

Abstract
Reversible lysine acetylation regulates the activity of cardiac metabolic enzymes, including those controlling fuel substrate metabolism. Mitochondrial-targeted GCN5L1 and SIRT3 have been shown to regulate the acetylation status of mitochondrial enzymes, but the role that lysine acetylation plays in driving metabolic differences between male and female hearts is not currently known. In this study, we describe a significant difference in GCN5L1 levels between male and female mouse hearts, and in the hearts of women between post- and premenopausal age. We further find that estrogen drives GCN5L1 expression in a cardiac cell line and uses pharmacological approaches to determine the mechanism to be G protein-coupled estrogen receptor (GPER) activation, via translational regulation.
NEW & NOTEWORTHY We demonstrate here for the first time that mitochondrial protein acetylation is increased in female hearts, associated with an increase in GCN5L1 levels through a GPER-dependent mechanism. These findings reveal a new potential mediator of divergent cardiac mitochondrial function between men and women.
Keywords: acetylation, estrogen, GCN5L1, GPER, SIRT3
INTRODUCTION
Improved understanding of the physiological and metabolic differences between men and women may allow us to develop new therapies that can address sex-based disparities in cardiac disease treatment outcomes. Sex hormones testosterone and estrogen, as well as chromosomal effects, may contribute to sex-based differences. Premenopausal women exhibit increased estrogen levels relative to men and postmenopausal women, which result in greater activation of estrogen receptors in the myocardium. These are comprised of the canonical estrogen receptors α and β (ERα and ERβ), and the G protein-coupled estrogen receptor (GPER, or GPR30) (1). Canonical ERs are targeted directly to the nucleus, and interact with ER responsive elements (EREs) within the genome to regulate transcription, whereas GPER activation results in a cascade of posttranslational modifications in the cell that may also ultimately drive genomic responses (2–4).
Estrogen receptor activation has been associated with changes in the abundance and activity of numerous enzymes involved in glucose and fatty acid energy metabolism, which result in significant sexual dimorphism in cardiac metabolic profiles. Of particular note, women exhibit greater cardiac fatty acid uptake and oxidation relative to men under both normal and pathophysiological conditions (5). Estrogen modulates the expression of cardiac metabolic proteins localized to the mitochondria, and upregulates proteins that impact fatty acid metabolism, including peroxisome proliferator-activated receptor-γ coactivator 1α (PGC-1α) and acyl-CoA dehydrogenases (ACADs) (6–8). Consequently, the presence of estrogen has a significant impact on fuel substrate utilization in the heart.
The posttranslational acetylation of non-nuclear targets has emerged as a critical regulator of metabolic activity in the heart. In mitochondria, GCN5L1 and SIRT3 have been reported to increase and decrease, respectively, the acetylation status of enzymes that metabolize fatty acids and glucose (9–15). However, sex differences in the acetylation of metabolic proteins in cardiac mitochondria have not been investigated. We hypothesized that differences in the expression of GCN5L1 and SIRT3 between men and women may change the acetylation status and activity of mitochondria-targeted enzymes, and that estrogen signaling may drive this process.
The studies presented here demonstrate that mitochondrial protein acetylation is increased in female mice relative to males, which is associated with sex-dependent elevations in GCN5L1 abundance. In addition, we show that estrogen directly increases GCN5L1 expression in human-derived cardiac cells, and that GCN5L1 is decreased in the hearts of postmenopausal women relative to younger women. Finally, we determine that the primary mechanism for estrogen-mediated GCN5L1 upregulation is identified as GPER activation, through a transcription-independent pathway.
METHODS
Human Tissues
Fresh human cardiac tissue samples were collected from the left ventricles of organ donors deemed not suitable for transplant, under a protocol approved by the University of Pittsburgh Committee for Oversight of Research and Clinical Training (CORID). Tissues were flash-frozen and stored at −80°C until processing. Postmenopause: range = 65–86 yr, median = 69 yr, n = 7. Premenopause: range = 22–39 yr, median = 36 yr, n = 5.
Animal Care and Use
All housing and experiments in mice were conducted in accordance with the guidelines established by the National Institutes of Health and approved by the University of Pittsburgh Institutional Animal Care and Use Committee. Male and female C57BL/6J mice (aged 8–10 wk) were purchased from The Jackson Laboratory and maintained on a regular chow diet with a 12-h:12-h light/dark cycle.
Cell Culture and Drug Treatments
AC16 cells (a proliferating cell line derived from female human cardiomyoctyes) (16, 17) were purchased from Millipore. Stable GCN5L1 knockdown was generated as previously described (18). Cells were treated with 10 nM 17β-estradiol (E2), 10 mM ICI 182, 780 (Fulvestrant; an ERα and ERβ antagonist with GPER agonist activity) (19–21), 110 nM G-1 (a selective GPER agonist), G-36 (a selective GPER antagonist), 10 µM MG-132 (a 26S protease inhibitor), and/or 10 ng/mL cycloheximide (CHX; a translation inhibitor). All treatments were administered for 24 h, except MG-132, which was administered for 4 h to avoid significant toxicity. Treatments were administered in media without phenol red and 2.5% charcoal-stripped FBS (Millipore Sigma) to minimize uncontrolled exposure to estrogens.
