

Abstract
The BCR comprises a membrane-bound Ig that is noncovalently associated with a heterodimer of CD79A and CD79B. While the BCR Ig component functions to sense extracellular Ag, CD79 subunits contain cytoplasmic ITAMs that mediate intracellular propagation of BCR signals critical for B cell development, survival, and Ag-induced activation. CD79 is therefore an attractive target for Ab and chimeric Ag receptor T cell therapies for autoimmunity and B cell neoplasia. Although the mouse is an attractive model for preclinical testing, due to its well-defined immune system, an obstacle is the lack of cross-reactivity of candidate therapeutic anti-human mAbs with mouse CD79. To overcome this problem, we generated knockin mice in which the extracellular Ig-like domains of CD79A and CD79B were replaced with human equivalents. In this study, we describe the generation and characterization of mice expressing chimeric CD79 and report studies that demonstrate their utility in preclinical analysis of anti-human CD79 therapy. We demonstrate that human and mouse CD79 extracellular domains are functionally interchangeable, and that anti-human CD79 lacking Fc region effector function does not cause significant B cell depletion, but induces 1) decreased expression of plasma membrane-associated IgM and IgD, 2) uncoupling of BCR-induced tyrosine phosphorylation and calcium mobilization, and 3) increased expression of PTEN, consistent with the levels observed in anergic B cells. Finally, anti-human CD79 treatment prevents disease development in two mouse models of autoimmunity. We also present evidence that anti-human CD79 treatment may inhibit Ab secretion by terminally differentiated plasmablasts and plasma cells in vitro.
Key Points
Anti-human CD79A prevents autoimmunity in mice without significant B cell depletion.
Anti-human CD79A induces a transient form of B cell anergy.
Anti-human CD79A may silence Ab production by plasmablasts and plasma cells.
Introduction
Cell surface expression of pre- and mature B cell receptors, as well as development and maturation of B cells in the bone marrow, is dependent on expression of CD79 (1). Similarly, productive signaling of these outcomes by the BCR requires the presence of ITAMs contained within the cytoplasmic domains of CD79A and CD79B (2, 3). Membrane-bound Ig molecules, for example, IgM, IgD, and IgG, pair with heterodimers of CD79A/B via noncovalent transmembrane domain interactions (4) (Fig. 1A). Aggregation of BCRs by multimeric Ags initiates phosphorylation of CD79 ITAM tyrosines, leading to recruitment and activation of proximal tyrosine kinases Lyn and spleen tyrosine kinase (Syk) that nucleate downstream signaling and ultimately drives B cell activation (5).
FIGURE 1.
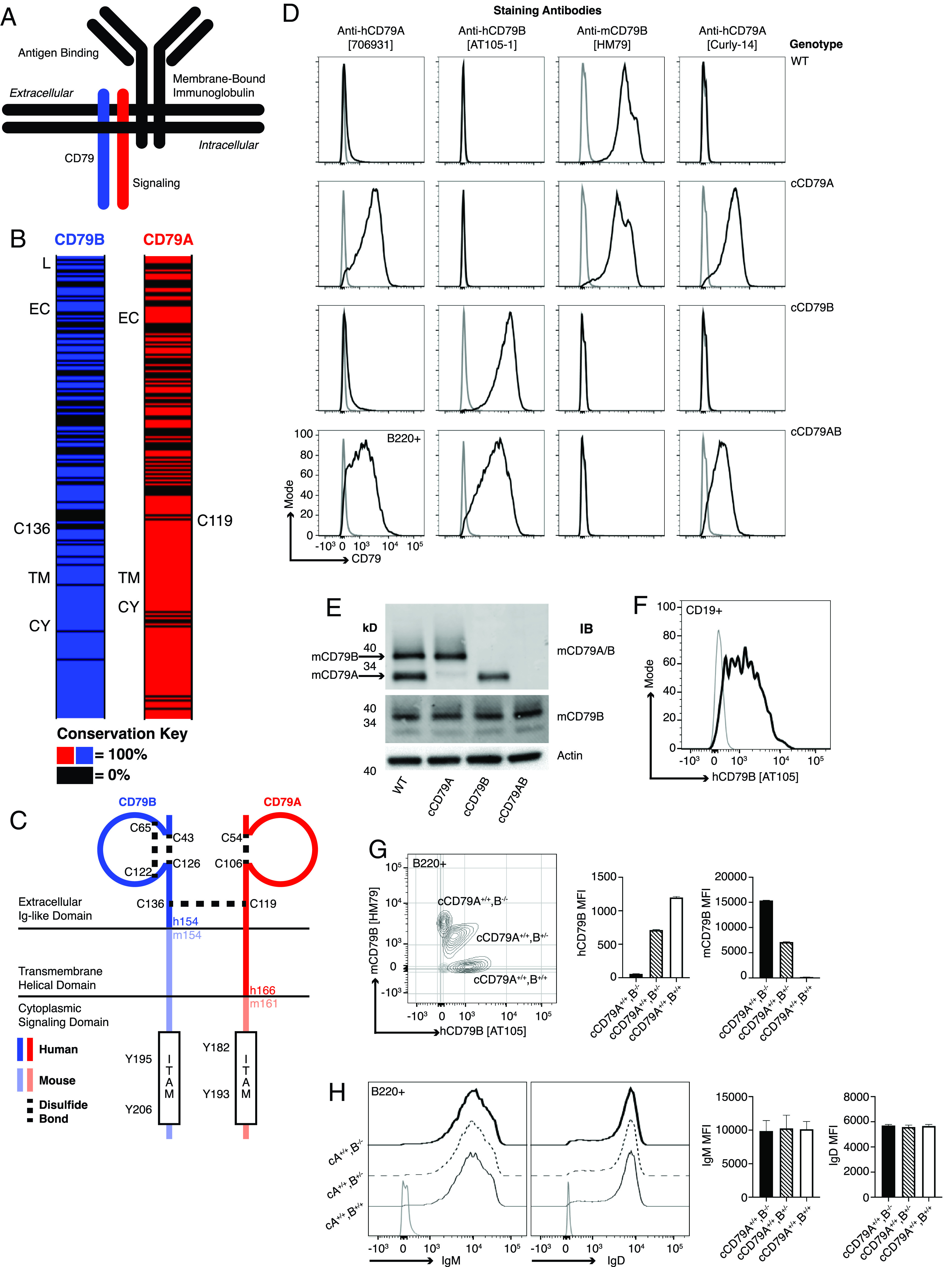
Generation of chimeric human/mouse CD79 knockin mice. (A) Schematic representation of B cell Ag receptor complex. (B) CD79 amino acid conservation between mice and humans. Black intervals represent regions of 0% amino acid conservation. Domain demarcation: CY, cytoplasmic; EC, extracellular; L, leader; TM, transmembrane. Conserved cysteines required for interchain disulfide bonding shown for each. (C) Schematic representation of cCD79. Human sequences comprise the CD79B extracellular domain and CD79A extracellular/transmembrane domains. (D) Surface staining of splenic B cells (B220+) from chimeric (h/m)CD79 knockin mice and control C57BL/6 mice. Gray lines show B220−. (E) Rabbit anti-mCD79 immunoblot analysis of whole-cell lysates (purified splenic B cells, CD43−) from cCD79 and WT mice. Upper membrane probed with a polyclonal rabbit Ab raised against a mCD79A and mCD79B extracellular domain fusion protein (Cambier laboratory). Middle membrane probed with a polyclonal rabbit Ab raised against the cytoplasmic domain of mCD79B (Cambier laboratory). An anti–β actin blot was used as a protein loading control. (F) Surface staining of B cell gated (CD19+) human PBMCs with anti-hCD79B (AT-105). Gray line shows CD19−. (G) Surface staining as a function of cCD79B allele dosage. Splenocytes from cCD79 mice of the indicated genotypes were stained with both anti-hCD79B (AT-105) and anti-mCD79B (HM79). Gray contour shows B220−. (H) IgM and IgD surface expression in chimeric mice described in (G). Gray line shows B220−. n = 4 female mice per group for (G) and (H). Error bars represent SEM. All data represent at least three independent experiments; representative data are shown.
CD79 expression is restricted to B lineage cells. CD79A (mb-1, Ig-α, Igα) and CD79B (B29, Ig-β, Igβ) are single-spanning, transmembrane proteins covalently associated by a disulfide bond (6, 7). The greatest conservation of amino acid sequences between human CD79 (hCD79) and mouse CD79 (mCD79) is found in the transmembrane and cytoplasmic domains (89% in CD79A, 96% in CD79B) (Fig. 1B). Reduced homology in the extracellular domains (57% in CD79A, 55% in CD79B) facilitates mouse immune responsiveness to non–cross-reactive hCD79 epitopes. Such murine Abs can be humanized for therapeutic application.
The potential utility of anti-CD79 Abs for therapeutic intervention has been demonstrated in mouse models of autoimmunity, including collagen-induced arthritis and MRL/lpr lupus (8–10). In 2014, Hardy et al. (9) described a form of polyclonal B cell anergy that can be induced using anti-mCD79 mAb and could be exploited for therapeutic purposes. Selected anti-CD79 Abs act as “reverse agonists,” inducing BCR desensitization. Importantly, mutant mIgG2a Ab lacking the ability to mediate Ab-dependent cellular cytotoxicity (ADCC) and complement-dependent cytotoxicity (CDC) was shown to prevent collagen-induced arthritis, while not inducing B cell depletion. This polyclonal anergy is lost upon decay of the Ab in vivo. Thus, the mechanism of action of ADCC- and CDC-incompetent anti-CD79 is distinct from anti-CD20, which acts by depleting B cells. The utility of B cell–depleting therapy in humans has been demonstrated using several iterations of CD20-directed therapies in indications, including, but not limited to, multiple sclerosis (11, 12), type 1 diabetes (13), rheumatoid arthritis (14), lupus (15), and neuromyelitis optica (16, 17). The success of anergizing CD79-targeted therapies is predicted by these examples. Because they are nondepleting and reversible, anti-CD79 therapies should have improved safety relative to anti-CD20 (18). A murine model in which to conduct preclinical testing of candidate anergizing anti-hCD79 Abs would be an invaluable tool for defining their mechanism of action and potential clinical utility.
In this study, we describe the generation and characterization of hCD79 extracellular domain knockin mice and validation of their use in studying anti-CD79 therapies for potential application in human autoimmune disease. B cell development and function is normal in these mice, and a desensitizing anti-hCD79 Ab treatment blocks pristane-induced production of anti-chromatin autoantibodies. Additionally, prophylactic treatment in the experimental autoimmune encephalomyelitis (EAE) model with non–B cell-depleting anti-hCD79 Ab significantly reduced disease severity and led to complete recovery.
Materials and Methods
Human and animal use
All experiments involving mice were performed in accordance with the regulations and with approval of the University of Colorado Denver Institutional Animal Care and Use Committee. Samples from human subjects were obtained with informed consent prior to inclusion in the study at the University of Colorado Anschutz Medical Center using protocols approved by the University of Colorado Institutional Review Board.
Chimeric CD79A-targeting construct design
Constructs were assembled using bacterial artificial chromosome (BAC) recombineering techniques including Red recombination and cleavage with homing endonuclease I-SceI. In order to generate mice expressing human-mouse chimeric CD79A (cCD79A), targeting constructs were designed in which aa 29–160 of mouse (mCD79A) were replaced by aa 34–166 of human (hCD79A). MCD79A BAC DNA was isolated from RP23-80P21 and RP23-284H16 bacteria and transformed into pGS1783 cells. The hCD79A fragment, aa 34–166 (Fig. 1C), was amplified from human genomic DNA using the following primers: 5′-ACGGTACCTGGATGCACAAGGTCCCAGCATCAT-3′, 5′-TGAATTCATTGAAGACACCCCCTGGGCTCAGCG-3′.
The resulting fragment was digested with KpnI and EcoRI and cloned into the PL452 (loxP-Neo-loxP; G418 selection) vector, digested with the same enzymes. The fragment containing hCD79A-loxP-Neo-loxP was amplified using the following primers: 5′-ACCCATCTGTCTCCTCTCCTCTCTCCACAGGTCCCGGATGCCAGGCCCTGTGGATGCACAAGGTCCCAGCA-3′, 5′-GAGGACACACCTTCTTCCCTTGTCTTGGCTGCATTTGCAGGGGACTCCCGATAACTTCGTATAGCATACATTATACGAAGTTATATTAAGGGTTCCGCAAG-3′.
The resulting PCR product was digested with DpnI and electroporated into pGS1783-RP2380P21 cells. A fragment containing pGK-DTA (negative selection in embryonic stem [ES] cells) was amplified from the pBSKS vector using the following primers: 5′-ACATCGGGCAAAACTTATCACTCCTATCTGCCTCTTTCTTTCTAACCCACCGGGTAGGGGAGGCGCTTTTCCCA-3′, 5′-GGAGTGGAGCTGGACGCAAAGGCGGCCTCTGGCTCGGCTAGAGCGACTCTATCGATGAATTCCTGCAGCCCGGGGGATCC-3′.
The resulting PCR product was digested with DpnI and electroporated into pGS1783-RP23-80P21-hCD79A-loxP-Neo-loxP cells. The final construct was sequence verified, maxi prepared, linearized with ClaI, and purified for ES cell targeting.
cCD79B construct design
To generate mice expressing human-mouse cCD79B, targeting constructs were designed in which aa 29–153 of mCD79A were replaced by aa 29–154 of hCD79B. The hCD79B fragment, aa 29–154 (Fig. 1C), was amplified from human genomic DNA using the following primers: 5′-GCCAGATCGGAGGACCGGTACCGGA-3′, 5′-TCTGCTTCAGCTGTGCCAAGGTGCT-3′.
The resulting PCR product was cloned into the pCR8/GW/TOPO vector. A fragment containing EPKanS was amplified using the following primers: 5′-TCAGTCGACCCCCTCCCTGCATGACTTCCCTCTATCATCCCCCTCTTCCCTCCTGCAGGTCGATTTATTCAACAAAGCCACG-3′, 5′-TCAGTCGACCGCGTATATCTGGCCCGTACATCG-3′.
The resulting product was digested with SalI and cloned into pCR8/GW/TOPO-hCD79B, also digested with SalI. MCD79B BAC DNA was isolated from RP23-386O11 and RP23-141A4 bacteria and then transformed into pGS1783 cells. A fragment containing hCD79B and EPKanS was amplified using the following primers: 5′-ACCTAGCCCTGCCCACTTCCTTCCACCCTCCAGGTGAGCCGGTACCAGCAGCCAGATCGGAGGACCGGTACCGGA-3′, 5′-GATGATGAGGAGGGTCTGGATCAAGATAATGCCATCTTTCAGTGTGTTCCTCTGCTTCAGCTGTGCCAAGGTGCT-3′.
The resulting PCR product, containing the Kan/I-SceI insert, was electroporated into pGS1783-RP23-386O11 cells. The Kan/I-SceI selectable marker was then removed from the mutant BAC DNA. A fragment containing loxP-PGK-Neo-loxP was amplified using the following primers: 5′-CCCAGTCCCTGCAGGAGGGAAGAGGGGGATGATAGAGGGAAGTCATGCAGAAGCTTATAACTTCGTATAATGTATGCTATACGAAGTTATTAGGTCTGAAGAGGAG-3′, 5′-GGCCAAATTCCTAGTCTGTCAACTGAGACCTTCCTCAGTCGACCCCCTCCATAACTTCGTATAGCATACATTATACGAAGTTATATTAAGGGTTCCGCAAG-3′.
The resulting PCR product containing loxP-PGK-Neo-loxP was electroporated into pGS1783-RP23-386O11-hCD79B cells. A fragment containing pGK-DTA was amplified from vector pBSKS using the following primers: 5′-GAGCACCCGACTTCTTTTCCAAAGGTCCGGAGTTCAAATCCCAGCAACCACGGGTAGGGGAGGCGCTTTTCCCA-3′, 5′-TTTAAAAGCAGCTATAAAGTTGGGCTGGGCATGGTCACTCACAGCTGTATATCGATGAATTCCTGCAGCCCGGGGGATCC-3′.
The resulting PCR product containing pGK-DTA was electroporated into pGS1783-RP23-386O11-hCD79B-loxP-PGK-Neo-loxP cells. The final construct was sequence verified, maxi prepared, linearized with ClaI, and purified for ES cell targeting.
ES cell targeting, blastocyst injection, and germline transmission
JM8A3 ES cells (19) were used for independent targeting of cCD79A and cCD79B constructs. Successfully targeted ES cells were selected using neomycin and screened to identify positive homologous recombinant clones using the loss of allele technique as previously described (20). Putative positive clones were independently verified by long-range PCR (Mouse Genetics Core Facility) and by sequencing (Cambier Lab). Positive ES cell clones were injected into blastocysts to create chimeras. The resulting chimeras were bred with C57BL/6J wild-type (WT) mice to establish germline transmission.
