

Abstract
T cells residing in mucosal tissues play important roles in homeostasis and defense against microbial pathogens. The gut and female reproductive tract (FRT) are both tolerogenic environments, but differ in the kinds of foreign antigens they need to tolerate. How these different environments influence the properties of their T cells is poorly understood, but important for understanding women’s health. We recruited ART-suppressed women living with HIV who donated, within one visit, blood and tissue samples from the ileum, colon, rectosigmoid, endometrium, endocervix, and ectocervix. With these samples, we conducted 36-parameter CyTOF phenotyping of T cells. Although gut and FRT T cells shared features discriminating them from their blood counterparts, they also harbored features distinguishing them from one another. These included increased proportions of CD69+ Trm cells of the Tem phenotype, and preferential co-expression of CD69 and CD103, on the gut-derived cells. By contrast, CD69+CD103+ Trm CD8+ T cells from FRT, but not those from gut, preferentially expressed PD1. We further determined that a recently described population of CXCR4+ T inflammatory mucosal cells differentially expressed multiple other chemokine receptors relative to their blood counterparts. Our findings suggest that T cells resident in different tolerogenic mucosal sites take on distinct properties.
Keywords: CyTOF, T cells, mucosa, HIV
INTRODUCTION
T cells play important roles in antiviral immunity, including within mucosal tissues where many viral pathogens initiate infection. While knowledge of human T cells has historically mostly derived from studies of peripheral blood for practical reasons, recent studies utilizing post-mortem specimens have markedly increased our understanding of T cells residing within mucosal tissues. For example, T cells within the gut have been shown to express the T resident memory (Trm) markers CD69 and CD103, to consist mostly of T effector memory (Tem) cells, and to exhibit other distinguishing phenotypic features such as low surface expression of the co-stimulatory molecule CD28 (1–3). Such post-mortem studies, however, have not interrogated T cell features within the female reproductive tract (FRT), the gateway tissue in women for sexually transmitted pathogens.
That being said, T cells from the lower FRT (cervical & vaginal tissues) of women undergoing hysterectomy were recently characterized. These studies, which used a combination of flow cytometry and RNAseq to characterize both CD4+ and CD8+ T cells, revealed unique features of multiple populations of genital T cells. For example, CD4+ T cells expressing the Trm markers CD69 and CD103 were shown to preferentially express the effector markers PD1 and CCR5 relative to their CD69-CD103- counterparts, and to harbor a Th17 signature (4). It was also demonstrated that cervicovaginal CD69-CD103- CD8+ T cells exhibit unique phenotypic, inflammatory, cytotoxic, and migratory features, and that these “T inflammatory mucosal” (Tim) cells expand during HIV infection (5).
These studies, in addition to revealing interesting features of mucosal T cells, also bring up the notion of whether there may be differences between the gut and FRT mucosa that may reflect the different foreign antigens they need to tolerate—the gastrointestinal microbiome and food antigens passing through the digestive system in the case of the gut, and the vaginal microbiome, male antigens, and an allogeneic fetus in the case of the FRT. At the same time, there are many parallels between these two mucosal sites: they are both colonized by endogenous flora, secrete mucus which contributes to barrier function, are responsive to sex steroids, and harbor mucosa-associated lymphoid tissue (MALT) structures (6). The aforementioned T cell characterization studies, unfortunately, did not allow for a direct in-depth comparison between the gut and FRT, because the gut and FRT specimens were not taken from the same women. In addition, previous studies of the FRT did not take into account cycle phase and menopausal status, which can very much affect immune responses, and did not include the upper FRT, in particular the endometrium, which is highly hormonally-responsive and serves as the site of embryo implantation but also as a potential site for viral invasion (7, 8).
Here, we performed a study where we recruited 5 HIV+, ART-suppressed women who donated, within the same study visit, blood together with biopsies from three gut sites (ileum, colon, and rectosigmoid) and samples from three areas of the FRT (biopsies from the endometrium and ectocervix, plus endocervical curettage). None of the women were post-menopausal, and they were all sampled during the midluteal phase of the ovulatory cycle. Cells from the specimens were all phenotyped without ever having gone through any cryopreservation. Phenotyping was conducted using cytometry by time of flight (CyTOF), enabling deep 36-parameter single-cell characterization of differentiation, activation, and homing properties of both CD4+ and CD8+ T cells from the specimens. Overall, we found expected phenotypic differences between blood and tissue T cells, and furthermore discovered both shared and unique features between T cells from the gut and FRT. Our results support the notion that T cells resident within different mucosal sites can take on distinct properties, presumably to help tailor their activities to their surrounding milieu.
METHODS
Study Participants.
Five participants were enrolled at UCSF from the Women’s Interagency HIV Study (WIHS), described elsewhere (9), and now known as the Multicenter AIDS Cohort Study (MACS)-WIHS Combined Cohort Study (10). Peripheral blood, and specimens from the gut (ileum, colon, rectosigmoid) and female reproductive tract (endometrium, endocervix, and ectocervix) were obtained during the midluteal phase of the ovulatory cycle, as established by urine luteinizing hormone detection. Hormone phase was further verified by blood progesterone levels. None of the women were post-menopausal as defined by the STRAW criteria (11). All blood, gut, and FRT specimens were obtained at the same study visit, between May 2017 and October 2019. Aliquots of these specimens were previously analyzed by CyTOF for HIV DNA and RNA levels (12). The study was approved by the University of California, San Francisco Institutional Review Board, and each participant provided written informed consent prior to participation in the study.
Specimen processing.
All specimens were processed the day of collection. Peripheral blood mononuclear cells (PBMCs) were isolated from blood specimens through Ficoll–Paque density gradient sedimentation. Whole blood was diluted with FACS buffer (PBS with 2% fetal bovine serum [FBS] and 2 mM EDTA), after which Ficoll (Stemcell Technologies) was slowly added to the bottom of the diluted specimen at a ratio (v/v) of 1 ficoll : 4 FACS buffer. Specimens were then centrifuged at 2,000 rpm at room temperature, without braking, using an Allegra X‐12 (Beckman Coulter, Brea, California, USA). The layer corresponding to PBMCs was transferred to a new tube, washed three times with FACS buffer, and treated for 10 minutes at room temperature with Red Lysis Buffer (BioLegend). The cells were then washed twice with RPMI 1640 media and prepared for CyTOF as described further below.
Tissue specimens were processed into single‐cell suspensions as previously described (12). Briefly, each tissue specimen was first incubated with intra‐epithelia lymphocyte (IEL) digestion buffer (PBS supplemented with 10 mM DTT (Sigma‐Aldrich), 5 mM EDTA (Thermo Fisher Scientific), 10 mM HEPES (Thermofisher), and 5% FBS) for 20 minutes at 37°C, to collect IELs from the supernatant fraction. The remaining tissues were then transferred to MACS C tubes (Miltenyi Biotec) in digestion solution (6 mL RPMI‐1640, 10 mM HEPES, 5% FBS, 6 mg CLSPA collagenase [Worthington‐Biochemical Corp] and 7.5 μg/mL DNAse [Sigma‐Aldrich]), and incubated for 30 minutes at 37°C. The tubes were then put through the gentleMACS™ Dissociator (Miltenyi, program: m_spleen), followed by further dissociation by 10 passes through a blunt 20G needle (Becton Dickinson). Cells were washed, treated for 30 minutes at 4°C with DNase solution (6 mL RPMI 1640, 10 mM HEPES, 5% FBS and 7.5 μg/mL DNAse [Sigma‐Aldrich]), and combined with the IEL fraction. All cells were filtered through 70 μm strainers (Fisher) prior to CyTOF staining.
Generation of in vitro HIV-infected cells.
To validate our ability to detect HIV-infected cells, we generated HIV-infected primary cells. CCR5-tropic F4.HSA virus was produced from F4.HSA plasmid (13) as described (14) and exposed at a final concentration of 100–200 ng/ml p24Gag to human lymphoid aggregate cultures (HLACs) generated from tonsils similar to recently described methods (15). Control cultures received media lacking F4.HSA. Infections were allowed to proceed for four days, after which cultures were processed for CyTOF analysis as described below.
Mass Cytometry (CyTOF).
CyTOF was conducted similar to methods recently described (14, 16, 17). Single-cell suspensions of the patient blood and tissue specimens, as well as in vitro-infected tonsils, were treated with 25 μM cisplatin (Sigma) for 60 seconds to enable subsequent live/dead discrimination. The cells were then quenched with CyFACS buffer (PBS supplemented with 0.1% BSA and 0.1% sodium azide) and fixed for 10 minutes with 2% paraformaldehyde (PFA; Electron Microscopy Sciences). Next, the fixed cells were washed twice with CyFACs, and then frozen at −80°C until CyTOF antibody staining. Of note, this processing protocol enables analysis of cells that never underwent cryopreservation.
