

Abstract
Dystonia is a disabling disease that manifests as prolonged involuntary twisting movements. DYT-THAP1 is an inherited form of isolated dystonia caused by mutations in THAP1 encoding the transcription factor THAP1. The phe81leu (F81L) missense mutation is representative of a category of poorly understood mutations that do not occur on residues critical for DNA binding. Here, we demonstrate that the F81L mutation (THAP1F81L) impairs THAP1 transcriptional activity and disrupts CNS myelination. Strikingly, THAP1F81L exhibits normal DNA binding but causes a significantly reduced DNA binding of YY1, its transcriptional partner that also has an established role in oligodendrocyte lineage progression. Our results suggest a model of molecular pathogenesis whereby THAP1F81L normally binds DNA but is unable to efficiently organize an active transcription complex.
Introduction
DYT-THAP1 or DYT6 is an inherited form of isolated dystonia resulting from dominantly inherited mutations in THAP1 (1). THAP1 is a ubiquitously expressed member of the THAP protein family of transcription factors that share an atypical zinc-finger DNA-binding domain, termed the ‘THAP’ domain (2–4). THAP1 was first described as having a role in regulating cell cycle and apoptotic genes in endothelial (HUVEC) cells (3,5). Subsequent to the discovery of THAP1 mutations in DYT6 dystonia (1), considerable work has focused upon identifying the THAP1 function and its regulated transcriptome in CNS tissue. This work demonstrates an essential role for THAP1 in CNS development and function (4,6–9). THAP1 plays a pronounced role in CNS myelination via a cell-autonomous role in the development of the oligodendrocyte progenitor cells (OPC) into mature myelinating oligodendrocytes (OLs) (4). In addition to its function in OLs, THAP1’s role in normal neuronal function has been explored. THAP1 loss-of-function models demonstrate dysregulated striatal eIF2α signaling (6), abnormal cerebellar physiology (9) and elevated extracellular striatal acetylcholine (7). These studies also demonstrate that the THAP1-regulated transcriptome is highly tissue dependent and likely dependent on additional transcriptional partners. The findings of Aguilo et.al emphasize this point, demonstrating that there is only an ~10% overlap between THAP1-bound genes (ChIP-Seq) in mouse embryonic stem (mES) cells and genes differentially regulated from Thap1 null mES (RNAseq) (10). THAP1 partners are therefore very likely to comprise a key regulatory element of THAP1 transcriptional activity in the CNS.
More than 100 putative THAP1 mutations have been reported; most (73/113) are missense mutations (11), while fewer are indel mutations or cause early truncation (11–13). Many missense mutations occur in the N-terminal THAP domain, but relatively few are predicted to impact DNA binding directly. Studies that combine structure–function analyses and NMR studies demonstrate that DNA binding is mediated directly by eight invariant residues (C5, C10, C54, H57, P26, W36, F58 and P78) and five additional residues (K24, R29, R42, F45 and T48) (3,14,15). Most of these residues reside on the DNA binding surface formed by the anti-parallel two-stranded β-sheet and the loop–helix–loop structure in the THAP domain (15).
Like all missense mutations that do not impact DNA binding residues, the mechanism whereby F81L impacts THAP1 function is poorly understood. Here, we demonstrate that THAP1F81L exhibits impaired transcriptional activity. Similar to THAP1 null mice, THAP1F81L mutant mice exhibit deficits in CNS myelination and abnormalities of compact myelin. Strikingly, the F81L mutation does not disrupt the DNA binding of THAP1. Rather, loss-of-function characteristic of THAP1F81L is caused by significantly reduced binding of its transcriptional partner YY1 and corresponding decreases in the epigenetic marker for active transcription, H3K9ac at its target loci. These observations support a model whereby THAP1F81L is normally bound to DNA but unable to organize an active transcription complex.
Results
The F81L DYT6 mutation disrupts THAP1 transcriptional activity
In prior work, we generated and characterized a knock-in mouse line containing a floxed Thap1F81L allele (4). The F81 residue does not bind DNA but is part of the invariantly conserved AVPTIF motif in the THAP domain (Fig. 1A and B) (15). These prior findings demonstrate that germline Thap1−/− mice exhibit embryonic lethality (4,10). In contrast, Thap1F81L/− mice are born at expected Mendelian ratios and do not exhibit significant abnormalities of growth or lifespan (4). Considered together, these observations demonstrate that the Thap1F81L allele complements Thap1 function but does not indicate whether F81L is a gain- or loss-of-function mutation.
Figure 1 .
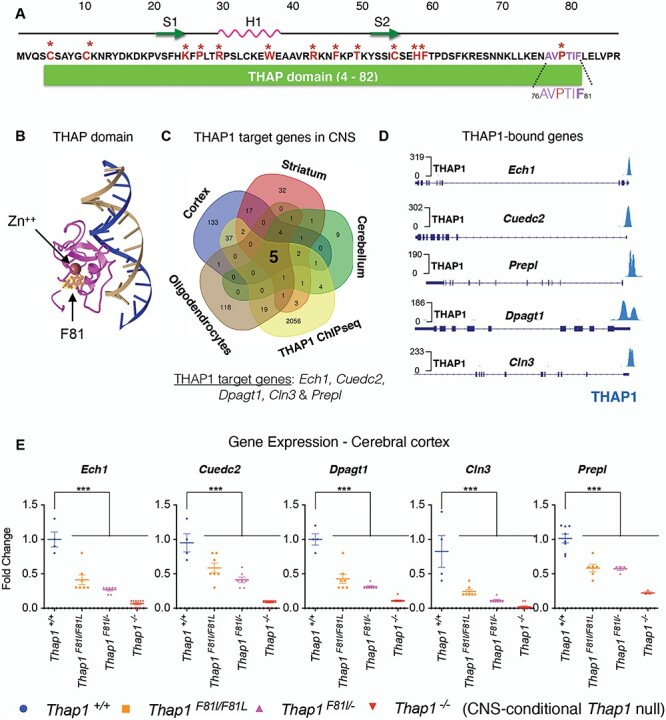
The DYT6 F81L mutation impairs THAP1 transcriptional activity. (A) Residues of the N-terminal THAP domain (residues 4-82). Residues involved directly in DNA binding are highlighted in red and starred. Also shown is the AVPTIF motif (highlighted in purple in inset) and the phe81 residue that is a focus of this study (highlighted in bold purple in the inset). Also illustrated is the location of the phe81 residue relative to the location of the DNA binding surface formed by the two β-strands (S1 and S2) and the α-helix motif (H1) forming the loop–helix–loop structure (15). (B) 3D model of the solution structure (15) of the complex between the THAP zinc finger of hTHAP1 and its specific Rrm1 DNA target highlighting the location of the phe81 residue (yellow) generated using Cn3D (https://www.ncbi.nlm.nih.gov/Structure/CN3D/cn3d.shtml). (C) Venn diagram depicting the identification of target genes that are bound (ChIP-Seq) and regulated (Thap1+/+ versus Thap1−/− differentially expressed genes (DEG) in CNS (cortex, striatum and cerebellum and OL lineage)) by THAP1. The intersection of all datasets identified five THAP1-target genes Ech1, Cuedc2, Dpagt1, Prepl and Cln3. (D) Genome browser track (Integrative Genomics Viewer; https://igv.org) demonstrating THAP1 enrichment at the promoter region of its target loci Ech1, Cuedc2, Dpagt1, Prepl and Cln3 (ENCODE dataset; K652 cells). (E) Significant reduction in the mRNA expression of Ech1, Cuedc2, Cln3 and Dpagt1 in Thap1F81L/F81L, Thap1F81L/− and N-CKO CNS (cerebral cortex) relative to control (Thap1+/+) as measured by qRT-PCR. mRNA expression for each individual gene was normalized to Rpl19 expression and represented in the bar graph (mean ± SEM) as fold change (y-axis) for all genotypes (x-axis) with respect to Thap1+/+. One-way ANOVA for Ech1 = F(3, 23) = 49.75, P < 0.0001; Cuedc2 = F(3, 23) = 35.55, P < 0.0001; Dpagt1 = F(3, 23) = 58.763, P < 0.0001; Cln3 = F(3, 23) = 20.53, P < 0.0001 and Prepl = F(3, 23) = 28.13, P < 0.0001; Dunnett’s multiple comparisons test: adjusted P-value < 0.0001.