Neo cassette removal and establishing cCD79 homozygosity
To remove the loxP-Neo-loxP selection cassette, cCD79A and cCD79B mice were crossed with B6.C-Tg(CMV-cre)1Cgn/J (CMV-cre) mice (The Jackson Laboratory). Resulting mice were PCR screened for cre recombination and cCD79 targeting with the following primers: cCD79A Cre, 5′-CTTGGGAGAATGAAAGCACCCACCA-3′, 5′-TGAGTTCAAGGCCAGCCAGGGCTA-3′; CD79A forward, 5′-GGTACGGCTCCACTCCTGATG-3′; hCD79A reverse, 5′-CATTCTGGATGATCAGCGTAC-3′; mCD79A reverse, 5′-GTGCTGCTCAGCATTCTGGTG-3′; cCD79B Cre, 5′-GTGCTCACTAAGAACAAGGCAGTCG-3′, 5′-CTGCACAGGCTTGAAATCCTCAGTC-3′; CD79B forward, 5′-CGGAAGGAGGAAGTAGCTCTG-3′; hCD79B reverse, 5′-CCTTCAGTGGCCAAGGCTGGA-3′; mCD79B reverse, 5′-ATCCTTAGCCTGTCACCCGAG-3′.
Mice positive for CD79A and CD79B cre recombination generated PCR bands at 322 and 254 bp, respectively. Positive targeting of cCD79A and cCD79B knockin sequences generated bands at 303 and 387 bp, respectively. WT mCD79A and mCD79B generated bands at 590 and 524 bp, respectively. Nonagouti (i.e., black), cre-recombined mice heterozygous for cCD79A or cCD79B were crossed to generate singly homozygous mice expressing cCD79A or cCD79B. To ensure accurate targeting of the cCD79, cDNA was purified from homozygous cCD79A or cCD79B knockin mice and sequenced using the following primers: cCD79A PCR, 5′-ACGGCTGAACAGGAAGTGAG-3′, 5′-AGGGATGCTGGAGTCAGACA-3′; cCD79A sequencing, 5′-CTGGTACCTCAAGGCTACGG-3′; cCD79B PCR, 5′-TCCTTGGGCTCAGAGACAGA-3′, 5′-TGATGGTCCAGCTTCAGACG-3′; cCD79B sequencing, 5′-GCCCAACAGTTGAGCAGGAT-3′.
Transcription of the cCD79 molecules in B cells was first confirmed via PCR analysis of splenic B cell–derived cDNA followed by alignment of sequences with those of the targeting constructs. Singly homozygous mice expressing cCD79A and cCD79B were intercrossed to generate the cCD79A/B doubly homozygous mouse.
Generation of anti-hCD79 Abs
Details about the generation of humanized anti-hCD79 Abs can be found in U.S. patent application 20200109198 “Anti-CD79 Abs and their uses” (21). To generate the surrogate mouse anti-hCD79 Ab (Curly-14), mice were immunized with hCD79 fusion proteins. The extracellular, Ig-like domains of hCD79A and hCD79B, joined by a peptide linker, were expressed via baculovirus and fused to a FLAG tag (Sigma-Aldrich). After expression in Sf9 cells, protein was purified using an anti-FLAG gravity column packed with anti-FLAG resin (Sigma-Aldrich). hCD79 fusion protein was eluted in buffer containing 0.01% sodium azide, PBS, and competing FLAG peptide (Sigma-Aldrich). The mouse deemed “Curly” was immunized i.p. with hCD79 in CFA and IFA according to the following schedule: day 0, CFA + 50 µg of hCD79; day 14, IFA + 50 µg of hCD79; day 24, serum tested for binding to hCD79 by ELISA; day 136, IFA + 50 µg of hCD79; day 199, IFA + 60 µg of hCD79; day 270, 50 µg of hCD79 in PBS. On day 273, after this immunization regimen, Curly was sacrificed and fused using standard hybridoma procedures (22). Curly-14 was humanized, mutated, and affinity matured, generating the anti-hCD79A clinical candidate. hIgG4 anti-hCD79A Abs contain the S228P mutation to stabilize the hinge region and prevent variable light Fab arm exchange. In addition to S228P, Abs were produced with either the D265A or F234A/L235A (“FALA”) mutations to reduce effector function. Experiments presented in the current study were performed with the hIgG4 S228P/D265A anti-hCD79A variant. Anti-hCD79 Abs were provided by Panorama (hIgG4) and Biogen (humanized variable heavy/variable light chimerized to mIgG1 bearing N297Q agly mutation as reduced effector function murine surrogate or mIgG2a as effector-competent comparator).
PBMC isolation
PBMCs were purified using cell prep tubes (Vacutainer CPT) according to the manufacturer’s protocol (BD Biosciences, 362753).
Mice
Unless otherwise noted, 8- to 16-wk-old male and female mice expressing cCD79 (cCD79A, cCD79B, cCD79AB) were used in all experiments. No differences were observed between males and females of any of the three genotypes. Control female, age-matched, WT C57BL/6J mice were purchased from The Jackson Laboratory. Formal nomenclature and informal names include C57BL/6-hCD79AB chimeric mice and ch-hCD79AB or cCD79AB mice, respectively.
Tissue harvesting and B cell isolation
Genomic DNA was prepared from ear clips for PCR and sequence analysis. A 1- to 2-mm ear notch was placed in 50 µl of alkaline lysis reagent (25 mM NaOH, 0.2 mM EDTA, pH ∼12) and incubated at 95°C overnight before neutralizing with 50 µl of 40 mM Tris-HCl (pH ∼5). Lymphocytes were prepared for analysis by multiple means. Spleens and bone marrow were harvested in IMDM supplemented with 5% FCS, 1 mM sodium pyruvate, 50 µg/ml gentamicin, 100 U/ml penicillin/streptomycin, 2 mM l-glutamine, and 5 × 10−5 M 2-ME; single-cell suspensions were prepared by mechanical disruption. RBCs were lysed with 1 ml of ACK (150 mM NH4Cl, 10 mM KHCO3, and 100 mM Na2EDTA) for 1 min at room temperature (RT). Cells were subsequently washed and resuspended in complete medium.
For B cell isolation, spleen cells were harvested, RBC depleted by ammonium chloride Tris lysis, and resuspended in 1 ml of medium together with 40 µl of anti-CD43 MicroBeads (Miltenyi Biotec) per mouse per spleen. The cell/bead mixture was rotated while incubating at 4°C for 10 min. LS MACS separation columns (Miltenyi Biotec) were washed with 5 ml of medium. The cell/bead mixture was flowed over the column, which was then washed with 5 ml of medium while collecting the flowthrough. These preparations were routinely >95% B cells. Serum was prepared from peripheral blood collected from the tail vein.
Flow cytometry
Cells were resuspended at 1 × 107 cells/ml in cold FACS buffer (1% BSA in PBS) containing 0.1% sodium azide. Then, 100-μl aliquots containing 1 × 106 cells were stained with fluorochrome-conjugated mAbs. After staining for 1 h in the dark at 4°C, cells were washed twice and resuspended in 300 µl of FACS buffer. Data were acquired on an LSRFortessa X-20 flow cytometer (Becton Dickinson) and analyzed with FlowJo software.
For intracellular detection of p-Syk and phosphatase and tensin homolog (PTEN), splenocytes were collected from mice that had received 250-µg i.p. injections of either control hIgG4 (VLN3G2), reactive with an irrelevant human carcinoma Ag, or hIgG4 D265A anti-hCD79A 24 h prior to harvest. Cells were rested at 37°C in serum-free medium for 1 h. To measure BCR-mediated induction of Syk phosphorylation, 1 × 106 splenocytes in 1 ml of serum-free medium were stimulated for 5 min with 10 µg/ml goat F(ab′)2 anti-mIgM (Jackson ImmunoResearch). To ensure that there were no interactions between Abs, both resting and stimulated cells were centrifuged at 4500 rpm for 20 s before aspirating supernatant and resuspending in 100 µl of fixation buffer (BD Cytofix, BD Biosciences) for 20 min at 4°C. Cells were washed three times in FACS buffer before being stained with fluorescent Fab anti-mIgG (H+L) and anti-B220. After staining, cells were washed three times in permeabilization buffer (BD Cytoperm, BD Biosciences). Rested and stimulated cells were then stained with anti–p-Syk (Y352) or isotype control Abs in 50 µl of permeabilization buffer. Rested cells were stained with anti-PTEN or isotype control. Cells were washed and data were collected as described above.
Abs
Bone marrow, peripheral blood, and splenic B cells from cCD79 and WT mice were analyzed using the following Abs: hCD79A-PE (R&D Systems, FAB69201P), hCD79B-PE (Abcam, ab33295), mCD79B-AF488 (HM79, made in-house), hCD79A-AF647 (Curly-14, made in-house), B220-BV786 (BD Biosciences, 563894), B220-allophycocyanin (BD Biosciences, 103212), B220-PE (BD Biosciences, 561878), CD43 allophycocyanin-Cy7 (BD Biosciences, 562866), BP1-PE (BD Biosciences, 553735), CD24-allophycocyanin (BioLegend, 101814), IgD-FITC (BioLegend, 405704), IgM-BUV396 (BD Biosciences, 564025), CD19-FITC (BioLegend, 152404), CD93-allopycocyanin (BioLegend, 136510), CD23-FITC (BD Biosciences, 561772), CD1d-PE (BD Biosciences, 553846), CD95-PE (BD Biosciences, 554258), GL7-FITC (BD Biosciences, 553666), Fab anti-mIgG (H+L)-AF647 (Jackson ImmunoResearch, 115-606-072), p-Syk (Y352)-PE (BD Biosciences, 557881), PTEN-PE (BD Biosciences, 560002), and mIgG1 isotype control-PE (BD Biosciences, 559320).
Immunohistochemistry
Spleens were flash-frozen in tissue casts on dry ice containing OCT embedding medium (Tissue-Tek) and stored at −80°C until sectioning. Then, 6-µm sections were cut using a cryostat and thaw-mounted on Superfrost Plus glass slides (Fisher Scientific), which were then air-dried for 3 h at RT and stored at −80°C. Frozen sections were fixed in ice-cold acetone for 5 min, air-dried, rehydrated with PBS, and then blocked for 20 min with PBS + 5% FCS. Blocking buffer was aspirated and sections were stained for 2 h in 100 µl of PBS containing fluorescent Abs. B cells were detected with B220-PE (BD Pharmingen). CD4 T cells were detected with CD4-Cy5 (Caltech). Slides were washed twice before being mounted in Fluoromount-G. Images were recorded on a Nikon Eclipse TE2000-E fluorescence microscope and further analyzed with SlideBook 6 software.
SRBC immunizations
Mice were immunized with SRBCs in accordance with standard procedures (23). One milliliter of citrated sheep’s blood (Colorado Serum) was washed twice in 50 ml of PBS and then resuspended 1:10 in PBS. Mice received 100 µl i.p. of this 0.1% suspension. Spleens were harvested at day 5 postimmunization and B cells were assayed for germinal center markers (CD95, GL7).
Immunoblotting
Isolated B cells were resuspended in serum-free medium and allowed to rest at 37°C for 30 min. B cells (4 × 106) in 1 ml of serum-free medium were stimulated for 5 min with 10 µg/ml rabbit F(ab′)2 anti-mIgG (H+L) (Jackson ImmunoResearch, 315-006-003). Stimulated cells were centrifuged at 4500 rpm for 20 s before aspiration of medium and addition of 30 µl lysis buffer (Nonidet P-40 + inhibitors [PMSF, protease inhibitors, NaVO3, NaF]). Cells were suspended by vortexing and incubated on ice for 10 min. After centrifugation of whole-cell lysates for 15 min, 13,000 rpm, 4°C, the supernatants were collected and to 30 µl, 10 µl of 4× reducing SDS loading buffer was added. Samples were then boiled at 100°C for 10 min and stored at −20°C until use.
Samples were thawed in boiling water before being loaded into a 10% SDS-PAGE Tris-HCl gel. Current was applied for ∼1.5 h at 110 V until the dye front reached the bottom of the gel. The separated proteins were then transferred to a polyvinylidene difluoride (PVDF) membrane using a semidry transfer apparatus and transfer buffer (25 mM Tris [pH 7.5], 192 mM glycine, 20% methanol, and 0.05% SDS). The transfer was run for 1.5 h, 25 V, and 0.15 A per gel.
Protein-transferred membranes were blocked in Odyssey blocking buffer (LI-COR Biosciences) while rocking overnight at 4°C. The next day primary Abs against intracellular signaling molecules, that is, phosphotyrosine (4G10)-AF647, actin (Santa Cruz, sc1616), p-Syk (Cell Signaling Technology, 2711), pIga (Cell Signaling Technology, 5713), and affinity-purified polyclonal rabbit Abs made in-house, including anti-Syk, mCD79B cytoplasmic tail, and mCD79 extracellular domains, were added in blocking buffer and incubated for 2 h while rocking at RT. After washing membranes three times in TBS + 1% Tween 20 for 10 min each, fluorochrome-conjugated secondary Abs, donkey anti-goat IgG IR800 (LI-COR Biosciences), goat anti-rabbit AF680 (Life Technologies), or SA-AF647 (made in-house) were added in blocking buffer and incubated for 2 h while rocking at RT. Membranes were washed three more times before being imaged on the Odyssey (LI-COR Biosciences). Image Studio software (LI-COR Biosciences) was used to quantify Western blot band densities.
Measurement of intracellular Ca2+ concentration
For measurement of relative intracellular Ca2+ concentration ([Ca2+]i), RBC-depleted splenocytes (1 × 107/ml in complete medium containing 2% FCS) were stained with anti-B220 and Fab anti-mIgG (H+L), while being loaded with 5 μM indo-1 acetoxymethyl ester (INDO-1 AM, Thermo Fisher Scientific) according to the manufacturer’s protocols. Both Indo loading and flow cytometry were performed at RT (∼22°C). After being washed once in medium, cells were resuspended at 5 × 106 cells/ml in RT medium in 500-μl aliquots. Indo-1 was excited with a 355-nm UV laser, and Ca2+-bound indo-1 was detected with a 379/28 bandpass filter; unbound indo-1 was detected with a 524/40 bandpass filter. Induced changes in relative [Ca2+]i was determined by calculating the ratio of Ca2+-bound/unbound indo-1 signals over time. After data were acquired for 30 s to establish [Ca2+]i baseline, cells were stimulated with the indicated dose of goat F(ab′)2 anti-mIgM, rabbit F(ab′)2 anti-mIgG (H+L), goat F(ab′)2 anti-hIgM Cµ5, or rat anti-mIgM (B-7-6) (Jackson ImmunoResearch) and data were collected for an additional 2 min 30 s. The relative [Ca2+]i was measured using an LSRFortessa X-20 flow cytometer and analyzed with FlowJo software.
Nitrophenyl-conjugated OVA immunizations
To prepare the immunogen, a 1:1 vol mixture of 1 mg/ml nitrophenyl (NP)4.5 OVA (Biosearch Technologies) and 10 mg/ml alum (Serva; Alu-Gel-S) was incubated, while rotating, for 3 h at RT. A total of 200 µl of the mixture was injected i.p. per mouse to achieve immunization with 100 µg of NP4.5OVA and 1 mg of alum.
NP-Ficoll immunizations
Mice were injected i.p. with 25 µg of NP59-Ficoll (Biosearch Technologies) in 200 µl of PBS. Spleens were harvested on day 7 postimmunization for analysis of NP-specific, IgM+ Ab-secreting cells (ASCs).
Measurement of IgM, IgG, NP-specific IgG, and chromatin-specific IgG
Enzyme immunoassay/RIA 96-well plates (Corning, CLS3590) were coated with 50 µl of 20 µg/ml NP20BSA or NP2BSA (Biosearch Technologies) or 1 µg/ml goat anti-mIgG or goat anti-mIgM (SouthernBiotech) in ELISA capture buffer (PBS + 150 mM NaCl). For detection of anti-chromatin IgG, plates were coated using 50 µl of 10 µg/ml calf chromatin (from Larry Wysocki, National Jewish Health) in ELISA buffer containing 1 mM EDTA. Plates were incubated overnight at 4°C. The following day, wells were aspirated, blocked with 2% BSA in PBS, and incubated overnight at 4°C. Serum was added and serially diluted in 3-fold increments across the plate. The initial serum dilutions used were 1:500 for total IgM, 1:1000 for total IgG, and 1:200 for NP-specific IgG. For detection of IgG anti-chromatin, serum was diluted 2-fold down the plate starting at a serum dilution of 1:20. Plates were incubated overnight at 4°C before being washed three times with PBS + 0.05% Tween 20. Secondary Abs, for respective detection, were added in 100 µl of 1% BSA in PBS + 0.05% Tween 20. These Abs included goat anti–mIgG1-HRP (SouthernBiotech), goat anti–mIgG-HRP (SouthernBiotech), or goat anti–mIgM-HRP (SouthernBiotech). After overnight incubation with secondary Abs, wells were washed and 50 µl of tetramethylbenzidine single solution (Life Technologies) was added for 5 min. Wells were quenched with 50 µl of 1 M H2SO4 before recording absorbance at 450 nm.
NP-specific ELISPOT
ELISPOT plates were prepared as above. For ELISPOT analysis, serial 2-fold dilutions of 1 × 106 splenocytes/100 µl of sample were plated in triplicate. Cells were allowed to settle to the well bottoms while tapping the edges of the plate. Plates were incubated at 37°C for 6 h, then washed three times, for 10 min each, with PBS + 0.05% Tween 20. Next, 50 µl of secondary goat anti–mIgG-alkaline phosphatase (AP) or IgM-AP (SouthernBiotech) in 2% BSA/PBS was added and incubated overnight at 4°C. After washing three times in 0.05% Tween 10/PBS, plates were incubated with ELISPOT development buffer (25 µM 5-bromo-4-chloro-3-indolyl phosphate p-toluidine, 100 mM NaCl, 100 mM Tris, 10 mM MgCl2 [pH 9.5]) for 1 h. The reaction was stopped by washing plates three times with tap water and once in deionized water. The number of spots at a cell dilution was determined. The frequency of NP-specific ASCs was calculated based on spot counts within the linear range.