Immediately prior to antibody staining, all blood and tissue cells from each participant were barcoded using the Cell‐ID™ 20‐Plex PD Barcoding kit (Fluidigm, South San Francisco, CA, USA) and combined into a single specimen. These cells were then then blocked for 15 minutes at 4°C with 1.5% mouse and rat sera (both from Thermo Fisher) and 0.3% human AB sera (Sigma‐Aldrich). The cells were then washed twice with CyFACS and stained for 45 minutes at 4°C with the cell surface antibodies listed in Table 2. Antibodies were either purchased pre-conjugated from Fluidigm, or self-conjugated using the MaxPAR conjugation kit (Fluidigm). Cells were then washed with CyFACS and fixed overnight at 4°C in 2% PFA. The next day, the cells were permeabilized for 30 minutes with Foxp3 Fix/Permeabilization Buffer (Fisher Scientific), washed twice with Permeabilization Buffer (Fisher Scientific), and blocked for 15 minutes at 4°C with mouse and rat sera diluted in Permeabilization Buffer. After two washes with Permeabilization Buffer, cells were stained for 45 minutes at 4°C with the intracellular antibodies listed in Table 2. Cells were then incubated for 20 minutes with a 1:500 dilution DNA intercalator (Fluidigm), washed twice with CyFACS, once with Cell Acquisition Solution (CAS, Fluidigm), and resuspended in 1x EQ™ Four Element Calibration Beads (Fluidigm) diluted in CAS. Cells were analyzed on a CyTOF 2 instrument (Fluidigm) at the UCSF Parnassus Flow Core.
Table 2.
List of antigens analyzed by CyTOF
| Antigen Target | Clone | Elemental Isotope | Vendor | Marker of |
|---|---|---|---|---|
| HLADR | TU36 | Qdot (112Cd) | Life Technolgies | Activation |
| RORγδ* | AFKJS-9 | 115In | In-house | Lineage |
| CD49d (α4) | 9F10 | 141Pr | Fluidigm | Trafficking |
| CD19 | HIB19 | 142Nd | Fluidigm | Lineage |
| CD57 | HNK-1 | 143Nd | In-house | Differentiation |
| CCR5 | NP6G4 | 144Nd | Fluidigm | Trafficking |
| CD8 | RPA-T8 | 146Nd | Fluidigm | Lineage |
| CD7 | CD76B7 | 147Sm | Fluidigm | Differentiation |
| ICOS | C398.4A | 148Nd | Fluidigm | Activation |
| Tbet* | 4B10 | 149Sm | In-house | Lineage |
| Gag (KC57)* | FH190-1-1 | 150Nd | In-house | HIV infection |
| CD103 | Ber-ACT8 | 151Eu | Fluidigm | Trafficking |
| TCRγδ | 11F2 | 152Sm | Fluidigm | Lineage |
| CD62L | DREG56 | 153Eu | Fluidigm | Trafficking |
| TIGIT | MBSA43 | 154Sm | Fluidigm | Exhaustion |
| CCR6 | 11A9 | 155Gd | In-house | Trafficking |
| CD29 (β1) | TS2/16 | 156Gd | Fluidigm | Trafficking |
| OX40 | ACT35 | 158Gd | Fluidigm | Activation |
| CCR7 | G043H7 | 159Tb | Fluidigm | Lineage/Trafficking |
| CD28 | CD28.2 | 160Gd | Fluidigm | Activation |
| CD45RO | UCHL1 | 161Dy | In-house | Lineage |
| CD69 | FN50 | 162Dy | Fluidigm | Activation/Residence |
| CRTH2 | BM16 | 163Dy | Fluidigm | Lineage |
| PD-1 | EH12.1 | 164Dy | In-house | Activation/Exhaustion |
| CD127 | A019D5 | 165Ho | Fluidigm | Differentiation |
| CXCR5 | RF8B2 | 166Er | In-house | Lineage/Trafficking |
| CD27 | L128 | 167Er | Fluidigm | Lineage |
| CD30 | BERH8 | 168Er | In-house | Activation |
| CD45RA | HI100 | 169Tm | Fluidigm | Lineage |
| CD3 | UCHT1 | 170Er | Fluidigm | Lineage |
| Gag (mixture)* | 71-31, 91-5, 241-D, AG3.0 | 171Yb | In-house | HIV infection |
| CD38 | HIT2 | 172Yb | Fluidigm | Activation |
| α4β7 | Act1 | 173Yb | In-house | Trafficking |
| CD4 | SK3 | 174Yb | Fluidigm | Lineage |
| CXCR4 | 12G5 | 175Lu | Fluidigm | Trafficking |
| CD25 | M-A251 | 176Yb | In-house | Activation |
Intracellular antibodies
Quantitation and correlation analysis of HIV DNA and RNA species.
The levels of HIV DNA and RNA were obtained from a recently published study (12), which describes the ddPCR-based approaches for quantitating these nucleic acid products. Spearman correlations were calculated using the scipy ‘spearmanr’ function (18) and depicted as heatmaps.
CyTOF Data Analysis.
CyTOF datasets were normalized to EQ calibration beads to minimize variability in intra-machine performance. The numbers of live, singlet T cells collected for each type of sample ranged from 70,177 – 315,125 for blood (median 95,927), from 1,484 – 10,439 for gut (median 3,894), and from 792 – 35,304 for FRT (median 5,835). T cells were manually gated using the FlowJO software (BD Biosciences) for CD4+ T cells (CD3+CD19-CD8- or CD3+CD19-CD4+CD8- cells as indicated), memory CD4+ T cells (CD3+CD19-CD4+CD8-CD45RO+CD45RA- cells), CD8+ T cells (CD3+CD19-CD4-CD8+ cells), or memory CD8+ T cells (CD3+CD19-CD4-CD8+CD45RO+CD45RA- cells), as well as other subsets defined within the Results section. tSNE visualizations of the datasets were performed in Cytobank, with the following settings: Iteration=16000; Perplexity=45; Theta=0.5. FlowSOM (19) was used to identify clusters from the high-dimensional CyTOF datasets. When comparing blood, gut and FRT, the following FlowSOM settings were used: hierarchical consensus; metaclusters=20; clusters=225, iterations=10. When clustering just the gut and FRT specimens together, FlowSOM was performed in Cytobank, using the following settings: hierarchical consensus; metaclusters=10; clusters=225, iterations=10.
RESULTS
Study participant specimens
The participants were five women living with HIV (WLWH) and on ART (Table 1). We used a cutoff of < 200 HIV-1 RNA copies / ml to include only participants without evidence of virological failure. The DHHS guidelines and the AIDS Clinical Trials Group (ACTG) define virological failure as a confirmed viral load of > 200 HIV-1 RNA copies / ml, a threshold that eliminates most cases of apparent viremia caused by viral load blips or assay variability. For each participant, whole blood, and ileum, colon, rectosigmoid, endometrium, endocervix, and ectocervix specimens were obtained. For the tissue specimens, we implemented a protocol that uses gentle digestion to maintain high lymphocyte recovery while preserving the integrity of many T cell surface antigens (20). Immediately after isolating the single-cell suspensions, we stained cells with cisplatin as a viability dye, fixed with paraformaldehyde (PFA), and then analyzed the cells by CyTOF. This approach enabled established the phenotypes of cells that had never undergone cryopreservation. Raw CyTOF datasets are available for download through the following public repository link: https://doi.org/10.7272/Q6HM56P8. Aliquots of these specimens were set aside for HIV RNA and DNA quantitation, which was published elsewhere (12).
Table 1.
Clinical Parameters by Donor
| HIV-positive ART-treated cisgender women | ||||||
|---|---|---|---|---|---|---|
| Donor A | Donor B | Donor C | Donor D | Donor E | Summary | |
| Age (years) | 43.2 | 51.4 | 48.5 | 40.8 | 52.3 | 48.5 [40.8 to 52.3] |
| Time since HIV diagnosis (years) | 23 | 15.5 | 7.4 | 7.4 | 23.5 | 15.5 [7.4 to 23.5] |
| Time on ART (years) | 18.4 | 13.5 | 7.3 | 7.3 | 23 | 13.5 [7.3 to 23] |
| Viral load (HIV-1 RNA cp/ml plasma) | 50 | 185 | 91 | 20 | 20 | 50 [20 to 185] |
| Nadir CD4 (cell/mm3) | 216 | 263 | 356 | 338 | 328 | 328 [216 to 356] |
| Absolute CD4 count (cell/mm3) | 682 | 878 | 998 | 1062 | 1092 | 998 [682 to 1092] |
| Percentage CD4 (%) | 55 | 42 | 43 | 37 | 39 | 42 [37 to 55] |
| CD4/CD8 ratio (median [range]) | 2.4 | 1.4 | 0.9 | 0.7 | 1.1 | 1.1 [0.7 to 2.4] |
Abbreviations: ART, antiretroviral therapy
Both shared and distinct features are observed between T cells from gut vs. FRT
Live, singlet cell events were exported from the CyTOF datasets and gated separately for CD4+ and CD8+ T cells. As an original goal of this study was to try to characterize HIV-infected cells that persist in WLWH (e.g., due to diminished ART drug concentrations within tissues), we first assessed whether we could identify any HIV-infected cells from our specimens. Although we could detect a population of HIV Gag+ cells among in vitro-infected T cells, which had downregulated cell-surface CD4 as expected (21) (Fig. 1A), Gag-expressing cells were absent from any of our patient specimens (Fig. 1B). These results suggest that ART fully suppressed HIV protein expression in our cohort of WLWH including within the tissue compartment (although HIV RNA was detectable (12)), and/or that the numbers of T cells we obtained from the specimens were too low to detect infected cells. Therefore, for the remainder of the study we focused on comparing the phenotypes of total CD4+ T cells (CD3+CD4+CD8-) or CD8+ T cells (CD3+CD4-CD8+) between the different sampling sites.
Figure 1. No HIV-infected cells were detectable in blood or tissue specimens from five ART-suppressed women living with HIV.
