

Abstract
Growing evidence demonstrates that circulating tumor DNA (ctDNA) minimal residual disease (MRD) following treatment for solid tumors predicts relapse. These results suggest that ctDNA MRD could identify candidates for adjuvant therapy and measure response to such treatment. Importantly, factors such as assay type, amount of ctDNA release, and technical and biological background can impact ctDNA MRD results. Furthermore, the clinical utility of ctDNA MRD for treatment personalization remains to be fully established. Here, we review the evidence supporting the value of ctDNA MRD in solid cancers and highlight key considerations in the application of this potentially transformative biomarker.
Introduction
Nearly two-thirds of patients with solid tumors present with locoregional disease and are amenable to curative therapies (1). Surgery, radiation therapy, systemic therapy, or a combination of these approaches can achieve disease remission in the majority of these cases when using conventional measures of response, such as functional body imaging. However, in a significant subset of patients, small numbers of remnant tumor cells, termed minimal residual disease (MRD), can persist at levels below the detection threshold of imaging or physical exam and ultimately lead to disease relapse (2,3).
Systemic therapy delivered after surgery (i.e. adjuvant therapy) or radiation therapy (i.e. consolidation therapy) has been shown to improve long-term survival in multiple cancer types (4–7), providing strong clinical evidence that eradication of MRD can improve rates of cure. However, for most solid cancers where adjuvant/consolidation therapy is currently part of the standard of care, the magnitude of benefit from adjuvant therapies is modest. This is likely in part because a significant subset (and sometimes the majority) of patients who receive adjuvant therapies are already cured by the preceding local therapy (4,6,8).
For several hematologic malignancies, detection of MRD via flow cytometry for tumor cells or quantitative molecular techniques for patient-specific aberrations has long been established as a poor prognostic factor following induction therapy. Accordingly, modification of therapy based on the presence of MRD has become a standard of care for acute lymphoblastic leukemia, acute promyelocytic leukemia, and chronic myelogenous leukemia (9,10). Given such actionability of MRD in blood neoplasms, biomarkers that can similarly identify which patients with solid tumors harbor MRD could also have significant utility in personalizing adjuvant/consolidation therapy. However, until recently approaches to detect MRD in solid cancers have lacked the sensitivity and specificity required for clinical application (11).
Recent work focused on circulating tumor DNA (ctDNA) has produced promising results suggesting that this analyte could serve as a generalizable biomarker for MRD in solid cancers. Tumors release DNA into the blood that can be isolated from plasma collected via routine blood draws (12–14). Despite generally representing a small fraction of cell-free DNA (cfDNA) in blood plasma, ctDNA can be detected via polymerase chain reaction (PCR) or next generation sequencing (NGS) assays targeting tumor-specific mutations, structural variants, copy number alterations, and epigenetic features (15,16).
Recent advances in molecular and computational biology have significantly improved the limit of detection (LOD) for ctDNA using somatic alterations (17,18). These improvements have raised the possibility that ctDNA analysis could be used to identify patients harboring MRD following curative therapy of solid cancers and to guide the administration of adjuvant or consolidation therapies (19). Here, we review the current evidence that detection of ctDNA following definitive therapy is prognostic in solid tumors and discuss the promise and limitations of ctDNA MRD testing.
Approaches for ctDNA MRD analysis
Digital PCR
Several approaches have been utilized to identify known or common tumor mutations in plasma cfDNA samples. Digital PCR (dPCR) improves on conventional allele-specific PCR amplification by partitioning a DNA sample into a large number of smaller reactions to provide an absolute quantification that improves sensitivity (20). Digital PCR primers and probes can be designed to achieve very high specificity, and droplet-based approaches such as BEAMing (beads, emulsion, amplification, and magnetics) have maximized the number of individual DNA molecules that can be analyzed from a single sample (21). As a result, the detection limit of such digital PCR assays is limited in practice by the amount of cfDNA that can be isolated from a blood draw (22). With DNA inputs routinely achievable from patient plasma samples (~30 ng), dPCR has been shown to have a detection limit of approximately 0.1% (23–25). However, due to differences in DNA input, sample quality, and analysis approaches, reported LODs vary substantially between studies. dPCR is very effective for tracking a small number of mutations identified from sequencing of tumor tissue or hot-spot mutations with a high prevalence in the cancer of interest, such as KRAS mutations in pancreatic cancer (26). Due to complexities of multiplexing a large number of dPCR assays, the extent of multiplexing varies widely between studies and dPCR generally has inferior clinical sensitivity for MRD than highly parallel NGS methods monitoring multiple mutations (27,28). Accordingly, dPCR is not a commonly preferred approach for solid tumor MRD detection in most contexts (29).
PCR amplicon-based NGS
NGS, also known as massively parallel sequencing, has been incorporated into several different ctDNA analysis approaches, enabling interrogation of millions to billions of DNA molecules from a biological sample. One approach to achieve sufficient sensitivity to detect rare ctDNA molecules uses gene-specific PCR amplicons to amplify one or more genomic regions expected to harbor tumor-derived mutations prior to NGS. Several approaches, including Safe-SeqS, introduce unique molecular identifier (UMI) sequences during preparation of DNA libraries for sequencing to reduce technical errors (17). Personalized multiplex PCR can be utilized to monitor multiple patient-specific mutations identified from sequencing of tumor tissue (30), and NGS sequencing of these lesions in cfDNA can achieve remarkable sensitivity levels (31). A related set of approaches uses a combination of ligation and gene specific PCR primers to partially preserve cfDNA fragment end information (32,33). Multiple commercial platforms have been developed based on these approaches, including Natera Inc.’s Signatera (34–36), ArcherDX’s personalized cancer monitoring assay (37), and Inivata’s RaDaR assay (38). Although beyond the scope of this review, numerous other PCR amplicon-based NGS methods have been used for ctDNA analysis in the past and have been reviewed in detail elsewhere (39–41).
Hybridization Capture-based NGS
DNA enrichment using hybrid capture with biotinylated oligonucleotides allows sequencing of larger targeted panels with better uniformity of coverage (42,43). Capture-based NGS approaches such as CAncer Personalized Profiling by deep Sequencing (CAPP-Seq) preserve cfDNA fragment size information and can also incorporate unique molecular identifiers to minimize technical background (18). Capture panels can be used as “off-the-shelf” tools designed to target frequently mutated genes in one or more cancers of interest (e.g. AVENIO assay from Roche Diagnostics) (44). Alternatively, personalized capture panels can be designed for each patient to enrich for patient-specific mutations identified from tumor sequencing (45,46) (Figure 1A). Recently, we developed a novel capture-based ctDNA MRD assay called Phased variant Enrichment and Detection Sequencing (PhasED-Seq), which leverages multiple somatic mutations within individual DNA fragments to decrease both technical and biological error rates (see below) and improves ctDNA MRD detection limits down to 1 part per million, 30–100 fold lower than previous approaches (47).
Figure 1:
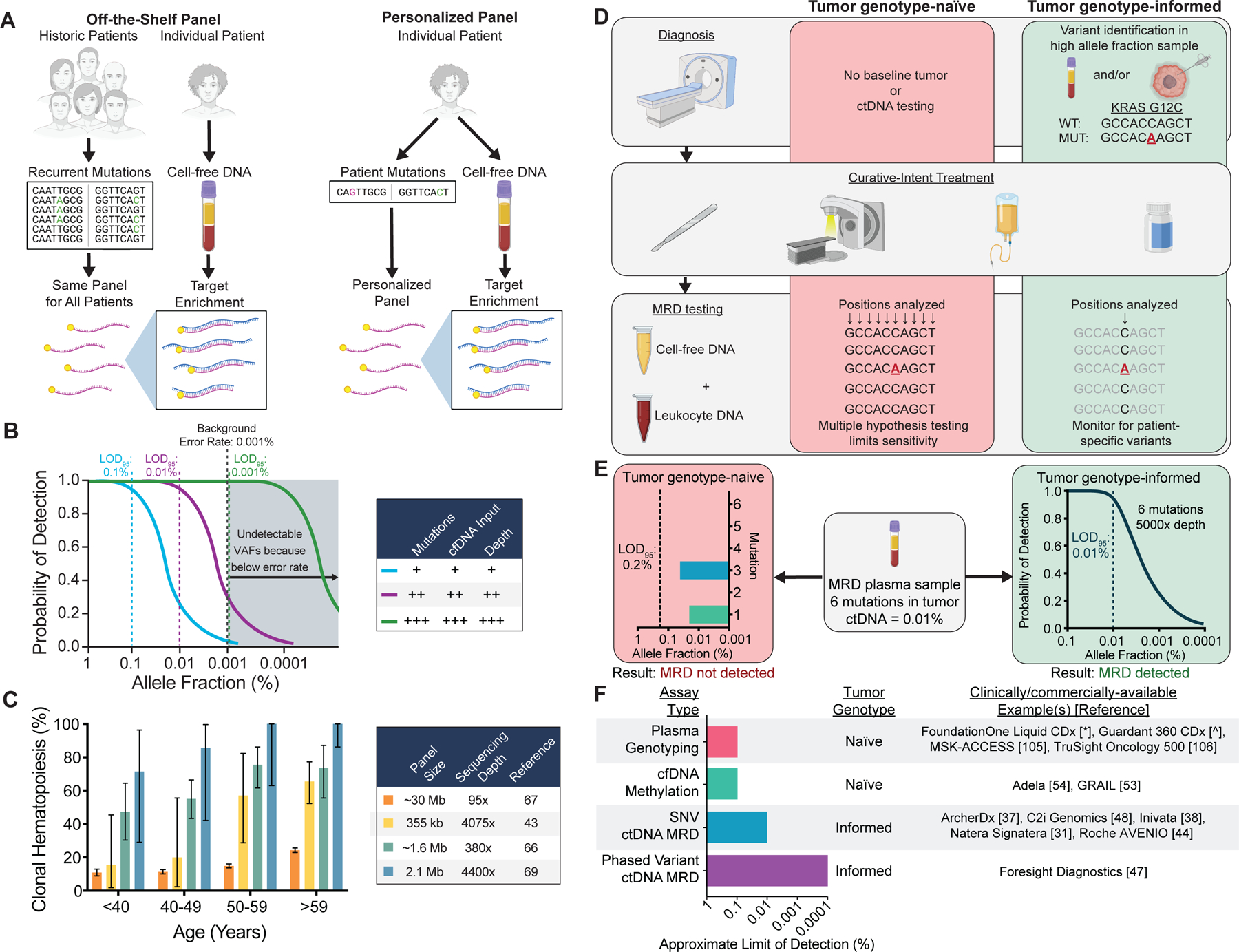
Technical approaches for ctDNA MRD detection and factors affecting assay sensitivity and specificity. A) Schematic comparing assays using off-the-shelf versus personalized sequencing panels. Off-the-shelf panels are designed to cover recurrently mutated genes in the cancer type(s) of interest. The same panel is applied to tumor tissue and plasma of every patient, and personalization is achieved by bioinformatically considering only the positions mutated in the matched tumor . Personalized panels are designed to cover patient-specific mutations identified through sequencing of their tumor DNA. In this approach, personalization is achieved by every patient having a unique panel. B) Factors affecting the probability of ctDNA detection. Increasing the number of mutations tracked, the sequencing depth at mutant positions, or the cfDNA input can increase the probability of detection. Technical background from sources such as polymerase errors and oxidative damage sets the lower limit of detection due to an inability to distinguish artifacts from true tumor variants. Limit of detection with 95% probability = LOD95. C) Prevalence of at least 1 clonal hematopoiesis variant detected in plasma as a function of the sequencing panel size and sequencing depth in published studies (46,75–77). Error bars represent binomial 95% confidence intervals. D) Schematic of tumor genotype-informed versus tumor genotype-naïve ctDNA analysis. In genotype-naïve analysis, the MRD sample is interrogated at all sequenced genomic positions, leading to reduced sensitivity due to multiple hypothesis testing. In tumor genotype-informed analysis, variants are identified from tumor tissue or pre-treatment plasma samples with high tumor allele fraction. Only patient-specific variants are monitored in the MRD sample. In both cases, genotyping DNA from leukocytes in the peripheral blood can improve specificity by identifying variants stemming from clonal hematopoiesis. E) Comparison of the LODs of tumor genotype-naïve and tumor genotype-informed ctDNA analysis at a median sequencing depth of 5000× for a patient with 6 tumor mutations and a ctDNA allele fraction of 0.01%. Due to multiple hypothesis testing with tumor genotype-naïve analysis, the LOD95 is 0.2%, and no mutations are detected above background despite mutations with allele fractions of 0.02% (1/5000 molecules) and 0.04% (2/5000 molecules) being present in the sample. In contrast, in the same sample tumor genotype-informed analysis at 5000× depth is associated with an LOD95 of 0.01% (approximated by a binomial distribution), and therefore ctDNA MRD has a 95% chance of being detected. F) Summary of assay types, tumor genotyping requirements, and approximate LODs (i.e. analytical sensitivity) for commercially/clinically available ctDNA analysis tests. Since no data comparing these methods on the same samples are available, the approximate limit of detection at which 95% of samples would be expected to be called positive for each group of assays is estimated from published manuscripts and/or conference abstracts and rounded to the nearest log (31,37,38,44,47,48,53,54,105,106).
