

Abstract
Non-obstructive azoospermia (NOA) is an important cause of male infertility, and the genetic pathogenesis is still incompletely understood. The previous study reported that heterozygous mutation of c.346-1G > A in spermatogenesis and oogenesis specific basic helix–loop–helix 1 (SOHLH1) was identified in two NOA patients and suggested it is the pathogenic factor for NOA. However, in our research, this heterozygous mutation was confirmed in three Chinese infertile patients who suffered from teratozoospermia, but they had normal sperm number. Intriguingly, a homozygous mutation of c.346-1G > A in SOHLH1 was detected in a severe oligozoospermia (SOZ) patient, characterized with severely decreased sperm count. Notably, we unprecedently revealed that this homozygous mutation of c.346-1G > A in SOHLH1 leads to the sharp decrease in various germ cells and spermatogenesis dysfunction, which is similar to the phenotype of SOHLH1 knockout male mice. Moreover, western blotting confirmed that the homozygous mutation declined SOHLH1 protein expression. Additionally, we correlated the good prognosis of intracytoplasmic sperm injection (ICSI) in the patients carrying the mutation of c.346-1G > A in SOHLH1. Thus, we suggested that the heterozygous mutation of c.346-1G > A in SOHLH1 is responsible for teratozoospermia, and this homozygous mutation in SOHLH1 impairs spermatogenesis and further leads to the reduced sperm count, eventually causing male infertility, which unveils a new recessive-inheritance pattern of SOHLH1-associated male infertility initially.
Introduction
Infertility is one of the major conditions that impairs human health, affecting an estimated 15% of couples worldwide (1). Factors leading to male infertility include urogenital infections, abnormal hormone levels and genetic causes (2). As whole-exome sequencing (WES) is now widely used, certain genetic causes responsible for male infertility have been uncovered. However, due to the complex and precise process of germ cell development—as well as the high genetic heterogeneity of male infertility—only a limited number of infertile male patients are diagnosed with a definite genetic etiology.
The spermatogenesis and oogenesis specific basic helix–loop–helix 1 (SOHLH1) gene is an important transcriptional regulatory factor in germ cell formation. It is principally expressed in spermatogonia and early oocytes and is involved in their maturation and differentiation (3). In males, SOHLH1 is primarily expressed in A1–A4 type spermatogonia, and it gradually decreases in Int and B type spermatogonia (3). Several previous findings showed that SOHLH1 regulates the spermatogonial stem cell (SSC) differentiation-related gene Ngn3 (3). In type A spermatogonia cells, SOHLH1 affects the maturation of spermatogonia by regulating c-Kit expression (4,5). Moreover, SOHLH1 can form a heterodimer with SOHLH2 to down-regulate the expression of Stra8, which is related to meiotic initiation (6). Therefore, SOHLH1 participates in important signaling pathways and plays an essential role in spermatogenesis. Intriguingly, in previous studies, Choi Y et al. reported three novel variations including one intronic variant (c.346-1G > A), and two non-synonymous exonic variants (c.91T4C and c.529C4A), and 11 single-nucleotide polymorphism (SNP) in SOHLH1 gene in 96 Korean patients with NOA (7). In 2017, Nakamura et al. (8) analyzed 25 azoospermia-related genes in 40 Japanese infertile men diagnosed as NOA and detected the previously reported heterozygous SOHLH1 c.346-1G-A mutation in two patients. However, male Sohlh1 homozygous null (Sohlh1−/−) mice presented disrupted spermatogonial differentiation, and there are no reports of SOHLH1 homozygous mutation in humans; thus, the effect of SOHLH1 on human spermatogenesis is ambiguous.
In the actual study, we identified a recurrent heterozygous mutation of c.346-1G > A in SOHLH1 in three infertile subjects, who exhibited normal sperm quantity but abnormal morphology. Interestingly, the homozygote of c.346-1G > A mutation was found in an infertile patient with an extremely low sperm count. The deleterious effect of the homozygous mutation on SOHLH1 expression was confirmed by western blotting. Hematoxylin and eosin (H&E) and immunofluorescence (IF) staining showed that the germ cells in the homozygous patient’s testis decreased in number and were disordered, indicating the impaired spermatogenesis. Collectively, we initially suggested that the heterozygous mutation of c.346-1G > A in SOHLH1 is associated with aberrant sperm morphology but does not play a role in diminished sperm count. Remarkably, a homozygous mutation of SOHLH1 c.346-1G > A was first unveiled, which disrupts sperm production. Therefore, we suggested that the contribution of SOHLH1 c.346-1G > A mutation in male infertility should be reconsidered, since the novel genotype and phenotype of this variant were discovered here.
