

Abstract
The COVID-19 pandemic emerged in 2019, bringing with it the need for greater stores of effective antiviral drugs. This paper deals with the conformation-independent, QSAR model, developed by employing the Monte Carlo optimization method, as well as molecular graphs and the SMILES notation-based descriptors for the purpose of modeling the SARS-CoV-3CLpro enzyme inhibition. The main purpose was developing a reproducible model involving easy interpretation, utilized for a quick prediction of the inhibitory activity of SAR-CoV-3CLpro. The following statistical parameters were present in the best-developed QSAR model: (training set) R2 = 0.9314, Q2 = 0.9271; (test set) R2 = 0.9243, Q2 = 0.8986. Molecular fragments, defined as SMILES notation descriptors, that have a positive and negative impact on 3CLpro inhibition were identified on the basis of the results obtained for structural indicators, and were applied to the computer-aided design of five new compounds with (4-methoxyphenyl)[2-(methylsulfanyl)-6,7-dihydro-1H-[1,4]dioxino[2,3-f]benzimidazol-1-yl]methanone as a template molecule. Molecular docking studies were used to examine the potential inhibition effect of designed molecules on SARS-CoV-3CLpro enzyme inhibition and obtained results have high correlation with the QSAR modeling results. In addition, the interactions between the designed molecules and amino acids from the 3CLpro active site were determined, and the energies they yield were calculated.
Supplementary Information
The online version contains supplementary material available at 10.1007/s11696-022-02170-8.
Keywords: 3CLpro inhibitors, COVID-19 therapy, QSAR, Molecular modeling, Drug design
Introduction
The period from the end of 2019 and the beginning of 2020 will be remembered as the onset of the most hazardous pandemic in modern history, i.e. the COVID-19 outbreak. COVID-19 is caused by coronavirus 2 (SARS-CoV-2), or by the positive-sense single-stranded RNA virus (Chen et al. 2020; Guo et al. 2020; Kumar et al. 2020; Rothan and Byrareddy 2020). The Center for Disease Control and Prevention (CDC) first established the existence of this virus in Wuhan, China in the final days of 2019, and from then on, the virus has spread to other countries, posing a considerable threat to global health (Lai et al. 2020; Pedersen and Ho 2020). The droplets produced when those infected exhale, cough or sneeze represent the main means of SARS-CoV-2 distribution. the crucial factor in spreading the virus is the ease with which it is transmitted, since breathing in the proximity of someone who has COVID-19, or touching a virus contaminated surface before making contact with one’s mouth, eyes, or nose, constitute the main routs of infection (Peng et al. 2020; Singhal 2020; Sohrabi et al. 2020). Conversely, the chief danger of contracting COVID-19 lies in the possibility of an asymptomatic person spreading the virus, since 10% of the infections originate from people who do not exhibit any symptoms whatsoever, in spite of the fact that COVID-19 is the most infectious once someone develops symptoms related to COVID-19 (Kampf et al. 2020; Xiao et al. 2020). The chief symptoms attributed to COVID-19 are the potential loss of smell, cough, or taste, as well as the occurrence of fever, or having trouble breathing (Shang et al. 2020; Wu et al. 2020). Fortunately, to date, proper care and treatment usually lead to the recovery of most people infected with COVID-19 (Jiang et al. 2020; Sanders et al. 2020; Vellingiri et al. 2020). The WHO (World Health Organization) has determined two groups which pose a greater risk of suffering from severe illness and even mortality caused by the SARS-CoV-2 infection—adults over 60 years of age, and people whose immune systems are compromised by various health issues (Xiao et al. 2020, Kampf et al. 2020). Since SARS-CoV-2 is a recently discovered virus, a lot of its characteristics are still unknown.
One promising approach for COVID-19 treatment is blocking the replication of the virus by inhibiting viral protease (Lin et al. 2005; Hoffmann et al. 2020; Jin et al. 2020). One of the key steps in viral replication, which also includes SARS-CoV-2, is the processing of replicase polypeptides 1a and 1ab into functional proteins, which is a process determined with 3C-like protease (3CLpro) (Chen et al. 2005; Deng et al. 2014; Ramajayam et al. 2010; Zhavoronkov et al. 2020). Two proteases, papain-like protease (PLpro) and main protease or 3-chymotrypsin-like protease (Mpro/3CLpro), are essential components for replication of SARS-CoV-2, whit main role related to cleaving the two polyproteins, i.e., PP1A and PP1AB into several functional components. In this process, N-terminal domain is split to viral precursor protein at the three sites by PLpro, while the C-terminal domain is split to precursor protein at the 11 sites by 3CLpro (Akaji and Konno 2020). Targeting these enzymes had been used for the treatment of other pathogenic coronaviruses (i.e., MERS-CoV) (Amin and Jha 2020) and since the sequence homology of SARS-CoV-2 3CLpro is 96% structurally closer to the SARS-CoV 3CLpro targeting SARS-CoV-2 3CLpro as a potential therapeutic target is quite a feasible approach for anti-CoVID-19 drug development (Amin et al. 2021; Zhang et al 2020).
Chemoinformatics and computational chemistry have developed methodologies which have proven to be of great importance when designing potential therapeutics. When it comes to drug development research, the most significant contributions made by these in silico methods are the search for new leading compounds, or the optimization of therapeutic activities (or pharmacokinetic properties) of the series of chemical compounds whose biological activities have already been determined (Ekins et al. 2007; Tabeshpour et al. 2018). Among the most used methodologies are the quantitative structure–activity relationship (QSAR) and molecular docking. The mathematical equation linking the biological activities of the studied molecules with their chemical characteristics, defined as molecular descriptors, represents one of the most widely used practices for QSAR model representation (Cherkasov et al. 2014). In current practice, the applied representation of molecule structure is used for the calculation of a considerable number of descriptors, calculated in such fashion that could be further utilized for the development of a relevant QSAR model (Liu and Long 2009; Pérez González et al. 2008). A novel approach involves the use of conformation-independent optimal descriptors which are based on the constitutional and topological molecular features, and the descriptors based on the Simplified Molecular Input Line Entry System (SMILES) notation for QSAR model development, where the Monte Carlo optimization approach is used for this purpose. One of the recently suggested approaches to the manner of overcoming issues related to mechanistic interpretation is the application of SMILES descriptors, since these have a physical meaning and can be associated with molecular fragments (Toropova et al. 2016; Veselinović et al. 2015; Zivkovic et al. 2020), which is why this type of QSAR modeling is conducted under the OECD (Organization for Economic Co-operation and Development) guidelines for QSAR model development.
Several in silico methods were administered for the purpose of revealing new compounds having a potential 3C-like protease (3CLpro) inhibition activity in the presented research. Since molecular descriptors based on the SMILES notation were used, along with local graph invariants, the developed QSAR models were conformation-independent. One of the chief aims of this study was to determine the structural requirements or molecular fragments responsible for the 3CLpro inhibition effect. Moreover, this research defined the structural attributes found in small molecules concerning ligand-receptor interactions, which could be used in designing and developing SARS-CoV infection treatment therapeutics. Since the SMILES notation is used for chemical representation by most of the currently utilized chemical databases, this type of modeling could be employed by other scientists for screening purposes, as the results of the developed QSAR models have displayed considerably good values for various validation metrics, both internal and external ones.
