

Abstract
Protein arginine methyltransferases (PRMTs) are a widely expressed class of enzymes responsible for catalyzing arginine methylation on numerous protein substrates. Among them, Type I PRMTs are responsible for generating asymmetric dimethylarginine. By controlling multiple basic cellular processes, such as DNA damage responses, transcriptional regulation, and mRNA splicing, Type I PRMTs contribute to cancer initiation and progression. A Type I PRMT inhibitor, GSK3368715, has been developed and has entered clinical trials for solid and hematological malignancies. Although Type I PRMTs have been reported to play roles in modulating immune cell function, the immunological role of tumor intrinsic pathways controlled by Type I PRMTs remains uncharacterized. Here, our TCGA dataset analysis revealed that expression of Type I PRMTs associated with poor clinical response and decreased immune infiltration in melanoma patients. In cancer cell lines, inhibition of Type I PRMTs induced an interferon gene signature, amplified responses to interferon and innate immune signaling, and decreased expression of the immunosuppressive cytokine VEGF (vascular endothelial growth factor). In immunocompetent mouse tumor models, including a model of T-cell exclusion that represents a common mechanism of anti-PD-1 resistance in human, Type I PRMT inhibition increased T-cell infiltration, produced durable responses dependent on CD8+ T cells, and enhanced efficacy of anti-PD-1 therapy. These data indicated that Type I PRMT inhibition exhibits immunomodulatory properties and synergizes with immune checkpoint blockade to induce durable antitumor responses in a T cell–dependent manner, suggesting that Type I PRMT inhibition can potentiate an antitumor immunity in refractory settings.
Keywords: Arginine methylation, T-cell infiltration, checkpoint blockade, cancer immunotherapy
Introduction
Immunotherapies using antibodies targeting immune checkpoint molecules expressed on exhausted T cells, such as programmed death 1 (PD-1) and cytotoxic T lymphocyte-associated protein 4 (CTLA-4), potentiate the activity of the immune system against tumor cells. These strategies have produced dramatic responses in reducing tumor growth and prolonging overall survival to unprecedented rates in melanoma, lung cancer, renal cancer, and mismatch repair-deficient colon cancer patients (1). Despite approval for immune checkpoint inhibitors in a wide range of cancers, the majority of patients relapse following initial responses to immunotherapy or fail to respond altogether. These failures can be attributed to an insufficient immune response against tumors and/or mechanisms co-opted by tumor cells to produce a suppressive tumor microenvironment (TME)(2). Therefore, identification of combination strategies to expand the clinical benefit of immunotherapies to a broader patient population is the current subject of intensive basic and clinical research.
Combination targeted strategies, including upregulation of tumor antigen expression (3) and sensitization of tumors to T-cell cytotoxicity (4,5), have been proposed with the intent to transform a refractory TME into one that favors antitumor immunity and improves the efficacy of the inhibition of the PD-(L)1 signaling axis. The most promising therapies exploit (i) activation of innate immune signaling within the tumor to stimulate an adaptive immune response or (ii) attenuation of immunosuppressive cytokines. Combinations of immune checkpoint inhibitors with agents that modulate either of these mechanisms are currently under clinical investigation. For example, atezolizumab (anti-PD-L1) is being tested in combination with carboplatin-paclitaxel and bevacizumab, a neutralization antibody for the immunosuppressive cytokine VEGF (vascular endothelial growth factor) and has shown progression-free survival benefit in non-small cell lung cancer patients (6).
Protein arginine methyltransferases (PRMTs) are a widely expressed class of enzymes responsible for catalyzing arginine methylation on numerous, diverse protein substrates. PRMTs can be classified into Type I, II, or III enzymes, based on their capacity to generate asymmetric dimethyl, symmetric dimethyl, or monomethylated forms of arginine, respectively. These modifications have distinct effects on protein function and impact interactions with other proteins and RNAs (7). Modulation of arginine methylation on PRMT substrates has essential roles in cell biology, including transcription, mRNA splicing, DNA damage response, and immune signaling (8,9). PRMTs have been shown to contribute to progression, proliferation, invasiveness, and survival of cancer cells by modulation of arginine residues on their substrates (8,10). For example, both runt-related transcription factor 1 (RUNX1) and mixed lineage leukemia (MLL) fusion proteins can recruit PRMT1 to methylate histones on target genes, promoting oncogenic transcriptional programs (11,12). PRMT1 has been also shown to methylate and potentiate the proliferative signaling of epidermal growth factor receptor (EGFR)(13). Consequently, inhibitors targeting either the Type I PRMT family or PRMT5 (the major Type II PRMT) have been developed and have entered clinical trials for a variety of human neoplasms (14).
PRMTs have been described as regulators of immune response pathways in several cell types via direct arginine methylation on specific substrates, as well as through indirect mechanisms (15,16). Although targeting Type II PRMTs has been reported to promote antitumor immune responses (17), the immunoregulatory role of Type I PRMTs in the TME remains poorly understood. Using The Cancer Genome Atlas (TCGA) of skin cutaneous melanoma (SKCM) dataset, we show that increased mRNA expression of several Type I PRMTs, including PRMT1, PRMT3, and PRMT4, associated with poor clinical response and decreased immune infiltration in melanoma patients. Using a series of preclinical tumor models with varying degrees of immune infiltration, we demonstrated that inhibition of Type I PRMTs increased hallmarks of an inflamed TME and sensitivity to inhibition of the PD-(L)1 signaling axis. Accordingly, Type I PRMT inhibition synergized with immune checkpoint blockade to induce durable antitumor responses in an array of immunocompetent tumor models. Overall, these data provide a rationale to combine Type I PRMT inhibition with immune checkpoint blockade to maximize clinical benefit in cancer patients.
Materials and Methods
Cell lines and mice
Cell lines were obtained from various repositories and licensed accordingly. Cell lines including NCI-H1975, NCI-H1563, NCI-H522, NCI-H520 and NCI-H226 were obtained from Drs. Gazdar and Minna at the Department of Health and Human Services (Washington, D.C.) and the University of Texas Southwestern Medical Center (Dallas, TX). EBC-1 and LK-2 cell lines were obtained from the Japanese Collection of Research Bioresources Cell Bank at the National Institutes of Biomedical Innovation, Health and Nutrition (Osaka, Japan). Capan-2 and SK-MES-1 cells were obtained from the Memorial Sloan Kettering Cancer Center (New York, NY). A549-Dual and A549-Dual-MAVS knockout (KO) cell lines with two discrete reporters for NFκB and IRF3 were obtained from Invivogen (San Diego, CA). HCC1395 was obtained from the University of Texas Southwestern Medical Center. SW900, BxPC3, COR-L105, PC-9, HupT4, Panc03.27, Panc08.13, A549, HCC827, Miapaca-2, Aspc-1, Panc02.03, HPAF-II, A427, CT26, EMT6, and A20 were obtained from the American Type Culture collection (ATCC, Manassas, VA). Cloudman S91 was obtained from the Sigma (St. Louis, MO). MC38 were obtained from the National Cancer Institute (NCI; Bethesda, MD). The MC38/gp100 cell line, pmel-1 T cells used in in vitro assays and luciferase-expressing pmel-1 T cells used for in vivo studies were established and generated as previously described without modification (18). Mel2357 and TIL2357 cells were derived from one metastatic melanoma patient enrolled in the ongoing clinical trial at the University of Texas MD Anderson Cancer Center (MDACC). Written consent was obtained to generate the cell line, and the study conducted in accordance to ethical standards.
NCI-H1975, NCI-H1563, NCI-H226, NCI-H520, SK-MES-1, SW900, BxPC3, COR-L105, PC-9, HupT4 HPAF-II, NCI-H522, LK-2, HCC827, Miapaca-2, Aspc-1, HCC1395, and CT26 were maintained in RPMI 1640 medium (Gibco, Carlsbad, CA; 11875093) supplemented with 10% fetal bovine serum (FBS; sigma, 12176C). Capan-2 was maintained in RPMI 1640 medium supplemented with 20% FBS. Panc03.27, Panc08.13 and Panc02.03 were maintained in RPMI 1640 medium supplemented with 15% FBS and 10 mg/L bovine insulin (Sigma, IO516–5mL). A20 was maintained in RPMI 1640 medium supplemented with 10% FBS and 0.05 M 2-mercaptoethanol (Sigma, 63689–100ml). A549 and MC38 were maintained in DMEM medium (Gibco, 11965–092) supplemented with 10% FBS. A549-Dual and A549-Dual-MAVS KO cell lines were maintained in DMEM medium supplemented with 10% FBS with 10 μg/mL blasticidin (Gibco, A1113903). A427 and EBC-1 was maintained in EMEM medium (Gibco, 31095–029) supplemented with 10% FBS. Cloudman S91 was maintained in F12K medium (ATCC, 30–2004) supplemented with 2.5% FBS and 15% horse serum (Gibco, 26050–088). EMT6 was maintained in Waymouth’s medium (Gibco, 11220–035) supplemented with 15% FBS. When necessary, the culture medium were supplemented with 1 mM sodium pyruvate (Gibco, 11360–070), 2 mM glutamax (Gibco, 35050–061), 1x non-essential amino acids (Gibco, 11400–050) and 100 μg/mL normocin (Invivogen, ant-nr-2). Mel2357 and TIL2357 cells were cultured as previously described without modification (19,20). All cell lines were maintained at 37°C and in 5% CO2. Identity of all cell lines was validated by short tandem repeat profiling, and all cell lines were routinely monitored for mycoplasma contamination by using the MycoAlert kit (Lonza, Basel, Switzerland; LT07–218). The maximum length of time of in vitro cell culture between thawing and use in the described experiments was two weeks.
Pmel-1 TCR/Thy1.1 transgenic mice on a C57BL/6 background were a generous gift from Dr. Nicholas Restifo (NCI, Bethesda, Maryland) and in-house bred. C57BL/6 mice, C57BL/6 albino mice, DBA/2N mice, and BALB/c mice were purchased from the Charles River Laboratories International, Inc. (Wilmington, MA). All mice were maintained in a specific pathogen-free barrier facility at either MDACC or GlaxoSmithKline (GSK). All studies were conducted in accordance with the MDACC and GSK Policy on the Care, Welfare, and Treatment of Laboratory Animals and were reviewed by the Institutional Animal Care and Use Committee either at GSK or by the ethical review process at the institution where the work was performed.
TCGA and cell line RNA-sequencing (RNA-seq) dataset analysis
For the analysis of Type I PRMT expression, The Cancer Genome Atlas (TCGA, https://tcga-data.nci.nih.gov) RNA-seq data and clinical information of skin cutaneous melanoma (SKCM, n=469 samples), pancreatic adenocarcinoma (PAAD, n=179 samples), and non-small cell lung cancer (NSCLC, n=1013 samples) cohorts were used for the analysis. The Genotype-Tissue Expression (GTEx, https://www.gtexportal.org/home) in normal skin tissues (n=557 samples), pancreatic tissues (n=167 samples) and lung tissues (n=288 samples) were used as the normal counterparts of tumors. All tissues from each cohort were pre-treatment tissues and were included without any exclusion. The recomputed RNA expression was obtained from the UCSC Toil RNA-seq Recompute Compendium (21). The gene-level transcripts per million (TPM) values were log2-transformed with pseudo-count 1 in Python Individual Edition (https://www.anaconda.com/products/individual, v4.11.0) for further analysis. The limma package (https://bioconductor.org/packages/release/bioc/html/limma.html, v 3.36.5) was used for the differential expression analysis (22). The intratumoral Cytolytic Activity (CYT) score was calculated by taking the geometric mean of granzyme A (GZMA) and perforin (PRF1) TPM values, and then the cohort was stratified into CYT score-low and -high groups based on the median of CTY Score. The differential expression of Type I PRMT genes was calculated between the two groups. The lymphocyte infiltration score (L Score) information was obtained from the TCGA-SKCM cohort clinical information. The cohort was stratified into L Score-low (0–3) and -high (4–6) groups, and the differential expression of Type I PRMT genes was calculated between low and high groups. Patients in TCGA-SKCM database were stratified into two groups based on the median expression of each Type I PRMT and the relapse-free survival (RFS) between these two subsets of patients were analyzed. We also further stratified patients in TCGA-SKCM database into four groups based on the median expression of PRMT1 and STING and compared the relapse-free survival (RFS) and L score among these four subsets of patients.
Bulk mRNA-seq expression data (upper-quantile normalized) in the Pan-Cancer Atlas, including bladder urothelial carcinoma (BLCA), esophageal carcinoma (ESCA), glioblastoma multiforme (GBM), head and neck squamous cell carcinoma (HNSC), brain lower grade glioma (LGG), lung adenocarcinoma (LUAD), lung squamous cell carcinoma (LUSC), mesothelioma (MESO), PAAD, sarcoma (SARC), SKCM, and stomach adenocarcinoma (STAD) generated by TCGA Program were downloaded from the NCI Cancer Genomic Data Commons (NCI-GDC: https://gdc.cancer.gov) to perform Immune deconvolution analysis. Immune deconvolution analysis was performed using the R package MCP-counter (https://github.com/cit-bioinfo/mMCP-counter). Pearson correlation coefficient (r) between the expression of PRMT1 and abundances of B, T, cytotoxic lymphocyte (CTL), and natural killer (NK) cell lineages was computed using R function stat_cor from the ggpubr package (https://github.com/kassambara/ggpubr) as previously described without modification (23). Cor.test R function was used to test the significance level followed by multiple test correction with the Benjamini-Hochberg method (24).
For the correlation between PRMT1 expression and the half maximal growth inhibitory concentration (gIC50) of PRMTi, PRMT1 expression values in 16 human cancer cell lines (HupT4, Miapaca-2, Panc08.13, BxPC3, Aspc-1, Panc02.03, Capan-2, HPAF-II,NCI-H1975, HCC827, NCI-H1563, COR-L105, A549, A427, PC-9, HCC1395) were retrieved from the 21Q2 version of the DepMap Public RNA-seq expression data available on the DepMap portal (https://depmap.org/portal/). The reported gene expression values were generated using RSEM and log2-transformed TPM counts were calculated with an added pseudo-count of 1.
Assay for Transposase-Accessible Chromatin using sequencing (ATAC-seq)
MC38 cells were treated with 0.1% DMSO (Fisher Scientific, Waltham, MA; BP231–100) or 2 μM GSK3368712 (Type I PRMT inhibitor, PRMTi, GSK, Collegeville, PA) for 4 days. After 4 days of treatment, cells were resuspended in 500 μL of RPMI 1640 medium with 10% FBS and 5% DMSO, and then transferred to a 1.7 mL tube for freezing using a Mr. Frosty container (Sigma, C1562–1EA) for DNA fragment preparation.
