org/Eagle/) to determine the haplotypes. Genotypes were imputed using Minimac4 (version1.0.0) (45). After imputation, variants with MAF < 0.005 and low imputation quality (R2 < 0.3) were excluded (Supplementary Material, Table S1).
GWAS in Japanese subjects
We used SLE_ON(+) samples collected from 2012 to 2016 as first GWAS as we previously published (17) and from 2017 to 2019 as second GWAS. The BBJ control samples were used for the first and second studies roughly proportionally to the case subjects. GWAS was conducted by logistic regression with the top 10 principal components and sex (only for variants in autosome) as covariates using PLINK2.0 software (46) for first and second GWAS, separately. For meta-analysis comprising first and second GWAS, an inverse-variance-based method was performed by METAL (version2011-03-25) (47). Variants satisfying P-value < 5 × 10−8 were regarded as significant for SLE_ON(+). We regarded multiple significant loci as independent when they were apart from each other at least 1Mbp. We conducted conditional analyses for all significant variants conditioned for a lead variant in the same locus using GCTA-COJO (version1.93.1) (48). Variants with P-value < 5 × 10−8 in the conditional analyses were regarded as independent from the lead variant. To distinguish confounding bias from polygenic effects, we performed LDSC with LD scores from East Asian population of 1000 Genomes Project descendants using LDSC software (version1.0.0) (22). Variants were restricted to Hapmap3 (49) as reliable variants for the test. An intercept nearby 1 indicated that inflation of the genomic control inflation factor was not derived from bias, but from polygenicity. We performed firth logistic regression analysis for significant variants with current sample size and down-sampled controls (case–control ratio of 1:19) by using PLINK2.0 software (50,51).
Comparison of effect size in with association studies using SLE_ON(−)
We excluded regions within 1Mbp from the significant variants (P-value < 5 × 10−8) in known SLE susceptibility loci using GWAS catalog (https://www.ebi.ac.uk/gwas/), GWASkb (http://gwaskb.stanford.edu) and significant loci in our recent SLE international meta-analysis (23) from significant loci. GWAS catalog was obtained on 13 November, 2020. We regarded remaining variants as candidates for SLE_ON(+) susceptibility. We compared the allele frequencies of candidate variants between SLE_ON(+) and SLE_ON(−) or BBJ controls. We conducted the association study for SLE_ON(+)-associated variants between SLE_ON(−) and SLE_ON(+) or BBJ using the same approach of the SLE_ON(+) GWAS (we took the two-staged GWAS depending on collection dates for SLE subjects, Supplementary Material, Fig S1). Meta-analysis of the first and second studies was conducted by METAL.
Statistical power analysis
We evaluated the statistical power of our datasets [SLE_ON(+) vs. BBJ controls and SLE_ON(+) vs. SLE_ON(−)] using the Genpwr package (version1.0.2) in R software as an additive model (https://cran.r-project.org/web/packages/genpwr/index.html). We set the type 1 error rate and statistical power to 5 × 10−8 and 0.99 or 0.5, respectively.
Korean association study and meta-analysis with Japanese GWAS
We conducted association study using Korean data to assess consistent associations of the SLE_ON(+) associated variants in East Asian populations. We collected 148 SLE_ON(+) and 1158 SLE_ON(−) in Hanyang University Hospital for Rheumatic Diseases (Seoul, Korea). Subjects were diagnosed as SLE by the 1997 ACR criteria (21). SLE_ON(+) was defined as ONFH findings on X-rays or MRI. SLE_ON(−) was defined as no hip pain. We used 37 015 samples in KoGES and the Hanyang University Hospital for Rheumatic Diseases database as population controls (24). Written informed consent was obtained from all patients and/or their guardians. The study was approved by the Institutional Review Board of Hanyang University Hospital. The procedures for genotyping, QC and imputation were described previously (24).
We focused on significant variants in the Japanese study and evaluated their associations in the Korean dataset using (1) 148 SLE_ON(+) and 37 015 population controls, (2) 148 SLE_ON(+) and 1158 SLE_ON(−) and (3) 1158 SLE_ON(−) and 37 015 population controls. We conducted logistic regression analysis with the top 10 principal components and sex (for only variants in autosome) as covariates using EPACTS software (v3.2.6 or 3.3; http://genome.sph.umich.edu/wiki/EPACTS) (52). Because Korean SLE_ON(+) and SLE_ON(−) genotyping data were composed of two datasets, the association studies were further adjusted for the datasets. For the meta-analysis comprising SLE_ON(+) and controls in Japan and Korea, an inverse-variance-based method was performed by METAL (version2011-03-25) (47). P-heterogeneity among Japanese 2 studies and Korean study was evaluated using Cochran Q test and PLINK1.90 software (46). Finally, variants satisfying P-value < 5 × 10−8 were regarded as significant for SLE_ON(+).
