

Abstract
Saving lives of wounded military warfighters often depends on the ability to resolve or mitigate the pathophysiology of hemorrhage, specifically diminished oxygen delivery to vital organs that leads to multiorgan failure and death. However, caring for hemorrhaging patients on the battlefield presents unique challenges that extend beyond applying a tourniquet and giving a blood transfusion, especially when battlefield care must be provided for a prolonged period. This review describes these challenges and potential strategies for treating hemorrhage on the battlefield in a prolonged casualty care situation.
Keywords: battlefield, hemorrhage, prolonged casualty care, trauma
Introduction
Traumatic injury occurs in both combat and civilian populations; it is the leading cause of death in the United States for people aged 1–44 (1). Many traumatic injuries result in hemorrhage. In the recent wars in Iraq and Afghanistan, the inability to control hemorrhage was the leading cause of potentially survivable death (2, 3), as it has been throughout history. Since 1990, United States forces have been engaged in counterinsurgency conflicts that produce relatively few casualties, who were easily evacuated to a surgical capability for definitive surgical care within a matter of minutes to hours. Indeed, Defense Secretary Robert Gates promulgated a 2009 policy that required prehospital evacuation of critically injured casualties in ≤60 min (“the Golden Hour”), producing a median transport time of 43 min and a significant decrease in mortality (4, 5). Additionally, these wars saw the use of advanced life-saving interventions, such as more liberal use of tourniquets to arrest severe extremity bleeding, blood products for resuscitation, hemostatic dressings, and improved training in combat casualty care (6). Together, these policies, tools, and education produced the lowest case fatality rates in history, despite increasing injury severity scores throughout the conflicts (7). Lessons learned have been adopted into civilian trauma practice as well.
Casualty care during the recent conflicts also benefited from the ability to expend tremendous resources to save severely injured patients. For example, it was not uncommon to massively transfuse (>10 units) blood into hemorrhaging patients before and during surgical procedures (6). But what if war reverts to those of the past, with advanced weaponry, mass casualties, limited resources, and the inability to freely move them from the battlefield? This is the issue posed as the United States and its allies contemplate the possibility of conflicts against near-peer adversaries that are expected