

Abstract
Ulcerative colitis (UC) is a recurrent and persistent nonspecific inflammatory bowel disease (IBD) that greatly affects human survival and social wealth. Despite the advances in the treatment of UC, there is still a high demand for novel therapeutic strategies for UC patients. Cell death is critical to the development and progression of UC. Understanding how intestinal cells die and how to prevent damage to intestinal cells is of great interest for the diagnosis and early treatment of UC. Ferroptosis, a novel form of regulated cell death (RCD) manifested by iron accumulation, lipid peroxidation, and excessive reactive oxygen species (ROS) production, has been shown to contribute to the development and progression of UC. Inhibitors of ferroptosis have been validated in models of UC. Here, we reviewed the mechanisms of initiation and control of ferroptosis and summarize the therapeutic activity of ferroptosis inhibitors in models of UC. We further discussed the possibility of inhibiting ferroptosis as a novel therapeutic target for UC. These findings revealed novel mechanisms to protect the colonic mucosa and highlighted the importance of ferroptosis in the disease process.
1. Introduction
Ulcerative colitis (UC) is a chronic nonspecific inflammatory bowel disease (IBD) characterized by abdominal pain, diarrhea, blood in the stool, and weight loss [1]. Worldwide, the prevalence of UC continues to rise, and its chronic recurrence and unpredictable nature lead to costly medical treatment, which imposes a huge economic burden on society [2]. The pathophysiological mechanisms of UC are still not well understood, and therefore, the efficacy of existing therapies is limited [3]. Further research on the pathogenesis of UC and the development of novel and effective therapeutic approaches are still urgently needed. Recently, regulated forms of cell death have been observed to be involved in the development and progression of IBD and have the potential to be novel therapeutic targets [4]. The first regulatory cell death modality identified was caspase-dependent apoptosis [5], which has accounted for the vast majority of cell death studies in recent decades. Autophagy, a process in which cells use lysosomes for self-digestion, is observed in both physiological and pathological processes of the organism, but whether it plays a positive or negative role has not been fully elucidated [6]. Ferroptosis is a novel form of programmed cell death mode distinguished from apoptosis, necrosis, and autophagy at the cellular morphology, biochemical characteristics, and genetic level, which is characterized by iron-dependent accumulation of lipid peroxidation to a lethal level [7–9].
As a unique and novel form of cell death, ferroptosis was initially observed in tumor cells [8]. As research has progressed, it has been found that the development and progression of a variety of diseases in addition to tumors are associated with ferroptosis [10–15]. Recent studies have identified ferroptosis as a key regulatory mechanism in intestinal diseases, and inhibition of ferroptosis is expected to be a new direction in the prevention and treatment of intestinal diseases [16–18]. Extensive ferroptosis has been reported to be observed in UC patients, and recent evidence has found that blocking ferroptosis significantly relieves UC symptoms and promotes intestinal repair [16, 19]. These findings imply a new understanding of the development of therapeutic strategies for UC. Therefore, to synthesize the role of ferroptosis in UC pathology and attempt to elaborate the possibility of targeting ferroptosis in UC therapeutic strategies, we conducted the present review.
2. Ferroptosis and Its Mechanism
Ferroptosis was formally defined in 2012 as nonapoptotic, iron-dependent cell death characterized by lipid peroxidation product accumulation and membrane polyunsaturated fatty acid (PUFA) depletion [9]. As an iron-catalyzed lipid peroxidation process, a large accumulation of reactive oxygen species (ROS) is the most prominent feature of ferroptosis, with sources including excessive production and insufficient scavenging [20, 21]. Details of the process of ferroptosis are shown in Figure 1, and markers that can be used for ferroptosis validation are outline in Table 1.
Figure 1.
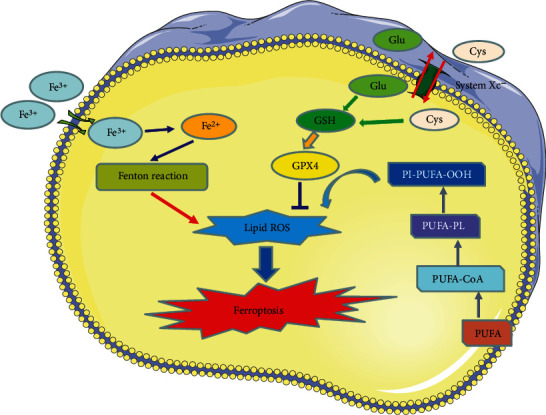
Ferroptosis is characterized by iron accumulation, lipid peroxidation, and excessive ROS production. The ferroptosis process is shown by this figure, including metabolic pathways, amino acid, lipid, and iron pathways which are showed in this figure.
Table 1.
Markers that can be used for ferroptosis validation.
| Metabolic processes | Markers |
|---|---|
| Iron metabolism pathway | Fe3+, Fe2+ |
| Oxidative stress | ROS, SOD, OH, H2O2 |
| Lipid metabolism pathway | MDA, LPO |
| Amino acid metabolism pathway | GPX4, GSH, GR, GLU, Cys |
2.1. Iron Metabolism Pathway
Iron is one of the intrinsic elements of the body, and proper iron levels are essential to maintain normal physiological functions of the body, while once the free iron level exceeds the normal range, it can damage cells. Under physiological conditions, extracellular iron is in dynamic equilibrium with intracellular iron [22, 23]. Extracellular iron is imported into the cell as ferric ions (Fe3+) and is converted to ferrous ions (Fe2+) by intracellular endosomes before being transported to the cell membrane [13, 20]. Once the equilibrium is disrupted, intracellular Fe2+ is then in excess, and the excess Fe2+ and hydroxyl radicals can be directly catalyzed by Fenton reaction to produce large amounts of ROS, which promotes lipid peroxidation and induces ferroptosis [24, 25].
Intracellular iron overload is central to the stimulation of oxidative damage and iron sagging. Specifically, causes of intracellular iron overload are attributed to increased iron uptake by TfR1, decreased iron excretion by ferroportin, and increased free iron due to ferritin degradation [26]. Iron overload can also induce nonclassical iron uptake pathways causing ferroptosis [27, 28]. For example, CDGSH iron-sulfur structural domain 1 in mitochondrial membranes may contribute to the reduction of iron content and ROS production in mitochondria, thereby inhibiting the development of iron prolapse. However, the role of mitochondria in ferroptosis remains to be clarified [29].