Evaluation for effect of significant variants to I-ONFH
We evaluated the associations of significant variants with 127 I-ONFH (17) cases using the same BBJ controls and statistical framework we used in SLE_ON(+) analysis.
Evaluation of significant associations and potentially causal genes
For each significant locus, we conducted bioinformatics analysis to identify potentially causal genes. At first, to identify candidates of causal markers, we performed fine-mapping analyses calculating PP of being causal variants using the FINEMAP software (25). LD structure of reference panel restricting to Japanese whole-genome sequence data and 1000 Genomes Project phase 3 (version 5) (44) was used for the analyses. We analyzed the two regions (the TRIM49/NAALAD2 and MIR4293/1265 regions) containing multiple GWAS significant variants in the current study. We regarded a variant as causal if it showed PP > 0.5. If none of variants showed PP > 0.5, a 95% credible set of variants was generated. We functionally annotated the causal or a 95% credible set of variants by using Annovar (version: 2017-07-17) (53). If the locus has no another significant variants, the lead variants is functionally annotated. We analyzed whether these variants were missense or loss-of-function variants. Then, we checked whether these variants have been reported as significant eQTL in 49 tissues in GTEx v8 (where >80% of its subjects are of European descent) (26). We checked the colocalization signal between eQTL and GWAS by Spearman’s rank correlation and Locus CompareR (54). If annotated variants were missense, loss-of-function or eQTL variants for surrounding genes, we regarded the genes as potentially causal genes. If not, we regarded the nearest genes from the lead variants as potentially causal genes.
Pathway enrichment analysis for target genes of microRNA
In general, a microRNA downregulates several genes by binding to their 3′UTRs (27). If microRNA was identified as potentially causal gene, we investigated genes regulated by microRNA using miRDB (version 6.0) (28), a computational prediction tool that searches for microRNA target genes. We defined genes satisfying target score ≥80 as target genes, representing reliable microRNA-regulated genes. To evaluate the pathway in which target genes are accumulated, we performed pathway enrichment analysis for target genes using FUMA (version 1.3.6) (29) and regarded pathways satisfying false discovery rate (FDR) < 0.05 as significant.
LDSC-heritability enrichment analysis
Partition heritability enrichment analysis for SLE_ON(+) was conducted in 10 specific cell groups and 220 combinations of cell types and histone marks using LDSC software with baseline model version 2.2 (32). Genetic correlation analyses between SLE_ON(+) and 99 other traits (59 complex traits (34) and 40 diseases (35)) were conducted using LDSC software (22). For both partition heritability enrichment and genetic correlation analysis, Japanese GWAS results for SLE_ON(+) and BBJ controls were used as association data for SLE_ON(+), and the significance threshold was set at FDR < 0.05. We defined significant results as SLE_ON(+)-specific heritability enrichment or genetic correlations only if observed in the SLE_ON(+) study and not observed in SLE susceptibility studies (23,33).
Supplementary Material
Acknowledgements
We acknowledge the participants in this study. We thank Edanz Group (https://en-author-services.edanzgroup.com/ac) for editing a draft of this manuscript. We appreciate members of Japanese Research Committee on Idiopathic Osteonecrosis of the Femoral Head: Yasuharu Nakashima, Kazuyuki Karasuyama, Kazuhiko Sonoda, Yusuke Kubo, Takeshi Utsunomiya, Hiroyuki Hatanaka, Shuji Baba, Koichiro Kawano, Noriko Yamamoto, Yukihide Iwamoto, Satoshi Ikemura and Ryosuke Yamaguchi (Department of Orthopaedic Surgery, Graduate School of Medical Sciences, Kyushu University), Hiroshi Kataoka and Makoto Kondo (Department of Rheumatology & Clinical Immunology, Sapporo City General Hospital), Tsuyoshi Asano, Tohru Irie and Norimasa Iwasaki (Department of Orthopaedic Surgery, Faculty of Medicine and Graduate School of Medicine, Hokkaido University), Tatsuya Atsumi (Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University), Shunji Kishida (Department of Orthopaedic Surgery, Seirei Sakura Citizen Hospital), Shigeo Hagiwara (Department of Orthopaedic Surgery, Graduate School of Medicine, Chiba University), Ichiei Narita (Division of Clinical Nephrology and Rheumatology, Kidney Research Center, Niigata University Graduate School of Medical and Dental Sciences), Koichi Akashi (Faculty of Medical Sciences, Kyushu University), Hiroshi Tsukamoto (Shin-Kokura Hospital), Yojiro Arinobu and Hiroki Mitoma (Kyushu University Hospital), Mitsuteru Akahoshi (Faculty of Medicine, Saga University), Masahiro Ayano (Graduate School of Medical Sciences, Kyushu University), Atsuhiro, Yoshiaki Toyama and Atsushi Funayama (Department of Orthopaedic Surgery, Keio University School of Medicine), Kunihiro Yamaoka (Department of Rheumatology and Infectious Diseases, Kitasato University School of Medicine), Hironari Hanaoka (Division of Rheumatology, Department of Internal Medicine, Keio University School of Medicine), Yasushi , Hisashi Yamanaka, Tetsuji Hosozawa and Shigeki Momohara (Institute of Rheumatology, Tokyo Women’s Medical University), Fumio Sekiya and Masakazu Matsushita (Department of Internal Medicine and Rheumatology, Juntendo University School of Medicine), Megumi Matsuhashi and Kazuhide Tanimura (Hokkaido Medical Center for Rheumatic Diseases), Takashi Sakai (Department of Orthopaedic Surgery, Yamaguchi University Graduate School of Medicine), Wataru Ando, Takashi Nishii and Masaki Takao (Department of Orthopaedic Medical Engineering, Osaka University Graduate School of Medicine), Ryosuke Nakanishi (Department of Orthopaedic Surgery, Fujigaoka Hospital, Showa University School of Medicine), Yukiharu Hasegawa (Department of Rehabilitation, Kansai University of Welfare Science), Kazuyoshi Saito (Tobata General Hospital), Kazuhisa Nakano (The First Department of Internal Medicine, University of Occupational and Environmental Health, Japan), Tetsuya Sakamoto (Department of Orthopaedic Surgery, Faculty of Medicine, Fukuoka University), Keiichiro Ueshima (Kyoto Interdisciplinary Institute Hospital of Community Medicine), Yoshitomo Kajino (Department of Orthopaedic Surgery, Graduate School of Medical Science, Kanazawa University), Yoshiharu Amasaki (Department of Rheumatology, Kuriyama Red Cross Hospital), Hiroaki Nakamura and Shigekazu Mizokawa (Department of Orthopaedic Surgery, Osaka City University Graduate School of Medicine), Shinichiro Kume (Orthopedis and Joint Surgery Center, Kurume Univ. Medical Center), Akihiro Sudo and Masahiro Hasegawa (Department of Orthopaedic Surgery, Mie University Graduate School), Toru Ichiseki (Department of Orthopaedic Surgery, Kanazawa Medical University), Takuma Yamasaki (Department of Orthopaedic Surgery, National Hospital Organization Kure Medical Center and Chugoku Cancer Center).
Conflict of Interest statement. The authors declare no conflicts of interest associated with this manuscript.
Contributor Information
Hiroyuki Suetsugu, Laboratory for Bone and Joint Diseases, RIKEN Center for Medical Sciences, Tokyo 108-8639, Japan; Laboratory for Statistical and Translational Genetics Analysis, RIKEN Center for Integrative Medical Sciences, Kanagawa 230-0045, Japan; Department of Orthopaedic Surgery, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan.
Kwangwoo Kim, Department of Biology, Kyung Hee University, Seoul 02447, Republic of Korea; Department of Biomedical and Pharmaceutical Sciences, Kyung Hee University, Seoul, Republic of Korea.
Takuaki Yamamoto, Department of Orthopaedic Surgery, Faculty of Medicine, Fukuoka University, Fukuoka 814-0180, Japan.
So-Young Bang, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea; Hanyang University Institute for Rheumatology Research, Seoul 04763, Korea.
Yuma Sakamoto, Koga Hospital 21, Fukuoka 839-0801, Japan.
Jung-Min Shin, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea.
Nobuhiko Sugano, Department of Orthopaedic Medical Engineering, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan.
Ji Soong Kim, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea.
Masaya Mukai, Department of Rheumatology & Clinical Immunology, Sapporo City General Hospital, Hokkaido 060-8604, Japan.
Yeon-Kyung Lee, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea.
Koichiro Ohmura, Department of Rheumatology and Clinical Immunology, Kyoto University Graduate School of Medicine, Kyoto 606-8507, Japan.
Dae Jin Park, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea.
Daisuke Takahashi, Department of Orthopaedic Surgery, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, Hokkaido 060-8638, Japan.
Ga-Young Ahn, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea.
Kohei Karino, Department of Rheumatology, Endocrinology and Nephrology, Faculty of Medicine and Graduate School of Medicine, Hokkaido University, Hokkaido 060-8638, Japan.
Young-Chang Kwon, Hanyang University Institute for Rheumatology Research, Seoul 04763, Korea.
Tomoya Miyamura, Department of Internal Medicine and Rheumatology, National Hospital Organization Kyushu Medical Center, Fukuoka 810-8563, Japan.
Jihye Kim, Hanyang University Institute for Rheumatology Research, Seoul 04763, Korea.
Junichi Nakamura, Department of Orthopaedic Surgery, Graduate School of Medicine, Chiba University, Chiba 260-8677, Japan.
Goro Motomura, Department of Orthopaedic Surgery, Graduate School of Medical Sciences, Kyushu University, Fukuoka 812-8582, Japan.
