

Abstract
Bottom-up proteomics provides peptide measurements and has been invaluable for moving proteomics into large-scale analyses. Commonly, a single quantitative value is reported for each protein coding gene by aggregating peptide quantities into protein groups following protein inference and/or parsimony. However, given the complexity of both RNA splicing and post-translational protein modification, it is overly simplistic to assume that all peptides that map to a singular protein coding gene will demonstrate the same quantitative response. By assuming all peptides from a protein coding sequence are representative of the same protein we may miss the discovery of important biological differences. To capture the contributions of existing proteoforms, we need to reconsider the practice of aggregating protein values to a single quantity per protein coding gene.
Keywords: Quantitative proteomics, proteoforms, post-translational modifications, quantitative analysis, protein grouping
Graphical Abstract
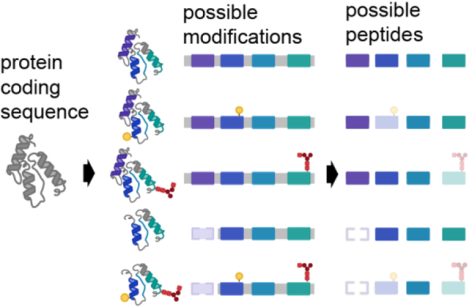
Mass spectrometry-based proteomics has become a key method for characterizing the protein composition of biological samples. The field of proteomics includes a diverse collection of data acquisition and analysis methods, but so-called “bottom-up” proteomics based on proteolysis of proteins into peptide fragments remains the primary strategy for robust surveys of complex protein mixtures. Mass spectra collected from these peptide fragments are then used to infer what proteins were present in the original sample. In the early 2000’s as large scale peptide identification took off, parsimony was used to assert the set of proteins that could give rise to the peptide data that was observed directly.(1,2) As data increased in scale, controlling for false discovery rate (FDR) at the protein level was determined to be a more conservative way to assert protein presence.(3)
With the rise in quantitative proteomics, it became desirable to summarize or aggregate peptide quantities into a single value on the protein level. Many strategies have been created to accomplish this, with most assuming that peptides belonging to the same protein will behave similarly. However, based on historical work in protein biochemistry, 2-dimensional gels, and top-down proteomics, it is estimated that there may be up to 100 proteoforms per protein on average.(4,5) This estimate is based on the possible variations that can occur to a protein’s coding sequence or by post-translational modifications (PTMs) to a protein. As most of the amino acid sequence is shared among related proteoforms, a given tryptic peptide can be derived from multiple different proteoforms (Figure 1). Once digested in a mixture, the direct connection between a peptide and its originating proteoform(s) is lost, such that the measurements of individual peptides are convolutions of the proteoforms the peptides are present. This issue of conflation is conceptually similar to the problem of haplotype phasing in genomics.(5)
Figure 1. Effect of proteoforms on possible peptide detection.

A single protein coding gene can be modified to give rise to dozens or many thousands of proteoforms, including those harboring multiple modifications. After proteolysis, proteoforms yield peptides that may be missed in bottom-up proteomics database searching and data processing.
Rationale for combining peptide measurements to a single protein quantity
The idea of aggregating peptide measurements to the protein level is appealing for interpretation and integration of proteomics data with other data types. Since the beginning of quantitative proteomics, scientists have compared the quantification and coverage of proteomics to the latest gene expression data.(6) Intuitively this practice makes sense based on the central-dogma of molecular biology. However, this comparison assumes that for each mRNA transcript there is a single protein quantity for comparison. Despite knowing that there may not be a single “protein” derived from the expressed gene, this analysis is standard practice in the field. Such comparisons have demonstrated that the correlation between gene expression and an individual protein measurement is relatively poor.(7) While several explanations have been proposed, it is important to note that most experiments were performed with bottom-up proteomics data that has been summarized to a single measurement per protein, even though it is likely that multiple proteoforms exist.
Beyond the proposed ease of biological interpretation, there are technical reasons that make aggregating peptides to a protein level measure attractive for quantification. In quantitative proteomics, our ability to find differences is affected by three parameters: 1) the size of the biological effect, 2) the biological and technical variability, and 3) the number of hypothesis tests that are made within the experiment. Thus, it is important to consider how summarizing peptide quantities at the protein level will affect these three parameters. By aggregating peptides mapping to a protein coding sequence into a single measure, especially by common methods that average or sum peptide measurements, outliers or noisy signals are suppressed. For example, we observe more variability in the peptide level values compared to the protein level values in technical replicate injections of cerebrospinal fluid (CSF) digests (Figure 2). In the case of replicate measures, the reduction in variability is viewed as a positive outcome. Additionally, by aggregating to a single protein measure we reduce the number of hypotheses tested, therefore making the analysis more sensitive to finding changes in protein abundance.