* = http://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032B.pdf
^ = https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010B.pdf
Whole genome sequencing (WGS)
In theory, application of WGS to post treatment plasma could allow detection of an even greater number of mutations than the aforementioned NGS approaches and not require custom panel design. However, due to assay background error rates, there are diminishing returns of increasing the number of tracked mutations past a certain point (see below). Therefore, recent work suggests that when combined with customized bioinformatics, direct WGS of plasma can potentially achieve similar but not superior limits of MRD detection as the other NGS approaches (48). Additionally, the amount of sequencing required for the much larger targeted genomic space makes the per sample costs of WGS significantly higher.
Emerging techniques
Although not a focus of this review, other analysis approaches based on DNA methylation or other epigenetic features reflecting chromatin state of tumor cells (termed “fragmentomics”) have been used to detect ctDNA (49). While these emerging techniques have not been extensively evaluated in the context of MRD, epigenetic features may be complementary to somatic mutation tracking and may enable tumor genotype-naïve MRD detection. For example, the Guardant Reveal assay integrates somatic mutation and “epigenomic” (i.e. methylation and fragmentomic) approaches to detect ctDNA without prior sequencing of tumor DNA (50,51). A recent study using this assay for detecting MRD in colorectal cancer demonstrated that incorporating epigenomic analysis improved sensitivity compared with tumor-naïve somatic alterations alone (52). However, methylation-based approaches have been reported to have LODs of approximately 0.1% (53,54) and rigorous LOD analyses for fragmentomic-based approaches in the context of ctDNA MRD have not been published to date. Therefore, while more studies are clearly needed, it seems unlikely that these approaches will be able to match the LODs of the most sensitive tumor genotype-informed, somatic mutation-based ctDNA MRD approaches.
Technical aspects of ctDNA MRD detection using NGS
Physical limits of ctDNA analysis
The LOD for an assay is the lowest quantity of an analyte that can be reliably detected (55). Unfortunately, most studies in the ctDNA field have not rigorously defined or established the LOD. To conform to clinical laboratory testing guidelines, the LOD should ideally be defined as the ctDNA concentration at which 95% of clinical samples will be called positive (56). For mutation-based NGS ctDNA MRD assays, the LOD depends on both biological and technical factors. In practice, reliable LODs have not been published for most ctDNA analysis approaches, and multiple definitions have been used, making it challenging to compare across studies. In order to be meaningful, LODs should be determined in settings that match clinical samples as closely as possible, including in the amount of cell-free DNA input (57).
Regardless of the detection limit, for non-invasive detection to be feasible using circulating nucleic acids in blood plasma, tumor DNA must first be released into the blood and collected in a blood draw. Although ctDNA is generally thought to be released from necrotic or apoptotic tumor cells (58–60), the exact mechanisms of ctDNA release into the bloodstream and the relative contributions of different cell death mechanisms, phagocytosis, exocytosis, active secretion, and other cellular processes have not been well characterized in human cancers (61). Across detection platforms, ctDNA levels generally correlate with tumor burden on imaging (28,31,46,62,63). Accordingly, the number of residual cells after therapy likely also correlates with ctDNA MRD levels. Prior studies have also demonstrated that ctDNA release varies within and between tumor types and histologies, with a recurring observation that squamous cell carcinomas tend to shed higher ctDNA levels than adenocarcinomas (31,46,64,65). Furthermore, levels of normal cfDNA increase with tissue injury from inflammation and ischemia related to non-malignant conditions, surgery, or even vigorous exercise (66), which can lower the allele fraction of ctDNA molecules below the LOD of an assay. Indeed, post-surgical inflammatory changes have been shown to induce a significant increase in cfDNA levels postoperatively for up to 3–4 weeks (67), suggesting that ctDNA MRD should not be measured immediately following surgery. Collectively and irrespective of the assay used, these biological factors can substantially impact the sensitivity of ctDNA MRD techniques for accurately detecting residual disease and predicting relapse.
Assuming that ctDNA molecules are present in a blood sample, detection of these molecules relies on their efficient profiling. Each of the ctDNA MRD assay approaches includes multiple molecular biology steps that have imperfect molecule recovery and at any of which rare mutant ctDNA molecules could potentially be lost. For instance, when interrogated by NGS profiling, mutant ctDNA molecules need to be incorporated into the sequencing library and this library needs to be sequenced deeply enough to observe these rare molecules in the final result. Due to losses of molecules during library preparation and sequencing, next-generation sequencing methods generally have cfDNA molecule recovery efficiencies ≤50% (44,68).
Lastly, the probability of detecting ctDNA MRD is a function of the ctDNA concentration, the number of mutations tracked, and the number of unique cfDNA molecules interrogated (Figure 1B) (18). For next-generation sequencing approaches, tracking multiple mutations, using more cell-free DNA input, and sequencing to a greater depth can improve the likelihood of identifying mutant ctDNA molecules in a given blood sample. Cell-free DNA input is ultimately limited by the amount of plasma that can be collected and analyzed. In patients with cancer, concerns for anemia limits the amount of blood that can be collected for ctDNA analysis (69). Although greater cfDNA input improves sensitivity across different methods, increasing input cannot increase the limit of detection beyond an assay’s background error rate.
Technical and biological sources of error
Even if ctDNA molecules are efficiently recovered by an NGS MRD assay, their subsequent detection relies on the successful identification and quantitation by the associated bioinformatics pipeline. Within these pipelines, a key challenge is resolving the desired ctDNA biological signal from the noise or background errors arising from various technical or biological sources. In this context, there are two major components that together determine the overall background error rate of ctDNA MRD assays: (1) technical errors leading to artifactual mutations occurring ex vivo during the various molecular biology steps, and (2) bona fide somatic variants stemming from non-tumor tissues that harbor mutations.
Technical errors can be introduced during sample processing from sources such as unrepaired DNA polymerase errors arising during PCR (17) and oxidative DNA damage (70,71). Because true tumor variants cannot be resolved below the error noise floor, these errors serve to limit the lowest possible ctDNA concentration that can be detected. To address this issue, multiple strategies have been developed to decrease technical errors. These strategies include the use ‘barcoding’ techniques that employ unique molecular identifiers (UMIs), in silico elimination of stereotypical background artifacts (i.e. “polishing”), and inclusion of free radical scavengers during library preparation to decrease oxidative damage (17,18,31,46,68). Collectively, these error-suppression strategies can dramatically reduce the technical errors arising ex vivo during cfDNA profiling.
Biological background secondary to somatic mutations found in cfDNA but not originating from tumor cells represents a second important source of potential false-positive mutations. Through a process called age-related clonal hematopoiesis (CH), hematopoietic stem cells can acquire mutations that can be found in both circulating peripheral blood cells and in cell-free DNA (72). When considering peripheral blood leukocytes, patients with clonal mutations above a VAF of 2% in genes that are canonically associated with hematologic malignancies, but who do not meet the criteria for diagnosis of leukemia, are considered to have clonal hematopoiesis of indeterminate potential (CHIP) (73). Because the majority of cfDNA arises from hematopoietic sources (74), CH represents a major contributor to biological background in cfDNA profiling exercises, including detection of ctDNA MRD. Importantly, the prevalence of CH variants increases with patient age, broader panels, and increasing sequencing depth (Figure 1C), and has been found to be as high as 100% in patients 60 years or older (46,75–77).
CH represents the dominant biological source of false positive mutations when identifying somatic mutations in cfDNA. Importantly, sequencing peripheral blood leukocytes can help to identify cfDNA mutations due to CH. In support of this notion, a recent ctDNA study of localized non-small cell lung cancer (NSCLC) found that up to 15% of TP53 mutations in the cfDNA were attributable to CH and found in matched leukocytes but not matched tumor (78). In a separate study, deeper sequencing of matched leukocytes in patients with NSCLC found that 40.6% of TP53 mutations present in cfDNA were also detected in matched leukocytes (46). TP53 mutations in cfDNA that were also found in leukocytes displayed less evidence of the smoking mutational signature than TP53 mutation also found in matched tumor tissue, supporting their disparate biological sources.
Although sequencing peripheral blood leukocytes is helpful for distinguishing tumor-derived mutations from CH, approximately 10% of mutations found in cfDNA of non-cancer controls are not found in matched leukocytes (46). This observation suggests that some CH mutations can be missed due to low prevalence of CH subclones in the peripheral blood, perhaps reflecting CH in non-circulating hematopoietic precursors. Additionally, it is possible that mutations from non-malignant, non-hematopoietic cells can be found in cfDNA. In support of this, recent studies have demonstrated that somatic mutations can be present in diverse non-malignant cell types, including epithelial (79,80), endothelial (81), stromal (82), and others (83). As detailed below, incorporating sequencing of tumor tissue to identify somatic mutations that are then monitored in plasma samples can guard against these sources of biological background.
Tumor genotype-informed versus tumor genotype-naïve ctDNA analysis
Due to the complications posed by technical and biological background mutations described above, most ctDNA MRD studies have performed “tumor genotype-informed” analyses to monitor known tumor variants in post-treatment plasma (Figure 1D). This approach includes the genotyping of tumor tissue to identify mutations that are then tracked in plasma, which lowers the risk of false positives due to technical and biological background sources of error. Additionally, such a tumor genotype-informed approach limits the number of genomic positions interrogated in cell free DNA, therefore decreasing multiple hypothesis testing. Accordingly, the tumor genotype-informed approach can be less demanding for blood sample volumes, since fewer unique mutant cfDNA molecules are required for detecting ctDNA as compared with a genotype-naive approach (Figure 1E).
Several commercially available ctDNA platforms support tumor genotype-informed MRD detection, including assays for solid tumors from Natera, Roche Diagnostics, Invitae (ArcherDx), Inivata, and Foresight Diagnostics, among others. Notably, these platforms are technically distinct from liquid biopsy panels such as Guardant360 (https://www.accessdata.fda.gov/cdrh_docs/pdf20/P200010A.pdf) and FoundationOne Liquid CDx (http://www.accessdata.fda.gov/cdrh_docs/pdf19/P190032A.pdf) that have been developed for non-invasive genotyping in the setting of advanced disease (84). Although such non-invasive genotyping assays are very useful for identifying actionable tumor mutations in patients with metastatic disease (85), a recent analysis by the SEQC2 Working Group led by the FDA found that detection of variants was less reliable below an allele fraction of 0.5% (86). Therefore, currently available assays designed for the primary purpose of non-invasive genotyping in advanced disease are not optimal for ctDNA MRD detection. This is mainly due to the low ctDNA allele fractions typically observed following definitive treatment of localized solid cancers in the absence of radiographic disease burden as well as the confounding effects of CH. In contrast, tumor genotype-informed MRD detection approaches can attain LODs of ≤0.01% which makes them preferrable for detection of minute amounts of MRD (18,37,47,87) (Figure 1F).
Evidence supporting the prognostic value of ctDNA MRD in solid cancers
Landmark versus surveillance ctDNA MRD analysis
In ctDNA MRD studies focused on solid tumors, two main types of analysis have generally been reported: 1) MRD landmark analysis and 2) surveillance analysis (Figure 2A). While these two types of analysis are related, their distinct features are relevant for the clinical application of ctDNA MRD. MRD landmark analysis determines the ctDNA status of patients at a single, pre-specified timepoint, which is typically shortly after completion of standard-of-care treatment (e.g. surgery, radiotherapy, etc.). In contrast, ctDNA surveillance analysis involves evaluating longitudinal blood draws at multiple time points during follow up, with ctDNA status determined by whether any blood draw (irrespective of time point) is positive.