Mitochondrial Isolation
Mitochondrial fractions were purified from tissue and cells using the Qproteome Mitochondrial Isolation Kit (Qiagen) according to the manufacturer’s instructions. Briefly, samples were homogenized in cold lysis buffer, and centrifuged at 1,000 g. Supernatant containing the cytosolic fraction was discarded, and the pellet was re-suspended and processed in cold Disruption Buffer by shearing through a 25-G needle and syringe. Samples were centrifuged at 1,000 g, the supernatant was collected, and centrifuged again at 6,000 g to pellet the mitochondrial fraction. The pellet was washed in Storage Buffer, and then used for subsequent immunoblot.
Immunoblotting
Tissue, cells, or purified mitochondria were lysed in 1% CHAPS buffer. Protein was quantitated using a BioDrop μLITE analyzer (BioDrop). For immunoprecipitation (IP) study, lysates were incubated with anti-AcK antibodies (1:100, Cell Signaling) overnight, and then incubated for 3 h with Protein-G and Protein-A beads. Beads were washed four times before elution at 95°C in SDS loading buffer. Lysates or IP samples were loaded on a 12% SDS-PAGE gel, before transfer to nitrocellulose membranes.
Membranes were blocked using Odyssey blocking buffer and incubated in primary antibodies overnight (α-tubulin, 1:1,000, Cell Signaling; SIRT3, 1:1,000, Cell Signaling; GAPDH,1:1,000, Cell Signaling; GDH, 1:1,000, Cell Signaling; MCAD, 1:1,000, Cell Signaling; GCN5L1 1:500, generated as previously described) (22), followed by incubation at room temperature with fluorescent secondary antibodies for 1 h (800 nm anti-rabbit, Li-Cor). Antibodies from Cell Signaling were validated according to their Western blotting validation protocol, including the examination of several cell lines and/or tissues of known expression levels allows accurate determination of species cross reactivity and verifies specificity, the use of siRNA transfection or knockout cell lines to verify specificity, and side-by-side comparison of lots to ensures lot-to-lot consistency. In-house GCN5L1 antibodies were routinely validated against tissue from knockout mice and knockdown AC16 cells as previously described (18). Bands were visualized using an Odyssey Imager, and quantitated using Image Studio Lite v. 5.2 (Li-Cor). All Western blot results were normalized to the appropriate loading controls as indicated.
Quantitative RT-PCR
RNA was isolated from tissue or cells using RNEasy kit (Qiagen). RNA was quantified and 500–1,000 ng was used to generate cDNA using Maxima Reverse Transcriptase (Thermo Fisher). Quantitative PCR was performed using SYBR-Green (Thermo Fisher) and primers for GCN5L1 or SIRT3. GAPDH or PPIA were used for normalization.
Statistical Analysis
Statistical analyses were performed using GraphPad Prism 8.3. Student’s t tests were used for simple comparisons between groups. One-way analyses of variance (ANOVA) was used to compare more than two groups, followed by post hoc Tukey’s multiple comparison’s test. For studies examining multiple time-points, a two-way ANOVA was used with post hoc Tukey’s multiple comparisons tests. A P value <0.05 was regarded as significant. All data are represented as the means ± SE.
RESULTS
Mitochondrial Acetylation and GCN5L1 Expression is Increased in the Hearts of Female Mice Compared with Male Mice
To determine whether mitochondrial protein acetylation status is altered between male and female mice, we isolated mitochondria from the hearts of male and female C57BL/6J mice and immunoblotted for acetylated lysine residues. We observed a modest but significant increase in the intensity of bands in female mice compared with male mice (Fig. 1A). We next examined whether the increase in mitochondrial acetylation is associated with changes in the expression of proteins that regulate mitochondrial protein acetylation. No changes were observed in the expression of the mitochondrial-targeted deacetylase SIRT3 (Fig. 1B). However, a significant increase in both GCN5L1 mRNA and protein was observed (Fig. 1, B and C). Based on these data, we conclude that increased acetylation in female cardiac mitochondria is driven by increased GCN5L1 abundance.
Figure 1.