to produce greater numbers of casualties (100s to 1,000s per day) but without air superiority facilitating evacuation (FIGURE 1). In current planning, the expectation is that future conflicts will be fought simultaneously in cyberspace, from the air and sea, and with ground large-scale combat operations (8). Scenarios in which a combat medic must provide prolonged casualty care with only the resources that s/he can carry have occurred (e.g., Black Hawk Down scenario); these are expected to increase in future conflicts but with many more casualties. In essence, the prolonged care scenarios envisioned are a return to those seen in the World Wars.
FIGURE 1.
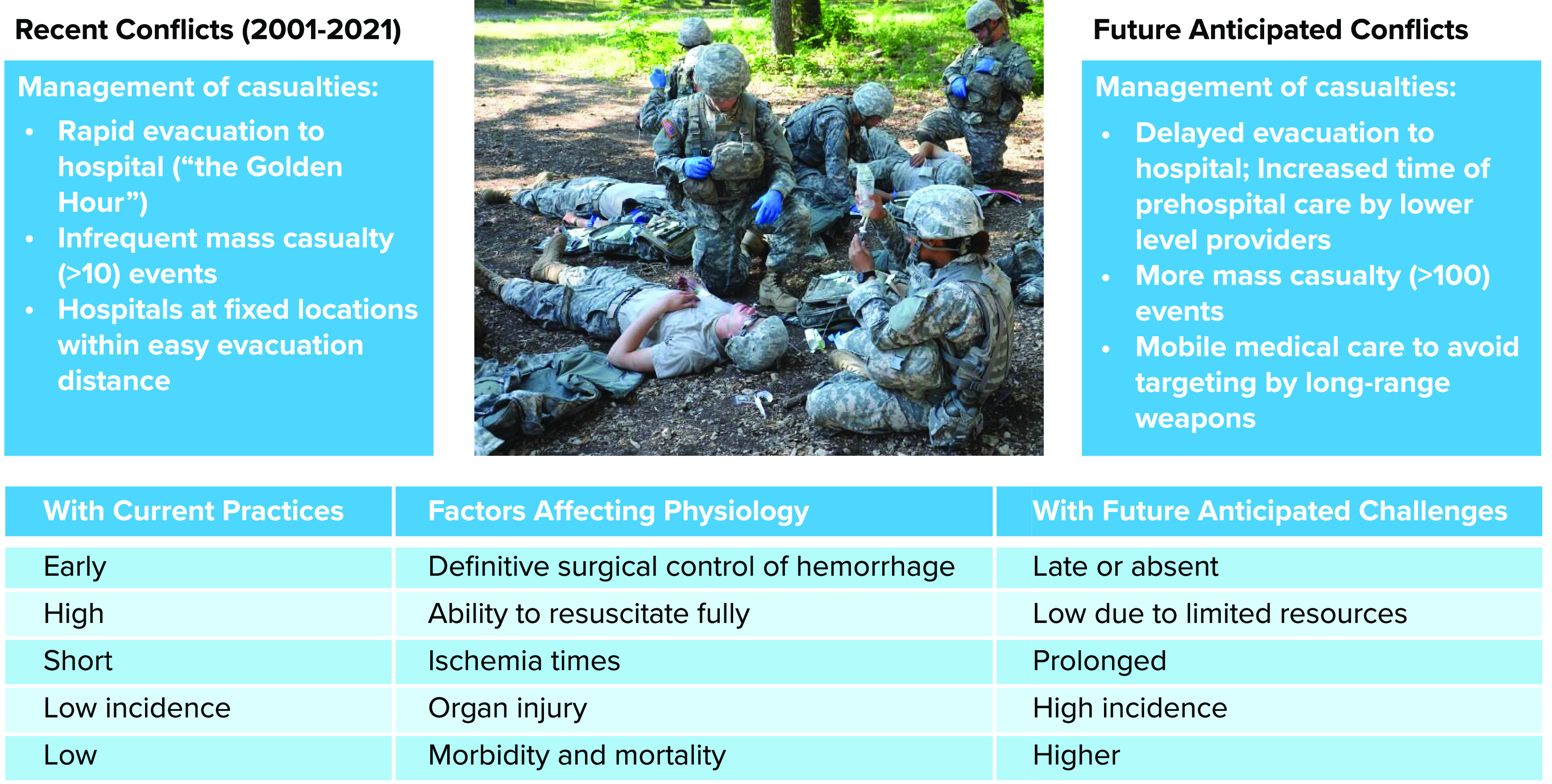
A comparison of casualty care management challenges during recent conflicts and future near-peer conflicts The table at bottom represents effects of these situational challenges on the development of pathophysiology.
Although we were successful in treating hemorrhagic shock in the conflicts of the 1990s to 2021 by relying on rapid evacuation and ample resources, treatment must evolve further to maintain recent case fatality rates under future conditions. However, the physiology underlying the development of hemorrhagic shock is unchanged; studying its pathophysiology will yield insights into future treatments that could be used to maintain patients even under conditions of delayed surgical repair. This review focuses on the pathophysiology of hemorrhage in a prolonged care scenario, with the assumption that initial hemorrhage control has been achieved but the medic lacks resources to provide full resuscitation. The intent here is to provide an overview of the multifactorial nature of the pathophysiology following hemorrhage; each subsection is worthy of its own detailed review, and references are provided to such reviews.
Hemorrhage Progression
There are four commonly accepted stages of hemorrhage, from minor blood loss to the development of shock, a condition in which tissue perfusion is too low to provide adequate tissue oxygenation and maintain normal metabolic functions (TABLE 1; Ref. 9). Class 1 and class 2 stages represent conditions of compensated blood loss because autonomic and neurohumoral compensatory responses maintain hemodynamic stability. During this period of mild to moderate hemorrhage, vital organ perfusion and oxygenation are preserved. Class 3 is considered decompensated shock because compensatory mechanisms are no longer effective to maintain vital organ oxygenation. Class 4 is the most severe level of hemorrhagic shock and is associated with impending cardiovascular collapse, vital organ injury, and death.
Table 1.