Catalytic iron, namely, ferrous iron, mediates lipid ROS production via Fenton reaction and promote lipid peroxidation directly. Markers like iron content and ROS can be used for ferroptosis validation in this process.
2.2. Lipid Metabolism Pathway
The lipid bilayer structure of the cell membrane directly determines biofilm properties and is essential for maintaining the integrity of membrane function. Increasing evidence suggest that lipid peroxidation is the driving force of ferroptosis [30, 31]. As the key component of cell membranes, polyunsaturated fatty acids (PUFAs), especially arachidonic acid (AA) and epinephrine (AdA), are preferentially oxidized by reactive free radicals [31]. Phospholipids containing PUFAs are lipid precursors for peroxidation reactions, and exposure to exogenous PUFAs can increase ferroptotic sensitivity [31]. Conversely, supplementation with deuterated PUFAs or exogenous MUFAs decelerated the accumulation of lipid peroxides and thus protected cells from ferroptosis [21, 31].
Phosphatidylethanolamine (PE) containing AA or AdA has been reported to be oxidized to phospholipid hydroperoxides (PE-AA/AdA-OOH) via a nonenzymatic reaction, which triggers ferroptosis [31]. In the presence of intracellular ferrous overload, ROS may be converted to hydroxyl radicals (HO˙), which subsequently affect PUFA on the cell membrane [32]. Free PUFAs are oxidized via the catalytic pathways of lysophosphatidylcholine acyltransferase 3 (LPCAT3), acyl-CoA synthetase long-chain family member 4 (ACSL4), and lipoxygenases (LOXs) [21, 33]. ACSL4 and LPCAT3 have been demonstrated to be key regulators of PUFA-PL biosynthesis [31]. PUFA can be acetylated by ACSL4 to form PUFA-CoA, followed by LPCAT3 insertion of PUFA-CoA into lysophospholipids to form PUFA-PL [34]. Enzymatic lipid peroxidation of ferroptosis is regulated by the LOXs and is predominantly dominated by LOX5 and LOX12/15 [35].
Notably, GPX enzymes inhibit the oxidation of lipids, particularly GPX4, which limits the formation of reactive lipid alkoxides and reduces phospholipid hydroperoxides to lipid alcohols [11].
Lipid peroxide-induced ferroptosis can be summarized in the following three procedures (Figure 1). First, ACSL4 catalyzes the esterification of AA or AdA to PE. Second, LPCAT3 generates PUFA-PE based on PE substrates. Finally, 15-LOX oxidizes AA-PE and AdA-PE to ferroptotic signals (PE-AA-OH and PE-AdA-OOH) [36].
2.3. Amino Acid Metabolism Pathway
As a member of the glutathione peroxidase family, GPX4 catalyzes the conversion of free hydrogen peroxide to water or the reduction of cytotoxic lipid hydroperoxides (L-OOH) to nontoxic lipid alcohols (L-OH), thereby inhibiting the production of lipid ROS [37, 38]. Thiol-containing tripeptide glutathione (GSH) is the major antioxidant in mammalian cells and the primary substrate of GPX4. The conditions leading to glutathione depletion directly affect the activity and stability of GPX4, thus rendering the cells more susceptible to ferroptosis [39]. GSH prevents lipid peroxidation of polyunsaturated fatty acids in cell membranes via GSH-Px, whereas inhibition of the uptake of GSH-generating substrates causes lipid peroxidation to occur [39].
Both the metabolism and synthesis of amino acids are associated with the process of ferroptosis [40]. Glutamine can be converted to glutamate by glutaminases (GLS1 and GLS2) [41]. High extracellular glutamate concentrations promote intracellular cystine depletion and eventually lead to GSH depletion and GPX4 inactivation, with ferroptosis triggered as a result [42] Α-ketopentanoic acid is a product of glutamine-driven intracellular metabolic pathways and can also have a glutamine-like effect when ferroptosis occurs [43]. In addition, the glutaminase GLS2, a transcriptional target of the tumor suppressor p53, can inhibit iron sag by limiting DPP4-mediated lipid peroxidation [44, 45].
3. Ferroptosis in Ulcerative Colitis
The basic features of iron sagging include iron deposition, lipid peroxidation accumulation, GSH depletion, GPX4 inactivation, and LOX upregulation, all of which have been elucidated to be associated with the pathogenesis of UC [16, 46–49]. Iron chelators have been reported to significantly reduce ROS accumulation, improve clinical symptoms, and promote intestinal epithelial cell repair in UC patients [50, 51], and conversely, high dietary iron supplementation can exacerbate UC symptoms [52, 53]. Sensitivity to erastin-induced ferroptosis was found in a UC cell model and could be rescued by a ferroptosis specific inhibitor [16, 46–49]. The ferroptosis phenomenon has also been observed in mouse models of UC and can reduce symptoms by targeted inhibition of ferroptosis [16, 46–49]. These findings further validate that inhibition of ferroptosis may be a novel target for alleviating UC. Details of the role of ferroptosis in UC are shown in Figure 2.
Figure 2.

Role of ferroptosis in UC. Ferroptosis promotes the release of damage-associated molecular patterns (DAMPs) from the intestinal epithelium. Subsequently, the immune response is hyperactivated, leading to intestinal inflammation and epithelial damage.
3.1. The Role of Iron in UC
In UC, excessive ROS production by the colorectal mucosa may cause changes in cellular proteins, lipids, and nucleic acids, leading to several cellular dysfunctions that may affect the disease process [54]. Excess-free iron can aggravate oxidative activity within the intestinal epithelium through multiple mechanisms. First, hydrogen peroxide and ferrous ions can generate large amounts of ROS directly through the Fenton reaction. Moreover, superoxide can promote the conversion or release of ferrous ions, which in turn promotes the Fenton reaction [51]. Recessive mutations in the hemochromatosis gene (Hfe) are strongly associated with iron overload [55]. Hfe knockout mice were observed to have increased MDA in colonic tissue [56] and exhibited more severe symptoms of colonic mucosal injury, such as hematochezia and diarrhea [57]. In addition, iron overload not only leads to dysregulated ROS generation and interferes with intestinal bacteria, which in turn aggravates enteritis [58]. While the phenomenon of ferroptosis has been found in UC, accompanied by iron overload, the use of iron chelators can significantly reduce ROS and improve colitis symptoms [16, 46–51]. Therefore, these studies all highlight the pathological role of iron overload in the development of UC; that is, the deposition of iron in the intestine leads to severe oxidative stress (OS), promotes the production of ROC through the Fenton reaction, triggers ferroptosis, and then stimulates the release of damage-associated molecular patterns (DAMPs) to cause intestinal immune and inflammatory responses. Iron chelation, on the other hand, may be a promising therapeutic strategy for UC.