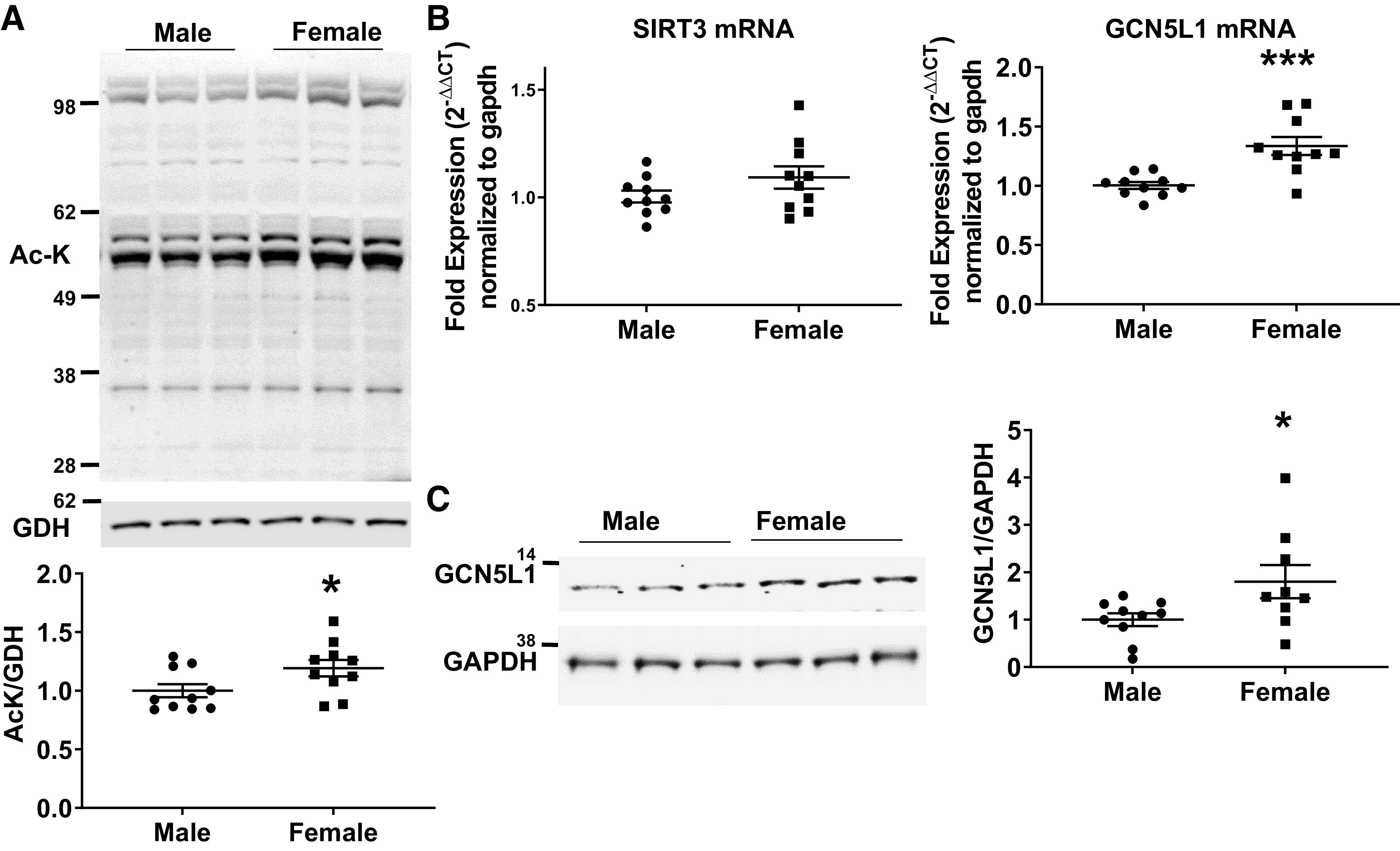
Mitochondrial protein acetylation and GCN5L1 are upregulated in female mice. A: immunoblotting of mitochondrial lysate fractions from male and female mice demonstrate that females exhibit modestly but significantly higher levels of total protein acetylation. n = 10. B: GCN5L1, but not SIRT3, mRNA is significantly increased in female hearts relative to males. C: GCN5L1 protein levels are significantly increased in the myocardium of female mice compared with male mice. n = 10, *P < 0.05, ***P < 0.001 vs. male (Student’s t test).
Estrogen Increases GCN5L1 Expression in Human Cardiomyocytes via GPER
To determine if changes in GCN5L1 abundance are present in a clinically relevant setting, we analyzed heart tissue obtained from female patients of pre- and postmenopausal age. Immunoblotting revealed that cardiac tissues from women after menopause, when estrogen levels are lower, have a significantly lower GCN5L1 protein abundance (Fig. 2A). As estrogen has been reported to mediate several of the sex differences observed in human myocardial tissue (4), we next determined whether estrogen induces GCN5L1 expression directly. Treatment of AC16 cells (derived from human ventricular cardiomyocytes) (16) with 17-β estradiol (E2) resulted in significantly increased levels of GCN5L1 protein (Fig. 2B). To determine whether signaling for increased GCN5L1 expression was through canonical estrogen receptors, we incubated AC16 cells with ICI 182, 780 (Fulvestrant), a potent inhibitor of ERα and ERβ. Surprisingly, rather than blocking GCN5L1 induction, ICI 182, 780 additively increased GCN5L1 levels, and produced a robust increase in GCN5L1 even in the absence of E2 (Fig. 3A). ICI 182, 780, in addition to blocking ERα and ERβ, also has been reported to act as a partial agonist for the G protein-coupled estrogen receptor (GPER) (20, 21, 23). We therefore examined whether GPER played a role in estrogen-mediated GCN5L1 induction using the GPER agonist G-1, and the GPER antagonist G-36. Incubation with G-1 significantly increased GCN5L1 protein abundance, whereas G-36 blocked E2-mediated GCN5L1 expression (Fig. 3B). From these data, we conclude that GCN5L1 abundance is increased by estrogen exposure via GPER-mediated signaling.