Experimental
Methods
Dataset selection
A group of 84 molecules encompassing a wide range of heterocyclic compound classes, with an established 3C-like protease inhibiting activity against the infectious bronchitis virus (SARS-CoV virus), were selected from the binding database, according to “The Scripps Research Institute Molecular Screening Center” (Gilson et al. 2015), and used for QSAR model development. What needs to be specially emphasized is that the 3CLpro protease of SARS-CoV-2 has a sequence similarity greater than 95% when compared with that of the bronchitis virus (SARS-CoV virus), which was the main reason for the use of the surrogate enzyme for developing inhibitors against SAR-CoV-2 3CLpro (Liu et al. 2020). All the used molecules have an activity in IC50 (nM) values, which was determined using the same bioassay protocol (QFRET-based dose–response biochemical high throughput screening assay) (Jacobs et al. 2013). The IC50 values were converted to pIC50 (pIC50 = − logIC50) values as a studied activity. Afterwards, Open Babel was utilized to canonize the SMILES notations obtained from the above-stated database. Table S1 in the Supplementary Material cites the SMILES notations of all the used molecules, with their pIC50. Furthermore, the obtained data set was divided randomly into the training set (63 compounds, 75%) and the test set (21 compounds, 25%) for three random splits. In addition, the published method was used to verify the normality of the activity distribution (Ojha and Roy 2011).
Descriptor calculation and QSAR model development
The Monte Carlo optimization method was applied as the main algorithm for the conformation-independent QSAR models development. A hybrid approach was employed for the selection of adequate molecular descriptors as this approach represents the combination of the best features both from the molecular graph and from the descriptors based on the SMILES notation.
Molecular graph-based descriptors were derived from local graph invariants, and the most elementary theoretical graph concepts, such as paths and walks, were used in this research. Literature contains detailed mathematical definitions and applications in QSAR model development (Stoičkov et al. 2018). The following local graph invariants were applied for the purpose of defining the optimal descriptors used in conformation-independent QSAR models: Morgan extended connectivity indices of increasing orders (EC0), path numbers with the length of 2 and 3 (p2, p3), valence shells in the range of 2 and 3 (s2, s3), as well as the number of carbon atom neighbors (NumberOfCarbon), and the non-carbon atom neighbors (NumberofNonCarbon).
Medicinal chemists find descriptors with mechanistic interpretations very appealing, in addition to those that could be correlated with molecular fragments. Unfortunately, most molecular graph-based descriptors do not have this feature, and the SMILES notation was used as a highly convenient alternative to them. One of the key features of the SMILES notation-based descriptors is the possibility of relating them to the appropriate molecular fragment. Data concerning the detailed description of the SMILES notation, as well as its application for defining molecule structure can be found in literature (Toropova et al. 2016; Veselinović et al. 2015). The descriptors based on the SMILES notation need a numerical value that is to be used in further QSAR model development, as is the case with all molecular descriptors. The numerical value used for this purpose is defined as correlation weight. Its mathematical representation is the sum of all the defined SMILES descriptors, according to Eq. 1.
| 1 |
Z, x, y, t, α, β and γ can have the values of 1 (yes) or 0 (no). These values represent coefficients based on which these descriptors are used in QSAR modeling, which is done by following a simple rule—if the coefficient is 1, the descriptor is used, and if it is 0, then it is discarded. In Eq. 1, the Sk symbol is related to the local descriptors which are associated with one SMILES notation symbol (or two that cannot be separated)—SMILES atoms. The new optimal descriptors, represented with the SSk and SSSk symbols, respectively, are defined by the linear combinations of two and three SMILES atoms. With the exception of the local ones used in the QSAR modeling, the global SMILES notation descriptors could be used. When compared to the local ones, the global descriptors differ since they are related to the global features of a studied molecule, not only its structure. The following global SMILES notation-based descriptors were used in the presented research: NOSP, HALO, BOND, and ATOMPAIR. These are defined on the basis of the presence or absence of the following chemical elements: nitrogen, oxygen, sulfur and phosphorus (NOSP); fluorine, chlorine and bromine (HALO); double, triple, or stereochemical bonds (BOND), as well as the presence of seven chemical elements: F, Cl, Br, N, O, S, and P (ATOMPAIR). The presence/absence of eight chemical elements (fluorine, chlorine, bromine, iodine, nitrogen, oxygen, sulfur and phosphorus) and different types of chemical bonds (stereo chemical bond, double bond and triple bond) is defined as with the HARD-index a global SMILES notation-based descriptor and represented as a line of eleven symbols. A combination of both SMILES notation (both local and global) and local graph invariant descriptors is utilized in the presented research for the QSAR model deDCW, and is defined as the sum of all the used optimal descriptors’ correlation weights, according to Eq. 2.
| 2 |
where Sk is one SMILES symbol, defined as the SMILES atom, SSk and SSSk are the linear combinations of two or three neighbor SMILES atoms, EC0k is the Morgan connectivity index of the zero order (a hydrogen suppressed graph was used in this research), PT2k and PT2k are paths in the length of 2 and 3, VS2k and VS3k are valence shell 2 and 3, and NNCk is Nearest Neighbors (Toropov et al. 2003).
The calculations of all the above mentioned molecular descriptors and the QSAR model development were conducted with the use of the Monte Carlo optimization method and its algorithm, implemented in CORAL (CORrelation and Logic) (http://www.insilico.eu/coral) software. The Monte Carlo optimization method depends on two essential parameters for the QSAR model development—the threshold (T), used for eliminating rare molecule features, and the number of epochs (Nepoch), which represents the number of iterative processes of the algorithm for the purpose of reaching the top correlation coefficient value. Within the QSAR model developing process, the Monte Carlo optimization method matches molecular descriptors with their numerical values (CW) until CWs are determined. In this manner, the Least Squares method can calculate the DCW (T,Nepoch) for the training and test set compounds, as defined in Eq. 3. A systematic review of the applied method is described in the literature (Toropova et al. 2016; Veselinović et al. 2015). The search for the best combination of T and Nepoch in the presented research was performed within the values of 0–10 for T, and 0–70 for Nepoch.
| 3 |
The quality of the developed QSAR model needs to be evaluated by employing different validation methods. The process should provide information on the benefits of the developed model and whether it could be used for future predictions of the studied activities. The methodology from the published paper was used entirely for this purpose (Veselinović et al. 2015; Ojha et al. 2011; Roy et al. 2008), which involved the calculation of various statistical parameter values, such as the regular and cross-validated correlation coefficient, standard estimation error, mean absolute error (MAE), the Fischer ratio, root-mean-square error, Rm2, and MAE-based metrics. What is more, the validation of the developed QSAR models in this research was achieved through data randomization (Y-scrambling test) and with the determination of concordance correlation coefficient (CCC), as well as the novel parameter, dubbed the index of ideality of correlation (IIC) (Stoičkov et al. 2018; Toropov and Toropova 2017; Veselinović et al. 2018).