To further assess the effect of the F81L mutation, we sought to identify genes that are highly transcriptionally sensitive to THAP1. We examined four datasets that include genes significantly altered by THAP1 loss in brain tissue (separate datasets for cortex, striatum and cerebellum), and in primary cultures of oligodendrocyte progenitor cells (16), as well as ChIP-seq data from ENCODE (Fig. 1C and Supplementary Material, Table S1). We identified five genes (Ech1, Cuedc2, Dpagt1, Prepl and Cln3) that were common to all of these datasets (Fig. 1C). THAP1 binds to the promoter of each of these genes (Fig. 1D), and the expression of these genes is reduced more than 10-fold in the cerebral cortex of CNS-conditional Thap1 null mice (Thap1flx/−; Nestin-Cre or ‘N-CKO’; N-CKO versus Thap1+/+; one-way ANOVA; P < 0.0001 for all genes; N = 4; Fig. 1E). The sensitivity of these genes to THAP1 in multiple contexts identified them as ideal candidates to explore the transcriptional impact of pathogenic Thap1 mutations.
We examined the effect of the F81L mutation on the transcription of these five target genes (Ech1, Cuedc2, Dpagt1, Cln3 and Prepl). We assayed the expression of these genes in cortical tissue from an allelic series of Thap1 mutant mice (Thap1+/+, Thap1F81L/F81L, Thap1F81L/− and Thap1−/−). Expression of all five genes was significantly decreased in both Thap1F81L/F81L and Thap1F81L/− compared with Thap1+/+ cortical tissue (Thap1F81L/F81L: >2-fold decrease for all genes; N = 4; one-way ANOVA: P < 0.001 for Ech1, Dpagt1, Prepl and Cln3 and P < 0.0016 for Cuedc2; Thap1F81L/−: >5-fold decrease for Ech1, Dpagt1 and Cln3; one-way ANOVA: p < 0.0001 and > 2-fold decrease for Cuedc2 and Prepl; N = 4; one-way ANOVA: P < 0.0001; Fig. 1E). Expression of all five genes was further reduced by THAP1 deletion in the CNS (N-CKO) (Fig. 1E). These results demonstrate that the F81L mutation significantly reduces THAP1 transcriptional activity, despite having no effect on THAP1 expression (Supplementary Material, Fig. S1). There were no significant differences in the expression of four (of five) of these genes between Thap1F81L/F81L, Thap1F81L/− cortical tissue, indicating that the F81 mutation has a marked impact on the transcription of these targets. These data, together with the viability of mice containing a single F81L allele, identify Thap1F81L as a hypomorphic allele.
The F81L DYT6 mutation causes CNS hypomyelination
We reported that conditional deletion of Thap1 either from the CNS (nestin-Cre; `N-CKO') or from oligodendrocytes (Olig2-Cre; ‘O-CKO’) delays myelination, overtly visible as hypomyelination in the juvenile CNS (4). We therefore explored whether the F81L loss-of-function mutation similarly causes myelination defects. We assessed the density of myelinated axons and myelin ultrastructure in the genu of the corpus callosum (CC) from P21 Thap1+/+, Thap1F81L/F81L, Thap1F81L/− and N-CKO mice using transmission electron microscopy (TEM). Mice carrying the Thap1F81L allele exhibited significant reductions in the density of myelinated axons in the CC (Fig. 2A and D; 25% decrease in Thap1F81L/F81L; one-way ANOVA; P = 0.007 and 35% decrease in Thap1F81L/−; one-way ANOVA; P = 0.0018 and 70% decrease in N-CKO; one-way ANOVA; P < 0.0001). The observed hypomyelination did not result from axonal degeneration, as total axonal density (myelinated and non-myelinated combined) did not differ significantly between control, F81L and N-CKO genotypes (Fig. 2E). The compact myelin formed also exhibited structural defects in DYT6 mutant CNS. The myelin sheaths of N-CKO, Thap1F81L/− and Thap1F81L/F81L mice were significantly thinner (larger g-ratio) (Fig. 2B, C, F; 15% increase in g-ratio for both Thap1F81L/F81L; and Thap1F81L/−; one-way ANOVA; P < 0.0001 and 25% increase in g-ratio for N-CKO; one-way ANOVA; P < 0.0001). Loss of THAP1 function also increased the average diameter of myelinated axons, suggesting preferential loss in the myelination of smaller caliber axons (Fig. 2B and G; ~20% increase in the caliber of axons myelinated in Thap1F81L/F81L, Thap1F81L/− and N-CKO tissue; one-way ANOVA; P < 0.0001).
Figure 2 .