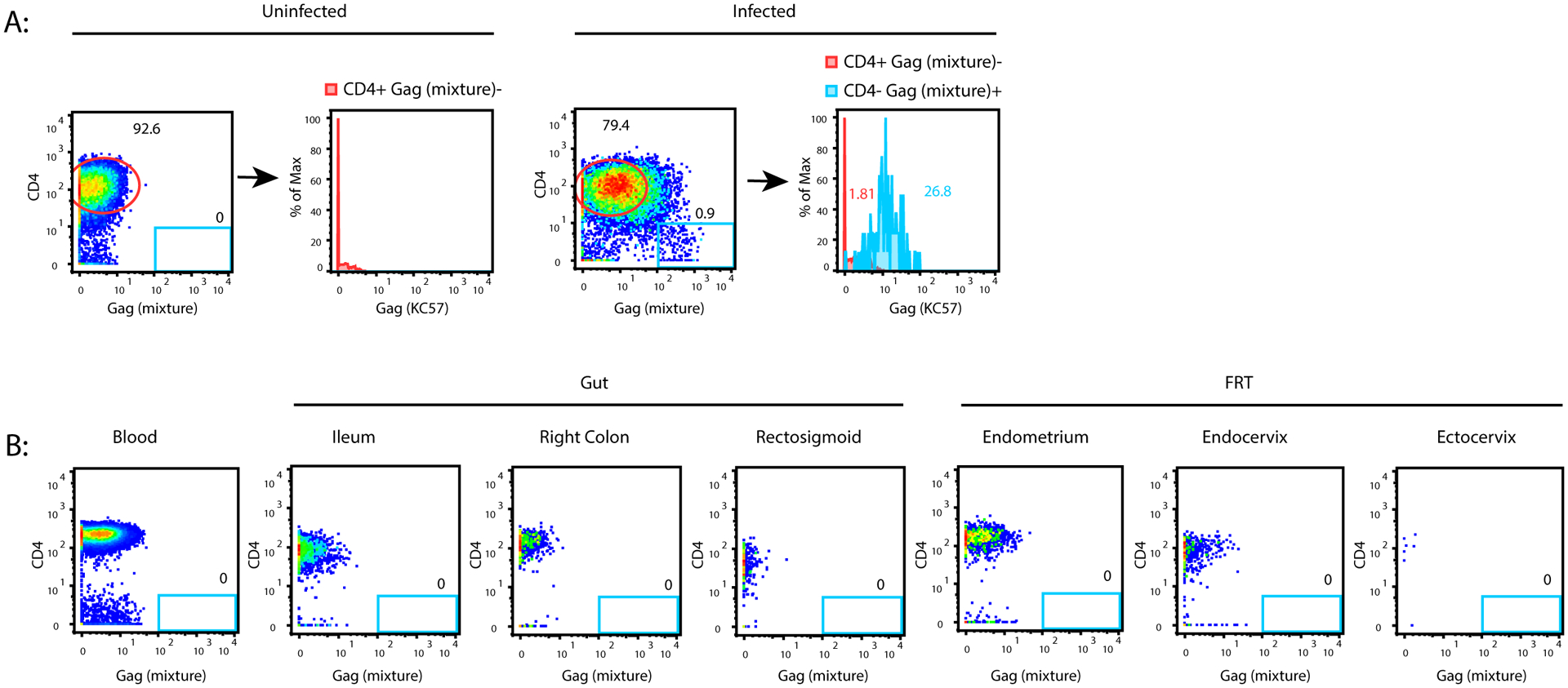
(A) Validation of HIV-1 Gag protein detection reagents. Tonsils from HIV-uninfected donors were mock-treated (left) or infected in vitro (right) with a CCR5-tropic HIV-1 reporter virus (F4.HSA) and analyzed by CyTOF. Events corresponding to live, singlet CD3+CD8- cells were then exported and visualized as 2D dot plots for expression of CD4 and Gag as assessed by a mixture of multiple anti-Gag antibody clones (Table 2). The blue gate shows a clear population of Gag+ cells that had downregulated cell-surface CD4 in the infected sample. That population is absent in the uninfected sample. The histogram plots on the right demonstrate that Gag+ cells identified in this manner also bound the anti-Gag antibody KC57 labeled for detection on a different CyTOF channel (blue histogram). The red histograms correspond to the circled populations of CD4+Gag- cells on the 2D dot plots, and serve as a negative control for populations that should not stain for KC57. (B) No Gag+ cells were detectable in blood or tissue specimens from HIV+ participants. Live, singlet CD3+CD8- cells from blood, ileum, right colon, rectosigmoid, endometrium, endocervix and ectocervix from a representative donor demonstrating no detectable Gag+ cells in these specimens. Similar results were observed for the remaining 4 donors.
We first visualized all the datasets together by tSNE. Not surprisingly, blood T cells resided in distinct areas of the tSNE relative to gut and FRT T cells, within both the CD4+ (Fig. 2A, left) and CD8+ (Fig. 2B, left) compartments. A large overlap between the gut and FRT T cells within this tSNE space suggested shared features between T cells from these mucosal sites. However, when the tSNE was repeated with only the tissue specimens, it became apparent that T cells from the gut and FRT segregated apart from each other, within both the CD4+ (Fig. 2A, right) and CD8+ (Fig. 2B, right) compartments. Of note, for both the blood and tissue T cells, some heterogeneity was also observed between donors. Since the 3 gut sites largely overlapped with one another, as did the 3 FRT sites, for the remainder of the study we combined the three sites from each tissue unless otherwise indicated, which gave us larger numbers of cells for our analyses. Overall, this initial view of the datasets suggests both shared and distinct features between T cells from the gut vs. FRT of WLWH, as well as some heterogeneity between donors.
Figure 2. T cells from gut and female reproductive tract (FRT) exhibit both common and distinguishing features, and are distinct from T cells from blood.

Donor-matched T cells from blood, three gut sites (ileum, right colon, and rectosigmoid) and three FRT sites (endometrium, endocervix, and ectocervix) were obtained from five ART-suppressed women living with HIV (WLWH) and phenotyped by CyTOF. Datasets corresponding to CD4+ (A) or CD8+ (B) T cells from each of the sites were visualized by tSNE. Analysis of the blood specimens within the same tSNE space as the tissue specimens (left side of panels) suggests shared features between gut and FRT T cells distinguishing them from blood T cells, as gut and FRT cells reside in a shared region of the tSNE distinct from the region occupied by the blood cells. By contrast, tSNE analysis of the tissue specimens alone (right side of panels), unveils phenotypic differences between gut and FRT cells, as gut and FRT cells reside in distinct areas. Each donor is shown in a separate column, but for each analysis all donors were analyzed within the same tSNE space. The source of cells from each donor is indicated in the colored key at the top. Missing entries correspond to tissue sites where not enough T cells were isolated for CyTOF analysis. As the phenotypes of T cells were overall similar between the three gut sites, and between the three FRT sites, for the remainder of the study we combined the datasets for the three gut sites together, and the three FRT sites together.
Gut and FRT T cells are predominantly memory T cells expressing the Trm marker CD69 and lacking expression of Tcm markers
To compare the subset distribution of the donor-matched T cells between the blood, gut, and FRT compartments, we first performed manual gating. CD4+ and CD8+ T cells were gated for T naïve (Tn), T central memory (Tcm), T effector memory (Tem), T transitional memory (Ttm), and two populations of T resident memory (Trm) cells: those expressing CD69 (CD69+ Trm), and those co-expressing CD69 and CD103 (CD69+CD103+ Trm) (Fig. S1). CD4+ T cells were additionally gated for T follicular helper (Tfh), Th1, Th17, and Th1Th17 cells (Fig. S1A). Memory CD4+ and CD8+ T cells were more frequent in both tissues relative to blood, with gut harboring the highest frequencies (Fig. 3). Correspondingly, naïve T cells were less frequent in the tissues, particularly the gut. Within both the CD4+ and CD8+ compartments, Tcm cells were more abundant in the blood than in either tissue, which harbored negligible numbers of these cells. Also, within both the CD4+ and CD8+ compartments, CD69+ Trm cells were more abundant in either tissue as compared to blood. Interestingly, while a subset of these CD69+ Trm cells from gut co-expressed CD103, such cells were rare in the FRT. These results overall suggest that relative to blood T cells, gut and FRT T cells are enriched for memory T cells including Trm cells, but disenriched for cells of the Tcm phenotype.
Figure 3. Frequencies of canonical T cell subsets in blood, gut, and FRT.