Molecular docking
For molecular docking studies were applied the Molegro Virtual Docker (MVD) software. The studies molecules were drawn using the Marvin sketch (Marvin 6.1.0, 2013, ChemAxon), whereas the MMFF94 force field was utilized to gain their optimal 3D geometry. The protein databank provided the structure of the studied enzyme with the PDB id: 6lu7 (the crystal structure of COVID-19 main protease in a complex with an inhibitor N3) (Jin et al. 2020). The purpose of MVD was obtaining an adequate geometrical orientation of the flexible ligand within the active site of the studied enzyme, surrounded by rigid amino acids, as well as identifying the hydrogen bonds and hydrophobic interactions between them. Finally, MVD was also used to calculate the relevant binding energies, also defined as “scoring” functions (Thomsen and Christensen 2006). The use of these functions is assessing the studied molecules’ inhibitory effect, and the following “scoring” functions were calculated in this research—Hbond, NoHbond, VdW, Steric, Pose energy, MolDock, and Rerank Score. The energy from hydrogen and no hydrogen bond interactions were calculated with HBond and NoHbond90, respectively; the energies from the Van der Walls and Steric interactions were calculated with VdW and Steric “scoring” functions; the overall energy of the best-calculated pose was calculated with the Pose energy. MolDock Score and Rerank Score were calculated as the final estimators of ligand and amino acids from the enzyme’s active site interaction energies, and the whole molecular docking protocol was validated in accordance with the published methodology (Amin et al. 2018, 2019; Jain et al. 2020; Zivkovic et al. 2020). All the crystallized water molecules were removed before the docking. The binding site was computed within spacing in such a way that it was well sampled with the grid resolution of 0.3 Å. The MolDock SE was used as a search algorithm, with the set number of runs being up to 100. The docking procedure parameters were: population size − 50; maximum number of iterations − 1.500; energy threshold − 100.00, and the maximum number of steps − 300. The maximum number of poses to generate was increased to 10 from a default value of 5. Discovery Studio Client v20.1.0.19 was used for showing two-dimensional representations of the interactions between the studied molecules and the amino acids 3CLpro active site.
Results and discussion
To consider any QSAR model for prediction purposes, its applicability domain (AD) should be defined prior to use (Gadaleta et al. 2016; Gramatica 2007). The methodology described in the literature was used to define the AD for all the developed QSAR models in this study (Toropova et al. 2016; Veselinović et al. 2015). Based on the results obtained, all the molecules fall within the defined AD, and there were no outliers. The numerical values for all the metrics used for the determination of the developed QSAR model’s quality are cited in Table 1. On the basis of the results presented, the Monte Carlo optimization method was able to produce a QSAR model which exhibited good reproducibility, and a high predictability potential. When it comes to the 3CLpro inhibitory activity, the second split yielded the best QSAR model, developed with the T value of 1, whereas the best Nepoch value was 20. Figure 1 presents the graphical representations of the best Monte Carlo optimization runs (the highest value for r2) for the developed QSAR models (all the three splits). Equation 4 contains the mathematical representation of the best-developed model. The reproducibility concordance correlation coefficient (CCC) was used for the assessment of the developed QSAR models, and the obtained numerical values for the CCC indicate that the presented QSAR models show high reproducibility values. The metrics results based on the MAE indicate a GOOD model, thus classifying the developed QSAR model as valid. The numerical values of the index of ideality of correlation (IIC) were calculated for the purpose of making a final estimation regarding the quality of the developed QSAR models, and the obtained results exhibit a high predictive potential for the developed QSAR models. The numerical values calculated for Y-randomization, in which the Y values were scrambled in 1000 trials in ten separate runs, are presented in Table 2, and these were used to evaluate the sturdiness of the developed QSAR models. The presented results indicate that the developed QSAR models were free from correlation by chance because the numerical value for CR2p was higher than 0.5.
| 4 |
Table 1.
The statistical quality of the developed QSAR models for 3CLpro inhibitory activity
| Run | Training set | Test set | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| R2 | CCC | IIC | Q2 | s | MAE | F | R2 | CCC | IIC | Q2 | Q2F1 | Q2F2 | Q2F3 | s | MAE | F | ||
| Split 1 | 1 | 0.9393 | 0.9687 | 0.8812 | 0.9353 | 0.082 | 0.066 | 944 | 0.8723 | 0.9151 | 0.9339 | 0.8499 | 0.8623 | 0.8523 | 0.9014 | 0.107 | 0.073 | 130 |
| 2 | 0.9294 | 0.9634 | 0.7712 | 0.9236 | 0.089 | 0.072 | 803 | 0.8841 | 0.8897 | 0.9402 | 0.8614 | 0.8384 | 0.8268 | 0.8843 | 0.115 | 0.086 | 145 | |
| 3 | 0.9351 | 0.9665 | 0.9367 | 0.9311 | 0.085 | 0.067 | 880 | 0.8862 | 0.8990 | 0.9413 | 0.8676 | 0.8476 | 0.8366 | 0.8909 | 0.112 | 0.092 | 148 | |
| Av | 0.9346 | 0.9662 | 0.863 | 0.9300 | 0.085 | 0.068 | 876 | 0.8809 | 0.9013 | 0.9385 | 0.8596 | 0.8494 | 0.8386 | 0.8992 | 0.111 | 0.084 | 141 | |
| Split 2 | 1 | 0.9326 | 0.9651 | 0.7242 | 0.9285 | 0.084 | 0.064 | 844 | 0.9042 | 0.9294 | 0.9456 | 0.8646 | 0.8913 | 0.8738 | 0.8923 | 0.108 | 0.087 | 161 |
| 2 | 0.9314 | 0.9645 | 0.9350 | 0.9271 | 0.085 | 0.066 | 828 | 0.9243 | 0.9414 | 0.9561 | 0.8986 | 0.9102 | 0.8958 | 0.9111 | 0.098 | 0.075 | 203 | |
| 3 | 0.9407 | 0.9695 | 0.9396 | 0.9368 | 0.079 | 0.064 | 968 | 0.9317 | 0.9417 | 0.9653 | 0.9160 | 0.9129 | 0.8989 | 0.9137 | 0.096 | 0.077 | 259 | |
| Av | 0.9349 | 0.9664 | 0.8663 | 0.9308 | 0.083 | 0.065 | 880 | 0.9201 | 0.9375 | 0.9557 | 0.8930 | 0.9048 | 0.8895 | 0.9057 | 0.107 | 0.080 | 208 | |
| Split 3 | 1 | 0.8685 | 0.9296 | 0.8472 | 0.8610 | 0.120 | 0.091 | 403 | 0.9077 | 0.9463 | 0.9527 | 0.8688 | 0.9054 | 0.9019 | 0.9235 | 0.093 | 0.073 | 187 |
| 2 | 0.8853 | 0.9391 | 0.8025 | 0.8792 | 0.112 | 0.086 | 471 | 0.9140 | 0.9172 | 0.9560 | 0.8952 | 0.8691 | 0.8642 | 0.8941 | 0.109 | 0.080 | 202 | |
| 3 | 0.9036 | 0.9493 | 0.8641 | 0.8981 | 0.102 | 0.080 | 572 | 0.9298 | 0.9600 | 0.9642 | 0.9100 | 0.9228 | 0.920 | 0.9376 | 0.084 | 0.063 | 252 | |
| Av | 0.8858 | 0.9393 | 0.8379 | 0.8794 | 0.111 | 0.086 | 482 | 0.9172 | 0.9412 | 0.9576 | 0.8913 | 0.8891 | 0.8954 | 0.9184 | 0.095 | 0.072 | 214 | |
R2 correlation coefficient, Q2 cross-validated correlation coefficient, s standard error of estimation, MAE mean absolute error, F Fischer ratio, Av average value for statistical parameters obtained from three independent Monte Carlo optimization runs
Fig. 1.

Graphical representation of the best Monte Carlo optimization runs (the highest value for r2) for the developed QSAR models
Table 2.