Figure 2. Technical variability is reduced when peptide measurements are combined to a protein measurement.
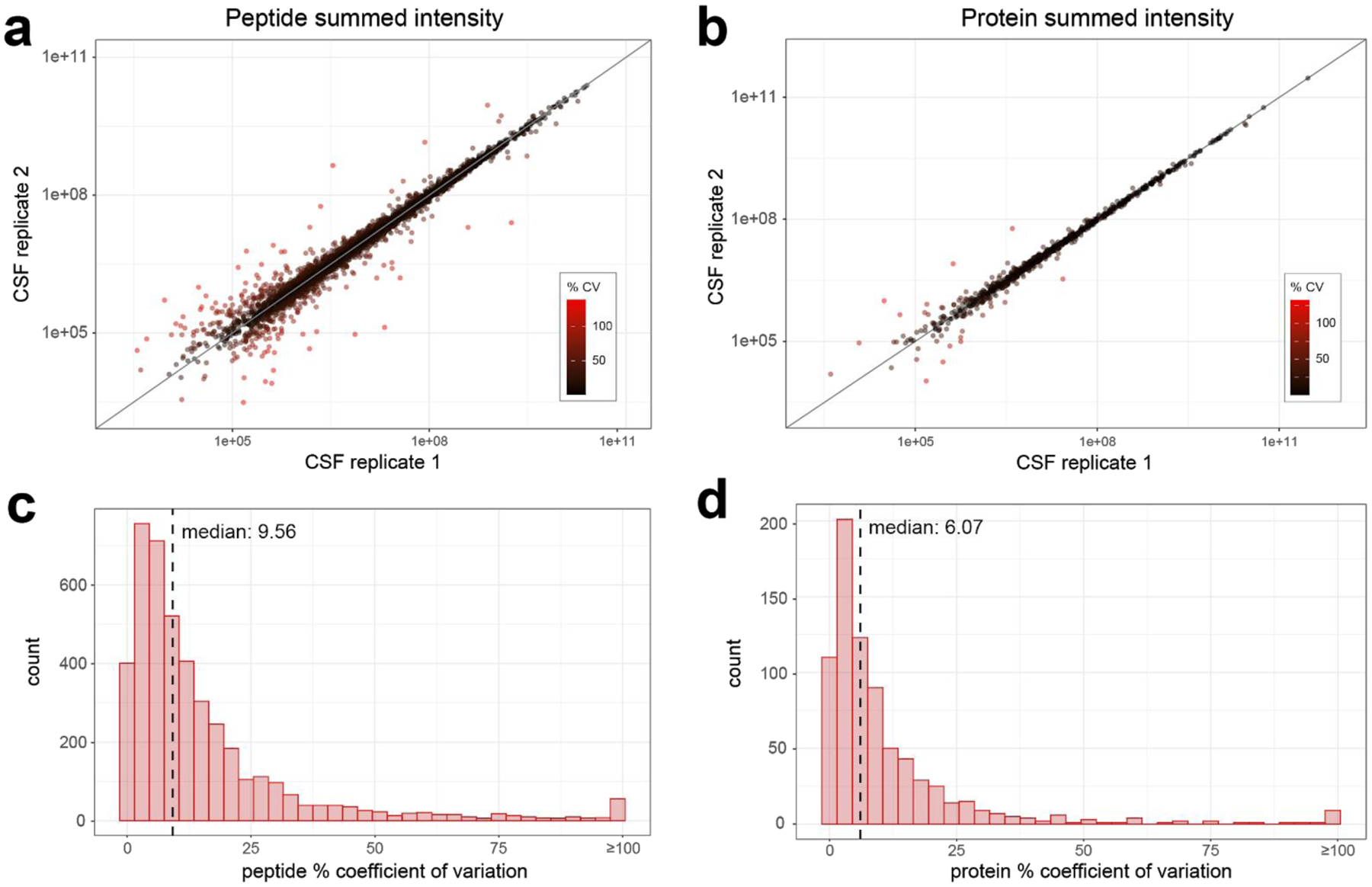
A human cerebrospinal fluid sample digest was analyzed by DIA-MS with 8 m/z staggered windows (4 m/z after demultiplexing). The relationship between a) peptide quantities, or b) summed protein quantities across two replicate instrument runs are plotted, with each peptide colored according to calculated percent coefficient of variation. The distribution of % coefficient of variation for c) peptides and d) summed protein quantities between replicate instrument runs, with the median % coefficient of variation for each indicated by the dashed line.