Results
Identified heterozygous c.346-1G > A mutation of SOHLH1 in patients with teratozoospermia
In this study, the heterozygous variation c.346-1G > A in SOHLH1 had been detected in three teratozoospermia patients (Patients A, B and C), and surprisingly, this variation has been considered as a pathogenic cause for NOA (7,8) (Fig. 1). The three patients showed normal sperm concentration and count, while almost all sperm were immotile (Table 1). Papanicolaou staining showed that a majority of sperm from the patients had amorphous heads and anomalies in their flagella compared to those from the normal control (Supplementary Material, Fig. S1a). SEM further confirmed that sperm of the patients possessed aberrant heads, which included round, double, tapered, pyriform and/or irregular heads, with short, bent or coiled flagella (Fig. 2A). We additionally analyzed sperm ultrastructure using TEM and showed that normal control spermatozoa had the proper ratio of head length to width and that the 9 + 2 configuration (nine peripheral microtubule doublets and two central microtubules, surrounded by an outer dense fiber and fibrous sheath) was integrated and regularly arranged (Fig. 2B and Fig. 3). In contrast, we observed in the patients that the sperm plasma membrane was swollen and damaged, the nucleus contained vacuoles, the mitochondria were empty and there was an atypical and irregular arrangement of axonemal microtubules in the sperm flagella (Fig. 3). Based on this evidence, we suggested that the heterozygous c.346-1G > A mutation in SOHIL1 is a pathogenic factor of teratozoospermia.
Figure 1.
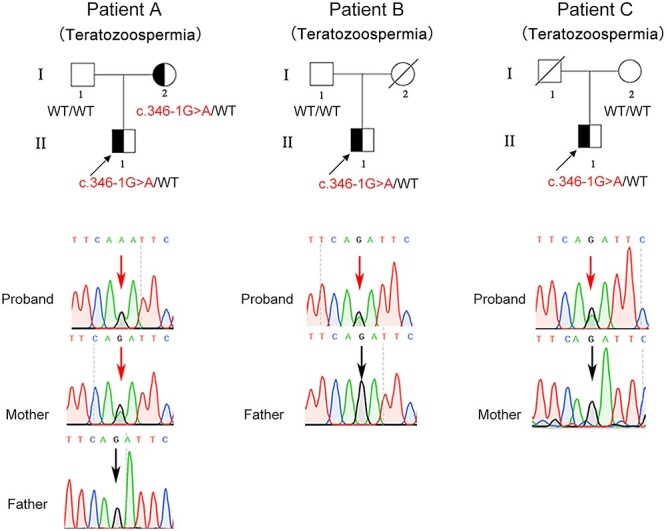
A recurrent heterozygous mutation in SOHLH1 identified in three teratozoospermia patients. Pedigrees of three families with the c.346-1G > A variant in SOHLH1. Sanger sequencing confirmed the SOHLH1 mutation in these families. Squares indicate males, and circles represent females; the mutation site is designated by a red arrow, and the wild-type site is designated by a black arrow.
Table 1.
Semen analysis of patients
| Sperm parameters | SOZ patient homozygous | Teratozoospermia patient A heterozygous | Teratozoospermia patient B heterozygous | Teratozoospermia patient C heterozygous | Normospermic parameters |
|---|---|---|---|---|---|
| Sperm volume (ml) | 3.6 | 3.2 | 4.7 | 4.9 | ≥ 1.5 |
| Sperm concentration (million/ml) | 4.3 | 29.6 | 65.1 | 20.5 | ≥ 15 |
| Motility sperm (%) | 15 | 7 | 28 | 3 | ≥ 40 |
| Vitality (%) | 47.0 | 21.0 | 39.0 | 32.0 | ≥ 58 |
| Normal morphology (%) | 1 | 1.4 | 2.5 | 0.9 | ≥ 4 |
Figure 2.
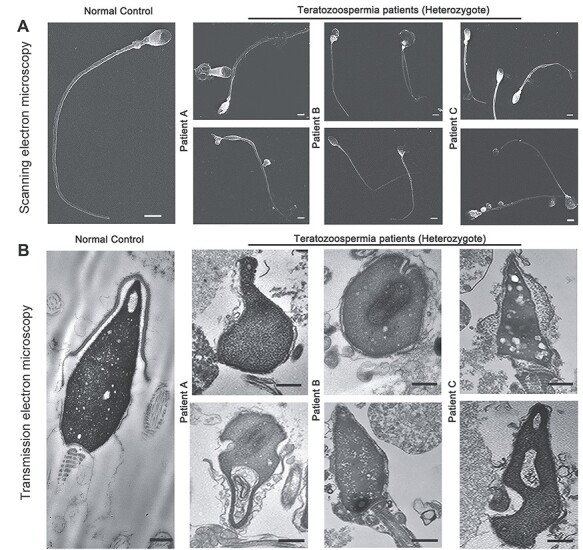
The morphology of spermatozoa from the teratozoospermia patients by electron microscopy. (A) The abnormal sperm phenotypes were observed using SEM (scale bars, 5 μm). (B) TEM shows abnormal ultrastructure of the head from the patients’ spermatozoa compared to normal control (scale bars, 100 nm).
Figure 3.