Figure 2.

Estrogen drives GCN5L1 expression. A: human tissues from female patients of premenopausal (PRE, n = 7) or postmenopausal (POST, n = 5) age. B: human-derived AC16 cells incubated with 17β-estradiol (E2) show an increase in GCN5L1. n = 4, *P < 0.05 (Student’s t test).
Figure 3.

Estrogen drives GCN5L1 expression via G protein-coupled estrogen receptor (GPER). A: GCN5L1 immunoblotting after incubation for 24 h with 17β-estradiol (E2) and or the estrogen receptor (ER)α/ERβ antagonist ICI 182, 780. n = 4, *P < 0.05, ***P < 0.001, ****P < 0.0001 vs. vehicle (one-way ANOVA with post hoc Tukey’s multiple comparison’s test). B: GCN5L1 is elevated after incubation for 24 h with E2 and the GPER agonist G-1, and is blocked in the presence of GPER antagonist G-36. n = 5 or 6, *P < 0.05, **P < 0.01 vs. vehicle (one-way ANOVA with post hoc Tukey’s multiple comparison’s test). C: acetylation of medium-chain acyl-CoA dehydrogenase (MCAD) is significantly reduced in the presence of E2 and G-1 when GCN5L1 is absent. n = 4, *P < 0.05 vs. control vehicle (one-way ANOVA with post hoc Tukey’s multiple comparison’s test).
Cardiac GCN5L1 has been reported to mediate the acetylation and activation of acyl-CoA dehydrogenases (ACADs), which mediate fatty acid catabolism (12, 24). Among the ACADs, MCAD has been repeatedly identified as a target regulated by estrogens in the heart (6–8). We therefore tested whether GCN5L1 may link estrogen receptor agonism to increases in MCAD acetylation and activity. When GCN5L1 was silenced as previously described (18), MCAD acetylation was significantly reduced in E2 and G-1-treated cells, relative to control cells under the same conditions (Fig. 3C). These data suggest that MCAD acetylation in the presence of E2 and GPER agonism is maintained via GCN5L1.
Estrogen Drives GCN5L1 Translation
To understand the mechanism of GPER-induced GCN5L1 elevations, we monitored mRNA levels in AC16 cells after exposure to E2 or G-1 using qPCR. Surprisingly, no change in mRNA was observed at any of the time points measured, suggesting that E2 control of GCN5L1 expression occurs downstream of transcription (Fig. 4A). To determine whether GCN5L1 protein elevation was due to an estrogen-induced reduction in protein degradation, the effects of 26S proteasomal inhibitor MG132 on GCN5L1 expression were evaluated. It was expected that if estrogen signaling elevates GCN5L1 levels by reducing protease activity, a blockade of protease activity would normalize protein levels in vehicle-treated cells, and the difference observed in E2-treated cells would disappear. Data showed that this was not the case, and rather MG132 amplified the increase in protein observed in the presence of E2 or G-1 (Fig. 4B). We next examined whether estrogen alters GCN5L1 mRNA translation. To test this hypothesis, E2 and G-1-treated AC16 cells were incubated with the translation inhibitor cycloheximide (CHX). CHX treatment effectively normalized GCN5L1 levels, with no significant difference between vehicle and E2 and G-1-treated cells in the presence of CHX (P = 0.58 and P = 0.16, respectively), indicating that differences in protein expression may be attributed to GCN5L1 translational regulation (Fig. 4C).
Figure 4.

Estrogen promotes GCN5L1 translation. A: expression of GCN5L1 mRNA levels determined by qPCR after treatment with 17β-estradiol (E2) or G-1. n = 3. B: GCN5L1 levels are significantly elevated by blocking 26S degradation, suggesting that G protein-coupled estrogen receptor (GPER) agonism does not increase GCN5L1 via impaired proteolysis. n = 4, *P < 0.05, ***P < 0.001 vs. control (two-way ANOVA with post hoc Tukey’s multiple comparison’s test). C: GCN5L1 induction is blocked by cycloheximide (CHX), a translation inhibitor. n = 4, *P < 0.05, ***P < 0.001 vs. control (two-way ANOVA with post hoc Tukey’s multiple comparison’s test). D: proposed mechanism of action.
DISCUSSION
Here we determine that mitochondrial protein acetylation, and GCN5L1 expression, is elevated in female mouse hearts compared with male. We find that estrogen upregulates GCN5L1 via GPER, and pharmacological block of GPER ablates induction. GCN5L1 is also elevated in premenopausal women, where estrogen levels are higher, compared with postmenopausal women. E2 and G-1 do not alter GCN5L1 gene transcription or proteasomal degradation; instead, translational blockade prevents GCN5L1 induction. These data point to a significant role for GCN5L1 in estrogen-mediated regulation of cardiac fuel metabolism (summarized in Fig. 4D).