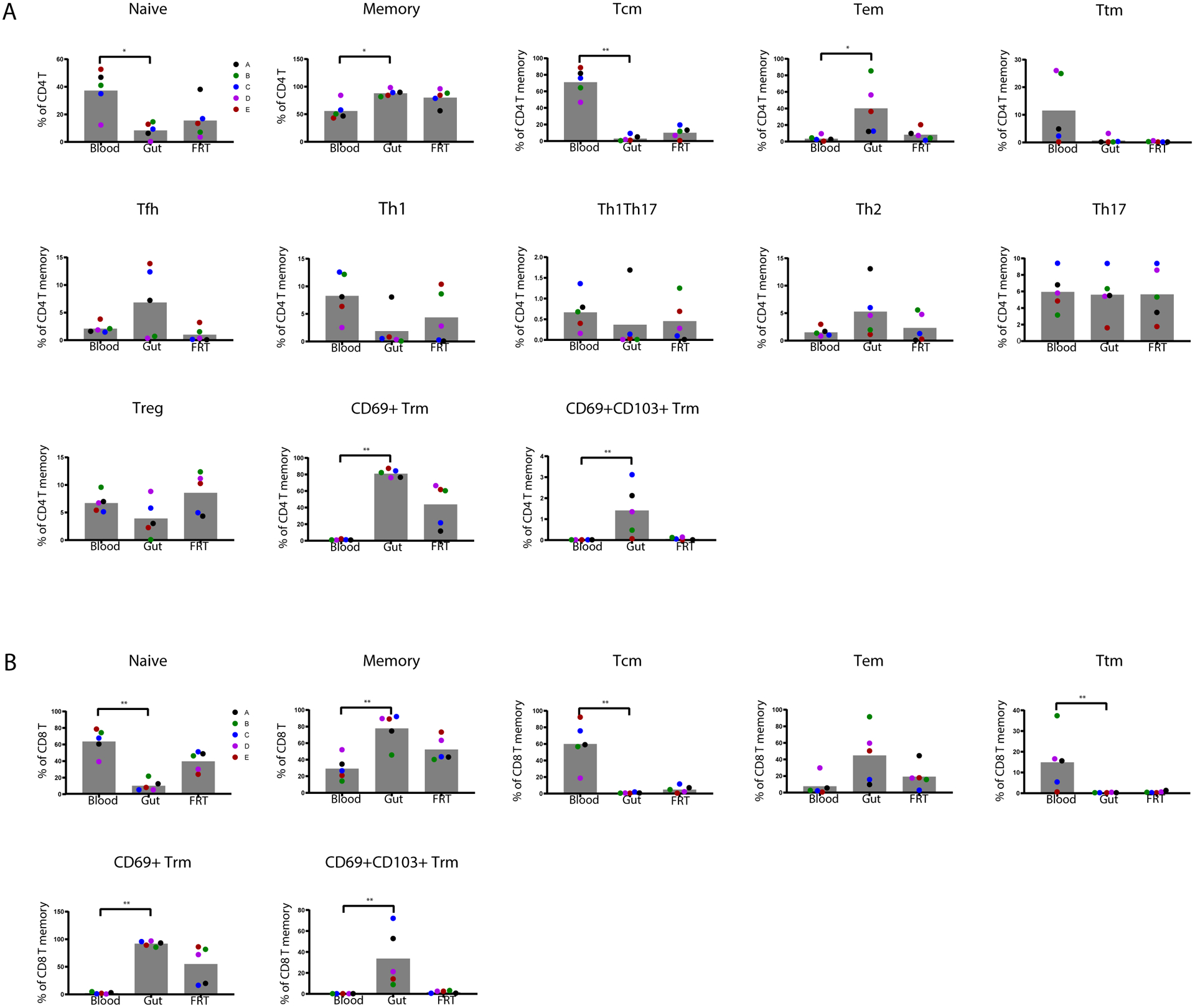
T cell subsets defined in Fig. S1 were assessed for relative frequencies among CD4+ (A) and CD8+ (B) T cells from the blood and indicated tissue compartments. * p < 0.05 and ** p < 0.01 as determined by a Kruskal-Wallis test with Dunn’s correction. Each colored dot corresponds to a different donor (n=5 donors).
We then delved deeper into the CyTOF datasets by taking advantage of phenotyping markers beyond those corresponding to the canonical subsets. Comparison of the mean signal intensity (MSI) of all the T cell antigens revealed some differentially expressed antigens between T cells from blood, gut, and FRT (Fig. S2). For example, among CD4+ T cells, CD27 and CD30 were both significantly elevated in the blood relative to both tissues, while CD69 was elevated within tissues, consistent with the manual gating data.
As the amount of HIV DNA and RNA within the blood, gut, and FRT of these donors had previously been quantified (12), we looked for potential correlations between the donors’ T cell populations and reservoir size. To this end, we compared frequencies of any manually gated subset or CyTOF phenotyping antigen levels (as assessed by MSI) to levels of total HIV DNA, and to levels of initiated (based on levels of HIV TAR) or 5’ elongated (based on levels of “Long-LTR” [R-U5-pre-Gag]) HIV RNA in the same tissues. No correlations were statistically significant, likely due to the limited number of participants analyzed. However, the specimens from the three gut sites tended to exhibit similar patterns: for example, the donors with the highest levels of HIV transcriptional elongation also had the lowest proportions of Tregs and of CD8+ CD69+ and CD69+CD103+ Trm cells in the rectosigmoid, as well as rectum and ileum (Fig. S3). These results suggest the possibility that within the gut, the transcriptionally-active reservoir resides outside the Treg population of memory CD4+ T cells (22), although further studies would be needed to directly test this.
Features of gut and FRT T cells distinguishing them from their blood counterparts
To distinguish the T cells from the three different sites in a more unbiased manner, we performed FlowSOM clustering (19). Depiction of the cluster distribution as pie graphs revealed limited variations between the donors relative to the tissue-associated signatures (Fig. 4A, B, pie charts). In particular, some clusters were over-represented in blood relative to tissues while others were over-represented in tissues (Fig. 4A, B, bar graphs).
Figure 4. Clustering reveals phenotypic differences between T cells from blood vs. tissues.
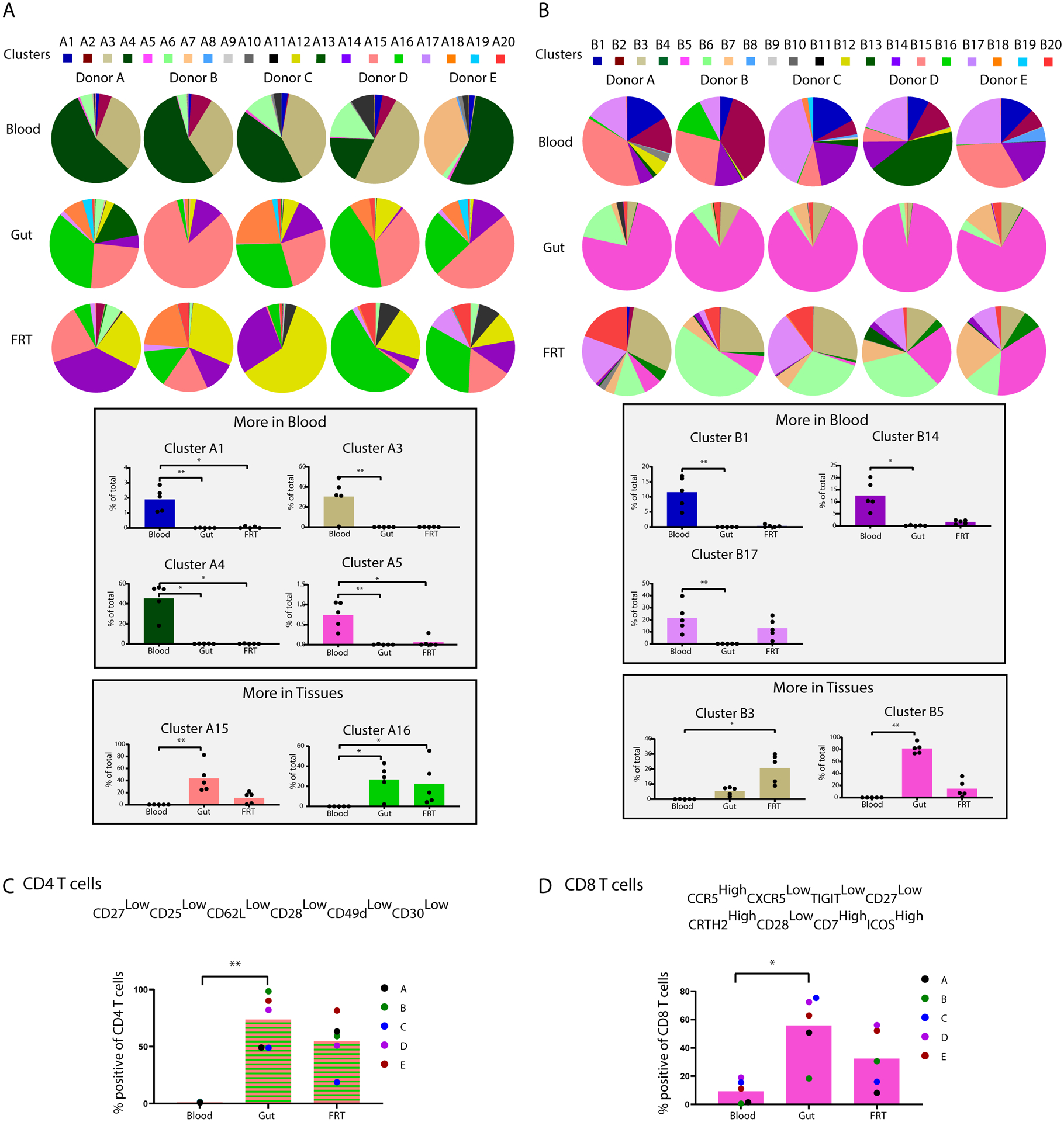
CD4+ (A) and CD8+ (B) T cells from blood, gut, and FRT were separated into 20 clusters each (A1-A20, and B1-B20) by FlowSOM. The top portions of the two panels show cluster distributions depicted as pie charts, separated by donor. The bottom portions of the two panels depict bar graphs of clusters that were significantly over-represented in the indicated blood or tissue compartment. Validation of markers defining tissue CD4+ (C) and CD8+ (D) T cells was then performed by selecting antigens differentially expressed in the clusters enriched in tissues and conducting manual gating. This analysis demonstrated that relative to blood, gut and FRT CD4+ T cells are preferentially CD27lowCD25lowCD62LlowCD28lowCD49dlowCD30low. Relative to blood, gut and FRT CD8+ T cells are preferentially CCR5highCXCR5lowTIGITlowCD27lowCRTH2highCD28lowCD7highICOShigh. * p < 0.05 and ** p < 0.01 as determined by a Kruskal-Wallis test with Dunn’s correction (n=5 donors).