Takeshi Kuroda, Niigata University Health Administration Center, Niigata 950-2181, Japan.
Hiroaki Niiro, Department of Medical Education, Kyushu University Graduate School of Medical Sciences, Fukuoka 812-8582, Japan.
Takeshi Miyamoto, Department of Orthopaedic Surgery, Faculty of Life Sciences, Kumamoto University, Kumamoto 860-8556, Japan.
Tsutomu Takeuchi, Division of Rheumatology, Department of Internal Medicine, Keio University School of Medicine, Tokyo 160-8582, Japan.
Katsunori Ikari, Institute of Rheumatology, Tokyo Women’s Medical University, Tokyo 162-8666, Japan.
Koichi Amano, Department of Rheumatology & Clinical Immunology, Saitama Medical Center, Saitama Medical University, Saitama 350-8550, Japan.
Yoshifumi Tada, Department of Rheumatology, Faculty of Medicine, Saga University, Saga 849-8501, Japan.
Ken Yamaji, Department of Internal Medicine and Rheumatology, Juntendo University School of Medicine, Tokyo 113-8421, Japan.
Masato Shimizu, Hokkaido Medical Center for Rheumatic Diseases, Hokkaido 063-0811, Japan.
Takashi Atsumi, Department of Orthopaedic Surgery, Showa University School of Medicine, Tokyo 142-8555, Japan.
Taisuke Seki, Department of Orthopaedic Surgery, Nagoya University Graduate School of Medicine, Nagoya 466-8550, Japan.
Yoshiya Tanaka, The First Department of Internal Medicine, School of Medicine, University of Occupational and Environmental Health, Fukuoka 807-8555, Japan.
Toshikazu Kubo, Graduate School of Medical Science, Kyoto Prefectural University of Medicine, Kyoto 602-8566, Japan.
Ryo Hisada, Obihiro Kosei Hospital, Hokkaido 080-0024, Japan.
Tomokazu Yoshioka, Division of Regenerative Medicine for Musculoskeletal System, Faculty of Medicine, University of Tsukuba, Ibaraki 305-8575, Japan.
Mihoko Yamazaki, Kido Hospital, Niigata 950-0862, Japan.
Tamon Kabata, Department of Orthopaedic Surgery, Graduate School of Medical Science, Kanazawa University, Kanazawa 920-8641, Japan.
Tomomichi Kajino, Department of Orthopaedic Surgery, Tonan Hospital, Hokkaido 060-0004, Japan.
Yoichi Ohta, Department of Orthopaedic Surgery, Osaka City University Graduate School of Medicine, Osaka 545-8585, Japan.
Takahiro Okawa, Orthopedis and Joint Surgery Center, Kurume University Medical Center, Fukuoka 839-0863, Japan.
Yohei Naito, Department of Orthopaedic Surgery, Mie University Graduate School of Medicine, Mie 514-8507, Japan.
Ayumi Kaneuji, Department of Orthopaedic Surgery, Kanazawa Medical University, Ishikawa 920-0293, Japan.
Yuji Yasunaga, Hiroshima Prefectural Rehabilitation Center, Hiroshima 739-0036, Japan.
Kenji Ohzono, Department of Orthopaedic Surgery, Amagasaki Chuo Hospital, Hyogo 661-0976, Japan.
Kohei Tomizuka, Laboratory for Statistical and Translational Genetics Analysis, RIKEN Center for Integrative Medical Sciences, Kanagawa 230-0045, Japan.
Masaru Koido, Laboratory for Statistical and Translational Genetics Analysis, RIKEN Center for Integrative Medical Sciences, Kanagawa 230-0045, Japan; Division of Molecular Pathology, Department of Cancer Biology, Institute of Medical Science, The University of Tokyo, Tokyo 108-8639, Japan.
Koichi Matsuda, Laboratory of Genome Technology, Human Genome Center, Institute of Medical Science, The University of Tokyo, Tokyo 108-8639, Japan; Laboratory of Clinical Genome Sequencing, Department of Computational Biology and Medical Sciences, Graduate School of Frontier Sciences, The University of Tokyo, Tokyo 108-8639, Japan.
Yukinori Okada, 44Department of Statistical Genetics, Osaka University Graduate School of Medicine, Osaka 565-0871, Japan; Laboratory of Statistical Immunology, Immunology Frontier Research Center (WPi-iFReC), Osaka University, Osaka 565-0871, Japan.
Akari Suzuki, Laboratory for Autoimmune Diseases, RIKEN Center for Integrative Medical Sciences, Kanagawa 230-0045, Japan.
Bong-Jo Kim, Division of Genome Research, Center for Genome Science, National Institute of Health, Osong Health Technology Administration Complex, Cheongju 28159, Korea.
Yuta Kochi, Department of Genomic Function and Diversity, Medical Research Institute, Tokyo Medical and Dental University, Tokyo 113-8510, Japan.