ATAC-seq was performed by Active Motif (Carlsbad, CA). Duplicates for each group were used. The raw fastq reads were mapped to mouse genome GRCm38/mm10 using HiSAT2 aligner (26). The peaks were detected with MACS2 package (27), and differentially enriched peaks were called using the DiffBind package (28). Supplementary Table S1 includes peaks identified from all the samples. The DESeq2 method was used to call differentially expressed peaks between DMSO and PRMTi groups. Peaks were visualized using the University of California, Santa Cruz (UCSC) genome browser (https://genome.ucsc.edu). The significant peak cut-offs were set as p≤0.05 and |Log2 fold-change| > 0.585. A peak was annotated by GENCODE V 23 (https://www.gencodegenes.org/) and assigned to a gene if it was located in its promoter region (upstream 1.5 kb to downstream 0.5kb of the transcription start site).
ChIP analysis
MC38 cells were treated with 0.1% DMSO or 2 μM GSK3368712 for 4 days, and then harvested after cells were crosslinked with 1% formaldehyde (Sigma, F8775–500ML). ChIP was performed using the Chromatin Immunoprecipitation (ChIP) Assay Kit (Sigma, 17–295) according to manufacturer’s protocol. Briefly, nuclei were isolated from cells, and Bioruptor Sonicator (Diagenode, Denville, NJ) was used to sonicate the chromatin DNA, which was sheared into ~500 bp DNA fragments. Protein-DNA complexes were immunoprecipitated with the appropriate antibodies overnight at 4°C. The complexes were then incubated with Pierce™ Protein A/G UltraLink™ Resin (Thermo Scientific, 53133) for 1 hour at 4°C. Bound DNA was eluted, reverse crosslinked, and purified using the PCR purification kit (Qiagen, 28106). The antibodies used for ChIP assay were anti-H3K4me3 (Abcam, Cambridge, UK; ab8580, 1:500) and anti-H3 (Abcam, ab1791, 1:500). Using 1% DNA purified after ChIP assay as the template, ChIP-qPCR was performed on a QuantStudio 7 (Applied Biosystems, Waltham, MA) according to manufacturer’s protocol to evaluate the protein’s occupancy at the Vegfa promoter region, which was one of the major peak regions shown in the ATAC-seq. The primers used for the ChIP-qPCR: forward: 5’-CATTTCGCGGTAGTGGCCTA-3’ and reverse: 5’-GAAATCTAGCTCCACCCCCA-3’ for the Vegfa promoter region. The relative abundance was calculated relative to input (N=3/group).
In vitro compound treatment
Type I PRMT inhibitors (Type I PRMTi), GSK3368712 and GSK3368715, were synthesized at GSK. To determine the dose effect of Type I PRMTi in interferon-stimulated gene (ISG) induction, Pan08.13 cells were seeded at 100,000 cells per well and treated with 7.8 nM, 31.3 nM, 125 nM, 500 nM, and 2000 nM GSK3368715 for 6 days. For the rest of in vitro treatments, all tested tumor cell lines were treated with either 2 μM GSK3368712 or 2 μM GSK3368715 for 4 or 6 days. Tumor cells treated with 0.1% DMSO were used as controls. After treating with Type I PRMTi, cells were co-treated with either 30 μg/mL 2’3’cGAMP (Invivogen, tlrl-nacga23–02), 1 μg/mL of Poly I:C/LMW LyoVec (Invivogen, tlrl-picwlv) or 1μg/mL of Poly I:C/HMW LyoVec (Invivogen, tlrl-piclv), 1000 U/mL recombinant human IFNβ (R&D Systems, Minneapolis, MN; 8499-IF-010) or 1.0 ng/mL recombinant mouse IFNβ (R&D Systems, 8234-mb), 100 ng/mL human recombinant human IL29 (IFNλ; R&D Systems, 1598-IL-025) or 10 ng/mL mouse IL28a (IFNλ; R&D Systems, 4635-ML-025), and 1000 U/mL human recombinant IFNγ (R&D Systems, 285-IF-100) or 10 ng/mL recombinant mouse IFNγ (R&D Systems, 485-MI-100).
For the recombinant mouse VEGF treatment, 4 ml of A20 mouse cells was plated in 6-well plates with a density of 2,500 cells/mL and pre-treated with 2 μM GSK3368712 or 0.1% DMSO for 6 days. After treatment, cells were treated in combination with 0 or 10 ng/mL of recombinant mouse VEGF (Gibco, PMG0114) for additional 6 hours, followed by the treatment of 1000 U/mL of recombinant mouse IFNβ (R&D Biosystems, 12400–1). Cells were lysed with Trizol (Life technologies, Carlsbad, CA; 15596018), and RNA was isolated using the 96-well Direct-Zol kit (Zymo Research, Irvine, CA; R2055) according to manufacturer’s protocol.
Gene-specific knockdown by siRNAs
Pan08.13, HupT4, Capan-2, A549, HCC827, and A549 Dual cells were seeded in 6-well plates at 1×105, 1×105, 1.5×105, 6×104, 1×105, and 6×104 cells/well, respectively. 24 hours after seeding, cells were transfected with either 25 pmol non-targeting siRNA (Dharmacon, Lafayette, CO; D-001810–10-20), STING-specific siRNA (Dharmacon, L-024333–02-0005), or cGAS-specific siRNA (Dharmacon, L-015607–02-0005) using Lipofectamine RNAi Max (Invitrogen, 13778–075), according to manufacturer’s protocols. 24 hours after transfection, cells were treated with either 0.1% DMSO or 2 μM GSK3368712 for 4 days. DMSO-treated cells were then treated with 500 ng/mL of the cGAS agonist, G3-YSD (Invivogen, tlrl-ydna), in LyoVec (Invivogen, lyec-1) for 24 hours.
Syngeneic mouse xenograft survival and tumor growth
Single-cell suspensions of Cloudman S91 (5×105 per injection), A20 (3×106 per injection), EMT6 (1×106 per injection), CT26 (3×105 per injection), and MC38 (5X105 cells per injection) were delivered subcutaneously in the rear flank of DBA/2N (for Cloudman S91), BALB/c (for A20, EMT6 and CT26) or C57BL/6 (for MC38) mice. Orthotopic EMT6 tumors were formed by injecting 2.5×105 EMT-6 cells suspended in a volume of 50 μL RPMI 1640 medium into the mammary fat pad (MFP, right upper udder) by means of a tuberculin syringe. Couldman S91 and A20 models were carried out at GSK. EMT6 and CT26 models were carried out by Oncodesign and Charles River Laboratories, respectively. MC38-related models were carried out at MDACC. All experiments were conducted in a blind, random manner. Once tumor growth was evident, tumor volumes were measured two to three times weekly by caliper and calculated based on the following formula: tumor volume = (Length x Width2)/2. Meanwhile, body weights were measured twice weekly. The number of days after tumor inoculation was used to plot the survival and tumor growth curves. Following randomization into study groups (N = 5–10 per group) when the mean tumor size reached ~ 150–250mm3, animals were orally dosed once daily with 75 mg/kg Type I PRMTi (GSK3368712 or GSK3368715 based on compound availability) for the PRMTi treatment group 3 weeks of bi-weekly intraperitoneal injection of 10 mg/kg IgG2a isotype control (BioXcell, Lebanon, NH; BE0089) for the vehicle group, 10 mg/kg anti-PD-1(BioXcell, BE0146) for the anti-PD-1 treatment group, or both 75 mg/kg PRMTi orally and 3 weeks of bi-weekly intraperitoneal injection of 10 mg/kg anti-PD-1 for the combination group. For Cloudman S91 xenografts, after 49 days of treatment, animals were observed for an additional 49 days post the last dose. On day 99, surviving animals were re-challenged with 5×105 Cloudman S91 tumor cells in the left flank. For CD8 depletion studies, male DBA/2N mice bearing Cloudman S91 allografts were randomized into study groups (N = 10 per group) when the mean tumor size reached ~ 150–250 mm3 and injected with 100 μg anti-mouse CD8α (BioXcell, BE0061) or saline once per week for 4 weeks during dosing.
To determine the synergistic effects of the combination of type I PRMT inhibition and anti-PD1 activity, first the relative survival was determined by dividing the percent survival of each treatment group by 100 at Day 133, Day 100, Day 40, day 60, and day 30 for the Cloudman S91, A20, MC38, EMT6, and CT26 models, respectively. The Bliss model calculation was employed by determining the relative survival of each combination concentration based on the relative survival obtained with the single agents according to the calculation Ea+Eb-Ea*Eb where E is the effect (relative survival), a is Type I PRMT inhibitor and b is anti-PD1. The highest over single-agent model (HSA) calculation was employed by subtracting the observed relative growth inhibition by the most potent growth effect of either single agent. The difference between the expected and observed from both models were determined and values ≥20 were considered to be strong synergy, values between 10 and 20 were considered synergy, −10 and 10 were additive effect, and values between −10 and −20 were considered antagonistic.
To determine the in vivo effect of Type I PRMTi on the function of tumor-reactive CD8+ T cells, an adoptive transfer model was used as previously described (18). Briefly, splenocytes harvested from pmel-1 TCR/Thy1.1 transgenic mice were cultured in RPMI 1640 medium with 10% FBS, 100 μg/mL normocin, 250 U/mL hIL2 (Prometheus Laboratories Inc., NDC Code 65483–116-07), and 0.3 μg/mL anti-mouse CD3 (BD Biosciences, San Jose, CA; 553058). 24 hours after culture, pmel-1 T cells were transduced with retrovirus expressing optimized firefly luciferase (OFL) fused with GFP, and GFP-positive cells were sorted by a FACSAria flow cytometer (BD Biosciences) after transduction. Sorted T cells were further expanded in RPMI 1640 medium with 10% FBS, 100 μg/mL normocin and 250 U/mL hIL2 for adoptive T-cell transfer. 1 × 106 of luciferase-expressing pmel-1 T cells were transferred into C57BL/6 albino mice bearing MC38/gp100 tumors. Mice were imaged using an IVIS 100 system (Xenogen) on day 6, 7, and 14 after T-cell transfer.
Gene expression analysis
Total RNAs were isolated from cell lines using RNeasy Mini Kit (Qiagen, 74134) according to manufacturer’s instructions. Both RNA-seq and quantitative real-time PCR (qRT-PCR) were used to determine the mRNA expression. For the RNA-seq analysis, RNA integrity of samples from all lung cancer cells and MC38 cells was determined using an Agilent Bioanalyzer. RNA integrity numbers (RIN) of all tested samples are greater than 8. RINs of samples from pancreatic cells are unavailable. 100–500ng RNA samples were converted into cDNA libraries using the Illumina TruSeq Stranded mRNA sample preparation kit (Illumina, San Diego, CA; 20020595). Duplicates for each group were used for the RNA-seq. Samples were sequenced at a depth of 100 million paired-end reads per sample at a 100 base-pair read length by Illumina HiSeq 2500 for Pancreatic cells, Illumina HiSeq 4000 for MC38 cells and Illumina NovaSeq 6000 for the other samples. Analysis of RNA-seq was performed as previously described without modification (25). For hallmark gene pathway analysis, a hypergeometric test of enrichment was used to obtain a set of p values for gene sets matching differential expression criteria. A gene needed to meet a cutoff of |log2(Fold Change) | ≥ 1 and an adjusted p-value ≤ 0.05 in a given comparison to be considered for a hypergeometric test of enrichment. P values were then adjusted for multiple hypothesis testing using the Benjamini and Hochberg method (24). A cut-off of 5% was used to determine significance.
Reverse transcription was carried out with 1–2.5 μg of RNA using the High-Capacity cDNA kit (Applied Biosystems, Waltham, MA; 4368814) or iScript™ Reverse Transcription Supermix (Bio-Rad, Hercules, CA; 1708840) following manufacturer’s instructions. Taqman or SYBR-green qRT-PCR was carried out using Fast Taqman master mix (Applied Biosystems, 4444557) or SsoAdvanced Universal SYBR Green Supermix (Bio-Rad,1725274), respectively, with 1μΛ cDNA as the template, and triplicate PCR reactions were run on an ABI ViiA7 or QuantStudio 7 according to manufacturer’s protocols. The SYBR-green and Taqman probes were normalized using the housekeeper genes, PPIB and/or GAPDH, and the average 2ΔΔCt values were calculated. Fold-change relative to the respective control was determined. The information of Taqman gene expression assay kits used in this study are summarized in Supplementary Table S2. The primers for mouse Vegfa expression detection using SYBR-green: forward: 5’-CTGTGCAGGCTGCTGTAACG-3’ and reverse: 5’-GTTCCCGAAACCCTGAGGAG-3’. In the experiments to determine whether the addition of PRR agonists or IFN synergizes ISG induction by Type I PRMTi, a gene was considered synergistic if the observed effect was greater than 2-fold and greater than the product of the combined fold-change of the agonist and Type I PRMTi treatment (the ‘expected’ effect). To evaluate the effect of Type I PRMTi on gene expression at single-cell level, we selected the A549-Dual cell line to represent cells derived from a single cell clone. The IRSE and NFκB reporters in A549-Dual and A549-Dual-MAVS KO cell lines were assayed using the Quanti-Luc™ (Invivogen, rep-qlc2) and Quanti-Blue™ (Invivogen, rep-qbs), respectively, as described in manufacturer’s protocol, and each reporter was normalized to cell growth using CellTiter-Glo (Promega, Madison, WI; G8462) as described in manufacturer’s instructions.
VEGF ELISA
MC38 cells were seeded in a 6-well plate at a density of 1X105 cells per well and treated with 0.1% DMSO or 2 μM GSK3368712 for 6 days. 1X104 treated MC38 cells were re-seeded into a 96-well plate and cultured overnight in 100 μΛ fresh growth medium. Undiluted conditioned media was used to determine VEGF production by tumor cells using Mouse VEGF Quantikine ELISA Kit (R&D system, MMV00) according to the manufacturer’s recommendation. The optical density of each sample at wavelengths 450nm was determined by the SYNERGY HT microplate reader (BioTek, Winooski, VT).
In vitro proliferation assays
Growth inhibition of tumor cells in response to Type I PRMTi was evaluated as previously described (25). A20, Cloudman S91, CT26, EMT6 were seeded at 31, 500, 62.5, and 125 cells per well, respectively, in 96-well plates. Data were fit with a four-parameter equation (y = (A+ ((B−A)/(1+(10^((C−x)8D))))), where A is the minimum response (ymin), B is the maximum response (ymax), C is the inflection point of the curve (EC50) and D is the Hill coefficient) to generate a concentration response curve. gIC50 of Type I PRMTi was determined. A minimum of two biological replicates were evaluated for each assay.