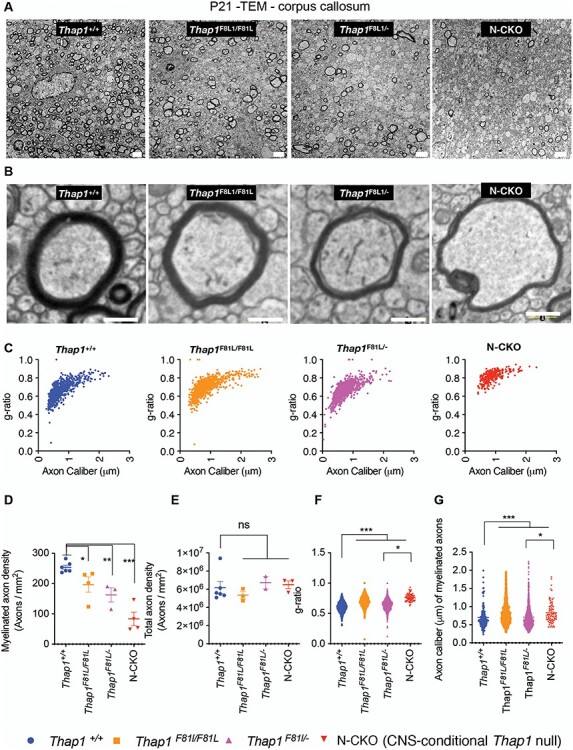
The DYT6 F81L mutation causes hypomyelination in vivo. (A, B) Representative transmission EM images at low (scale bar 4 μm) (A) and high magnification (scale bar 200 nm) (B) from the genu of corpus callosum (CC) at P21 from control (Thap1+/+), DYT6 homozygous (Thap1F81L/F81L), DYT6 hemizygous (Thap1F81L/−) and N-CKO (Thap1F81L/−; Nestin-Cre+) mice. (C) Graph depicting g-ratio (y-axis) represented in relation to axon caliber (μm; x-axis) for Thap1+/+, Thap1F81L/F81L, Thap1F81L/− and Thap1F81L/−; Nestin-Cre+. (D) Quantification of the density of myelinated axons (y-axis; number of axons/mm2) represented as mean ± SEM for all genotypes (x-axis). Thap1+/+ = 253.8 ± 7.11, Thap1F81L/F81L = 196.8 ± 26.33; Thap1F81L/− = 162.6 ± 23.6; N-CKO = 83.5 ± 22.39; one-way ANOVA F(3, 13) = 16.73, P < 0.0001, Dunnett’s multiple comparisons test: adjusted P-value < 0.0001. (E) Quantification of the density of total axons (y-axis; number of axons/mm2) represented as mean ± SEM for all genotypes (x-axis). Thap1+/+ = 6.18 × 106 ± 6.6 × 105, Thap1F81L/F81L = 5.35 × 106 ± 3.86 × 105; Thap1F81L/− = 6.73 × 106 ± 6.87 × 105; N-CKO = 6.50 × 106 ± 4.31 × 105; one-way ANOVA F(3, 10) = 16.73, P = 0.619, Dunnett’s multiple comparisons test: adjusted P-value = 0.7183. (F) g-ratio represented in relation to genotype (x-axis) as mean ± SEM. Thap1+/+ = 0.60 ± 0.002, Thap1F81L/F81L = 0.698 ± 0.0028; Thap1F81L/− = 0.652 ± 23.6; N-CKO = 0.76 ± 0.007 one-way ANOVA F(3,3035) = 205.1, P < 0.0001, Dunnett’s multiple comparisons test: adjusted P-value < 0.0001. (G) Axon caliber of myelinated axons (y-axis; μm of individual axons) represented as mean ± SEM for all genotypes (x-axis). Thap1+/+ = 0.69 μm ± 0.025, Thap1F81L/F81L = 0.828 μm ± 0.010; Thap1F81L/− = 0.71 μm ± 0.008; N-CKO = 0.83 μm ± 0.03. One-way ANOVA F(3,3035) = 205.1, P < 0.0001, Dunnett’s multiple comparisons test: adjusted P-value < 0.0001.
The F81L mutation does not disrupt endogenous THAP1 binding at target loci
Having established that the F81L mutation impairs THAP1 function both transcriptionally and functionally at the level of myelination, we next explored the molecular mechanism responsible for this effect. Available evidence from structural analyses (3,14,15) (Fig. 1B) indicates that the F81 residue does not participate directly in DNA binding. We assessed whether the mutation may nevertheless affect, perhaps indirectly, DNA occupancy of THAP1. Using quantitative chromatin immunoprecipitation (qChIP), we compared the binding of THAP1 and THAP1F81L at several target loci. We performed qChIP analyses in clonal neural stem cell (NSC) lines because they are a uniform cell source common to the neural lineage. NSC was derived from Thap1+/+, Thap1F81L/− and Thap1−/− (Thap1flx/−; Nestin-Cre) mouse lines. Prior to qChIP, we assessed whether the transcription of our established target genes remained sensitive to THAP1 loss in these cells. Of the five THAP1 target genes, four (Ech1, Cuedc2, Dpagt1 and Prepl) exhibited significantly decreased expression in Thap1−/− NSC cells (>10-fold decreased expression for Ech1 and Cuedc2 and > 5-fold decreased expression for Dpagt1 and Prepl in Thap1−/− versus Thap1+/+; ANOVA: P < 0.0001 for Ech1 and Cuedc2; Fig. 3A) confirming that THAP1 is required for the expression of these genes in NSC. The other core THAP1 target gene, Cln3, showed negligible expression in NSC, so it was not further studied (data not shown).
Figure 3 .
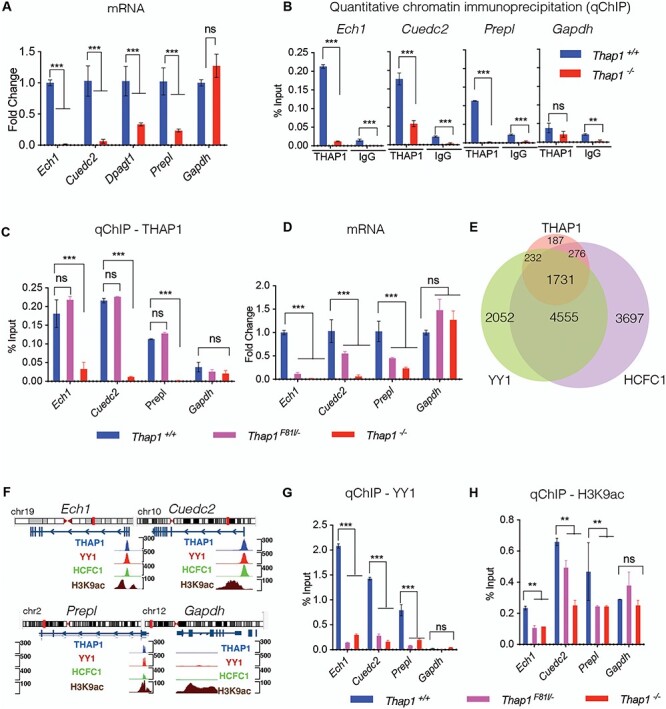
The DYT6 F81L mutation disrupts endogenous YY1 binding. (A) Significant reduction in the mRNA expression of Ech1, Cuedc2, Dpagt1 and Prepl in Thap1−/− NSC relative to Thap1+/+ NSC as measured by qRT-PCR (Gapdh expression was unchanged). mRNA expression for each individual gene (x-axis) was normalized to Rpl19 expression and represented in the bar graph (mean ± SEM) as fold change (y-axis) for Thap1−/− relative to Thap1+/+. t-test; P < 0.0001 for Ech1, Cuedc2, Dpagt1 and Prepl; P = 0.37 for Gapdh. (B) Quantitative ChIP (qChIP) demonstrating the binding of THAP1 and isotype-matched IgG antibody (x-axis) at the promoter region of target genes in NSC. Binding, represented as % input (y-axis), is demonstrated for THAP1 and normalized anti-goat IgG for chromatin isolated from Thap1+/+ and Thap1−/− NSC cells for Ech1, Ceudc2, Prepl and Gapdh loci; ANOVA; P < 0.0001 for Ech1 and Cuedc2 and P = 0.342 for Gapdh. (C) Quantitative ChIP (qChIP) demonstrating the binding of THAP1 at the promoter region of Ech1, Cuedc2, Prepl and Gapdh loci in NSC. Binding, represented as % input (y-axis), is demonstrated for THAP1 for chromatin isolated from Thap1+/+, Thap1F81L/− and Thap1−/− NSC cells for the promoter region of Ech1, Cuedc2, Prepl