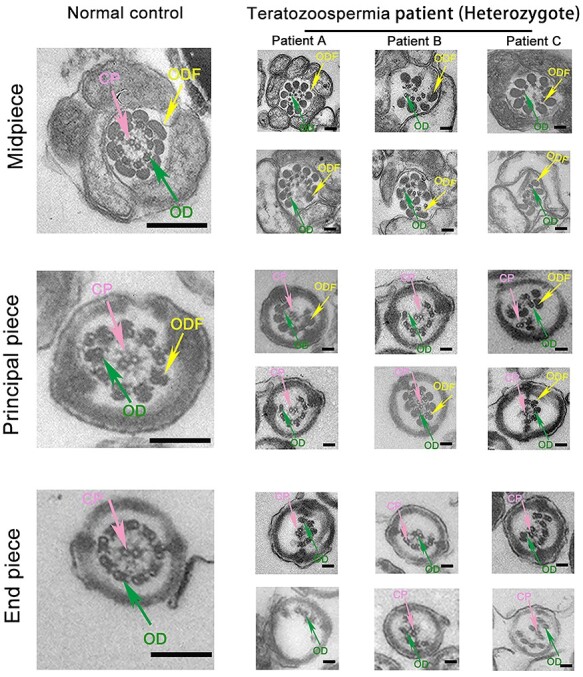
Ultrastructure of the spermatozoa flagella of teratozoospermiac patients by TEM. In cross sections, the ultrastructural abnormalities were observed in the patients compared with the normal control by TEM (scale bars, 100 nm).
The homozygous mutation of c.346-1G > A in SOHLH1 detected in an infertile patient with severe oligozoospermia
Remarkably, we also detected a homozygous mutation of c.346-1G > A in SOHLH1 in a severe oligozoospermia (SOZ) patient (Fig. 4A). Sanger sequencing identified his father did not carry this mutation and his mother’s DNA was not available. For this patient, no spermatozoa were initially observed in the replicated wet preparations. After centrifugation at 3000 g for 15 min, we finally located only a few sperm with abnormal morphology (Supplementary Material, Fig. S1a).
Figure 4.
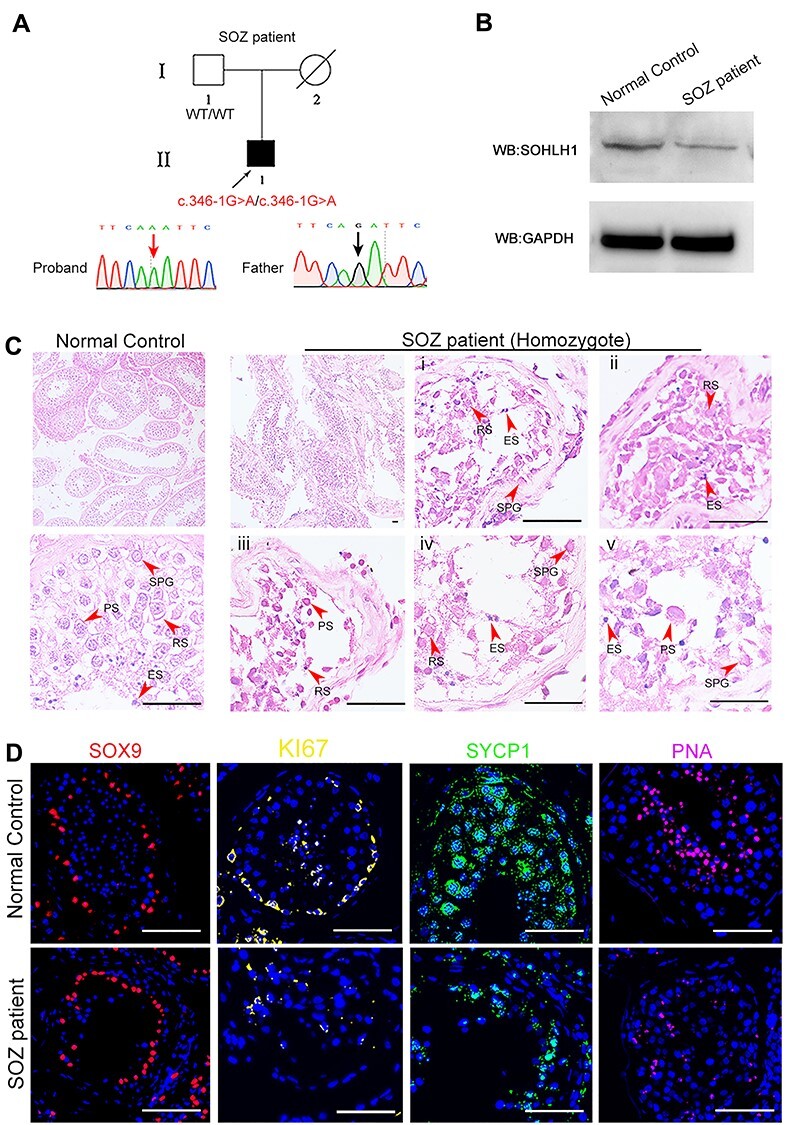
The role of the homozygous c.346-1G > A mutation in SOHLH1 in male infertility. (A) The homozygous variant in SOZ patient was confirmed by Sanger sequencing. (B) Western immunoblotting indicated the decrease of SOHLH1 expression in the testicular tissues of the SOZ patient compared to the normal control. (C) H&E staining of testicular sections from a normal control and SOZ patient. Compared with the normal control, we observed sparse cellular arrangements in patient’s sperm at different spermatogenic stages. i, ii, iii, iv and v show testicular sections from SOZ patient at higher magnification. Red arrowheads denote various germ cells. SPG, spermatogonia; PS, primary spermatocytes; RS, round spermatids; ES, elongated spermatids (scale bar, 20 μm). (D) Staining of SOX9, KI67, SYCP1 and PNA in the testes from a normal control and SOZ patient. The results showed an obvious signal decline in KI67, SYCP1 and PNA in the testis of patient A, while the SOX9 signal was indistinguishable from that of the normal control. SOX9 marks Sertoli cells; KI67 marks spermatogonia; SYCP1 marks spermatocytes and PNA marks round spermatids (red, SOX9; yellow, KI67; green, SYCP1; pink, PNA; blue, DAPI; scale bar, 50 μm). SOZ, Severe oligozoospermia.