Cytotoxicity assays and immunophenotyping by flow cytometry
A flow cytometry-based cytotoxicity assay was used. 1X105 MC38/gp100 cells and Mel2357 cells were pre-treated by 0.1% DMSO or 2 μM GSK3368712 for 6 days. Treated tumor cells were labeled with CellTrace™ Far Red Cell Proliferation Kit (Thermo Fisher Scientific, Waltham, MA; C34564) as described in the manufacturer’s protocol and co-cultured with paired tumor-reactive T cells (pmel-1 T cells were paired with MC38/gp100 cells, and TIL2357 cells were paired with Mel2357 cells) at 37°C for three hours. The cells were fixed with Fix/Perm solution (BD Biosciences, 554714) for 20 minutes at room temperature and stained with PE-conjugated anti-cleaved caspase-3 (BD Biosciences, 550821), an apoptotic marker, in PBS (Corning, 21–040-CV) supplied with 2% FBS. The percentage of cleaved caspase-3+ cells in total labeled tumor cells was assessed by FACSCanto II (BD Biosciences) cytometer, and data was analyzed by DIVA software and FlowJo from BD Biosciences.
To characterize tumor immune microenvironment in Cloudman S91, A20, EMT6, CT26, and MC38 mouse models, blood, spleen and tumor were harvested from tumor-bearing mice after 7 days of tumor inoculation and from MC38 tumor-bearing mice after 14 days of tumor inoculation. Spleens and tumors were excised and placed in RPMI 1640 medium with 10% FBS on ice until processing. Spleens were mechanically grinded into single-cell suspensions with the rough end of a syringe plunger rod (BD, Franklin Lakes, NJ; 309626) in 5 mL of RPMI 1640 medium with 10% FBS. Tumors were manually minced, and single-cell suspensions were then generated using a tumor dissociation kit (Miltenyi biotec, Bergisch Gladbach, Germany; 130–096-730), M-tubes (Miltenyi biotec, 130–093-236), and the Miltenyi Octo-dissociator. The program setting as “m_imp_tumor_02” was used, followed by a 45-minute incubation at 37 oC, and then the “m-impTumor_03” program was repeated. Single cell suspensions were then pelleted, washed with PBS, and filtered through a 30 μm pre-separation filter (Miltenyi biotec, 130–041-407). After lysing red blood cells with ACK lysis buffer (Gibco, A1049201), samples were incubated with Fc receptor blocking buffer (Miltenyi biotec, 130–092-575) for 15 minutes at 4°C. Suspensions were stained with LIVE/DEAD Fixable Aqua Dead Cell Stain (Invitrogen, L34957) and surface marker antibodies together for 30 minutes at 4°C protected from light, washed, fixed and permeabilized using the Fixation/ Permeabilization kit (BD Biosciences, 554714; eBioscience, San Diego, CA; 00–5523-00), and then stained with intracellular marker antibodies at 4°C for 30 minutes protected from light using BD Fix/Perm solution. The antibodies used for staining include anti-CD45 (BD Biosciences, 563053), anti-CD3 (TONBO biosciences, San Diego, CA; 20–0031-U100; BD Biosciences, 553062), anti-CD4 (eBioscience, 48–0042-82; BD Biosciences, 550954), anti-CD8 (TONBO biosciences, 60–0081-U025; BD Biosciences, 560182), anti-CD25 (eBioscience, 45–0251-82), anti-FoxP3 (eBioscience, 12–5773-82 and 17–5773-82), anti-CD19 (TONBO biosciences, 35–0193-U500; BioLegend, San Diego, CA; 115543), anti-CD49B (BioLegend, 108922), anti-NK1.1 (BD Biosciences, 550627), anti-CD11b (eBioscience, 48–0112-82; BD Biosciences, 561098), anti-CD11c (TONBO biosciences, 20–0114-U100; BD Biosciences, 560584), and anti-Ki67 (eBioscience, 25–5698-82 and 11–5698-82), anti-CD103 (BD Biosciences, 560570). An LSRFortessa X-20 (BD Biosciences) and FACSCanto II cytometers were used for acquisition. Lymphocytes were gated either by the combination of forward scatter and side scatter or CD45 expression. CD8+ T cells were defined by CD3+ CD8+. CD4+ T cells were defined by CD3+ CD4+, and Tregs were defined by CD3+ CD4+ FoxP3+. NK cells were defined by CD3− CD19− NK1.1+ or CD3− CD19− CD49+. Dendritic cells were defined by CD11b− CD11c+.
NanoString analysis of Cloudman S91 xenografts
Whole Cloudman S91 tumors were lysed with Trizol, and RNA was isolated using RNeasy Mini Kit according to manufacturer’s protocol. RNA integrity was determined using an Agilent Bioanalyzer (Agilent Technologies), and 100ng of RNA (RIN 9.4–10) was used for assessing immune gene expression by the NanoString nCounter PanCancer Immune Profiling Panel (NanoString, Seattle, WA; XT-CSO-MIP1–12) according to manufacturer’s protocol. Data analysis was performed using the nCounter Advanced Analysis Module, using the default settings in the normalization module. Z-scores of gene expression were calculated for a set of genes associated with the T cell-inflamed phenotype.
Immunohistochemical analysis of CD8 localization in EMT6 tumors
Orthotopically implanted EMT6 tumors were excised after 7 days of dosing and embedded in Tissue-Tek® O.C.T. Compound (Sakura, Torrance, CA; 4583), snapped frozen in isopentane cooled over liquid nitrogen and stored at −80°C, and used for CD8 immunohistochemistry (IHC). 10-μM sections were treated with peroxidase and non-specific protein block (Dako, Santa Clara, CA; X0909) and then incubated with anti-CD8 primary antibody (Abcam, ab23378; 1:250). Goat anti-Rat IgG (H&L) secondary antibodies (Immunoreagents, Raleigh, NC; GtxRt-003-E2HRPX) were added to sections, and CD8 staining was developed using Dako EnVision®+ System-HRP (Dako, K400111–2). Sections were counterstained using Mayer’s hematoxylin (Abcam, ab220365). Whole slide digital scans were obtained on the Hamamatsu Nanozoomer (Shizuoka, Japan) at 20x magnification. Images were scored for CD8 staining at the periphery (< 150 μm from tumor margin), core (> 150 μm from tumor margin), and the entire section on a 0–4 scale, where 0 reflects no CD8 staining and 4 reflects highest CD8 staining. Double-blind scoring was repeated twice for each image, and the average score was determined.
Statistical analyses
Summary statistics (e.g., mean, standard error of the mean (SEM)) of the data are reported. Assessments of differences in continuous measurements between two groups were made using unpaired two-tailed t-test posterior to data transformation (typically logarithmic, if necessary). Differences in tumor size and gene expression among different treatments were evaluated using two-way repeated measures ANOVA (analysis of variance). The Cox Proportional Hazards (Cox PH) method and log-rank test were used to compare survival between groups. P≤0.05 was considered significant. Graph generation and statistical analyses were performed using Prism software (GraphPad Software) and R software programming language (V.3.1.0).
Data Availability Statement
The raw data files and analyzed data files from RNA-seq and ATAC-seq have been deposited in the NCBI GEO database, including 1) GSE161910: RNA-seq and ATAC-seq of murine tumor cells with or without Type I PRMT inhibitor treatment; 2) GSE126651: RNA-seq of human tumor cells with or without Type I PRMT inhibitor treatment.
Results
Increased Type I PRMTs associate with an immunologically ‘cold’ tumor microenvironment
To evaluate the relationship between Type I PRMTs and cancer immune evasion and disease progression, the expression of the six Type I PRMTs was analyzed in tumors and normal tissues using TCGA and GTEx datasets, respectively. All Type I PRMTs, except PRMT8 and PRMT2, were significantly overexpressed in skin cutaneous melanoma (SKCM), pancreatic adenocarcinoma (PAAD), and non-small cell lung cancer (NSCLC) relative to normal tissues (Fig. 1A–C). Melanoma patients with high expression of PRMT1 (stratified by the median expression in tumor tissues), displayed significantly reduced relapse-free survival (RFS, Supplementary Fig. S1A). A weaker association between poor RFS and increased expression of PRMT3 and PRMT4 was found (HR=1.381, p=0.054; HR=1.635, p=0.056, respectively, Fig. 1D). Tumor samples with low CYT score, an index of antitumor immunity (29), had increased PRMT1, PRMT2, PRMT3, or PRMT4 mRNA expression (Supplementary Fig. S1B). Increased expression of PRMT1 or PRMT4 was observed in tumors with reduced numbers of tumor-infiltrating immune cells determined by histopathology analysis (30) (Fig. 1E).
Fig. 1: Association of Type I PRMT expression with clinical outcome and immune characteristics in melanoma patients.
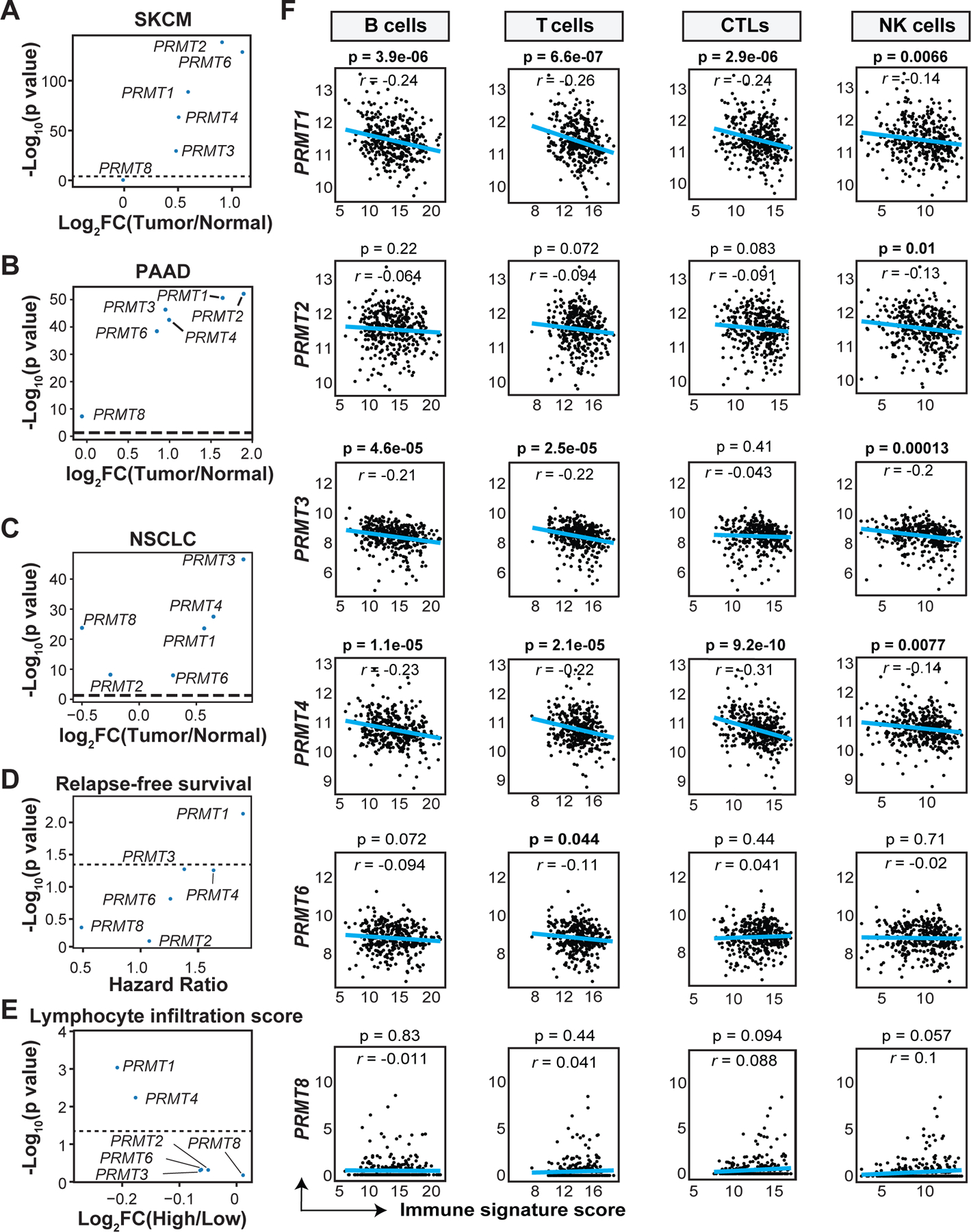
A-C, Comparison of the expression of Type I PRMTs between the tumor tissues and normal tissues in (A) skin cutaneous melanoma (SKCM, 469 tumor samples and 557 normal samples), (B) pancreatic adenocarcinoma (PAAD, 179 tumor samples and 167 normal samples) and (C) non-small cell lung cancer (NSCLC, 1013 tumor samples and 288 normal samples) from the TCGA and GTEx databases respectively. Fold-changes (FC) for the mean of each PRMT in tumors versus normal tissues and -Log10 p values are shown. Statistical significance for panels A-C was determined by the empirical Bayes moderated t-statistics test implemented in the limma package. D, Correlation of Type I PRMT expression with relapse-free survival in melanoma patients from the TCGA-SKCM cohort. Patients were stratified in two groups based on the median expression of each Type I PRMT gene. Hazard ratios (HRs) and -Log10 p values of relapse-free survival for patients with different PRMT expression were calculated by Cox Proportion Hazards Regression analysis. E, Comparison of Type I PRMT expression in melanoma patients (TCGA-SKCM) with varying tumor immune infiltration, determined by the lymphocytic infiltration score (L score) based on pathological assessment of immune cell abundances at the tumor site. Fold-changes for the mean of each PRMT between melanoma patients with high (L>3, N=130) and low (L≤3, N=198) L scores and -Log10 p values are shown. Statistical significance was determined by the empirical Bayes moderated t-statistics test implemented in the limma package. F, Correlation of Type I PRMT expression with different immune signature score. Bulk RNA-seq data from the TCGA-SKCM cohort were used to determine the association between Type I PRMT expression and immune cell components at tumor site. Statistical significance was determined by Pearson correlation. Values of Pearson correlation coefficient (r) and p values are shown.
To further examine the effects of Type I PRMTs on the composition of immune cells in the TME in human tumors, immune deconvolution analysis by the R package MCP-counter was performed using bulk RNA-seq data from the TCGA-SKCM cohort. Expression of PRMT1 or PRMT4 inversely correlated with tumor infiltration of B cells, T cells, cytotoxic lymphocytes (CTLs), and natural killer (NK) cells (Fig. 1F). In addition to melanoma, PRMT1 expression consistently showed a negative correlation with the immune scores among patients with bladder urothelial carcinoma (BLCA) and PAAD (Supplementary Fig. S1C). Taken together, these observations indicate that increased Type I PRMTs, particularly PRMT1, associate with an immunologically ‘cold’ tumor microenvironment.