and Gapdh loci (x-axis). One-way ANOVA for THAP1 qChIP—Ech1—F(2, 6) = 18,65, P = 0.0027; Cuedc2—F(2, 6) = 1354, P value < 0.0001 and Gapdh—F(2, 6) = 11.72, P = 0.038; Dunnett’s multiple comparisons test: adjusted P-value < 0.0001. (D) Significant reduction in the mRNA expression of Ech1, Cuedc2 and Prepl but not Gapdh in Thap1F81L/− and Thap1−/− NSC relative to Thap1+/+ NSC as measured by qRT-PCR. mRNA expression for each individual gene (x-axis) was normalized to Rpl19 expression and represented in the bar graph (mean ± SEM) as fold change (y-axis) for all genotypes with respect to Thap1+/+. One-way ANOVA for Ech1—F(3, 23), P < 0.0001; Cuedc2—F(3, 23) = 22.36, P < 0.0001 and Gapdh—F(3, 23) = 22.44 P = 0.0016; Dunnett’s multiple comparisons test: adjusted P-value < 0.0001. (E) Venn diagram depicting the number of genes co-bound by THAP1, YY1 and HCFC1 (ChIP-Seq K652 cells, ENCODE). (F) Genome browser track showing CHIP-Seq signals (peaks) for THAP1, YY1, HCFC1 and H3K9ac (ENCODE datasets in K652 cells) at Ech1, Ceudc2, Prepl and Gapdh loci. The top panel shows idiogram and the exon–intron structure for all the loci of interest. (G, H) Quantitative ChIP (qChIP) demonstrating the binding of YY1, and H3K9ac at the promoter region of Ech1, Cuedc2, Prepl and Gapdh loci in NSC. Binding, represented as % input (y-axis), is demonstrated for (G) YY1 and (H) H3k9ac for chromatin isolated from Thap1+/+, Thap1F81L/− and Thap1−/− NSC cells for the promoter region of Ech1, Cuedc2, Prepl and Gapdh loci (x-axis); one-way ANOVA for YY1 qChIP—Ech1—F(2, 3) = 1877, P < 0.0001; Cuedc2—F(2, 3) = 624.9, P < 0.0001 and Gapdh—F(2, 3) = 12.48, P = 0.0351; Dunnett’s multiple comparisons test: adjusted P-value < 0.0001. One-way ANOVA for H3K9ac qChIP—Ech1—F(2, 3) = 35.78, P = 0.0081; Cuedc2—F(2, 3) = 35.32, P = 0.0082 and Gapdh—F(2, 3) = 11.72, P = 0.038; Dunnett’s multiple comparisons test: adjusted P-value < 0.0001.
qChIP analyses confirmed that THAP1 is bound to the promoter region of Ech1, Cuedc2 and Prepl loci in the NSC lineage, but not in THAP1 null NSC (Fig. 3B). We detected very low amounts of THAP1 (~10-fold lower) bound to the promoter of Dpagt1 loci in NSC, leading us to focus on Ech1, Cuedc2 and Prepl for further mechanistic studies in NSC. Specificity of qChIP was further confirmed by lack of signal from the promoter of the active housekeeping gene Gapdh (Fig. 3B) and lack of binding by an isotype-specific negative control antibody (Goat IgG) to the target loci (Fig. 3B). We next measured endogenous THAP1F81L binding at Ech1, Cuedc2 and Prepl loci in Thap1F81L/− NSC and compared these data to THAP1 binding in Thap1+/+ NSC. In striking contrast to chromatin derived from Thap1−/− NSC, F81L-THAP1 binding in Thap1F81L/− NSC is comparable to that of Thap1+/+ cultures at the promoter regions of Ech1, Cuedc2 and Prepl loci (Fig. 3C), whereas no binding was detected at the control Gapdh loci (Fig. 3C). Thus, the F81L DYT6 mutation does not significantly disrupt THAP1 DNA binding.
Given these observations, we tested whether the F81L mutation impairs THAP1 function in NSC, as it does in CNS tissue. We compared the expression of the THAP1 target genes Ech1, Cuedc2, Prepl and Gapdh in Thap1F81L/−,Thap1+/+ and Thap1−/− NSCs. Compared with controls, Thap1F81L/− NSCs exhibited significantly lower expression of all THAP1 target genes assessed (>2-fold decrease in Thap1F81L/− for Ech1, Prepl and Cuedc2; ANOVA: P < 0.0001; Fig. 3D). These findings are consistent with the findings of those in DYT6 cerebral cortex (Fig. 1E) and indicate that the F81L mutation impairs the transcriptional function of THAP1 via a mechanism distinct from DNA binding.
DYT6 mutations disrupt the THAP1-mediated transcriptional complex at target loci
Significantly reduced transcription of core THAP1 target genes despite normal Thap1F81L DNA binding at their promoters suggests that the F81L mutation impairs the binding of THAP1 transcriptional partners. We used in silico analyses to identify the candidate THAP1 transcriptional partners. We utilized ChromNet, which uses a conditional-dependence network among regulatory factors from ENCODE ChIP-seq datasets to identify closely related datasets (17). A network of YY1, HCFC1 and H3K9ac were the strongest predicted THAP1 interactors, based on shared genome location (Supplementary Material, Fig. S2). These analyses demonstrate that 80.9 and 82.7% of the promoter regions of all THAP1-target genes are co-bound with YY1 and HCFC1, with 71.3% of genes co-bound with both YY1 and HCFC1 (Venn diagram, Fig. 3E). Consistent with our findings, these datasets predicted strong enrichment of YY1, HCFC1 and H3K9ac specifically for all our core THAP1 target genes (Ech1, Cuedc2, Dpagt1 and Prepl) (Fig. 3F).