To better define the phenotypic features of the tissue T cells, we assessed which antigens were differentially expressed among the tissue-enriched CD4+ (A15 and A16) and CD8+ (B3 and B5) clusters. Consistent with the notion that T cells within tissues are primarily memory cells, all four of these clusters expressed high levels of the memory marker CD45RO and low levels of the naïve marker CD45RA (Fig. S4A–D). Clusters A15 and B3 preferentially expressed CD69 but not CD103, while clusters A16 and B5 included both cells expressing CD69 in the absence of CD103, as well as CD69+CD103+ cells (Fig. S4A–D). These results suggest enrichment of multiple types of Trm cells within the tissues.
Multiple chemokine receptors as well as other surface antigens were also differentially expressed among these clusters as compared to total T cells. For instance, markers differentially expressed in both of the tissue-associated CD4+ T cell clusters (A15 and A16) included CD27, CD25, CD62L, CD28, CD49d, and CD30, all of which were expressed at lower levels in these two clusters compared to total CD4+ T cells from all sampled sites (Fig. S4A, B). To validate whether cells expressing low levels of these antigens were indeed preferentially present within the tissues, we gated on CD4+ T cells that were CD27lowCD25lowCD62LlowCD28lowCD49dlowCD30low. The frequency of these cells was indeed lower in the blood than in either tissue compartment (Fig. 4C), thereby validating these phenotypic features as shared features of gut/FRT T cells that distinguish them from their blood counterparts. A similar analysis among the tissue-associated CD8+ T cell cluster B5 revealed CCR5, CRTH2, CD7, and ICOS to be expressed at higher levels, while CXCR5, TIGIT, CD27, and CD28 were expressed at lower levels, in these two clusters as compared to total CD8+ T cells (Fig. S4C, D). Manual gating validation demonstrated that CD8+ T cells that were CCR5highCXCR5lowTIGITlowCD27lowCDTh2highCD28lowCD7highICOShigh were indeed more frequent in both gut and FRT as compared to blood (Fig. 4D).
Features of gut and FRT T cells distinguishing them from each other
Having demonstrated and described some phenotypic features of T cells shared between the gut and FRT, we next wanted to better characterize the differences between these two compartments’ T cells. We repeated the FlowSOM clustering of the CD4+ and CD8+ T cells using just the tissue specimens and found different cluster distributions between the two tissue sites, along with some donor-dependent features (Figure 5A, B, pie charts). Moreover, for both CD4+ and CD8+ T cells, some clusters were enriched in the gut, and others in the FRT (Figure 5A, B, bar graphs). The gut-associated clusters from CD4+ and CD8+ T cells (C2 and D2) shared phenotypic features of memory (CD45RO+) cells expressing high levels of Trm marker CD69 and low levels of Tcm markers CCR7 and CD27 (Fig. S4E, F). Manual gating of CD45ROhighCD69highCCR7lowCD27low cells within both the CD4+ and CD8+ T cell subsets revealed these cells to be significantly enriched in the gut as compared to the FRT (Fig. 5C, D). These results suggest that CD69+ Trm cells of the effector memory (CCR7lowCD27low) phenotype are more prevalent in the gut than FRT.
Figure 5. Clustering reveals phenotypic differences between T cells from gut vs. FRT.
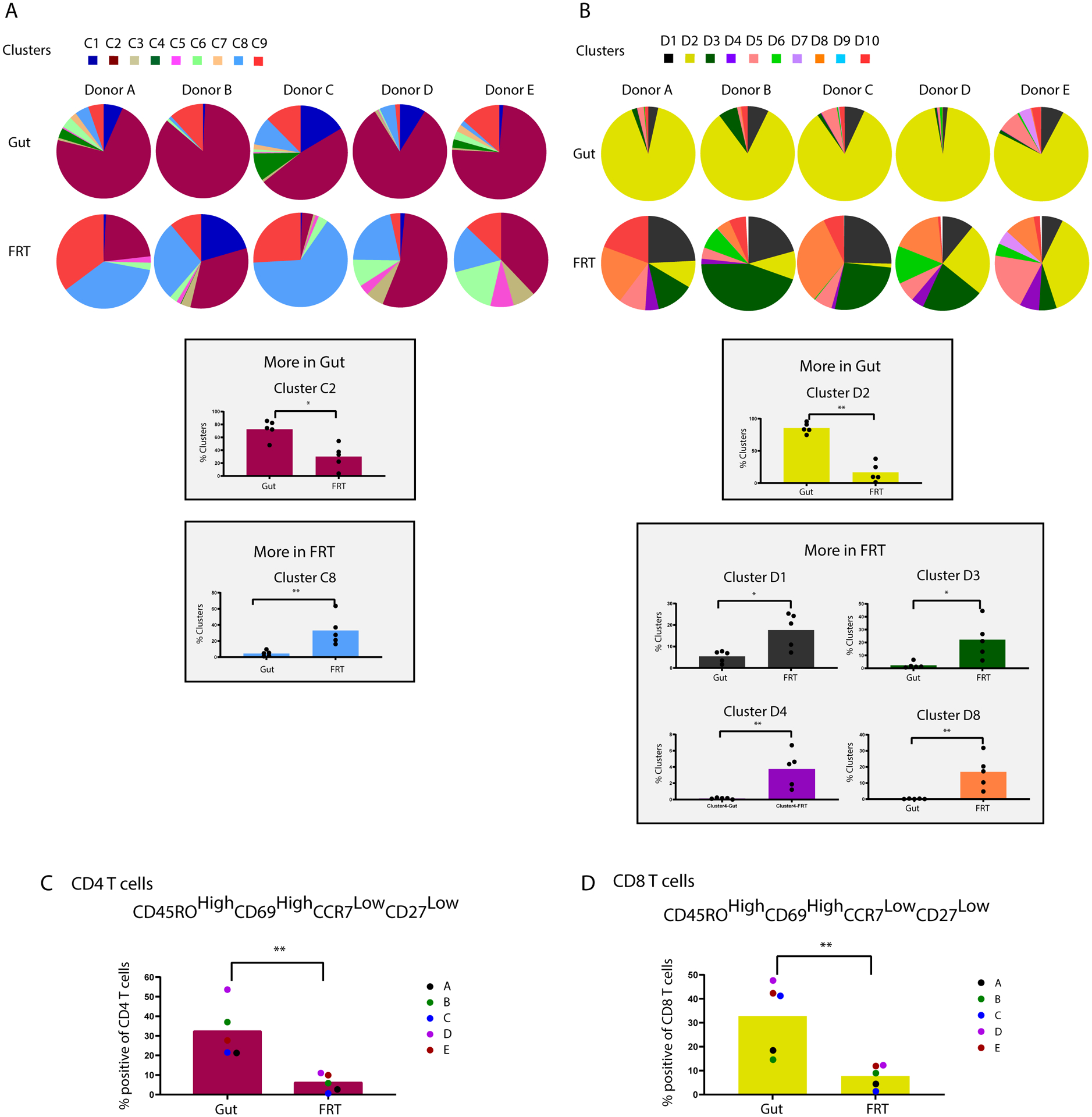
CD4+ (A) and CD8+ (B) T cells from gut and FRT were reclustered (C1-C9, and D1-D10) by FlowSOM, after removal of the blood specimens’ data. The top portions of the two panels show cluster distributions depicted as pie charts, separated by donor. The bottom portions of the two panels depict bar graphs of clusters that were significantly over-represented in gut or FRT. Validation of markers preferentially expressed on gut CD4+ (C) and CD8+ (D) T cells was then performed by selecting antigens differentially expressed in the clusters enriched in the gut as compared to FRT (C2 and D2), and conducting manual gating. This analysis demonstrated that relative to their FRT counterparts, both CD4+ and CD8+ T cells from gut are preferentially CD45ROhighCD69highCCR7lowCD27low. * p < 0.05 and ** p < 0.01 as determined by a Mann-Whitney U test (n=5 donors).
CD69+CD103+ Trm cells exhibit defining features in a manner dependent on tissue site
A recent study reported that among CD4+ T cells within the lower FRT, CD69+CD103+ Trm cells exhibited unique transcriptional and phenotypic features, including preferential expression of PD1 and CCR5, relative to their CD69-CD103- counterparts (4). We confirmed within our FRT datasets that CD69+CD103+ CD4+ Trm cells expressed higher levels of both PD1 and CCR5 than CD69-CD103- CD4+ T cells did, and furthermore discovered this to be true among CD8+ T cells as well (Fig. 6A, C). When we assessed the gut compartment in a similar manner, we found that like in the FRT, CD69+CD103+ Trm cells preferentially expressed CCR5 (Fig. 6B, D). Surprisingly, however, PD1 was not preferentially expressed on CD69+CD103+ Trm cells, and in fact was expressed at lower levels relative to their CD69-CD103- counterparts (Fig. 6B, D). These results suggest that the unique phenotypic features of CD69+CD103+ Trm cells are dependent on the mucosal site.
Figure 6. Phenotypic features of CD69+CD103+ Trm cells in tissues.