Type I PRMT inhibition promotes immune-stimulated gene expression in cancer cells
To evaluate the involvement of Type I PRMTs in antitumor immune responses in both immunogenic and non-immunogenic cancer types, we selected melanoma, lung cancer, and lymphoma to represent immunogenic tumors, and pancreatic and colon cancers to represent non-immunogenic tumors. Because both tumor intrinsic and extrinsic mechanisms contribute to the inflammatory state of the TME (31), gene expression changes induced by Type I PRMT inhibitors (Type I PRMTi) GSK3368712 or GSK3368715 in a panel of human lung and pancreatic cancer cell lines were evaluated by RNA-seq. Both compounds show similar potency against all Type I PRMTs tested, with minimal activity against other PRMTs and histone methyltransferases (25). Analysis of gene expression changes indicated that pathways related to immune signaling were enriched upon treatment with Type I PRMTi across cell lines representing immunogenic (NSCLC) and non-immunogenic (PAAD) tumors (Fig. 2A). Specifically, hallmark gene pathways for interferon (IFN) signaling were significantly altered in 10 of 15 cell lines analyzed, with most genes showing increased expression with Type I PRMTi treatment (Fig. 2A; Supplementary Fig. S2A). In Panc08.13 cells, Type I PRMTi induced high CXCL10 expression at both 4 and 6 days, with a dose-dependent induction of gene expression (Supplementary Fig. S2B–C).
Fig. 2: Induction of interferon stimulated genes (ISGs) in cancer cell lines with Type I PRMT inhibition.
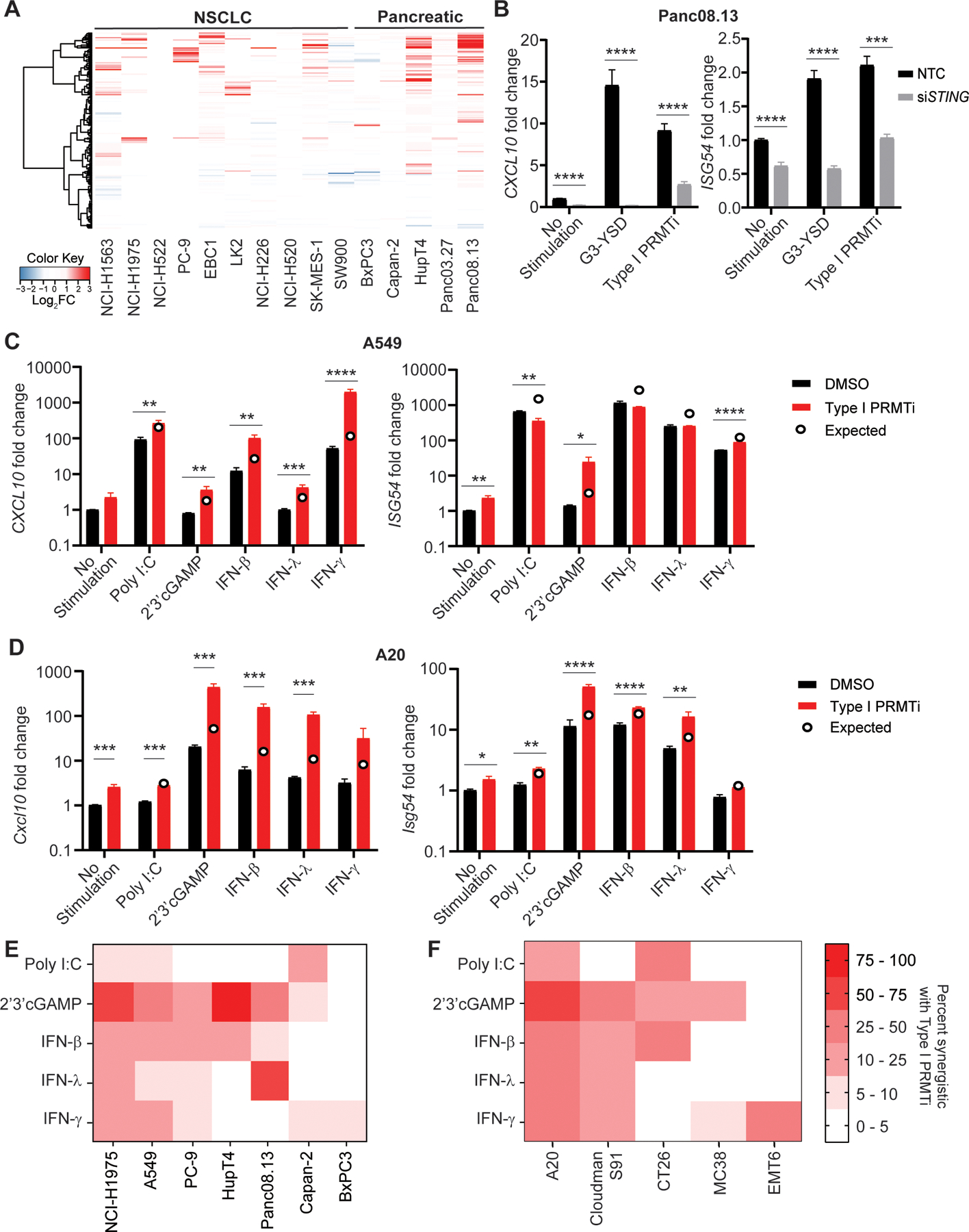
A, Heatmap of genes in the IFNγ response pathway in the indicated tumor cell lines with/without Type I PRMTi treatment. Tumor cells were treated with 0.1% DMSO or 2 μM Type I PRMTi for 4 days. RNA-seq was used to determine gene expression. Log2-fold differential expression of genes was plotted. B, Human Panc08.13 pancreatic tumor cells were transfected with either pooled non-targeting control (NTC) or pooled STING-specific siRNAs (siSTING), and transfected cells were then treated with either 0.1% DMSO or 2 μM Type I PRMTi for 6 days. Treatment with 24 hours of cGAS (G3-YSD, 500 ng/mL) agonist was served as positive control. qRT-PCR was used to determine the expression of CXCL10 or ISG54 in treated tumor cells (N≥2). C-D, Type I PRMTi, PRR agonists, and interferon effects on CXCL10 and ISG54 expression in tumor cells. (C) Human A549 lung cancer cells and (D) mouse A20 lymphoma cells were treated with 0.1% DMSO or 2 μM Type I PRMTi for 6 days with/without 24-hour exposure to PRR agonists or interferons. qRT-PCR was used to determine the expression of CXCL10 and ISG54 in treated tumor cells (N≥2). Open circles: predicted additive effect of the combination treatment. E-F, Heatmap of percentage of ISGs’ synergistic effects with Type I PRMTi and PRR agonists or interferons in a panel of (E) human and (F) mouse cell lines. Cells were treated with 0.1% DMSO or 2 μM Type I PRMTi for 6 days with/without 24-hour exposure to PRR agonists or interferons. The percentages of ISGs synergistic for the combination of Type I PRMTi with individual PRR agonist were calculated based on the expression of ISGs induced by the indicated treatment. All data are presented as mean±SEM. N indicates number of biological replicates for each group. Three technical replicates were performed for each biological replicate. P values were calculated by student’s 2-tailed t-test; *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤0.0001.
Induction of ISGs in tumors associates with better immune infiltration and response to immunotherapy (32,33). In addition to IFNs present in the TME, ISG induction can occur through the activation of pattern recognition receptors (PRRs) on tumor cells in response to evolutionarily conserved hallmarks of bacterial or viral infection (34) or cytosolic self-DNA due to genomic instability in cancer (35). To functionally investigate whether PRR activation contributed to ISG induction by Type I PRMTi, siRNAs were utilized to test the requirement of DNA sensing pathways in a series of human pancreatic and non-small cell lung cancer cell lines (Supplementary Fig. S2D). Type I PRMTi treatment upregulated the expression of ISGs, including CXCL10 and ISG54, whereas knockdown of STING, a component of the DNA sensing pathway, significantly suppressed the expression of ISGs in tumor cells (Supplementary Fig. S2E). Knockdown of either STING or cGAS diminished the induction of ISGs or reporter gene activity by Type I PRMTi to near baseline levels in all tested human cancer cell lines (Fig. 2B; Supplementary Fig. S3A–H). In contrast, knockout of mitochondrial anti-viral signaling protein (MAVS), a critical component of the dsRNA sensing pathway, had no effect on ISG induction or reporter gene activity by Type I PRMTi in A549-Dual KO reporter cell line (Supplementary Fig. S3I–J). Knockdown of STING or cGAS decreased basal expression of ISGs in tumor cells (Supplementary Fig. S2E, Supplementary Fig. S3G), suggesting that Type I PRMT activity may function to suppress intracellular responses to cytosolic DNA. To determine whether the correlation of PRMT1 expression with clinical outcome and immune score was independent on STING expression, we stratified patients in TCGA-SKCM cohort into four groups based on the median expression of PRMT1 and STING. Significantly shorter RFS was observed in patients with high PRMT1 expression than those with low PRMT1 expression, regardless the STING expression (Supplementary Fig. S4A). The inverse correlation between PRMT1 expression and tumor lymphocyte infiltration was also independent of STING expression (Supplementary Fig. S4B).
To determine whether inhibition of Type I PRMTs in cancer cells could enhance the induction of ISGs by immune stimuli encountered by tumor cells in the TME, the effect of Type I PRMTi on ISG induction in human pancreatic and lung cancer lines and mouse cell lines were systematically examined using the addition of PRR agonists or interferons. Although the magnitude and number of changes in response to treatment varied among tested human and mouse cell lines, synergistic effects on ISG induction were observed by combining Type I PRMTi with all immune stimuli tested (Fig. 2C–F, Supplementary Fig. S5). Addition of 2’3’-cGAMP produced the most robust induction of gene expression with Type I PRMTi across cell lines, with the significant synergistic effects observed in NCI-H1975, HupT4, and A20 (Fig. 2E–F). These observations suggest that inhibition of Type I PRMT activity can further stimulate gene expression induced by interferon signaling.
Type I PRMT inhibition downregulates expression of VEGF in cancer cells
To understand the mechanistic effects of Type I PRMT inhibition on gene expression, the global chromatin accessibility and transcriptional profile of tumor cells after treatment with Type I PRMTi were assessed using ATAC-seq and RNA-seq, respectively. Although global changes induced by Type I PRMTi were limited, both chromatin accessibility and mRNA expression of 23 genes were significantly changed after Type I PRMTi treatment (Fig. 3A). Among them, a 19% decrease in chromatin accessibility at the Vegfa promoter, together with lower Vegfa mRNA, were observed (Fig. 3A). Beyond its angiogenic role, VEGF can promote an immunosuppressive TME and repress activity of T cells (4,36). Quantitative real-time PCR (qRT-PCR) validated that VEGFA expression decreased by 25% or more in 7/8 human and 3/5 mouse cancer cell lines upon Type I PRMT inhibitor treatment (Fig. 3B). To validate the ATAC-seq results, ChIP assays using antibody against the transcriptional activation marker, methylated histone H3 (H3K4me3), were performed to determine H3K4me3 modification on the promoter region of Vegfa. Occupancy of H3K4me3, but not histone H3, at Vegfa promoter region was reduced after Type I PRMT inhibition (Fig. 3C), further confirming that Vegfa was less transcriptionally active in Type I PRMTi-treated tumors. Consistent with this decrease in mRNA, reduced secretion of VEGF was detected by ELISA in the media from MC38 mouse colon cells treated with Type I PRMTi (Fig. 3D). Together, these data indicate that Type I PRMT inhibition alters chromatin accessibility at the Vegfa promoter region and decreases Vegfa expression.
Fig. 3: Reduction of VEGFA mRNA in tumors with Type I PRMT inhibition.
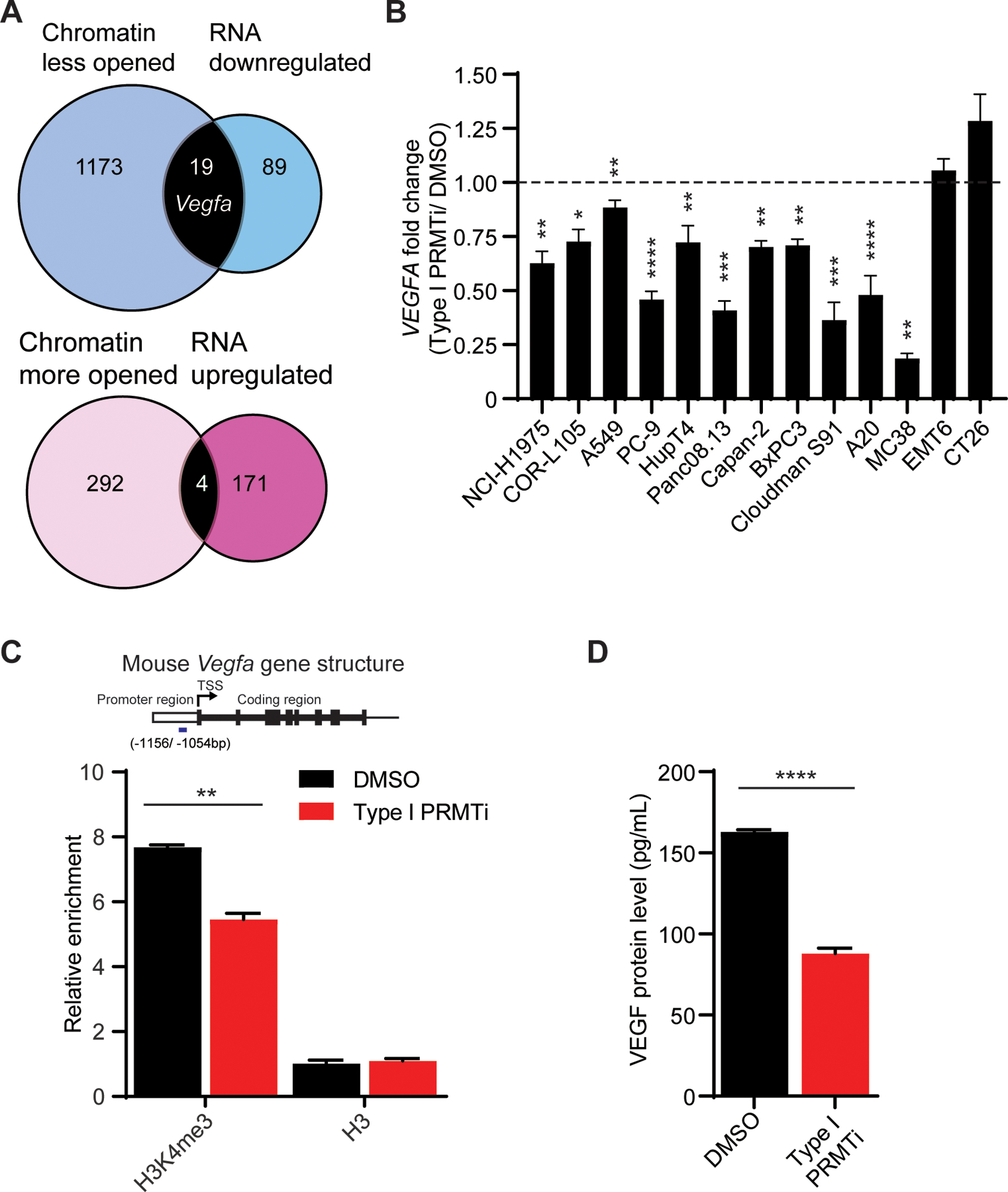
A, VENN diagrams showing the overlap of significantly changed genes revealed by ATAC-seq or RNA-seq in Type I PRMTi-treated MC38 tumors. MC38 cells were treated with 0.1% DMSO or 2 μM Type I PRMTi for 4 days. Chromatin and RNA were harvested for ATAC-seq and RNA-seq, respectively. The p value of peak difference between DMSO and PRMTi groups in ATAC-seq was calculated by negative binomial model implemented in DESeq2. Statistical significance for RNA-seq analysis was determined by the empirical Bayes moderated t-statistics test implemented in the limma package. Genes whose chromatin accessibility and expression were significantly changed by Type I PRMTi treatment (p≤0.05) were selected for the analysis. B, Type I PRMTi effects on VEGFA mRNA in human and murine cancer cell lines. Fold-change of VEGFA expression was determined by qRT-PCR in cell lines treated with 0.1% DMSO or 2 μM Type I PRMTi for 6 days (N≥2). N indicates number of biological replicates for each group. Three technical replicates were performed for each biological replicate. C, Abundance of tri-methylation of histone H3K4 (H3K4me3) at the Vegfa promoter region with Type I PRMT inhibition. MC38 tumor cells were treated with 0.1% DMSO or 2 μM Type I PRMTi for 4 days. The occupancy of H3K4me3 and H3 at the major peak region within the Vegfa promoter region shown in the ATAC-seq was determined by ChIP-qPCR. The relative abundance was calculated relative to input (N=3). Mouse Vegfa gene structure and the relative enrichment in each treatment group were shown. Short blue bar: Vegfa promoter region corresponding to the ChIP-qPCR products (−1156/ −1054 bp from transcription start site, TSS). D, Production of VEGF by tumors treated with Type I PRMTi. MC38 cells were treated with either 0.1% DMSO or 2 μM Type I PRMTi for 6 days. Treated cells were re-seeded and cultured overnight in fresh medium. VEGF in conditioned media was measured by ELISA (N=4). All data are presented as mean±SEM. At least two independent experiments were performed for all the assays except RNA-seq and ATAC-seq. P values were calculated by student’s 2-tailed t-test for panels B-D; *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤ 0.0001.