Hye-Soon Lee, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea; Hanyang University Institute for Rheumatology Research, Seoul 04763, Korea.
Shiro Ikegawa, Laboratory for Bone and Joint Diseases, RIKEN Center for Medical Sciences, Tokyo 108-8639, Japan.
Sang-Cheol Bae, Department of Rheumatology, Hanyang University Hospital for Rheumatic Diseases, Seoul 04763, Korea; Hanyang University Institute for Rheumatology Research, Seoul 04763, Korea.
Chikashi Terao, Laboratory for Statistical and Translational Genetics Analysis, RIKEN Center for Integrative Medical Sciences, Kanagawa 230-0045, Japan; Clinical Research Center, Shizuoka General Hospital, Shizuoka 420-8630, Japan; The Department of Applied Genetics, The School of Pharmaceutical Sciences, University of Shizuoka, Shizuoka 422-8526, Japan.
Funding
This study was supported in part by the Practical Research Project for Rare/Intractable Diseases from Japan Agency for Medical Research and development, AMED, by RIKEN Junior Research Associate Program (for H.S.), by Grant of Japan Orthopaedics and Traumatology Research Foundation, No. 350 (for Y.S.), by Center for Genome Science, Korea National Institute of Health, Republic of Korea (4845-301, 3000-3031) (for B-J Kim), by Basic Science Research Program through the National Research Foundation of Korea (NRF) funded by the Ministry of Education (NRF-2021R1A6A1A03038899) (for S-C.B) and Hanyang University Institute for Rheumatology Research (for S-C.B). The study sponsors had no involvement in this study.
Patient consent for publication
Not required.
Ethics approval
The study was approved by the Institutional Review Boards at RIKEN Center for Medical Sciences and by the Hanyang University Hospital of Rheumatic Diseases.
Data sharing statement
The meta-analysis summary association statistics in the present study are available from the corresponding author on reasonable request.
Authors’ contributions
H.S. and K.K. contributed equally to this work. C.T. conceived the study design. S.-C.B. and C.T. acquainted the financial support. H.S., K.K., S.-C.B. and C.T. wrote the manuscript. H.S. and K.K. conducted all of the analyses with the help of M.K., S.I., Y.K. and C.T. T.T., Y.S., N.S., M.M., K.O., D.T., K.K., T.M., J.N., G.M., T.K., H.N., T.M. K.K., S.-Y.B., J.-M.S., J.S.K., Y.-K.L., D.J.P., G.-Y.A., T.T., Y.-C.K., J.K., K.I., K.A., Y.T., K.Y., M.S., T.A., T.S., Y.T., T.K., R.H., T.Y., M.Y., T.K., T.K., K.O., T.O., Y.N., Y.O., A.K., Y.Y., K.O., B.-J.K., H.-S.L., S.I., S.-C.B., and the Japanese Research Committee on Idiopathic Osteonecrosis of the Femoral Head collected case samples and clinical information, or generated genetic data. K.M. provided genetic data of BioBank Japan. T.K. made Japanese reference panel for imputation. S.-C.B. and C.T. managed the cohort data. S.-C.B. and A.S. conducted procedure of Institutional Review Board. All authors reviewed and approved the manuscript.
References
- 1. Mankin, H.J. (1992) Nontraumatic necrosis of bone (osteonecrosis). N. Engl. J. Med., 326, 1473–1479. [DOI] [PubMed] [Google Scholar]
- 2. Fukushima, W., Fujioka, M., Kubo, T., Tamakoshi, A., Nagai, M. and Hirota, Y. (2010) Nationwide epidemiologic survey of idiopathic osteonecrosis of the femoral head. Clin. Orthop. Relat. Res., 468, 2715–2724. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Yamaguchi, R., Yamamoto, T., Motomura, G., Ikemura, S. and Iwamoto, Y. (2011) Incidence of nontraumatic osteonecrosis of the femoral head in the Japanese population. Arthritis Rheum., 63, 3169–3173. [DOI] [PubMed] [Google Scholar]
- 4. Mont, M.A. and Hungerford, D.S. (1995) Non-traumatic avascular necrosis of the femoral head. J. Bone Joint Surg. Am., 77, 459–474. [DOI] [PubMed] [Google Scholar]
- 5. Nagasawa, K., Tada, Y., Koarada, S., Horiuchi, T., Tsukamoto, H., Murai, K., Ueda, A., Yoshizawa, S. and Ohta, A. (2005) Very early development of steroid-associated osteonecrosis of femoral head in systemic lupus erythematosus: prospective study by MRI. Lupus, 14, 385–390. [DOI] [PubMed] [Google Scholar]