Y-randomization of the best QSAR model (best optimization run) for three independent splits
| Split 1 | Split 2 | Split 3 | ||||
|---|---|---|---|---|---|---|
| Training | Test | Training | Test | Training | Test | |
| 0.9351 | 0.8862 | 0.9407 | 0.9317 | 0.9036 | 0.9298 | |
| 1 | 0.0064 | 0.0340 | 0 | 0.0197 | 0 | 0.0726 |
| 2 | 0.0009 | 0.0019 | 0.0090 | 0.0202 | 0.0235 | 0.0823 |
| 3 | 0.0935 | 0.0152 | 0.0196 | 0.0042 | 0.0151 | 0.0708 |
| 4 | 0.0087 | 0.0193 | 0.0309 | 0.0281 | 0.0422 | 0.1384 |
| 5 | 0.0248 | 0.0008 | 0.0853 | 0.0098 | 0.0061 | 0.0009 |
| 6 | 0.0035 | 0.0028 | 0.0007 | 0 | 0.0207 | 0.0129 |
| 7 | 0.0051 | 0.0149 | 0.0004 | 0.0215 | 0.0041 | 0.0002 |
| 8 | 0.0020 | 0.0096 | 0.0006 | 0.0368 | 0.0145 | 0.0237 |
| 9 | 0.0061 | 0.0410 | 0.0015 | 0.0006 | 0.0103 | 0.0134 |
| 10 | 0.0040 | 0.1760 | 0.0246 | 0.0965 | 0.0684 | 0.0780 |
| Rr2 | 0.0155 | 0.0316 | 0.0173 | 0.0237 | 0.0205 | 0.0493 |
| CRp2 | 0.9274 | 0.8703 | 0.932 | 0.9198 | 0.8933 | 0.9048 |
CRp2 = R × (R2-Rr2)1/2 should be > 0.5 (Ojha and Roy 2011)
A QSAR model was developed by Kumar and Roy for the same activity, with the use of 2D molecular descriptors and the multiple linear regression (MLR) technique (Kumar and Roy 2020). When compared with this research, where the authors used 69 molecules (16 molecules with outlier behavior were omitted from the original database), all the molecules were used in the presented research as they all fall into the defined AD. The following validation parameters were present in the QSAR model reported by Kumar and Roy: Internal validation) R2 = 0.764, Q2 = 0:627; External) Q2F1 = 0,727, Q2F2 = 0,652, Avgrm2 = 0.610, Δrm2 = 0.110, MAE = 0.127. In addition to the validation parameters cited in Table 1, the QSAR models developed with the application of the Monte Carlo optimization method had Avgrm2 and Δrm2 values of 0.8966–0.6097 and 0.1885–0.0191, respectively. When comparing the two approaches, by applying solely the reported statistical parameters, in terms of predictability, the QSAR model developed with the application of the Monte Carlo optimization method is superior.
The determination of molecular fragments, defined as the SMILES notation optimal descriptors having a positive and negative impact on the studied activity, was one of the chief aims of this research (Halder 2018; Kumar et al. 2019; Manisha et al. 2019; Toropov et al. 2019). The full list of molecular descriptors, both based on the molecular graph and the SMILES notation, are shown in Table S2 (Supplementary material), while the selected ones are detailed in Table 3. The calculation example is presented in Table S2, where molecular graph-based descriptors were omitted with the aim of achieving an easier interpretation.
Table 3.
Selected promoters of inhibition activity increase/decrease
| Promoters of inhibition activity increase | Promoters of inhibition activity decrease |
|---|---|
| (…(…… | (…Br..(… |
| (…N…(… | (…Cl..(… |
| (…O…(… | = …1…… |
| + + + + CL–N = = = | C…2…(… |
| + + + + N–-O = = = | N………. |
| + + + + O–-B2 = = | N…2…… |
| C…/…… | N… = …… |
| C…/…C… | N… = …C… |
| C…C…C… | N…C…(… |
| C…O…C… | N…C…… |
| BOND10000000 | N…1…… |
| N…C…C… | O… = …… |
| O…1…… | O… = …C… |
| O…C…… | O…C…N… |
| O…C…C… | n…n…… |
| s…c…… | s…n…… |
| s………. | s…n…n… |
| c…c…… | C…(…… |
| c…c…c… | N…(…… |
The selected molecular fragments, defined as the SMILES notation descriptors which have a positive influence on the studied activity (a lower concentration of the compound is required to achieve IC50) are as follows: “(…(…….”, “(…N…(…” and “(…O…(…”—additional branching in the molecule, and additional branching in the molecule realized by adding aliphatic nitrogen and oxygen atom; “ + + + + CL–N = = = ”, “ + + + + N–-O = = = ” and “ + + + + O–-B2 = = ”—the presence of chlorine and nitrogen atoms in the molecule, the presence of oxygen and nitrogen atoms in the molecule, and the presence of oxygen and a double bond in the molecule; “BOND10000000”, “C…/…….”, and “C…/…C…”—double bond between two carbon atoms; “C…C…C…”—propyl group; “N…C…C…”—ethyl amine group; “O…1…….” oxygen atom inside the ring; “O…C…….”, “O…C…C…” and “C…O…C…”—methoxy, ethoxy and dimethoxy groups, respectively; “s…c…….” and “s………..”—aromatic sulfur atom; “c…c…….” and “c…c…c…”—aromatic carbon atoms. The same analysis may be performed for molecular fragments, defined as the SMILES notation descriptors with a negative impact on the studied activity. The selected molecular fragments are: “C…(…….”, “N…C…(…” and “N…(…….”—simple molecular branching on the carbon and nitrogen atom; “(…Br..(…” and “(…Cl..(…”—additional molecular branching with bromine and chlorine atoms; “C…2…(…” and “N…2…….”—second ring inside the molecule, and second ring inside the molecule with a nitrogen atom; “ = …1…….”—double bond within the ring; “N………..” and “– aliphatic nitrogen; “N… = …….”, “N… = …C…”, “O… = …….” and “O… = …C…”—the double bond between the nitrogen or oxygen atom and the carbon atom; “n…n…….”, “s…n…….” and “s…n…n…”—aromatic nitrogen atoms and the presence of both aromatic nitrogen and sulfur atoms. The graphical representation of the impact of molecular fragments on the selected molecule activity is displayed in Fig. 2. The green color in Fig. 2 indicates a positive influence, while red indicates a negative impact on the studied activity.
Fig. 2.
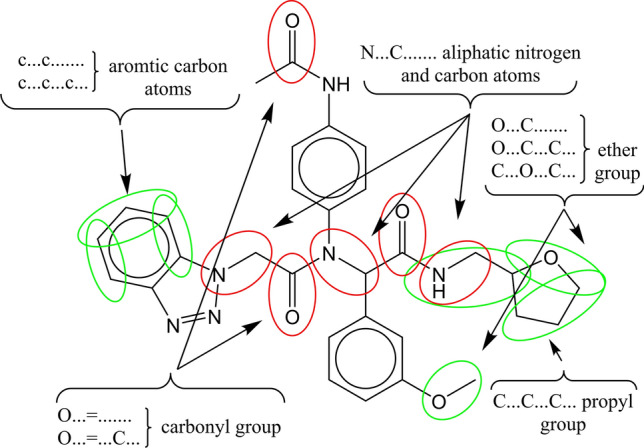
Identified molecular fragments with their impact on 3CLpro inhibitory activity
Identified molecular fragments were used for the computer-aided design of novel 3CLpro inhibitors. The molecule with the highest inhibitory effect (compound 1 from the dataset) was used as a starting/template molecule for the purpose of producing molecules with a higher inhibition potential. Table 4 indicates the SMILES notation of all the designed molecules, along with their calculated values for the -pIC50, utilizing the best-developed QSAR model, while their chemical structures are presented in Fig. 3.
Table 4.