Stages of hemorrhage
| Class Designation | Blood Volume Lost, mL | % of Total Blood Volume Lost | MAP/SP | Pulse Pressure | HR | RR | Urinary Output | Perfusion in Vital Organs |
|---|---|---|---|---|---|---|---|---|
| 1 | <750 | <15 | ― | ― | ― | ― | ― | Compensated |
| 2 | 750–1,500 | 15–30 | ―↓ | ↓ | ↑ | ↑ | ―↓ | Compensated |
| 3 | 1,500–2,000 | 30–40 | ↓ | ↓ | ↑ | ↑ | ↓ | Starting decompensation |
| 4 | >2,000 | >40 | ↓↓ | ↓↓ | ↑↓ | ↑↓ | ↓↓ | Decompensated |
HR, heart rate; MAP, mean arterial pressure; RR, respiratory rate; SP, systolic pressure. Table adapted from Ref. 9 with permission.
The onset of hemorrhage produces an immediate decrease in blood pressure that stimulates an important compensatory mechanism, the arterial baroreflex. Baroreflex-mediated autonomic mechanisms inhibit the cardioinhibitory center, resulting in sympathetic nervous system activation and parasympathetic inhibition; this produces an increase in peripheral vascular resistance and an increase in heart rate (FIGURE 2; Refs. 10, 11). Sympathetic activation also produces constriction of major capacitance vessels, resulting in increased venous return (12). In addition, baroreflex activation of the sympathetic nervous system stimulates the adrenal gland to secrete epinephrine and norepinephrine to augment vasoconstriction and increases in heart rate. Baroreflex activation also stimulates the release of various neuroendocrine hormones such as renin from the kidney to activate the renin-angiotensin system and the secretion of vasopressin from the hypothalamus to mediate vasoconstriction and increase intravascular volume (11, 13–16). Other neuroendocrine hormones released during hemorrhage include aldosterone (17, 18) and neuropeptide Y (15), which also contribute to the maintenance of vascular volume and organ perfusion. These compensatory baroreflex responses and regional autoregulation act to maintain blood pressure and blood flow to vital organs (heart and brain) despite blood volume loss. Although respiratory compensatory responses to hemorrhage and the mechanisms that regulate these responses are not well understood, measurements in conscious rabbits revealed that during the early stage of hemorrhage arterial partial pressure of carbon dioxide () decreases but respiration rate does not change (19).
FIGURE 2.

Flow diagram of early compensation of vital organ perfusion after hemorrhage Activation is represented by solid black lines, and inhibition is represented by dashed red lines. Therapeutic interventions and their targets are shown in green. CO, cardiac output; MAP, mean arterial pressure; SNS, sympathetic nerve system; TPR, total peripheral resistance.
Capillary fluid shift is the major mechanism for volume expansion in early hemorrhage before resuscitation or reestablishment of water balance via the renin-angiotensin system. The first step of capillary fluid shift is rapid movement of protein-free fluid from the interstitium into capillaries. The second step is a slow shift of proteins from tissue into the circulation to support plasma oncotic pressure (20). However, the driving force for movement of protein depends on movement of fluid from cells to the interstitium, which may be a “bottleneck” in severe or prolonged hemorrhage because of ATP depletion and cellular edema. Establishment of an improved osmotic gradient across pericapillary interstitial spaces that facilitates this fluid shift may therefore be a therapeutic target.
The stress response and rapid release of catecholamines alter systemic and local metabolism, with the end result of providing more fuel (glucose) to tissues. Stress hyperglycemia can be detected within minutes after injury because of inhibited insulin secretion and elevated glycogenolysis and glucagon release or cortisol-mediated gluconeogenesis (21–23). Elevated glucose can have both deleterious and beneficial effects during the early stages of hemorrhage; therefore, treatments to prevent early stress hyperglycemia must be considered with caution. For example, stress hyperglycemia can increase endothelial damage, endoplasmic reticulum stress, acidosis, inflammatory responses, and reactive oxygen species (ROS) (23–25). Meanwhile, stress hyperglycemia may facilitate catabolism shift from peripheral tissue to the vital organs, such as the brain and the heart, the two organs most vulnerable to ischemia. Attempts to interfere with this exceedingly complex multisystem adaptive response may be harmful (26).