- 6. Oinuma, K., Harada, Y., Nawata, Y., Takabayashi, K., Abe, I., Kamikawa, K. and Moriya, H. (2001) Osteonecrosis in patients with systemic lupus erythematosus develops very early after starting high dose corticosteroid treatment. Ann. Rheum. Dis., 60, 1145–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Zhao, D.W., Yu, M., Hu, K., Wang, W., Yang, L., Wang, B.J., Gao, X.H., Guo, Y.M., Xu, Y.Q., Wei, Y.S. et al. (2015) Prevalence of nontraumatic osteonecrosis of the femoral head and its associated risk factors in the Chinese population: results from a nationally representative survey. Chin. Med. J., 128, 2843–2850. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Liu, Y.F., Chen, W.M., Lin, Y.F., Yang, R.C., Lin, M.W., Li, L.H., Chang, Y.H., Jou, Y.S., Lin, P.Y., Su, J.S. et al. (2005) Type II collagen gene variants and inherited osteonecrosis of the femoral head. N. Engl. J. Med., 352, 2294–2301. [DOI] [PubMed] [Google Scholar]
- 9. Song, Y., Du, Z., Chen, B., Ren, M., Yang, Q., Sui, Y., Wang, Q., Wang, A., Zhao, H., Qin, Y. et al. (2017) Association of SREBP2 gene polymorphisms with the risk of osteonecrosis of the femoral head relates to gene expression and lipid metabolism disorders. Mol. Med. Rep., 16, 7145–7153. [DOI] [PubMed] [Google Scholar]
- 10. An, F., Zhang, L., Gao, H., Wang, J., Liu, C., Tian, Y., Ma, C., Zhao, J., Wang, K. and Wang, J. (2020) Variants in RETN gene are associated with steroid-induced osteonecrosis of the femoral head risk among Han Chinese people. J. Orthop. Surg. Res., 15, 96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Kim, T.H., Baek, S.H., Lim, J.O., Lee, S.H. and Kim, S.Y. (2015) Genetic variation in the coagulation factor V gene and risk of femoral head osteonecrosis. Mol. Med. Rep., 12, 4434–4440. [DOI] [PubMed] [Google Scholar]
- 12. Zhao, X., Yang, F., Sun, L. and Zhang, A. (2019) Association between NOS3 polymorphisms and osteonecrosis of the femoral head. Artif Cells Nanomed Biotechnol, 47, 1423–1427. [DOI] [PubMed] [Google Scholar]
- 13. Ma, W., Xin, K., Chen, K., Tang, H., Chen, H., Zhi, L. and Liu, H. (2018) Relationship of common variants in VEGFA gene with osteonecrosis of the femoral head: a Han Chinese population based association study. Sci. Rep., 8, 16221. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Karol, S.E., Yang, W., Van Driest, S.L., Chang, T.Y., Kaste, S., Bowton, E., Basford, M., Bastarache, L., Roden, D.M., Denny, J.C. et al. (2015) Genetics of glucocorticoid-associated osteonecrosis in children with acute lymphoblastic leukemia. Blood, 126, 1770–1776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Karol, S.E., Mattano, L.A., Jr., Yang, W., Maloney, K.W., Smith, C., Liu, C., Ramsey, L.B., Fernandez, C.A., Chang, T.Y., Neale, G. et al. (2016) Genetic risk factors for the development of osteonecrosis in children under age 10 treated for acute lymphoblastic leukemia. Blood, 127, 558–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Zhang, Y., Bowen, T.R., Lietman, S.A., Suk, M., Williams, M.S. and Lee, M.T.M. (2020) PPARGC1B is associated with nontraumatic osteonecrosis of the femoral head: a genomewide association study on a chart-reviewed cohort. J. Bone Joint Surg. Am., 102, 1628–1636. [DOI] [PubMed] [Google Scholar]
- 17. Sakamoto, Y., Yamamoto, T., Sugano, N., Takahashi, D., Watanabe, T., Atsumi, T., Nakamura, J., Hasegawa, Y., Akashi, K., Narita, I. et al. (2017) Genome-wide association study of idiopathic osteonecrosis of the femoral head. Sci. Rep., 7, 15035. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Hirata, M., Kamatani, Y., Nagai, A., Kiyohara, Y., Ninomiya, T., Tamakoshi, A., Yamagata, Z., Kubo, M., Muto, K., Mushiroda, T. et al. (2017) Cross-sectional analysis of BioBank Japan clinical data: a large cohort of 200,000 patients with 47 common diseases. J. Epidemiol., 27, S9–S21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Nagai, A., Hirata, M., Kamatani, Y., Muto, K., Matsuda, K., Kiyohara, Y., Ninomiya, T., Tamakoshi, A., Yamagata, Z., Mushiroda, T. et al. (2017) Overview of the BioBank Japan Project: study design and profile. J. Epidemiol., 27, S2–S8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Tan, E.M., Cohen, A.S., Fries, J.F., Masi, A.T., McShane, D.J., Rothfield, N.F., Schaller, J.G., Talal, N. and Winchester, R.J. (1982) The 1982 revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum., 25, 1271–1277. [DOI] [PubMed] [Google Scholar]
- 21. Hochberg, M.C. (1997) Updating the American College of Rheumatology revised criteria for the classification of systemic lupus erythematosus. Arthritis Rheum., 40, 1725. [DOI] [PubMed] [Google Scholar]