3.2. The Role of Oxidative Stress in UC
OS is considered to be a potential driver of UC induction and progression and has been well reported in both patients and animal models of UC [60–63]. A higher OS status is thought to be a cause of altered immune and inflammatory responses that contribute to the development of UC [64, 65]. With the development of UC, the activity of inflammatory cells in the colon is greatly increased, leading to increased production of pro-oxidant molecules [60]. OS is attributed to redox imbalance, which is caused by excessive production of ROS and inadequate response of the antioxidant system to eliminate ROS [66]. Cytokine-induced elevated levels of myeloperoxidase also lead to ROS production [67]. Excessive ROS production leads to changes in cellular proteins, lipids, and nucleic acids, resulting in several cellular dysfunctions that may affect the course of UC. Colonic epithelial cells contain several antioxidant systems, such as antioxidant enzymes, namely, GSH, GPX4, and LOXs [68], but they are usually dysregulated in UC pathological conditions. As mentioned above, dysregulation of OS function is also observed in iron hypoplasia; it is hard to believe that the broad similarities between UC pathology and aspects of the ferroptosis cell death pathway are merely coincidental.
GSH can directly scavenge ROS and enhance cellular antioxidant capacity [69]. However, higher OS can promote GSH depletion and reduce GSH synthesis [70]. The depletion of GSH is widely observed in UC patients and experimental animal models of UC [71, 72]. Inhibition of the synthesis of GSH has been found to result in intestinal epithelial cell injury, while GSH supplementation significantly improves colonic health [73]. GPX4 is an important antioxidant enzyme that plays an important regulatory role in ferroptosis [74]. Inactivation of GPX4 promotes lipid peroxidation and induces ferroptosis [42]. Studies have reported that Nrf2-Gpx4 signaling pathway can significantly inhibit ferroptosis [75, 76], but Nrf2-Gpx4 signaling pathway is inhibited in UC [16, 46, 77]. Activation of Gpx4 can markedly inhibit ferroptosis and improve UC symptoms [46, 47]. LOXs are able to promote lipid hydroperoxide production and drive ironophilic cell death [31]. Alox15 deletion has been reported to promote inflammation suppression, intestinal barrier integrity maintenance, and colonic injury, while Alox15 overexpression exhibits more severe symptoms of colitis [78]. As a master regulatory molecule of 15-LOX, it was observed that the loss of phosphatidylethanolamine-binding protein 1 (PEBP1) is beneficial to reduce colitis symptoms and accelerate mucosal recovery [79]. Similar effects have been observed with other selective inhibitors acting on 5-LOX [80, 81].
3.3. The Role of Other Ferroptosis Regulators in UC
Nrf2, a major regulator of the antioxidant response, induces the expression of endogenous antioxidant proteins responsible for blocking lipid peroxidation to alleviate OS. Recent studies have demonstrated that Nrf2 can protect UC by inhibiting ferroptosis [19, 46, 49, 77]. Nrf2 has been reported to be involved in the regulation of ferroptosis by controlling the expression of quinone oxidoreductase 1, iron metabolism proteins, GPX4, and GSH production [82, 83]. Nrf2 has been widely demonstrated to be involved in the pathophysiological processes of UC. In NRF2 knockout UC mice, a significant increase in the severity of colitis and risk of colitis-associated colorectal cancer was observed [84, 85]. The regulatory effect of NRF2 on ferroptosis was also observed, with activation of NRF2 inhibiting ferroptosis and knockdown of NRF2 increasing sensitivity to various ferroptosis inducers [86, 87]. Notably, compounds that activate NRF2 inhibit ferroptosis and attenuate colitis-related mucosal damage and colonic inflammation [88]. However, the response of Nrf2 to OS is not specific to ferroptosis, as Nrf2 is also associated with the regulation of pyroptosis [89]. This suggests that the protective effect of Nrf2 on UC may be related to the regulation of multiple forms of cell death.
Mutations in the tumor suppressor P53 have been associated with the development of UC and UC-associated colorectal cancer [90]. Furthermore, P53 transcription inhibits the expression of the cystine/glutamate reverse transporter protein subunit SLC7A11, which in turn disrupts GSH production, thereby sensitizing cells to ferroptosis [73]. It has been observed that P53 is also involved in the regulation of ferroptosis by a mechanism that lies downstream of activation of arginine/arginine N1-acetyltransferase 1 and Alox15 or through transcriptional upregulation of mitochondrial glutaminase 2 [43, 91].
4. Conclusion and Future Perspectives
Patients with UC have a high demand for novel therapeutic strategies. Ferroptosis is a form of RCD characterized by iron overload, lipid peroxidation GPX4 inactivation, and GSH depletion. Based on these features, it is difficult to believe that the broad similarities in UC pathological features and ferroptosis pathways are merely coincidental. Ferroptosis plays an important role in the pathogenesis and progression of UC. The main features of ferroptosis have been widely observed in the colonic tissue of UC patients and animal models. By using UC mouse models, genetic or pharmacological manipulation of ferroptosis-associated genes can improve symptoms and promote recovery from experimental colitis. More specifically, a number of potent ferroptosis regulators (Table 2) can resist lipid oxidation and promote repair of intestinal damage in UC. Targeted inhibition of iron sagging may be a potential novel therapeutic strategy for UC.
Table 2.