The list of all designed molecules with their SMILES notation and calculated activities
| Molecule | SMILES notation | Ac(calc.) |
|---|---|---|
| A | CSc1nc2c(n1C(=O)c1ccc(cc1)OC)cc1c(c2)OCCO1 | − 3.0608 |
| A1 | CSc1nc2c(n1C(=O)c1ccc(cc1)OC(C)C)cc1c(c2)OCCO1 | − 2.8396 |
| A2 | CSc1nc2c(n1C(=O)c1ccc(cc1)OCC1CC1)cc1c(c2)OCCO1 | − 2.7599 |
| A3 | COc1ccc(cc1)C(=O)n1c(SC(C)C)nc2c1cc1OCCOc1c2 | − 2.8704 |
| A4 | COCOc1ccc(cc1)C(=O)n1c(SC)nc2c1cc1OCCOc1c2 | − 2.801 |
| A5 | COCSc1nc2c(n1C(=O)c1ccc(cc1)OC)cc1c(c2)OCCO1 | − 2.791 |
Ac(calc.) calculated values for -pIC50 with the application of the best QSAR model
Fig. 3.
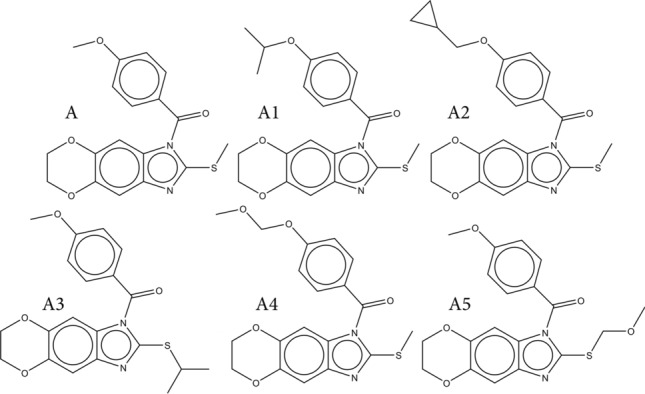
Chemical structures of all designed molecules
Added molecular fragments were identified as structural alerts that have a positive effect on the studied activity in all the designed molecules (A1-A5). Molecules A1-A3 had the following molecular fragments added—the isopropyl and cyclopropyl group on the different parts of template molecule A. The groups are defined as the following SMILES notation descriptors, all promoters of the studied activity increase—“(…(…….”; “1………..”; “C………..”; “C…C…….”; “C…C…C…”. Molecules A4 and A5 have added methyl ether group. They all have a positive impact on the studied activity and are defined with the following SMILES notation descriptors – “C…O…C…”; “O…C…….”; “O…C…C…”; “O………..”.
The approach suggested in the literature was taken to assess the predictability of the developed QSAR model, and to further estimate inhibitory potential of designed molecules (Zivkovic et al. 2020). Within this approach all designed molecules and template molecule A were subjected to molecular docking studies with SARS-CoV-3CLpro enzyme to assess their binding potential and numerical values for all calculated “scoring” functions are presented in Table 5. One of assumption is that the more molecule is bounded to enzyme, indicated with higher binding energies between molecule and amino acids form active site and calculated in regards to appropriate “scoring” functions, the higher inhibition would be, resulting to higher values of pIC50, smaller concentrations are needed to achieve inhibition. Also, different ligand-amino acids interactions could be associated with various scoring functions, so everything should be taken into consideration when performing the assessment of the inhibitory potency. Literature contains the detailed definitions of other “scoring” functions (Thomsen and Christensen 2006, Zivkovic et al. 2020). Calculated values for energies with the application of defined “scoring functions” are much higher in comparison to real binding energies and most likely they are unrealistic. However, since same approach was used for all molecules, obtained values could be compared to assess the binding preferences, meaning the higher the binding energy, the higher binding preferences, leading to higher inhibitory potential. According to the obtained results for MolDock and ReRank, molecule A5 has the potentially highest inhibitory activity and molecule A the lowest. This result is in correlation with the results from the QSAR modeling since the calculated values for -pIC50 using the best QSAR model obtained with the Monte Carlo optimization method are the most preferable for molecule A5 and the least preferable for molecule A. All the interactions between the selected molecules and amino acids from the 3CLpro enzyme’s active site have been identified, and Figures S1-S6 in the Supplementary Information section present the 2D representation of hydrogen bonds, in addition to hydrophobic, and hydrophilic interactions inside the 3CLpro binding pocket. The best-calculated poses for all studied molecules inside the active site of 3CLpro are cited in Fig. 4.
Table 5.
Score values (kcal/mol) for studied molecules
| Molecule | HBond | NoHBond 90 |
Steric | VdW | Energy | MolDock Score |
Rerank Score |
|---|---|---|---|---|---|---|---|
| A | − 9.95391 | − 12.2513 | − 124.08 | − 36.6289 | − 126.41 | − 120.905 | − 100.528 |
| A1 | − 2.28481 | − 2.28481 | − 139.612 | − 45.7151 | − 134.173 | − 125.953 | − 108.021 |
| A2 | − 6.02484 | − 6.80429 | − 138.895 | − 38.4165 | − 134.373 | − 127.337 | − 103.769 |
| A3 | − 2.29903 | − 2.29903 | − 148.374 | − 41.8895 | − 138.963 | − 130.133 | − 113.401 |
| A4 | − 12.39 | − 14.89 | − 133.877 | − 45.782 | − 137.214 | − 130.258 | − 111.3548 |
| A5 | − 7.19636 | − 7.7621 | − 142.37 | − 49.4728 | − 145.603 | − 137.197 | − 122.4118 |
Fig. 4.
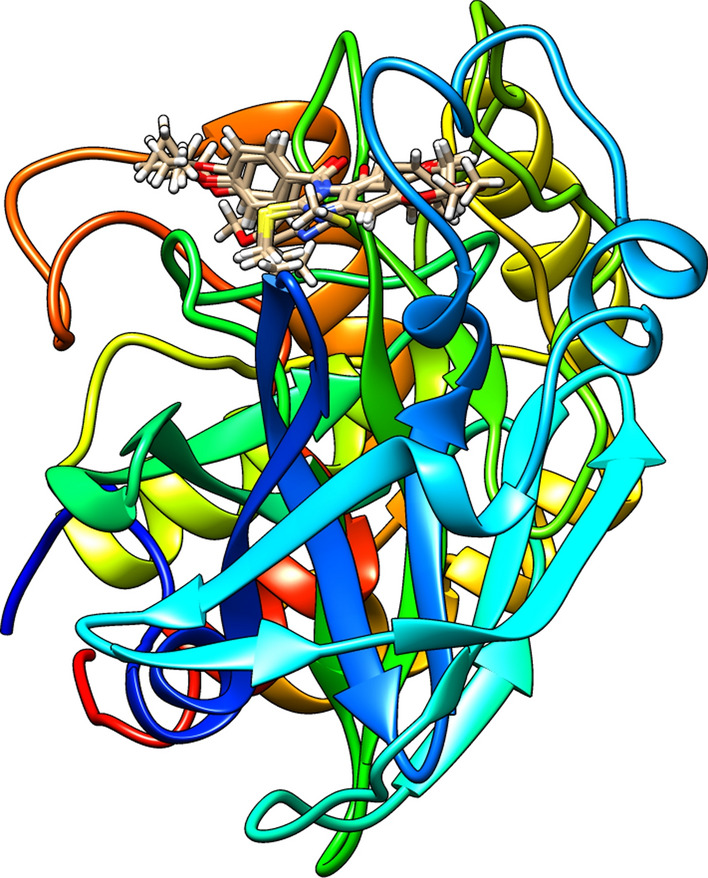
The best-calculated poses for designed molecules (A1-A5) and template molecule A inside the active site of 3CLpro
It is of extreme importance to determine the studied compounds’ drug-likeness when the aim is for them to become therapeutics. Compounds should possess physical and chemical parameters enabling them to be considered as potential medication. The application of Lipinski’s “Rule of Five” represents one of the extensively used approaches. These rules determine the absorption/permeation of the compound, and if the compound possesses a molecular weight higher than 500, logP over 5, and the number of hydrogen bond donors and acceptors is higher than 5 and 10, respectively, it will most likely have poor absorption/permeation. In addition, the number of rotatable bonds, associated with conformational molecular flexibility, is used to assess the efficacy of binding to receptors/channels, as well as their bioavailability. The molecule should have a satisfactory number of rotatable bonds, i.e. 10 or fewer (Lipinski et al. 2001). In addition to Lipinski’s “Rule of Five”, Veber proposed having an additional set of rules for a molecule’s drug-likeness assessment (Veber et al. 2002), and they can be summarized as acceptable when the oral bioavailability is more likely to occur when the molecule has 10 or fewer rotatable bonds, a polar surface area equal to or less than 140 Å2, and 12 or fewer hydrogen donors and acceptors. For template molecule A and all the designed molecules, the physical and chemical parameters defined above are calculated using the Molinspiration Cheminformatics software (www.molinspiration.com), and presented in Table 6. In accordance with the obtained results, all the molecules were in accordance with Lipinski’s “Rule of Five”, and the Veber rules, which may be associated with their potentially good solubility and permeability through biological membranes, thus leading to satisfactory bioavailability, which is why they may be considered as potential therapeutics.