- 22. Bulik-Sullivan, B.K., Loh, P.R., Finucane, H.K., Ripke, S., Yang, J., Schizophrenia Working Group of the Psychiatric Genomics Consortium, Patterson, N., Daly, M.J., Price, A.L. and Neale, B.M. (2015) LD Score regression distinguishes confounding from polygenicity in genome-wide association studies. Nat. Genet., 47, 291–295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Yin, X., Kim, K., Suetsugu, H., Bang, S.Y., Wen, L., Koido, M., Ha, E., Liu, L., Sakamoto, Y., Jo, S. et al. (2021) Meta-analysis of 208370 East Asians identifies 113 susceptibility loci for systemic lupus erythematosus. Ann. Rheum. Dis., 80, 632–640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Kwon, Y.C., Lim, J., Bang, S.Y., Ha, E., Hwang, M.Y., Yoon, K., Choe, J.Y., Yoo, D.H., Lee, S.S., Lee, J. et al. (2020) Genome-wide association study in a Korean population identifies six novel susceptibility loci for rheumatoid arthritis. Ann. Rheum. Dis., 79, 1438–1445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Benner, C., Spencer, C.C., Havulinna, A.S., Salomaa, V., Ripatti, S. and Pirinen, M. (2016) FINEMAP: efficient variable selection using summary data from genome-wide association studies. Bioinformatics, 32, 1493–1501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. GTEx Consortium (2020) The GTEx Consortium atlas of genetic regulatory effects across human tissues. Science (New York, N.Y.), 369, 1318–1330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bartel, D.P. (2018) Metazoan MicroRNAs. Cell, 173, 20–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Chen, Y. and Wang, X. (2020) miRDB: an online database for prediction of functional microRNA targets. Nucleic Acids Res., 48, D127–D131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Watanabe, K., Taskesen, E., van Bochoven, A. and Posthuma, D. (2017) Functional mapping and annotation of genetic associations with FUMA. Nat. Commun., 8, 1826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Wyles, C.C., Paradise, C.R., Houdek, M.T., Slager, S.L., Terzic, A., Behfar, A., van Wijnen, A.J. and Sierra, R.J. (2019) CORR® ORS Richard A. Brand award: disruption in peroxisome proliferator-activated receptor-γ (PPARG) increases osteonecrosis risk through genetic variance and pharmacologic modulation. Clin. Orthop. Relat. Res., 477, 1800–1812. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Miyanishi, K., Yamamoto, T., Irisa, T., Yamashita, A., Jingushi, S., Noguchi, Y. and Iwamoto, Y. (2002) Bone marrow fat cell enlargement and a rise in intraosseous pressure in steroid-treated rabbits with osteonecrosis. Bone, 30, 185–190. [DOI] [PubMed] [Google Scholar]
- 32. Gazal, S., Finucane, H.K., Furlotte, N.A., Loh, P.R., Palamara, P.F., Liu, X., Schoech, A., Bulik-Sullivan, B., Neale, B.M., Gusev, A. et al. (2017) Linkage disequilibrium-dependent architecture of human complex traits shows action of negative selection. Nat. Genet., 49, 1421–1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Akizuki, S., Ishigaki, K., Kochi, Y., Law, S.M., Matsuo, K., Ohmura, K., Suzuki, A., Nakayama, M., Iizuka, Y., Koseki, H. et al. (2019) PLD4 is a genetic determinant to systemic lupus erythematosus and involved in murine autoimmune phenotypes. Ann. Rheum. Dis., 78, 509–518. [DOI] [PubMed] [Google Scholar]
- 34. Terao, C., Momozawa, Y., Ishigaki, K., Kawakami, E., Akiyama, M., Loh, P.R., Genovese, G., Sugishita, H., Ohta, T., Hirata, M. et al. (2019) GWAS of mosaic loss of chromosome Y highlights genetic effects on blood cell differentiation. Nat. Commun., 10, 4719. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Ishigaki, K., Akiyama, M., Kanai, M., Takahashi, A., Kawakami, E., Sugishita, H., Sakaue, S., Matoba, N., Low, S.K., Okada, Y. et al. (2020) Large-scale genome-wide association study in a Japanese population identifies novel susceptibility loci across different diseases. Nat. Genet., 52, 669–679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Zhang, L.D., Kang, J.F. and Xue, H.L. (1990) Distribution of lesions in the head and neck of the humerus and the femur in dysbaric osteonecrosis. Undersea Biomed. Res., 17, 353–358. [PubMed] [Google Scholar]
- 37. Adesina, O.O. and Neumayr, L.D. (2019) Osteonecrosis in sickle cell disease: an update on risk factors, diagnosis, and management. Hematology Am. Soc. Hematol. Educ. Program, 2019, 351–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Dubois, E.L. and Cozen, L. (1960) Avascular (aseptic) bone necrosis associated with systemic lupus erythematosus. JAMA, 174, 966–971. [DOI] [PubMed] [Google Scholar]