Tetanus toxoid ASC ELISPOT
Detection of Ag-specific B cells was carried out as previously described (24). Briefly, PBMCs were collected from consenting volunteers at 7–10 d after boost with tetanus, diphtheria, and pertussis vaccine (Tdap). PBMCs (5 × 106) were incubated overnight in 1 ml of medium containing 25 µg of either hIgG4 D265A anti-hCD79A, mouse anti-hCD20 (Panorama), or control hIgG4. Following overnight incubations, cells were washed, counted, and loaded onto ELISPOT plates that had been previously coated with tetanus toxoid (100 µL at 10 µg/ml). Cell suspensions were diluted 2-fold across the plate starting with 1 × 106 cells in the first column. After a 6-h incubation, plates were washed, and ASCs were detected with biotinylated anti-hIgG (H+L) and streptavidin-AP. ELISPOT plates were further developed and analyzed as described above.
Pristane induction of autoantibody formation
Female and male cCD79A knockin mice, aged 8–12 wk, were injected i.p. with 500 µl of pristane (Sigma-Aldrich) on day 0. Weekly injections (250 µg, i.p.) of either hIgG4 anti-hCD79 D265A or isotype control hIgG4 (VLN3G2) (American Type Culture Collection, HB-8636) were given on days 0, 7, 14, 21, 28, 35, 42, 49, and 56. Serum was collected for anti-chromatin autoantibody analysis prior to any injections and on days 30 and 50 and at the conclusion of the experiment at day 64. Pooled sera from diseased systemic lupus erythematosus (SLE)1.2.3 mice (Raul Torres, University of Colorado Anschutz School of Medicine) and serum from WT C57BL/6 mice were used as positive and negative controls, respectively, in IgG anti-chromatin assays.
EAE
EAE was induced by following standard protocols (25). All reagents, including Abs, for this experiment were provided by Biogen. Serum ELISAs and spleen flow cytometry were also performed at Biogen. Briefly, 16-wk-old male and female cCD79A+/+B+/− mice were immunized s.c. with 50 µg of human recombinant myelin oligodendrocyte glycoprotein (hMOG1–125) emulsified in 200 µg of CFA on day 0. Mice also received i.p. injections containing 200 ng of pertussis toxin in PBS on days 0 and 2. Weekly Ab injections were given on days 3, 4, 11, 18, and 25, relative to immunization with hMOG. Treatment arms were blinded, only to be revealed after mice had received scores for the duration of the experiment. Mice received 20 mg/kg i.p. injections of either mIgG2a anti-hCD79, mIgG1 aglycosylated anti-hCD79, or anti–hen egg lysozyme (HEL) isotype controls. Mice were weighed and scored daily using a 5-point scale (0, no disease; 1, complete tail paralysis; 2, hind leg weakness; 2.5, complete paralysis of one hind limb; 3, complete bilateral hind leg paralysis; 4, front limb weakness with complete bilateral hind limp paralysis; 5, complete bilateral hind limb and fore limb paralysis, moribund, or dead). Sera were collected on days 7, 15, 22, and 30 for analysis of anti-MOG IgG and phosphorylated neurofilament H chain (pNfH). At takedown on day 30, spleens were harvested to measure B cell depletion, IgD expression, and other immune cells. Spinal cords were also harvested for histological examination of disease pathology.
For detection of anti-MOG autoantibodies, serum samples were diluted 1:5000 in casein blocking buffer before loading 100 µl onto plates that had previously been coated with 100 µl of 1.0 µg/ml recombinant mouse MOG (R&D Systems, 8536-MO-050). After incubation and washing, anti-MOG Abs were detected with goat anti–mIgG-HRP followed by tetramethylbenzidine substrate. Plates were developed for 5–10 min before quenching with 1.0 N sulfuric acid solution and reading plates at 450 nm.
Neurofilament assays were run as follows: briefly, sera were thawed on ice and then diluted 1:6 by adding 12 µl of serum to 60 µl of dilution reagent from the Simple Plex human NF-H cartridge (ProteinSimple, SPCKB-PS-000519) in a V-bottom 96-well plate. Fifty microliters of each sample was then loaded into the sample wells of the cartridge, and the plate was prepared with wash buffer as per the protocol and then run on the Bio-Techne (ProteinSimple) Ella automated immunoassay system (product no. 600-100).
For flow analysis of spleen cells, RBC-lysed splenocytes were suspended at 5 × 106 cells/ml and fixed by adding 0.5 ml of 6% paraformaldehyde to 1 ml of splenocytes. After a 20-min fixation, cells were washed and frozen. Thawed samples were resuspended in Fc Block (BD Biosciences, 553142) prior to staining with the following Abs: CD45-AF488 (30-F11) (BioLegend, 103122), CD8a-PerCP-Cy5.5 (53-6.7) (BioLegend, 100734), CD4-BV421 (GK1.5) (BioLegend, 100443), CD19-allophycocyanin (1D3) (BioLegend, 152410), B220-BV785 (RA3-6B2) (BioLegend, 103246), Ly6G-AF700 (1A8) (BioLegend, 127622), Ly6C-BV605 (HK1.4) (BioLegend, 128035), CD11b-PE (M1/70) (BioLegend, 101208), and IgD-BV510 (11-26c.2a) (BD Biosciences, 563110). Data were acquired on a Bio-Rad ZE5 flow cytometer and analyzed using FlowJo software.
Tamoxifen induction of PTEN knockout
B cell–specific knockout of PTEN was achieved with a single i.p. injection of 2 mg of tamoxifen, dissolved in corn oil, into hCD20-CreTAM × ROSA26-STOPflox-YFP × PTENflox/flox or control hCD20-CreTAM × ROSA 26 STOPflox YFP × PTENWT mice (26). On day 7 after tamoxifen, mice are injected with anti-CD79 or isotype control Abs. After 18 h, YFP+ B220+ splenic B cells were analyzed on PTEN expression, Ab coating, and BCR-mediated calcium mobilization.
Statistical analysis
GraphPad Prism software was used to generate graphs and perform statistical tests. Unless otherwise noted, statistical significance was calculated by a Student t test to make pairwise comparisons between control and experimental groups as well as between experimental groups. For statistical analysis of EAE disease scores and time to EAE onset, Kruskal–Wallis with a Dunn multiple comparison test and Wilcoxon log-rank test were employed, respectively. Statistical significance was designated as follows: *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
Results
B cells in chimeric human-mouse CD79 knockin mice express fully functional chimeric Ag receptors and undergo normal development
To develop mice expressing extracellular hCD79 domains, the Ig-like domains of mCD79 were replaced by genomic DNA knockin of human homologs, independently creating cCD79A and cCD79B mice (Fig. 1C).
The rationale for construction of cCD79 mice is as follows: epitopes recognized by anti-hCD79 Abs must be expressed without disturbing the signaling capacity of mouse BCRs. Therefore, we replaced only the extracellular portions of mCD79 A and B molecules. Conserved cysteines, and therefore the supportive disulfide bridges, are maintained in the extracellular Ig-like domains of human and mouse CD79A and CD79B. This suggests that heterodimer pairings of cCD79 with endogenous mCD79 proteins should be expressed on the cell surface. This was an important consideration due to the fact that cCD79A and cCD79B knockin mice were made independently, later to be interbred to create doubly homozygous cCD79AB animals. High conservation of mouse and human transmembrane domains suggest uninterrupted association of the cCD79 with mIg in B cell plasma membranes (27). Cytoplasmic domains containing ITAMs are entirely mouse derived. Therefore, interaction of cCD79 with intracellular signaling effectors should be intact. Thus, we hypothesized that these constructions would allow cCD79 binding of anti-hCD79 Abs while preserving interactions among mCD79 chains and membrane-bound Ig. Furthermore, cCD79 should interact faithfully with downstream signaling intermediaries. The former interactions are critical for BCR transport to and expression on the cell surface and the latter for BCR signaling, both required for B cell development and function.
Before engaging in the study of the effects of candidate anti-hCD79 therapeutics, it was important to verify normalcy of cCD79 expression, association with murine partners, and function in B cell development and Ab responses. Knockin mice homozygous for cCD79A, cCD79B, or cCD79A and cCD79B were observed to be viable, fertile, and develop normally with no grossly apparent signs of developmental defect nor immunodeficiency. As depicted in the first two columns of (Fig. 1D, anti-hCD79A (706931) is reactive with cCD79A and cCD79AB mice, but not fully murine CD79 or cCD79B mice. Conversely, anti-hCD79B (AT105-1) stained B cells from cCD79B and cCD79AB mice, but not fully murine CD79 or cCD79A mice. B cells from cCD79A mice also stained with the mouse anti-hCD79 (Curly-14) and the previously described hamster anti-mCD79 (HM79) allowed assignment of their specificities for hCD79A and mCD79B, respectively. Intensity of staining further suggests that the epitope recognized by Curly-14 is contained entirely within the A chain of hCD79. Importantly, the cCD79A homozygous mouse conveniently allows head-to-head comparison of biological effects of the anti-mCD79B and the anti-hCD79A clinical candidate in the same cell.
Loss of the extracellular portions of mCD79 proteins was confirmed by immunoblot analysis of whole-cell lysates (Fig. 1E). B cells (CD43−) from the spleens of cCD79 and WT mice were lysed and subjected to protein fractionation by SDS-PAGE followed by transfer to PVDF membranes. Protein-adsorbed membranes were probed with polyclonal rabbit Abs recognizing mCD79. The upper panel was blotted with affinity-purified polyclonal rabbit Abs reactive with Sf9-expressed mCD79AB extracellular domain heterodimers. Bands representing mCD79A (∼34 kDa) and CD79B (∼37 kDa) are seen in WT B cell lysates. As seen in the cCD79A lane, the rabbit anti-mCD79AB recognizes mCD79B but does not recognize hCD79A. Similarly, this polyclonal Ab does not recognize hCD79B when expressed as a heterodimer with WT mCD79A, or a heterodimer of cCD79A. The middle panel shows the membrane blotted with polyclonal rabbit Ab raised against the mCD79B cytoplasmic domain, the sequence of which is found in both mCD79B and cCD79B, illustrating equivalent lane loading. These data, together with the B cell staining in (Fig. 1D, suggest not only successful expression of chimeric receptors but also successful paring of chimeric and endogenous mCD79 molecules. The presence of B cells in the spleens of cCD79 mice is also suggestive of successful selection, egress from the bone marrow, and survival in the periphery. These observations further demonstrate a lack of polyclonal rabbit Ab cross-reactivity between mouse and human CD79A and CD79B extracellular domains.
To further examine in vivo coexpression of cCD79 and mCD79, we analyzed B cell surface staining in animals homozygous at cCD79A containing no, one, or two copies of the cCD79B encoding gene. Staining of CD19+ human PBMCs by anti-hCD79B (AT105) was first confirmed (Fig. 1F) before costaining cCD79 B cells with anti-mCD79B (HM79) (Fig. 1G). cCD79A+/+cCD79B+/− B cells stained positively for both mouse and human CD79B, with intensities consistent with cCD79B gene dose. This suggests similar efficiency of cCD79A pairing with either mCD79B or cCD79B. However, mCD79B appears to be favored slightly in pairing with cCD79A, as shown by dual staining of mCD79B and hCD79B in cCD79A+/+B+/− cells (Fig. 1G). This is suggested by off-diagonal staining of mCD79B. This finding is consistent with the reduced signal seen in (Fig. 1D; that is, in some B cells hCD79A staining is reduced in cCD79AB B cells relative to cCD79A animals. In contrast, gain of hCD79B staining and coincident loss of mCD79B further demonstrate near equivalent pairing and expression of both chimeric and mCD79 proteins. Near equivalent surface levels of both IgM and IgD on B cells from these genotypes suggests that there is no defect in BCR expression as a consequence of pairing of cCD97 and mCD79 chains (Fig. 1H).
Taken together, these data demonstrate successful generation of the chimeric human-mouse CD79 extracellular domain knockin mice. Furthermore, the ability to detect nearly normal expression of the cCD79 molecules, as well as identical expression of IgM and IgD, demonstrates effective pairing and suggests that no defect exists in B cell development and maturation as a consequence of CD79 extracellular domain swapping. Formal analysis of B cell development is described in Supplemental Figs. 1–4. Briefly, no significant deviations in B cell developmental populations in the bone marrow (i.e., Hardy fractions) or peripheral B cell populations in the spleen were observed in cCD79 mice as compared with C57BL/6 WT animals (Supplemental Figs. 1–3). Additionally, B cell activation in vivo and steady-state serum Ig concentrations are unaffected by expression of cCD79 (Supplemental Fig. 4).
Expression of cCD79 does not affect BCR signaling function
Data presented thus far suggest that function of the murine BCR is unaffected by replacement of CD79A and CD79B extracellular domains with their human counterparts; B cell development and survival are dependent on surface expression and tonic BCR signaling (28–30). In order to properly assess effects of cCD79 on BCR signaling, we measured tyrosine phosphorylation of membrane-proximal signaling effectors and calcium mobilization following receptor stimulation.
Initial steps in BCR signaling include the tyrosine phosphorylation of Lyn, Syk, Btk, PLCγ, and, most proximally, CD79 receptor components on which conserved tyrosine residues in ITAMs are phosphorylated to initiate downstream signaling (31). Phosphorylation of these signaling intermediaries propagates the BCR signal, leading to the eventual activation of the B cell. To assess total BCR-mediated tyrosine phosphorylation in cCD79 animals, we prepared whole-cell lysates of splenic B cells that had been stimulated in vitro. B cells from WT and cCD79 animals were purified by CD43 negative selection and subjected to BCR stimulation with F(ab′)2 fragments of rabbit anti-mIg containing reactivity to H chains and L chains (H+L), and therefore reactive with all BCR. Whole-cell lysates from stimulated B cells were fractionated by SDS-PAGE and transferred to PVDF membranes, which were subsequently blotted with phosphotyrosine/context-dependent Abs, that is, p-Tyr (Fig. 2A), p-CD79A (Fig. 2B), and p-Syk (Fig. 2C). Whole-cell lysates from resting cells demonstrate equivalent levels of basal phosphorylation. Membranes were reprobed with anti-actin, anti-mCD79B, or anti-Syk Abs, respectively, to control for equivalent loading. Images obtained from these blots demonstrate a similar degree of tyrosine phosphorylation in cCD79 B cells compared with WT. This includes global phosphotyrosine (Fig. 2A), p-CD79A (Fig. 2B) and, p-Syk (Fig. 2C).
FIGURE 2.