Table 6.
Calculated physico-chemical properties of the designed molecules used for drug-likeness determination
| Molecule | miLogP | TPSA | natoms | MW | nON | nOHNH | nviolations | nrotb | volume |
|---|---|---|---|---|---|---|---|---|---|
| A | 3.43 | 62.6 | 25 | 356.4 | 6 | 0 | 0 | 3 | 300.61 |
| A1 | 4.17 | 62.6 | 27 | 384.46 | 6 | 0 | 0 | 4 | 333.99 |
| A2 | 4.3 | 62.6 | 28 | 396.47 | 6 | 0 | 0 | 5 | 340.44 |
| A3 | 4.17 | 62.6 | 27 | 384.46 | 6 | 0 | 0 | 4 | 333.99 |
| A4 | 3.4 | 71.83 | 27 | 386.43 | 7 | 0 | 0 | 5 | 326.39 |
| A5 | 3.4 | 71.83 | 27 | 386.43 | 7 | 0 | 0 | 5 | 326.39 |
miLogP octanol–water partition coefficient, TPSA molecular polar surface area, natoms number of atoms in molecule, MW molecular weight, nON number of nitrogen and oxygen atoms in molecule, nOHNH number of amino and hydroxyl groups, nviolations number of “Rule of five” violations, nrotb number of rotable bonds, volume volume of molecule
Conclusion
The present research demonstrates the development of new robust and reliable QSAR models for 3CLpro inhibition based on the Monte Carlo optimization method as the main model, and developed on the basis of optimal descriptors derived both from a local graph and the SMILES notation invariants. The assessment of the developed QSAR model’s robustness and its predictive potential was achieved by applying various statistical techniques and obtained numerical values, used to validate the developed QSAR model, indicating that it has high applicability. The applied methodology was able to determine the molecular fragments, used as the SMILES notation fragments in QSAR modeling, which have a positive and negative impact on 3CLpro inhibition. They were used for the computer-aided design of novel 3CLpro inhibitors with a potentially higher activity when compared to template molecule A. To assess the developed QSAR models' predictability all designed molecules and template molecule A were subjected to molecular docking studies with 3CLpro. Their binding potential was assessed according to the comparison of numerical values for all calculated “scoring” functions, related to energies from the interactions between the molecules and the amino acids within the active site of the 3CLpro, and calculated pIC50 from the best QSAR model developed with the Monte Carlo optimization method. Values for all defined “scoring” functions calculated for each designed molecule and calculated values for pIC50 show high inter-correlation. Also, all designed molecules have potentially good pharmacokinetic properties and possess drug-likeness. The methodology presented in this research paper can be applied in the search for novel therapeutics for the treatment of COVID-19 based on 3CLpro inhibition.
Supplementary Information
Below is the link to the electronic supplementary material.
Acknowledgements
This work is supported by the Ministry of Education and Science, the Republic of Serbia and the Faculty of Medicine, University of Niš, Republic of Serbia (project No. 70). The authors would like to thank the Ministry of Education, Science and Technological Development of Republic of Serbia (Grant No: 451-03-9/2021-14/200113) for financial support.
Declarations
Conflict of interest
We have no conflicts of interest to disclose.
Footnotes
Publisher's Note
Springer Nature remains neutral with regard to jurisdictional claims in published maps and institutional affiliations.
References
- Akaji K, Konno H. Design and evaluation of anti-SARS-coronavirus agents based on molecular interactions with the viral protease. Molecules. 2020;25:3920. doi: 10.3390/molecules25173920. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SA, Jha T. Fight against novel coronavirus: a perspective of medicinal chemists. Eur J Med Chem. 2020 doi: 10.1016/j.ejmech.2020.112559. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amin SA, Adhikari N, Gayen S, Jha T. Exploring pyrazolo[3,4-d]pyrimidine phosphodiesterase 1 (PDE1) inhibitors: a predictive approach combining comparative validated multiple molecular modelling techniques. J Biomol Struct Dyn. 2018;36:590–608. doi: 10.1080/07391102.2017.1288659. [DOI] [PubMed] [Google Scholar]
- Amin SA, Adhikari N, Gayen S, Jha T. Reliable structural information for rational design of benzoxazole type potential cholesteryl ester transfer protein (CETP) inhibitors through multiple validated modelling techniques. J Biomol Struct Dyn. 2019;37:4528–4541. doi: 10.1080/07391102.2018.1552895. [DOI] [PubMed] [Google Scholar]
- Amin SA, Banerjee S, Gayen S, Jha T. Protease targeted COVID-19 drug discovery: what we have learned from the past SARS-CoV inhibitors? Eur J Med Chem: 2021 doi: 10.1016/j.ejmech.2021.113294. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen S, Chen LL, Luo HB, Sun T, Chen J, Ye F, Cai JH, Shen JK, Shen X, Jiang HL. Enzymatic activity characterization of SARS coronavirus 3C-like protease by fluorescence resonance energy transfer technique 1. Acta Pharmacol Sin. 2005;26:99–106. doi: 10.1111/j.1745-7254.2005.00010.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen Y, Liu Q, Guo D. Emerging coronaviruses: genome structure, replication, and pathogenesis. J Med Virol. 2020;92:418–423. doi: 10.1002/jmv.25681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherkasov A, Muratov EN, Fourches D, Varnek D, Baskin A, Cronin II, Dearden MJ, Gramatica P, Martin YC, Todeschini R, Consonni V, Kuz'min VE, Cramer R, Benigni R, Yang C, Rathman J, Terfloth L, Gasteiger J, Richard A, Tropsha A. QSAR modeling: Where have you been? Where are you going to? J Med Chem. 2014;57:4977–5010. doi: 10.1021/jm4004285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Deng X, St John SE, Osswald HL, O’Brien A, Banach BS, Sleeman K, Ghosh AK, Mesecar AD, Baker SC. Coronaviruses resistant to a 3C-like protease inhibitor are attenuated for replication and pathogenesis, revealing a low genetic barrier but high fitness cost of resistance. Int J Virol. 2014;20:11886–11898. doi: 10.1128/JVI.01528-14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ekins S, Mestres J, Testa B. In silico pharmacology for drug discovery: applications to targets and beyond. Br J Pharmacol. 2007;152:9–20. doi: 10.1038/sj.bjp.0707306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gadaleta D, Mangiatordi GF, Catto M, Carotti A, Nicolotti O. Applicability domain for QSAR models: where theory meets reality. IJQSPR. 2016;1:45–63. doi: 10.4018/IJQSPR.2016010102. [DOI] [Google Scholar]