Validated ferroptosis regulators in the UC model.
| Gene/axis/compound | Mechanism | Function |
|---|---|---|
| Ferrostatin-1 [19, 92] | Downregulation of PTGS2 levels, MDA levels, and iron content | Inhibition |
| Furin [46] | Activation of Nrf2 and upregulation of Gpx4 expression | Inhibition |
| MELK [48] | Inhibited ferritin formation in intestinal tissues | Inhibition |
| Lip-1 [19] | Blocking Nrf2/HO-1 and reducing the levels of COX2, ACSL4, FTH1 | Inhibition |
| Deferprone [19] | Blocking Nrf2/HO-1 and reducing the levels of COX2, ACSL4, FTH1 | Inhibition |
| NF-κBp65 [16] | Inhibition of endoplasmic reticulum stress | Inhibition |
| Curculigoside [47] | Induction of GPX4 | Inhibition |
| Fer-1 [16] | Reduces MDA, iron, and FTH levels | Inhibition |
| Astragalus polysaccharide [49] | Inhibiting NRF2/HO-1 pathway | Inhibition |
| RSL3 [16] | Accumulation of ROS | Induction |
Notably, targets involved in the regulation of ferroptosis are continuously being explored. Frataxin protein, which localizes to mitochondria and is involved in the biosynthesis of iron-sulfur clusters, has recently received the attention of investigators. Decreased frataxin expression can lead to iron accumulation at the mitochondrial level, uncontrolled production of reactive oxygen species, and lipid peroxidation [93]. These features are also common to ferroptosis. It has been reported that suppression of frataxin expression specifically activated iron starvation stress, accelerated free iron accumulation, enhanced lipid peroxidation, and resulted in ferroptosis [94–96]. Conversely, enforced expression of frataxin blocked the iron starvation response and erastin-induced ferroptosis [94, 95]. Hence, frataxin is considered to be a key regulator of ferroptosis by modulating iron homeostasis and mitochondrial function. There is a lack of evidence for the role of frataxin in UC. Since frataxin has a key regulatory role on ferroptosis, it may affect UC by regulating ferroptosis, but this hypothesis needs to be further validated by future studies.
Ferroptosis mainly occurs in IE mediating the pathogenesis and development of UC. Based on the currently available evidence, we believe that ferroptosis is a negative regulator of UC, and inhibition of ferroptosis is expected to be a novel approach for UC therapeutics. Although various inhibitors for ferroptosis have been observed to have positive effects in attenuating tissue damage associated with colitis, the underlying molecular mechanisms remain elusive, and further molecular mechanism studies are important for identifying more selective ferroptosis modulators. Furthermore, it is uncertain whether intestinal immune cells undergo ferroptosis in addition to IECs, and exploring ferroptosis in specific types of epithelial cells and intestinal immune cells will help to comprehensively understand the effects of ferroptosis on UC and provide new evidence for clinical decision-making in UC.
Acknowledgments
This work was supported by the National Natural Science Foundation of China (No. 81830118), China Academy of Chinese Medical Sciences Innovation Fund (No. CI 2021A01012), China Academy of Chinese Medical Sciences Excellent Young Talent Cultivation Fund (No. ZZ 15-YQ-002), and Administration of Traditional Chinese Medicine Digestive Refractory Disease Inheritance and Innovation Team Project (No. ZYYCXTD-C-C202010).
Contributor Information
Fengyun Wang, Email: wfy811@163.com.
Xudong Tang, Email: txdly@sina.com.
Data Availability
All data obtained or analyzed during this work are included within the article.
Conflicts of Interest
The authors declare that there is no conflict of interest.
Authors' Contributions
Jinke Huang initiated the study design and drafted the manuscript. Jiaqi Zhang, Jinxin Ma, Jing Ma, and Jiali Liu searched the literature. Fengyun Wang and Xudong Tang contributed to review and editing. All authors read and approved the final manuscript.
References
- 1.de Souza H. S., Fiocchi C. Immunopathogenesis of IBD: current state of the art. Nature Reviews Gastroenterology & Hepatology . 2016;13(1):13–27. doi: 10.1038/nrgastro.2015.186. [DOI] [PubMed] [Google Scholar]
- 2.Ng S. C., Shi H. Y., Hamidi N., et al. Worldwide incidence and prevalence of inflammatory bowel disease in the 21st century: a systematic review of population-based studies. Lancet . 2017;390(10114):2769–2778. doi: 10.1016/S0140-6736(17)32448-0. [DOI] [PubMed] [Google Scholar]
- 3.Ooi C. J., Hilmi I., Banerjee R., et al. Best practices on immunomodulators and biologic agents for ulcerative colitis and Crohn’s disease in Asia. Journal of Gastroenterology and Hepatology . 2019;34(8):1296–1315. doi: 10.1111/jgh.14648. [DOI] [PubMed] [Google Scholar]
- 4.Patankar J. V., Becker C. Cell death in the gut epithelium and implications for chronic inflammation. Nature Reviews Gastroenterology & Hepatology . 2020;17(9):543–556. doi: 10.1038/s41575-020-0326-4. [DOI] [PubMed] [Google Scholar]
- 5.Kerr J. F., Wyllie A. H., Currie A. R. Apoptosis: a basic biological phenomenon with wideranging implications in tissue kinetics. British Journal of Cancer . 1972;26(4):239–257. doi: 10.1038/bjc.1972.33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Eskelinen E. L., Saftig P. Autophagy: a lysosomal degradation pathway with a central role in health and disease. Biochimica et Biophysica Acta . 2009;1793(4):664–673. doi: 10.1016/j.bbamcr.2008.07.014. [DOI] [PubMed] [Google Scholar]