With continued or more severe hemorrhage, stroke volume and cardiac output continue to decrease, physiological mechanisms that attempt to maintain homeostasis eventually fail, resulting in a dramatic and sudden decrease in total peripheral resistance, mean arterial pressure, and heart rate (10, 11, 27). If severe and prolonged, hemorrhage results in a mismatch between tissue oxygen delivery () and oxygen consumption (). When the body does not have an adequate oxygen supply, cells rely on anaerobic glycolysis, which results in increased levels of blood lactate and decreases in blood pH resulting in metabolic acidosis. The increases in blood hydrogen ion and carbon dioxide levels stimulate the chemoreceptor reflex, resulting in an activation of central respiratory centers to compensate for the metabolic acidosis (28). Ultimately, the presence of continued tissue hypoxia and metabolic acidosis produces progressive cellular deterioration and cell death and multiorgan failure (29). Eventually, with prolongation of this uncompensated phase of blood loss, irreversible hemorrhagic shock occurs, which is fatal (30). For a more in-depth review of compensatory responses to hemorrhage, see Ref. 31.
Mitochondrial and Cellular Function
Decreased stalls forward electron flux through the mitochondrial electron transport chain, leading to the loss of proton pumping and the loss of inner membrane potential, which halts ATP synthesis (FIGURE 3). If the inner membrane potential rises high enough during hypoxia, ATP synthase reverses, hydrolyzing ATP generated via anaerobic mechanisms to regenerate membrane potential; estimates suggest that during hypoxia at least 35% and as much as 90% of all anaerobically generated ATP is hydrolyzed by the reversed ATP synthase (32–37).
FIGURE 3.
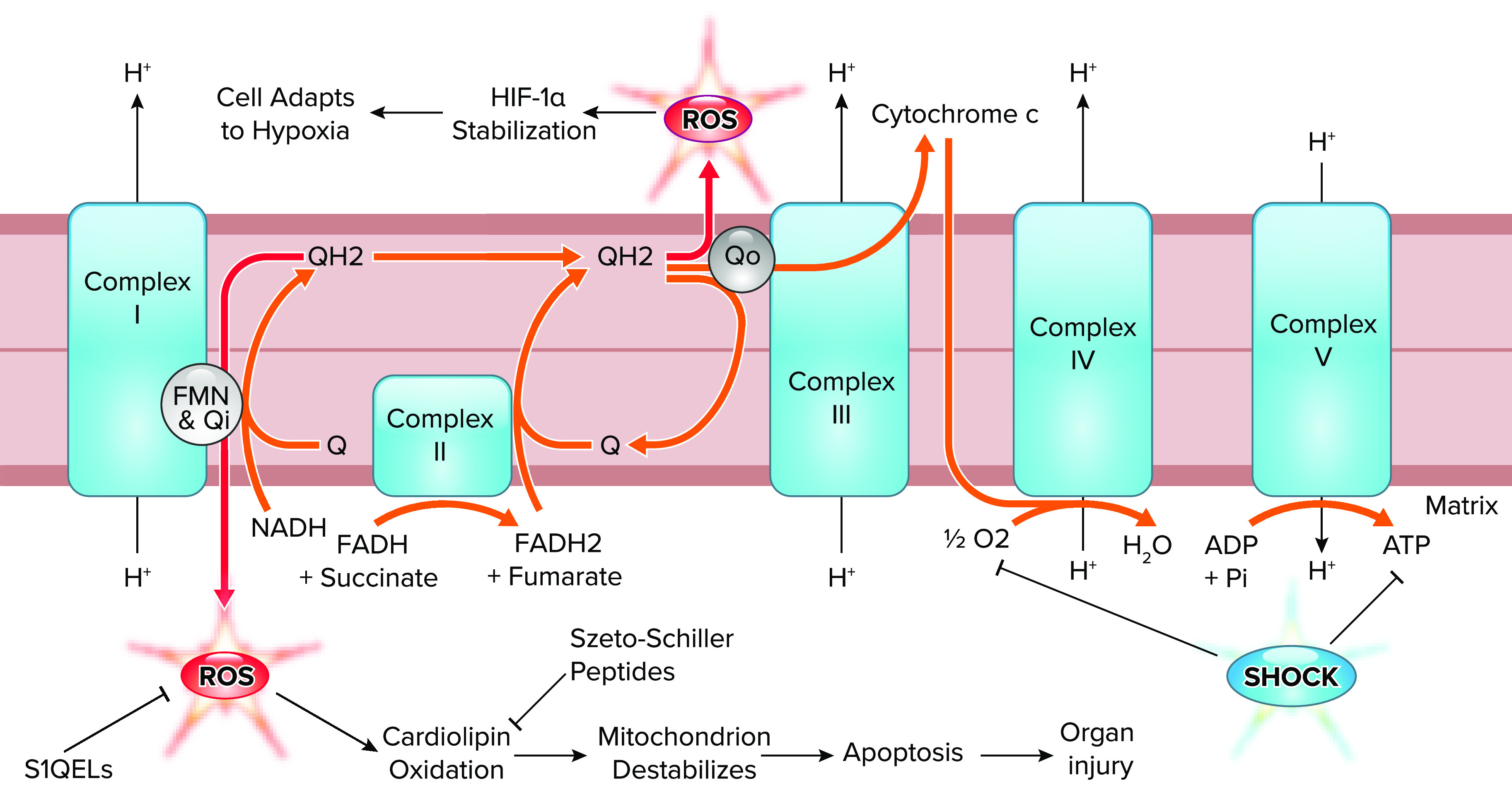