Figure 2:
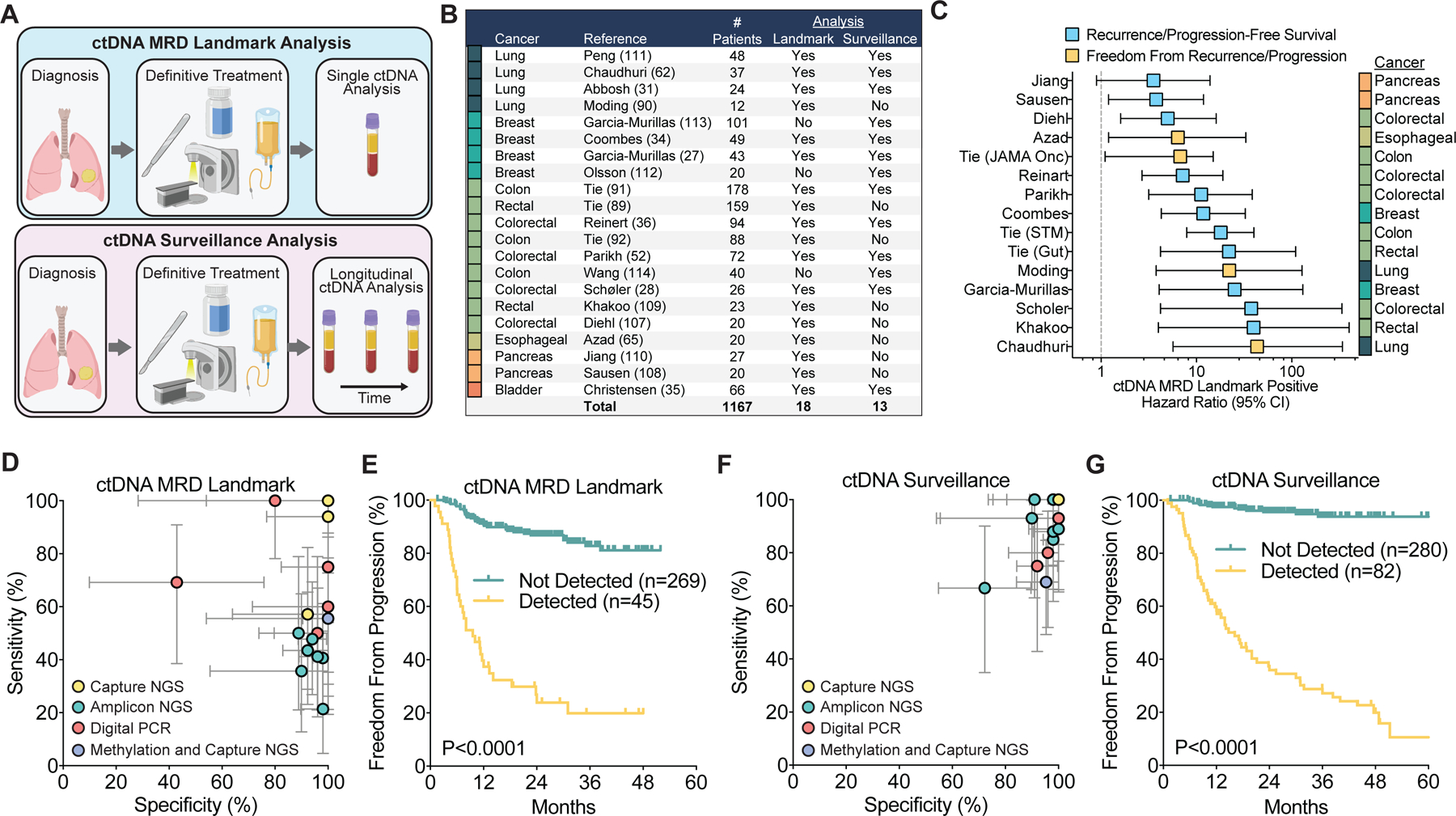
Performance of ctDNA analysis approaches for detecting MRD in solid tumors. A) Schematic comparing MRD landmark and surveillance ctDNA analysis. MRD landmark analysis determines the ctDNA status of a patient at one defined timepoint, shortly after completing curative therapy. Surveillance analysis evaluates multiple post-treatment blood draws over time, and patients are considered positive if ctDNA is detected in any sample. B) Summary table of studies included in the analysis. C) Summary of hazard ratios with 95% confidence intervals for freedom from recurrence/progression or recurrence/progression-free survival in published studies using MRD landmark analysis. D) Clinical sensitivity and specificity for ctDNA detection at the first post-treatment time point (ctDNA MRD landmark) (27,28,31,34–36,52,62,65,89–92,107–111). Clinical sensitivity is defined as the percentage of patients who relapsed in the follow up period and who were ctDNA positive at the landmark. Clinical specificity is defined as the percentage of patients who did not relapse in the follow up period who were ctDNA negative at the landmark. Patients who received adjuvant/consolidation therapy after ctDNA testing were excluded. Each study is colored based on the ctDNA analysis approach utilized. Error bars represent binomial 95% confidence intervals. E) Freedom from progression from the start of therapy based on ctDNA detection using MRD landmark analysis combining studies with individual patient survival reported (35,90,91,108,111). Studies with a median time of first blood collection after completing surgery or radiation therapy of greater than 20 weeks were excluded to minimize guarantee-time bias. Median time of first blood draw after completing surgery or radiation therapy for included studies was 7 weeks (range 2 hours to 11 weeks). P-value calculated using a two-sided log-rank test. F) Clinical sensitivity and specificity for ctDNA detection with longitudinal monitoring post-treatment (ctDNA Surveillance) (27,28,31,34–36,52,62,91,111–114). Error bars represent binomial 95% confidence intervals. G) Freedom from progression from the start of therapy based on ctDNA detection using surveillance analysis across studies with individual patient survival reported (35,91,111,112,114). P-value calculated using a two-sided log-rank test.
From a clinical perspective, determining MRD status at an early post-treatment landmark time is attractive because it would facilitate immediate decision-making about adjuvant or consolidation therapies, while minimizing the costs from testing serial blood samples. However, one could also envision performing repeat MRD surveillance over time and initiating adjuvant or consolidation therapy at the first positive blood draw. This approach has the potential advantage of decreasing false negatives. For either the landmark or surveillance approaches to offer the potential of meaningful improvement over current standard of care, MRD must be detectable with a significant lead time over conventional imaging. Given the differences in these two approaches, the type of analysis must be considered when comparing across studies and techniques.
Clinical sensitivity and specificity of current approaches
More than 20 studies to date have demonstrated the prognostic value of ctDNA MRD detection in multiple solid cancers with a variety of technical approaches (Table 1). We attempted to summarize the performance of current ctDNA analysis approaches from some of these seminal studies, which we selected from the literature based on inclusion of at least 10 patients with ctDNA analysis performed after completion of curative-intent therapy for non-metastatic solid tumors (Figure 2B). Studies and/or patients were excluded if adjuvant therapy was given after ctDNA analysis, but this information was not available for every study. It is important to note that different methodologies for ctDNA MRD analysis have not been tested side-by-side on the same set of samples, and these analyses would be challenging due to the large amounts of plasma that would be required. Additionally, the same method may perform differently across laboratories or in samples with different quality. Our goal in this review is to synthesize the published data rather than to rigorously compare diverse methods.
Table 1:
Studies evaluating circulating tumor DNA minimal residual disease in solid cancers.
| Study | Tumor Type | Methodology | Mutations Monitored | Total Patients | ctDNA MRD Landmark | ctDNA Surveillance | ||
|---|---|---|---|---|---|---|---|---|
| Sensitivity | Specificity | Sensitivity | Specificity | |||||
| Peng et al. 2020 (111) | Lung | Amplicon NGS | Multiple | 48 | 50.0% | 88.9% | 66.7% | 72.2% |
| Chaudhuri et al. 2017 (62) | Lung | Capture NGS | Multiple | 37 | 94.0% | 100.0% | 100.0% | 100.0% |
| Abbosh et al. 2017 (31) | Lung | PCR NGS | Multiple | 24 | 35.7% | 90.0% | 92.9% | 90.0% |
| Moding et al. 2020 (90) | Lung | Capture NGS | Multiple | 12 | 100.0% | 100.0% | - | - |
| Garcia-Murillas et al. 2019 (113) | Breast | Digital PCR | Single | 101 | - | - | 75.0% | 92.1% |
| Coombes et al. 2019 (34) | Breast | Amplicon NGS | Multiple | 49 | 55.6% | 100.0% | 88.9% | 100.0% |
| Garcia-Murillas et al. 2015 (27) | Breast | Digital PCR | Single | 43 | 50.0% | 96.0% | 80.0% | 96.4% |
| Olsson et al. 2015 (112) | Breast | Digital PCR | Multiple | 20 | - | - | 92.8% | 100.0% |
| Tie et al. 2016 (91) | Colon | Amplicon NGS | Single | 178 | 40.7% | 98.0% | 85.2% | 98.0% |
| Tie et al. 2019 (92) | Colon | Amplicon NGS | Single | 88 | 43.5% | 92.3% | - | - |
| Wang et al. 2019 (114) | Colon | Amplicon NGS | Single | 40 | - | - | 100.0% | 90.6% |
| Reinert et al. 2019 (36) | Colorectal | Amplicon NGS | Multiple | 94 | 41.2% | 96.1% | 87.5% | 98.3% |
| Parikh et al. 2021 (52) | Colorectal | Methylation and Capture NGS | Multiple | 72 | 55.6% | 100.0% | 69.0% | 95.3% |
| Scholer et al. 2017 (28) | Colorectal | Digital PCR | Single | 26 | 60.0% | 100.0% | 100.0% | 100.0% |
| Diehl et al. 2008 (107) | Colorectal | Digital PCR | Single | 20 | 100.0% | 80.0% | - | - |
| Tie et al. 2018 (89) | Rectal | Amplicon NGS | Single | 159 | 47.8% | 94.1% | - | - |
| Khakoo et al. 2019 (109) | Rectal | Digital PCR | Multiple | 23 | 75.0% | 100.0% | - | - |
| Azad et al. 2020 (65) | Esophageal | Capture NGS | Multiple | 15 | 55.6% | 100.0% | - | - |
| Jiang et al. 2020 (110) | Pancreas | Capture NGS | Multiple | 27 | 57.1% | 92.3% | - | - |
| Sausen et al. 2015 (108) | Pancreas | Digital PCR | Single | 20 | 69.2% | 42.9% | - | - |
| Christensen et al. 2019 (35) | Bladder | Amplicon NGS | Multiple | 66 | 21.4% | 98.1% | 100.0% | 97.9% |
Abbreviations: ctDNA: circulating tumor DNA, MRD: minimal residual disease, NGS: next-generation sequencing, - : not reported or unable to be calculated.
Clinical sensitivity (percentage of patients who relapsed in the follow up period who were ctDNA positive) and clinical specificity (percentage of patients who did not relapse in the follow up period who were ctDNA negative) were calculated for the first follow up sample after completing definitive therapy (ctDNA MRD Landmark) or for repeated ctDNA analysis during follow up (ctDNA Surveillance). Patients who received additional therapy after ctDNA analysis were excluded if noted in the study.
Despite variable study designs, different tumor types, and inconsistent study endpoints, detection of ctDNA MRD at the post-treatment landmark was consistently associated with inferior prognosis (Figure 2C). Hazard ratios for progression-free survival or freedom from progression in ctDNA MRD positive patients ranged from 3.5 to 43.3, demonstrating the strong prognostic value of ctDNA MRD analysis. To summarize results in more detail, for each study we calculated the clinical sensitivity for detection of residual disease (i.e., the percentage of patients who recurred who were ctDNA-positive after therapy), as well as the clinical specificity (i.e., the percentage of patients without recurrence who were ctDNA-negative). Studies where clinical sensitivity and specificity could not be determined from the reported results and published supplementary materials could not be included.
To compare across studies employing an MRD landmark analysis, we considered the first blood draw after completion of all therapy. Of note, blood draw timing was heterogenous across studies, and the majority did not report the length of time between completing therapy and ctDNA analysis for individual samples. Given that limitation, at the MRD landmark, clinical specificity was greater than 90% in the majority of studies. However, clinical sensitivity was more modest and ctDNA was detected in only about half of subjects who ultimately relapsed (median 56%; range 21–100%; Figure 2D). Therefore, patients who are ctDNA positive by landmark analysis appear to have a very high likelihood of relapse (i.e. high positive-predictive value). However, since many patients who ultimately recur are missed by current approaches, there is significant room for improving both clinical sensitivity and the negative-predictive value of ctDNA MRD. Despite this limitation, freedom from progression based on ctDNA MRD landmark analysis was strongly significant when combining data across studies (HR 8.9, 95% CI 4.0–19.9; Figure 2E).
For summarizing surveillance analysis across studies, we considered patients as positive for MRD if ctDNA was detected in any longitudinal sample obtained prior to relapse on imaging. As compared with landmark analysis, extending ctDNA testing to longitudinal samples with surveillance analysis substantially improved sensitivity, approaching 100% in most studies (median 89%; range 67–100%; Figure 2F). Importantly, specificity remained high despite analyzing multiple samples. Combining data across studies and compared with ctDNA MRD landmark analysis, ctDNA surveillance analysis improved stratification of freedom from progression (HR 23.6, 95% CI 13.0–42.9; Figure 2G).
Summarizing across studies, residual disease was identified at a median of 5.8 months earlier using ctDNA MRD than by standard of care radiologic imaging. This diagnostic lead time could provide a substantial head start for initiating adjuvant or consolidation therapies and potentially lead to improved outcomes. When comparing between ctDNA analysis approaches, surveillance analysis counterintuitively had an improved lead time over the landmark approach (Figure 3A). Furthermore, more than half of patients who ultimately developed recurrence had false negative MRD tests at the post-treatment landmark immediately after completing therapy (Figure 3B). Of note, it is possible that this result is affected by guarantee-time bias, since complete raw data are not available for most of the studies included in these analyses (88). This caveat aside, the apparently longer lead time in the surveillance analyses is most likely due to patients with initially false-negative MRD results having lower microscopic disease burden and/or more indolent tumors with lower ctDNA shedding that takes longer to manifest radiologically. Indeed, among patients who ultimately progressed, freedom from progression from the start of treatment was significantly longer in patients with ctDNA not detected in the first post-treatment sample compared with patients who had ctDNA detected at that timepoint (Figure 3C). Thus, there is an opportunity for MRD assays with higher sensitivity to detect residual disease in patients with the lowest disease burden immediately after therapy. This would provide the longest window of opportunity before radiologic progression in which to intervene and potentially improve outcomes. Additionally, decreasing the number of false negative MRD results immediately after local therapy could facilitate sparing patients who are already cured from unnecessary adjuvant/consolidation therapy.