The heterozygous mutation c.346-1G > A in the SOHLH1 gene caused the original splicing-site disruption and partial deletion within exon 4, demonstrating that this mutation was detrimental (7). To investigate the influence of a homozygous mutation on spermatogenesis, we characterized the effects of the homozygous mutation c.346-1G > A of the SOHIL1 gene in greater detail. Western blotting indicated the decrease of the SOHLH1 protein in patient’s testicular tissues compared to normal control (Fig. 4B). Dysfunctional spermatogenesis was observed in the SOZ patient, showing a sharp decline in the sperm count, and also displayed in the disarray and irregularities within various germ cells using IF and H&E staining (Fig. 4C and D). Collectively, our findings proved that the homozygous mutation was the direct genetic factor causing reduced sperm production and suggested that the c.346-1G > A mutation in SOHLH1 impaired sperm production in a recessive-inheritance pattern.
The special expression pattern of SOHLH1 during spermatogenesis
To highlight the role of SOHLH1 in the different stages of sperm development, we used the STA-PUT velocity sedimentation method to isolate the different stages of germ cells in mouse testes. We noted that SOHLH1 protein was principally distributed in the cytoplasm of spermatogonia and round spermatids (steps 1–8) (Supplementary Material, Fig. S1b and c). During sperm formation, SOHLH1 is highly expressed in spermatogonia, and in spermatocytes, the expression began to drop off. After entering the spermatids stage, the expression of SOHLH1 decreased sharply and finally disappeared, suggesting that it was a major morphogenetic participant in the early spermatogenic process (Supplementary Material, Fig. S1b and c).
Favorable prognosis with ICSI in patients harboring the mutation in SOHLH1
Given that ICSI is a useful technology that allows conception to occur for sterile couples, ICSI cycles were attempted for three patients (teratozoospermia patients A, B and SOZ patient), and written informed consent was obtained for the procedure. This is the first study that aimed to discern whether infertility patients with a mutation in SOHLH1 exhibited a favorable prognosis following ICSI. After careful examination, all three patients’ wives demonstrated normal basal hormone levels (Table 2) and had undergone a long gonadotrophin-releasing hormone (GnRH) agonist protocol (Table 2). Patient with SOZ carried the homozygous mutation, and his wife was followed up for one ICSI cycle. Eleven oocytes were retrieved, and 10 mature oocytes were microinjected with sperm; seven were ultimately fertilized. Following extended culture, we obtained four available D3 embryos, including one at 8 II, one at 10 II, one at 4AB and one at 4 BC. Although the 10 II embryo was transferred immediately, it failed to develop; however, one 4AB embryo was then transferred successfully. Ultimately, SOA patient’s wife achieved a clinical pregnancy and gave birth to a healthy baby. For teratozoospermia patient A’s wife, five metaphase II oocytes were retrieved and injected in the first cycle. A resulting embryo, however, failed to develop after reaching the available D3 stage. In the second cycle, we retrieved five metaphase II oocytes and injected them, obtaining five available D3 embryos, and one 4BB embryo was chosen for transplantation that resulted in teratozoospermia patient A’s wife becoming pregnant in the same cycle (Table 2). Teratozoospermia patient B was a heterozygous carrier, and the couple was followed up for one ICSI cycle—with 16 oocytes retrieved after GnRH treatment. Following extended culture, we obtained three available D3 embryos, and two 8-cell blastocysts were subsequently transferred into the uterus with no obvious complications—resulting in a pregnancy (Table 2). With this study, we are the first to demonstrate that male sterility associated with SOHLH1 mutation—whether heterozygous or homozygous—can be treated using ICSI.
Table 2.