Forward and reversed electron transport in shock The forward flow of electrons through the electron transport chain (orange arrows) is decreased by shock, which causes a loss of O2 delivery and a loss of ATP synthesis, leading to reverse electron transport (red arrows) at the flavin mononucleotide (FMN) and Qi site of complex I and the Qo site of complex III. These reversed electrons generate reactive oxygen species (ROS), which can be both destructive (complex I) and adaptive (complex III). Potential therapeutic interventions and their targets are suppressors of site 1Q electron leak (S1QEL) and Szeto-Schiller peptides. HIF-1α, hypoxia-inducible factor 1 alpha. Figure modified from Sabiston Textbook of Surgery (21st ed.) (162) with permission.
As forward electron flux through the electron transport chain slows, demand for high-energy electrons from NADH falls, leading to NADH accumulation (37, 38), while succinate accumulates from forward tricarboxylic acid (TCA) cycle activity (39–41). Because the NADH-to-NAD+ ratio controls the redox state of complex I’s flavin mononucleotide (FMN) site, the hypoxic accumulation of NADH favors the generation of ROS at complex I via slip of electrons from the FMN site onto oxygen (42). Similarly, reoxygenation causes the rapid oxidation of accumulated succinate, which drives ROS generation through complex I via reversed electron transport (43).
Cellular ATP depletion is therefore the trigger of ischemic injury (FIGURE 4). ATPase inactivation reduces sodium and calcium efflux and limits the reuptake of calcium by the endoplasmic reticulum, thereby producing increases in both intracellular calcium and sodium. In addition, accumulation of hydrogen ions secondary to anaerobic metabolism further exacerbates these processes by decreasing sodium-potassium-ATPase, enhancing sodium/hydrogen exchanges, and thus reversing transport of sodium/calcium. Calcium-mediated opening of mitochondrial permeability transition pores (mPTPs) and initiation of apoptosis cascades further dampen ATP production and lead to cell death (44–46).
FIGURE 4.