- 7.Galluzzi L., Vitale I., Aaronson S. A., et al. Molecular mechanisms of cell death: recommendations of the Nomenclature Committee on Cell Death 2018. Cell Death and Differentiation . 2018;25(3):486–541. doi: 10.1038/s41418-017-0012-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stockwell B. R., Friedmann Angeli J. P., Bayir H., et al. Ferroptosis: a regulated cell death nexus linking metabolism, redox biology, and disease. Cell . 2017;171(2):273–285. doi: 10.1016/j.cell.2017.09.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dixon S. J., Lemberg K. M., Lamprecht M. R., et al. Ferroptosis: an iron-dependent form of nonapoptotic cell death. Cell . 2012;149(5):1060–1072. doi: 10.1016/j.cell.2012.03.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Liang C., Zhang X., Yang M., Dong X. Recent progress in ferroptosis inducers for cancer therapy. Advanced Materials . 2019;31(51):p. e1904197. doi: 10.1002/adma.201904197. [DOI] [PubMed] [Google Scholar]
- 11.Ren J. X., Sun X., Yan X. L., Guo Z. N., Yang Y. Ferroptosis in neurological diseases. Frontiers in Cellular Neuroscience . 2020;14 doi: 10.3389/fncel.2020.00218. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Yan H. F., Tuo Q. Z., Yin Q. Z., Lei P., Department of Neurology and State Key Laboratory of Biotherapy, West China Hospital, Sichuan University, and Collaborative Center for Biotherapy, Chengdu, Sichuan 610041, China, Chengdu University of Traditional Chinese Medicine, Chengdu, Sichuan 610041, China The pathological role of ferroptosis in ischemia/reperfusion-related injury. Zoological Research . 2020;41(3):220–230. doi: 10.24272/j.issn.2095-8137.2020.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Wu X., Li Y., Zhang S., Zhou X. Ferroptosis as a novel therapeutic target for cardiovascular disease. Theranostics . 2021;11(7):3052–3059. doi: 10.7150/thno.54113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Belavgeni A., Meyer C., Stumpf J., Hugo C., Linkermann A. Ferroptosis and necroptosis in the kidney. Cell Chemical Biology . 2020;27(4):448–462. doi: 10.1016/j.chembiol.2020.03.016. [DOI] [PubMed] [Google Scholar]
- 15.Capelletti M. M., Manceau H., Puy H., Peoc'h K. Ferroptosis in liver diseases: an overview. International Journal of Molecular Sciences . 2020;21(14):p. 4908. doi: 10.3390/ijms21144908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Xu M., Tao J., Yang Y., et al. Ferroptosis involves in intestinal epithelial cell death in ulcerative colitis. Cell Death & Disease . 2020;11(2):p. 86. doi: 10.1038/s41419-020-2299-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Li Y., Feng D., Wang Z., et al. Ischemia-induced ACSL4 activation contributes to ferroptosis-mediated tissue injury in intestinal ischemia/reperfusion. Cell Death and Differentiation . 2019;26(11):2284–2299. doi: 10.1038/s41418-019-0299-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Song Y., Yang H., Lin R., Jiang K., Wang B. M. The role of ferroptosis in digestive system cancer (review) Oncology Letters . 2019;18:2159–2164. doi: 10.3892/ol.2019.10568. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chen Y., Zhang P., Chen W., Chen G. Ferroptosis mediated DSS-induced ulcerative colitis associated with Nrf2/HO-1 signaling pathway. Immunology Letters . 2020;225:9–15. doi: 10.1016/j.imlet.2020.06.005. [DOI] [PubMed] [Google Scholar]
- 20.Xie Y., Hou W., Song X., et al. Ferroptosis: process and function. Cell Death and Differentiation . 2016;23(3):369–379. doi: 10.1038/cdd.2015.158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Yang W. S., Kim K. J., Gaschler M. M., Patel M., Shchepinov M. S., Stockwell B. R. Peroxidation of polyunsaturated fatty acids by lipoxygenases drives ferroptosis. Proceedings of the National Academy of Sciences of the United States of America . 2016;113(34):E4966–E4975. doi: 10.1073/pnas.1603244113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Lane D. J., Merlot A. M., Huang M. L. H., et al. Cellular iron uptake, trafficking and metabolism: key molecules and mechanisms and their roles in disease. Biochimica et Biophysica Acta . 2015;1853(5):1130–1144. doi: 10.1016/j.bbamcr.2015.01.021. [DOI] [PubMed] [Google Scholar]
- 23.Coffey R., Ganz T. Iron homeostasis: an anthropocentric perspective. The Journal of Biological Chemistry . 2017;292(31):12727–12734. doi: 10.1074/jbc.R117.781823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dixon S. J., Stockwell B. R. The role of iron and reactive oxygen species in cell death. Nature Chemical Biology . 2014;10(1):9–17. doi: 10.1038/nchembio.1416. [DOI] [PubMed] [Google Scholar]
- 25.Stoyanovsky D. A., Tyurina Y. Y., Shrivastava I., et al. Iron catalysis of lipid peroxidation in ferroptosis: regulated enzymatic or random free radical reaction? Free Radical Biology & Medicine . 2019;133:153–161. doi: 10.1016/j.freeradbiomed.2018.09.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Yang W. S., Stockwell B. R. Synthetic lethal screening identifies compounds activating iron-dependent, nonapoptotic cell death in oncogenic-RAS-harboring cancer cells. Chemistry & Biology . 2008;15(3):234–245. doi: 10.1016/j.chembiol.2008.02.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Hassannia B., Wiernicki B., Ingold I., et al. Nano-targeted induction of dual ferroptotic mechanisms eradicates high-risk neuroblastoma. Journal of Clinical Investigation . 2018;128(8):3341–3355. doi: 10.1172/JCI99032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Li Q., Han X., Lan X., et al. Inhibition of neuronal ferroptosis protects hemorrhagic brain. JCI Insight . 2017;2(7):p. e90777. doi: 10.1172/jci.insight.90777. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Gao M., Yi J., Zhu J., et al. Role of mitochondria in ferroptosis. Molecular Cell . 2019;73(2):354–363.e3. doi: 10.1016/j.molcel.2018.10.042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Kajarabille N., Latunde-Dada G. O. Programmed cell-death by ferroptosis: antioxidants as mitigators. International Journal of Molecular Sciences . 2019;20(19):p. 4968. doi: 10.3390/ijms20194968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Kagan V. E., Mao G., Qu F., et al. Oxidized arachidonic and adrenic PEs navigate cells to ferroptosis. Nature Chemical Biology . 2017;13(1):81–90. doi: 10.1038/nchembio.2238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Berson E. L., Rosner B., Sandberg M. A., et al. Further evaluation of docosahexaenoic acid in patients with retinitis pigmentosa receiving vitamin A treatment: subgroup analyses. Archives of Ophthalmology . 2004;122(9):1306–1314. doi: 10.1001/archopht.122.9.1306. [DOI] [PubMed] [Google Scholar]