Clinical features of the patients with ICSI treatment
| SOZ patient | Teratozoospermia patient A | Teratozoospermia patient B | ||
|---|---|---|---|---|
| Age (y) | 41 | 32 | 30 | |
| Length of primary infertility history (y) | 3 | 8 | 6 | |
| BMI | 18.5 | 18.2 | 17.9 | |
| Basal hormones | FSH (IU/l) | 6.3 | 5.5 | 7.4 |
| LH (IU/l) | 6.7 | 1.3 | 0.9 | |
| E2 (pg/ml) | 47.5 | 18.9 | 68.9 | |
| PRL (ng/ml) | 8.3 | 8.6 | 7.9 | |
| Prog (ng/ml) | 0.57 | 0.78 | 0.6 | |
| Testo (ng/ml) | 0.23 | 0.18 | 0.21 | |
| Cycle 1 | Protocol | Long | Long | Long |
| E2 level on the trigger day (pg/ml) | 2849 | 3168 | 2648 | |
| No. of follicles ≥14 mm on the trigger day | 8 | 7 | 7 | |
| No. of follicles ≥18 mm on the trigger day | 3 | 2 | 4 | |
| No. of oocytes retrieved | 11 | 16 | 5 | |
| ICSI progress | Oocytes injected | 10 | 14 | 5 |
| Fertilization rate (%) | 70% (7/10) | 42.85% (6/14) | 80% (4/5) | |
| Cleavage rate (%) | 100% (7/7) | 100% (6/6) | 50% (2/4) | |
| Available D3 embryos | 5 | 42.85% (3/14) | 0 | |
| Blastocyst formation rate (%) | 25% (1/4) | 28.5% (2/7) | 0 | |
| Protocol | Long | |||
| E2 level on the trigger day (pg/ml) | 785 | |||
| Cycle 2 | No. of follicles ≥14 mm on the trigger day | 5 | ||
| No. of follicles ≥18 mm on the trigger day | 0 | |||
| No. of oocytes retrieved | 5 | |||
| ICSI progress | Oocytes injected | 5 | ||
| Fertilization rate (%) | 100% (5/5) | |||
| Cleavage rate (%) | 100% (5/5) | |||
| Available D3 embryos | 5 | |||
| Blastocyst formation rate (%) | 40% (2/5) | |||
Discussion
Spermatogenesis is a dynamic process and requires precise regulation of mitotic of spermatogonia, meiotic division of spermatocytes and maturation of spermatids. Demolitions of these phases can cause spermatogenic arrest and lead to male sterility. Mutations in genes responsible for SOZ, such as TEX15, SYCP2, USP9Y and M1AP, have been demonstrated that are required for DNA double-strand break repair, chromosome synapsis and meiotic recombination in spermatocytes (10–14). However, only a small minority of SOZ cases could be elucidated. In the present study, we identified a novel recessively inherited pattern of c.346-1G > A mutation in the SOHLH1 gene that caused SOZ and described the monoallelic mutation in SOHLH1 as contributing to teratozoospermia. SEM and TEM results showed severe abnormalities in the morphology and ultrastructure of the heterozygous patients’ sperm, while IF staining results depicted an obvious reduction in the number of germ cells at various stages and western blotting confirmed the disrupted SOHLH1 expression in the homozygous patient’s testis. Furthermore, the clinical outcome of the patients—whether heterozygote or homozygote—who underwent ICSI was entirely successful. Collectively, these data strongly indicate that the splice-acceptor site, bi-allelic mutation in SOHLH1 leads to male infertility via the absence of normal spermatogenesis.
The heterozygous c.346-1G > A mutation in SOHLH1 is currently recognized as a pathogenic gene for NOA, and some researchers have focused on the relationship between SOHLH1 and male infertility (7,8). Male SOHLH1-knockout mice (SOHLH1−/−) possessed a large number of apoptotic spermatocytes at the meiotic stage, resulting in spermatogenic failure and leading to male infertility (15). Similarly, our patient with SOZ who carried the homozygous SOHLH1 mutation lacked spermatogenic cells and manifested a severe reduction in sperm number. Considering that the SOHLH1 gene shares low sequence identity between humans and mice (https://www.ncbi.nlm.nih.gov/homologene/), it is reasonable to posit that there exist differences in phenotype between humans and mice manifesting the homozygous variation in SOHLH1; nevertheless, the variations might still contribute to the diminution in the number of spermatozoa. Although Li et al. did not mention any sperm parameter with respect to their heterozygous male mice (15), we speculate that the heterozygotes showed normal fertility and could sire the homozygous mice. In addition, two men harboring a frameshift mutation and a non-sense mutation in SOHLH1, respectively, showed normal reproduction (16). Moreover, Song et al. performed SNP linkage analysis of SOHLH1 and discovered that there was no relationship between the rs558113 in the SOHLH1 gene and susceptibility to NOA in the Chinese population—suggesting that the heterozygous mutation in SOHLH1 was not associated with NOA (17). More importantly, our patients with the heterozygous mutation showed normal sperm number. All of these observations might be explained by another normal allele of SOHLH1 being completely translated into the intact protein and performing normal biological functions, while the homozygous mutation led to the complete absence of SOHLH1 expression. In addition, there is no evidence to suggest that the SOHLH1 gene shows haploinsufficiency (https://clinicalgenome.org/). We indicated that the heterozygous mutation of SOHLH1 exhibits a certain degree of determination toward male infertility, and it may combine with other gene mutations or factors to drive the occurrence and development of NOA or epigenetic modifications that affect the gene at the transcriptional level. In addition, in public database (http://dgv.tcag.ca/dgv/app/home), partial deletions of SOHLH1 have been identified in human population (such as deletion of chr9:138587981–138 588 037 and chr9:138589985–138 653 652), which do not cover the SOHLH1 c.346-1G > A position. We therefore supposed that the combination of partial deletion and heterozygous mutation c.346-1G > A in SOHLH1 might be the cause of severe dyszoospermia phenotype in previous reports. Regrettably, this study includes a cohort of 150 infertile patients, a more comprehensive investigation including more patients is needed to further validate our findings and evaluate the contribution of SOHLH1 mutation to male infertility.