Significant differences in cardiac physiology and pathology between men and women are well established. Premenopausal women are largely protected from cardiovascular disease (CVD) compared with men, but this advantage is reduced with age (25). Estrogen loss has been suggested to be a major mediator of this effect. Postmenopausal women become more susceptible to left ventricular (LV) diastolic dysfunction, and hormone replacement therapy mitigates this effect (26). Ovariectomized mice and rats are similarly more susceptible to insults to the heart. Mice lacking estrogen receptors are found to be more sensitive to ischemia-reperfusion (I/R) injury and hypertensive cardiomyopathy (27, 28). Although less well studied, testosterone also plays a role in driving sexual dimorphism between male and female individuals. Since estrogen was sufficient to reproduce an increase in GCN5L1 production, we have not evaluated the effects of testosterone. However, we cannot discount the possibility of an effect, and further studies are required to make this determination.
In recent years, GPER has taken a central role in our understanding of how estrogen impacts cardiac function and resiliency (29). GPER activation in cardiomyocytes lacking classical ERα and ERβ receptors was reported to alter intracellular calcium influx (30). In addition, GPER activation is associated with protection from ischemia-reperfusion injury, dependent on PI3K activation (31). Agonism of GPER with G-1 is reported to protect estrogen-deficient rats from LV remodeling (32). G-1 is also reported to inhibit the opening of the mitochondrial membrane permeability pore (mPTP) (33), and reduces the upregulation of inflammatory cytokines TNF-α, IL-1β, and IL-6 (34). Our earlier observation that GCN5L1 protects the heart from I/R injury (18) raises the possibility that GPER-mediated upregulation of GCN5L1 may be an additional mechanism by which estrogen protects the heart.
We demonstrate here that GCN5L1 is required for estrogen and GPER agonism to maintain the acetylation of medium-chain acyl-CoA dehydrogenase (MCAD) in cardiac-derived cells. MCAD is a mitochondria-localized enzyme that catalyzes the rate-limiting step in the β-oxidation of medium-chain fatty acids, the α,β-dehydrogenation of fatty acyl-CoA. MCAD plays a key role in the progression of myocardial metabolic dysregulation induced by heart failure (35–37). In addition, MCAD has been reported previously to be regulated by estrogen (6–8, 38, 39). Our laboratory has shown that increased acetylation of ACADs, through high-fat feeding-driven upregulation of GCN5L1, increases their activity in the heart (12). It may therefore be expected that the GCN5L1-mediated acetylation of MCAD may lead its activation, and thus improved fatty acid metabolism, in the presence of GPER agonism by estrogens in female hearts.
GPER agonism drives translation and production of GCN5L1 protein, but at no time point after GPER activation GCN5L1 message was elevated in cells, suggesting a posttranscriptional regulation mechanism. This is in agreement with a recent publication suggesting posttranscriptional regulation of GCN5L in diabetic kidney cells, via ubiquitination (40), although we did not observe evidence that G-1 blocks the degradation of GCN5L1 by the 26S proteasome. An alternate means of GCN5L1 may be miRNA control of GCN5L1 translation. Interestingly, there are reports of GCN5L1 acting as a translational coactivator to ERα in HeLa cells, binding directly to both the receptor and the corepressor element MTA1 (41). That study did not evaluate a direct interaction between GCN5L1 and GPER, and although a mechanism by which GCN5L1 might interact directly with GPER was briefly considered, our immunoprecipitation studies did not support the direct binding of GPER to GCN5L1 (data not shown). We did observe both increased message and increased protein levels of GCN5L1 in female mouse hearts compared with male, suggesting a potentially complimentary transcriptional mechanism in the whole organism after long-term exposure to estrogens, an area that will require further study to fully elucidate.
In summary, we demonstrate here for the first time that GCN5L1 is upregulated by estrogen signaling in both mouse and human myocardium, and in cultured cardiomyocytes. The mechanism of upregulation is identified as GPER-mediated signaling. These findings shed new light on the role that posttranslational acetylation, mediated by GCN5L1, may play in the differences observed between men and women in cardiac metabolism.
GRANTS
This work was supported by National Heart, Lung, and Blood Institute Grants HL132917, HL147861, and HL156874 (to I. Scott) and a University of Pittsburgh Medical Center Competitive Medical Research Fund Grant 2019 (to J. R. Manning).
DISCLOSURES
No conflicts of interest, financial or otherwise, are declared by the authors.
AUTHOR CONTRIBUTIONS
J.R.M. and I.S. conceived and designed research; J.R.M., D.T., M.Z., M.W.S., J.C.S., and M.R. performed experiments; J.R.M. analyzed data; J.R.M. and I.S. interpreted results of experiments; J.R.M. and I.S. prepared figures; J.R.M. drafted manuscript; J.R.M. and I.S. edited and revised manuscript; J.R.M., D.T., M.Z., M.W.S., J.C.S., M.R., and I.S. approved final version of manuscript.