- 33.Doll S., Proneth B., Tyurina Y. Y., et al. ACSL4 dictates ferroptosis sensitivity by shaping cellular lipid composition. Nature Chemical Biology . 2017;13(1):91–98. doi: 10.1038/nchembio.2239. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Magtanong L., Ko P. J., Dixon S. J. Emerging roles for lipids in non-apoptotic cell death. Cell Death and Differentiation . 2016;23(7):1099–1109. doi: 10.1038/cdd.2016.25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Gaschler M. M., Stockwell B. R. Lipid peroxidation in cell death. Biochemical and Biophysical Research Communications . 2017;482(3):419–425. doi: 10.1016/j.bbrc.2016.10.086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D'Herde K., Krysko D. V. Ferroptosis: oxidized PEs trigger death. Nature Chemical Biology . 2017;13(1):4–5. doi: 10.1038/nchembio.2261. [DOI] [PubMed] [Google Scholar]
- 37.Ursini F., Maiorino M., Valente M., Ferri L., Gregolin C. Purification from pig liver of a protein which protects liposomes and biomembranes from peroxidative degradation and exhibits glutathione peroxidase activity on phosphatidylcholine hydroperoxides. Biochimica et Biophysica Acta . 1982;710(2):197–211. doi: 10.1016/0005-2760(82)90150-3. [DOI] [PubMed] [Google Scholar]
- 38.Brigelius-Flohé R., Maiorino M. Glutathione peroxidases. Biochimica et Biophysica Acta . 2013;1830(5):3289–3303. doi: 10.1016/j.bbagen.2012.11.020. [DOI] [PubMed] [Google Scholar]
- 39.Martin H. L., Teismann P. Glutathione—a review on its role and significance in Parkinson’s disease. The FASEB Journal . 2009;23(10):3263–3272. doi: 10.1096/fj.08-125443. [DOI] [PubMed] [Google Scholar]
- 40.Angeli J. P. F., Shah R., Pratt D. A., Conrad M. Ferroptosis inhibition: mechanisms and opportunities. Trends in Pharmacological Sciences . 2017;38(5):489–498. doi: 10.1016/j.tips.2017.02.005. [DOI] [PubMed] [Google Scholar]
- 41.Cassago A., Ferreira A. P., Ferreira I. M., et al. Mitochondrial localization and structure-based phosphate activation mechanism of glutaminase C with implications for cancer metabolism. Proceedings of the National Academy of Sciences of the United States of America . 2012;109(4):1092–1097. doi: 10.1073/pnas.1112495109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Yang W. S., SriRamaratnam R., Welsch M. E., et al. Regulation of ferroptotic cancer cell death by GPX4. Cell . 2014;156(1-2):317–331. doi: 10.1016/j.cell.2013.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Gao M., Monian P., Quadri N., Ramasamy R., Jiang X. Glutaminolysis and transferrin regulate ferroptosis. Molecular Cell . 2015;59(2):298–308. doi: 10.1016/j.molcel.2015.06.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Xie Y., Zhu S., Song X., et al. The tumor suppressor p53 limits ferroptosis by blocking DPP4 activity. Cell Reports . 2017;20(7):1692–1704. doi: 10.1016/j.celrep.2017.07.055. [DOI] [PubMed] [Google Scholar]
- 45.Jennis M., Kung C. P., Basu S., et al. An African-specific polymorphism in the TP53 gene impairs p53 tumor suppressor function in a mouse model. Genes & Development . 2016;30(8):918–930. doi: 10.1101/gad.275891.115. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Dong S., Lu Y., Peng G., et al. Furin inhibits epithelial cell injury and alleviates experimental colitis by activating the Nrf2-Gpx4 signaling pathway. Digestive and Liver Disease . 2021;53(10):1276–1285. doi: 10.1016/j.dld.2021.02.011. [DOI] [PubMed] [Google Scholar]
- 47.Wang S., Liu W., Wang J., Bai X. Curculigoside inhibits ferroptosis in ulcerative colitis through the induction of GPX4. Life Sciences . 2020;259:p. 118356. doi: 10.1016/j.lfs.2020.118356. [DOI] [PubMed] [Google Scholar]
- 48.Tang B., Zhu J., Fang S., et al. Pharmacological inhibition of MELK restricts ferroptosis and the inflammatory response in colitis and colitis-propelled carcinogenesis. Free Radical Biology & Medicine . 2021;172:312–329. doi: 10.1016/j.freeradbiomed.2021.06.012. [DOI] [PubMed] [Google Scholar]
- 49.Chen Y., Wang J., Li J., et al. Astragalus polysaccharide prevents ferroptosis in a murine model of experimental colitis and human Caco-2 cells via inhibiting NRF2/HO-1 pathway. European Journal of Pharmacology . 2021;911:p. 174518. doi: 10.1016/j.ejphar.2021.174518. [DOI] [PubMed] [Google Scholar]
- 50.Minaiyan M., Mostaghel E., Mahzouni P. Preventive therapy of experimental colitis with selected iron chelators and anti-oxidants. International Journal of Preventive Medicine . 2012;3:S162–S169. [PMC free article] [PubMed] [Google Scholar]
- 51.Millar A. D., Rampton D. S., Blake D. R. Effects of iron and iron chelation in vitro on mucosal oxidant activity in ulcerative colitis. Alimentary pharmacology & therapeutics . 2000;14(9):1163–1168. doi: 10.1046/j.1365-2036.2000.00828.x. [DOI] [PubMed] [Google Scholar]
- 52.Kobayashi Y., Ohfuji S., Kondo K., et al. Association between dietary iron and zinc intake and development of ulcerative colitis: a case–control study in Japan. Journal of Gastroenterology and Hepatology . 2019;34(10):1703–1710. doi: 10.1111/jgh.14642. [DOI] [PubMed] [Google Scholar]
- 53.Seril D. N., Liao J., Ho K. L. K., Warsi A., Yang C. S., Yang G. Y. Dietary iron supplementation enhances DSS-induced colitis and associated colorectal carcinoma development in mice. Digestive Diseases and Sciences . 2002;47(6):1266–1278. doi: 10.1023/A:1015362228659. [DOI] [PubMed] [Google Scholar]