VEGF signaling is reported to repress transcriptional activation of the interferon response (37), so the effect of Type I PRMTi on ISG induction could be secondary to the suppression of VEGF. However, amplification of ISGs by Type I PRMTi in murine tumor cells was not attenuated by adding exogenous recombinant mouse VEGF, and decreases in VEGFA expression by Type I PRMTi were not affected by knockdown of cytosolic DNA sensing components in human tumor cells (Supplementary Fig. S6A–B). These data indicate that inhibition of Type I PRMT activity results in decreased VEGF expression and secretion from cancer cells through a mechanism that does not require cytosolic DNA sensing components, nor does VEGF impair Type I PRMTi mediated ISG induction. Moreover, the expression of PRMT1, the degree of ISG induction, or decrease in VEGFA expression in cell lines do not correlate with sensitivity to GSK3368715 (Supplementary Fig. S6C), suggesting that the immunomodulatory effects of Type I PRMT inhibition are distinct from the mechanisms of growth inhibition.
Type I PRMTs regulate the rate-limiting steps of T cell-mediated antitumor immune responses
ISGs and VEGFA expression changes induced by Type I PRMTi in cancer cells suggest the capacity of PRMTs to modulate the two rate-limiting steps of T cell-mediated antitumor immunity: T-cell trafficking to tumors and T cell-mediated cell killing. To evaluate the effect of Type I PRMTi on the trafficking of T cells into tumors, an established adoptive T cell transfer (ACT) mouse model (18) was used to visualize the localization of tumor-reactive T cells following addition of a Type I PRMT inhibitor (Fig. 4A). Although this model is based on gp100-expressing MC38 (MC38/gp100), a carcinogen-induced colorectal cancer cell line, MC38 tumor cells have been reported to display more immune-related features of human melanoma cells, such as the expression of MHC molecules and cytokine production (18). Our previous genome-wide immune screen using MC38 cells revealed the contribution of PRMT1 to immune resistance (38), further supporting the selection of this model to evaluate the in vivo effect of Type I PRMTi on T-cell function. Based on transferred T-cell luciferase intensity, treatment with Type I PRMTi significantly increased trafficking of tumor-reactive T cells into tumor tissues on day 6 and day 7 after T-cell transfer (Fig. 4B–C).
Fig. 4: Enhanced T cell-mediated antitumor activity with Type I PRMT inhibitor treatment.
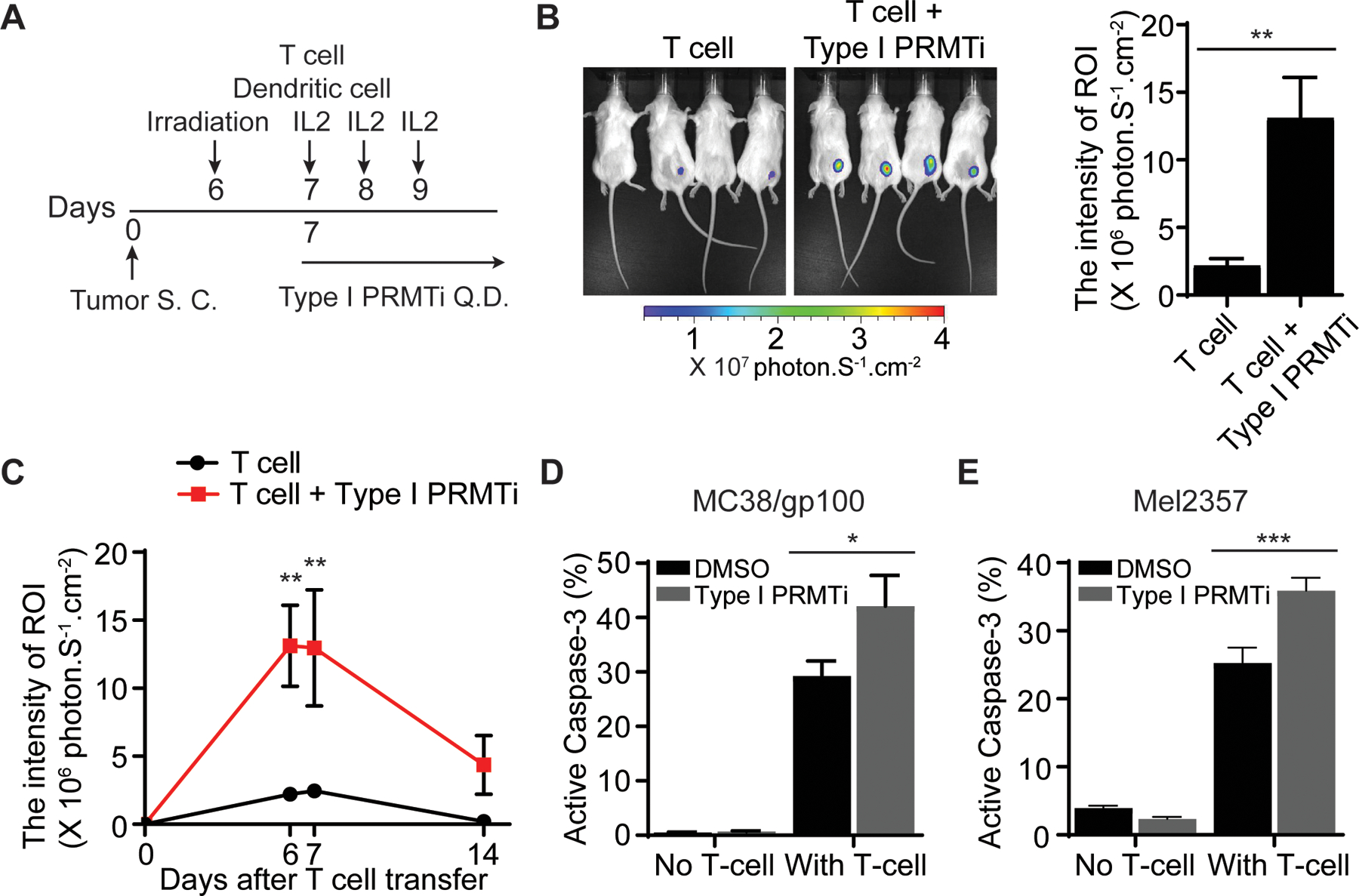
A, Schematic of the adoptive transfer model to evaluate the trafficking of tumor-reactive T cells in tumors. Luciferase-expressing pmel-1 T cells (1×106) were transferred into C57BL/6 albino mice bearing MC38/gp100 tumors (N=4–5). A DC vaccine and IL-2 treatment were administered. Either vehicle or Type I PRMTi (75 mg/kg/day) was orally administrated after T-cell transfer until the end of experiment. Luciferase intensity at the tumor sites was used to determine the trafficking of transferred tumor-reactive T cells. B, Representative images and quantitative imaging analysis showing the luciferase intensity of transferred T cells at tumor sites 6 days after T-cell transfer. Quantification was performed as the average of photon flux within region of interest (ROI). C, Time course of quantitative imaging analysis of tumor trafficking of transferred T cells. D-E, Type I PRMTi treatment increased sensitivity to T cell-mediated killing in vitro in (D) mouse MC38/gp100 tumor cells and (E) patient-derived melanoma Mel2357 cells. MC38/gp100 and Mel2357 tumor cells were pretreated with 0.1% DMSO or 2 μM Type I PRMTi for 6 days, re-seeded in fresh culture medium, followed by co-culture with pmel-1 T cells or autologous T cells (TIL2357), respectively. Tumor cell apoptosis was determined by the percentage of cleaved caspase-3+ tumor cells (N=3). All data are presented as means±SEM. Statistical significance was determined by student’s 2-tailed t-test (for panels B, D and E) or two-way repeated measures ANOVA (for panel C). *p≤0.05, **p≤0.01; ***p≤0.001.
To further examine the relationship between Type I PRMT activity and antitumor immunity, the impact of Type I PRMT inhibition on sensitizing tumor cells to T cell-mediated killing was evaluated in cancer cell lines in vitro. Mouse MC38/gp100 cells were pre-treated with Type I PRMTi for 6 days, and then co-cultured with pmel-1 T cells expressing a gp100-specific T-cell receptor. Co-culture with pmel-1 T cells increased apoptosis in tumor cells treated with Type I PRMTi relative to DMSO controls (Fig. 4D). Pretreatment of patient-derived melanoma cells (Mel2357 cells) with Type I PRMTi enhanced the sensitivity of tumor cells to apoptosis by autologous tumor-reactive T cells (TIL2357 cells) (Fig. 4E). Together with our previous studies demonstrating that inhibiting VEGF can improve tumor trafficking of T cell in vivo (4) and that upregulating ISGs in tumor cells can sensitize tumors to T cell-mediated killing in vitro (5), these results indicate that Type I PRMT inhibition can enhance key steps of T cell-mediated antitumor responses by induction of ISG and reduction of VEGF.
Type I PRMT inhibition enhances the efficacy of anti-PD-1 in immunocompetent tumor models
To determine whether inhibition of Type I PRMT activity could contribute to the establishment of a TME favoring T cell-mediated antitumor activity, the efficacy of Type I PRMTi was evaluated in multiple syngeneic tumor models. Although derived from various tissue types, these models have been suggested to better represent a range of different TMEs, rather than their histology of origin (39). Therefore, we used these models to represent varying degrees of responsiveness to checkpoint blockade and, by extension, antitumor immune activity. In vitro Type I PRMT inhibition demonstrated minimal anti-proliferative activity against a panel of mouse cancer cell lines, suggesting these cell lines do not have intrinsic sensitivity to Type I PRMTi (Supplementary Fig. S7A; Supplementary Table S3). Although Cloudman S91 cells showed limited sensitivity to Type I PRMT inhibition in vitro, Type I PRMTi alone significantly increased survival of mice bearing subcutaneous Cloudman S91 melanoma allografts (Fig. 5A). Survival benefit was reduced when CD8+ T cells were depleted, with 0/10 animals surviving beyond day 25 (p=0.086; Type I PRMTi-treated versus Type I PRMTi-treated with CD8+ T-cell depletion), suggesting that the efficacy of Type I PRMTi in this model is dependent on cytotoxic CD8+ T cells. This is consistent with our previous findings observed in mice bearing PRMT1-knockout tumors (38).
Fig. 5: Type I PRMT inhibitor treatment synergizes with PD-1 inhibition in murine subcutaneous tumor models.