Another reason to aggregate to a protein level has been to reduce the amount of missing data. With data-dependent acquisition the sampling is stochastic, leading to more missing data at a peptide level if the same precursor is not selected in all the experimental runs. This missing data can have serious implications for the quality of quantitative data. One method to combat this problem is sample multiplexing by isobaric labeling, such as tandem mass tagging peptides. Evaluating large, multiplexed experiments in comparison to label-free approaches, the pattern of the missing data appears to be very distinct, but the macrostructure overall is similar in regard to the relationship between abundance and missing data.(8) These multiplexing methods are still limited in the number of samples that can be uniquely tagged, combined, and analyzed at once, and while multiple batches of samples can be acquired, the same peptides are less frequently sampled in different batches compared to proteins.(9)
Interestingly, protein groups with greater numbers of peptides observed tend to be statistically different less often than protein groups with fewer peptides (Figure 3). Despite the different types of proteomics data, the difference in scale of the data, and using either a sum-based or reference-based quantification, the fold-change consistently trends towards zero. The loss in quantitative significance in proteins with greater coverage is initially counterintuitive. While the decreased magnitude of change can still be statistically significant, it doesn’t mean those differences are representative of every peptide measured from that protein. Greater peptide coverage will likely span more proteoforms, meaning a measured peptide quantity could be derived from multiple proteoforms containing that peptide. Unless all those proteoforms change similarly among conditions, aggregating more peptides to a single protein value can average away the biological effect. Conversely, if coverage is low, differences in peptides specific to a subset of proteoforms may or may not be captured. If a value is reported at a protein level it makes these differences difficult to compare across studies since the peptides measured may be important for interpreting the results.
Figure 3. The effect size on the protein level is minimized for proteins with greater numbers of peptides.

An isobaric-labeled dataset associated with the Clinical Proteomics Tumor Analysis Consortium (CPTAC),(41) consists of 181,389 peptides mapped to 10,495 unique protein identifiers; proteins ranged from having 1 to 563 peptides associated with them. The a) log2 fold-change and b) log10 p-value is based on a comparison of tumor residual disease. The second dataset is label free and smaller, based on a Calu-3 cell culture experiment, also publicly available (MSV000079152).(42) This dataset has 15,953 unique protein identifiers, with proteins represented by 1 to 311 peptides. In this dataset the a) log2 fold-change and b) log10 p-value is based on a Middle East Respiratory Syndrome (MERS) infection to a sham control. Protein sum-based quantification sums all peptide measures per protein coding gene. For b) and d) the red line indicates the significance cutoff corresponding to p=0.05, with significantly different proteins falling below the line. Figures are truncated to 50 for ease of visualization.
The problem of reducing the biological effect when aggregating peptide quantities into a single protein value is analogous to single cell versus bulk tissue analysis. It is well known that tissues are heterogeneous and that you could have a large change occurring in a single cell or a small change occurring across all cells -- these would be indistinguishable in a bulk analysis but clearly very different results biologically. By averaging the results from bulk tissue, the ability to assess the degree of heterogeneity on the effect will be lost. Furthermore, differences could disappear entirely in the bulk sample because the effect on each cell could be very different. The same is true with reporting protein level quantities from peptides. A change might only be reflected in a proteoform that is best reflected in the quantity of a single peptide. By aggregating the peptides into a single protein level measurement this difference will be 1) misinterpreted as an effect of the entire protein or 2) averaged away and missed entirely.
Limitations of assuming a single quantity per protein coding gene
The fact that many proteins we are detecting are modified cannot be ignored. The estimate of an average of 100 proteoforms per protein coding gene may seem large until one investigates just how many modified peptides have been detected for most proteins.(4) Some notable examples can be seen with clinical biomarkers derived from post-translational modification which are further described below. These examples highlight that just because peptides map to a protein coding sequence, it does not mean that the peptides will be present at the same quantity in a biological system. To the extent this is true, statistical power will be reduced in connecting phenotype to proteomic data.
Amyloid-beta is a peptide derived from the amyloid precursor protein gene.In Alzheimer’s disease a series of cleavage events lead to several shorter soluble forms of amyloid precursor protein (sAPPα, sAPPβ), C-terminal fragments (AICD50, CTF 83, CTF 89, CTF 99, p3) and amyloid-beta peptides, which contribute to forming the characteristic plaques observed in the brains of diseased individuals.(10) The amyloid-beta peptides can be variable lengths depending on specific cleavage site, but commonly occur as a peptide of either 40 or 42 amino acids.(11) In addition to the widely known amyloid-beta 40 and amyloid-beta 42 peptides, over 20 additional amyloid-beta proteoforms have been detected in samples of Alzheimer’s brain samples arising from endogenous cleavage and post-translational modifications.(12,13) Knowing that the amyloid precursor protein is heavily processed, it is difficult to determine the origin of many of its tryptic peptides - whether they are derived from an unprocessed amyloid precursor protein, or from one of many processed forms.