- 54.Wan Y., Yang L., Jiang S., Qian D., Duan J. Excessive Apoptosis in Ulcerative Colitis: Crosstalk Between Apoptosis, ROS, ER Stress, and Intestinal Homeostasis. Inflammatory bowel diseases . 2021 doi: 10.1093/ibd/izab277. [DOI] [PubMed] [Google Scholar]
- 55.Feder J. N., Gnirke A., Thomas W., et al. A novel MHC class I-like gene is mutated in patients with hereditary haemochromatosis. Nature Genetics . 1996;13(4):399–408. doi: 10.1038/ng0896-399. [DOI] [PubMed] [Google Scholar]
- 56.Stevens R. G., Morris J. E., Cordis G. A., Anderson L. E., Rosenberg D. W., Sasser L. B. Oxidative damage in colon and mammary tissue of the HFE-knockout mouse. Free Radical Biology and Medicine . 2003;34(9):1212–1216. doi: 10.1016/S0891-5849(03)00072-8. [DOI] [PubMed] [Google Scholar]
- 57.Sivaprakasam S., Ristic B., Mudaliar N., et al. Hereditary hemochromatosis promotes colitis and colon cancer and causes bacterial dysbiosis in mice. Biochemical Journal . 2020;477(19):3867–3883. doi: 10.1042/BCJ20200392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Constante M., Fragoso G., Lupien-Meilleur J., Calve A., Santos M. M. Iron supplements modulate colon microbiota composition and potentiate the protective effects of probiotics in dextran sodium sulfate-induced colitis. Inflammatory Bowel Diseases . 2017;23(5):753–766. doi: 10.1097/MIB.0000000000001089. [DOI] [PubMed] [Google Scholar]
- 59.Ettreiki C., Gadonna-Widehem P., Mangin I., Coëffier M., Delayre-Orthez C., Anton P. M. Juvenile ferric iron prevents microbiota dysbiosis and colitis in adult rodents. World Journal of Gastroenterology: WJG . 2012;18(21):2619–2629. doi: 10.3748/wjg.v18.i21.2619. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Rana S. V., Sharma S., Kaur J., et al. Relationship of cytokines, oxidative stress and GI motility with bacterial overgrowth in ulcerative colitis patients. Journal of Crohn's & Colitis . 2014;8(8):859–865. doi: 10.1016/j.crohns.2014.01.007. [DOI] [PubMed] [Google Scholar]
- 61.Rana S. V., Sharma S., Prasad K. K., Sinha S. K., Singh K. Role of oxidative stress & antioxidant defence in ulcerative colitis patients from North India. The Indian Journal of Medical Research . 2014;139(4):568–571. [PMC free article] [PubMed] [Google Scholar]
- 62.Reissig K., Silver A., Hartig R., et al. Chk1 promotes DNA damage response bypass following oxidative stress in a model of hydrogen peroxide-associated ulcerative colitis through JNK inactivation and chromatin binding. Oxidative Medicine and Cellular Longevity . 2017;2017:20. doi: 10.1155/2017/9303158.9303158 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Elmaksoud H. A. A., Motawea M. H., Desoky A. A., Elharrif M. G., Ibrahimi A. Hydroxytyrosol alleviate intestinal inflammation, oxidative stress and apoptosis resulted in ulcerative colitis. Biomedicine & Pharmacotherapy . 2021;142:p. 112073. doi: 10.1016/j.biopha.2021.112073. [DOI] [PubMed] [Google Scholar]
- 64.Jena G., Trivedi P. P., Sandala B. Oxidative stress in ulcerative colitis: an old concept but a new concern. Free Radical Research . 2012;46(11):1339–1345. doi: 10.3109/10715762.2012.717692. [DOI] [PubMed] [Google Scholar]
- 65.Wang Z., Li S., Cao Y., et al. Oxidative stress and carbonyl lesions in ulcerative colitis and associated colorectal cancer. Oxidative Medicine and Cellular Longevity . 2016;2016:15. doi: 10.1155/2016/9875298.9875298 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Schieber M., Chandel N. S. ROS function in redox signaling and oxidative stress. Current Biology . 2014;24(10):R453–R462. doi: 10.1016/j.cub.2014.03.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Wu C. C., Chen J. S., Wu W. M., et al. Myeloperoxidase serves as a marker of oxidative stress during single haemodialysis session using two different biocompatible dialysis membranes. Nephrology, Dialysis, Transplantation . 2005;20(6):1134–1139. doi: 10.1093/ndt/gfh764. [DOI] [PubMed] [Google Scholar]
- 68.Holmes E. W., Yong S. L., Eiznhamer D., Keshavarzian A. Glutathione content of colonic mucosa (evidence for oxidative damage in active ulcerative colitis) Digestive Diseases and Sciences . 1998;43(5):1088–1095. doi: 10.1023/A:1018899222258. [DOI] [PubMed] [Google Scholar]
- 69.Kim C. J., Kovacs-Nolan J., Yang C., Archbold T., Fan M. Z., Mine Y. L-cysteine supplementation attenuates local inflammation and restores gut homeostasis in a porcine model of colitis. Biochimica et Biophysica Acta (BBA)-Gen- eral Subjects . 2009;1790(10):1161–1169. doi: 10.1016/j.bbagen.2009.05.018. [DOI] [PubMed] [Google Scholar]
- 70.Sido B., Hack V., Hochlehnert A., Lipps H., Herfarth C., Dröge W. Impairment of intestinal glutathione synthesis in patients with inflammatory bowel disease. Gut . 1998;42(4):485–492. doi: 10.1136/gut.42.4.485. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Buffinton G. D., Doe W. F. Depleted mucosal antioxidant defences in inflammatory bowel disease. Free Radical Biology and Medicine . 1995;19(6):911–918. doi: 10.1016/0891-5849(95)94362-H. [DOI] [PubMed] [Google Scholar]
- 72.Goldin E., Ardite E., Elizaldeetal J. I. Gastric mucosal damage in experimental diabetes in rats: role of endogenous glutathione. Gastroenterology . 1997;112(3):855–863. doi: 10.1053/gast.1997.v112.pm9041247. [DOI] [PubMed] [Google Scholar]
- 73.Gao W., Zhang T., Wu H. Emerging pathological engagement of ferroptosis in gut diseases. Oxidative Medicine and Cellular Longevity . 2021;2021:16. doi: 10.1155/2021/4246255.4246255 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Yang W. S., Stockwell B. R. Ferroptosis: death by lipid peroxidation. Trends in Cell Biology . 2016;26(3):165–176. doi: 10.1016/j.tcb.2015.10.014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Dai C., Chen X., Li J., Comish P., Kang R., Tang D. Transcription factors in ferroptotic cell death. Cancer Gene Therapy . 2020;27(9):645–656. doi: 10.1038/s41417-020-0170-2. [DOI] [PubMed] [Google Scholar]