Figure 3:
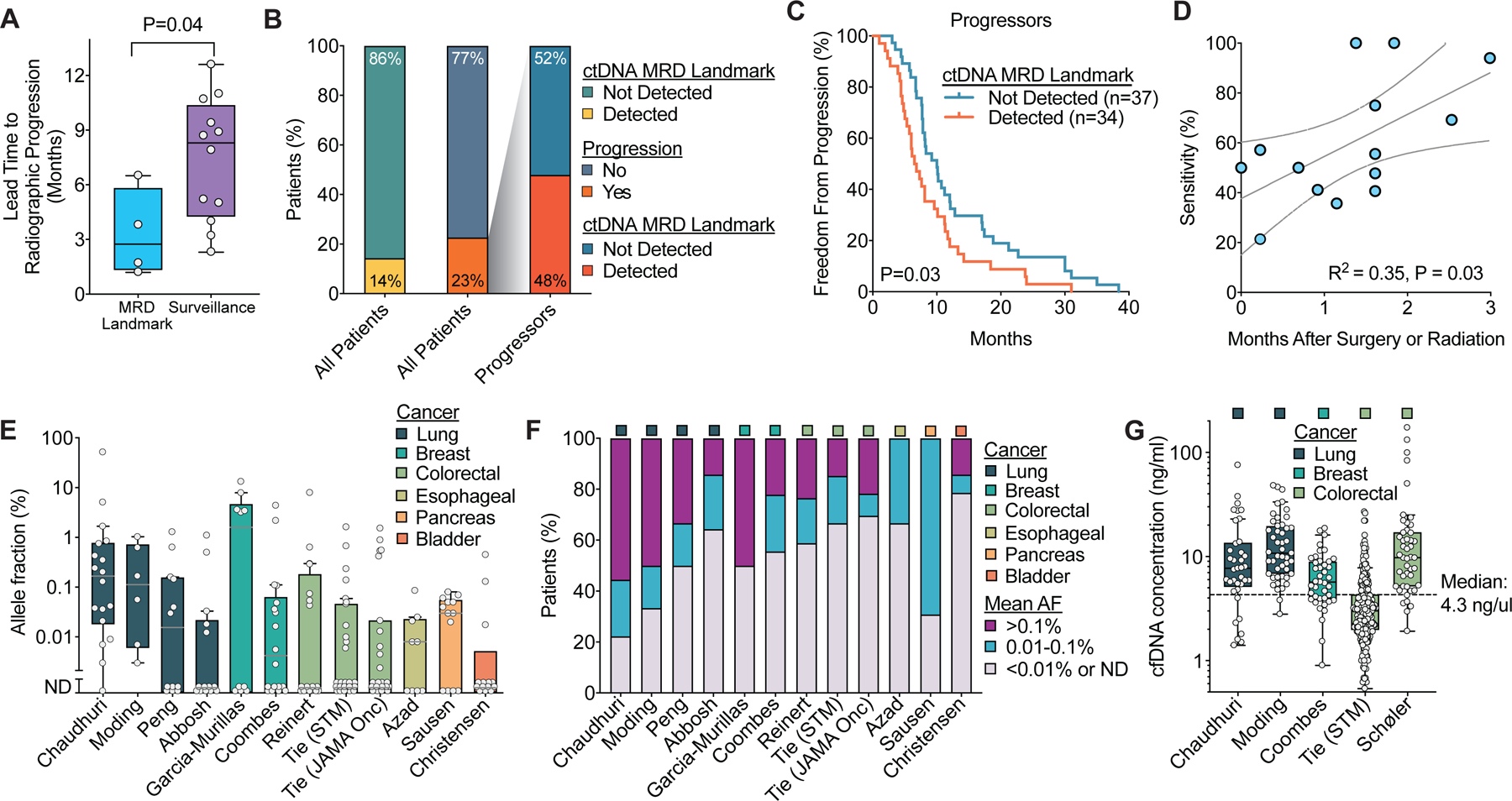
Summary of lead times and ctDNA levels after curative-intent therapy in solid tumors. A) Comparison of lead time from blood draw to progression on standard of care imaging for ctDNA MRD landmark (65,90,92,108) and ctDNA surveillance studies (27,28,31,34–36,62,91,111–114). Boxes represent median and interquartile range (IQR) and whiskers represent 1.5 times the IQR per the Tukey method. B-C) Analysis of outcomes by ctDNA status at the MRD landmark among patients who ultimately developed progressive disease using the studies from Figure 2E to assess the possibility that patients who are ctDNA negative at the MRD landmark but ultimately progress have more indolent disease. B) Stacked box plot summarizing the proportion of all patients and proportion of progressors with ctDNA MRD detected. C) Freedom from progression from the start of therapy based on ctDNA MRD detection only in patients who developed progressive disease. P-value calculated using a two-sided log-rank test. D) Linear correlation of clinical sensitivity for landmark MRD studies with the median time after completing surgery or radiation therapy for the ctDNA landmark blood draw. Studies with a median time of greater than 20 weeks were excluded. Line of best fit is shown with 95% confidence intervals. E) Box plots of mean ctDNA allele fraction at the first post-treatment time point across studies in patients who ultimately relapsed. Only studies with allele fractions reported are included. Boxes represent median and IQR and whiskers represent 1.5 times the IQR per the Tukey method. F) Distribution of mean allele fractions at the first post-treatment time point in the studies from E. G) Box plots of cfDNA concentration at the first blood draw after completing local therapy across studies. Boxes represent median and IQR and whiskers represent 1.5 times the IQR per the Tukey method.
As discussed above, false negatives at the MRD landmark could result from elevated normal cfDNA after surgery lowering the allele fraction of ctDNA molecules or few residual tumor cells. Consistent with the potential adverse impact of tissue injury associated with treatment and/or a progressive increase in the amount of residual disease over time, clinical sensitivity increased with a larger gap between local therapy and ctDNA analysis (Figure 3D). Additionally, certain clinical scenarios consisting of low burden recurrence may be particularly challenging for ctDNA MRD detection. For example, isolated local recurrences after radiation therapy have been recurrently associated with false negative MRD testing in lung, esophageal, and rectal cancers (62,65,89).
To help determine the LOD required for ctDNA MRD landmark analysis, we analyzed the mean ctDNA allele fraction in the first post-treatment sample for patients who developed recurrent disease in published data (Figure 3E). The median ctDNA allele fraction post-therapy ranged from not detected to 1.6% across the studies. Across studies and tumor types, the majority of patients experiencing recurrence had ctDNA allele fractions less than 0.01% at the MRD landmark (Figure 3F). This suggests that in order to minimize false negatives in most clinical scenarios, ctDNA MRD assay will require LODs substantially below 0.01%.
Because cfDNA input directly impacts the LOD for ctDNA analysis, we also examined the cfDNA concentration at the first blood draw after completing local therapy across studies that provided this information (Figure 3G) (28,34,62,90,91). The median plasma cfDNA concentration ranged from 3.0 to 10.8 ng/ml across studies. Combining the data from these studies, the overall median cfDNA concentration was 4.3 ng/ml. Because one haploid human genome has a mass of ~3.3 pg, this is equivalent to ~1303 haploid genomes per ml of plasma.
Adjuvant therapy to improve outcomes in patients with ctDNA MRD
The ultimate goal of ctDNA MRD analysis is to allow personalization of adjuvant/consolidation therapy to increase the probability of cure and/or reduce toxicity. However, there is limited evidence that treatment escalation or de-escalation based on ctDNA MRD status can improve patient outcomes. A growing number of published cases have suggested that adjuvant chemotherapy can lower ctDNA levels in patients with MRD following definitive therapy (31,36,91). In the largest study, 88 patients with stage III colon cancer underwent ctDNA testing with Safe-SeqS post-operatively and following adjuvant chemotherapy (92). Among 18 patients with evidence of ctDNA MRD positivity after surgery, 9 patients converted to ctDNA MRD negative after chemotherapy, and 6 of these patients remained disease-free at last follow up. This suggests that ctDNA MRD can be used to track response to adjuvant systemic therapy.
A critical open question is whether adjuvant/consolidation therapy can improve clinical outcomes in patients with ctDNA MRD. Given the significant false negative rates of ctDNA MRD landmark analysis in most studies, concerns have been raised that ctDNA MRD might mainly detect patients with the highest residual disease burden who might not be able to benefit from early initiation of additional therapy. To address this question, we recently leveraged a change in the standard of care for locally advanced NSCLC to compare outcomes based on ctDNA MRD status using CAPP-Seq in patients treated with and without consolidation immunotherapy (90). Patients who were ctDNA MRD positive (but not patients who were ctDNA MRD negative) after chemoradiation and who received consolidation immunotherapy had significantly better freedom from progression than patients treated with chemoradiation therapy alone. This is the first evidence we are aware of that additional therapy can not only reduce ctDNA levels but may improve clinical outcomes in patients who are ctDNA MRD positive. In further support of the idea that adjuvant/consolidation immunotherapy can improve outcomes in ctDNA MRD positive patients, a recent exploratory analysis of the IMvigor010 study evaluating adjuvant atezolizumab in muscle invasive urothelial cancer demonstrated that although there was no overall survival benefit in the intent to treat population, adjuvant atezolizumab improved overall survival in ctDNA positive patients (93). These two studies suggest that personalized therapy based on ctDNA MRD status could result in improved patient outcomes.
Lastly, it is possible that new mutations detected at the time of MRD analysis but that were absent pre-treatment could inform therapy. Evidence from chronic myelogenous leukemia and acute lymphoblastic leukemia where molecular monitoring has become standard of care (94–96) suggests mutations identified during MRD monitoring could be used to personalize additional therapy. In solid cancers, the identification of resistant or emergent clones with actionable mutations via ctDNA analysis could potentially similarly guide the selection of adjuvant therapy (62). Indeed, non-invasive genotyping has been shown to identify actionable mutations that predict response to therapy in solid cancers (85,97,98). The main limitations of this approach are the low ctDNA allele fractions usually observed after definitive therapy and the relatively low detection limits of tumor-naïve ctDNA analyses, which will likely hinder the identification of clinically actionable variants in most settings.
Future directions
Prospective clinical trials
Although many retrospective studies have demonstrated that strong prognostic power of ctDNA MRD over other disease risk factors, the clinical utility added by such assays in solid tumors remains to be determined. For example, in contrast to the established impact of MRD for outcomes in leukemias, specific evidence for the clinical benefit of ctDNA MRD analysis for personalizing adjuvant/consolidation therapy in solid tumors is scant. To firmly establish the clinical utility of ctDNA MRD for treatment personalization, prospective clinical trials will be critical. Auspiciously, ctDNA analysis has opened the door to several novel clinical trial designs in which treatment decisions are based on ctDNA MRD status (Figure 4A) (99). As of April 2021, at least 19 interventional trials are under way exploring the ability of ctDNA MRD to guide adjuvant/consolidation therapy in a variety of solid tumors using diverse randomized and non-randomized schemas (Table 2).