If we aggregate all the tryptic peptide measures, we are assuming they are all derived from the unprocessed state, which may not be the most accurate assumption for peptides mapping to amyloid precursor protein. If we look at data from tryptic peptides, we see that some biologically relevant differences would not be accurately represented if our peptide measures are combined to a singular protein level (Figure 4). Specifically, in tryptic peptides mapping to the region of the amyloid beta sequence we observe a different abundance profile compared to tryptic peptides mapping to other regions of the protein. In addition to amyloid beta, phosphorylated tau proteoforms in the cerebrospinal fluid of patients have also gained acceptance as diagnostic biomarkers of disease.(14) Additional studies indicate that specific tau phosphosites may be better indicators of disease progression, emphasizing the importance of distinguishing between different pTau isoforms and proteoforms.(15,16)
Figure 4. Differential abundance profiles of tryptic peptides mapping to amyloid precursor protein.
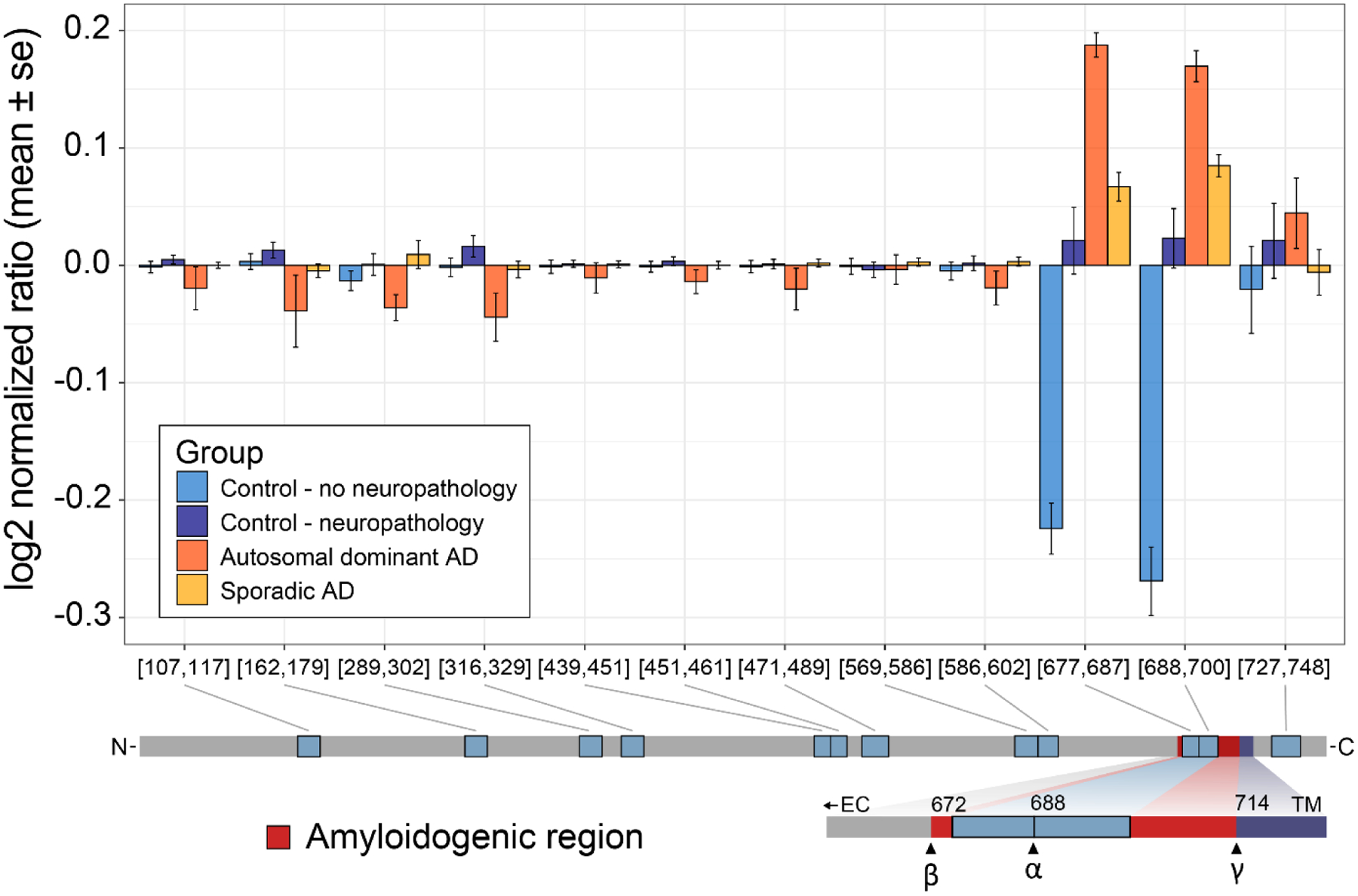
Hippocampus tissue from four experimental groups of patients were analyzed by DIA-MS; Control/No Neuropath with normal cognitive function and no neuropathologic changes of Alzheimer’s disease including no amyloid accumulation, Control/Neuropath with normal cognitive function and intermediate or severe level of neuropathologic changes of Alzheimer’s disease, Sporadic AD with dementia and intermediate or severe level of neuropathologic changes of Alzheimer’s disease, and Autosomal dominant AD with dementia and intermediate or severe level of neuropathologic changes and an autosomal dominant mutation. For all unique peptides mapping to the amyloid precursor protein sequence, peptide measures are normalized to the mean and the mean & standard error are plotted by group. Based on known protein processing we see that the two peptides with large differences map to the amyloidogenic Aβ polypeptide.