Several lines of evidence led us to focus on YY1 as potentially impacted by the F81L mutation. YY1 is a transcription factor known to interact with THAP1 (4), has a known role in OPC differentiation (18) and is implicated in dystonia (19–22). We also examined THAP1-dependent changes in H3K9ac, an epigenetic marker of active transcription that is highly associated with THAP1-bound chromatin. qChIP in NSC confirmed YY1 binding and H3K9ac enrichment at the promoter regions of Ech1, Cuedc2 and Prepl in NSC (Fig. 3G and H). A signal for H3K9ac, but not YY1, was detected at the Gapdh locus (Fig. 3G and H), validating the in silico analyses. Strikingly, there was a significant loss of YY1 binding (Fig. 3G) from the promoters of Ech1, Cuedc2 and Prepl in Thap1F81L/− and Thap1−/− NSC (>75% decrease in Thap1F81L/− and >90% decrease in Thap1−/− for Ech1, Cuedc2 and Prepl relative to Thap1+/+; ANOVA; p < 0.001) that correlated with decreased transcription from these loci (Fig. 3A). As with YY1, we observed a significant reduction of H3K9ac enrichment (Fig. 3H) in DYT6 Thap1F81L/− NSC (>50% decrease in Thap1F81L/− and >75% loss in Thap1−/− NSC relative to Thap1+/+ for Ech1 and Cuedc2; ANOVA; P < 0.001; for Prepl; ANOVA; P < 0.05). No differences were observed for enrichment of H3K9ac at the Gapdh loci in Thap1F81L/− or Thap1−/− NSC, demonstrating specificity at THAP1 target genes. These findings suggest strongly that the F81L mutation acts by impairing the ability of THAP1 to normally assemble an active transcriptional complex that includes YY1.
Discussion
Our studies establish that the F81L mutation impairs THAP1 transcriptional function and causes in vivo defects in myelination similar to those observed in CNS conditional THAP1 knockout mice. We demonstrate that the F81L missense mutation prevents THAP1 from organizing a YY1-containing co-activator complex but does not significantly alter DNA binding. The myelination abnormalities we observed in the CNS of mice with the Thap1F81L disease mutation and the white matter microstructure abnormalities previously described in DYT-THAP1 (23,24) and other forms of dystonia (25–30) add to the growing evidence of abnormal myelination in the pathogenesis of dystonia.
The THAP1 F81L substitution mutation was first reported in an Amish–Mennonite family with mixed-onset primary torsion dystonia (1), with an onset age ranging from 9 to 13 years (1). The F81L mutation was present in four affected individuals across two generations. Prior structural studies of the THAP domain predict that the F81 residue does not participate in DNA binding (6,15), while gel shift assays suggested F81L mutation could affect THAP1 binding affinity (1) and another study reporting it could affect THAP1 stability (31). Our qChIP data are consistent with the prediction made from structural data, demonstrating no change in binding of endogenous levels of F81L mutant THAP1 at its target loci. This behavior appears distinct from that caused by the C54Y missense mutation, which mutates the Zn2+ binding C2CH motif of the THAP domain. In contrast to Thap1F81L/− mutants, Thap1C54Y/− mutant mice exhibit embryonic lethality (32), because of a marked impairment of DNA binding (10,14). Our studies of the F81L mutation highlights that DNA binding ability does not fully predict disease relevance.
The significant reduction in DNA binding of YY1 was paralleled by the loss of H3K9ac histone modification. These observations support the conclusion that F81L acts by impairing the ability of THAP1 to organize an active transcriptional complex. In prior studies, we and others (4,10) have identified several transcription factors and epigenetic modifiers that are significantly co-enriched with THAP1 on the genome. Most prominent among them are YY1 and HCFC1 (Fig. 3E), proteins that interact with THAP1 (4,33,34) and co-regulate THAP1-bound genes (4,35).
Multiple aspects of YY1 function are consistent with a key role in DYT6 pathogenesis. There is extensive co-binding of YY1 and THAP1 at the genomic level (4), and these proteins play a co-regulatory role in transcription (4). Both proteins also participate in CNS myelination via cell autonomous effects in the oligodendrocyte lineage (18). Several recent studies report YY1 loss-of-function mutations in dystonia patients (19–22). Considered together, these findings suggest that these two transcription factors operate in a shared pathway impacting CNS myelination and motor function. The established role of THAP1 and YY1 in neurodevelopment (4,16,18) and the recent identification of several monogenic variants with high diagnostic value in early onset dystonias (36,37) offer strong support to the premise that dysregulation of neurodevelopmental mechanisms is a key feature of dystonia pathophysiology. Indeed, pathogenic mechanisms are observed during CNS development and maturation in multiple animal models and appear to be directly linked to the onset of abnormal movements (4,38–40).
Prior work identifying the relationship between THAP1 and YY1 was performed in THAP1 null mice (4). Our new findings extend that work, linking abnormalities of YY1 binding to a pathogenic, disease-causing DYT6 mutation. We expect that the binding of additional THAP1 co-factors (e.g. HCFC1) are also impaired by THAP1F81L. Indeed, prior studies demonstrate that when expressed in SH-SY5Y, several DYT6 mutants (p.N136S, p.N136K, p.Y137C) exhibit functional deficits in recruiting HCFC1 (34).
Identification of the F81L-THAP1 as a hypomorphic protein has important implications for understanding the dominant inheritance of the disease. As THAP1 functions as a dimer (41–43), our findings raise the possibility that THAP1F81L exerts a dominant negative effect on the wild-type protein. Exploring whether F81L and wild-type THAP1 interact, the transcriptional impact of such a heterodimer is an important future consideration that will further advance our understanding of the molecular pathogenesis of DYT6 dystonia.
Materials and Methods
Generation and maintenance of mice
Animal research was conducted in accordance with the NIH laboratory animal care guidelines and with the Institutional Animal Care and Use Committee (IACUC) at the University of Michigan. Generation, characterization and genotyping of knock-in mice containing a floxed Thap1F81L allele in exon 2 have been previously described (4). Nestin-Cre+ was purchased from Jackson Laboratory (Stock # 003771). The breeding strategy used to derive all primary NSC cells and conditional null mice was as follows: Thap1F81L/−, Nestin-Cre+ was crossed with Thap1F81L/+ or Thap1F81L/+ to produce the following genotypes: Thap1+/+; Thap1+/−; Thap1F81L/−; Thap1F81L/+; Nestin-Cre+; Thap1F81L/−; Nestin-Cre+ (N-CKO). Age- and sex-matched littermate mice were used for all experiments. Primers used for genotyping in this study (Thap1 and Cre) are listed in Supplementary Material, Table S2.
RNA extraction and qRT-PCR
Total RNA extraction from NSC cultures for qRT-PCR analysis was done using NucleoSpin® RNA (Takara) and TRIzol (Thermo Fisher) for mouse cerebral cortex as per manufacturer’s instructions. cDNA synthesis from total RNA was done using MMLV Reverse Transcriptase (Takara) as per manufacturer’s instructions. Quantitative real-time PCR (qRT–PCR) was performed with the StepOnePlus System (ABI) and 2× SYBR Power Mix (ABI). Primers used for gene expression analyses in this study are listed in Supplementary Material, Table S2.