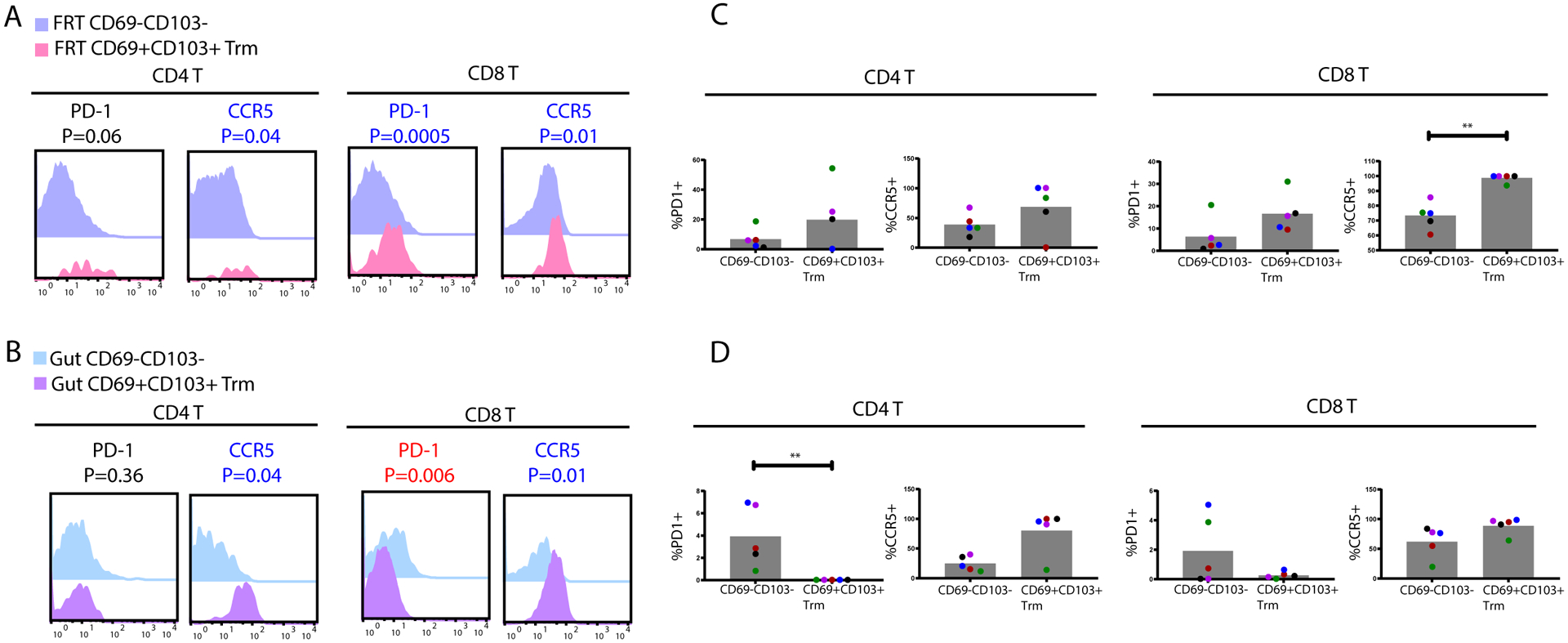
FRT (A) and gut (B) CD69+CD103+ Trm cells express higher levels of PD1 and CCR5 relative to their respective CD69-CD103- T memory (Tm) counterparts. Shown are overlaid histograms from the CD4+ (left) and CD8+ (right) T cell subsets, corresponding to concatenated events from all donors. Antigens significantly (p < 0.05) overexpressed among the CD69+CD103+ populations are in blue font, while those underexpressed are in red font. P-values were determined by a Student’s paired t-test and adjusted for multiple testing using the Benjamini-Hochberg for FDR. The proportions of CD69-CD103- vs. CD69+CD103+ Tm cells from FRT (C) or gut (D) were compared by manual gating for the percentages of cells expressing PD1 or CCR5. * p < 0.05 and ** p < 0.01 as determined by a Mann-Whitney U test (n=5 donors).
Distinguishing features of Tim cells are shared between the CD4/CD8 subsets and tissue sites
FRT-derived CD69-CD103- CD8+ T cells, termed Tim cells, were recently reported to harbor unique phenotypic features relative to their blood counterparts (5). More specifically, these cells were shown to exhibit inflammatory and cytotoxic features, and to preferentially express CXCR4, a chemokine receptor that can direct immune cells to tissue sites of inflammation. We confirmed preferential expression of CXCR4 on CD8+ Tim cells from the FRT, and further demonstrated that this preferential expression occurred among CD69-CD103- T cells from both the CD4+ and CD8+ compartments, and in a manner also observed in the gut (Fig. 7A–D). As Tim cells have been suggested to have distinct migratory patterns, we further assessed whether other chemokine receptors were differentially expressed on these cells. Like CXCR4, CCR5 was over-expressed on CD69-CD103- T cells in both tissue sites as compared to blood, and among both CD4+ and CD8+ T cells (Fig. 7A–D). By contrast, CCR6 was under-expressed on CD69-CD103- T cells in both tissue sites as compared to blood, and this was true among both CD4+ and CD8+ T cells (Fig. 7A–D). Taken together, these results suggest that CD69-CD103- T cells from tissues are preferentially CCR5+CCR6-, which we confirmed by manual gating (Fig. 7E). We conclude that Tim cells may not be unique to the CD8 compartment or to the FRT, but rather may be a common subset persisting in multiple mucosal tissues.
Figure 7. Differential expression of chemokine receptors in tissue- as compared to blood-derived CD69-CD103- T cells.
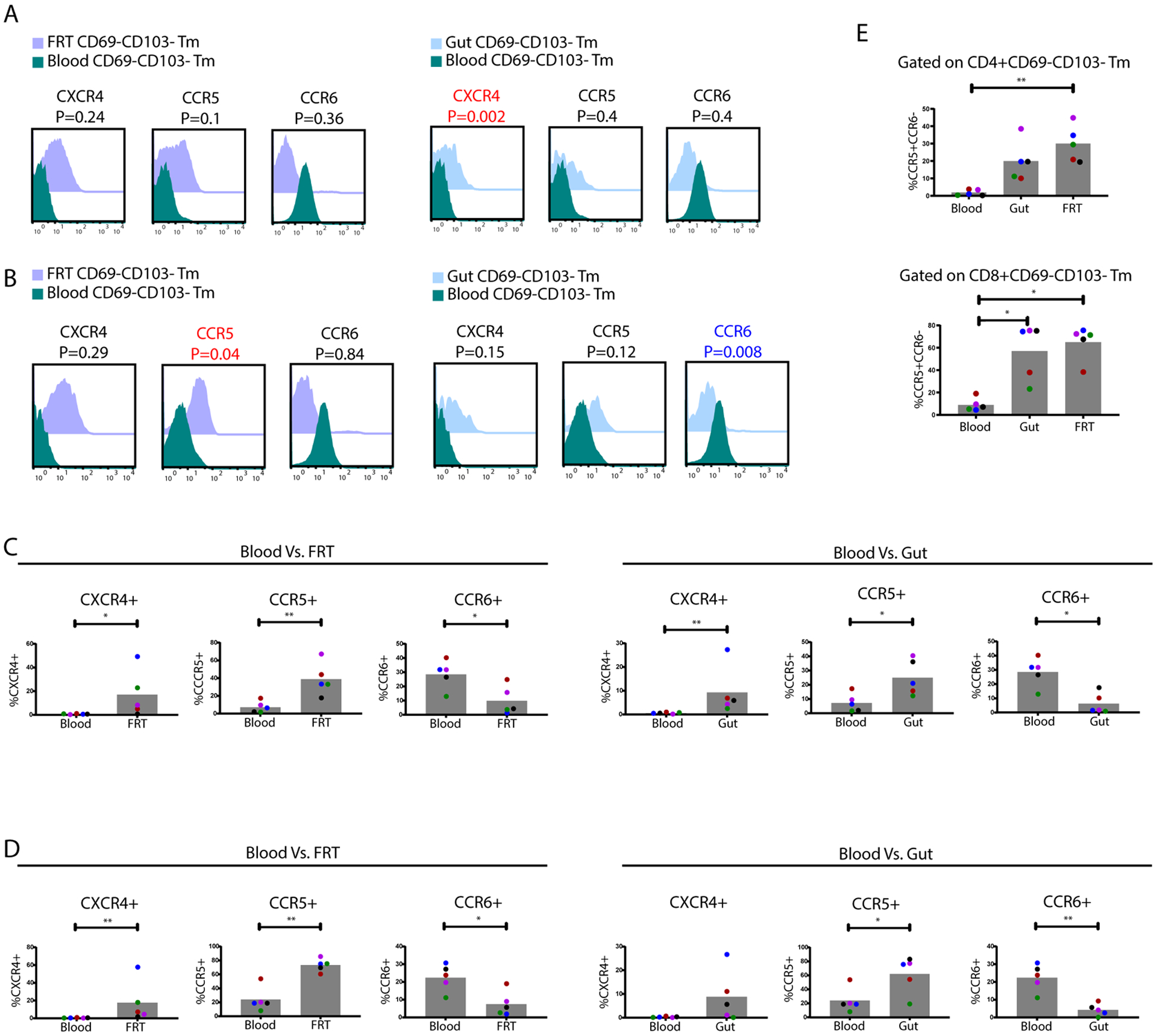
CD4+ (A) and CD8+ (B) CD69-CD103- Tm cells from FRT and gut were compared to those in blood for expression levels of CXCR4, CCR5, and CCR6, and depicted as overlaid histograms concatenated from all donors. Antigens significantly (p < 0.05) overexpressed among the gut/FRT relative to blood CD69-CD103- Tm cells are in red font, while those underexpressed are in blue font. P-values were determined by a Student’s paired t-test and adjusted for multiple testing using the Benjamini-Hochberg for FDR. The proportions of CD4+ (C) and CD8+ (D) CD69-CD103- Tm cells from FRT and gut were compared by manual gating to their counterparts in blood for percentages of cells expressing CXCR4, CCR5, or CCR6. P-values were determined by a Mann-Whitney U test. * p < 0.05 and ** p < 0.01 (n=5 donors). (E) The over-representation of CCR5+CCR6- cells among CD69-CD103- Tm cells in tissues as compared to blood was validated by manual gating. P values were determined by a Kruskal-Wallis test with Dunn’s correction. * p < 0.05 and ** p < 0.01 (n=5 donors).