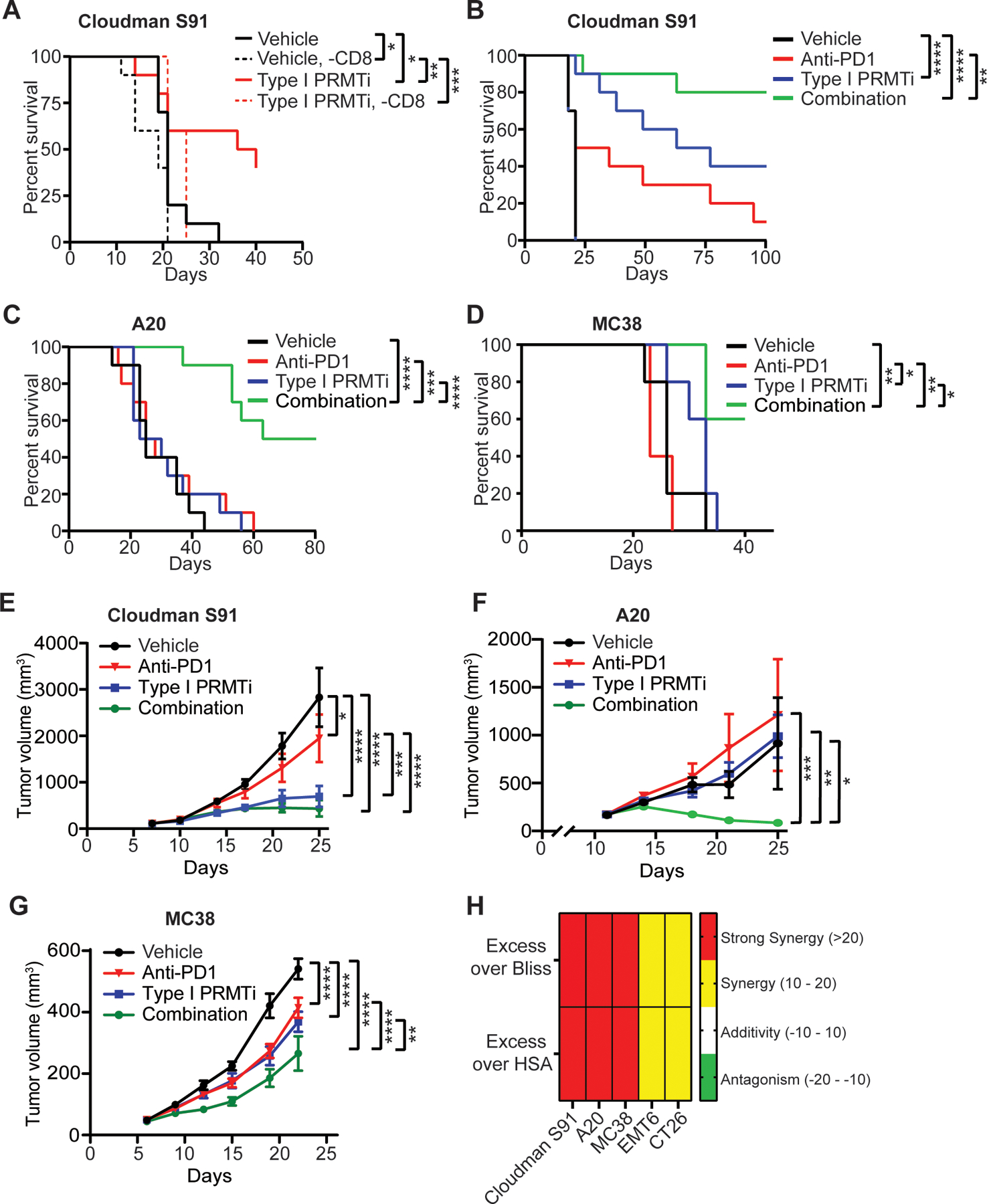
A, Survival curves of tumor bearing mice in response to Type I PRMTi and CD8 depletion treatment. C57BL/6 mice bearing established Cloudman S91 tumors were treated with vehicle, Type I PRMTi, vehicle with CD8 depletion, or Type I PRMTi with CD8 depletion (N=10). B-D, Survival curves for (B) Cloudman S91, (C) A20, or (D) MC38 tumor models treated with vehicle, Type I PRMTi, anti-PD-1, or combination Type I PRMTi and anti-PD-1 (N=5–10). E-G, Average tumor volumes in multiple models: (E) Cloudman S91, (F) A20, and (G) MC38 for animals from (B-D) (N = 5–10). H, The evaluation for the synergistic effect of combination with Type I PRMTi and anti-PD-1 treatment (see Methods for calculation). All data are presented as mean±SEM. For all the tumor models, Type I PRMTi was orally administrated at a dose of 75 mg/kg once per day. Statistical significance was determined by log-rank test (for panels A-D) or two-way repeated measures ANOVA (for panels E-G); *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤0.0001.
To determine if this activity could be further potentiated by immune checkpoint blockade, the antitumor efficacy of Type I PRMTi was evaluated in combination with anti-PD-1 in multiple subcutaneous, syngeneic tumor models. Although efficacy of single-agent anti-PD-1 or Type I PRMTi varied between models, the best responses to Type I PRMTi alone were observed in mice bearing Cloudman S91 tumors, with other models showing little to no treatment effect on tumor growth or survival (Fig. 5B–G, Supplementary Fig. S7B–E). However, combination treatment significantly extended survival of tumor-bearing mice and delayed tumor growth in Cloudman S91, A20, and MC38 tumor models (Fig. 5B–G). Combining Type I PRMTi with anti-PD-1 suppressed in vivo growth of EMT6 and CT26 tumors, with limited effect on survival of tumor-bearing mice (Supplementary Fig. S7B–E). No significant changes in body weights or other health-related signs were observed in the tumor models or in the three strains of mice (C57BL/6, DBA/2N and BALB/c) (Supplementary Fig. S7F–H). To determine whether the effects of the combination treatment on survival were synergistic, we estimated additivity using both the bliss and HSA approaches as descried in the method section. Although these approaches are typically used for in vitro combination studies where ranges of concentrations can be evaluated, they have been applied to clinical data, where doses are more limited (40). Combined treatment with Type I PRMTi and anti-PD-1 significantly extended survival in each model relative to either single agent, and to a greater degree than would have been predicted by an additive effect of the two agents (Fig. 5H). Responding animals remained tumor-free beyond the completion of dosing, demonstrating a durable effect (Supplementary Fig. S8A). In the Cloudman S91 model, animals that were tumor-free for 7 weeks after the final dose of Type I PRMTi were re-challenged with Cloudman S91 tumor cells injected on the opposite flank of the original inoculation. Tumors failed to grow in 4/4 mice previously treated with Type I PRMTi, whereas tumor growth occurred in 5/5 naïve mice (Supplementary Fig. S8B), suggesting Type I PRMTi-induced immune memory against Cloudman S91 tumors.
To understand which immune cell types may be participating in Type I PRMT inhibition to modulate antitumor immunity, we performed flow cytometric and transcriptomic profiling of the periphery and tumors in syngeneic tumor models. Type I PRMTi treatment led to significant increases in intratumoral total (CD3+) T cells in 3 of the mouse models and increased intratumoral cytotoxic (CD8+) T cells in two models (Fig. 6A–B). Of note, significant T-cell responses in the A20 and MC38 models is consistent with the significant synergistic antitumor effect of Type I PRMTi and PD-1 checkpoint blockade. Regulatory T cells (Tregs) were significantly increased in the Cloudman S91 and MC38 models, whereas Tregs remained unchanged in A20 and CT26 models (Fig. 6C). NK cells were only significantly increased in the Cloudman S91 model (Fig. 6D). In addition to changes within the tumor, dendritic cells (DCs) were significantly increased in the peripheral blood of mice bearing Cloudman S91 and A20 tumors (Supplementary Fig. S9A–B). In tumors, only Cloudman S91 showed a significant increase in DCs, whereas DCs were decreased in the A20 model, which had the lowest proportion of DCs among the 4 models analyzed (Supplementary Fig. S9B–C). Type I PRMTi had no detrimental effects on the proliferation of either CD4+ T cells, CD8+ T cells, or DCs in the MC38 model (Supplementary Fig. S10). Together, inhibition of Type I PRMT activity has the potential to increase T cell-mediated antitumor immune responses.
Fig. 6: Type I PRMT inhibitor treatment increases the infiltration of CD8+ T cells into tumor tissues.
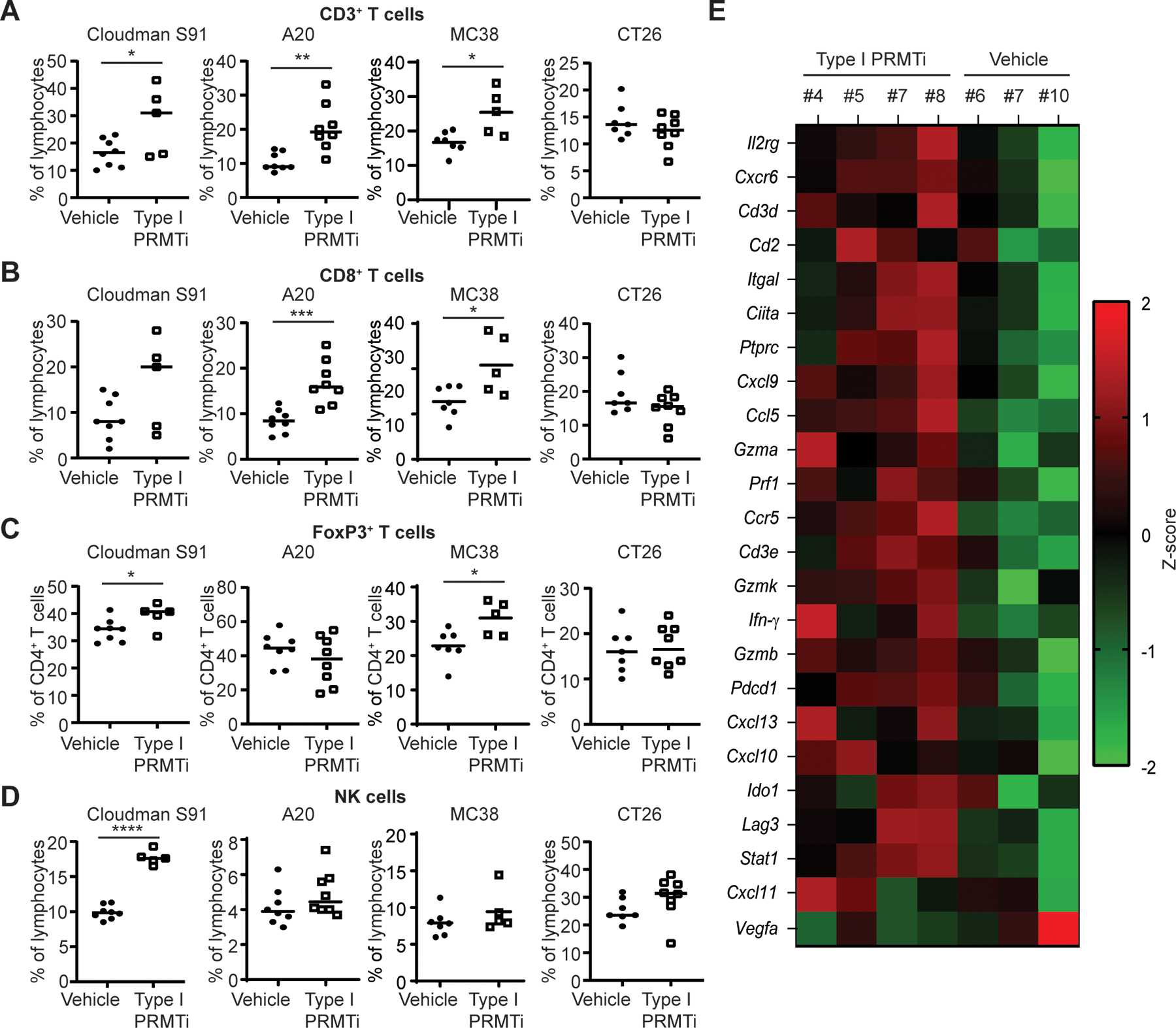
A-B, Percentage of (A) CD3+ T cells and (B) CD8+ T cells in Cloudman S91, A20, MC38 and CT26 tumor models. Mice were treated with vehicle or Type I PRMTi (N≥5). C, Percentage of FoxP3+ T cells within the CD4+ T-cell population in Cloudman S91, A20, MC38 and CT26 tumor-bearing mice treated with vehicle or Type I PRMTi (N≥5). D, Percentage of NK cells in Cloudman S91, A20, MC38 and CT26 tumor-bearing mice treated with vehicle or Type I PRMTi (N≥5). Representative gating strategy is shown in Supplementary Figure S12. Individual values and mean of each group are plotted. Statistical significance was determined by student’s 2-tailed t-test. *p≤0.05, **p≤0.01; ***p≤0.001; ****p≤0.0001. E, Z-scores of gene expression associated with the antitumor activity of PD-1 inhibition in Cloudman S91 tumors treated with Type I PRMTi. For all the tumor models, Type I PRMTi was orally administrated at a dose of 75 mg/kg once per day for 7–14 days.
To relate efficacy in vivo to gene expression changes in vitro, the effects of Type I PRMT inhibition on ISG induction and Vegfa expression in cell culture were compared to the efficacy of the Type I PRMTi and anti-PD-1 combination in mouse models for each cell line (Supplementary Fig. S11). The A20, Cloudman S91, and MC38 models had the highest synergy for ISG induction and/or greatest decreases in Vegfa expression and the most durable responses. In contrast, the CT26 and EMT6 models exhibited the lowest survival and lowest response to the Type I PRMTi in the context of both ISG synergy and Vegfa decrease. These results underscore the involvement of ISG induction and Vegfa suppression in enhanced antitumor immune responses by Type I PRMTi.
A study has identified a predictive gene expression signature of clinical response to anti-PD-1 therapy, including a set of genes associated with the T cell-inflamed phenotype (41). Genes in this signature include T-cell markers and genes related to IFNγ pathway activation. Given the effect of Type I PRMTi at inducing ISG expression and reducing VEGF production in vitro, we determined whether expression of this gene set would be affected in vivo. Using the NanoString platform, the expression levels of a panel of mouse orthologous to the human genes in the T cell-inflamed gene signature were higher in Cloudman S91 tumors with Type I PRMTi treatment relative to tumors from vehicle-treated mice (Fig. 6E). Vegfa expression was decreased in Type I PRMTi-treated Cloudman S91-bearing mice, consistent with previous in vitro results (Fig. 6E). Together, the increased expression of a T cell-inflamed signature and VEGF reduction, along with increased T-cell number in tumors, indicate that Type I PRMTi modulates these known biomarkers predictive of clinical response to PD-(L)1 inhibition.
Type I PRMT inhibition increases T-cell infiltration into the core of T cell-excluded tumors
Mechanisms that restrict T cells to the tumor periphery are a major source of resistance to immune checkpoint blockade in human patients across multiple tumor types, including melanoma and NSCLC (32). To determine whether Type I PRMTi can potentiate a response to immunotherapy in a T cell-excluded microenvironment, the activity of Type I PRMTi alone and in combination with anti-PD-1 was evaluated in a mouse model that recapitulates many of the features of T-cell exclusion. The orthotopically implanted EMT6 breast cancer model recapitulates many features of an excluded TME, including localization of T cells to the stromal boundary of the tumor and increased markers of TGFβ signaling activity (42). Whereas anti-PD-1 had no effect on survival or tumor growth in this model, Type I PRMTi alone significantly extended survival with one mouse showing a durable regression (Fig. 7A–B). The combination treatment showed significant improvement over either single agent, with four animals showing regressions that were durable 2 and 5 weeks after the final dose of Type I PRMTi and anti-PD-1, respectively (Fig. 7B). To determine effects of the inhibitor on the TME in this model, the localization of CD8+ T cells was evaluated by immunohistochemistry. T cells in vehicle-treated animals were limited to the tumor periphery, whereas tumors from Type I PRMTi-treated animals showed a significant increase of intratumoral CD8+ T cells (Fig. 7C–E). Therefore, inhibition of Type I PRMT activity can promote T-cell infiltration in a model of T-cell exclusion and sensitizes immune resistant tumors to PD-1 inhibition.
Fig. 7: Type I PRMT inhibitor synergizes with anti-PD-1 treatment to inhibit tumor progression in a murine orthotopic tumor model.