Ambiguity due to modified or processed protein biomarkers is not a problem unique to Alzheimer’s disease, but rather is general to human biology and therefore human disease. The products of processing a precursor protein into polypeptides are important markers in diabetes. C-peptide and insulin are both derived from proinsulin, with C-peptide being a valuable measure of insulin secretion and therefore pancreatic beta cell function.(17) Proglucagon is processed to form up to nine different polypeptide products, including the better-known glucagon and GLP1. Both polypeptides have distinct roles in metabolism, and both are drug targets for diabetes and obesity.(18) Additional examples of this type of processing can be found in the kallikrein-kinin system and coagulation pathways.(19) While these examples are well studied, we should not assume that these types of modifications leading to unique biologically relevant proteoforms are uncommon among other less studied proteins.
When interpreting bottom-up proteomics data we are only able to make conclusions about the peptides we detect, not the proteoforms from which they originate. For example, a study of cerebrospinal fluid in Parkinson’s disease found that specific tryptic peptides are differentially abundant in affected individuals compared to healthy, age-matched controls. Specifically, peptides in the C-terminal or N-terminal regions of granin family proteins were found to be decreased in Parkinson’s.(20) Importantly, the granin family of proteins is known to play a role in regulating secretion and delivery of peptides and neurotransmitters and are known to be processed into a number of derived bioactive peptides (Figure 5). As demonstrated in figure 5, if we sum all peptide measures that map to the protein coding sequence of secretogranin 2, then we miss the differences between experimental groups for several of the individual peptides. Instead, aggregating peptides to a single measure per protein coding sequence only accurately reflects the peptide level measurements if all peptides are in agreement (Figure 5). In contrast, if we look at peptides detected and quantified from GAPDH protein in the same CSF experiment we observe the same trend across peptides. Interestingly, GAPDH has been observed not to have many proteoforms by top-down analysis.(21) Although there are known proteoforms, from the peptides we detect we cannot conclude that only one proteoform of GAPDH is present in our samples. Instead, we can only conclude that all the peptides we detect share the same abundance trend.
Figure 5. Abundance profiles of tryptic peptides mapping to a) GAPDH and b) SCG2 proteins in cerebrospinal fluid.

Three groups of human cerebrospinal fluid samples were analyzed by DIA-MS: Alzheimer’s disease, Parkinson’s disease, and healthy age and sex-matched controls. Unique peptides mapping to the proteins a) GAPDH and c) SCG2 report quantitatively on their relative expression ratios. The protein-level display integrates the mean values from all peptide-level results (box-and-whisker plot at left), with the expression ratio for each individual peptide and the group shown in the bar graphs at right. b) GAPDH has been observed as three proteoforms which form homo-tetramers from human cell lines including HEK-tsa. Intact mass spectra of the monomeric form reveal a canonical form, a persulfide-modified form, and a glutathione-modified form. Reported masses represent average masses and ppm mass error from the calculated theoretical average mass. d) SCG2 is proteolytically processed to produce several peptides, has a sulfotyrosine, and can be phosphorylated at several serine residues.