Figure 4:
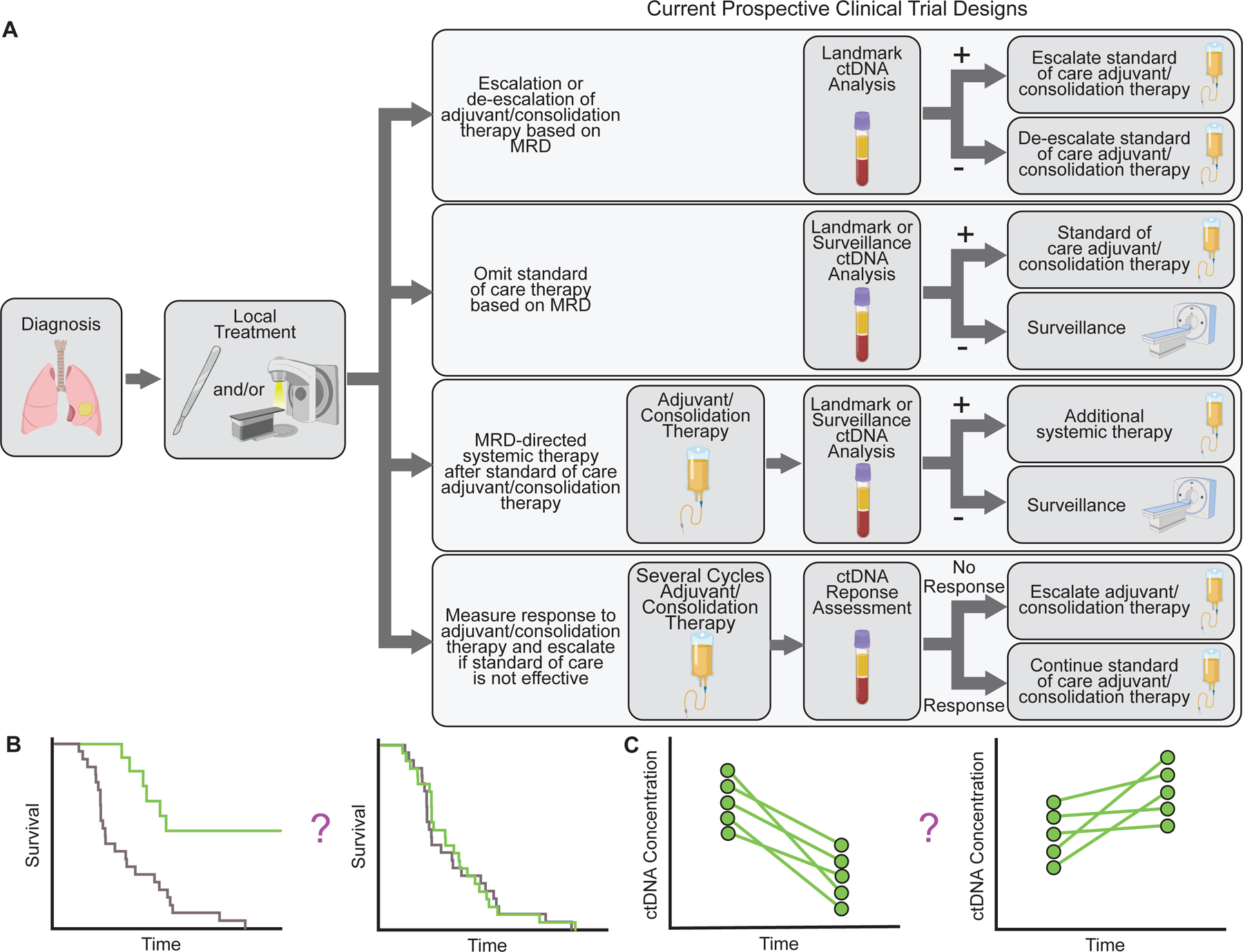
Examples of designs of ongoing interventional clinical trials testing personalization of adjuvant/consolidation therapy based on ctDNA MRD. A) Schematic depicting examples of ongoing interventional clinical trials using ctDNA MRD to guide treatment. B-C) Potential clinical trial endpoints for ctDNA MRD studies. B) Survival (overall survival, disease free survival, or event free survival) for patients managed based on ctDNA MRD can be compared with a control arm or historical cohort or patients managed according to standard of care. C) For patients with ctDNA MRD, ctDNA clearance or change in ctDNA concentration could be used as a surrogate endpoint.
Table 2:
Ongoing Clinical Trials Using ctDNA MRD to Guide Adjuvant/Consolidation Therapy
| Number | Name | Cancer | Stage | Patients | Drug | Phase | Trial Type | Randomized | Description | Primary outcome(s) |
|---|---|---|---|---|---|---|---|---|---|---|
| NCT04089631 | Circulating Tumour DNA Based Decision for Adjuvant Treatment in Colon Cancer Stage II Evaluation (CIRCULATE) | Colorectal cancer | II | 4812 | Capecitabine | III | 1 | Yes | Post-resection ctDNA positive randomized to Arm 1: chemotherapy or Arm 2: follow-up. Post-resection ctDNA negative patients are randomized to Arm 1: follow-up within study or Arm 2: follow-up off-study. | DFS |
| NCT04120701 | Circulating Tumour DNA Based Decision for Adjuvant Treatment in Colon Cancer Stage II Evaluation (CIRCULATE) | Colorectal cancer | II | 1980 | FOLFOX | III | 1 | Yes | Post-resection ctDNA positive randomized to Arm 1: chemotherapy or Arm 2: follow-up. Post-resection ctDNA negative patients are randomized to Arm 1: follow-up within study or Arm 2: follow-up off-study. | DFS |
| Not available | Colon Adjuvant Chemotherapy Based on Evaluation of Residual Disease (CIRCULATE-US) | Colon cancer | II-III | 1800 | FOLFOX, CAPOX, or FOLFOXIRI | II/III | 2/4 | Yes | Post-resection ctDNA positive randomized to Arm 1: FOLFOX/CAPOX or Arm 2: FOLFOXIRI. Post-resection ctDNA negative randomized to Arm 1: CAPOX/FOLFOX or Arm 2: ctDNA surveillance. | DFS and time to ctDNA positivity |
| EUCTR2018–000070-30-DK | IMPROVE Intervention Trial: Implementing non-invasive circulating tumor DNA analysis to optimize the operative and postoperative treatment for patients with colorectal cancer (IMPROVE IT) | Colorectal Cancer | I-II | 70 | FOLFOX or CAPOX | II | 1 | Yes | Post resection ctDNA positive randomized to Arm 1: chemotherapy or Arm 2: observation. | DFS |
| NCT04457297 | Initial Attack on Latent Metastasis Using TAS-102 for ctDNA Identified Colorectal Cancer Patients After Curative Resection (ALTAIR) | Colorectal cancer | III | 240 | Trifluridine and tipiracil | III | 1 | Yes | Post-resection and adjuvant chemotherapy ctDNA positive randomized to Arm 1: chemotherapy or Arm 2: placebo. | DFS |
| NCT03803553 | Identification and Treatment Of Micrometastatic Disease in Stage III Colon Cancer | Colorectal cancer | III | 500 | FOLFIRI, nivolumab, or encorafenib/ binimetinib/ cetuximab |
III | 1 | Yes | Post-resection and adjuvant chemotherapy ctDNA positive randomized to Arm 1: FOLFIRI or Arm 2: active surveillance, ctDNA negative receives active surveillance. MSI-H and ctDNA positive receives nivolumab. BRAF mutant and ctDNA positive receive encorafenib/binimetinib/ cetuximab. |
DFS and ctDNA clearance |
| ACTRN12617001566325 | Circulating Tumour DNA Analysis Informing Adjuvant Chemotherapy in Stage III Colon Cancer: A Multicentre Phase II/III Randomised Controlled Study (DYNAMIC-III) | Colorectal cancer | III | 1000 | Flurouracil, capecitabine, FOLFOX, CAPOX, or FOLFOXIRI | II/III | 2 | Yes | Post-resection randomized to Arm 1: clinician’s choice of standard of care or Arm 2: ctDNA-informed escalation or de-escalation of therapy. | RFS |
| NCT04068103 | Phase II/III Study of Circulating Tumor DNA as a Predictive Biomarker in Adjuvant Chemotherapy in Patients With Stage IIA Colon Cancer (COBRA) | Colon cancer | IIA | 1408 | FOLFOX or CAPOX | II/III | 1 | Yes | Post-resection randomized to Arm 1: Active surveillance with blood stored for ctDNA testing or Arm 2: ctDNA positive patients receive chemotherapy until progression, ctDNA negative patients undergo active surveillance. | ctDNA clearance and RFS |
| NTR6455 | Prevention of recurrent disease by additional chemotherapy in patients with detectable circulating tumor DNA in the blood after surgery for stage II (lymph nodes unaffected) colon cancer. | Colon Cancer | II | 1320 | FOLFOX or CAPOX | III | 1 | Yes | Post-resection randomized to Arm 1: adjuvant chemotherapy if ctDNA positive or Arm 2: observation with ctDNA analyzed at end of trial. | Recurrence rate |
| NCT04259944 | Post-surgical Liquid Biopsy-guided Treatment of Stage III and High-risk Stage II Colon Cancer Patients: the PEGASUS Trial | Colon cancer | II-III | 140 | CAPOX, capecitabine, or FOLFIRI | II | 2/3 | No | Post-resection ctDNA positive patients will receive CAPOX for 3 months and ctDNA negative patients will receive capecitabine for 6 months. ctDNA positive patients who stay positive post-adjuvant will escalate to FOLFIRI, ctDNA negative patients who become positive will escalate to CAPOX and then FOLFIRI if they continue to be positive. ctDNA positive patients who become negative post-adjuvant will de-escalate to capecitabine. ctDNA negative patients who remain negative post-adjuvant will be monitored and switch to CAPOX if positive. | Number of cases that become ctDNA positive or experience radiologic relapse after negative ctDNA post-surgery and post-adjuvant. |
| JPRN-jRCT1031200006 | A Randomized, Phase III Controlled Study to Compare CAPOX Therapy as Post-operative Adjuvant Chemotherapy with Surgery Alone in Patients with Completely Resected Circulating Tumor DNA-negative High-risk Stage II and Low-risk Stage III Colon Cancer | Colon cancer | II-III | 1240 | Observation | III | 1/4 | Yes | Post-resection ctDNA negative randomized to Arm 1: CAPOX or Arm 2: observation. | DFS |
| NCT04585477 | Adjuvant Durvalumab for Early Stage NSCLC Patients With ctDNA Minimal Residual Disease | NSCLC | I-III | 80 | Durvalumab | II | 1 | No | Post-resection or SBRT and adjuvant chemotherapy ctDNA positive receive adjuvant durvalumab. ctDNA negative receive no treatment. | ctDNA > 3 fold decrease |
| NCT04625699 | Study of Durvalumab + Tremelimumab in NSCLC Patients After Adjuvant Treatment | NSCLC | II-IIIB | 15 | Durvalumab/ tremelimumab |
II | 1 | No | Post-resection and adjuvant ctDNA positive receive durvalumab/tremelimumab. | Number of evaluable patients enrolled |
| NCT04385368 | Phase III Study to Determine the Efficacy of Durvalumab in Combination With Chemotherapy in Completely Resected Stage II-III Non-small Cell Lung Cancer (NSCLC) (MERMAID-1) | NSCLC | II-III | 332 | Durvalumab | III | 1 | Yes | Post-resection and adjuvant ctDNA positive randomized to Arm 1: placebo or Arm 2: durvalumab. | DFS |
| NCT04585490 | Personalized Escalation of Consolidation Treatment Following Chemoradiotherapy and Immunotherapy in Stage III NSCLC in Stage III NSCLC | NSCLC | III | 48 | Durvalumab/ platinum doublet |
III | 3 | No | Post-chemoradiation and 2 cycles consolidation durvalumab ctDNA positive escalated to durvalumab/chemotherapy. ctDNA negative continue on durvalumab. | ctDNA > 3 fold decrease |
| NCT03145961 | A Trial Using ctDNA Blood Tests to Detect Cancer Cells After Standard Treatment to Trigger Additional Treatment in Early Stage Triple Negative Breast Cancer Patients (c-TRAK-TN) | Triple negative breast | II-III | 208 | Pembrolizumab | II | 1 | Yes | Post-primary treatment ctDNA testing every 3 months for 1 year. Positive patients randomized 2:1 to Arm 1: pembrolizumab or Arm 2: observation. | Proportion ctDNA positive and ctDNA clearance or recurrence |
| NCT04849364 | Circulating Tumor DNA Enriched, Genomically Directed Post-neoadjuvant Trial for Patients With Residual Triple Negative Breast Cancer (PERSEVERE) | Triple negative breast cancer | I-III | 197 | Capecitabine and talazoparib, atezolizumab, inavolisib, or talazoparib/ atezolizumab | II | 2 | No | Post-neoadjuvant therapy and surgery +/− radiation therapy ctDNA positive receive genomically targeted treatment. ctDNA negative receive physician’s choice. | DFS |
| NCT04660344 | A Study of Atezolizumab Versus Placebo as Adjuvant Therapy in Patients With High-Risk Muscle-Invasive Bladder Cancer Who Are ctDNA Positive Following Cystectomy (IMvigor011) | Muscle-invasive bladder cancer | III | 495 | Atezolizumab | III | 1 | Yes | Post-cystectomy ctDNA positive randomized to Arm 1: atezolizumab or Arm 2: placebo. | DFS |
| NCT04510285 | Study of Pembrolizumab Plus Trastuzumab or Trastuzumab Alone After Surgery in Patients With Esophagogastric Tumors | Esophago-gastric cancer |
I-III | 48 | Pembrolizumab/trastuzumab or trastuzumab | II | 1 | Yes | Post-curative resection and chemotherapy HER2 positive and ctDNA positive randomized to Arm 1: trastuzumab/placebo or Arm 2: trastuzumab/pembrolizumab | ctDNA clearance |
| NCT03832569 | Study of Pembrolizumab Following Surgery in Patients With Microsatellite Instability High (MSI-H) Solid Tumors | MSI-H solid tumors | I-III | 10 | Pembrolizumab | I | 1 | Yes | Post-resection and standard of care adjuvant therapy ctDNA positive randomized to Arm 1: pembrolizumab or Arm 2: placebo. | ctDNA clearance |
Trial Type: 1) Post-standard of care therapy, 2) Escalate/de-escalate standard of care therapy, 3) Measure response to adjuvant/consolidation therapy and escalate treatment if standard of care is not effective, 4) Omit standard of care therapy.
Abbreviations: DFS: disease-free survival, RFS: recurrence-free survival, MSI-H: microsatellite instability-high, NSCLC: non-small cell lung cancer, SBRT: stereotactic body radiation therapy, FOLFOX: folinic acid, fluorouracil, and oxaliplatin, CAPOX: capecitabine and oxaliplatin, FOLFIRI: folinic acid, fluorouracil, and irinotecan, FOLFOXIRI: folinic acid, fluorouracil, oxaliplatin, and irinotecan.