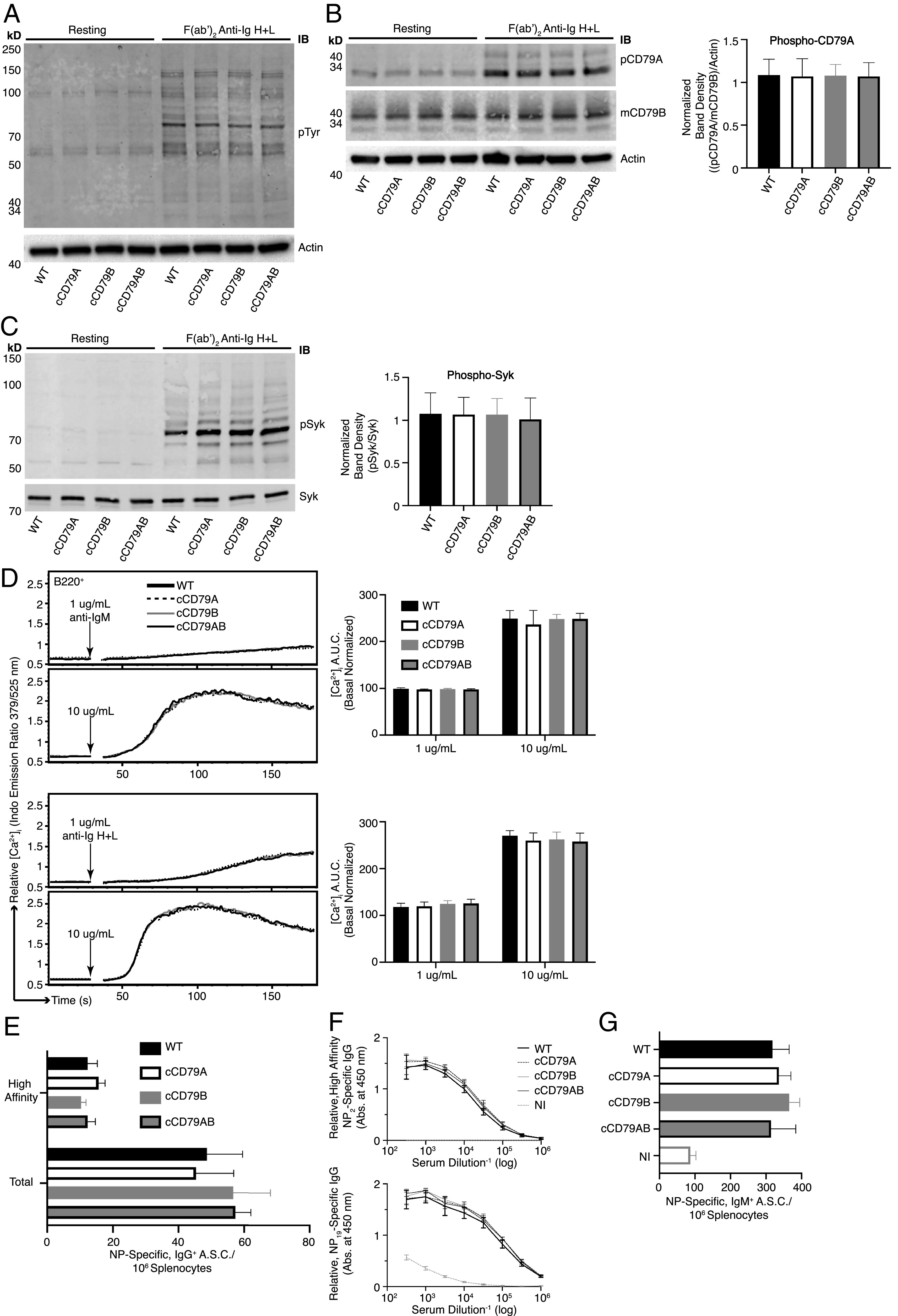
BCR signaling and B cell immune responses are unaffected by expression of cCD79. (A) BCR-mediated global tyrosine phosphorylation in chimeric and control B cells. Cell equivalents (2 × 106; CD43−) per lane, resting or stimulated with 10 µg/ml rabbit F(ab′)2 anti-mIg (H+L) for 5 min. Unstimulated cells were run in parallel (left four lanes) to show basal phosphorylation levels. Protein-laden PVDF membranes probed with Abs against p-Tyr (4G10) and actin. (B) BCR-mediated CD79A phosphorylation. Prepared as in (A), probed with anti–p-CD79A (Y182) (rabbit polyclonal anti-mouse pCD79A [Y182]). An anti-mCD79B (see (Fig. 1E), which recognizes the cytoplasmic tail of mCD79B, was used together with actin to normalize the relative abundance of phosphorylated mCD79A (p-CD79A/p-CD79B/actin). Normalized band densities are depicted to the right. (C) BCR-mediated Syk phosphorylation. Prepared as in (A), probed with anti–p-Syk (Y252) (polyclonal rabbit anti–p-Syk [Y525/526]) and biotinylated anti-Syk (in-house) followed by fluorescently conjugated streptavidin. Relative, normalized band densities (pSyk/Syk) are depicted to the right. (D) Representative relative intracellular free calcium before and after BCR stimulation. Splenocytes, stained with anti-B220 and loaded with Indo-1 AM, were stimulated with 1 or 10 µg/ml F(ab′)2 of either goat anti-mIgM (upper traces) or rabbit anti-mIg (H+L) (lower), approximating IgM-only and total BCR stimulation, respectively. Poststimulation, basal-normalized area under the curve (AUC) is depicted on the right. (E) Total (NP19-binding) and high affinity (NP2-binding) IgG+ ELISPOT quantification, 16 d postimmunization with NP-conjugated OVA in alum. (F) Relative serum concentrations of IgG anti-NP Abs from mice immunized in (E) at 16 d postimmunization. NI, not immunized. (G) Total (NP19 binding) IgM+ ELISPOT quantification, at 7 days postimmunization with NP59-Ficoll. n = 3 male and 3 female mice per group. Error bars show SEM. All data represent at least three independent experiments; representative data are shown.
Calcium mobilization, along with parallel activation of other downstream pathways, is important in BCR signaling (32). Tyrosine phosphorylation leads to activation of PLCγ, which cleaves phosphatidylinositol 4,5-bisphosphate (PIP2) into diacylglycerol (DAG) and inositol 1,4,5-trisphosphate (IP3). Stimulation of IP3 receptors in the endoplasmic reticulum membrane initiates release of intracellular calcium stores, and this activates entry of extracellular calcium. Ultimately, calcium-dependent nuclear translocation of the transcription factor NFAT contributes significantly to B cell activation. Thus, elevation of [Ca+2]i in BCR-stimulated cells is a simple measure of complex signaling events that occur proximal to the receptor. To assess calcium mobilization in B cells expressing cCD79, we recorded fluorescent emissions of an intracellular calcium reporter (INDO-1) before and after stimulation with Abs recognizing membrane Igs. Splenocytes from cCD79 and WT control animals were simultaneously stained for B cells (B220) and loaded with the calcium-sensing dye. When bound to cytoplasmic Ca2+ ions, the emission of INDO shifts from 525 (unbound) to 379 nm. A ratio of 379/525 is used as a measure of relative [Ca+2]i. After 30 s of data acquisition, to establish basal [Ca+2]i, cells are stimulated with anti-BCR Abs (e.g., anti-IgM, IgG) and [Ca+2]i is recorded for an additional 150 s. As seen in (Fig. 2D, stimulation of cCD79-containing BCRs results in similar amplitude changes in [Ca2+]i, time to maximal calcium, and attenuation of the calcium signal. These observations suggest that circuitry involved in both activation and regulation of BCR signaling is operative in cCD79 B cells.
Taken together, these data demonstrate normal signaling functionality of the BCRs comprising mIgs and human/mouse cCD79A and cCD79B. To further interrogate BCR function in cCD79 animals, we analyzed B cell Ab responses following immunization.
WT and cCD79 mice respond equivalently to immunization with thymus-dependent and thymus-independent immunogens
B cell immune responses to both thymus-dependent (TD) and thymus-independent (TI) immunogens, as measured by generation of ASCs and serum Abs, reflect a complex orchestration of both intercellular and intracellular interactions (33). To test the capabilities of cCD79-containing BCRs to participate in these responses, we first immunized mice i.p. with NP4.5-conjugated OVA, in alum, and measured the primary immune response via ELISPOT and ELISA (Fig. 2E, 2F). At takedown, 16 d after immunization with NP-Ova + alum, we observed equivalent numbers of both total and high-affinity IgG ASCs specific for NP (Fig. 2E). Similarly, accumulated IgG Ab in peripheral blood serum of immunized animals was found to be relatively identical between control and humanized animals (Fig. 2F). We also measured responses that are independent of T cell help, which rely more heavily on the signaling efficiency of the BCR. To this end, we immunized control and knockin animals with high-avidity NP59-conjugated Ficoll. Seven days later, near equivalent numbers of IgM anti-NP ASCs were seen in all groups of immunized animals (Fig. 2G).
In conclusion, in addition to functioning at the level of molecular signaling, activation of cCD79+ B cells results in immune responses that are indistinguishable from WT animals. Confirmation of B cell functionality in cCD79-expressing animals permitted us to explore the treatment effects of candidate anti-huCD79 therapeutic Abs in vivo. This includes effects of treatment on BCR signaling, immune responsiveness, and modulation of murine autoimmune disease models.
In vivo treatment with anti-hCD79 Ab induces changes similar to those observed in anergic B cells
Ultimately, cCD79 mice could serve as an important tool for validation of therapeutic Abs and chimeric Ag receptor T cells targeting hCD79. Thus, we wanted to explore the biologic activity of anti-hCD79A. Both human peripheral B cells (CD19+) (Fig. 3A, left) and Ramos cells (Fig. 3A, middle) were recognized by the Curly-14 anti-hCD79A Ab. At the same dilution of staining Ab, Curly-14 bound cCD79A B cells, which are dually stained with anti-mCD79B (HM79) (Figs. 1D and 3A, right). The mouse anti-hCD79A monoclonal (Curly-14) was further humanized (hIgG4), Fc mutated (S228P/D265A or S228P/F234A/L235A), and affinity matured in preparation for preclinical analysis. The candidate hIgG4 S228P/D265A anti-hCD79A, referred to as hIgG4 D265A anti-hCD79A, was used for all experiments presented herein. This mAb was first validated based on its ability to bind cCD79A B cells in vivo (Fig. 3B). cCD79A mice were injected i.p. with 250 µg of either hIgG4 D265A anti-hCD79A, isotype control hIgG4 (VLN3G2), or hamster anti-mCD79B (HM79). After 18 h, splenic B cells (B220+) were analyzed by flow cytometry for staining with anti-hCD79A (Curly-14-AF647) and anti-hIgG4 (Fig. 3B). The first panel of (Fig. 3B shows that previous injection with hIgG4 D265A anti-hCD79A completely blocks binding of Curly-14, the parental anti-hCD79A; neither HM79 nor control hIgG4 blocks Curly-14 binding. The second panel of (Fig. 3B shows coating of B cells with hIgG4 in mice previously injected with hIgG4 D265A anti-hCD79A but not in mice injected with hIgG4 isotype or HM79. These data confirm that Curly-14’s specificity is maintained in the humanized clinical candidate. The first observable in vivo effects of the anti-hCD79A clinical candidate are seen at the B cell surface in splenocytes taken from cCD79A mice injected i.p. with 250 µg of Ab 18 h prior (Fig. 3C). In comparison with hIgG4 isotype–treated animals, anti-hCD79A treatment leads to a highly significant decrease in both IgM and IgD on the surface of splenic B cells. This is similar to the observations of Hardy et al. (as shown in figure 5B in Ref. 9) using surrogate anti-mCD79B. This effect is different from that seen upon induction of B cell anergy, wherein cells express normal levels of IgD and decreased levels of IgM on their surface. Given the confirmation of binding and the initial cues from in vivo biologic activity, we wanted to further characterize BCR signaling in animals treated with anti-hCD79A.
FIGURE 3.
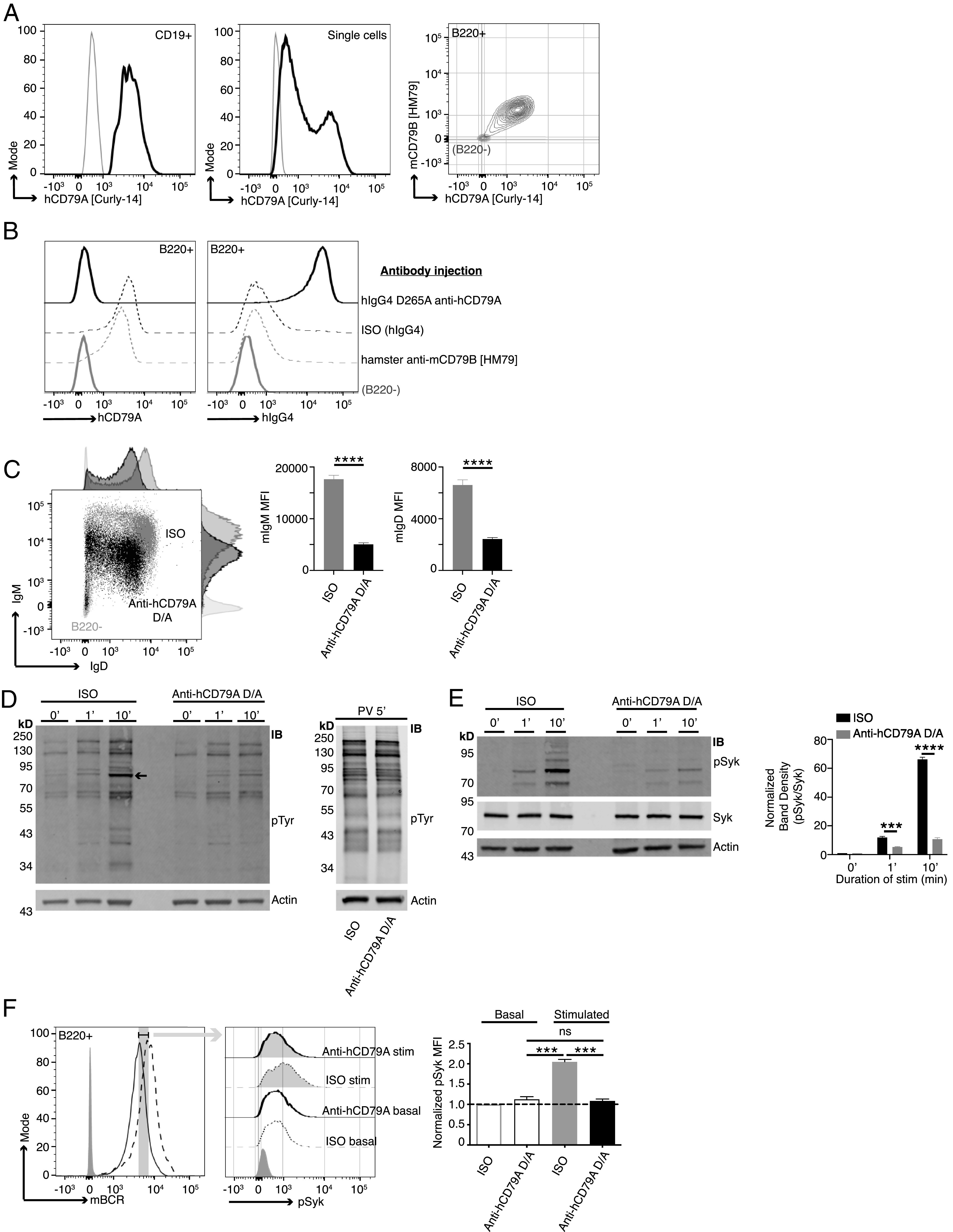
Anti-hCD79A treatment induces an anergic-like phenotype in B cells. (A) Surface staining of PBMCs (left) and Ramos cells (middle) with anti-hCD79A (Curly-14). Dual staining of cCD79A+/+B−/− knockin splenocytes (right) with anti-hCD79A (Curly-14) and anti-mCD79B (HM79). Gray lines indicate CD19−, ns, and B220− for PBMCs, Ramos cells, and cCD79A knockin splenocytes, respectively. (B) Binding of the humanized anti-hCD79A clinical candidate to cCD79A/cCD79B cells. Eighteen hours prior to assay, cCD79A knockin mice received 250-µg i.p. injections of either hIgG4 D265A anti-hCD79A (solid black line), hIgG4 isotype control (dashed black line), or hamster anti-mCD79B (HM79) (dashed gray line). RBC-lysed splenocytes were stained with anti-B220 and either anti-hCD79A (Curly-14) (left) or anti-hIgG4 (right). Solid gray lines represent B220−. (C) Surface expression of IgM and IgD on cCD79 B cells (B220+) 18 h after i.p injection with 250 µg of either hIgG4 D265A anti-hCD79A (black) or isotype control hIgG4 (gray). Light gray histograms show B220−. (D) Western blot analysis of global tyrosine phosphorylation. Eighteen hours prior to assay, cCD79A mice were injected i.p. with 250 µg of either anti-hCD79A D265A or hIgG4 isotype control. Splenic B cells were purified by CD43 exclusion and stimulated with 10 µg/ml F(ab′)2 goat anti-mIgM for the indicated number of minutes (left) or stimulated with 1× pervanadate for 5 min (right). Whole-cell lysates of 2 × 106 B cell (CD43−) equivalents, both stimulated and not, were separated by SDS-PAGE and transferred to PVDF membranes. Protein-transferred membranes were blotted with Abs against p-Tyr (4G10) and actin. (E) Western blot analysis of Syk phosphorylation. Eighteen hours prior to assay, cCD79A mice were injected i.p. with 250 µg of either anti-hCD79A D265A or hIgG4 isotype control. Splenic B cells were purified by CD43 exclusion and stimulated with 10 µg/ml F(ab′)2 goat anti-mIgM for the indicated number of minutes. Whole-cell lysates of 2 × 106 B cell (CD43−) equivalents, both stimulated and not, were separated by SDS-PAGE and transferred to PVDF membranes. Protein-transferred membranes were blotted with Abs against p-Syk (Y525), total Syk, and actin. A densitometric summary of p-Syk/Syk is shown on the right. (F) Flow cytometric analysis of Syk phosphorylation as a function of mBCR expression. Mice were treated as in (E) before RBC-lysed splenocytes were stimulated as above for 5 min. Fixed cells were stained with fluorescent Abs against B220 and mBCR. After permeabilization, cells were stained with anti–p-Syk (Y525). B cells were gated on equivalent mBCR expression (left) before comparing p-Syk mean fluorescence intensities (MFIs) (middle and right). Error bars show SEM. All data represents at least three independent experiments; representative data are shown. A Student t test used to evaluate statistical significance. ***p < 0.001, ****p < 0.0001.
To interrogate membrane-proximal BCR signaling events in anti-hCD79A–treated animals, we first assayed global BCR-induced tyrosine phosphorylation by Western blot (Fig. 3D). As before, 18 h prior to the assay, cCD79A mice received 250-µg i.p. injections of either hIgG4 D265A anti-hCD79A or control hIgG4. Splenic B cells were harvested via CD43 negative cell selection and rested in serum-free medium at 37°C for 1 h before stimulation with either goat F(ab′)2 anti-mIgM or pervanadate. Whole–B cell lysates were fractionated by SDS-PAGE and electrophoretic transfer to PVDF membranes. These were blotted with anti–p-Tyr (4G10) and anti-actin. Unstimulated cells (0′) serve to demonstrate basal tyrosine phosphorylation. Whereas actin blots control for equal protein loading, stimulation with the protein-tyrosine phosphatase inhibitor pervanadate controls for kinase activity and substrate availability (Fig. 3D, right). Most notably, global BCR-mediated tyrosine phosphorylation is impaired in B cells from anti-hCD79A–treated animals. The arrow-indicated band between the 70 and 95 kDa markers most likely represents Syk and appears to be significantly diminished in the corresponding anti-hCD79A–treated lanes, at 1 and 10 min after BCR stimulation. To this end, identical sets of cell lysates were used to confirm Syk phosphorylation directly (Fig. 3E). Having confirmed that BCR-induced Syk phosphorylation is significantly reduced in anti-hCD79A–treated B cells, we next wanted to reconcile this signaling defect with decreased mIgM and mIgD expression. To do this we harvested spleens from cCD79A mice, treated as before, and stimulated RBC-lysed splenocytes with goat F(ab′)2 anti-mIgM for 5 min. Cells were immediately fixed before surface staining with anti-B220 and a nonstimulating, fluorescently conjugated Fab anti-mIg (H+L). This method of staining allowed us to gate on cells bearing equivalent levels of mBCRs while analyzing Syk phosphorylation. Cells that had been fixed and surface stained were permeabilized and stained for intracellular p-Syk (Y525). As shown in (Fig. 3F, when we normalized for mBCR surface expression, anti-hCD79A–treated cells showed decreased phosphorylation of Syk in response to IgM stimulation.