ACKNOWLEDGMENTS
Present address of D. Thapa: Dept. of Exercise Physiology, West Virginia University, Morgantown, West Virginia.
REFERENCES
- 1.Aryan L, Younessi D, Zargari M, Banerjee S, Agopian J, Rahman S, Borna R, Ruffenach G, Umar S, Eghbali M. The role of estrogen receptors in cardiovascular disease. Int J Mol Sci 21: 4314–4326, 2020. doi: 10.3390/ijms21124314. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Meyer MR, Prossnitz ER, Barton M. The G protein-coupled estrogen receptor GPER/GPR30 as a regulator of cardiovascular function. Vascul Pharmacol 55: 17–25, 2011. doi: 10.1016/j.vph.2011.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Menazza S, Murphy E. The expanding complexity of estrogen receptor signaling in the cardiovascular system. Circ Res 118: 994–1007, 2016. doi: 10.1161/CIRCRESAHA.115.305376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Ueda K, Adachi Y, Liu P, Fukuma N, Takimoto E. Regulatory actions of estrogen receptor signaling in the cardiovascular system. Front Endocrinol (Lausanne) 10: 909, 2019. doi: 10.3389/fendo.2019.00909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Mather KJ, DeGrado TR. Imaging of myocardial fatty acid oxidation. Biochim Biophys Acta 1861: 1535–1543, 2016. doi: 10.1016/j.bbalip.2016.02.019. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Schreiber SN, Knutti D, Brogli K, Uhlmann T, Kralli A. The transcriptional coactivator PGC-1 regulates the expression and activity of the orphan nuclear receptor estrogen-related receptor α (ERRα). J Biol Chem 278: 9013–9018, 2003. doi: 10.1074/jbc.M212923200. [DOI] [PubMed] [Google Scholar]
- 7.Hsieh Y-C, Choudhry MA, Yu H-P, Shimizu T, Yang S, Suzuki T, Chen J, Bland KI, Chaudry IH. Inhibition of cardiac PGC-1α expression abolishes ERβ agonist-mediated cardioprotection following trauma-hemorrhage. FASEB J 20: 1109–1117, 2006. doi: 10.1096/fj.05-5549com. [DOI] [PubMed] [Google Scholar]
- 8.Kim MS, Shigenaga JK, Moser AH, Feingold KR, Grunfeld C. Suppression of estrogen-related receptor α and medium-chain acyl-coenzyme A dehydrogenase in the acute-phase response. J Lipid Res 46: 2282–2288, 2005. doi: 10.1194/jlr.M500217-JLR200. [DOI] [PubMed] [Google Scholar]
- 9.Kendrick AA, Choudhury M, Rahman SM, McCurdy CE, Friederich M, Van Hove JLK, Watson PA, Birdsey N, Bao J, Gius D, Sack MN, Jing E, Kahn CR, Friedman JE, Jonscher KR. Fatty liver is associated with reduced SIRT3 activity and mitochondrial protein hyperacetylation. Biochem J 433: 505–514, 2011. doi: 10.1042/BJ20100791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Alrob OA, Sankaralingam S, Ma C, Wagg CS, Fillmore N, Jaswal JS, Sack MN, Lehner R, Gupta MP, Michelakis ED, Padwal RS, Johnstone DE, Sharma AM, Lopaschuk GD. Obesity-induced lysine acetylation increases cardiac fatty acid oxidation and impairs insulin signalling. Cardiovasc Res 103: 485–497, 2014. doi: 10.1093/cvr/cvu156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Koentges C, Pfeil K, Schnick T, Wiese S, Dahlbock R, Cimolai MC, Meyer-Steenbuck M, Cenkerova K, Hoffmann MM, Jaeger C, Odening KE, Kammerer B, Hein L, Bode C, Bugger H. SIRT3 deficiency impairs mitochondrial and contractile function in the heart. Basic Res Cardiol 110: 36, 2015. doi: 10.1007/s00395-015-0493-6. [DOI] [PubMed] [Google Scholar]
- 12.Thapa D, Zhang M, Manning JR, Guimarães DA, Stoner MW, O'Doherty RM, Shiva S, Scott I. Acetylation of mitochondrial proteins by GCN5L1 promotes enhanced fatty acid oxidation in the heart. Am J Physiol Heart Circ Physiol 313: H265–H274, 2017. doi: 10.1152/ajpheart.00752.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Manning JR, Thapa D, Zhang M, Stoner MW, Traba J, Corey C, Shiva S, Sack MN, Scott I. Loss of GCN5L1 in cardiac cells disrupts glucose metabolism and promotes cell death via reduced Akt/mTORC2 signaling. Biochem J 476: 1713–1724, 2019. doi: 10.1042/BCJ20190302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Thapa D, Stoner MW, Zhang M, Xie B, Manning JR, Guimaraes D, Shiva S, Jurczak MJ, Scott I. Adropin regulates pyruvate dehydrogenase in cardiac cells via a novel GPCR-MAPK-PDK4 signaling pathway. Redox Biol 18: 25–32, 2018. doi: 10.1016/j.redox.2018.06.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Carnevale I, Pellegrini L, D'Aquila P, Saladini S, Lococo E, Polletta L, Vernucci E, Foglio E, Coppola S, Sansone L, Passarino G, Bellizzi D, Russo MA, Fini M, Tafani M. SIRT1-SIRT3 axis regulates cellular response to oxidative stress and etoposide. J Cell Physiol 232: 1835–1844, 2017. doi: 10.1002/jcp.25711. [DOI] [PubMed] [Google Scholar]