The most straightforward trial design consists of treating patients who are ctDNA positive after completing the current standard of care (be it local therapy alone or a combination of local and systemic therapy) with additional systemic therapy. This trial design is attractive because it does not withhold standard of care treatment, and it can be implemented in solid cancers with and without a proven benefit for adjuvant therapy. Furthermore, such trials leverage the generally high PPV of ctDNA MRD assays. ctDNA negative patients can be followed on study with active surveillance or excluded from the study. Several clinical trials are currently testing the utility of this approach in colorectal cancers (NCT04089631, NCT04120701, NCT04068103, NCT04457297, NCT03803553, EUCTR2018–000070-30-DK, and NTR6455), esophagogastric cancers (NCT04510285), NSCLC (NCT04385368, NCT04585477 and NCT04625699), triple negative breast cancers (NCT03145961), muscle-invasive bladder cancers (NCT04660344), and solid tumors with high microsatellite instability (MSI-high) levels (NCT03832569).
In cancers with a proven benefit for adjuvant/consolidation therapy, ctDNA testing can potentially be used to escalate or de-escalate the standard of care adjuvant/consolidation therapy. Such strategies are currently being tested in colorectal cancer (ACTRN12617001566325) and triple negative breast cancer (NCT04849364). Alternatively, ctDNA testing could be used to measure the response to adjuvant/consolidation therapy and escalate treatment if standard of care is not effective, as is currently being tested in NSCLC (NCT04585490). These approaches can also be combined into composite trials, as is currently being done in stage II-III colon cancers (NCT04259944). Although intensification of adjuvant therapy has often failed to improve outcomes in unselected patients when rates of cure are high with local therapy (100,101), escalation of adjuvant therapy can have a dramatic effect on patients at high risk of developing metastatic disease (102). As a result, escalating adjuvant therapy could improve outcomes when applied to patients with ctDNA MRD who are at very high risk of relapse.
Another ctDNA MRD-driven personalization strategy that is currently being explored in low-risk stage III colon cancer (JPRN-jRCT1031200006) is the use of ctDNA to guide the omission of adjuvant/consolidation therapy in patients who are MRD negative. Such trials will need to carefully consider the false negative rate of the ctDNA MRD assays being used to ensure that it is sufficiently low to justify such an approach. Surveillance ctDNA testing could also be incorporated into this type of trial to trigger salvage systemic therapy if patients become ctDNA positive during follow up. For all of these studies, patients would ideally be randomized to the standard of care or a ctDNA-guided approach. Concerns have been raised by multiple stake holders, including both patients and providers, that the high positive predictive value of ctDNA MRD detection for recurrence makes equipoise challenging for randomizing ctDNA MRD positive patients to no further treatment. One approach that is being used to address this issue is randomizing patients prior to ctDNA analysis, with patients on the control arm having plasma samples banked for retrospective analysis and therefore not receiving real-time ctDNA MRD results (e.g. NCT04068103).
Although the majority of ongoing interventional studies focus on traditional clinical outcomes such as recurrence-free survival (Figure 4B), clinical trials can also be designed to use ctDNA changes as a primary or secondary outcome measure (Figure 4C). Several trials have implemented ctDNA clearance or decrease after starting adjuvant/consolidation therapy as a disease endpoint (e.g. NCT03803553, NCT04068103, NCT04585477, NCT04585490, NCT03145961, NCT04510285, and NCT03832569). If demonstrated to correlate with traditional patient outcomes, ctDNA changes could eventually serve as surrogate for clinical benefit. Because ctDNA changes can be assessed substantially earlier than traditional outcome measures, this approach could help accelerate testing of new adjuvant/consolidation therapies.
The technical and biological aspects of ctDNA MRD testing described above are important considerations for the success of such clinical trials. The ability to improve outcomes with ctDNA-guided therapy will likely depend of the amount of residual disease that can be identified and the lead-time prior to clinical recurrence. As a result, it will likely be critical to utilize ctDNA MRD assays with maximal sensitivity. Because ctDNA shedding varies between patients, it may be reasonable to exclude patients without ctDNA detected prior to treatment from clinical trials. However, these patients have been shown to have low risks of recurrence in some contexts, suggesting they might benefit from treatment de-intensification analogously to patients who become ctDNA negative after initial therapy (46,53). Clinical trials will also need to be powered sufficiently to account for only a subset of patients being either ctDNA MRD positive or negative. Furthermore, biomarker analysis must be completed in a timely manner to inform therapeutic decisions because delaying adjuvant therapy may lead to worse outcomes (103,104). To allow intervention as early as possible, studies investigating whether MRD landmark analysis can be performed closer to the end of definitive therapy or even during standard-of-care treatment in the case of radiation therapy and systemic therapy are warranted.
Medicare coverage of ctDNA MRD assays
A key barrier to the clinical implementation of ctDNA MRD testing in the U.S. will be reimbursement by payors. Based on prospective non-randomized evidence (36), the first local coverage determination (LCD) was recently finalized by the Centers for Medicare and Medicaid Services to provide coverage for ctDNA MRD testing. The initial LCD focuses on the application of Natera’s Signatera MRD test to patients with colorectal cancers. However, a broader draft LCD was recently released to enable coverage of ctDNA MRD across tumor types. The draft LCD outlines the data required for ctDNA tests to be covered by Medicare, including: 1) the ctDNA test identifies recurrence prior to clinical exams or radiographical imaging with similar sensitivity and specificity to radiology, 2) the test must identify recurrence or progression with considerably more accuracy than other established forms of surveillance, 3) the test must have comparable performance to other covered ctDNA MRD tests, and 4) the test must complete a technical assessment that confirms its analytical and clinical validity. Notably absent is a requirement for randomized prospective data demonstrating improved outcomes. Thus, it is likely that an increasing number of ctDNA MRD assays for a variety of tumor types will be covered by Medicare in the near future.
Conclusion
Multiple studies have demonstrated that detection of ctDNA MRD following definitive therapy for solid cancers is strongly prognostic and has extremely high positive predictive value for risk of recurrence. Due to the low allele fractions at post-treatment timepoints, optimizing cfDNA recovery, tracking multiple tumor genotype-informed mutations, and minimizing the effects of technical and biological background are critical to achieve high sensitivity for identifying patients at risk of progressive disease. Several interventional clinical trials testing the utility of ctDNA MRD for personalization of adjuvant/consolidation therapy are now underway, and it is likely that multiple ctDNA MRD assays could soon be used in routine clinical care. Therefore, we appear to be at the cusp of a revolution in personalized management of adjuvant/consolidation therapies in solid tumors.
Significance.
Circulating tumor DNA analysis enables detection of minimal residual disease and predicts relapse after definitive treatment for solid cancers, thereby promising to revolutionize personalization of adjuvant and consolidation therapies.
Acknowledgments
E.J. Moding is supported by an Conquer Cancer Young Investigator Award, supported by GO2 Foundation for Lung Cancer and the My Blue Dots organization. B.Y. Nabet is a Stanford Cancer Systems Biology Scholar and supported by the NIH (5R25CA180993) and by the Postdoctoral Research Fellowship (134031-PF-19–164-01-TBG) from the American Cancer Society. A.A. Alizadeh is a Scholar of the Leukemia & Lymphoma Society, and supported by the National Cancer Institute (R01CA257655, R01CA233975, R01CA188298) and the Virginia and D.K. Ludwig Fund for Cancer Research. M. Diehn is supported by the National Cancer Institute (R01CA188298, R01CA233975, R01CA244526, and R01CA254179), Stand Up To Cancer, the Tobacco Related Disease Research Program, the Virginia and D.K. Ludwig Fund for Cancer Research, and the CRK Faculty Scholar Fund. The schematics in Figures 1, 2, and 4 were created with BioRender.com.
Footnotes
Competing Interests
E.J.M. has served as a paid consultant for DeciBio. B.Y.N. is currently an employee and stockholder at Roche/Genentech and has patent filings related to immunomodulatory RNA and cancer biomarkers. A.A.A. and M.D. are co-inventors on patent applications related to CAPP-Seq and have patent filings related to other cancer biomarkers. A.A.A. reports ownership interest in CiberMed and Foresight Diagnostics, paid consultancy from Roche/Genentech and Gilead, and research support from BMS. M. Diehn reports research funding from Varian Medical Systems, AstraZeneca, and Illumina, ownership interest in CiberMed and Foresight Diagnostics, patent filings related to cancer biomarkers, paid consultancy from AstraZeneca, Genentech, Novartis, Boehringer Ingelheim, Illumina, Roche Sequencing Solutions, Gritstone Oncology, and BioNTech, and travel/honoraria from RefleXion.
References
- 1.Siegel RL, Miller KD, Fuchs HE, Jemal A. Cancer Statistics, 2021. CA Cancer J Clin. 2021;71:7–33. [DOI] [PubMed] [Google Scholar]
- 2.Luskin MR, Murakami MA, Manalis SR, Weinstock DM. Targeting minimal residual disease: a path to cure? Nat Rev Cancer. 2018;18:255–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Goddard ET, Bozic I, Riddell SR, Ghajar CM. Dormant tumour cells, their niches and the influence of immunity. Nat Cell Biol. 2018;20:1240–9. [DOI] [PubMed] [Google Scholar]
- 4.Pignon J-P, Tribodet H, Scagliotti GV, Douillard J-Y, Shepherd FA, Stephens RJ, et al. Lung adjuvant cisplatin evaluation: a pooled analysis by the LACE Collaborative Group. J Clin Oncol. 2008;26:3552–9. [DOI] [PubMed] [Google Scholar]
- 5.Sargent D, Sobrero A, Grothey A, O’Connell MJ, Buyse M, Andre T, et al. Evidence for cure by adjuvant therapy in colon cancer: observations based on individual patient data from 20,898 patients on 18 randomized trials. J Clin Oncol. 2009;27:872–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Early Breast Cancer Trialists’ Collaborative Group (EBCTCG), Peto R, Davies C, Godwin J, Gray R, Pan HC, et al. Comparisons between different polychemotherapy regimens for early breast cancer: meta-analyses of long-term outcome among 100,000 women in 123 randomised trials. Lancet. 2012;379:432–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Antonia SJ, Villegas A, Daniel D, Vicente D, Murakami S, Hui R, et al. Overall Survival with Durvalumab after Chemoradiotherapy in Stage III NSCLC. N Engl J Med. 2018;379:2342–50. [DOI] [PubMed] [Google Scholar]
- 8.André T, de Gramont A, Vernerey D, Chibaudel B, Bonnetain F, Tijeras-Raballand A, et al. Adjuvant Fluorouracil, Leucovorin, and Oxaliplatin in Stage II to III Colon Cancer: Updated 10-Year Survival and Outcomes According to BRAF Mutation and Mismatch Repair Status of the MOSAIC Study. J Clin Oncol. 2015;33:4176–87. [DOI] [PubMed] [Google Scholar]
- 9.Pieters R, de Groot-Kruseman H, Van der Velden V, Fiocco M, van den Berg H, de Bont E, et al. Successful Therapy Reduction and Intensification for Childhood Acute Lymphoblastic Leukemia Based on Minimal Residual Disease Monitoring: Study ALL10 From the Dutch Childhood Oncology Group. J Clin Oncol. 2016;34:2591–601. [DOI] [PubMed] [Google Scholar]
- 10.Hourigan CS, Karp JE. Minimal residual disease in acute myeloid leukaemia. Nat Rev Clin Oncol. 2013;10:460–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Mordant P, Loriot Y, Lahon B, Castier Y, Lesèche G, Soria J-C, et al. Minimal residual disease in solid neoplasia: New frontier or red-herring? Cancer Treat Rev. 2012;38:101–10. [DOI] [PubMed] [Google Scholar]