Electron microscopy
EM was performed as previously described (4). P21 mice were anesthetized and perfused with 3% paraformaldehyde/2.5% glutaraldehyde in 0.1 M phosphate buffer. Brains were dissected and post fixed at 4°C overnight. Tissues were dissected, processed and sectioned at Emory University EM core facility. EM image acquisition was done at the University of Michigan, MIL core services using JEOL JSM 1400.
Analysis of g-ratio and axon caliber
Axon caliber (average of the major and minor axes of the axonal perimeter) and g-ratio (ratio of the inner axonal diameter to total (including myelin) outer diameter) were calculated using an automated approach as follows: for each EM image acquired at 20 000× magnification, the inner and outer perimeters of myelinated axons were manually traced and saved as binary masks using ImageJ. The g-ratio and axon caliber of each myelinated axon were calculated using CellProfiler 3.1.9 (44). For each axon, the major axis, minor axis and length of inner and outer perimeter were measured using the manually traced binary masks. G-ratio was calculated by dividing the inner perimeter by the outer perimeter, and axon caliber was calculated by taking the average of the major and minor axes of the inner perimeter.
Derivation of NSC cells
NSC was isolated from the sub-ventricular zone (SVZ) of P7 mouse pups corresponding to Thap1+/+; Thap1F81L/− or Thap1F81L/−; Nestin-Cre+(N-CKO) genotypes as previously described (4,45) to be propagated and maintained as clonal lines. SVZ-derived neurospheres were propagated in NSC growth media (Neurobasal media supplemented with 1× B27, 1× Antibiotic-Antimycotic, 1× GlutaMAX, 20 ng/μl Fgf2 and 20 ng/μl Egf2) in low attachment T-25 flasks for the first two passages. NSC clonal lines were further expanded as a monolayer on laminin-coated dishes in NSC expansion media (DMEM/F12 media supplemented with 1× N2, 1× AA, 1× GlutaMAX, 20 ng/μl Fgf2 and 20 ng/μl Egf2).
Quantitative chromatin immunoprecipitation (qCHIP)
qCHIP was performed as previously described (46). Sheared chromatin (sonicated to 200–500 bp) from 2 × 106 mouse NSCs was incubated with 4 μg of Goat THAP1 (sc-98174, Santa Cruz), 4 μg of Rabbit YY1 (sc-98174, Santa Cruz), 2 μg of Rabbit H3K9ac (sc-98174, Activemotif) or 4 μg of normalized Goat IgG (Santa Cruz), or normalized 4 μg of Rabbit IgG (Santa Cruz) using Dynabeads (Thermo Fisher Scientific). After washing, elution and cross-link reversal, DNA from each ChIP sample and the corresponding input sample was purified (PCR Cleanup, Takara) and analyzed further using qPCR. Each ChIP sample and a range of dilutions of the corresponding input sample (0.01—2% input) were quantitatively analyzed with gene-specific primers using the StepOnePlus System (ABI) and SYBR qPCR Powermix (ABI). Primers used for qChIP analyses in this study are listed in Supplementary Material, Table S2.
Statistics
All data are reported as mean ± SEM. All statistical tests reported (Student’s t-tests, one-way ANOVAs) were performed using GraphPad Prism software (V9).
Data and software availability
The GEO accession number for CHIP-seq data used in manuscript for THAP1 is GSM803408, YY1 is GSM803446 and H3K9ac is GSM788082. The GEO accession number for gene expression data for Thap1 cKO CNS tissue is GSE97372 and Thap1 cKO oligodendrocyte lineage is GSE161556. The code utilized for automated analysis of g-ratio and axon caliber using CellProfiler 3.1.9 is available at https://github.com/suminkim/Analysis_CellProfiler.
Supplementary Material
Acknowledgements
We thank Haoran Huang and Jack Koulos for technical assistance. We thank Dr Vikram Shakkottai and Dr Roman Giger for assistance with laboratory equipment and microscope. We thank Cathy Collins for critical reading of manuscript. We thank the staff of University of Michigan’s Core Facilities (DNA Sequencing Core, Microscopy and Image Analysis Laboratory and Unit of Laboratory Animal Medicine) and Hong Yi (EM Core, Emory University). This research was supported in part by the following grants: to W.T.D. (1R01NS109227 NINDS). Conflict of Interest statement. The authors declare that they have no competing interests.
Contributor Information
Dhananjay Yellajoshyula, Department of Neurology, University of Michigan, Ann Arbor, MI 48109, USA.
Abigail E Rogers, Molecular Cellular and Developmental Biology, University of Michigan, Ann Arbor, MI 48109, USA.
Audrey J Kim, Peter O’Donnell Jr. Brain Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Sumin Kim, Department of Neurology, University of Michigan, Ann Arbor, MI 48109, USA; Cellular and Molecular Biology Graduate Program, University of Michigan, Ann Arbor, MI 48109, USA.
Samuel S Pappas, Peter O’Donnell Jr. Brain Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Neurology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
William T Dauer, Peter O’Donnell Jr. Brain Institute, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Neurology, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA; Department of Neuroscience, University of Texas Southwestern Medical Center, Dallas, TX 75390, USA.
Funding
NATIONAL INSTITUTE OF NEUROLOGICAL DISORDERS AND STROKE (1R01NS109227 and 1R01NS110853).