While bottom-up proteomics is arguably the most common method for characterizing protein mixtures, alternative methods are gaining interest. These include methods that use antibodies and aptamer affinity to recognize a specific protein or protein domain.(22–24) These methods usually rely on either a single affinity reagent or paired reagents per protein coding gene. It should be noted that any method that constrains complex proteoforms into a single quantitative value per protein coding gene may miss many of the underlying differences. Even assays that use multiple affinity reagents or many tryptic peptides to different domains or modified sites of a protein will likely provide an undersampling of the proteoform species in the sample. For example, the microtubule-associated protein tau is often measured using antibodies that represent phosphorylation at threonine 181 and one that measures so called “total-tau”. However, at least 95 post-translational modifications have been discovered on tau with potentially many 100s of possible proteoforms resulting from the combinations.(16)
The examples given are just a subset of possible causes of differential peptide signals that would generally get aggregated to a singular value. Genomic sequence variation can lead to differing peptide sequences in the population.(25–27) The detection of this sequence variation by bottom-up methods relies on examining individual peptide precursors and fragments. All possible sequence variation and modifications can occur in numerous combinations, further complicating the interpretation of bottom-up data (Figure 1). Thus, even the measurement of every unmodified and modified tryptic peptide along the predicted protein coding gene sequence using either a mass spectrometer or a sequence specific affinity reagent can’t put Humpty Dumpty back together again.
Outlook and Future directions
Over the years, this challenge that not all peptides from the same gene or protein group have the same differential abundance has been an important area of research. The approach of several proposed methods focuses on the exclusion of peptide measurements from inclusion in the aggregate protein quantity if they are outliers from other peptide measurements mapping to the same protein coding gene.(28–33) While these all demonstrate improved protein concentration estimates, they still only report a single protein quantity and ignore peptides that do not agree with that single value. If those outlier peptide measurements are discarded, then true biological signals may be lost. Alternatively, signal could be kept and a weighted distribution applied across all matching isoforms.(34) Another approach taken in previous methods is to try and identify the specific proteoforms present based on peptide quantification across conditions.(35–38) These methods are tolerant to having multiple proteoforms present in a sample, however, once a molecule is digested to peptides it is impossible to track the peptide-protein molecule relationship.
While the challenge of aggregating peptide measurements may not be solved yet, one thing that is apparent is that we should no longer blindly merge all peptides into a single gene level quantity. A solution to the presence of discordant peptides could be to keep all peptides as independent measurements because it is impossible to accurately merge peptides without detailed knowledge of all proteoforms in the sample. While remaining as true to the acquired data as possible, this strategy may prove to be difficult for interpretation of experiments because the role of individual tryptic peptides in biology may be difficult to infer, especially in less studied systems. Additionally, reduced statistical power for differential abundance testing on tens of thousands of peptides compared to thousands of protein groups will also likely result in fewer significant differences. However, there has been recent work towards integrating top-down proteomics with bottom-up proteomic measurements.(39) This strategy could provide higher resolution information about the protein quantity resulting from specific proteoforms present in a sample, which then can be used to determine how peptides could be combined to more accurately reflect those proteoforms present.
An alternative approach could be to combine peptides that both map to the same gene and co-vary across a diverse set of biological groups or conditions, without designating them as specific proteoforms. We need the ability to generate multiple “peptide groups” for each protein group -- resulting in 1 to N quantities for each protein where N is the number of peptides. This grouping would require a method that minimized variance and multiple testing while maximizing the biological effect. This approach would not require knowing which proteoforms were present but would still capture quantitative differences observed at the peptide level that would otherwise be eliminated by combining those differences with non-changing peptides within the same gene product. However, this approach could be heavily dependent on having multiple conditions with enough biological replicates and high reproducibility. Additionally, the approach may not be suitable for proteins with low peptide coverage.(35) Regardless of how we choose to analyze and report our proteomics data, if peptides are aggregated to a protein quantity, it should be transparent which peptides were used, how they were combined, and the individual peptide quantities should remain accessible. Furthermore, for a specific “protein” it is critical that the same peptides are used to create the protein level quantity for all samples as different peptides will likely reflect different combinations of proteoforms.
While bottom-up proteomics is still the preferred method for characterizing proteomes due to its coverage, robustness across diverse protein physiochemical properties, sensitivity, and quantitative capabilities -- there remain challenges. Moving forward we will need new or repurposed methods, tools, and datasets to better interpret peptide level measurements. Datasets with known differences in peptide measurements will be crucial for validating any new approaches that are proposed to deal with peptide level differences. Additionally, improved data visualization tools are necessary to better distinguish changes inclusive of conserved domains, known PTMs, and structural features within a protein coding gene in the context of a global proteome. Finally, a compiled reference or “atlas” of experimentally-observed proteoforms presents a major opportunity for future algorithm development, which the Human Proteoform Atlas recently framed.(40) As the technology has advanced, so too has our ability to obtain robust measurements across many samples without lots of missing data. We now need to move towards understanding why these peptide measurements may be different instead of simply forcing our data into a format in which it may not be best served and instead into a format in which it fits.