- 16.Davidson MM, Nesti C, Palenzuela L, Walker WF, Hernandez E, Protas L, Hirano M, Isaac ND. Novel cell lines derived from adult human ventricular cardiomyocytes. J Mol Cell Cardiol 39: 133–147, 2005. doi: 10.1016/j.yjmcc.2005.03.003. [DOI] [PubMed] [Google Scholar]
- 17.Mahmoodzadeh S, Pham TH, Kuehne A, Fielitz B, Dworatzek E, Kararigas G, Petrov G, Davidson MM, Regitz-Zagrosek V. 17β-Estradiol-induced interaction of ERα with NPPA regulates gene expression in cardiomyocytes. Cardiovasc Res 96: 411–421, 2012. doi: 10.1093/cvr/cvs281. [DOI] [PubMed] [Google Scholar]
- 18.Manning JR, Thapa D, Zhang M, Stoner MW, Traba J, McTiernan CF, Corey C, Shiva S, Sack MN, Scott I. Cardiac-specific deletion of GCN5L1 restricts recovery from ischemia-reperfusion injury. J Mol Cell Cardiol 129: 69–78, 2019. doi: 10.1016/j.yjmcc.2019.02.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Robertson JF. ICI 182, 780 (Fulvestrant)—the first oestrogen receptor down-regulator—current clinical data. Br J Cancer 85, Suppl 2: 11–14, 2001. doi: 10.1054/bjoc.2001.1982. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Thomas P, Pang Y, Filardo EJ, Dong J. Identity of an estrogen membrane receptor coupled to a G protein in human breast cancer cells. Endocrinology 146: 624–632, 2005. doi: 10.1210/en.2004-1064. [DOI] [PubMed] [Google Scholar]
- 21.Filardo EJ, Quinn JA, Bland KI, Frackelton J. Estrogen-induced activation of Erk-1 and Erk-2 requires the G protein-coupled receptor homolog, GPR30, and occurs via trans-activation of the epidermal growth factor receptor through release of HB-EGF. Mol Endocrinol 14: 1649–1660, 2000. doi: 10.1210/mend.14.10.0532. [DOI] [PubMed] [Google Scholar]
- 22.Scott I, Webster BR, Li JH, Sack MN. Identification of a molecular component of the mitochondrial acetyltransferase programme: a novel role for GCN5L1. Biochem J 443: 655–661, 2012. doi: 10.1042/BJ20120118. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Prossnitz ER, Barton M. Signaling, physiological functions and clinical relevance of the G protein-coupled estrogen receptor GPER. Prostaglandins Other Lipid Mediat 89: 89–97, 2009. doi: 10.1016/j.prostaglandins.2009.05.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Thapa D, Manning JR, Stoner MW, Zhang M, Xie B, Scott I. Cardiomyocyte-specific deletion of GCN5L1 in mice restricts mitochondrial protein hyperacetylation in response to a high fat diet. Sci Rep 10: 10665, 2020. doi: 10.1038/s41598-020-67812-x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Cho L, Davis M, Elgendy I, Epps K, Lindley KJ, Mehta PK, Michos ED, Minissian M, Pepine C, Vaccarino V, Volgman AS; ACC CVD Womens Committee Members. Summary of updated recommendations for primary prevention of cardiovascular disease in women: JACC state-of-the-art review. J Am Coll Cardiol 75: 2602–2618, 2020. doi: 10.1016/j.jacc.2020.03.060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Pines A, Fisman EZ, Drory Y, Levo Y, Shemesh J, Ben-Ari E, Ayalon D. Menopause-induced changes in doppler-derived parameters of aortic flow in healthy women. Am J Cardiol 69: 1104–1106, 1992. doi: 10.1016/0002-9149(92)90877-2. [DOI] [PubMed] [Google Scholar]
- 27.Pedram A, Razandi M, Lubahn D, Liu J, Vannan M, Levin ER. Estrogen inhibits cardiac hypertrophy: role of estrogen receptor-β to inhibit calcineurin. Endocrinology 149: 3361–3369, 2008. doi: 10.1210/en.2008-0133. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Pelzer T, Loza P-AA, Hu K, Bayer B, Dienesch C, Calvillo L, Couse JF, Korach KS, Neyses L, Ertl G. Increased mortality and aggravation of heart failure in estrogen receptor-β knockout mice after myocardial infarction. Circulation 111: 1492–1498, 2005. doi: 10.1161/01.CIR.0000159262.18512.46. [DOI] [PubMed] [Google Scholar]