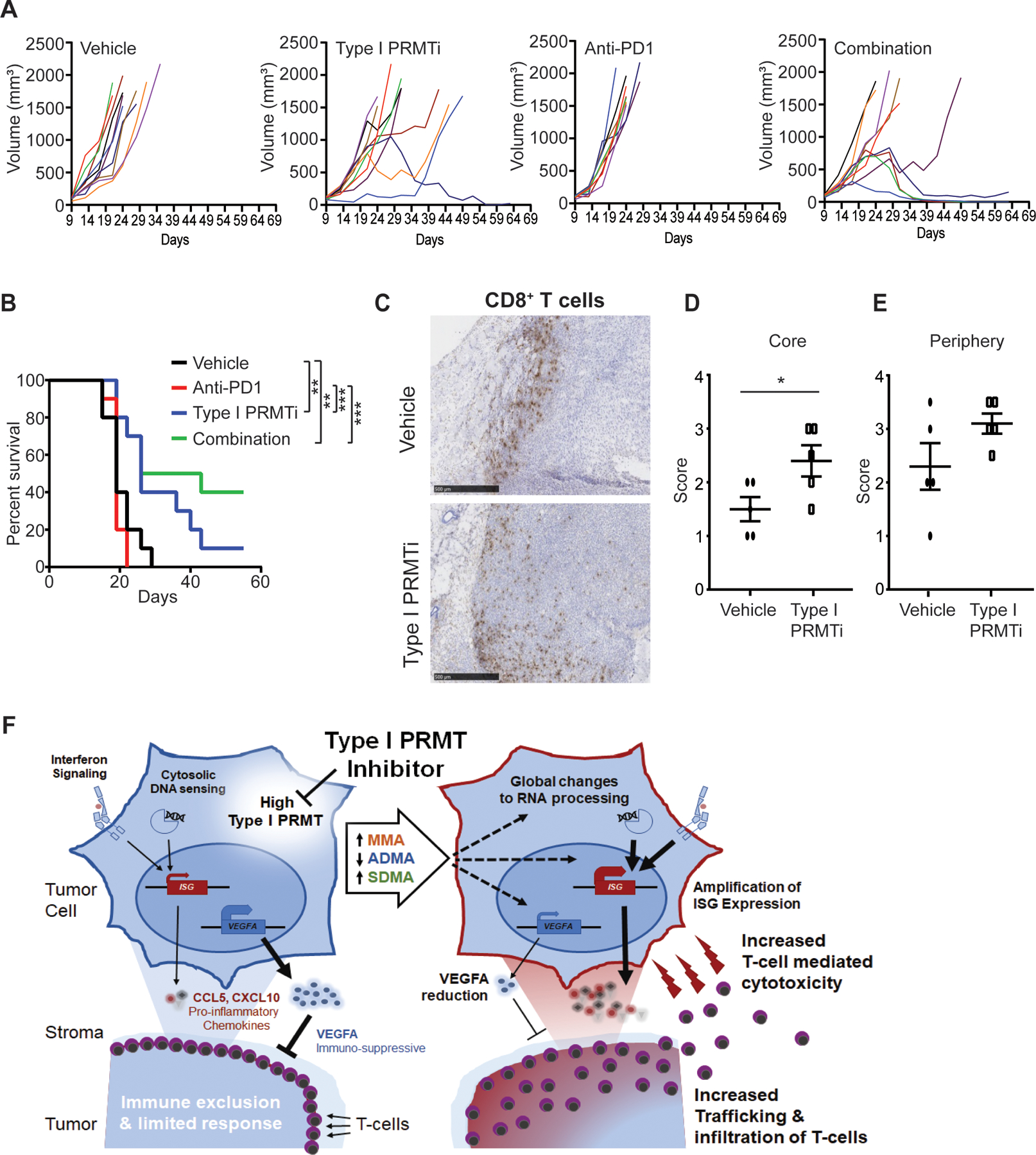
A-B, Mice bearing EMT6 orthotopic tumors were treated with vehicle, Type I PRMTi, anti-PD-1, or the combination of both agents (N=10). (A) Individual growth and (B) survival curves are shown. Type I PRMTi was orally administrated at a dose of 75 mg/kg once per day. Statistical significance was determined by log-rank test. C, Representative images of CD8 immunohistochemistry (IHC) on EMT6 orthotopic tumors from (A). D-E, Localization CD8+ T cells in the tumor core (D) and periphery (E) for animals in (A). The score was determined using the CD8 IHC results (N≥5). All data are presented as mean±SEM. Statistical significance was determined by student’s 2-tailed t-test. *p≤0.05, **p≤0.01; ***p≤0.001. F, The tumor intrinsic and extrinsic immunomodulatory effects of Type I PRMT inhibition. High Type I PRMTs associate with an immunosuppressive state, characterized by lower tumor inflammation (right). Left, global changes to monomethylation (MMA), asymmetric and symmetric dimethylation (ADMA, SDMA, respectively) induced by Type I PRMT inhibition have broad effects on splicing and transcription (dashed arrows). Interferon-stimulated genes (ISGs) are potentiated by Type I PRMT inhibition, promoting T-cell recruitment and infiltration and a more inflamed tumor microenvironment. Moreover, Type I PRMTi treatment attenuates the immunosuppressive function of VEGFA by lowering VEGFA transcript levels.
Discussion
Growing appreciation of the importance of aberrant arginine methylation in promoting tumor growth has resulted in the development of more than 20 inhibitors of PRMT activity (43). Among them, four PRMT5 inhibitors (JNJ-64619178, GSK3326595, PF-06939999, and PRT811) and one Type I PRMT inhibitor (GSK3368715) display antitumor activity in preclinical models and have entered Phase I clinical trials in patients with solid tumors and hematopoietic malignancies (NCT03573310, NCT02783300, NCT03614728, NCT03854227, NCT04089449, and NCT03666988). Many oncology agents require combinations in order to reveal maximal efficacy. In this regard, immune checkpoint therapy has been transformational, yet many patients do not respond. Here, we further characterized the immunomodulatory properties of Type I PRMT inhibition that create an environment to support antitumor immune activity (Fig. 7F). In cancer lines, inhibition of Type I PRMT activity decreased expression of VEGFA and increased expression of ISGs. Type I PRMTi treatment also enhanced infiltration and cytotoxic activity of T cells into the TME in immunocompetent tumor models. Consistent with increased hallmarks of an inflamed TME, Type I PRMT inhibition reduced tumor growth in vivo in a T cell-dependent manner, and combination with PD-1 inhibition produced synergistic activity and durable responses in multiple models. In addition to subcutaneous tumor models, Type I PRMT inhibition led to robust combination activity in a model refractory to anti-PD-1, in which T cells are restricted to the tumor periphery (42). The pro-inflammatory changes and activity observed in this diverse range of TME reflected by these subcutaneous and orthotropic models suggest that Type I PRMT inhibition can overcome numerous barriers to antitumor immunity. Our previous genome-wide CRISPR/cas9-based immune screen led to the discovery of PRMT1 as a tumor intrinsic factor governing immune resistance (38). Because PRMT1 catalyzes more than 80% of cellular asymmetric dimethylarginine modification (ADMA) of proteins (44), these studies further confirm a link between dysregulated Type I PRMT activity and immunosuppressive mechanisms within the TME.
ISG expression in human cancers correlates both with T-cell infiltration and clinical benefit to anti-PD-1 (41). Therapeutics that increase the expression of ISGs are considered a viable strategy for combination with anti-PD-1, particularly in patients that have an insufficient antitumor immune response. ISGs comprise a set of molecules that stimulate adaptive immunity, including T-cell chemoattractants and components of the antigen processing and presentation machinery. ISGs can be activated both directly by STING and TLR9 agonists, and indirectly through induction of DNA damage and subsequent activation of STING/cGAS DNA sensing pathways (i.e. radiation, chemotherapy, and PARP inhibitors). Both mechanisms are currently an area of active investigation in multiple clinical trials (45). Inhibition of Type I PRMTs increased basal expression of ISGs in cancer cells, which was predominantly dependent upon the DNA sensors STING and cGAS. This dependence indicates that Type I PRMTs may either directly regulate components of the DNA sensing signaling pathway or increase cytosolic DNA. Although arginine methylation of STING or cGAS has not been reported, a number of Type I PRMT substrates are involved in the DNA damage response, and a subset of these have been proposed to participate in the sensing of cytosolic DNA, including MRE11, HNRPA2B1 and DNAPK (46). Changes in asymmetric arginine methylation on these proteins may increase cytosolic DNA by compromising DNA repair activity, or sensitize their activity as DNA sensors to potentiate detection of basal cytosolic DNA. Beyond increasing basal expression of STING-dependent genes in cancer cells, Type I PRMTi amplified transcriptional responses to cytosolic DNA and dsRNA sensing pathways and interferons. The amplification effect of Type I PRMT inhibition was most pronounced upon stimulation of the DNA sensing pathway, but synergistic effects were achieved in combination with other stimuli, and the extent was variable among cell lines. This may be due to basal responsiveness of a cell line to each stimulus, both in number of genes and magnitude of transcriptional effect, and whether additional increases may be achieved by a combination. A similar priming effect has been described for deficiency of the ATM DNA damage response kinase, where increased DNA damage activates cytosolic DNA sensing and subsequently enhances responses to a number of PRR and TLR stimuli (47). Because these represent stimuli that cancer cells may encounter in the TME, Type I PRMT inhibition can potentiate multiple nodes important for ISG induction and offer the potential to prime the TME for responding to anti-PD-1.
In addition to ISG modulation, Type I PRMT inhibition reduced expression of the pro-angiogenic and immunosuppressive cytokine, VEGF. In the TME, VEGF can suppress an antitumor immune responses through inducing exhaustion of effector T cells (48) and inhibiting the activation and maturation of antigen-presenting DCs (49). VEGF can expand immunosuppressive immune cell populations in the TME, including Tregs and myeloid-derived suppressor cells (MDSCs)(50). Similar to Type I PRMTi, inhibiting the VEGF signaling axis increases T-cell infiltration and efficacy when combined with anti-PD-1 in preclinical models (50), and VEGF inhibitors are currently being evaluated with anti-PD-1 in numerous clinical trials (6). Combinations of anti-PD-1 with inhibitors of VEGFR kinase activity have shown benefit in early phase trials and appear greater than what would be predicted by the additive effect of each agent acting through independent mechanisms (6). These data support VEGF modulation as an approach to increasing the clinical activity of immune checkpoint inhibition and suggest that Type I PRMT inhibition may provide a novel strategy of engaging both VEGF and innate immune signaling pathways in cancer cells to promote an inflamed TME. Together, Type I PRMT inhibitors represent a class of agents that act pleiotropically to engage multiple nodes of tumor inflammation, producing a synergistic effect to sensitize patients to immunotherapy.
In addition to tumor intrinsic mechanisms, systemic Type I PRMT inhibition may influence the inflammatory state of the TME through other mechanisms, including direct effects on the function of immune cells within the TME. For example, PRMT1 activity has been linked to Th17 and Treg differentiation (51). Complicating the understanding of the molecular mechanisms by which Type I PRMT activity modulates the inflammatory state of the TME are hundreds of substrates and thousands of splicing changes induced by Type I PRMTi in cancer cell lines (25). Notably, these widespread alterations in arginine methylation and splicing may themselves produce novel, immunogenic peptides (52). Although these drug-induced immunogens would only be present in the presence of sufficient compound, their induction could lead to antigen spreading and lead to production of long-lasting immunological memory. Additional studies are required to evaluate the contribution of these mechanisms to the immunomodulatory effects of Type I PRMT inhibition.
Type I PRMT inhibition modulates a number of biomarkers associated with antitumor immunity and clinical response to PD-1 inhibitors, including increasing ISG expression and T-cell number within the TME. In the cancer types for which anti-PD-1 is approved, these biomarkers associate with better individual responses to PD-1 inhibition (53). Therefore, inhibition of Type I PRMT activity may increase the overall responses to PD-1 inhibition in these tumor types. Likewise, Type I PRMT inhibition may be utilized in tumor types where combinations of PD-1 and VEGF inhibitors have produced responses, with the added benefit of Type I PRMTi increasing ISG expression and T-cell infiltration. Type I PRMTi increased localization of T cells into the tumor core of an anti-PD-(L)1-resistant model, where T cells were restrained at the tumor periphery. This immune-excluded microenvironment accounts for a substantial proportion of human tumors, where it associates with lower response rates to PD-(L)1 inhibitors (32). Therefore, Type I PRMT inhibition may provide a mechanism to overcome clinically relevant barriers of resistance to immunotherapy and sensitize tumors with refractory microenvironments to immune checkpoint blockade. Collectively, Type I PRMT inhibition is a promising therapeutic strategy to extend the transformative benefit of immune checkpoint inhibitors to a larger patient population.
Supplementary Material
Synopsis.
Type I PRMT inhibition can override tumor immune evasion mechanisms and sensitize tumors to immune checkpoint blockade (ICB). The data highlight and provide support for the combination of Type I PRMT inhibition and ICB to enhance antitumor responses.
Grant Support and Acknowledgments
This work was supported in part by the following National Cancer Institute grants: R01CA187076 (PH&MD), P50CA221703 and P30CA016672 (MDACC Melanoma SPORE, MDACC CCSG for Flow facility), by philanthropic contributions to MDACC Melanoma Moon Shots Program; Melanoma Research Alliance Young Investigator Award (WP, 558998); Dr. Miriam and Sheldon G. Adelson Medical Research Foundation; Aim at Melanoma Foundation, Miriam and Jim Mulva research funds; and by Cancer Prevention and Research Institute of Texas (CPRIT RP170401, PH&WP; RP200520, WP). MTB is supported by the NIH (R01GM126421). The authors would like to thank past and present members of the MDACC TIL lab for tumor/TIL processing and banking.
Footnotes
Conflict of Interest:
The authors of this publication have research support from GlaxoSmithKline (GSK). The terms of this agreement have been reviewed and approved by the University of Texas MD Anderson Cancer Center in accordance with its policy on objectivity in research. W. Peng received honoraria and travel support from Bristol-Myers Squibb (BMS), and served as an advisor for Fresh wind biotechnologies. A. Fedoriw, S. O’Brien, K. N. Smitheman, C. Sherk, S. Rajapurkar, J. Laraio, O. Barbash, R. G. Kruger and H. P. Mohammad are full-time employees of GSK. P. Hwu is on the scientific advisory boards for Immatics, Dragonfly, Sanofi, and GSK. M. T. Bedford is a co-founder of EpiCypher. No potential conflicts of interest were disclosed by other authors.