Considering the above observation, we next analyzed calcium mobilization as a function of BCR surface expression. As before, cCD79A animals received an anti-hCD79A i.p. injection 18 h prior to spleen excision. RBC-lysed splenocytes were stained with anti-B220 and Fab anti-mIg (H+L) while being loaded with the ratiometric calcium indicator, INDO. On a UV laser-equipped flow cytometer, indo emissions were collected for 30 s to establish relative basal intracellular calcium concentrations, before stimulating cells with either 1 or 10 µg/ml rat anti-mIgM (B76) and acquiring data for an additional 150 s. Similar to Syk phosphorylation, when we analyzed BCR-induced calcium mobilization by first gating on cells bearing equivalent mBCRs, the suppressive effects of anti-hCD79A treatment remained (Fig. 4A). Ionomycin stimulation induces calcium flux in anti-hCD79A–treated cells to the level of isotype control–treated cells (Fig 4B). BCR-mediated calcium mobilization is also suppressed in Ramos B cells after overnight culture with hIgG4 D265A anti-hCD79A (Fig 4C). Taken together, these observations imply that the inhibition of BCR signaling in anti-hCD79A–treated B cells is, at least partially, independent of the coincident reduction in mBCRs and suggests that regulatory circuitry and/or receptor destabilization may be elicited by the treatment (34–37).
FIGURE 4.
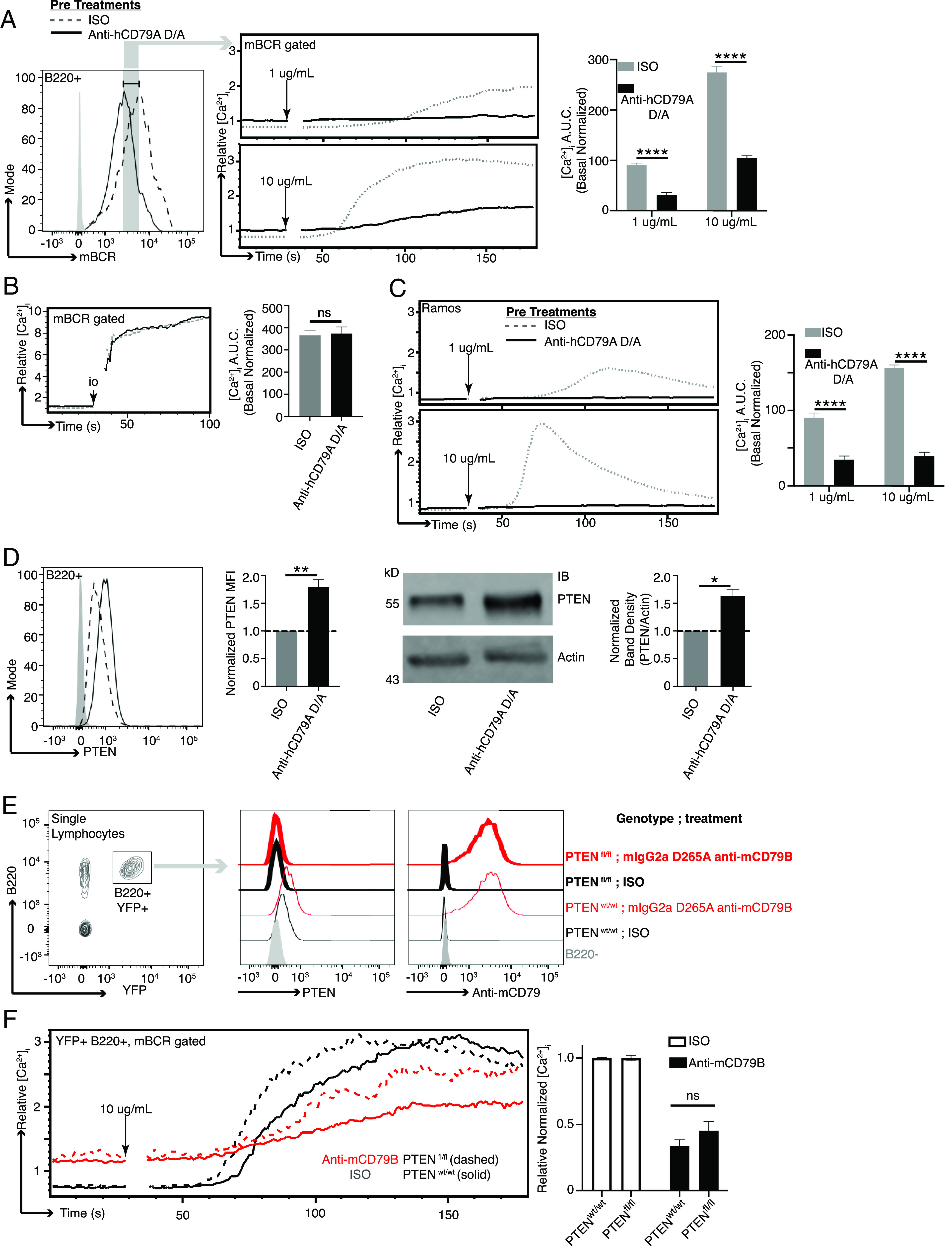
Anti-hCD79A treatment induces suppression of BCR-mediated calcium mobilization. (A) BCR-mediated calcium signaling in B cells from cCD79A mice receiving a 250-µg i.p. injection of anti-hCD79A D265A or control hIgG4 24 h prior. mBCR expression (left) was measured and gated by staining with a polyclonal goat Fab anti-mIgG (H+L). Anti-hCD79A, solid line; control hIgG4, dashed line. Cells were restimulated with 1 or 10 µg/ml rat anti-mIgM (B76). The poststimulation area under the curve (AUC) was normalized to basal calcium levels (right). (B) Cells prepared as in (A) stimulated with 10 µM ionomycin. (C) BCR-mediated calcium signaling in Ramos cells after overnight in vitro incubation with 25 µg/ml hIgG4 anti-hCD79A D265A (solid line) or isotype control hIgG4 (dashed line). Cells were restimulated with either 1 or 10 µg/ml goat F(ab′)2 anti-hIgM Cµ5. (D) Flow cytometric (left and mean fluorescence intensity [MFI] bar graph) and Western blot (right and densitometry) analysis of PTEN expression in B cells from cCD79A animals receiving a 250-µg i.p. injection of either anti-hCD79A D265A or control hIgG4, 24 h prior. For flow cytometry, RBC-lysed splenocytes were fixed and permeabilized before staining with Abs against B220 and PTEN. Gray histogram represents staining isotype control Ab. hIgG4, dashed line; anti-hCD79A D265A, solid line. For Western blot, membranes were prepared as in (Fig. 3D, without BCR stimulation, before being probed with Abs against PTEN or actin. (E) hCD20-CreTAM × ROSA26-STOPflox-YFP × PTENflox/flox or hCD20-CreTAM × ROSA-26-STOPflox-YFP × PTENWT mice were given 2-mg i.p. injections of tamoxifen (TAM) on day 0. On day 7 after TAM, mice were injected i.p. with 0.25 mg of either mIgG2a D265A anti-mCD79B or control mIgG2a anti-HEL. Eighteen hours after Ab injection, B220+YFP+ splenocytes were analyzed by flow for PTEN expression and Ab coating. (F) As before, calcium was measured as a function of equal mBCR expression. n = 3 mice per group. Error bars show SEM. All data represents at least three independent experiments; representative data are shown. A Student t test was used to evaluate statistical significance. *p < 0.05, **p < 0.01, ****p < 0.0001.
Of particular interest in the study of anergic B cells, and their activity in health and disease, is the inhibitory phosphatase PTEN (38). PTEN is an inositol lipid phosphatase that opposes the activity of PI3K and therefore regulates BCR signaling. PTEN can be observed at increased levels in anergic (BND) cells in humans and mice, and its dysregulation, that is, reduction, is associated with autoimmune pathogenesis (26, 38–40). Furthermore, targeted overexpression of PTEN in mouse B cells causes refractory BCR signaling and inhibition of B cell immune responses in vivo (26). For these reasons we assayed intracellular PTEN expression in B cells taken from animals injected with anti-hCD79A. (Fig. 4D demonstrates increased PTEN expression in cCD79A B cells at 18 h postinjection. Importantly, the level of increase is consistent with those seen in human anergic B cells and in mouse models of anergy. To formally test the contribution of PTEN to anti-CD79–induced anergy, we used a conditional and tissue-specific mouse model of PTEN deletion, that is, hCD20-CreTAM × ROSA26-STOPflox-YFP × PTENflox/flox (or PTENWT). These mice do not express hCD79 extracellular domains, which required that we use the surrogate anti-mCD79B Ab. Mice were first injected i.p. with 2 mg of tamoxifen on day 0 to activate CRE and delete PTEN (26). On day 7, when intracellular PTEN becomes undetectable, mice were injected i.p. with 250 µg of either mIgG2a D265A anti-mCD79B or control mIgG2a anti-HEL. After 18 h, we analyzed PTEN expression, Ab coating, and calcium mobilization in B220+YFP+ splenocytes. (Fig. 4E confirms the PTEN knockout was successful in both treated and control-treated animals. An anti-CD79–induced PTEN increase in WT animals can also be seen. Furthermore, we demonstrated that B cells in anti-mCD79B–treated animals are coated with the therapeutic as shown by surface staining with an anti-mIgG2a. When these cells are subjected to BCR stimulation and calcium analysis, we see that the removal of PTEN leads to increased maximal intracellular calcium in B cells from anti-mCD79B–treated animals but not isotype control–treated animals. This noted loss of PTEN does accelerate response kinetics in isotype control–treated B cells (Fig. 4F). These findings suggest that PTEN elevation plays a role in the reduced responsiveness of B cells exposed to anti-mCD79B in vivo. Preliminary observations in SHP1 KO and SHIP-1 KO experiments showed no requirement for these molecules in anti-mCD79B modulation of BCR signaling (S.M. Wemlinger and J.C. Cambier, unpublished observations). These results led us to believe that anti-CD79–induced desensitization of the BCR may involve multiple, partially redundant mechanisms in addition to receptor modulation. Moving forward, we wanted to investigate the effects of anti-hCD79A on B cell immune responses.
Anti-hCD79 treatment modulates primary B cell Ab responses to Ag and reduces Ab secretion by terminally differentiated plasmablasts and plasma cells
The ability to inhibit a TD Ab response to immunization with NP-conjugated OVA was previously demonstrated in Hardy et al.’s (9) anti-mCD79B experiments. In the current study, we chose to first assess the impact of anti-hCD79 treatment on TI Ab responses, which rely on high-valency BCR stimulation without T cell help. Ags with highly repetitive BCR epitopes are capable of stimulating B cells to produce low-affinity IgM Abs that provide early protection from infection. To test the impact of anti-hCD79A treatment in this regard, cCD79A animals received either hIgG4 D265A anti-hCD79A or control hIgG4 24 h before being immunized with NP59-Ficoll. (Fig. 5A shows a significant reduction (∼80%) in the number of NP-specific IgM+ ASCs detectable at 7 d postimmunization in animals injected with anti-hCD79A. The development of high-affinity IgG Abs is a function of complex cellular interactions and processes that take place within germinal centers. These are specialized structures within secondary immune tissues that promote B cell proliferation, class switching, and affinity maturation of their BCRs (41). Before confirming the findings involving anti-mCD79B, we looked at the effect of anti-hCD79A treatment on germinal center formation. To accomplish this, we treated cCD79A mice as before and then immunized with SRBCs (23). At 5 d postinjection, splenic B cells from immunized mice were analyzed by flow cytometry for germinal center B cell markers CD95/Fas and GL7. (Fig. 5B shows significant reduction (>50%) in both the proportion and number of splenic B cells expressing these markers. These results provide at least a partial explanation for the ability of anti-CD79 to inhibit TD Ab responses. To confirm and translate the effects of anti-mCD79B on TD Ab responses, we immunized cCD79A mice, injected 24 h prior with anti-hCD79A or control Ab, with NP-OVA in alum and enumerated NP-specific ASCs 16 d later. (Fig. 5C shows a significant reduction (∼60%) of IgM+ anti-NP ASCs found in the anti-hCD79A–injected mice. In addition, both total and high-affinity IgG+ anti-NP ASCs were significantly reduced (>75%) in the same mice. These observations corroborate the activities of the surrogate anti-mCD79B and extend them to include suppressive effects on immune responses to immunization with TI Ags.
FIGURE 5.
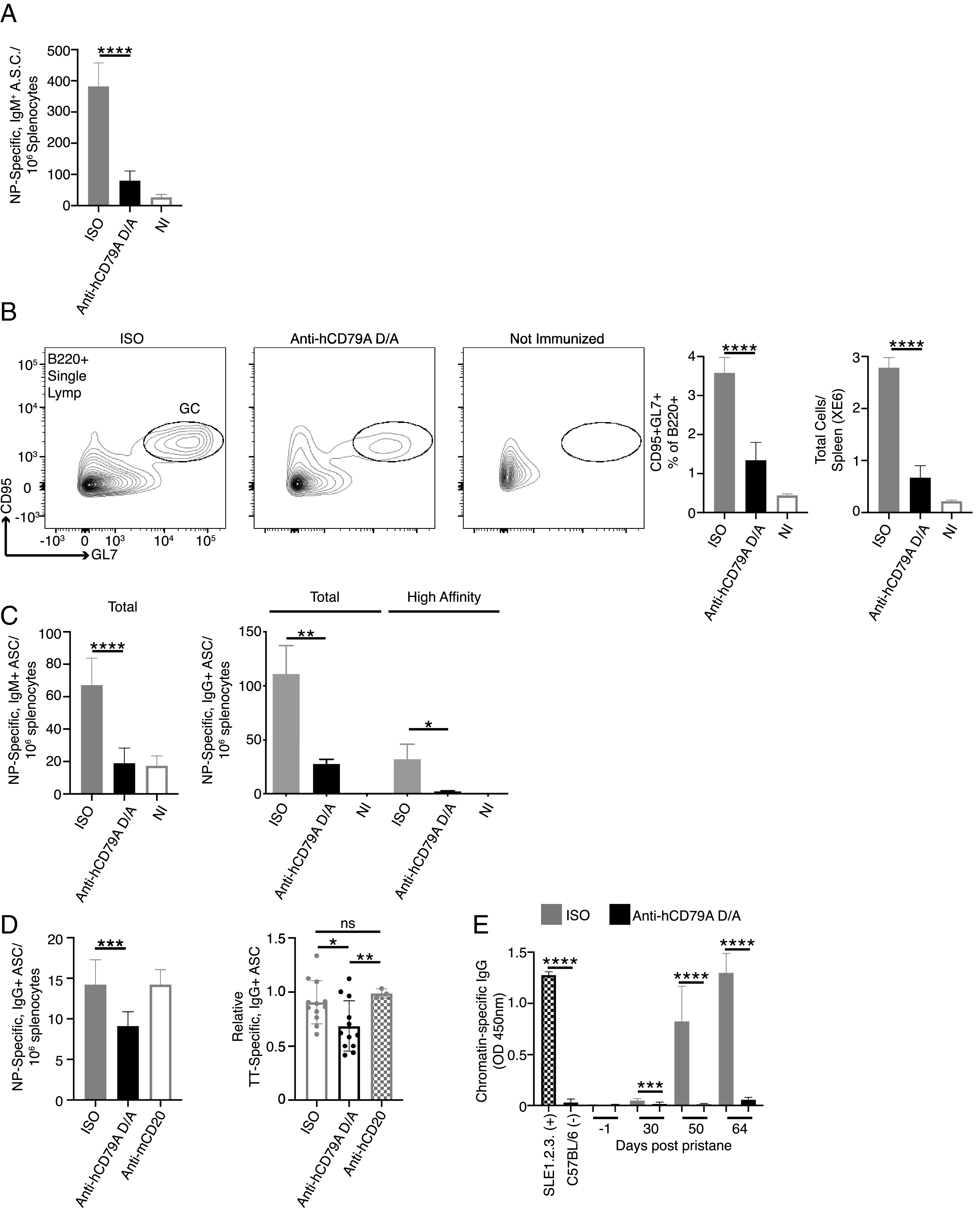
Anti-hCD79A treatment inhibits B cell immune responses. (A) ELISPOT analysis of NP-specific, IgM+ ASCs 7 d after immunization with NP59-Ficoll. Twenty-four hours prior to immunization, cCD79A mice received 250-µg i.p. injections of either anti-hCD79A D265A or control hIgG4. (B) Flow cytometric analysis of germinal center B cell induction 5 d after immunization with 0.1% SRBCs in PBS. Twenty-four hours prior to immunization, cCD79A animals received 250-µg i.p. injections of either anti-hCD79A D265A or control hIgG4. On day 5 postimmunization, RBC-lysed splenocytes were stained with Abs recognizing B220, CD95, and GL7. (C) ELISPOT analysis of NP-specific IgM+ (left) and IgG+ (right) ASCs at 16 d postimmunization with NP-OVA in alum. Twenty-four hours prior to immunization, cCD79A mice received 250-µg i.p. injections of either anti-hCD79A D265A or control hIgG4. For detection of total IgM and IgG anti-NP ASCs, plates were coated with NP19-BSA. For detection of high-affinity IgG anti-NP ASCs, plates were coated with NP2-BSA. (D) ELISPOT analysis of NP- (left) and tetanus toxoid–specific (right) IgG+ ASCs following overnight incubation with anti-hCD79A D265A. NP-specific ASCs were generated by immunizing cCD79A knockin mice with NP-OVA in alum and harvesting spleens 16 d later. RBC-lysed splenocytes (1 × 107) were put into 1-ml cultures containing 25 µg of either anti-hCD79A D265A, anti-mCD20, control hIgG4, or control mIgG2a. After 18-h incubations, cells were washed and loaded onto ELISPOT plates coated with NP19-BSA. n = 3 wells per condition. This experiment was repeated three times. PBMCs were isolated from peripheral blood obtained from subjects receiving Tdap booster vaccines 7–10 days prior. PBMCs (5 × 106) were put into 1-ml cultures containing 25 µg of either anti-hCD79A D265A, anti-hCD20, or control hIgG4. After overnight incubation, cells were washed and loaded onto tetanus toxoid–coated ELISPOT plates. n = 3 subjects whose PBMCs were incubated with anti-hCD79; n = 1 subject whose PBMCs were also incubated with anti-hCD20. Points represent individual counted wells. (E) Effects of serial, weekly treatments with hIgG4 D265A anti-hCD79A in pristane-induced development of autoantibodies (anti-chromatin IgG ELISAs). Pooled sera from aged/diseased SLE1.2.3 mice and wild-type C57BL/6 served as positive and negative controls, respectively. n = 20 male and 20 female cCD79A mice per treatment arm. Error bars show SEM. All data represents at least three independent experiments; representative data are shown. A Student t test was used to evaluate statistical significance. *p < 0.05, **p < 0.01, ***p < 0.001, ****p < 0.0001.