References
- 1. Fuchs, T., Gavarini, S., Saunders-Pullman, R., Raymond, D., Ehrlich, M.E., Bressman, S.B. and Ozelius, L.J. (2009) Mutations in the THAP1 gene are responsible for DYT6 primary torsion dystonia. Nat. Genet., 41, 286–288. [DOI] [PubMed] [Google Scholar]
- 2. Roussigne, M., Kossida, S., Lavigne, A.C., Clouaire, T., Ecochard, V., Glories, A., Amalric, F. and Girard, J.P. (2003) The THAP domain: a novel protein motif with similarity to the DNA-binding domain of P element transposase. Trends Biochem. Sci., 28, 66–69. [DOI] [PubMed] [Google Scholar]
- 3. Clouaire, T., Roussigne, M., Ecochard, V., Mathe, C., Amalric, F. and Girard, J.P. (2005) The THAP domain of THAP1 is a large C2CH module with zinc-dependent sequence-specific DNA-binding activity. Proc. Natl. Acad. Sci. USA, 102, 6907–6912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Yellajoshyula, D., Liang, C.C., Pappas, S.S., Penati, S., Yang, A., Mecano, R., Kumaran, R., Jou, S., Cookson, M.R. and Dauer, W.T. (2017) The DYT6 dystonia protein THAP1 regulates myelination within the oligodendrocyte lineage. Dev. Cell, 42, 52, e54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Cayrol, C., Lacroix, C., Mathe, C., Ecochard, V., Ceribelli, M., Loreau, E., Lazar, V., Dessen, P., Mantovani, R., Aguilar, L. et al. (2007) The THAP-zinc finger protein THAP1 regulates endothelial cell proliferation through modulation of pRB/E2F cell-cycle target genes. Blood, 109, 584–594. [DOI] [PubMed] [Google Scholar]
- 6. Zakirova, Z., Fanutza, T., Bonet, J., Readhead, B., Zhang, W., Yi, Z., Beauvais, G., Zwaka, T.P., Ozelius, L.J., Blitzer, R.D. et al. (2018) Mutations in THAP1/DYT6 reveal that diverse dystonia genes disrupt similar neuronal pathways and functions. PLoS Genet., 14, e1007169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Eskow Jaunarajs, K.L., Scarduzio, M., Ehrlich, M.E., McMahon, L.L. and Standaert, D.G. (2019) Diverse mechanisms lead to common dysfunction of striatal cholinergic interneurons in distinct genetic mouse models of dystonia. J. Neurosci., 39, 7195–7205. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Frederick, N.M., Shah, P.V., Didonna, A., Langley, M.R., Kanthasamy, A.G. and Opal, P. (2019) Loss of the dystonia gene Thap1 leads to transcriptional deficits that converge on common pathogenic pathways in dystonic syndromes. Hum. Mol. Genet., 28, 1343–1356. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. van der Heijden, M.E., Kizek, D.J., Perez, R., Ruff, E.K., Ehrlich, M.E. and Sillitoe, R.V. (2021) Abnormal cerebellar function and tremor in a mouse model for non-manifesting partially penetrant dystonia type 6. J. Physiol., 599, 2037–2054. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Aguilo, F., Zakirova, Z., Nolan, K., Wagner, R., Sharma, R., Hogan, M., Wei, C., Sun, Y., Walsh, M.J., Kelley, K. et al. (2017) THAP1: role in mouse embryonic stem cell survival and differentiation. Stem Cell Rep., 9, 92–107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Domingo, A., Yadav, R. and Ozelius, L.J. (2020) Isolated dystonia: clinical and genetic updates. J. Neural Transm. (Vienna), 128, 405–416. [DOI] [PubMed] [Google Scholar]
- 12. Blanchard, A., Ea, V., Roubertie, A., Martin, M., Coquart, C., Claustres, M., Beroud, C. and Collod-Beroud, G. (2011) DYT6 dystonia: review of the literature and creation of the UMD locus-specific database (LSDB) for mutations in the THAP1 gene. Hum. Mutat., 32, 1213–1224. [DOI] [PubMed] [Google Scholar]
- 13. Bragg, D.C., Armata, I.A., Nery, F.C., Breakefield, X.O. and Sharma, N. (2011) Molecular pathways in dystonia. Neurobiol. Dis., 42, 136–147. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Bessiere, D., Lacroix, C., Campagne, S., Ecochard, V., Guillet, V., Mourey, L., Lopez, F., Czaplicki, J., Demange, P., Milon, A. et al. (2008) Structure-function analysis of the THAP zinc finger of THAP1, a large C2CH DNA-binding module linked to Rb/E2F pathways. J. Biol. Chem., 283, 4352–4363. [DOI] [PubMed] [Google Scholar]
- 15. Campagne, S., Saurel, O., Gervais, V. and Milon, A. (2010) Structural determinants of specific DNA-recognition by the THAP zinc finger. Nucleic Acids Res., 38, 3466–3476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Yellajoshyula, D., Pappas, S.S., Rogers, A.E., Choudhury, B., Reed, X., Ding, J., Cookson, M.R., Shakkottai, V.G., Giger, R.J. and Dauer, W.T. (2021) THAP1 modulates oligodendrocyte maturation by regulating ECM degradation in lysosomes. Proc. Natl. Acad. Sci. USA., 118, e2100862118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Lundberg, S.M., Tu, W.B., Raught, B., Penn, L.Z., Hoffman, M.M. and Lee, S.I. (2016) Chrom net: learning the human chromatin network from all ENCODE ChIP-seq data. Genome Biol., 17, 82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. He, Y., Dupree, J., Wang, J., Sandoval, J., Li, J., Liu, H., Shi, Y., Nave, K.A. and Casaccia-Bonnefil, P. (2007) The transcription factor Yin Yang 1 is essential for oligodendrocyte progenitor differentiation. Neuron, 55, 217–230. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Gabriele, M., Vulto-van Silfhout, A.T., Germain, P.L., Vitriolo, A., Kumar, R., Douglas, E., Haan, E., Kosaki, K., Takenouchi, T., Rauch, A. et al. (2017) YY1 Haploinsufficiency causes an intellectual disability syndrome featuring transcriptional and chromatin dysfunction. Am. J. Hum. Genet., 100, 907–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Carminho-Rodrigues, M.T., Steel, D., Sousa, S.B., Brandt, G., Guipponi, M., Laurent, S., Fokstuen, S., Moren, A., Zacharia, A., Dirren, E. et al. (2020) Complex movement disorder in a patient with heterozygous YY1 mutation (Gabriele-de Vries syndrome). Am. J. Med. Genet. A, 182, 2129–2132. [DOI] [PubMed] [Google Scholar]
- 21. Keller Sarmiento, I.J. and Mencacci, N.E. (2021) Genetic Dystonias: update on classification and new genetic discoveries. Curr. Neurol. Neurosci. Rep., 21, 8. [DOI] [PubMed] [Google Scholar]
- 22. Zorzi, G., Sarmiento, I.J.K., Danti, F.R., Bustos, B.I., Invernizzi, F., Panteghini, C., Reale, C., Garavaglia, B., Chiapparini, L., Lubbe, S.J. et al. (2021) YY1-related dystonia: clinical aspects and long-term response to deep brain stimulation. Mov. Disord., 36, 1461–1462. [DOI] [PubMed] [Google Scholar]
- 23. Cheng, F.B., Wan, X.H., Feng, J.C., Ma, L.Y., Hou, B., Feng, F., Wang, L. and Yang, Y.M. (2012) Subcellular distribution of THAP1 and alterations in the microstructure of brain white matter in DYT6 dystonia. Parkinsonism Relat. Disord., 18, 978–982. [DOI] [PubMed] [Google Scholar]