- Gilson MK, Liu T, Baitaluk MG, Nicola G, Hwang L, Chong J. BindingDB: a public database for medicinal chemistry, computational chemistry and systems pharmacology. Nucleic Acids Res. 2015;19:1045–1053. doi: 10.1093/nar/gkv1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gramatica P. Principles of QSAR models validation: internal and external. QSAR Comb Sci. 2007;26:694–701. doi: 10.1002/qsar.200610151. [DOI] [Google Scholar]
- Guo Y-R, Cao Q-D, Hong Z-S, Tan Y-Y, Chen S-D, Jin H-J, Tan K-S, Wang D-Y, Yan Y. The origin, transmission and clinical therapies on coronavirus disease 2019 (COVID-19) outbreak- An update on the status. Mil Med Res. 2020 doi: 10.1186/s40779-020-00240-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Halder A. Finding the structural requirements of diverse HIV-1 protease inhibitors using multiple QSAR modelling for lead identification. SAR QSAR Environ Res. 2018;29:911–933. doi: 10.1080/1062936X.2018.1529702. [DOI] [PubMed] [Google Scholar]
- Hoffmann M, Kleine-Weber H, Schroeder S, Krüger N, Herrler T, Erichsen S, Schiergens TS, Herrler G, Wu N-H, Nitsche A, Müller MA, Drosten C, Pöhlmann S. SARS-CoV-2 cell entry depends on ACE2 and TMPRSS2 and is blocked by a clinically proven protease inhibitor. Cell. 2020;181:271–280. doi: 10.1016/j.cell.2020.02.052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jacobs J, Zhou S, Dawson E, Daniels JS, Hodder P, Tokars V, Mesecar A, Lindsley CW, Stauffer SR (2013) Discovery of non-covalent inhibitors of the SARS main proteinase 3CLpro, In Probe Reports from the NIH Molecular Libraries Program [Internet], National Center Biotechnol Inform (US), Available at https://www.ncbi.nlm.nih.gov/books/NBK133447 [PubMed]
- Jain S, Amin SA, Adhikari N, Jha T, Gayen S. Chemical-informatics approach to COVID-19 drug discovery: Monte Carlo based QSAR, virtual screening and molecular docking study of some in-house molecules as papain-like protease (PLpro) inhibitors. J Biomol Struct Dyn. 2020;38:66–77. doi: 10.1080/07391102.2020.1780946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jiang F, Deng L, Zhang L, Cai Y, Cheung CW, Xia Z. Review of the clinical characteristics of coronavirus disease 2019 (COVID-19) J Gen Inter Med. 2020;35:1545–1549. doi: 10.1007/s11606-020-05762-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin Z, Du X, Xu Y, Deng Y, Liu M, Zhao Y, Zhang B, Li X, Zhang L, Peng C, Duan Y, Yu J, Wang L, Yang K, Liu F, Jiang R, Yang X, You T, Liu X, Yang X, Bai F, Liu H, Liu X, Guddat LW, Xu W, Xiao G, Qin C, Shi Z, Jiang H, Rao Z, Yang H. Structure of Mpro from SARS-CoV-2 and discovery of its inhibitors. Nature. 2020;582:289–293. doi: 10.1038/s41586-020-2223-y. [DOI] [PubMed] [Google Scholar]
- Kampf G, Todt D, Pfaender S, Steinmann E. Persistence of coronaviruses on inanimate surfaces and its inactivation with biocidal agents. J Hosp Infect. 2020;104:246–251. doi: 10.1016/j.jhin.2020.01.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumar V, Roy K. Development of a simple, interpretable and easily transferable QSAR model for quick screening antiviral databases in search of novel 3Clike protease (3CLpro) enzyme inhibitors against SARS-CoV diseases. SAR QSAR Environ Res. 2020;31:511–526. doi: 10.1080/1062936X.2020.1776388. [DOI] [PubMed] [Google Scholar]
- Kumar P, Kumar A, Sindhu J. In silico design of diacylglycerol acyltransferase-1 (DGAT1) inhibitors based on SMILES descriptors using Monte-Carlo method. SAR QSAR Environ Res. 2019;30:525–541. doi: 10.1080/1062936X.2019.1629998. [DOI] [PubMed] [Google Scholar]
- Kumar D, Malviya R, Sharma PK. Corona virus: A review of COVID-19. EJMO. 2020;4:8–25. doi: 10.14744/ejmo.2020.51418. [DOI] [Google Scholar]
- Lai C-C, Shih T-P, Ko W-C, Tang H-J, Hsueh P-R. Severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) and coronavirus disease-2019 (COVID-19): the epidemic and the challenges. Int J Antimicrob Ag. 2020 doi: 10.1016/j.ijantimicag.2020.105924. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lin CW, Tsai FJ, Tsai CH, Lai CC, Wan L, Ho TY, Hsieh CC, Lee Chao PD. Anti-SARS coronavirus 3C-like protease effects of Isatis indigotica root and plant-derived phenolic compounds. Antivir Res. 2005;68:36–42. doi: 10.1016/j.antiviral.2005.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lipinski CA, Lombardo F, Dominy BW, Feeney PJ. Experimental and computational approaches to estimate solubility and permeability in drug discovery and development settings. Adv Drug Deliver Rev. 2001;46:3–26. doi: 10.1016/s0169-409x(00)00129-0. [DOI] [PubMed] [Google Scholar]
- Liu P, Long W. Current mathematical methods used in QSAR/QSPR studies. Int J Mol Sci. 2009;10:1978–1998. doi: 10.3390/ijms10051978. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu C, Zhou Q, Li Y, Garner LV, Watkins SP, Carter LJ, Smoot J, Albaiu D. Research and development on therapeutic agents and vaccines for COVID-19 and related human coronavirus diseases. ACS Cent Sci. 2020;6:315–331. doi: 10.1021/acscentsci.0c00272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Manisha CS, Kumar P, Kumar A. Development of prediction model for fructose-1,6-bisphosphatase inhibitors using the Monte Carlo method. SAR QSAR Environ Res. 2019;30:145–159. doi: 10.1080/1062936X.2019.1568299. [DOI] [PubMed] [Google Scholar]
- Ojha PK, Roy K. Comparative QSARs for antimalarial endochins: importance of descriptor-thinning and noise reduction prior to feature selection. Chemometr Intell Lab. 2011;109:146–161. doi: 10.1016/j.chemolab.2011.08.007. [DOI] [Google Scholar]
- Ojha PK, Mitra I, Das RN, Roy K. Further exploring rm2 metrics for validation of QSPR models. Chemometr Intell Lab. 2011;107:194–205. doi: 10.1016/j.chemolab.2011.03.011. [DOI] [Google Scholar]