So far, we have focused on the effects of anti-CD79 treatment as a preventative measure. Pretreatment with anti-CD79 strongly inhibits BCR signaling and primary B cell Ab responses. In other words, anti-CD79 is shown to act early in B cell activation by BCR stimulation or immunization. From a potentially therapeutic standpoint, it might be important to know the effects of anti-CD79 on terminally differentiated autoantibody-secreting cells. The ability to suppress pathogenic autoantibody secretion would be beneficial in diseases such as SLE and would further differentiate anti-CD79 from anti-CD20, which does not target these cells (i.e., plasmablasts and plasma cells). As a pioneering step toward this possibility, we generated NP-specific ASCs by immunizing cCD79A mice, as before, with NP-OVA in alum and harvested spleens 16 d later. RBC-lysed splenocytes were then cultured overnight with either anti-hCD79A D265A, anti-mCD20 (18B12), or isotype control Ab (hIgG4 or mIgG2a). Following overnight incubation, cells were washed, counted, and loaded onto NP-coated ELISPOT plates. The left panel of (Fig. 5D shows a significant decrease in ASCs in overnight cultures containing anti-hCD79A, but not in cultures with anti-mCD20, as compared with control Ab cultures. Importantly, note that no differences in splenocyte viability, that is, trypan blue staining, were observed following overnight culture. As an additional experiment, we obtained PBMCs from subjects who received a tetanus booster (Tdap) 7–10 d prior to isolation. These PBMCs were cultured as before with either anti-hCD79A D265A, anti-hCD20 (rituximab), or control hIgG4. Following overnight incubation, cells were washed and counted before being loaded onto tetanus toxoid–coated ELISPOT plates. The right panel of (Fig. 5D shows a similar decrease in tetanus toxoid–specific ASCs when cells were cultured with Fc effector function–incompetent anti-hCD79A but not with anti-hCD20. These preliminary findings suggest that anti-CD79 may be capable of targeting and suppressing Ab secretion by ASCs. Importantly, suppression of Ab secretion was not induced by anti-CD20, suggesting a potentially distinct range of use of anti-CD79 therapy. Further experimentation is needed to understand the exact mechanism of ASC modulation by anti-CD79 and whether the treatment effects are preserved in vivo. Given these results we wanted to look at anti-hCD79A treatment effects on autoantibody production.
To assess the potential impact of anti-hCD79A on the development of autoimmunity, we next explored the in vivo prophylactic effects of anti-hCD79A in the pristane induction model of SLE (42). cCD79A mice were pretreated on day 0 with 250 µg i.p. of either hIgG4 isotype control Ab or hIgG4 anti-hCD79A D265A (Fig. 5E). Mice then received a single 0.5-ml i.p. dose of pristane and were injected with Ab every 7 d thereafter. Normal mortality was observed in the first 30 d of the experiment, most likely due to pulmonary hemorrhage. There was no significant difference in survival rate between the groups, and autoantibody was not detectable during this time. This suggests that anti-hCD79A treatment does not modulate pristane-induced inflammatory processes operating early in the pristane model. Importantly, anti-hCD79A–treated mice failed to accumulate chromatin-specific IgG autoantibodies during the course of the experiment. We hypothesize that, in addition to preventing anti-chromatin autoantibody formation, extended prophylactic treatment with anti-hCD79A prevents the development of lupus-associated pathology in pristane-induced mice. Further experimentation is needed to measure the effects of anti-CD79 on autoantibody production to Ags other than chromatin as well as modulation of disease in fully therapeutic application.
Anti-hCD79 treatment prevents autoimmune disease development in EAE
Early validation of the potential therapeutic efficacy of CD79 targeting was carried out by independent laboratories using surrogate anti-mCD79B Abs in multiple disease models (8–10). First described in the MRL/lpr model of SLE, anti-mCD79B prevented onset of spontaneous lupus-like disease in these mice, which was reportedly dependent on B cell depletion (8). Importantly, treated mice accumulated far less chromatin-reactive IgG Abs in their serum and displayed improved survival and lower mean skin scores. More recently, it has been found that targeting mCD79B prevents disease independent of its abilities to deplete B cells, rather acting by induction of reversible anergy (9). In 2014, Hardy et al. (9) prevented development of severe arthritic disease in collagen-immunized mice. Importantly, these studies used mIgG2a anti-mCD79B Abs that contained the D265A mutation, significantly reducing ADCC and CDC effector functions, thus preventing B cell depletion (43). In contrast, when formulated in this fashion, anti-CD20 no longer induced B cell depletion and did not protect mice from developing anti-collagen autoantibodies and consequent joint inflammation. The effects of nondepleting anti-mCD79B on B cells were maintained as long as ∼20% of BCRs were engaged by Ab, as B cells recover their signaling ability after anti-mCD79B levels decay. Use of nondepleting therapy has the potential to mitigate safety concerns associated with long-term B cell depletion. This mode of protection from collagen-induced arthritis was confirmed by Brühl et al. (10) the following year. Their findings suggest that the tolerizing effects of anti-mCD79B extend beyond the BCR, potentially modulating signals emanating from CD40 and CD180. Apart from inducing anergy in B cells, other investigators are exploring CD79 targeting with chimeric Ag receptor T cells and drug–Ab conjugates for treatment of B cell malignancies (44, 45).
We sought additional evidence of clinical utility of anti-hCD79A Abs using the B cell–dependent EAE model of multiple sclerosis (46, 47). Male and female cCD79A+/+cCD79B+/− mice were immunized i.p. with 50 µg of recombinant hMOG1–125 in 200 µg of CFA on day 0. Mice also received i.p. injections containing 200 ng of pertussis toxin in PBS on days 0 and 2. Mice were given weekly i.p injections of Ab at 20 mg/kg of body weight, beginning on day −3 and continuing for the duration of the experiment. Ab treatment arms consisted of aglycosyl-mIgG1 anti-hCD79A (anergizing), mIgG2a anti-hCD79A (depleting), and both aglycosyl-mIgG1 and mIgG2a anti-HEL isotype controls. (Fig. 6A demonstrates that the anti-hCD79A formulation that is nondepleting is, nonetheless, efficacious. In this experiment aglycosylated mIgG1 anti-hCD79A Ab was used to prevent ADCC and CDC leading to B cell killing, thus isolating anergizing function, while anti-hCD79 Ab in IgG2a format was used for B cell depletion. Depleting and anergizing anti-hCD79A Abs reduced EAE disease severity similarly, to a degree equivalent to depleting anti-CD20 Abs (48, 49). The incidence of EAE was drastically reduced for the anergy and depletion groups compared with the isotype groups in this study (anergy, 56.3%; depletion, 58.8%; pooled isotype, 94.7%) (Fig. 6B). A significant delay in disease onset was also observed in the anti-hCD79A treatment groups (median day of onset: anergy, day 14; depletion, day 13.5; pooled isotype, day 12). Surprisingly, mice treated with anergizing anti-hCD79A tended to be protected from weight loss for the duration of the experiment (Fig. 6C). Importantly, B cell depletion was seen in EAE mice receiving WT mIgG2a anti-hCD79A whereas B cells were not significantly depleted in mice receiving the aglycosylated mIgG1 anti-hCD79A (Fig. 6D, left). Both anti-hCD79A Ab treatments caused reduced expression of surface IgD, a unique property of anti-CD79–induced B cell anergy, as it is not a characteristic of anergic B cells (Fig. 6D, right). No significant differences were observed in other immune cells (i.e., CD4, CD8, myeloid) in response to either anti-hCD79A treatments (Fig. 6E). (Fig. 6F shows a highly significant reduction in relative serum concentrations of anti-MOG IgG in mice receiving either anti-hCD79A treatment. Isotype treatment groups display rapid onset of anti-MOG Ab production whereas minimal levels are found in the anti-hCD79A treatment groups throughout the duration of the experiment. At the peak of disease (day 15) significantly decreased anti-MOG IgG in the anergized mice suggests inhibited B cell activation leading up to the peak of exacerbation in EAE-associated paralysis. Significantly reduced serum pNfH, a marker of neurodegeneration, at the peak of disease is also seen in mice treated with anergizing anti-hCD79A (Fig. 6G).
FIGURE 6.
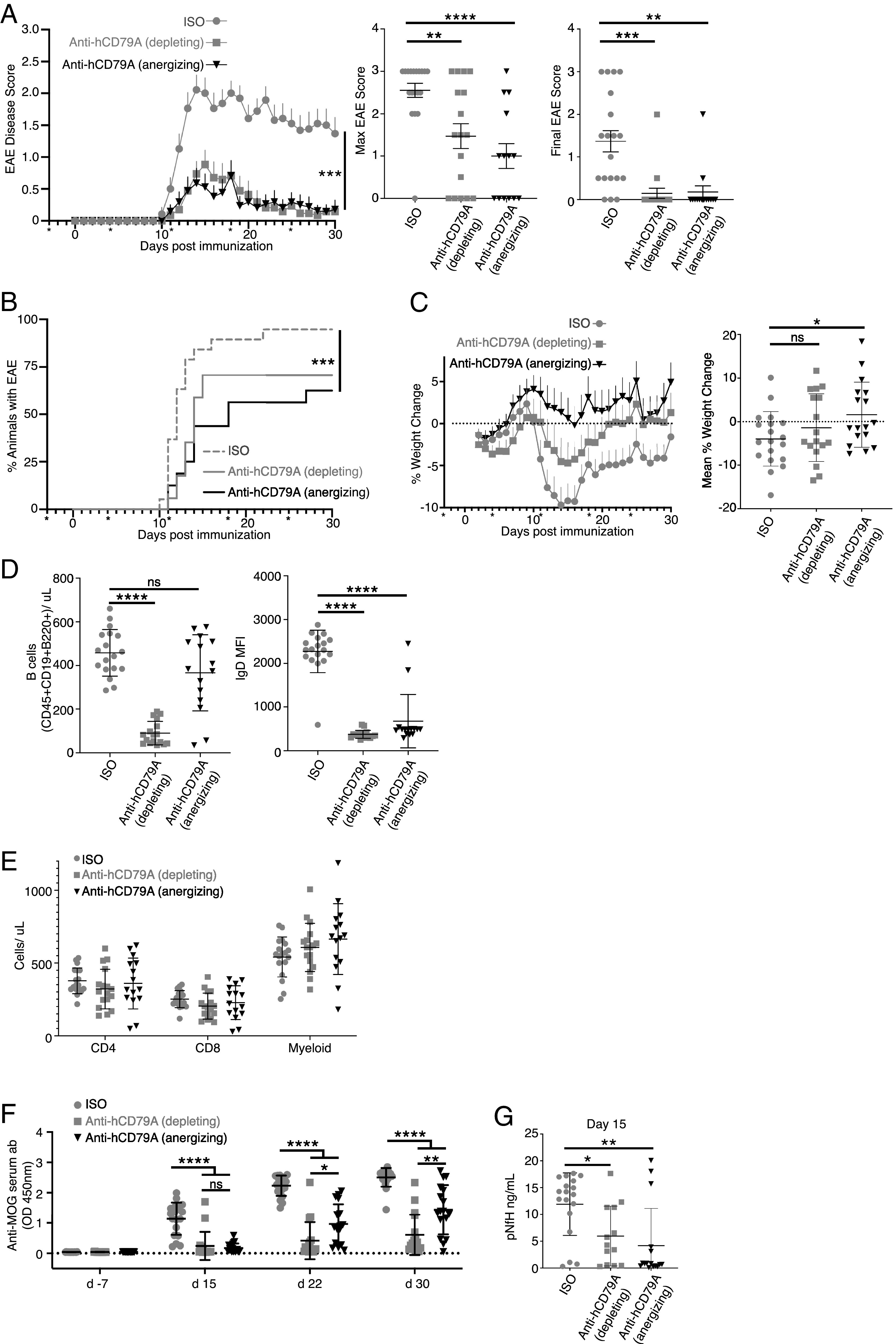
Anti-hCD79 treatment prevents the development of MOG1–125-induced EAE. (A) Prophylactic treatment of EAE in mice immunized with hMOG1–125. Anti-hCD79A mIgG2a (depleting; 20 mg/kg; n = 17 female and male mice) and aglycosyl-mIgG1 anti-hCD79A (anergizing; 20 mg/kg; n = 16 female and male mice) were compared with their respective anti-HEL isotype controls (mIgG2a, n = 9; mIgG1 agly, n = 10 female and male mice). Asterisks indicate day of Ab treatment. Error bars show SEM. Kruskal–Wallis test with a Dunn multiple comparison test. ***p < 0.001. (B) Time to EAE onset and EAE incidence based on scores only. Wilcoxon log-rank test. ***p < 0.001. (C) Weight change of EAE mice over time shown as percentage of initial weight (left). Mean weight change shown as percentage of initial weight (right). (D) Splenic B cell counts (CD45+CD19+B220+, left) and IgD expression (right) at takedown (day 30) of hMOG1–125-induced EAE. (E) Splenocyte counts of non–B cells in EAE spleens at takedown (CD45+CD4+ T cells, CD45+CD8+ T cells, Ly6G+Ly6C+CD11B+ myeloid cells). (F) Relative abundance (OD 450 nm) of anti-MOG Abs before EAE, during peak (day 15) and chronic (day 22) phases of disease, and at takedown (day 30). (G) Serum concentrations (ng/ml) of phosphorylated neurofilament H chain (pNfH) in EAE mice at peak of disease (day 15).
In conclusion, we have confirmed the ability of anti-hCD79A Abs to prevent autoimmune disease development in humanized cCD79 mice. Paramount in these experiments is demonstration that the ability of anti-hCD79A Ab to prevent development of autoimmunity is independent of its ability to deplete B cells. In the pristane model, anti-hCD79A was rendered Fc effector compromised by virtue of the D265A mutation. Similar Fc mutational approaches are used in therapeutic Abs currently being administered to patients (50). Blocking the development of anti-chromatin, and potentially other autoantibodies induced by pristane, suggests prevention of outright disease. In the EAE model of multiple sclerosis, anti-hCD79A Abs also prevented disease in the absence of B cell depletion. Non-depleting therapies that target B cells represent a potentially safer alternative to CD20-mediated B cell depletion.
Discussion
In this study, we describe the generation and application of a humanized mouse platform for preclinical validation of candidate anti-hCD79 therapeutic Abs. This approach is advantageous in situations in which the therapeutic target dictates the need for an in-depth understanding of the immunological impact of the intervention that is not achievable in nonhuman primates. In this case, knockin mice were created that express, in context, the extracellular domains of either or both hCD79A and CD79B. These mice were used to study the functional interchangeability of human and mouse CD79 extracellular domains, and the activity of a humanized IgG4 D265A anti-hCD79A mAb hypothesized to suppress immune responses, including autoimmune responses, by inducing an anergic-like state without significant B cell depletion.