Supplementary Material
Supporting Material S1. Frequently asked questions and responses based on discussion and feedback with reviewers and colleagues.
Figure S1. APOE tryptic peptide abundances in a human CSF Neurodegenerative disease cohort.
Figure S2. Observed post-translational modifications on human tubulin beta from Global Proteome Machine DB.
Figure S3. Observed post-translational modifications on human beta-actin from Global Proteome Machine DB.
Acknowledgements
This work was supported in part by National Institute of Health grants U19AG065156 (MJM, TJM, DLP), RF1AG053959 (MJM, TJM, DLP), P41GM103533 (MJM, DLP), F31AG069420 (DLP), P41GM108569 (NLK), UH3CA246635 (NLK), R35GM126914 (LMS), by Swedish Research Council grant 2017-04030 (LK), and U01-1CA184783 (BMW, LB) at PNNL, a multiprogram national laboratory operated by Battelle for the U.S. Department of Energy under contract DE-AC06-76RL01830. Some of the tissues used to produce the proteomic data in Figure 4 were provided by the Adult Changes in Thought study (R13AG057087).
References
- 1.Tabb DL, McDonald WH & Yates JR DTASelect and Contrast: Tools for Assembling and Comparing Protein Identifications from Shotgun Proteomics. J. Proteome Res 1, 21–26 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ma Z-Q et al. IDPicker 2.0: Improved Protein Assembly with High Discrimination Peptide Identification Filtering. J. Proteome Res 8, 3872–3881 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Nesvizhskii AI, Keller A, Kolker E & Aebersold R A Statistical Model for Identifying Proteins by Tandem Mass Spectrometry. Anal. Chem 75, 4646–4658 (2003). [DOI] [PubMed] [Google Scholar]
- 4.Aebersold R et al. How many human proteoforms are there? Nature Chemical Biology 14, 206–214 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Smith LM & Kelleher NL Proteoforms as the next proteomics currency. Science 359, 1106–1107 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Gygi SP, Rochon Y, Franza BR & Aebersold R Correlation between Protein and mRNA Abundance in Yeast. Mol. Cell. Biol 19, 1720–1730 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Liu Y, Beyer A & Aebersold R On the Dependency of Cellular Protein Levels on mRNA Abundance. Cell 165, 535–550 (2016). [DOI] [PubMed] [Google Scholar]
- 8.Bramer LM, Irvahn J, Piehowski PD, Rodland KD & Webb-Robertson B-JM A Review of Imputation Strategies for Isobaric Labeling-Based Shotgun Proteomics. J. Proteome Res 20, 1–13 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Brenes A, Hukelmann J, Bensaddek D & Lamond AI Multibatch TMT Reveals False Positives, Batch Effects and Missing Values. Molecular & Cellular Proteomics 18, 1967–1980 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.O’Brien RJ & Wong PC Amyloid Precursor Protein Processing and Alzheimer’s Disease. Annu. Rev. Neurosci 34, 185–204 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Ling Y, Morgan K & Kalsheker N Amyloid precursor protein (APP) and the biology of proteolytic processing: relevance to Alzheimer’s disease. The International Journal of Biochemistry & Cell Biology 35, 1505–1535 (2003). [DOI] [PubMed] [Google Scholar]
- 12.Portelius E, Westman-Brinkmalm A, Zetterberg H & Blennow K Determination of β-Amyloid Peptide Signatures in Cerebrospinal Fluid Using Immunoprecipitation-Mass Spectrometry. J. Proteome Res 5, 1010–1016 (2006). [DOI] [PubMed] [Google Scholar]
- 13.Wildburger NC et al. Diversity of Amyloid-beta Proteoforms in the Alzheimer’s Disease Brain. Scientific Reports 7, 9520 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Holtzman DM et al. Tau: From research to clinical development. Alzheimer’s & Dementia 12, 1033–1039 (2016). [DOI] [PubMed] [Google Scholar]
- 15.Barthélemy NR et al. A soluble phosphorylated tau signature links tau, amyloid and the evolution of stages of dominantly inherited Alzheimer’s disease. Nature Medicine 26, 398–407 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Wesseling H et al. Tau PTM Profiles Identify Patient Heterogeneity and Stages of Alzheimer’s Disease. Cell 183, 1699–1713.e13 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Leighton E, Sainsbury CA & Jones GC A Practical Review of C-Peptide Testing in Diabetes. Diabetes Ther 8, 475–487 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sandoval DA & D’Alessio DA Physiology of Proglucagon Peptides: Role of Glucagon and GLP-1 in Health and Disease. Physiological Reviews 95, 513–548 (2015). [DOI] [PubMed] [Google Scholar]
- 19.Weitz JI, Fredenburgh JC & Eikelboom JW A Test in Context: D-Dimer. Journal of the American College of Cardiology 70, 2411–2420 (2017). [DOI] [PubMed] [Google Scholar]