- 76.Xie L. W., Cai S., Zhao T. S., Li M., Tian Y. Green tea derivative (−)-epigallocatechin-3-gallate (EGCG) confers protection against ionizing radiation-induced intestinal epithelial cell death both in vitro and in vivo_. Free Radical Biology & Medicine . 2020;161:175–186. doi: 10.1016/j.freeradbiomed.2020.10.012. [DOI] [PubMed] [Google Scholar]
- 77.Mei Y., Wang Z., Zhang Y., et al. FA-97, a new synthetic caffeic acid phenethyl ester derivative, ameliorates DSS-induced colitis against oxidative stress by activating Nrf2/HO-1 pathway. Frontiers in Immunology . 2020;10:p. 2969. doi: 10.3389/fimmu.2019.02969. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kroschwald S., Chiu C. Y., Heydeck D., et al. Female mice carrying a defective Alox15 gene are protected from experimental colitis via sustained maintenance of the intestinal epithelial barrier function. Biochimica et Biophysica Acta (BBA)-Molecular and Cell Biology of Lipids . 2018;1863(8):866–880. doi: 10.1016/j.bbalip.2018.04.019. [DOI] [PubMed] [Google Scholar]
- 79.Lin W., Ma C., Su F., et al. Raf kinase inhibitor protein mediates intestinal epithelial cell apoptosis and promotes IBDs in humans and mice. Gut . 2017;66(4):597–610. doi: 10.1136/gutjnl-2015-310096. [DOI] [PubMed] [Google Scholar]
- 80.Laursen L. S., Naesdal J., Bukhave K., Lauritsen K., Rask-Madsen J. Selective 5-lipoxygenase inhibition in ulcerative colitis. Lancet . 1990;335(8691):683–685. doi: 10.1016/0140-6736(90)90803-d. [DOI] [PubMed] [Google Scholar]
- 81.Hillingso J., Kjeldsen J., Laursen L. S., et al. Blockade of leukotriene production by a single oral dose of MK-0591 in active ulcerative colitis. Clinical Pharmacology & Therapeutics . 1995;57(3):335–341. doi: 10.1016/0009-9236(95)90159-0. [DOI] [PubMed] [Google Scholar]
- 82.Kovac S., Angelova P. R., Holmström K. M., Zhang Y., Dinkova-Kostova A. T., Abramov A. Y. Nrf2 regulates ROS production by mitochondria and NADPH oxidase. Biochimica et Biophysica Acta . 2015;1850(4):794–801. doi: 10.1016/j.bbagen.2014.11.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kerins M. J., Ooi A. The roles of NRF2 in modulating cellular iron homeostasis. Antioxidants & Redox Signaling . 2018;29(17):1756–1773. doi: 10.1089/ars.2017.7176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Khor T. O., Huang M. T., Kwon K. H., Chan J. Y., Reddy B. S., Kong A. N. Nrf 2-deficient mice have an increased susceptibility to dextran sulfate sodium-induced colitis. Cancer Research . 2006;66(24):11580–11584. doi: 10.1158/0008-5472.CAN-06-3562. [DOI] [PubMed] [Google Scholar]
- 85.Khor T. O., Huang M. T., Prawan A., et al. Increased susceptibility of Nrf2 knockout mice to colitis-associated colorectal cancer. Cancer Prevention Research . 2008;1(3):187–191. doi: 10.1158/1940-6207.CAPR-08-0028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Roh J. L., Kim E. H., Jang H., Shin D. Nrf 2 inhibition reverses the resistance of cisplatin-resistant head and neck cancer cells to artesunate-induced ferroptosis. Redox Biology . 2017;11:254–262. doi: 10.1016/j.redox.2016.12.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Dodson M., Castro-Portuguez R., Zhang D. D. NRF2 plays a critical role in mitigating lipid peroxidation and ferroptosis. Redox Biology . 2019;23:p. 101107. doi: 10.1016/j.redox.2019.101107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Yang Y., Cai X., Yang J., et al. Chemoprevention of dietary digitoflavone on colitis-associated colon tumorigenesis through inducing Nrf2 signaling pathway and inhibition of inflammation. Molecular Cancer . 2014;13(1):1–14. doi: 10.1186/1476-4598-13-48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Ran X., Yan Z., Yang Y., et al. Dioscin improves pyroptosis in LPS-induced mice mastitis by activating AMPK/Nrf2 and inhibiting the NF-κB signaling pathway. Oxidative Medicine and Cellular Longevity . 2020;2020:25. doi: 10.1155/2020/8845521.8845521 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Lu X., Yu Y., Tan S. p53 expression in patients with ulcerative colitis - associated with dysplasia and carcinoma: a systematic meta-analysis. BMC Gastroenterology . 2017;17(1):p. 111. doi: 10.1186/s12876-017-0665-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Ou Y., Wang S. J., Li D., Chu B., Gu W. Activation of SAT1engages polyamine metabolism with p53-mediated ferroptotic responses. Proceedings of the National Academy of Sciences . 2016;113(44) doi: 10.1073/pnas.1607152113. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Xu J., Liu S., Cui Z., et al. Ferrostatin-1 alleviated TNBS induced colitis via the inhibition of ferroptosis. Biochemical and Biophysical Research Communications . 2021;573:48–54. doi: 10.1016/j.bbrc.2021.08.018. [DOI] [PubMed] [Google Scholar]
- 93.Turchi R., Faraonio R., Lettieri-Barbato D., Aquilano K. An overview of the ferroptosis hallmarks in Friedreich’s ataxia. Biomolecules . 2020;10(11):p. 1489. doi: 10.3390/biom10111489. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Du J., Zhou Y., Li Y., et al. Identification of frataxin as a regulator of ferroptosis. Redox Biology . 2020;32:p. 101483. doi: 10.1016/j.redox.2020.101483. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Liu J., He H., Wang J., et al. Oxidative stress-dependent frataxin inhibition mediated alcoholic hepatocytotoxicity through ferroptosis. Toxicology . 2020;445:p. 152584. doi: 10.1016/j.tox.2020.152584. [DOI] [PubMed] [Google Scholar]
- 96.La Rosa P., Petrillo S., Turchi R., et al. The Nrf2 induction prevents ferroptosis in Friedreich’s Ataxia. Redox Biology . 2021;38:p. 101791. doi: 10.1016/j.redox.2020.101791. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
All data obtained or analyzed during this work are included within the article.