CD79 heterodimers associate noncovalently with Igs and are required for membrane expression of the BCR; without CD79 expression, B cell development is arrested at the pro– to pre–B cell transition (1). In addition to its roles in B cell development, CD79 ITAMs are responsible for initiating intracellular signaling cascades subsequent to aggregation of the BCR (51). These signals ultimately drive B cell activation, proliferation, and differentiation in immune responses (52). HCD79 heterodimers are similar structurally to those found in mice. Both consist of CD79A-CD79B disulfide-linked heterodimers that associate with surface Ig in B cell plasma membranes. CD79A and CD79B each have a single extracellular Ig-like domain, a single-pass transmembrane domain, and cytoplasmic signaling domains. CD79 sequence homology between mice and humans is lowest in the extracellular Ig-like domains, suggesting a potential lack of cross-reactivity between anti-hCD79 and anti-mCD79 Abs. The highest conservation between these species can be found in the transmembrane and cytoplasmic domains. The transmembrane domain of CD79A is 100% conserved in mice and humans. This is also true of the membrane spanning domains of membrane-bound IgM, which is believed to contain the site of interaction with CD79A (53). To this end, surface expression of IgM in mouse B cells is achievable with expression of the hCD79A encoding gene mb-1 (54). Furthermore, humans expressing mutated CD79A, which resulted in a loss of the CD79A transmembrane domain, lack peripheral B cells and are agammaglobulinemic (55, 56). Thus, it was predicted that expression of our cCD79, in which only the extracellular domain sequences are human, would not disrupt BCR assembly in mice. Data presented in the current study confirm this prediction. Due to the low sequence homology between mCD79 and hCD79 extracellular Ig-like domains, we hypothesize that interactions between the Ig domains of CD79 and membrane Ig are much less important than those within the transmembrane domains of each.
Our findings led to a number of conclusions. Because CD79 plays a critical role in B cell development and function, and because hCD79 could disrupt these functions, we characterized the cCD79 knockin mouse lines in comparison with WT C57BL/6 mice. The results indicate that hCD79 and mCD79 extracellular domains are interchangeable in all regards. Our analyses show B cell developmental populations within the bone marrow and the spleen to be within normal ranges. Similarly, B cell activation and differentiation in the periphery is normal. BCR signaling and B cell immune responses were found to be unaffected by CD79 extracellular domain replacement. Although cCD79 B cells behave in a nearly identical fashion as WT B cells expressing mCD79, we addressed several interesting findings that came to light in our studies. First, the observed relative mass of our cCD79B molecule is closer to the known mass of mCD79B (Fig. 1E, middle membrane). Specifically, mCD79B migrates at 37 kDa whereas fully hCD79B migrates at 34 kDa (57, 58). Our chimeric human-mouse CD79B migrates at ∼35 kDa, suggesting that the differences may be associated with the cellular context and/or glycosylation. Another puzzling observation is the apparent preference of cCD79A for pairing with the endogenous mCD79B molecule on the surface of B cells in cCD79A+/+B+/− mice. However, there are no discrepancies in IgM or IgD expression levels between cCD79A mice expressing either no, one, or both copies of cCD79B. In addition, no discrepancies in B cell development or BCR function were observed in singly homozygous, for example, cCD79A and cCD79B mice, suggesting that the staining seen in (Fig. 1D and 1G is due to the relative affinities of the mAbs.
We further report that in WT and knockin mice, respectively, anti-mCD79 and anti-hCD79 Abs have similar inhibitory effects on BCR signaling and on immune responses. Chronic stimulation of the BCR by autoantigen in the absence of secondary signals induces a form of peripheral B cell tolerance known as anergy (59–62). Chronic engagement of the BCR via anti-CD79 Abs elicits a signaling phenotype similarly observed in anergic B cells (63). Hardy et al.’s (9) anti-mCD79B experiments demonstrated decreased IgM and IgD expression in response to treatment in vivo. BCR-mediated calcium signaling was also observed to be suppressed in these B cells. However, reduced calcium signaling was not shown to be independent of decreased membrane BCR. In the current study, we have demonstrated that the humanized IgG4 D265A anti-hCD79A clinical candidate induces inhibition of BCR-mediated Syk phosphorylation and calcium mobilization that is independent of changes in IgM and IgD surface expression. In comparison with anergic B cells, anti-CD79 treatment induces downmodulation of both IgM- and IgD-containing B cell Ag receptors. The ability to affect mIgD expression, a unique trait of anti-CD79 treatment, could be due to the expression pattern of IgM and IgD on peripheral B cells that are likely the primary target of the therapeutic, for example, mature naive B cells in peripheral blood and spleen. Developing autoreactive, and potentially anergic, B cells in the bone marrow encounter autoantigen upon expression of their IgM pre-BCR, prior to expression of IgD. This difference suggests that mIgD on anergic B cells may not bind Ag with the same efficiency as mIgM. This would be consisted with the greater length of IgM H chains (five Ig domains) relative to IgD H chains (three Ig domains). In addition, we confirm that ionomycin-induced calcium mobilization is unaffected by prior treatment with anti-CD79. These observations suggest that similar to anergic B cells, anti-CD79 treatment targets signaling events proximal to the BCR and may also rely on nondurable mechanisms.
In addition to effects on BCR signaling, in vivo treatment with anti-hCD79 induces increased intracellular expression of the regulatory phosphatase PTEN. PTEN expression in cCD79A B cells is increased by anti-hCD79A treatment to levels consistent with those observed in human anergic BND cells (38). However, targeted genetic ablation of PTEN in mice resulted in modest increases in BCR-mediated calcium flux in cells treated with anti-CD79A. We hypothesize that PTEN may play a partial role in the mechanism; suppression of signaling may involve multiple, partially redundant mechanisms, including mBCR modulation, regulatory signaling, and potential disassociation of the BCR signaling complex. Another possibility includes preferential monophosphorylation of CD79 ITAMs in cells treated with anti-CD79 (35). The known consequence of ITAM monophosphorylation is activation of Lyn-mediated inhibitory signaling pathways, but not Syk (64). Considering what is known about PTEN-mediated regulation of PI3K signaling, anti-CD79 treatment may also affect signaling by other activating receptors, for example, CD40, TLRs, and cytokine/chemokine receptors (65). Data presented by Brühl et al. (10) suggest inhibition of CD40 and CD180 signaling by treatment with anti-mCD79. Furthermore, because signaling by the receptor for B cell activating factor (BAFFR) has been shown to involve subunits of the BCR as adaptor molecules (66), anti-CD79 treatment may also modulate BAFF signaling. Additional experimentation is needed to fully define the molecular mechanisms operative in anti-CD79–induced B cell anergy.
We confirm and extend anti-CD79 effects on B cell immune responses to include those induced by immunization with TI and TD Ags. Not surprisingly, pretreatment with anti-CD79 resulted in significantly diminished recovery of CD95+GL7+ germinal center B cells in response to immunization with SRBCs. Perhaps more interesting from a therapeutic standpoint is the potential for anti-CD79 treatment to effect Ab secretion by terminally differentiated B cells. There is currently a need for therapies targeting secreting cells in diseases characterized by pathogenic autoantibodies. Precedent for this mode of inhibition was demonstrated in an IgM-secreting hybridoma specific for the hapten fluorescein (67). In 1980, Boyd and Schrader (67) showed that Ag binding to the surface of these cells induced inhibition of protein synthesis and Ab secretion. More recently it has been demonstrated that IgM+ and IgA+ human plasma cells express surface BCRs and that high doses of BCR-crosslinking Ab leads to significantly impaired recovery of ASCs (68). Importantly, it was shown that all human plasma cells, that is, IgM+, IgA+, and IgG+, stained positive for the intracellular portions of CD79 (Igα and Igβ), suggesting that surface expression of CD79 may be maintained irrespective of surface Ig. In this study, we show that in vitro anti-CD79 treatment results in significantly reduced recovery of Ag-specific ASCs. Importantly, Abs targeting CD20 did not induce this effect. Based on the level of inhibition, we hypothesize that at least some ASCs express CD79 and that treatment with anti-CD79 negatively affects Ab secretion. At this time we are unsure whether anti-CD79 treatment affects short-lived plasmablasts, long-lived plasma cells, or both. It is also unclear whether treatment suppresses Ab secretion and/or induces cell death. Further experimentation is needed to better understand this phenomenon and whether treatment effects are maintained in vivo. We are excited about the possibility that CD79 targeting may be successful at both preventing autoimmune B cell responses and at disrupting those responses that are already underway.
Most promising from the preclinical testing perspective were the effects of anti-hCD79A on pristane-induced autoantibody production and EAE disease development. In pristane-injected mice receiving anti-hCD79A, little to no anti-chromatin autoantibody is detected after 2 mo, unlike isotype control mice. Currently, our research efforts include validating fully therapeutic application of anti-hCD79A in this model. We anticipate that this approach will also help to clarify the therapeutic effect of anti-CD79 on terminally differentiated ASCs specific for lupus-associated autoantigens. In a preventative treatment of EAE, anti-hCD79A greatly reduced disease severity and incidence and led to near complete recovery. The magnitude of this effect was equal to or greater than that induced by B cell depletion using either anti-CD20 or fully competent anti-hCD79A. Similar to previous reports using effector function–compromised anti-mCD79B, disease modulation by anti-hCD79A was achieved without significant B cell depletion, which is the main mechanism of action exploited by current B cell–targeting clinical therapies (69). At takedown, 30 d postimmunization with hMOG1–125, we observed a nonsignificant reduction in splenic B cells in mice receiving anergizing anti-hCD79A. However, there is a downward trend that is reminiscent of B cell counts in the peripheral blood of Hardy et al.’s mice treated with an effector function–compromised anti-mCD79B (as shown in figure 4B in Ref. 9). By 4 wk postinjection, peripheral blood B cell numbers matched those of isotype control animals. Although there was a transient reduction in peripheral blood B cells for ∼3 wk following a single injection, B cells in mice treated with mutated (D265A) anti-mCD79B did not label with BrdU within this timeframe (as shown in figure 7 in Ref. 9). It was concluded that treatment with the surrogate, nondepleting anti-mCD79B induced a transient relocation of peripheral blood B cells. These observations support the potential impact of anti-CD79 on other receptors expressed by B cells, for example, chemokine receptors. At the peak of EAE disease (day 15), mice in the anergizing treatment arm were observed to have significantly reduced serum concentrations of pNfH, a marker of neurodegeneration (70). This measure is a correlate of reduced paralysis and reduced relative serum anti-MOG autoantibodies. Taken together, these observations suggest that anergizing anti-hCD79A treatment inhibits activation of autoreactive anti-MOG B cells, which prevents the development of severe EAE. We hypothesize that inhibiting B cells in this model modulates disease in part by downregulating B cell Ag presentation to CD4 T cells. It has previously been shown that expansion of Ag-specific (MOG) B cells in mice expressing MHC class II on B cells alone, and therefore the sole APC, drives EAE-associated neuroinflammation (71). Further experimentation is needed to directly assess the impact of anergizing anti-CD79A treatment on B cell Ag presentation. We also want to test this treatment in a fully therapeutic mode, administering Ab at the peak of clinical disease.
We conclude that anti-CD79 functions by inducing transient anergy in autoreactive B cells. Not only does this predict success of CD79-targeted therapies, but it also addresses potential safety concerns associated with depleting therapies (18). Historically, anti-CD20–mediated B cell depletion leads to a significant reduction of B cells in mouse peripheral blood for >40 d postinjection (as shown in figure 4A in Ref. 9). Preliminary experiments demonstrate that B cell functionality is fully restored at 35 d postinjection with Fc effector function–compromised anti-hCD79 (S.M. Wemlinger and J.C. Cambier, unpublished observations). Confirmation of these findings would support reversibility of anti-hCD79 treatment effects, offering an additional potential advantage over current B cell depleting therapies. Finally, the potential to inhibit autoantibody secretion by terminally differentiated plasmablasts and/or plasma cells could further differentiate CD79 targeting from CD20, which is not expressed on these cells.
Because anti-CD79 therapy relies on modulation of BCR signaling and B cell immune responses, it is important to define the mechanism of action in mouse B cells. The advantages of a murine preclinical model revolve around the more advanced state of our knowledge of and ability to manipulate the mouse immune system, as well as the prohibitive expense of preclinical studies in monkeys. Furthermore, non–cross-reactivity of anti-hCD79 Abs in cynomolgus monkeys presents a significant obstacle to preclinical validation of candidate therapeutic monoclonals. If necessary, a surrogate anti-cyno CD79 Ab can be used to assess toxicology in nonhuman primates (45, 72). We believe that our humanized mouse line represents an ideal preclinical model.
Supplementary Material
Acknowledgments
We thank the following groups: the Mouse Genetics Core Facility at National Jewish Health (Denver, CO) for producing singly heterozygous cCD79A and cCD79B mice; the Office of Laboratory Animal Resources at the University of Colorado Anschutz Medical Campus (Aurora, CO) for housing and caring for our mouse colony; the CU|AMC ImmunoMicro Flow Cytometry Shared Resource for providing and maintaining flow cytometry equipment (RRID:SCR_021321); Panorama Research (Sunnyvale, CA) for humanization and affinity maturation of anti-hCD79 Abs; the Raul Torres Lab at CU Anschutz for providing positive control sera from diseased SLE1.2.3 mice; the Lawrence Wysocki Lab at National Jewish Health for providing purified calf chromatin; Gisela Vaitaitis from the David Wagner laboratory at CU Anschutz for assisting with EAE induction; G. Campbell Kaynor and Kathryn Pellerin with the Multiple Sclerosis and Neurorepair Research Unit at Biogen for phospho-NF-H assays and anti-MOG ELISAs, respectively; and Mia Rushe and Joseph Amatucci with the Biologics Drug Discovery Unit at Biogen for preparation of the dosing solutions.
This work was supported by National Institute of Allergy and Infectious Diseases Grants R01 AI124487 and R44 AI120433 and by National Institute of Diabetes and Digestive and Kidney Diseases Grant DP3 DK110845.
John C. Cambier is a Distinguished Fellow of AAI.
The online version of this article contains supplemental material.
- ADCC
- Ab-dependent cellular cytotoxicity
- AP
- alkaline phosphatase
- ASC
- Ab-secreting cell
- BAC
- bacterial artificial chromosome
- [Ca2+]i
- intracellular Ca2+ concentration
- cCD79
- chimeric CD79
- CDC
- complement-dependent cytotoxicity
- EAE
- experimental autoimmune encephalomyelitis
- ES cell
- embryonic stem cell
- h
- human
- hCD79
- human CD79
- HEL
- hen egg lysozyme
- m
- mouse
- mCD79
- mouse CD79
- MOG
- myelin oligodendrocyte glycoprotein
- NP
- nitrophenyl
- pNfH
- phosphorylated neurofilament H chain
- PTEN
- phosphatase and tensin homolog
- PVDF
- polyvinylidene difluoride
- RT
- room temperature
- SLE
- systemic lupus erythematosus
- Syk
- spleen tyrosine kinase
- TD
- thymus-dependent
- TI
- thymus-independent
- WT
- wild-type
Disclosures
C.R.P.H., M.M., K.A.S., and T.O.C. are employees and shareholders of Biogen. The EAE portion of this project was funded by Biogen. S.M.W., J.W.L., and J.C.C. are stakeholders in Nepenthe Biosciences, an LLC whose interests lie in development of CD79-targeted therapies.