ICSI is a useful treatment for infertility, but it is not always successful. The current success rate is 30–40% due to technological limitations or certain genetic mutations that damage embryonic development (18). Patients with morphological abnormalities of the sperm flagella (MMAF) due to mutations in DNAH1, DNAH8, TTC29, CFAP44, CFAP74 or CFAP43 exhibit a good prognosis following ICSI (19–23). However, DNAH17, CEP135 and FSIP2 have also been associated with ICSI failure (21,24,25). Previous studies have revealed that infertility-affected males harboring specific gene variants acquired favorable prognoses following ICSI. In our study, ICSI was performed using sperm from SOHLH1-mutated males, and their wives became pregnant and underwent successful deliveries. This suggested that patients with the c.346-1G > A mutation in SOHLH1 produced sperm that completed fertilization and that the embryos were able to develop properly despite morphological abnormalities in the sperm. Therefore, with these findings, we recommend that ICSI can be used successfully for SOHLH1-associated male infertility.
Conclusions
In summary, our work identified a recessively inherited model of a SOHLH1 mutation in human male infertility that was associated with SOZ and also suggested that the SOHLH1 heterozygous c.346-1G > A mutation was associated with teratozoospermia but not a direct cause for NOA. This report thus corrected previous work stating that the heterozygous SOHLH1 mutation of c.346-1G > A led to NOA in a dominantly inherited manner (7). Therefore, we posit that the heterozygous mutation of c.346-1G > A in SOHLH1 might be a facilitator of NOA, but the homozygous status with regard to this mutation is the primary factor in sperm production. Our findings thus update the genetic mutation spectrum of male infertility and provide strong support toward clinical diagnosis.
Materials and Methods
Study participants
The infertile patients were enrolled at the West China Second University Hospital of Sichuan University, and these patients all had normal somatic karyotypes (46, XY). This study was conducted following the tenets of the Declaration of Helsinki, and ethical approval was obtained from the Ethical Review Board of West China Second University Hospital, Sichuan University. Informed consent was obtained from each study participant.
Genetic analysis
Genomic DNA was isolated from peripheral blood samples of subjects using a whole-blood DNA-purification kit (QIAGEN). We performed WES on the patient samples. One microgram of genomic DNA was utilized for exon capture using the Agilent SureSelect Human All Exon V6 Kit and sequenced on the Illumina HiSeq X system. Targeted testing of the potentially pathogenic variants in the patients’ parents was performed by Sanger sequencing. The primers used in the PCR analysis were as follows: F, 5′-CTCGAGCCTGGGGTAGCAGTCTTG-3′ and R, 5′-GGATCCACACACAGGAGCAGAAGA-3′.
Western blotting
Proteins were extracted from testicular tissue. Denatured proteins were separated on 10% SDS-polyacrylamide gels and transferred onto a polyvinylidene difluoride (PVDF) membrane (Millipore, MA, USA) for immunoblot analysis. The primary antibodies used were anti-SOHLH1 (1:1000, Proteintech, Chicago, USA) and GAPDH (1:5000, Abcam, MA, USA).
IF staining
Sperm and testicular tissues from patients and the normal controls and mouse sperm cells were fixed in 4% paraformaldehyde, permeabilized with 0.3% Triton X-100 and blocked with 1% BSA. The slides were then sequentially incubated with primary antibodies at 4°C overnight. The primary antibodies used were anti-SOHLH1 (1:50, Proteintech), anti-SOX9 (1:50, Abcam), anti-KI67 (1:50, Proteintech), anti-SYCP1 (1:50, Abcam), PNA (1:50, Vector, MA, USA) and α-tubulin (1:1000, Abcam).
Electron microscopy and concentrated Papanicolaou staining
Spermatozoal morphology was obtained using concentrated Papanicolaou staining and scanning electron microscopy (SEM). Ultrastructural assessments of flagellar cross sections were performed with transmission electron microscopy (TEM). Papanicolaou staining, SEM and TEM were performed as described in our previous study (9).
H&E for histologic staining
Testicular samples from patients and normal control were fixed with 4% paraformaldehyde overnight, dehydrated in ethanol, embedded in paraffin and sectioned at 5 μm. The sections then underwent routine staining with H&E for histological examination.
Flow cytometric analysis and cell sorting
The testes from 8-week-old male mice were removed and decapsulated. Two million single testicular cells diluted with 2 ml of 1× PBS buffer were stained with Hoechst 33342 (5 μg/ml; Sigma-Aldrich, St Louis, MO, USA) for 1 h at 32°C. Then, propidium iodide (PI, 2 μg/ml; Sigma) was added to exclude the dead cells. Finally, cell analysis and sorting were carried out on an FACS-Calibur flow cytometer (Beckman Coulter, CA, USA) equipped with a cell-sorting system.
Supplementary Material
{kind=link}
Acknowledgements
High tribute shall be paid to the patient and his family for their interest and cooperation. The authors would like to thank the Analytical and Testing Center of Sichuan University for the morphology characterization and the authors would be grateful to Guiping Yuan for her help with TEM images and Yi He for his help with SEM images.