- 24. Vo, A., Argyelan, M., Eidelberg, D. and Ulug, A.M. (2013) Early registration of diffusion tensor images for group tractography of dystonia patients. J. Magn. Reson. Imaging, 37, 67–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Carbon, M., Kingsley, P.B., Su, S., Smith, G.S., Spetsieris, P., Bressman, S. and Eidelberg, D. (2004) Microstructural white matter changes in carriers of the DYT1 gene mutation. Ann. Neurol., 56, 283–286. [DOI] [PubMed] [Google Scholar]
- 26. Blood, A.J., Tuch, D.S., Makris, N., Makhlouf, M.L., Sudarsky, L.R. and Sharma, N. (2006) White matter abnormalities in dystonia normalize after botulinum toxin treatment. Neuroreport, 17, 1251–1255. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Bonilha, L., de Vries, P.M., Vincent, D.J., Rorden, C., Morgan, P.S., Hurd, M.W., Besenski, N., Bergmann, K.J. and Hinson, V.K. (2007) Structural white matter abnormalities in patients with idiopathic dystonia. Mov. Disord., 22, 1110–1116. [DOI] [PubMed] [Google Scholar]
- 28. Auvin, S. (2018) Abnormal white matter: expanding the GLUT1-D phenotype. Eur. J. Paediatr. Neurol., 22, 345. [DOI] [PubMed] [Google Scholar]
- 29. Baizabal-Carvallo, J.F. and Alonso-Juarez, M. (2018) Generalized dystonia associated with mutation in the histone methyltransferase gene KMT2B (DYT28) and white matter abnormalities. Parkinsonism Relat. Disord., 49, 116–117. [DOI] [PubMed] [Google Scholar]
- 30. Blood, A.J., Kuster, J.K., Waugh, J.L., Levenstein, J.M., Multhaupt-Buell, T.J., Sudarsky, L.R., Breiter, H.C. and Sharma, N. (2019) White matter changes in cervical dystonia relate to clinical effectiveness of botulinum toxin treatment. Front. Neurol., 10, 265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Cheng, F., Walter, M., Wassouf, Z., Hentrich, T., Casadei, N., Schulze-Hentrich, J., Barbuti, P., Krueger, R., Riess, O., Grundmann-Hauser, K. et al. (2020) Unraveling molecular mechanisms of THAP1 missense mutations in DYT6 dystonia. J. Mol. Neurosci., 70, 999–1008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ruiz, M., Perez-Garcia, G., Ortiz-Virumbrales, M., Meneret, A., Morant, A., Kottwitz, J., Fuchs, T., Bonet, J., Gonzalez-Alegre, P., Hof, P.R. et al. (2015) Abnormalities of motor function, transcription and cerebellar structure in mouse models of THAP1 dystonia. Hum. Mol. Genet., 24, 7159–7170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Mazars, R., Gonzalez-de-Peredo, A., Cayrol, C., Lavigne, A.C., Vogel, J.L., Ortega, N., Lacroix, C., Gautier, V., Huet, G., Ray, A. et al. (2010) The THAP-zinc finger protein THAP1 associates with coactivator HCF-1 and O-GlcNAc transferase: a link between DYT6 and DYT3 dystonias. J. Biol. Chem., 285, 13364–13371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Hollstein, R., Reiz, B., Kotter, L., Richter, A., Schaake, S., Lohmann, K. and Kaiser, F.J. (2017) Dystonia-causing mutations in the transcription factor THAP1 disrupt HCFC1 cofactor recruitment and alter gene expression. Hum. Mol. Genet., 26, 2975–2983. [DOI] [PubMed] [Google Scholar]
- 35. Shinoda, K., Zong, D., Callen, E., Wu, W., Dumitrache, L.C., Belinky, F., Chari, R., Wong, N., Ishikawa, M., Stanlie, A. et al. (2021) The dystonia gene THAP1 controls DNA double-strand break repair choice. Mol. Cell, 81, 2611, e2610–2624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Wirth, T., Tranchant, C., Drouot, N., Keren, B., Mignot, C., Cif, L., Lefaucheur, R., Lion-Francois, L., Meneret, A., Gras, D. et al. (2020) Increased diagnostic yield in complex dystonia through exome sequencing. Parkinsonism Relat. Disord., 74, 50–56. [DOI] [PubMed] [Google Scholar]
- 37. Zech, M., Jech, R., Boesch, S., Skorvanek, M., Weber, S., Wagner, M., Zhao, C., Jochim, A., Necpal, J., Dincer, Y. et al. (2020) Monogenic variants in dystonia: an exome-wide sequencing study. Lancet Neurol., 19, 908–918. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Pappas, S.S., Liang, C.C., Kim, S., Rivera, C.O. and Dauer, W.T. (2018) TorsinA dysfunction causes persistent neuronal nuclear pore defects. Hum. Mol. Genet., 27, 407–420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Li, J., Kim, S., Pappas, S.S. and Dauer, W.T. (2021) CNS critical periods: implications for dystonia and other neurodevelopmental disorders. JCI Insight, 6, e142483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Li, J., Levin, D.S., Kim, A.J., Pappas, S.S. and Dauer, W.T. (2021) TorsinA restoration in a mouse model identifies a critical therapeutic window for DYT1 dystonia. J. Clin. Invest., 131, e139606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Sengel, C., Gavarini, S., Sharma, N., Ozelius, L.J. and Bragg, D.C. (2011) Dimerization of the DYT6 dystonia protein, THAP1, requires residues within the coiled-coil domain. J. Neurochem., 118, 1087–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Richter, A., Hollstein, R., Hebert, E., Vulinovic, F., Eckhold, J., Osmanovic, A., Depping, R., Kaiser, F.J. and Lohmann, K. (2017) In-depth characterization of the homodimerization domain of the transcription factor THAP1 and dystonia-causing mutations therein. J. Mol. Neurosci., 62, 11–16. [DOI] [PubMed] [Google Scholar]
- 43. Sanghavi, H.M., Mallajosyula, S.S. and Majumdar, S. (2019) Classification of the human THAP protein family identifies an evolutionarily conserved coiled coil region. BMC Struct. Biol., 19, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Carpenter, A.E., Jones, T.R., Lamprecht, M.R., Clarke, C., Kang, I.H., Friman, O., Guertin, D.A., Chang, J.H., Lindquist, R.A., Moffat, J. et al. (2006) Cell profiler: image analysis software for identifying and quantifying cell phenotypes. Genome Biol., 7, R100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Guo, W., Patzlaff, N.E., Jobe, E.M. and Zhao, X. (2012) Isolation of multipotent neural stem or progenitor cells from both the dentate gyrus and subventricular zone of a single adult mouse. Nat. Protoc., 7, 2005–2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Yellajoshyula, D., Patterson, E.S., Elitt, M.S. and Kroll, K.L. (2011) Geminin promotes neural fate acquisition of embryonic stem cells by maintaining chromatin in an accessible and hyperacetylated state. Proc. Natl. Acad. Sci. USA., 108, 3294–3299. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The GEO accession number for CHIP-seq data used in manuscript for THAP1 is GSM803408, YY1 is GSM803446 and H3K9ac is GSM788082. The GEO accession number for gene expression data for Thap1 cKO CNS tissue is GSE97372 and Thap1 cKO oligodendrocyte lineage is GSE161556. The code utilized for automated analysis of g-ratio and axon caliber using CellProfiler 3.1.9 is available at https://github.com/suminkim/Analysis_CellProfiler.