- 39. Liu, Y., Zong, Y., Shan, H., Lin, Y., Xia, W., Wang, N., Zhou, L., Gao, Y., Ma, X. and Jiang, C. (2020) MicroRNA-23b-3p participates in steroid-induced osteonecrosis of the femoral head by suppressing ZNF667 expression. Steroids, 163, 108709. [DOI] [PubMed] [Google Scholar]
- 40. Fang, S.H., Chen, L., Chen, H.H., Li, Y.F., Luo, H.B., Hu, D.Q. and Chen, P. (2019) MiR-15b ameliorates SONFH by targeting Smad7 and inhibiting osteogenic differentiation of BMSCs. Eur. Rev. Med. Pharmacol. Sci., 23, 9761–9771. [DOI] [PubMed] [Google Scholar]
- 41. Xiang, S., Li, Z. and Weng, X. (2019) The role of lncRNA RP11-154D6 in steroid-induced osteonecrosis of the femoral head through BMSC regulation. J. Cell. Biochem., 120, 18435–18445. [DOI] [PubMed] [Google Scholar]
- 42. Okada, Y., Hirota, T., Kamatani, Y., Takahashi, A., Ohmiya, H., Kumasaka, N., Higasa, K., Yamaguchi-Kabata, Y., Hosono, N., Nalls, M.A. et al. (2011) Identification of nine novel loci associated with white blood cell subtypes in a Japanese population. PLoS Genet., 7, e1002067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Liu, Y.F., Sowell, S.M., Luo, Y., Chaubey, A., Cameron, R.S., Kim, H.G. and Srivastava, A.K. (2015) Autism and intellectual disability-associated KIRREL3 interacts with neuronal proteins MAP1B and MYO16 with potential roles in neurodevelopment. PLoS One, 10, e0123106. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Auton, A., Brooks, L.D., Durbin, R.M., Garrison, E.P., Kang, H.M., Korbel, J.O., Marchini, J.L., McCarthy, S., McVean, G.A. and Abecasis, G.R. (2015) A global reference for human genetic variation. Nature, 526, 68–74. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Das, S., Forer, L., Schönherr, S., Sidore, C., Locke, A.E., Kwong, A., Vrieze, S.I., Chew, E.Y., Levy, S., McGue, M. et al. (2016) Next-generation genotype imputation service and methods. Nat. Genet., 48, 1284–1287. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Chang, C.C., Chow, C.C., Tellier, L.C., Vattikuti, S., Purcell, S.M. and Lee, J.J. (2015) Second-generation PLINK: rising to the challenge of larger and richer datasets. Gigascience, 4, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Willer, C.J., Li, Y. and Abecasis, G.R. (2010) METAL: fast and efficient meta-analysis of genomewide association scans. Bioinformatics, 26, 2190–2191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Yang, J., Ferreira, T., Morris, A.P., Medland, S.E., Genetic Investigation of ANthropometric Traits (GIANT) Consortium, DIAbetes Genetics Replication and Meta-analysis (DIAGRAM) Consortium, Madden, P.A., Heath, A.C., Martin, N.G., Montgomery, G.W., Weedon, M.N. et al. (2012) Conditional and joint multiple-SNP analysis of GWAS summary statistics identifies additional variants influencing complex traits. Nat. Genet., 44, 369–375 S361-363. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Altshuler, D.M., Gibbs, R.A., Peltonen, L., Dermitzakis, E., Schaffner, S.F., Yu, F., Dermitzakis, E., Schaffner, S.F., Yu, F., Peltonen, L. et al. (2010) Integrating common and rare genetic variation in diverse human populations. Nature, 467, 52–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Zhou, W., Nielsen, J.B., Fritsche, L.G., Dey, R., Gabrielsen, M.E., Wolford, B.N., LeFaive, J., VandeHaar, P., Gagliano, S.A., Gifford, A. et al. (2018) Efficiently controlling for case-control imbalance and sample relatedness in large-scale genetic association studies. Nat. Genet., 50, 1335–1341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Wang, X. (2014) Firth logistic regression for rare variant association tests. Front. Genet., 5, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52. Kang, H.M. Efficient and parallelizable association container toolbox (EPACTS).
- 53. Wang, K., Li, M. and Hakonarson, H. (2010) ANNOVAR: functional annotation of genetic variants from high-throughput sequencing data. Nucleic Acids Res., 38, e164. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Liu, B., Gloudemans, M.J., Rao, A.S., Ingelsson, E. and Montgomery, S.B. (2019) Abundant associations with gene expression complicate GWAS follow-up. Nat. Genet., 51, 768–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.