Contributor Information
Mohan Liu, Department of Obstetrics/Gynecology, Joint Laboratory of Reproductive Medicine, Key Laboratory of Obstetric, Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, China; State Key Laboratory of Biotherapy and Cancer Center, West China Hospital, Sichuan University and Collaborative Innovation Center, Chengdu, China.
Yihong Yang, Reproduction Medical Center of West China Second University Hospital, Key Laboratory of Obstetric, Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, Sichuan University, Chengdu, China.
Yan Wang, Reproduction Medical Center of West China Second University Hospital, Key Laboratory of Obstetric, Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, Sichuan University, Chengdu, China.
Suren Chen, Education Key Laboratory of Cell Proliferation and Regulation Biology, College of Life Sciences, Beijing Normal University, Beijing, China.
Ying Shen, Department of Obstetrics/Gynecology, Joint Laboratory of Reproductive Medicine, Key Laboratory of Obstetric, Gynecologic and Pediatric Diseases and Birth Defects of Ministry of Education, West China Second University Hospital, Sichuan University, Chengdu, China.
Authors’ Roles
Y.S. and M.L. were involved in the study design, execution and analysis, article drafting and critical discussion. Y.Y. and S.C. performed the experiments and collected the data and drafted the manuscript. Y.Y., Y.W. and S.C. were involved in the sample collection, whole-exome sequencing and screening for the mutations. All authors approved the final version to be published.
Conflict of Interest statement. The authors declare no conflict of interest related to this study.
Funding
National Key Research and Development Project (2019YFA0802101); Key Research Project of Science &Technology Department of Sichuan Province (2020YJ0291).
References
- 1. Jiao, S.Y., Yang, Y.H. and Chen, S.R. (2021) Molecular genetics of infertility: loss-of-function mutations in humans and corresponding knockout/mutated mice. Hum. Reprod. Update, 27, 154–189. [DOI] [PubMed] [Google Scholar]
- 2. Tournaye, H., Krausz, C. and Oates, R.D. (2017) Novel concepts in the aetiology of male reproductive impairment. Lancet Diabetes Endocrinol., 5, 544–553. [DOI] [PubMed] [Google Scholar]
- 3. Ballow, D., Meistrich, M.L., Matzuk, M. and Rajkovic, A. (2006) Sohlh1 is essential for spermatogonial differentiation. Dev. Biol., 294, 161–167. [DOI] [PubMed] [Google Scholar]
- 4. Barrios, F., Filipponi, D., Campolo, F., Gori, M., Bramucci, F., Pellegrini, M., Ottolenghi, S., Rossi, P., Jannini, E.A. and Dolci, S. (2012) SOHLH1 and SOHLH2 control kit expression during postnatal male germ cell development. J. Cell Sci., 125, 1455–1464. [DOI] [PubMed] [Google Scholar]
- 5. Suzuki, H., Ahn, H.W., Chu, T., Bowden, W., Gassei, K., Orwig, K. and Rajkovic, A. (2012) SOHLH1 and SOHLH2 coordinate spermatogonial differentiation. Dev. Biol., 361, 301–312. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Anderson, E.L., Baltus, A.E., Roepers-Gajadien, H.L., Hassold, T.J., de Rooij, D.G., van Pelt, A.M. and Page, D.C. (2008) Stra8 and its inducer, retinoic acid, regulate meiotic initiation in both spermatogenesis and oogenesis in mice. Proc. Natl. Acad. Sci. U. S. A., 105, 14976–14980. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Choi, Y., Jeon, S., Choi, M., Lee, M.H., Park, M., Lee, D.R., Jun, K.Y., Kwon, Y., Lee, O.H., Song, S.H. et al. (2010) Mutations in SOHLH1 gene associate with nonobstructive azoospermia. Hum. Mutat., 31, 788–793. [DOI] [PubMed] [Google Scholar]
- 8. Nakamura, S., Miyado, M., Saito, K., Katsumi, M., Nakamura, A., Kobori, Y., Tanaka, Y., Ishikawa, H., Yoshida, A., Okada, H. et al. (2017) Next-generation sequencing for patients with non-obstructive azoospermia: implications for significant roles of monogenic/oligogenic mutations. Andrology., 5, 824–831. [DOI] [PubMed] [Google Scholar]
- 9. Shen, Y., Zhang, F., Li, F., Jiang, X., Yang, Y., Li, X., Li, W., Wang, X., Cheng, J., Liu, M. et al. (2019) Loss-of-function mutations in QRICH2 cause male infertility with multiple morphological abnormalities of the sperm flagella. Nat. Commun., 10, 433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Reijo, R., Lee, T.Y., Salo, P., Alagappan, R., Brown, L.G., Rosenberg, M., Rozen, S., Jaffe, T., Straus, D. and Hovatta, O. (1995) Diverse spermatogenic defects in humans caused by Y chromosome deletions encompassing a novel RNA-binding protein gene. Nat. Genet., 10, 383–393. [DOI] [PubMed] [Google Scholar]