DISCUSSION
By recruiting five well-matched, ART-suppressed WLWH to donate blood and tissue specimens from six sites of the gut and FRT in the same study visit for CyTOF phenotyping, we were able to conduct an in-depth comparison of T cells from circulation to T cells from these two major mucosal sites. Despite being generated from a relatively small cohort of women, we believe that our reference dataset – downloadable via a link provided in the methods section – will be of use to the field because 1) our participants were clinically well-matched (all women not on hormonal contraceptives, and all sampled during the midluteal phase of the menstrual cycle); 2) each set of participant specimens came from the same study visit thus excluding differences due to longitudinal sampling; 3) our specimens were obtained purely for research purposes and therefore tissues were not removed due to disease or a treatment-related procedure; and 4) our specimens were all analyzed without ever having gone through any cell cryopreservation. Implementation of our 36-parameter T cell-centric CyTOF panel on these specimens allowed for an in-depth comparison of T cells from the sampled sites. Overall, we found both common and distinguishing T cell features between the gut and FRT mucosa, as well as between CD4+ and CD8+ T cells within each site.
That commonalities exist between gut and FRT T cells was evidenced by these cells residing in overlapping areas of the tSNE distinct from their blood counterparts, by their sharing expression patterns of multiple T cell antigens, and by their harboring different distributions of T cell subsets relative to blood T cells. For example, relative to T cells in circulation, those from gut and FRT both expressed low levels of CD28, and manually-identified subsets of CD4+ and CD8+ T cells validated to be enriched within mucosal sites, were CD28low. T cells expressing low levels of CD28 were previously described from the gut mucosa as those that had recently undergone signaling through the TCR (2), and our data suggest that such sustained activation through the TCR likely also occurs in the FRT.
A second conserved feature between T cells from the two mucosal sites was the population of Tim cells, recently-described CD8+ T cells exhibiting a CD69-CD103- phenotype. These pro-inflammatory cells identified from the FRT were reported to express CXCR4 at higher levels than CD69-CD103- CD8+ T cells from blood (5), data which we confirmed. We further demonstrated that preferential expression of CXCR4 also occurred on CD8+ Tim cells from the gut, and that within both FRT and gut, CD69-CD103- CD4+ T cells also preferentially expressed CXCR4. Interestingly, other chemokine receptors were also differentially expressed on mucosal CD4+ and CD8+ Tim cells, including higher expression of CCR5 and lower expression of CCR6 relative to their blood CD69-CD103- counterparts. As CXCR4 and CCR5 are co-receptors for HIV, these observations also suggest that CD4+ Tim cells may be important targets for HIV infection, and may help explain the high susceptibility of mucosal CD4+ T cells to HIV infection (17).
We also found that relative to blood, gut and FRT preferentially harbored Trm cells and were devoid of Tcm cells, consistent with prior studies (1–5, 23, 24). The tissue Trm cells from gut and FRT shared phenotypic features including preferential expression of CCR5, consistent with the notion of a “core signature” of Trm cells shared between different tissue sites (3). At the same time, we also found interesting differences in the quantity and quality of Trm cells from the gut vs. FRT. While CD69+ Trm cells were present in both gut and FRT, they were more abundant in the gut. These cells were of the Tem phenotype (CCR7lowCD27low), and indeed memory CD69+ Tem cells were significantly higher in gut as compared to FRT, among both CD4+ and CD8+ T cells. Furthermore, while the subset of CD69+ Trm cells expressing CD103 were clearly present in gut, they were detected at very low levels in the FRT. The reason for the overrepresentation of CD69+CD103+ Trm cells in the gut as compared to FRT is unclear, but may in part be due to the high abundance of TGF-β1 in the gut, which upregulates CD103 expression (25). Of note, however, CD69+CD103+ CD4+ Trm cells are more abundant in vaginal than cervical sites of the FRT (4). Given that the vaginal tract (not examined in our study) and gut are both sites highly colonized by microbiota, it is possible that CD69+CD103+ Trm cells are elevated in tissues with high microbiome load. Interestingly, CD69+CD103+ CD4+ Trm cells exhibit Th17 signatures and can induce IL17 (4), which can elicit anti-bacterial responses (26). Together with observations that IL17 levels associate with responses to intestinal bacteria (27, 28), these results suggest a potential role for CD69+CD103+ Trm cells in tolerating or responding to commensal microbiota, although future studies will be necessary to directly test this hypothesis.
In addition to quantitative differences in Trm cells between the gut and FRT, we also found phenotypic differences. In the FRT, CD69+CD103+ Trm cells preferentially expressed PD1 relative to CD69-CD103- cells, consistent with what was previously reported (4). In the gut, however, we found that CD69+CD103+ CD4+ Trm cells do not preferentially express PD1, and that CD69+CD103+ CD8+ Trm cells in fact express lower levels of PD1 relative to their CD69-CD103- counterparts. The molecular basis and functional outcome of this observation is unclear, but it could suggest a higher need for PD1-mediated checkpoint inhibition in CD69+CD103+ Trm cells in the FRT than is necessary in the gut. As the organ supporting the development of an allogeneic fetus, we postulate that perhaps the FRT requires more tolerogenic mechanisms as compared to the gut, and that PD1-mediated tolerance may be particularly important.
Our study has limitations. We only analyzed a small cohort of women, although we made efforts to clinically match them including by cycle phase. Despite that, some heterogeneity was observed between T cells within each site. This highlights that even attempts to match participants can still result in phenotypic heterogeneity, a challenge that stems from the diversity of even well-matched participants. As sex steroids can influence immune cell distribution and functions, future studies should investigate to what extent the T cell phenotypes we described in the gut and FRT differ during other phases of the menstrual cycle, as well as after menopause and in women taking oral contraceptives. In addition, to what extent the phenotypes described here among ART-suppressed WLWH are generalizable to HIV-uninfected women should be established. A second limitation of our study is that limited numbers of T cells could be isolated for immunophenotyping. This may have been one reason for our inability to identify any productively-infected cells in the specimens, although we cannot rule out complete suppression of viral protein expression by ART as another possibility. The low cell yield in some specimens also meant that some biopsies did not have enough cells for meaningful T cell analysis by CyTOF. Although most donors had enough cells from all three gut sites, the endometrium was the only site of the FRT that yielded enough cells from all donors for both CD4+ and CD8+ T cell analysis. The limited numbers of cells from some of the mucosal sites, particularly the endocervix and ectocervix, may have limited our ability to detect phenotypic differences between different sampling sites within each type of mucosa (gut vs. FRT). Future studies using specimens allowing for higher cell yields, for example from post-mortem HIV+ donors participating in end-of-life studies (29), would be highly valuable in that regard.
Supplementary Material
KEY POINTS.
CyTOF on matched blood, gut, and genital T cells from ART-suppressed HIV+ women
Shared and distinct phenotypic features of T cells from gut and genital tracts
Gut has more T resident memory (Trm), but genital Trm preferentially express PD1
ACKNOWLEDGEMENTS
The authors gratefully acknowledge the contributions of the study participants and dedication of the staff at the MWCCS sites. We thank N. Lazarus and E. Butcher for the Act1 antibody; S. Tamaki, T.K. Peech, and C. Bispo for CyTOF assistance at the Parnassus Flow Core; T. Roberts and G. Maki for Graphics assistance; F. Chanut for editorial assistance; and R. Givens for administrative assistance.
FINANCIAL ACKNOWLEDGEMENTS
This work was supported by the National Institutes of Health (R01AI127219, R01AI147777, UM1AI164567, P01AI131374, and UM1AI164559 to N.R.R.; U01HL146242, to N.R.R. and P.C.T.; and W16036 to N.R.R. and S.A.Y.; U01AI034989 to R.M.G. and S.A.Y.; and R01DK108349, R01AI132128, and R01DK120387 to S.A.Y.) and the amfAR Institute for HIV Cure Research (109301). We acknowledge support from CFAR (P30AI027763) and the James B. Pendleton Charitable Trust. We thank the PFCC (RRID:SCR_018206) for assistance in CyTOF data acquisition, enabled by an instrument that was supported in part by the DRC Center Grant NIH P30 DK063720 and NIH S10 1S10OD018040. The funders had no role in study design, data collection and analysis, decision to publish, or preparation of the manuscript. The contents of this publication are solely the responsibility of the authors and do not represent the official views of the NIH. We acknowledge the MWCCS Data Analysis and Coordination Center (Gypsyamber D’Souza, Stephen Gange and Elizabeth Golub), U01-HL146193; and the MWCCS Northern California site (Bradley Aouizerat, Jennifer Price, and Phyllis Tien), U01-HL146242.