- Pedersen SF, Ho Y-C. SARS-CoV-2: a storm is raging. J Clin Invest. 2020;130:2202–2205. doi: 10.1172/JCI137647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Peng X, Xu X, Li Y, Cheng L, Zhou X, Ren B. Transmission routes of 2019-nCoV and controls in dental practice. Int J Oral Sci. 2020 doi: 10.1038/s41368-020-0075-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pérez González M, Terán C, Saíaz-Urra L, Teijeira M. Variables selection methods in QSAR: an overview. Curr Top Med Chem. 2008;8:1606–1627. doi: 10.2174/156802608786786552. [DOI] [PubMed] [Google Scholar]
- Ramajayam R, Tan KP, Liu HG, Liang PH. Synthesis and evaluation of pyrazolone compounds as SARS-coronavirus 3C-like protease inhibitors. Bioorg Med Chem. 2010;18:7849–7854. doi: 10.1016/j.bmc.2010.09.050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rothan HA, Byrareddy SN. The epidemiology and pathogenesis of coronavirus disease (COVID-19) outbreak. J Autoimm. 2020 doi: 10.1016/j.jaut.2020.102433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Roy PP, Leonard JT, Roy K. Exploring the impact of size of training sets for the development of predictive QSAR models. Chemometr Intell Lab. 2008;90:31–42. doi: 10.1016/j.chemolab.2007.07.004. [DOI] [Google Scholar]
- Sanders JM, Monogue ML, Jodlowski TZ, Cutrell JB. Pharmacologic treatments for coronavirus disease 2019 (COVID-19): a review. JAMA J Am Med Assoc. 2020 doi: 10.1001/jama.2020.6019. [DOI] [PubMed] [Google Scholar]
- Shang J, Ye G, Shi K, Wan Y, Luo C, Aihara H, Geng Q, Auerbach A, Li F. Structural basis of receptor recognition by SARS-CoV-2. Nature. 2020;30:1–8. doi: 10.1038/s41586-020-2179-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Singhal T. A review of coronavirus disease-2019 (COVID-19) Indian J Pediatr. 2020;87:281–286. doi: 10.1007/s12098-020-03263-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sohrabi C, Alsafi Z, O'Neill N, Khan M, Kerwan A, Al-Jabir A, Iosifidis C, Agha R. World Health Organization declares global emergency: a review of the 2019 novel coronavirus (COVID-19) Int J Surg. 2020;76:71–76. doi: 10.1016/j.ijsu.2020.02.034. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stoičkov V, Stojanović D, Tasić I, Šarić S, Radenković D, Babović P, Sokolović D, Veselinović AM. Development of non-peptide ACE inhibitors as novel and potent cardiovascular therapeutics: an in silico modelling approach. Struct Chem. 2018;29:441–449. doi: 10.1080/1062936X.2018.1485737. [DOI] [PubMed] [Google Scholar]
- Tabeshpour J, Sahebkar A, Zirak MR, Zeinali M, Hashemzaei M, Rakhshani S, Rakhshani S. Computer-aided drug design and drug pharmacokinetic prediction: a mini-review. Curr Pharm Design. 2018;24:3014–3019. doi: 10.2174/1381612824666180903123423. [DOI] [PubMed] [Google Scholar]
- Thomsen R, Christensen MH. MolDock: a new technique for high-accuracy molecular docking. J Med Chem. 2006;49:3315–3321. doi: 10.1021/jm051197e. [DOI] [PubMed] [Google Scholar]
- Toropov AA, Toropova AP. The index of ideality of correlation: a criterion of predictive potential of QSPR/QSAR models? Mutat Res-Gen Tox En. 2017;819:31–37. doi: 10.1016/j.mrgentox.2017.05.008. [DOI] [PubMed] [Google Scholar]
- Toropov AA, Duchowicz P, Castro EA. Structure-toxicity relationships for aliphatic compounds based on correlation weighting of local graph invariants. Int J Mol Sci. 2003;4:272–283. doi: 10.3390/i4050272. [DOI] [Google Scholar]
- Toropov AA, Toropova AP, Selvestrel G, Benfenati E. Idealization of correlations between optimal simplified molecular input-line entry system-based descriptors and skin sensitization. SAR QSAR Environ Res. 2019;30:447–455. doi: 10.1080/1062936X.2019.1615547. [DOI] [PubMed] [Google Scholar]
- Toropova MA, Raška I, Jr, Toropov AA, Raškova M. The utilization of the Monte Carlo technique for rational drug discovery. Comb Chem High T Sc. 2016;19:676–687. doi: 10.2174/1386207319666160725145852. [DOI] [PubMed] [Google Scholar]
- Veber DF, Johnson SR, Cheng HY, Smith BR, Ward KW, Kopple KD. Molecular properties that influence the oral bioavailability of drug candidates. J Med Chem. 2002;45:2615–2623. doi: 10.1021/jm020017n. [DOI] [PubMed] [Google Scholar]
- Vellingiri B, Jayaramayya K, Iyer M, Narayanasamy A, Govindasamy V, Giridharan B, Ganesan S, Venugopal A, Venkatesan D, Ganesan H, Rajagopalan K, Rahman PKSM, Cho S-G, Kumar NS, Subramaniam MD. COVID-19: a promising cure for the global panic. Sci Total Environ. 2020 doi: 10.1016/j.scitotenv.2020.138277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Veselinović AM, Veselinović JB, Živković JV, Nikolić GM. Application of smiles notation based optimal descriptors in drug discovery and design. Curr Top Med Chem. 2015;15:1768–1779. doi: 10.2174/1568026615666150506151533. [DOI] [PubMed] [Google Scholar]
- Veselinović AM, Toropov A, Toropova A, Stanković-Dordević D, Veselinović JB. Design and development of novel antibiotics based on FtsZ inhibition—in silico studies. New J Chem. 2018;42:10976–10982. doi: 10.1039/C8NJ01034J. [DOI] [Google Scholar]
- Wu D, Wu T, Liu Q, Yang Z. The SARS-CoV-2 outbreak: what we know. Int J Infect Dis. 2020;94:44–48. doi: 10.1016/j.ijid.2020.03.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiao WX, Wu XX, Jiang XG, Xu KJ, Ying LJ, Ma CL, Li SB. Clinical findings in a group of patients infected with the 2019 novel coronavirus (SARS-Cov-2) outside of Wuhan, China: retrospective case series. BMJ. 2020;368:606. doi: 10.1136/bmj.m606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang L, Lin D, Sun X, Curth U, Drosten C, Sauerhering L, Becker S, Rox K, Hilgenfeld R. Crystal structure of SARS-CoV-2 main protease provides a basis for design of improved α-ketoamide inhibitors. Science. 2020;368:409–412. doi: 10.1126/science.abb3405. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhavoronkov A, Aladinskiy V, Zhebrak A, Zagribelnyy B, Terentiev V, Bezrukov DS, Polykovskiy D. Potential COVID-2019 3C-like protease inhibitors designed using generative deep learning approaches. Insilico Med Hong Kong Ltd A. 2020;307:E1. doi: 10.26434/chemrxiv.11829102.v2. [DOI] [Google Scholar]
- Zivkovic M, Zlatanovic M, Zlatanovic N, Golubović M, Veselinović AM. The application of the combination of Monte Carlo optimization method based QSAR modeling and molecular docking in drug design and development. Mini-Rev Med Chem. 2020;20:1389–1402. doi: 10.2174/1389557520666200212111428. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.