References
- 1. Pelanda R., Braun U., Hobeika E., Nussenzweig M. C., Reth M.. 2002. B cell progenitors are arrested in maturation but have intact VDJ recombination in the absence of Ig-α and Ig-β. J. Immunol. 169: 865–872. [DOI] [PubMed] [Google Scholar]
- 2. Cambier J. C. 1995. Antigen and Fc receptor signaling. The awesome power of the immunoreceptor tyrosine-based activation motif (ITAM). J. Immunol. 155: 3281–3285. [PubMed] [Google Scholar]
- 3. Reth M. 1989. Antigen receptor tail clue. Nature 338: 383–384. [PubMed] [Google Scholar]
- 4. Van Noesel C. J., Brouns G. S., van Schijndel G. M., Bende R. J., Mason D. Y., Borst J., van Lier R. A.. 1992. Comparison of human B cell antigen receptor complexes: membrane-expressed forms of immunoglobulin (Ig)M, IgD, and IgG are associated with structurally related heterodimers. J. Exp. Med. 175: 1511–1519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Flaswinkel H., Reth M.. 1994. Dual role of the tyrosine activation motif of the Ig-alpha protein during signal transduction via the B cell antigen receptor. EMBO J. 13: 83–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Sakaguchi N., Kashiwamura S., Kimoto M., Thalmann P., Melchers F.. 1988. B lymphocyte lineage-restricted expression of mb-1, a gene with CD3-like structural properties. EMBO J. 7: 3457–3464. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Hermanson G. G., Eisenberg D., Kincade P. W., Wall R.. 1988. B29: a member of the immunoglobulin gene superfamily exclusively expressed on beta-lineage cells. Proc. Natl. Acad. Sci. USA 85: 6890–6894. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Li Y., Chen F., Putt M., Koo Y. K., Madaio M., Cambier J. C., Cohen P. L., Eisenberg R. A.. 2008. B cell depletion with anti-CD79 mAbs ameliorates autoimmune disease in MRL/lpr mice. J. Immunol. 181: 2961–2972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Hardy I. R., Anceriz N., Rousseau F., Seefeldt M. B., Hatterer E., Irla M., Buatois V., Chatel L. E., Getahun A., Fletcher A., et al. 2014. Anti-CD79 antibody induces B cell anergy that protects against autoimmunity. J. Immunol. 192: 1641–1650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Brühl H., Cihak J., Talke Y., Rodriguez Gomez M., Hermann F., Goebel N., Renner K., Plachý J., Stangassinger M., Aschermann S., et al. 2015. B-cell inhibition by cross-linking CD79b is superior to B-cell depletion with anti-CD20 antibodies in treating murine collagen-induced arthritis. Eur. J. Immunol. 45: 705–715. [DOI] [PubMed] [Google Scholar]
- 11. Hauser S. L., Waubant E., Arnold D. L., Vollmer T., Antel J., Fox R. J., Bar-Or A., Panzara M., Sarkar N., Agarwal S., et al. HERM Trial Group . 2008. B-cell depletion with rituximab in relapsing-remitting multiple sclerosis. N. Engl. J. Med. 358: 676–688. [DOI] [PubMed] [Google Scholar]
- 12. Florou D., Katsara M., Feehan J., Dardiotis E., Apostolopoulos V.. 2020. Anti-CD20 agents for multiple sclerosis: spotlight on ocrelizumab and ofatumumab. Brain Sci. 10: 758. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Herold K. C., Pescovitz M. D., McGee P., Krause-Steinrauf H., Spain L. M., Bourcier K., Asare A., Liu Z., Lachin J. M., Dosch H. M., Type 1 Diabetes TrialNet Anti-CD20 Study Group . 2011. Increased T cell proliferative responses to islet antigens identify clinical responders to anti-CD20 monoclonal antibody (rituximab) therapy in type 1 diabetes. J. Immunol. 187: 1998–2005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Korhonen R., Moilanen E.. 2010. Anti-CD20 antibody rituximab in the treatment of rheumatoid arthritis. Basic Clin. Pharmacol. Toxicol. 106: 13–21. [DOI] [PubMed] [Google Scholar]
- 15. Shah K., Cragg M., Leandro M., Reddy V.. 2021. Anti-CD20 monoclonal antibodies in systemic lupus erythematosus. Biologicals 69: 1–14. [DOI] [PubMed] [Google Scholar]
- 16. Tahara M., Oeda T., Okada K., Kiriyama T., Ochi K., Maruyama H., Fukaura H., Nomura K., Shimizu Y., Mori M., et al. 2020. Safety and efficacy of rituximab in neuromyelitis optica spectrum disorders (RIN-1 study): a multicentre, randomised, double-blind, placebo-controlled trial. Lancet Neurol. 19: 298–306. [DOI] [PubMed] [Google Scholar]
- 17. Du F. H., Mills E. A., Mao-Draayer Y.. 2017. Next-generation anti-CD20 monoclonal antibodies in autoimmune disease treatment. Auto Immun. Highlights 8: 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Luna G., Alping P., Burman J., Fink K., Fogdell-Hahn A., Gunnarsson M., Hillert J., Langer-Gould A., Lycke J., Nilsson P., et al. 2020. Infection risks among patients with multiple sclerosis treated with fingolimod, natalizumab, rituximab, and injectable therapies. JAMA Neurol. 77: 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Pettitt S. J., Liang Q., Rairdan X. Y., Moran J. L., Prosser H. M., Beier D. R., Lloyd K. C., Bradley A., Skarnes W. C.. 2009. Agouti C57BL/6N embryonic stem cells for mouse genetic resources. Nat. Methods 6: 493–495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Valenzuela D. M., Murphy A. J., Frendewey D., Gale N. W., Economides A. N., Auerbach W., Poueymirou W. T., Adams N. C., Rojas J., Yasenchak J., et al. 2003. High-throughput engineering of the mouse genome coupled with high-resolution expression analysis. [Published erratum appears in 2003 Nat. Biotechnol. 21: 822.] Nat. Biotechnol. 21: 652–659. [DOI] [PubMed] [Google Scholar]
- 21. Larrick J., Yu B., Mendelsohn A., Cambier J., inventors; Nepenthe Bioscience LLC, assignee. Anti-CD79 antibodies and their uses. United States patent application 20200109198. Publication No. WO/2020/072705. 2020 Sept 4.
- 22. Zhang C. 2012. Hybridoma technology for the generation of monoclonal antibodies. Methods Mol. Biol. 901: 117–135. [DOI] [PubMed] [Google Scholar]
- 23. McAllister E. J., Apgar J. R., Leung C. R., Rickert R. C., Jellusova J.. 2017. New methods to analyze B cell immune responses to thymus-dependent antigen sheep red blood cells. J. Immunol. 199: 2998–3003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Smith M. J., Packard T. A., O’Neill S. K., Hinman R. M., Rihanek M., Gottlieb P. A., Cambier J. C.. 2017. Detection and enrichment of rare antigen-specific B cells for analysis of phenotype and function. J. Vis. Exp. (120): e55382. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Racke M. K. 2001. Experimental autoimmune encephalomyelitis (EAE). Curr. Protoc. Neurosci. Chapter 9:Unit 9.7. [DOI] [PubMed] [Google Scholar]
- 26. Getahun A., Wemlinger S. M., Rudra P., Santiago M. L., van Dyk L. F., Cambier J. C.. 2017. Impaired B cell function during viral infections due to PTEN-mediated inhibition of the PI3K pathway. J. Exp. Med. 214: 931–941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Radaev S., Zou Z., Tolar P., Nguyen K., Nguyen A., Krueger P. D., Stutzman N., Pierce S., Sun P. D.. 2010. Structural and functional studies of Igαβ and its assembly with the B cell antigen receptor. Structure 18: 934–943. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Lam K. P., Kühn R., Rajewsky K.. 1997. In vivo ablation of surface immunoglobulin on mature B cells by inducible gene targeting results in rapid cell death. Cell 90: 1073–1083. [DOI] [PubMed] [Google Scholar]
- 29. Meffre E., Nussenzweig M. C.. 2002. Deletion of immunoglobulin beta in developing B cells leads to cell death. Proc. Natl. Acad. Sci. USA 99: 11334–11339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Kraus M., Alimzhanov M. B., Rajewsky N., Rajewsky K.. 2004. Survival of resting mature B lymphocytes depends on BCR signaling via the Igα/β heterodimer. Cell 117: 787–800. [DOI] [PubMed] [Google Scholar]
- 31. Dal Porto J. M., Gauld S. B., Merrell K. T., Mills D., Pugh-Bernard A. E., Cambier J.. 2004. B cell antigen receptor signaling 101. Mol. Immunol. 41: 599–613. [DOI] [PubMed] [Google Scholar]
- 32. Scharenberg A. M., Humphries L. A., Rawlings D. J.. 2007. Calcium signalling and cell-fate choice in B cells. Nat. Rev. Immunol. 7: 778–789. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Nutt S. L., Hodgkin P. D., Tarlinton D. M., Corcoran L. M.. 2015. The generation of antibody-secreting plasma cells. Nat. Rev. Immunol. 15: 160–171. [DOI] [PubMed] [Google Scholar]
- 34. Getahun A., Beavers N. A., Larson S. R., Shlomchik M. J., Cambier J. C.. 2016. Continuous inhibitory signaling by both SHP-1 and SHIP-1 pathways is required to maintain unresponsiveness of anergic B cells. J. Exp. Med. 213: 751–769. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. O’Neill S. K., Getahun A., Gauld S. B., Merrell K. T., Tamir I., Smith M. J., Dal Porto J. M., Li Q. Z., Cambier J. C.. 2011. Monophosphorylation of CD79a and CD79b ITAM motifs initiates a SHIP-1 phosphatase-mediated inhibitory signaling cascade required for B cell anergy. Immunity 35: 746–756. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Vilen B. J., Famiglietti S. J., Carbone A. M., Kay B. K., Cambier J. C.. 1997. B cell antigen receptor desensitization: disruption of receptor coupling to tyrosine kinase activation. J. Immunol. 159: 231–243. [PMC free article] [PubMed] [Google Scholar]
- 37. Vilen B. J., Nakamura T., Cambier J. C.. 1999. Antigen-stimulated dissociation of BCR mIg from Ig-α/Ig-β: implications for receptor desensitization. Immunity 10: 239–248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Smith M. J., Ford B. R., Rihanek M., Coleman B. M., Getahun A., Sarapura V. D., Gottlieb P. A., Cambier J. C.. 2019. Elevated PTEN expression maintains anergy in human B cells and reveals unexpectedly high repertoire autoreactivity. JCI Insight 4: e123384. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Browne C. D., Del Nagro C. J., Cato M. H., Dengler H. S., Rickert R. C.. 2009. Suppression of phosphatidylinositol 3,4,5-trisphosphate production is a key determinant of B cell anergy. Immunity 31: 749–760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Wu X. N., Ye Y. X., Niu J. W., Li Y., Li X., You X., Chen H., Zhao L. D., Zeng X. F., Zhang F. C., et al. 2014. Defective PTEN regulation contributes to B cell hyperresponsiveness in systemic lupus erythematosus. Sci. Transl. Med. 6: 246ra99. [DOI] [PubMed] [Google Scholar]
- 41. Victora G. D., Nussenzweig M. C.. 2012. Germinal centers. Annu. Rev. Immunol. 30: 429–457. [DOI] [PubMed] [Google Scholar]
- 42. Freitas E. C., de Oliveira M. S., Monticielo O. A.. 2017. Pristane-induced lupus: considerations on this experimental model. Clin. Rheumatol. 36: 2403–2414. [DOI] [PubMed] [Google Scholar]
- 43. Baudino L., Shinohara Y., Nimmerjahn F., Furukawa J., Nakata M., Martínez-Soria E., Petry F., Ravetch J. V., Nishimura S., Izui S.. 2008. Crucial role of aspartic acid at position 265 in the CH2 domain for murine IgG2a and IgG2b Fc-associated effector functions. J. Immunol. 181: 6664–6669. [DOI] [PubMed] [Google Scholar]
- 44. Zhang L., French R. R., Chan H. T., O’Keefe T. L., Cragg M. S., Power M. J., Glennie M. J.. 1995. The development of anti-CD79 monoclonal antibodies for treatment of B-cell neoplastic disease. Ther. Immunol. 2: 191–202. [PubMed] [Google Scholar]
- 45. Li D., Lee D., Dere R. C., Zheng B., Yu S. F., Fuh F. K., Kozak K. R., Chung S., Bumbaca Yadav D., Nazzal D., et al. 2019. Evaluation and use of an anti-cynomolgus monkey CD79b surrogate antibody-drug conjugate to enable clinical development of polatuzumab vedotin. Br. J. Pharmacol. 176: 3805–3818. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Oliver A. R., Lyon G. M., Ruddle N. H.. 2003. Rat and human myelin oligodendrocyte glycoproteins induce experimental autoimmune encephalomyelitis by different mechanisms in C57BL/6 mice. J. Immunol. 171: 462–468. [DOI] [PubMed] [Google Scholar]
- 47. Marta C. B., Oliver A. R., Sweet R. A., Pfeiffer S. E., Ruddle N. H.. 2005. Pathogenic myelin oligodendrocyte glycoprotein antibodies recognize glycosylated epitopes and perturb oligodendrocyte physiology. Proc. Natl. Acad. Sci. USA 102: 13992–13997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Monson N. L., Cravens P., Hussain R., Harp C. T., Cummings M., de Pilar Martin M., Ben L. H., Do J., Lyons J. A., Lovette-Racke A., et al. 2011. Rituximab therapy reduces organ-specific T cell responses and ameliorates experimental autoimmune encephalomyelitis. PLoS One 6: e17103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Weber M. S., Prod’homme T., Patarroyo J. C., Molnarfi N., Karnezis T., Lehmann-Horn K., Danilenko D. M., Eastham-Anderson J., Slavin A. J., Linington C., et al. 2010. B-cell activation influences T-cell polarization and outcome of anti-CD20 B-cell depletion in central nervous system autoimmunity. Ann. Neurol. 68: 369–383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kang T. H., Jung S. T.. 2019. Boosting therapeutic potency of antibodies by taming Fc domain functions. Exp. Mol. Med. 51: 1–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51. Johnson S. A., Pleiman C. M., Pao L., Schneringer J., Hippen K., Cambier J. C.. 1995. Phosphorylated immunoreceptor signaling motifs (ITAMs) exhibit unique abilities to bind and activate Lyn and Syk tyrosine kinases. J. Immunol. 155: 4596–4603. [PubMed] [Google Scholar]
- 52. Packard T. A., Cambier J. C.. 2013. B lymphocyte antigen receptor signaling: initiation, amplification, and regulation. F1000Prime Rep. 5: 40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Reth M., Hombach J., Wienands J., Campbell K. S., Chien N., Justement L. B., Cambier J. C.. 1991. The B-cell antigen receptor complex. Immunol. Today 12: 196–201. [DOI] [PubMed] [Google Scholar]
- 54. Hombach J., Tsubata T., Leclercq L., Stappert H., Reth M.. 1990. Molecular components of the B-cell antigen receptor complex of the IgM class. Nature 343: 760–762. [DOI] [PubMed] [Google Scholar]
- 55. Minegishi Y., Coustan-Smith E., Rapalus L., Ersoy F., Campana D., Conley M. E.. 1999. Mutations in Igα (CD79a) result in a complete block in B-cell development. J. Clin. Invest. 104: 1115–1121. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56. Wang Y., Kanegane H., Sanal O., Tezcan I., Ersoy F., Futatani T., Miyawaki T.. 2002. Novel Igα (CD79a) gene mutation in a Turkish patient with B cell-deficient agammaglobulinemia. Am. J. Med. Genet. 108: 333–336. [DOI] [PubMed] [Google Scholar]
- 57. Friedrich R. J., Campbell K. S., Cambier J. C.. 1993. The gamma subunit of the B cell antigen-receptor complex is a C-terminally truncated product of the B29 gene. J. Immunol. 150: 2814–2822. [PubMed] [Google Scholar]
- 58. Clark M. R., Friedrich R. J., Campbell K. S., Cambier J. C.. 1992. Human pre-B and B cell membrane mu-chains are noncovalently associated with a disulfide-linked complex containing a product of the B29 gene. J. Immunol. 149: 2857–2863. [PubMed] [Google Scholar]
- 59. Cyster J. G., Hartley S. B., Goodnow C. C.. 1994. Competition for follicular niches excludes self-reactive cells from the recirculating B-cell repertoire. Nature 371: 389–395. [DOI] [PubMed] [Google Scholar]
- 60. Cooke M. P., Heath A. W., Shokat K. M., Zeng Y., Finkelman F. D., Linsley P. S., Howard M., Goodnow C. C.. 1994. Immunoglobulin signal transduction guides the specificity of B cell-T cell interactions and is blocked in tolerant self-reactive B cells. J. Exp. Med. 179: 425–438. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Eris J. M., Basten A., Brink R., Doherty K., Kehry M. R., Hodgkin P. D.. 1994. Anergic self-reactive B cells present self antigen and respond normally to CD40-dependent T-cell signals but are defective in antigen-receptor-mediated functions. Proc. Natl. Acad. Sci. USA 91: 4392–4396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. Goodnow C. C., Crosbie J., Adelstein S., Lavoie T. B., Smith-Gill S. J., Brink R. A., Pritchard-Briscoe H., Wotherspoon J. S., Loblay R. H., Raphael K., et al. 1988. Altered immunoglobulin expression and functional silencing of self-reactive B lymphocytes in transgenic mice. Nature 334: 676–682. [DOI] [PubMed] [Google Scholar]
- 63. Yarkoni Y., Getahun A., Cambier J. C.. 2010. Molecular underpinning of B-cell anergy. Immunol. Rev. 237: 249–263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64. Pao L. I., Famiglietti S. J., Cambier J. C.. 1998. Asymmetrical phosphorylation and function of immunoreceptor tyrosine-based activation motif tyrosines in B cell antigen receptor signal transduction. J. Immunol. 160: 3305–3314. [PubMed] [Google Scholar]
- 65. Donahue A. C., Fruman D. A.. 2004. PI3K signaling controls cell fate at many points in B lymphocyte development and activation. Semin. Cell Dev. Biol. 15: 183–197. [DOI] [PubMed] [Google Scholar]
- 66. Schweighoffer E., Vanes L., Nys J., Cantrell D., McCleary S., Smithers N., Tybulewicz V. L.. 2013. The BAFF receptor transduces survival signals by co-opting the B cell receptor signaling pathway. Immunity 38: 475–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Boyd A. W., Schrader J. W.. 1980. Mechanism of effector-cell blockade. I. Antigen-induced suppression of Ig synthesis in a hybridoma cell line, and correlation with cell-associated antigen. J. Exp. Med. 151: 1436–1451. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Pinto D., Montani E., Bolli M., Garavaglia G., Sallusto F., Lanzavecchia A., Jarrossay D.. 2013. A functional BCR in human IgA and IgM plasma cells. Blood 121: 4110–4114. [DOI] [PubMed] [Google Scholar]
- 69. Lee D. S. W., Rojas O. L., Gommerman J. L.. 2021. B cell depletion therapies in autoimmune disease: advances and mechanistic insights. Nat. Rev. Drug Discov. 20: 179–199. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Gresle M. M., Butzkueven H., Shaw G.. 2011. Neurofilament proteins as body fluid biomarkers of neurodegeneration in multiple sclerosis. Mult. Scler. Int. 2011: 315406. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71. Parker Harp C. R., Archambault A. S., Sim J., Ferris S. T., Mikesell R. J., Koni P. A., Shimoda M., Linington C., Russell J. H., Wu G. F.. 2015. B cell antigen presentation is sufficient to drive neuroinflammation in an animal model of multiple sclerosis. J. Immunol. 194: 5077–5084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72. Zheng B., Fuji R. N., Elkins K., Yu S. F., Fuh F. K., Chuh J., Tan C., Hongo J. A., Raab H., Kozak K. R., et al. 2009. In vivo effects of targeting CD79b with antibodies and antibody-drug conjugates. Mol. Cancer Ther. 8: 2937–2946. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.