- 20.Rotunno MS et al. Cerebrospinal fluid proteomics implicates the granin family in Parkinson’s disease. Scientific Reports 10, 2479 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Skinner OS et al. Top-down characterization of endogenous protein complexes with native proteomics. Nature Chemical Biology 14, 36–41 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Gold L et al. Aptamer-Based Multiplexed Proteomic Technology for Biomarker Discovery. PLOS ONE 5, e15004 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Assarsson E et al. Homogenous 96-Plex PEA Immunoassay Exhibiting High Sensitivity, Specificity, and Excellent Scalability. PLOS ONE 9, e95192 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Ngo D et al. Aptamer-Based Proteomic Profiling Reveals Novel Candidate Biomarkers and Pathways in Cardiovascular Disease. Circulation 134, 270–285 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Johansson A et al. Identification of genetic variants influencing the human plasma proteome. Proceedings of the National Academy of Sciences 110, 4673–4678 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Hoofnagle AN, Eckfeldt JH & Lutsey PL Vitamin D–Binding Protein Concentrations Quantified by Mass Spectrometry. N Engl J Med 373, 1480–1482 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Blanchard V et al. Kinetics of plasma apolipoprotein E isoforms by LC-MS/MS: a pilot study. Journal of Lipid Research 59, 892–900 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Forshed J et al. Enhanced Information Output From Shotgun Proteomics Data by Protein Quantification and Peptide Quality Control (PQPQ). Mol Cell Proteomics 10, M111.010264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Goeminne LudgerJ. E., Gevaert K & Clement L Peptide-level Robust Ridge Regression Improves Estimation, Sensitivity, and Specificity in Data-dependent Quantitative Label-free Shotgun Proteomics. Molecular & Cellular Proteomics 15, 657–668 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Zhang B, Pirmoradian M, Zubarev R & Käll L Covariation of Peptide Abundances Accurately Reflects Protein Concentration Differences. Mol Cell Proteomics 16, 936–948 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.The M & Käll L Integrated Identification and Quantification Error Probabilities for Shotgun Proteomics. Molecular & Cellular Proteomics 18, 561–570 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Tsai T-H et al. Selection of Features with Consistent Profiles Improves Relative Protein Quantification in Mass Spectrometry Experiments. Molecular & Cellular Proteomics 19, 944–959 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Dermit M, Peters-Clarke TM, Shishkova E & Meyer JG Peptide Correlation Analysis (PeCorA) Reveals Differential Proteoform Regulation. J. Proteome Res 20, 1972–1980 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Saltzman AB et al. gpGrouper: A Peptide Grouping Algorithm for Gene-Centric Inference and Quantitation of Bottom-Up Proteomics Data. Molecular & Cellular Proteomics 17, 2270–2283 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Webb-Robertson B-JM et al. Bayesian Proteoform Modeling Improves Protein Quantification of Global Proteomic Measurements. Molecular & Cellular Proteomics 13, 3639–3646 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bamberger C et al. Deducing the presence of proteins and proteoforms in quantitative proteomics. Nature Communications 9, 2320 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Malioutov D et al. Quantifying Homologous Proteins and Proteoforms. Molecular & Cellular Proteomics 18, 162–168 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bludau I et al. Systematic detection of functional proteoform groups from bottom-up proteomic datasets. Nat Commun 12, 3810 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Schaffer LV, Millikin RJ, Shortreed MR, Scalf M & Smith LM Improving Proteoform Identifications in Complex Systems Through Integration of Bottom-Up and Top-Down Data. J. Proteome Res 19, 3510–3517 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Smith LM et al. The Human Proteoform Project: Defining the human proteome. Sci Adv 7, eabk0734 (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Arshad OA et al. An Integrative Analysis of Tumor Proteomic and Phosphoproteomic Profiles to Examine the Relationships Between Kinase Activity and Phosphorylation. Molecular & Cellular Proteomics 18, S26–S36 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Matzke MM et al. A comparative analysis of computational approaches to relative protein quantification using peptide peak intensities in label-free LC-MS proteomics experiments. PROTEOMICS 13, 493–503 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supporting Material S1. Frequently asked questions and responses based on discussion and feedback with reviewers and colleagues.
Figure S1. APOE tryptic peptide abundances in a human CSF Neurodegenerative disease cohort.
Figure S2. Observed post-translational modifications on human tubulin beta from Global Proteome Machine DB.
Figure S3. Observed post-translational modifications on human beta-actin from Global Proteome Machine DB.