REFERENCES
- 1.Sathaliyawala T, Kubota M, Yudanin N, Turner D, Camp P, Thome JJ, Bickham KL, Lerner H, Goldstein M, Sykes M, Kato T, and Farber DL. 2013. Distribution and compartmentalization of human circulating and tissue-resident memory T cell subsets. Immunity 38: 187–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Thome JJ, Yudanin N, Ohmura Y, Kubota M, Grinshpun B, Sathaliyawala T, Kato T, Lerner H, Shen Y, and Farber DL. 2014. Spatial map of human T cell compartmentalization and maintenance over decades of life. Cell 159: 814–828. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Kumar BV, Ma W, Miron M, Granot T, Guyer RS, Carpenter DJ, Senda T, Sun X, Ho SH, Lerner H, Friedman AL, Shen Y, and Farber DL. 2017. Human Tissue-Resident Memory T Cells Are Defined by Core Transcriptional and Functional Signatures in Lymphoid and Mucosal Sites. Cell Rep 20: 2921–2934. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Woodward Davis AS, Vick SC, Pattacini L, Voillet V, Hughes SM, Lentz GM, Kirby AC, Fialkow MF, Gottardo R, Hladik F, Lund JM, and Prlic M. 2021. The human memory T cell compartment changes across tissues of the female reproductive tract. Mucosal Immunol 14: 862–872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Pattacini L, Woodward Davis A, Czartoski J, Mair F, Presnell S, Hughes SM, Hyrien O, Lentz GM, Kirby AC, Fialkow MF, Hladik F, Prlic M, and Lund JM. 2019. A pro-inflammatory CD8+ T-cell subset patrols the cervicovaginal tract. Mucosal Immunol 12: 1118–1129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Shacklett BL, and Greenblatt RM. 2011. Immune responses to HIV in the female reproductive tract, immunologic parallels with the gastrointestinal tract, and research implications. Am. J. Reprod. Immunol 65: 230–241. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neidleman JA, Chen JC, Kohgadai N, Muller JA, Laustsen A, Thavachelvam K, Jang KS, Sturzel CM, Jones JJ, Ochsenbauer C, Chitre A, Somsouk M, Garcia MM, Smith JF, Greenblatt RM, Munch J, Jakobsen MR, Giudice LC, Greene WC, and Roan NR. 2017. Mucosal stromal fibroblasts markedly enhance HIV infection of CD4+ T cells. PLoS Pathog 13: e1006163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Stieh DJ, Maric D, Kelley ZL, Anderson MR, Hattaway HZ, Beilfuss BA, Rothwangl KB, Veazey RS, and Hope TJ. 2014. Vaginal challenge with an SIV-based dual reporter system reveals that infection can occur throughout the upper and lower female reproductive tract. PLoS Pathog 10: e1004440. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Adimora AA, Ramirez C, Benning L, Greenblatt RM, Kempf MC, Tien PC, Kassaye SG, Anastos K, Cohen M, Minkoff H, Wingood G, Ofotokun I, Fischl MA, and Gange S. 2018. Cohort Profile: The Women’s Interagency HIV Study (WIHS). Int. J. Epidemiol 47: 393–394i. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.D’Souza G, Bhondoekhan F, Benning L, Margolick JB, Adedimeji AA, Adimora AA, Alcaide ML, Cohen MH, Detels R, Friedman MR, Holman S, Konkle-Parker DJ, Merenstein D, Ofotokun I, Palella F, Altekruse S, Brown TT, and Tien PC. 2021. Characteristics of the MACS/WIHS Combined Cohort Study: Opportunities for Research on Aging With HIV in the Longest US Observational Study of HIV. Am. J. Epidemiol 190: 1457–1475. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Harlow SD, Gass M, Hall JE, Lobo R, Maki P, Rebar RW, Sherman S, Sluss PM, de Villiers TJ, and Group SC. 2012. Executive summary of the Stages of Reproductive Aging Workshop + 10: addressing the unfinished agenda of staging reproductive aging. Menopause 19: 387–395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Moron-Lopez S, Xie G, Kim P, Siegel DA, Lee S, Wong JK, Price JC, Einachef N, Greenblatt RM, Tien PT, Roan NR, and Yukl SA. 2021. Tissue-specific differences in HIV DNA levels and mechanisms that govern HIV transcription in blood, gut, genital tract, and liver in ART-treated women. J. Int. AIDS Soc In press. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cavrois M, Banerjee T, Mukherjee G, Raman N, Hussien R, Rodriguez BA, Vasquez J, Spitzer MH, Lazarus NH, Jones JJ, Ochsenbauer C, McCune JM, Butcher EC, Arvin AM, Sen N, Greene WC, and Roan NR. 2017. Mass Cytometric Analysis of HIV Entry, Replication, and Remodeling in Tissue CD4+ T Cells. Cell Rep 20: 984–998. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Xie G, Luo X, Ma T, Frouard J, Neidleman J, Hoh R, Deeks SG, Greene WC, and Roan NR. 2021. Characterization of HIV-induced remodeling reveals differences in infection susceptibility of memory CD4(+) T cell subsets in vivo. Cell Rep 35: 109038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Hsiao F, Frouard J, Gramatica A, Xie G, Telwatte S, Lee GQ, Roychoudhury P, Schwarzer R, Luo X, Yukl SA, Lee S, Hoh R, Deeks SG, Jones RB, Cavrois M, Greene WC, and Roan NR. 2020. Tissue memory CD4+ T cells expressing IL-7 receptor-alpha (CD127) preferentially support latent HIV-1 infection. PLoS Pathog 16: e1008450. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Neidleman J, Luo X, Frouard J, Xie G, Hsiao F, Ma T, Morcilla V, Lee A, Telwatte S, Thomas R, Tamaki W, Wheeler B, Hoh R, Somsouk M, Vohra P, Milush J, James KS, Archin NM, Hunt PW, Deeks SG, Yukl SA, Palmer S, Greene WC, and Roan NR. 2020. Phenotypic analysis of the unstimulated in vivo HIV CD4 T cell reservoir. Elife 9: e55487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Ma T, Luo X, George AF, Mukherjee G, Sen N, Spitzer TL, Giudice LC, Greene WC, and Roan NR. 2020. HIV efficiently infects T cells from the endometrium and remodels them to promote systemic viral spread. Elife 9: e55487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, Burovski E, Peterson P, Weckesser W, Bright J, van der Walt SJ, Brett M, Wilson J, Millman KJ, Mayorov N, Nelson ARJ, Jones E, Kern R, Larson E, Carey CJ, Polat I, Feng Y, Moore EW, VanderPlas J, Laxalde D, Perktold J, Cimrman R, Henriksen I, Quintero EA, Harris CR, Archibald AM, Ribeiro AH, Pedregosa F, van Mulbregt P, and SciPy C. 2020. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat. Methods 17: 261–272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Van Gassen S, Callebaut B, Van Helden MJ, Lambrecht BN, Demeester P, Dhaene T, and Saeys Y. 2015. FlowSOM: Using self-organizing maps for visualization and interpretation of cytometry data. Cytometry A 87: 636–645. [DOI] [PubMed] [Google Scholar]
- 20.Trapecar M, Khan S, Roan NR, Chen TH, Telwatte S, Deswal M, Pao M, Somsouk M, Deeks SG, Hunt PW, Yukl S, and Sanjabi S. 2017. An Optimized and Validated Method for Isolation and Characterization of Lymphocytes from HIV+ Human Gut Biopsies. AIDS Res Hum Retroviruses 33: S31–S39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Garcia JV, and Miller AD. 1991. Serine phosphorylation-independent downregulation of cell-surface CD4 by nef. Nature 350: 508–511. [DOI] [PubMed] [Google Scholar]
- 22.Yukl SA, Khan S, Chen TH, Trapecar M, Wu F, Xie G, Telwatte S, Fulop D, Pico AR, Laird GM, Ritter KD, Jones NG, Lu CM, Siliciano RF, Roan NR, Milush JM, Somsouk M, Deeks SG, Hunt PW, and Sanjabi S. 2020. Shared Mechanisms Govern HIV Transcriptional Suppression in Circulating CD103+ and Gut CD4+ T Cells. J. Virol [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Yukl SA, Shergill AK, Ho T, Killian M, Girling V, Epling L, Li P, Wong LK, Crouch P, Deeks SG, Havlir DV, McQuaid K, Sinclair E, and Wong JK. 2013. The distribution of HIV DNA and RNA in cell subsets differs in gut and blood of HIV-positive patients on ART: implications for viral persistence. J. Infect. Dis 208: 1212–1220. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Yukl SA, Shergill AK, Girling V, Li Q, Killian M, Epling L, Li P, Kaiser P, Haase A, Havlir DV, McQuaid K, Sinclair E, and Wong JK. 2015. Site-specific differences in T cell frequencies and phenotypes in the blood and gut of HIV-uninfected and ART-treated HIV+ adults. PLoS One 10: e0121290. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Zhang N, and Bevan MJ. 2013. Transforming growth factor-beta signaling controls the formation and maintenance of gut-resident memory T cells by regulating migration and retention. Immunity 39: 687–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.McGeachy MJ, Cua DJ, and Gaffen SL. 2019. The IL-17 Family of Cytokines in Health and Disease. Immunity 50: 892–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ivanov II, Frutos Rde L, Manel N, Yoshinaga K, Rifkin DB, Sartor RB, Finlay BB, and Littman DR. 2008. Specific microbiota direct the differentiation of IL-17-producing T-helper cells in the mucosa of the small intestine. Cell Host Microbe 4: 337–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ivanov II, Atarashi K, Manel N, Brodie EL, Shima T, Karaoz U, Wei D, Goldfarb KC, Santee CA, Lynch SV, Tanoue T, Imaoka A, Itoh K, Takeda K, Umesaki Y, Honda K, and Littman DR. 2009. Induction of intestinal Th17 cells by segmented filamentous bacteria. Cell 139: 485–498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaillon A, Gianella S, Dellicour S, Rawlings SA, Schlub TE, De Oliveira MF, Ignacio C, Porrachia M, Vrancken B, and Smith DM. 2020. HIV persists throughout deep tissues with repopulation from multiple anatomical sources. J. Clin. Invest 130: 1699–1712. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.