- 11. Wang, X., Jin, H.R., Cui, Y.Q., Chen, J., Sha, Y.W. and Gao, Z.L. (2018) Case study of a patient with cryptozoospermia associated with a recessive TEX15 nonsense mutation. Asian J. Androl., 20, 101–102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Schilit, S., Menon, S., Friedrich, C., Kammin, T., Wilch, E., Hanscom, C., Jiang, S., Kliesch, S., Talkowski, M.E., Tüttelmann, F. et al. (2020) SYCP2 translocation-mediated dysregulation and frameshift variants cause human male infertility. Am. J. Hum. Genet., 106, 41–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Luddi, A., Margollicci, M., Gambera, L., Serafini, F., Cioni, M., De Leo, V., Balestri, P. and Piomboni, P. (2009) Spermatogenesis in a man with complete deletion of USP9Y. N. Engl. J. Med., 360, 881–885. [DOI] [PubMed] [Google Scholar]
- 14. Arango, N.A., Li, L., Dabir, D., Nicolau, F., Pieretti-Vanmarcke, R., Koehler, C., McCarrey, J.R., Lu, N. and Donahoe, P.K. (2013) Meiosis I arrest abnormalities lead to severe oligozoospermia in meiosis 1 arresting protein (M1ap)-deficient mice. Biol. Reprod., 88, 76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Li, Y., Qi, W., Liu, G., Du, B., Sun, Q., Zhang, X., Jin, M., Dong, W., Liu, J. and Zheng, Z. (2019) Sohlh1 is required for synaptonemal complex formation by transcriptionally regulating meiotic genes during spermatogenesis in mice. Mol. Reprod. Dev., 86, 252–264. [DOI] [PubMed] [Google Scholar]
- 16. Bayram, Y., Gulsuner, S., Guran, T., Abaci, A., Yesil, G., Gulsuner, H.U., Atay, Z., Pierce, S.B., Gambin, T., Lee, M. et al. (2015) Homozygous loss-of-function mutations in SOHLH1 in patients with nonsyndromic hypergonadotropic hypogonadism. J. Clin. Endocrinol. Metab., 100, E808–E814. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Song, B., Zhang, Y., He, X.J., Du, W.D., Ruan, J., Zhou, F.S., Wu, H., Zha, X., Xie, X.S., Ye, L. et al. (2015) Association of genetic variants in SOHLH1 and SOHLH2 with non-obstructive azoospermia risk in the Chinese population. Eur. J. Obstet. Gynecol. Reprod. Biol., 184, 48–52. [DOI] [PubMed] [Google Scholar]
- 18. Lepine, S., McDowell, S., Searle, L.M., Kroon, B., Glujovsky, D. and Yazdani, A. (2019) Advanced sperm selection techniques for assisted reproduction. Cochrane Database Syst. Rev., 7, CD010461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Wambergue, C., Zouari, R., Fourati Ben Mustapha, S., Martinez, G., Devillard, F., Hennebicq, S., Satre, V., Brouillet, S., Halouani, L., Marrakchi, O. et al. (2016) Patients with multiple morphological abnormalities of the sperm flagella due to DNAH1 mutations have a good prognosis following intracytoplasmic sperm injection. Hum. Reprod., 31, 1164–1172. [DOI] [PubMed] [Google Scholar]
- 20. Liu, C., He, X., Liu, W., Yang, S., Wang, L., Li, W., Wu, H., Tang, S., Ni, X., Wang, J. et al. (2019) Bi-allelic mutations in TTC29 cause male subfertility with asthenoteratospermia in humans and mice. Am. J. Hum. Genet., 105, 1168–1181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Liu, C., Miyata, H., Gao, Y., Sha, Y., Tang, S., Xu, Z., Whitfield, M., Patrat, C., Wu, H., Dulioust, E. et al. (2020) Bi-allelic DNAH8 variants lead to multiple morphological abnormalities of the sperm flagella and primary male infertility. Am. J. Hum. Genet., 107, 330–341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Sha, Y.W., Wang, X., Su, Z.Y., Mei, L.B., Ji, Z.Y., Bao, H. and Li, P. (2019) Patients with multiple morphological abnormalities of the sperm flagella harbouring CFAP44 or CFAP43 mutations have a good pregnancy outcome following intracytoplasmic sperm injection. Andrologia, 51, e13151. [DOI] [PubMed] [Google Scholar]
- 23. Sha, Y., Wei, X., Ding, L., Ji, Z., Mei, L., Huang, X., Su, Z., Wang, W., Zhang, X. and Lin, S. (2020) Biallelic mutations of CFAP74 may cause human primary ciliary dyskinesia and MMAF phenotype. J. Hum. Genet., 65, 961–969. [DOI] [PubMed] [Google Scholar]
- 24. Sha, Y.W., Xu, X., Mei, L.B., Li, P., Su, Z.Y., He, X.Q. and Li, L. (2017) A homozygous CEP135 mutation is associated with multiple morphological abnormalities of the sperm flagella (MMAF). Gene, 633, 48–53. [DOI] [PubMed] [Google Scholar]
- 25. Liu, M., Sun, Y., Li, Y., Sun, J., Yang, Y. and Shen, Y. (2021) Novel mutations in FSIP2 lead to multiple morphological abnormalities of the sperm flagella and poor ICSI prognosis. Gene, 781, 145536. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.