References
- 1.Postow MA, Callahan MK, Wolchok JD. Immune Checkpoint Blockade in Cancer Therapy. J Clin Oncol 2015;33(17):1974–82 doi 10.1200/JCO.2014.59.4358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Horn LA, Fousek K, Palena C. Tumor Plasticity and Resistance to Immunotherapy. Trends Cancer 2020;6(5):432–41 doi 10.1016/j.trecan.2020.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Frederick DT, Piris A, Cogdill AP, Cooper ZA, Lezcano C, Ferrone CR, et al. BRAF inhibition is associated with enhanced melanoma antigen expression and a more favorable tumor microenvironment in patients with metastatic melanoma. Clin Cancer Res 2013;19(5):1225–31 doi 10.1158/1078-0432.CCR-12-16301078-0432.CCR-12-1630 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Peng W, Chen JQ, Liu C, Malu S, Creasy C, Tetzlaff MT, et al. Loss of PTEN Promotes Resistance to T Cell-Mediated Immunotherapy. Cancer Discov 2016;6(2):202–16 doi 10.1158/2159-8290.CD-15-0283. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mbofung RM, McKenzie JA, Malu S, Zhang M, Peng W, Liu C, et al. HSP90 inhibition enhances cancer immunotherapy by upregulating interferon response genes. Nature communications 2017;8(1):451 doi 10.1038/s41467-017-00449-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Socinski MA, Jotte RM, Cappuzzo F, Orlandi F, Stroyakovskiy D, Nogami N, et al. Atezolizumab for First-Line Treatment of Metastatic Nonsquamous NSCLC. The New England journal of medicine 2018;378(24):2288–301 doi 10.1056/NEJMoa1716948. [DOI] [PubMed] [Google Scholar]
- 7.Bedford MT, Richard S. Arginine methylation an emerging regulator of protein function. Mol Cell 2005;18(3):263–72 doi 10.1016/j.molcel.2005.04.003. [DOI] [PubMed] [Google Scholar]
- 8.Yang Y, Bedford MT. Protein arginine methyltransferases and cancer. Nat Rev Cancer 2013;13(1):37–50 doi 10.1038/nrc3409. [DOI] [PubMed] [Google Scholar]
- 9.Guccione E, Richard S. The regulation, functions and clinical relevance of arginine methylation. Nat Rev Mol Cell Biol 2019;20(10):642–57 doi 10.1038/s41580-019-0155-x. [DOI] [PubMed] [Google Scholar]
- 10.Wei H, Mundade R, Lange KC, Lu T. Protein arginine methylation of non-histone proteins and its role in diseases. Cell Cycle 2014;13(1):32–41 doi 10.4161/cc.27353. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Shia WJ, Okumura AJ, Yan M, Sarkeshik A, Lo MC, Matsuura S, et al. PRMT1 interacts with AML1-ETO to promote its transcriptional activation and progenitor cell proliferative potential. Blood 2012;119(21):4953–62 doi 10.1182/blood-2011-04-347476. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Cheung N, Chan LC, Thompson A, Cleary ML, So CW. Protein arginine-methyltransferase-dependent oncogenesis. Nat Cell Biol 2007;9(10):1208–15 doi 10.1038/ncb1642. [DOI] [PubMed] [Google Scholar]
- 13.Liao HW, Hsu JM, Xia WY, Wang HL, Wang YN, Chang WC, et al. PRMT1-mediated methylation of the EGF receptor regulates signaling and cetuximab response. Journal of Clinical Investigation 2015;125(12):4529–43 doi 10.1172/Jci82826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Li X, Wang C, Jiang H, Luo C. A patent review of arginine methyltransferase inhibitors (2010-2018). Expert Opin Ther Pat 2019;29(2):97–114 doi 10.1080/13543776.2019.1567711. [DOI] [PubMed] [Google Scholar]
- 15.Zhang H, Han C, Li T, Li N, Cao X. The methyltransferase PRMT6 attenuates antiviral innate immunity by blocking TBK1-IRF3 signaling. Cell Mol Immunol 2019;16(10):800–9 doi 10.1038/s41423-018-0057-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Zhu W, Mustelin T, David M. Arginine methylation of STAT1 regulates its dephosphorylation by T cell protein tyrosine phosphatase. The Journal of biological chemistry 2002;277(39):35787–90 doi 10.1074/jbc.C200346200. [DOI] [PubMed] [Google Scholar]
- 17.Kim H, Kim H, Feng Y, Li Y, Tamiya H, Tocci S, et al. PRMT5 control of cGAS/STING and NLRC5 pathways defines melanoma response to antitumor immunity. Sci Transl Med 2020;12(551) doi 10.1126/scitranslmed.aaz5683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Peng W, Ye Y, Rabinovich BA, Liu C, Lou Y, Zhang M, et al. Transduction of tumor-specific T cells with CXCR2 chemokine receptor improves migration to tumor and antitumor immune responses. Clin Cancer Res 2010;16(22):5458–68 doi 10.1158/1078-0432.CCR-10-07121078-0432.CCR-10-0712 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Chacon JA, Wu RC, Sukhumalchandra P, Molldrem JJ, Sarnaik A, Pilon-Thomas S, et al. Co-stimulation through 4-1BB/CD137 improves the expansion and function of CD8(+) melanoma tumor-infiltrating lymphocytes for adoptive T-cell therapy. PLoS One 2013;8(4):e60031 doi 10.1371/journal.pone.006003PONE-D-12-34281 [pii]. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Forget MA, Tavera RJ, Haymaker C, Ramachandran R, Malu S, Zhang M, et al. A Novel Method to Generate and Expand Clinical-Grade, Genetically Modified, Tumor-Infiltrating Lymphocytes. Frontiers in immunology 2017;8:908 doi 10.3389/fimmu.2017.00908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol 2017;35(4):314–6 doi 10.1038/nbt.3772. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Law CW, Chen Y, Shi W, Smyth GK. voom: Precision weights unlock linear model analysis tools for RNA-seq read counts. Genome Biol 2014;15(2):R29 doi 10.1186/gb-2014-15-2-r29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Becht E, Giraldo NA, Lacroix L, Buttard B, Elarouci N, Petitprez F, et al. Estimating the population abundance of tissue-infiltrating immune and stromal cell populations using gene expression. Genome Biol 2016;17(1):218 doi 10.1186/s13059-016-1070-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Benjamini Y, Hochberg Y. Controlling the False Discovery Rate: A Practical and Powerful Approach to Multiple Testing. Journal of the Royal Statistical Society: Series B (Methodological) 1995;57(1):289–300 doi 10.1111/j.2517-6161.1995.tb02031.x. [DOI] [Google Scholar]
- 25.Fedoriw A, Rajapurkar SR, O’Brien S, Gerhart SV, Mitchell LH, Adams ND, et al. Anti-tumor Activity of the Type I PRMT Inhibitor, GSK3368715, Synergizes with PRMT5 Inhibition through MTAP Loss. Cancer Cell 2019;36(1):100-+ doi 10.1016/j.ccell.2019.05.014. [DOI] [PubMed] [Google Scholar]
- 26.Kim D, Langmead B, Salzberg SL. HISAT: a fast spliced aligner with low memory requirements. Nature methods 2015;12(4):357–60 doi 10.1038/nmeth.3317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, et al. Model-based analysis of ChIP-Seq (MACS). Genome Biol 2008;9(9):R137 doi 10.1186/gb-2008-9-9-r137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ross-Innes CS, Stark R, Teschendorff AE, Holmes KA, Ali HR, Dunning MJ, et al. Differential oestrogen receptor binding is associated with clinical outcome in breast cancer. Nature 2012;481(7381):389–93 doi 10.1038/nature10730. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Rooney MS, Shukla SA, Wu CJ, Getz G, Hacohen N. Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 2015;160(1–2):48–61 doi 10.1016/j.cell.2014.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Network. CGA. Genomic Classification of Cutaneous Melanoma. Cell 2015;161(7):1681–96 doi 10.1016/j.cell.2015.05.044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Binnewies M, Roberts EW, Kersten K, Chan V, Fearon DF, Merad M, et al. Understanding the tumor immune microenvironment (TIME) for effective therapy. Nat Med 2018;24(5):541–50 doi 10.1038/s41591-018-0014-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Hegde PS, Karanikas V, Evers S. The Where, the When, and the How of Immune Monitoring for Cancer Immunotherapies in the Era of Checkpoint Inhibition. Clin Cancer Res 2016;22(8):1865–74 doi 10.1158/1078-0432.CCR-15-1507. [DOI] [PubMed] [Google Scholar]
- 33.Fuertes MB, Woo SR, Burnett B, Fu YX, Gajewski TF. Type I interferon response and innate immune sensing of cancer. Trends Immunol 2013;34(2):67–73 doi 10.1016/j.it.2012.10.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Takeuchi O, Akira S. Pattern recognition receptors and inflammation. Cell 2010;140(6):805–20 doi 10.1016/j.cell.2010.01.022. [DOI] [PubMed] [Google Scholar]
- 35.Kwon J, Bakhoum SF. The Cytosolic DNA-Sensing cGAS-STING Pathway in Cancer. Cancer Discov 2020;10(1):26–39 doi 10.1158/2159-8290.CD-19-0761. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Tamura R, Tanaka T, Akasaki Y, Murayama Y, Yoshida K, Sasaki H. The role of vascular endothelial growth factor in the hypoxic and immunosuppressive tumor microenvironment: perspectives for therapeutic implications. Med Oncol 2019;37(1):2 doi 10.1007/s12032-019-1329-2. [DOI] [PubMed] [Google Scholar]
- 37.Arulanandam R, Batenchuk C, Angarita FA, Ottolino-Perry K, Cousineau S, Mottashed A, et al. VEGF-Mediated Induction of PRD1-BF1/Blimp1 Expression Sensitizes Tumor Vasculature to Oncolytic Virus Infection. Cancer Cell 2015;28(2):210–24 doi 10.1016/j.ccell.2015.06.009. [DOI] [PubMed] [Google Scholar]
- 38.Hou J, Wang Y, Shi L, Chen Y, Xu C, Saeedi A, et al. Integrating genome-wide CRISPR immune screen with multi-omic clinical data reveals distinct classes of tumor intrinsic immune regulators. J Immunother Cancer 2021;9(2) doi 10.1136/jitc-2020-001819. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Zhong W, Myers JS, Wang F, Wang K, Lucas J, Rosfjord E, et al. Comparison of the molecular and cellular phenotypes of common mouse syngeneic models with human tumors. BMC Genomics 2020;21(1):2 doi 10.1186/s12864-019-6344-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Palmer AC, Sorger PK. Combination Cancer Therapy Can Confer Benefit via Patient-to-Patient Variability without Drug Additivity or Synergy. Cell 2017;171(7):1678–91 e13 doi 10.1016/j.cell.2017.11.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Ayers M, Lunceford J, Nebozhyn M, Murphy E, Loboda A, Kaufman DR, et al. IFN-gamma-related mRNA profile predicts clinical response to PD-1 blockade. J Clin Invest 2017;127(8):2930–40 doi 10.1172/JCI91190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Mariathasan S, Turley SJ, Nickles D, Castiglioni A, Yuen K, Wang Y, et al. TGFbeta attenuates tumour response to PD-L1 blockade by contributing to exclusion of T cells. Nature 2018;554(7693):544–8 doi 10.1038/nature25501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Jarrold J, Davies CC. PRMTs and Arginine Methylation: Cancer’s Best-Kept Secret? Trends in Molecular Medicine 2019;25(11):993–1009 doi 10.1016/j.molmed.2019.05.007. [DOI] [PubMed] [Google Scholar]
- 44.Tang J, Frankel A, Cook RJ, Kim S, Paik WK, Williams KR, et al. PRMT1 is the predominant type I protein arginine methyltransferase in mammalian cells. Journal of Biological Chemistry 2000;275(11):7723–30 doi doi 10.1074/jbc.275.11.7723. [DOI] [PubMed] [Google Scholar]
- 45.Le Naour J, Zitvogel L, Galluzzi L, Vacchelli E, Kroemer G. Trial watch: STING agonists in cancer therapy. Oncoimmunology 2020;9(1):1777624 doi 10.1080/2162402X.2020.1777624. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Musiani D, Giambruno R, Massignani E, Ippolito MR, Maniaci M, Jammula S, et al. PRMT1 Is Recruited via DNA-PK to Chromatin Where It Sustains the Senescence-Associated Secretory Phenotype in Response to Cisplatin. Cell Rep 2020;30(4):1208–22 e9 doi 10.1016/j.celrep.2019.12.061. [DOI] [PubMed] [Google Scholar]
- 47.Hartlova A, Erttmann SF, Raffi FA, Schmalz AM, Resch U, Anugula S, et al. DNA damage primes the type I interferon system via the cytosolic DNA sensor STING to promote anti-microbial innate immunity. Immunity 2015;42(2):332–43 doi 10.1016/j.immuni.2015.01.012. [DOI] [PubMed] [Google Scholar]
- 48.Kim CG, Jang M, Kim Y, Leem G, Kim KH, Lee H, et al. VEGF-A drives TOX-dependent T cell exhaustion in anti-PD-1-resistant microsatellite stable colorectal cancers. Sci Immunol 2019;4(41) doi 10.1126/sciimmunol.aay0555. [DOI] [PubMed] [Google Scholar]
- 49.Long J, Hu Z, Xue H, Wang Y, Chen J, Tang F, et al. Vascular endothelial growth factor (VEGF) impairs the motility and immune function of human mature dendritic cells through the VEGF receptor 2-RhoA-cofilin1 pathway. Cancer Sci 2019;110(8):2357–67 doi 10.1111/cas.14091. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Fukumura D, Kloepper J, Amoozgar Z, Duda DG, Jain RK. Enhancing cancer immunotherapy using antiangiogenics: opportunities and challenges. Nat Rev Clin Oncol 2018;15(5):325–40 doi 10.1038/nrclinonc.2018.29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kagoya Y, Saijo H, Matsunaga Y, Guo TX, Saso K, Anczurowski M, et al. Arginine methylation of FOXP3 is crucial for the suppressive function of regulatory T cells. J Autoimmun 2019;97:10–21 doi 10.1016/j.jaut.2018.09.011. [DOI] [PubMed] [Google Scholar]
- 52.Shen L, Zhang J, Lee H, Batista MT, Johnston SA. RNA Transcription and Splicing Errors as a Source of Cancer Frameshift Neoantigens for Vaccines. Sci Rep 2019;9(1):14184 doi 10.1038/s41598-019-50738-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Woo SR, Corrales L, Gajewski TF. The STING pathway and the T cell-inflamed tumor microenvironment. Trends Immunol 2015;36(4):250–6 doi 10.1016/j.it.2015.02.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The raw data files and analyzed data files from RNA-seq and ATAC-seq have been deposited in the NCBI GEO database, including 1) GSE161910: RNA-seq and ATAC-seq of murine tumor cells with or without Type I PRMT inhibitor treatment; 2) GSE126651: RNA-seq of human tumor cells with or without Type I PRMT inhibitor treatment.