- 29.Groban L, Tran Q-K, Ferrario CM, Sun X, Cheng CP, Kitzman DW, Wang H, Lindsey SH. Female heart health: is GPER the missing link? Front Endocrinol (Lausanne) 10: 919, 2019. doi: 10.3389/fendo.2019.00919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Dennis MK, Field AS, Burai R, Ramesh C, Petrie WK, Bologa CG, Oprea TI, Yamaguchi Y, Hayashi S, Sklar LA, Hathaway HJ, Arterburn JB, Prossnitz ER. Identification of a GPER/GPR30 antagonist with improved estrogen receptor counterselectivity. J Steroid Biochem Mol Biol 127: 358–366, 2011. doi: 10.1016/j.jsbmb.2011.07.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Deschamps AM, Murphy E. Activation of a novel estrogen receptor, GPER, is cardioprotective in male and female rats. Am J Physiol Heart Circ Physiol 297: H1806–H1813, 2009. doi: 10.1152/ajpheart.00283.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Wang H, Sun X, Lin MS, Ferrario CM, Van Remmen H, Groban L. G protein-coupled estrogen receptor (GPER) deficiency induces cardiac remodeling through oxidative stress. Transl Res 199: 39–51, 2018. doi: 10.1016/j.trsl.2018.04.005. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Bopassa JC, Eghbali M, Toro L, Stefani E. A novel estrogen receptor GPER inhibits mitochondria permeability transition pore opening and protects the heart against ischemia-reperfusion injury. Am J Physiol Heart Circ Physiol 298: H16–H23, 2010. doi: 10.1152/ajpheart.00588.2009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Weil BR, Manukyan MC, Herrmann JL, Wang Y, Abarbanell AM, Poynter JA, Meldrum DR. Signaling via GPR30 protects the myocardium from ischemia/reperfusion injury. Surgery 148: 436–443, 2010. doi: 10.1016/j.surg.2010.03.011. [DOI] [PubMed] [Google Scholar]
- 35.Wu J, Lu J, Huang J, You J, Ding Z, Ma L, Dai F, Xu R, Li X, Yin P, Zhao G, Wang S, Yuan J, Yang X, Ge J, Zou Y. Variations in energy metabolism precede alterations in cardiac structure and function in hypertrophic preconditioning. Front Cardiovasc Med 7: 602100, 2020. doi: 10.3389/fcvm.2020.602100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Bernardo BC, Weeks KL, Pongsukwechkul T, Gao X, Kiriazis H, Cemerlang N, Boey EJH, Tham YK, Johnson CJ, Qian H, Du XJ, Gregorevic P, McMullen JR. Gene delivery of medium chain acyl-coenzyme A dehydrogenase induces physiological cardiac hypertrophy and protects against pathological remodelling. Clin Sci (Lond) 132: 381–397, 2018. doi: 10.1042/CS20171269. [DOI] [PubMed] [Google Scholar]
- 37.Rennison JH, McElfresh TA, Okere IC, Patel HV, Foster AB, Patel KK, Stoll MS, Minkler PE, Fujioka H, Hoit BD, Young ME, Hoppel CL, Chandler MP. Enhanced acyl-CoA dehydrogenase activity is associated with improved mitochondrial and contractile function in heart failure. Cardiovasc Res 79: 331–340, 2008. doi: 10.1093/cvr/cvn066. [DOI] [PubMed] [Google Scholar]
- 38.Maher AC, Akhtar M, Vockley J, Tarnopolsky MA. Women have higher protein content of β-oxidation enzymes in skeletal muscle than men. PLoS One 5: e12025, 2010. doi: 10.1371/journal.pone.0012025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maher AC, Akhtar M, Tarnopolsky MA. Men supplemented with 17β-estradiol have increased β-oxidation capacity in skeletal muscle. Physiol Genomics 42: 342–347, 2010. doi: 10.1152/physiolgenomics.00016.2010. [DOI] [PubMed] [Google Scholar]
- 40.Lv T, Lu Y, Liu Y, Feng H, Li C, Sheng W, Cui Z, Zhu S, Gu X, Yang Z, Wan Q. General control of amino acid synthesis 5-like 1-mediated acetylation of manganese superoxide dismutase regulates oxidative stress in diabetic kidney disease. Oxid Med Cell Longev 2021: 6691226, 2021. doi: 10.1155/2021/6691226. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Mishra SK, Mazumdar A, Vadlamudi RK, Li F, Wang R-A, Yu W, Jordan VC, Santen RJ, Kumar R. MICoA, a novel metastasis-associated protein 1 (MTA1) interacting protein coactivator, regulates estrogen receptor-α transactivation functions. J Biol Chem 278: 19209–19219, 2003. doi: 10.1074/jbc.M301968200. [DOI] [PubMed] [Google Scholar]