- 12.Schwarzenbach H, Hoon DSB, Pantel K. Cell-free nucleic acids as biomarkers in cancer patients. Nat Rev Cancer. 2011;11:426–37. [DOI] [PubMed] [Google Scholar]
- 13.Pantel K, Alix-Panabières C. Liquid biopsy and minimal residual disease - latest advances and implications for cure. Nat Rev Clin Oncol. 2019;16:409–24. [DOI] [PubMed] [Google Scholar]
- 14.Crowley E, Di Nicolantonio F, Loupakis F, Bardelli A. Liquid biopsy: monitoring cancer-genetics in the blood. Nature Reviews Clinical Oncology. 2013;10:472–84. [DOI] [PubMed] [Google Scholar]
- 15.Alix-Panabières C, Pantel K. Clinical Applications of Circulating Tumor Cells and Circulating Tumor DNA as Liquid Biopsy. Cancer Discov. 2016;6:479–91. [DOI] [PubMed] [Google Scholar]
- 16.Abbosh C, Birkbak NJ, Swanton C. Early stage NSCLC - challenges to implementing ctDNA-based screening and MRD detection. Nat Rev Clin Oncol. 2018;15:577–86. [DOI] [PubMed] [Google Scholar]
- 17.Kinde I, Wu J, Papadopoulos N, Kinzler KW, Vogelstein B. Detection and quantification of rare mutations with massively parallel sequencing. Proc Natl Acad Sci U S A. 2011;108:9530–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Newman AM, Lovejoy AF, Klass DM, Kurtz DM, Chabon JJ, Scherer F, et al. Integrated digital error suppression for improved detection of circulating tumor DNA. Nat Biotechnol. 2016;34:547–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Wan JCM, Massie C, Garcia-Corbacho J, Mouliere F, Brenton JD, Caldas C, et al. Liquid biopsies come of age: towards implementation of circulating tumour DNA. Nat Rev Cancer. 2017;17:223–38. [DOI] [PubMed] [Google Scholar]
- 20.Zonta E, Garlan F, Pécuchet N, Perez-Toralla K, Caen O, Milbury C, et al. Multiplex Detection of Rare Mutations by Picoliter Droplet Based Digital PCR: Sensitivity and Specificity Considerations. PLoS ONE. 2016;11:e0159094. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Dressman D, Yan H, Traverso G, Kinzler KW, Vogelstein B. Transforming single DNA molecules into fluorescent magnetic particles for detection and enumeration of genetic variations. Proc Natl Acad Sci U S A. 2003;100:8817–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Milbury CA, Zhong Q, Lin J, Williams M, Olson J, Link DR, et al. Determining lower limits of detection of digital PCR assays for cancer-related gene mutations. Biomol Detect Quantif. 2014;1:8–22. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Corless BC, Chang GA, Cooper S, Syeda MM, Shao Y, Osman I, et al. Development of Novel Mutation-Specific Droplet Digital PCR Assays Detecting TERT Promoter Mutations in Tumor and Plasma Samples. J Mol Diagn. 2019;21:274–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Dong L, Wang S, Fu B, Wang J. Evaluation of droplet digital PCR and next generation sequencing for characterizing DNA reference material for KRAS mutation detection. Scientific Reports. Nature Publishing Group; 2018;8:9650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Vessies DCL, Greuter MJE, van Rooijen KL, Linders TC, Lanfermeijer M, Ramkisoensing KL, et al. Performance of four platforms for KRAS mutation detection in plasma cell-free DNA: ddPCR, Idylla, COBAS z480 and BEAMing. Scientific Reports. Nature Publishing Group; 2020;10:8122. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pietrasz D, Pécuchet N, Garlan F, Didelot A, Dubreuil O, Doat S, et al. Plasma Circulating Tumor DNA in Pancreatic Cancer Patients Is a Prognostic Marker. Clinical Cancer Research. 2017;23:116–23. [DOI] [PubMed] [Google Scholar]
- 27.Garcia-Murillas I, Schiavon G, Weigelt B, Ng C, Hrebien S, Cutts RJ, et al. Mutation tracking in circulating tumor DNA predicts relapse in early breast cancer. Science Translational Medicine. 2015;7:302ra133–302ra133. [DOI] [PubMed] [Google Scholar]
- 28.Schøler LV, Reinert T, Ørntoft M-BW, Kassentoft CG, Árnadóttir SS, Vang S, et al. Clinical Implications of Monitoring Circulating Tumor DNA in Patients with Colorectal Cancer. Clinical Cancer Research. 2017;23:5437–45. [DOI] [PubMed] [Google Scholar]
- 29.Christensen E, Nordentoft I, Vang S, Birkenkamp-Demtröder K, Jensen JB, Agerbæk M, et al. Optimized targeted sequencing of cell-free plasma DNA from bladder cancer patients. Sci Rep. 2018;8:1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Leary RJ, Kinde I, Diehl F, Schmidt K, Clouser C, Duncan C, et al. Development of Personalized Tumor Biomarkers Using Massively Parallel Sequencing. Sci Transl Med. 2010;2:20ra14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Abbosh C, Birkbak NJ, Wilson GA, Jamal-Hanjani M, Constantin T, Salari R, et al. Phylogenetic ctDNA analysis depicts early-stage lung cancer evolution. Nature. 2017;545:446–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.McDonald BR, Contente-Cuomo T, Sammut S-J, Odenheimer-Bergman A, Ernst B, Perdigones N, et al. Personalized circulating tumor DNA analysis to detect residual disease after neoadjuvant therapy in breast cancer. Science Translational Medicine. 2019;11:eaax7392. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Zheng Z, Liebers M, Zhelyazkova B, Cao Y, Panditi D, Lynch KD, et al. Anchored multiplex PCR for targeted next-generation sequencing. Nature Medicine. Nature Publishing Group; 2014;20:1479–84. [DOI] [PubMed] [Google Scholar]
- 34.Coombes RC, Page K, Salari R, Hastings RK, Armstrong A, Ahmed S, et al. Personalized Detection of Circulating Tumor DNA Antedates Breast Cancer Metastatic Recurrence. Clin Cancer Res. 2019;25:4255–63. [DOI] [PubMed] [Google Scholar]
- 35.Christensen E, Birkenkamp-Demtröder K, Sethi H, Shchegrova S, Salari R, Nordentoft I, et al. Early Detection of Metastatic Relapse and Monitoring of Therapeutic Efficacy by Ultra-Deep Sequencing of Plasma Cell-Free DNA in Patients With Urothelial Bladder Carcinoma. J Clin Oncol. 2019;37:1547–57. [DOI] [PubMed] [Google Scholar]
- 36.Reinert T, Henriksen TV, Christensen E, Sharma S, Salari R, Sethi H, et al. Analysis of Plasma Cell-Free DNA by Ultradeep Sequencing in Patients With Stages I to III Colorectal Cancer. JAMA Oncol. 2019;5:1124–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Abbosh C, Frankell A, Garnett A, Harrison T, Weichert M, Licon A, et al. Abstract CT023: Phylogenetic tracking and minimal residual disease detection using ctDNA in early-stage NSCLC: A lung TRACERx study. Cancer Res. American Association for Cancer Research; 2020;80:CT023–CT023. [Google Scholar]
- 38.Heider K, Gale D, Ruiz-Valdepenas A, Marsico G, Sharma G, Perry M, et al. Abstract 735: Sensitive detection of ctDNA in early stage non-small cell lung cancer patients with a personalized sequencing assay. Cancer Res. 2020;80:735–735. [Google Scholar]
- 39.Chin R-I, Chen K, Usmani A, Chua C, Harris PK, Binkley MS, et al. Detection of Solid Tumor Molecular Residual Disease (MRD) Using Circulating Tumor DNA (ctDNA). Mol Diagn Ther. 2019;23:311–31. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Milbury CA, Li J, Makrigiorgos GM. PCR-based methods for the enrichment of minority alleles and mutations. Clin Chem. 2009;55:632–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Salk JJ, Schmitt MW, Loeb LA. Enhancing the accuracy of next-generation sequencing for detecting rare and subclonal mutations. Nat Rev Genet. 2018;19:269–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Shendure J, Ji H. Next-generation DNA sequencing. Nat Biotechnol. 2008;26:1135–45. [DOI] [PubMed] [Google Scholar]
- 43.Clark MJ, Chen R, Lam HYK, Karczewski KJ, Chen R, Euskirchen G, et al. Performance comparison of exome DNA sequencing technologies. Nat Biotechnol. 2011;29:908–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Newman AM, Bratman SV, To J, Wynne JF, Eclov NCW, Modlin LA, et al. An ultrasensitive method for quantitating circulating tumor DNA with broad patient coverage. Nat Med. 2014;20:548–54. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Wan JCM, Heider K, Gale D, Murphy S, Fisher E, Mouliere F, et al. ctDNA monitoring using patient-specific sequencing and integration of variant reads. Sci Transl Med. 2020;12:eaaz8084. [DOI] [PubMed] [Google Scholar]
- 46.Chabon JJ, Hamilton EG, Kurtz DM, Esfahani MS, Moding EJ, Stehr H, et al. Integrating genomic features for non-invasive early lung cancer detection. Nature. 2020;580:245–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Kurtz DM, Soo J, Co Ting Keh L, Alig S, Chabon JJ, Sworder BJ, et al. Enhanced detection of minimal residual disease by targeted sequencing of phased variants in circulating tumor DNA. Nat Biotechnol. 2021;doi: 10.1038/s41587-021-00981-w. [DOI] [PMC free article] [PubMed]
- 48.Zviran A, Schulman RC, Shah M, Hill STK, Deochand S, Khamnei CC, et al. Genome-wide cell-free DNA mutational integration enables ultra-sensitive cancer monitoring. Nature Medicine. 2020;26:1114–24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Lo YMD, Han DSC, Jiang P, Chiu RWK. Epigenetics, fragmentomics, and topology of cell-free DNA in liquid biopsies. Science. 2021;372:eaaw3616. [DOI] [PubMed] [Google Scholar]
- 50.Kim S-T, Raymond VM, Park JO, Zotenko E, Park YS, Schultz M, et al. Abstract 916: Combined genomic and epigenomic assessment of cell-free circulating tumor DNA (ctDNA) improves assay sensitivity in early-stage colorectal cancer (CRC). Cancer Res. American Association for Cancer Research; 2019;79:916–916. [Google Scholar]
- 51.Westesson O, Axelrod H, Dean J, He Y, Sample P, Zotenko E, et al. Abstract 2316: Integrated genomic and epigenomic cell-free DNA (cfDNA) analysis for the detection of early-stage colorectal cancer. Cancer Res. American Association for Cancer Research; 2020;80:2316–2316. [Google Scholar]
- 52.Parikh AR, Van Seventer EE, Siravegna G, Hartwig AV, Jaimovich A, He Y, et al. Minimal Residual Disease Detection using a Plasma-only Circulating Tumor DNA Assay in Patients with Colorectal Cancer. Clin Cancer Res. 2021;doi: 10.1158/1078-0432.CCR-21-0410. [DOI] [PMC free article] [PubMed]
- 53.Chen X, Dong Z, Hubbell E, Kurtzman KN, Oxnard GR, Venn O, et al. Prognostic Significance of Blood-Based Multi-cancer Detection in Plasma Cell-Free DNA. Clin Cancer Res. 2021;doi: 10.1158/1078-0432.CCR-21-0417. [DOI] [PMC free article] [PubMed]
- 54.Burgener JM, Zou J, Zhao Z, Zheng Y, Shen SY, Huang SH, et al. Tumor-Naïve Multimodal Profiling of Circulating Tumor DNA in Head and Neck Squamous Cell Carcinoma. Clin Cancer Res. 2021;doi: 10.1158/1078-0432.CCR-21-0110. [DOI] [PMC free article] [PubMed]
- 55.Armbruster DA, Pry T. Limit of Blank, Limit of Detection and Limit of Quantitation. Clin Biochem Rev. 2008;29:S49–52. [PMC free article] [PubMed] [Google Scholar]
- 56.Pierson-Perry JF, Vaks JE, Durham AP. Evaluation of detection capability for clinical laboratory measurement procedures; approved guideline - second edition. Wayne, Pa., U.S.A: CLSI; 2012. [Google Scholar]
- 57.Alcaide M, Cheung M, Hillman J, Rassekh SR, Deyell RJ, Batist G, et al. Evaluating the quantity, quality and size distribution of cell-free DNA by multiplex droplet digital PCR. Scientific Reports. Nature Publishing Group; 2020;10:12564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Thierry AR, Mouliere F, Gongora C, Ollier J, Robert B, Ychou M, et al. Origin and quantification of circulating DNA in mice with human colorectal cancer xenografts. Nucleic Acids Research. 2010;38:6159–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Jahr S, Hentze H, Englisch S, Hardt D, Fackelmayer FO, Hesch RD, et al. DNA fragments in the blood plasma of cancer patients: quantitations and evidence for their origin from apoptotic and necrotic cells. Cancer Res. 2001;61:1659–65. [PubMed] [Google Scholar]
- 60.Rostami A, Lambie M, Yu CW, Stambolic V, Waldron JN, Bratman SV. Senescence, Necrosis, and Apoptosis Govern Circulating Cell-free DNA Release Kinetics. Cell Reports. 2020;31:107830. [DOI] [PubMed] [Google Scholar]
- 61.Thierry AR, El Messaoudi S, Gahan PB, Anker P, Stroun M. Origins, structures, and functions of circulating DNA in oncology. Cancer Metastasis Rev. 2016;35:347–76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Chaudhuri AA, Chabon JJ, Lovejoy AF, Newman AM, Stehr H, Azad TD, et al. Early Detection of Molecular Residual Disease in Localized Lung Cancer by Circulating Tumor DNA Profiling. Cancer Discov. 2017;7:1394–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.McEvoy AC, Warburton L, Al-Ogaili Z, Celliers L, Calapre L, Pereira MR, et al. Correlation between circulating tumour DNA and metabolic tumour burden in metastatic melanoma patients. BMC Cancer. 2018;18:726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Bettegowda C, Sausen M, Leary RJ, Kinde I, Wang Y, Agrawal N, et al. Detection of circulating tumor DNA in early- and late-stage human malignancies. Sci Transl Med. 2014;6:224ra24. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Azad TD, Chaudhuri AA, Fang P, Qiao Y, Esfahani MS, Chabon JJ, et al. Circulating Tumor DNA Analysis for Detection of Minimal Residual Disease After Chemoradiotherapy for Localized Esophageal Cancer. Gastroenterology. 2020;158:494–505.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Vittori LN, Tarozzi A, Latessa PM. Circulating Cell-Free DNA in Physical Activities. Methods Mol Biol. 2019;1909:183–97. [DOI] [PubMed] [Google Scholar]
- 67.Henriksen TV, Reinert T, Christensen E, Sethi H, Birkenkamp-Demtröder K, Gögenur M, et al. The effect of surgical trauma on circulating free DNA levels in cancer patients—implications for studies of circulating tumor DNA. Molecular Oncology. 2020;14:1670–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Phallen J, Sausen M, Adleff V, Leal A, Hruban C, White J, et al. Direct detection of early-stage cancers using circulating tumor DNA. Sci Transl Med. 2017;9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Thavendiranathan P, Bagai A, Ebidia A, Detsky AS, Choudhry NK. Do blood tests cause anemia in hospitalized patients? The effect of diagnostic phlebotomy on hemoglobin and hematocrit levels. J Gen Intern Med. 2005;20:520–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Chen G, Mosier S, Gocke CD, Lin M-T, Eshleman JR. Cytosine Deamination is a Major Cause of Baseline Noise in Next Generation Sequencing. Mol Diagn Ther. 2014;18:587–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Costello M, Pugh TJ, Fennell TJ, Stewart C, Lichtenstein L, Meldrim JC, et al. Discovery and characterization of artifactual mutations in deep coverage targeted capture sequencing data due to oxidative DNA damage during sample preparation. Nucleic Acids Res. 2013;41:e67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Xie M, Lu C, Wang J, McLellan MD, Johnson KJ, Wendl MC, et al. Age-related cancer mutations associated with clonal hematopoietic expansion. Nat Med. 2014;20:1472–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Steensma DP, Bejar R, Jaiswal S, Lindsley RC, Sekeres MA, Hasserjian RP, et al. Clonal hematopoiesis of indeterminate potential and its distinction from myelodysplastic syndromes. Blood. 2015;126:9–16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Lui YYN, Chik K-W, Chiu RWK, Ho C-Y, Lam CWK, Lo YMD. Predominant hematopoietic origin of cell-free DNA in plasma and serum after sex-mismatched bone marrow transplantation. Clin Chem. 2002;48:421–7. [PubMed] [Google Scholar]
- 75.Genovese G, Kähler AK, Handsaker RE, Lindberg J, Rose SA, Bakhoum SF, et al. Clonal Hematopoiesis and Blood-Cancer Risk Inferred from Blood DNA Sequence. New England Journal of Medicine. 2014;371:2477–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Liu J, Chen X, Wang J, Zhou S, Wang CL, Ye MZ, et al. Biological background of the genomic variations of cf-DNA in healthy individuals. Annals of Oncology. 2019;30:464–70. [DOI] [PubMed] [Google Scholar]
- 77.Razavi P, Li BT, Brown DN, Jung B, Hubbell E, Shen R, et al. High-intensity sequencing reveals the sources of plasma circulating cell-free DNA variants. Nat Med. 2019;25:1928–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Hu Y, Ulrich BC, Supplee J, Kuang Y, Lizotte PH, Feeney NB, et al. False-Positive Plasma Genotyping Due to Clonal Hematopoiesis. Clin Cancer Res. 2018;24:4437–43. [DOI] [PubMed] [Google Scholar]
- 79.Martincorena I, Roshan A, Gerstung M, Ellis P, Van Loo P, McLaren S, et al. High burden and pervasive positive selection of somatic mutations in normal human skin. Science. 2015;348:880–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Martincorena I, Fowler JC, Wabik A, Lawson ARJ, Abascal F, Hall MWJ, et al. Somatic mutant clones colonize the human esophagus with age. Science. 2018;362:911–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Nikolaev SI, Vetiska S, Bonilla X, Boudreau E, Jauhiainen S, Rezai Jahromi B, et al. Somatic Activating KRAS Mutations in Arteriovenous Malformations of the Brain. New England Journal of Medicine. Massachusetts Medical Society; 2018;378:250–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Mäkinen N, Mehine M, Tolvanen J, Kaasinen E, Li Y, Lehtonen HJ, et al. MED12, the mediator complex subunit 12 gene, is mutated at high frequency in uterine leiomyomas. Science. 2011;334:252–5. [DOI] [PubMed] [Google Scholar]
- 83.Pollock PM, Harper UL, Hansen KS, Yudt LM, Stark M, Robbins CM, et al. High frequency of BRAF mutations in nevi. Nat Genet. 2003;33:19–20. [DOI] [PubMed] [Google Scholar]
- 84.Moding EJ, Diehn M, Wakelee HA. Circulating tumor DNA testing in advanced non-small cell lung cancer. Lung Cancer. 2018;119:42–7. [DOI] [PubMed] [Google Scholar]
- 85.Aggarwal C, Thompson JC, Black TA, Katz SI, Fan R, Yee SS, et al. Clinical Implications of Plasma-Based Genotyping With the Delivery of Personalized Therapy in Metastatic Non-Small Cell Lung Cancer. JAMA Oncol. 2019;5:173–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Deveson IW, Gong B, Lai K, LoCoco JS, Richmond TA, Schageman J, et al. Evaluating the analytical validity of circulating tumor DNA sequencing assays for precision oncology. Nature Biotechnology. Nature Publishing Group; 2021;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Sethi H, Salari R, Navarro S, Natarajan P, Srinivasan R, Dashner S, et al. Abstract 4542: Analytical validation of the SignateraTM RUO assay, a highly sensitive patient-specific multiplex PCR NGS-based noninvasive cancer recurrence detection and therapy monitoring assay. Cancer Res. American Association for Cancer Research; 2018;78:4542–4542. [Google Scholar]
- 88.Giobbie-Hurder A, Gelber RD, Regan MM. Challenges of guarantee-time bias. J Clin Oncol. 2013;31:2963–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Tie J, Cohen JD, Wang Y, Li L, Christie M, Simons K, et al. Serial circulating tumour DNA analysis during multimodality treatment of locally advanced rectal cancer: a prospective biomarker study. Gut. 2019;68:663–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Moding EJ, Liu Y, Nabet BY, Chabon JJ, Chaudhuri AA, Hui AB, et al. Circulating tumor DNA dynamics predict benefit from consolidation immunotherapy in locally advanced non-small-cell lung cancer. Nature Cancer. 2020;1:176–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Tie J, Wang Y, Tomasetti C, Li L, Springer S, Kinde I, et al. Circulating tumor DNA analysis detects minimal residual disease and predicts recurrence in patients with stage II colon cancer. Sci Transl Med. 2016;8:346ra92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Tie J, Cohen JD, Wang Y, Christie M, Simons K, Lee M, et al. Circulating Tumor DNA Analyses as Markers of Recurrence Risk and Benefit of Adjuvant Therapy for Stage III Colon Cancer. JAMA Oncol. 2019;5:1710–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Powles T, Assaf ZJ, Davarpanah N, Banchereau R, Szabados BE, Yuen KC, et al. ctDNA guiding adjuvant immunotherapy in urothelial carcinoma. Nature. 2021;1–6. [DOI] [PubMed]
- 94.Yeung CCS, Egan D, Radich JP. Molecular monitoring of chronic myeloid leukemia: present and future. Expert Rev Mol Diagn. 2016;16:1083–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Short NJ, Jabbour E. Minimal Residual Disease in Acute Lymphoblastic Leukemia: How to Recognize and Treat It. Curr Oncol Rep. 2017;19:6. [DOI] [PubMed] [Google Scholar]
- 96.Branford S, Rudzki Z, Parkinson I, Grigg A, Taylor K, Seymour JF, et al. Real-time quantitative PCR analysis can be used as a primary screen to identify patients with CML treated with imatinib who have BCR-ABL kinase domain mutations. Blood. 2004;104:2926–32. [DOI] [PubMed] [Google Scholar]
- 97.Oxnard GR, Thress KS, Alden RS, Lawrance R, Paweletz CP, Cantarini M, et al. Association Between Plasma Genotyping and Outcomes of Treatment With Osimertinib (AZD9291) in Advanced Non-Small-Cell Lung Cancer. J Clin Oncol. 2016;34:3375–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Kim ST, Banks KC, Lee S-H, Kim K, Park JO, Park SH, et al. Prospective Feasibility Study for Using Cell-Free Circulating Tumor DNA–Guided Therapy in Refractory Metastatic Solid Cancers: An Interim Analysis. JCO Precision Oncology. 2017;1:1–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Coakley M, Garcia-Murillas I, Turner NC. Molecular Residual Disease and Adjuvant Trial Design in Solid Tumors. Clin Cancer Res. 2019;25:6026–34. [DOI] [PubMed] [Google Scholar]
- 100.de Gramont A, Van Cutsem E, Schmoll H-J, Tabernero J, Clarke S, Moore MJ, et al. Bevacizumab plus oxaliplatin-based chemotherapy as adjuvant treatment for colon cancer (AVANT): a phase 3 randomised controlled trial. Lancet Oncol. 2012;13:1225–33. [DOI] [PubMed] [Google Scholar]
- 101.Alberts SR, Sargent DJ, Nair S, Mahoney MR, Mooney M, Thibodeau SN, et al. Effect of oxaliplatin, fluorouracil, and leucovorin with or without cetuximab on survival among patients with resected stage III colon cancer: a randomized trial. JAMA. 2012;307:1383–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Conroy T, Hammel P, Hebbar M, Ben Abdelghani M, Wei AC, Raoul J-L, et al. FOLFIRINOX or Gemcitabine as Adjuvant Therapy for Pancreatic Cancer. New England Journal of Medicine. Massachusetts Medical Society; 2018;379:2395–406. [DOI] [PubMed] [Google Scholar]
- 103.Gagliato D de M, Gonzalez-Angulo AM, Lei X, Theriault RL, Giordano SH, Valero V, et al. Clinical impact of delaying initiation of adjuvant chemotherapy in patients with breast cancer. J Clin Oncol. 2014;32:735–44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Biagi JJ, Raphael MJ, Mackillop WJ, Kong W, King WD, Booth CM. Association between time to initiation of adjuvant chemotherapy and survival in colorectal cancer: a systematic review and meta-analysis. JAMA. 2011;305:2335–42. [DOI] [PubMed] [Google Scholar]
- 105.Brannon AR, Jayakumaran G, Diosdado M, Patel J, Razumova A, Hu Y, et al. Enhanced specificity of high sensitivity somatic variant profiling in cell-free DNA via paired normal sequencing: design, validation, and clinical experience of the MSK-ACCESS liquid biopsy assay. bioRxiv. Cold Spring Harbor Laboratory; 2020;2020.06.27.175471. [Google Scholar]
- 106.Verhein KC, Hariani G, Hastings SB, Hurban P. Abstract 3114: Analytical validation of Illumina’s TruSight Oncology 500 ctDNA assay. Cancer Res. American Association for Cancer Research; 2020;80:3114–3114. [Google Scholar]
- 107.Diehl F, Schmidt K, Choti MA, Romans K, Goodman S, Li M, et al. Circulating mutant DNA to assess tumor dynamics. Nature Medicine. 2008;14:985–90. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Sausen M, Phallen J, Adleff V, Jones S, Leary RJ, Barrett MT, et al. Clinical implications of genomic alterations in the tumour and circulation of pancreatic cancer patients. Nat Commun. 2015;6:7686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Khakoo S, Carter PD, Brown G, Valeri N, Picchia S, Bali MA, et al. MRI Tumor Regression Grade and Circulating Tumor DNA as Complementary Tools to Assess Response and Guide Therapy Adaptation in Rectal Cancer. Clin Cancer Res. 2020;26:183–92. [DOI] [PubMed] [Google Scholar]
- 110.Jiang J, Ye S, Xu Y, Chang L, Hu X, Ru G, et al. Circulating Tumor DNA as a Potential Marker to Detect Minimal Residual Disease and Predict Recurrence in Pancreatic Cancer. Front Oncol. 2020;10:1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Peng M, Huang Q, Yin W, Tan S, Chen C, Liu W, et al. Circulating Tumor DNA as a Prognostic Biomarker in Localized Non-small Cell Lung Cancer. Front Oncol. 2020;10:561598. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Olsson E, Winter C, George A, Chen Y, Howlin J, Tang ME, et al. Serial monitoring of circulating tumor DNA in patients with primary breast cancer for detection of occult metastatic disease. EMBO Molecular Medicine. 2015;7:1034–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Garcia-Murillas I, Chopra N, Comino-Méndez I, Beaney M, Tovey H, Cutts RJ, et al. Assessment of Molecular Relapse Detection in Early-Stage Breast Cancer. JAMA Oncol. 2019;5:1473–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Wang Y, Li L, Cohen JD, Kinde I, Ptak J, Popoli M, et al. Prognostic Potential of Circulating Tumor DNA Measurement in Postoperative Surveillance of Nonmetastatic Colorectal Cancer. JAMA Oncology. 2019;5:1118. [DOI] [PMC free article] [PubMed] [Google Scholar]