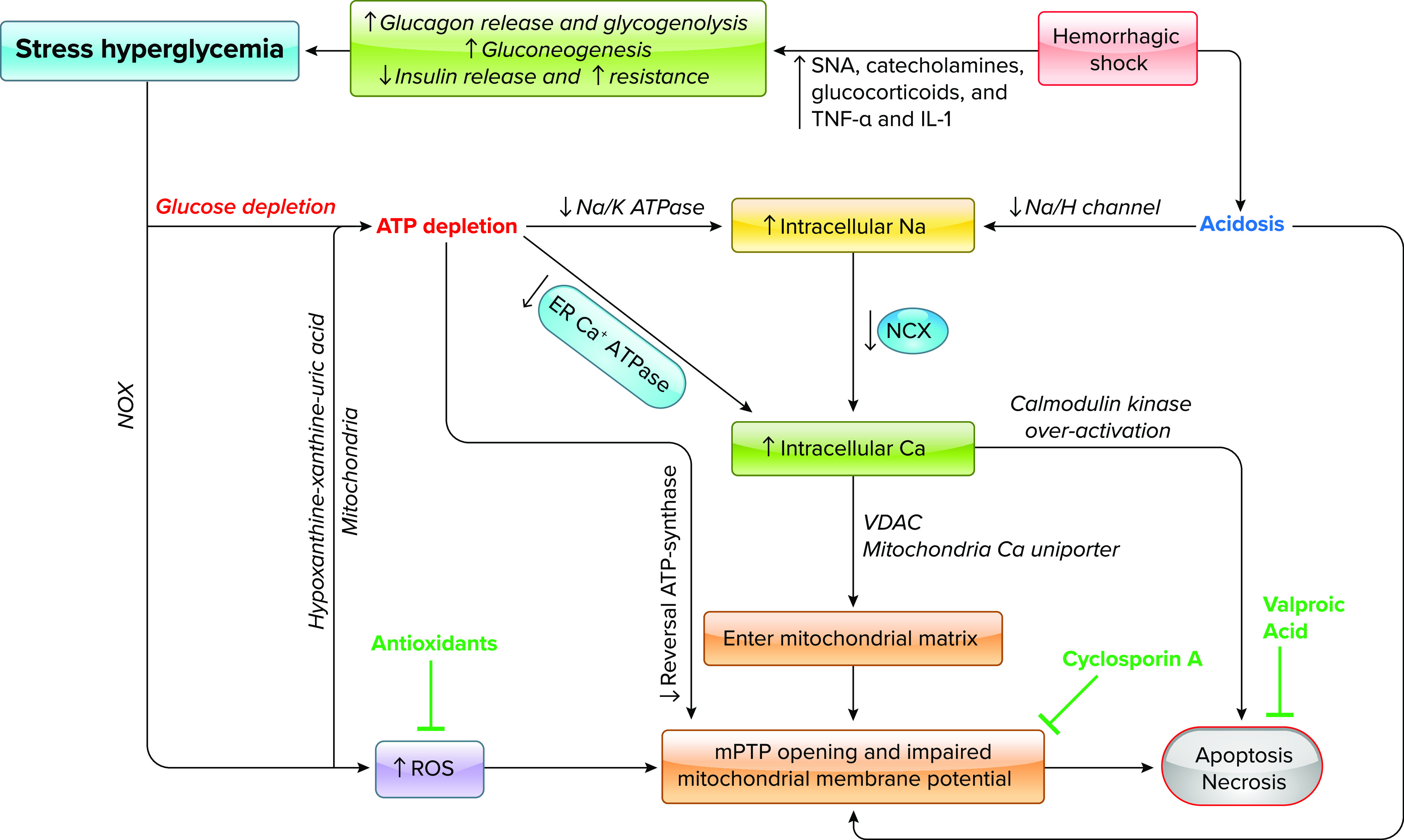
Flow diagram of ischemia-induced cell death Therapeutic interventions and their targets are shown in green. Ca, calcium; ER, endoplasmic reticulum; H, hydrogen; IL-1, interleukin-1; K, potassium; mPTP, mitochondrial permeability transition pore; Na, sodium; NCX, sodium/calcium exchanger; NOX; nicotinamide adenine dinucleotide phosphate oxidases; ROS, reactive oxygen species; SNA, sympathetic nerve activation; TNF-α, tumor necrosis factor-alpha; VDAC, voltage-dependent anion channel.
During ischemia, mitochondria are an early source of ROS due to generation of superoxide mainly from complex I, translocation of P66shc (a protein regulator of cellular redox state and apoptosis) to the outer membrane, and deactivation of antioxidative systems. Knockout mice missing P66shc exhibit enhanced resistance to oxidative stress and ischemic injury, suggesting P66shc as a potential therapeutic target (47). With progressive hypoxia and ATP depletion, generation of superoxide from the hypoxanthine-xanthine-uric acid pathway becomes dominant (44, 48, 49). In addition, stress hyperglycemia and immune cells via nicotinamide adenine dinucleotide phosphate oxidase/myeloperoxidase pathways have been shown to be important sources of free radicals.
Regulated Cell Death
Into this century, cell death was generally grouped into apoptotic (programmed) and necrotic (uncontrolled) (50), but we have come to recognize numerous types of nonapoptotic cell death that operate via regulated pathways. There are several excellent reviews on known forms of regulated cell death (RCD) (51–53). Certain pathways have greater battlefield relevance by relation to ischemia-reperfusion injury with attendant low-energy states and oxidative stress. Briefly, these are pyroptosis (caspase-1-mediated oncotic/inflammatory cell death) (54); necroptosis [damage-associated molecular patterns (DAMPs)-triggered RIPK complex leads to MLKL necrosome] (55, 56); parthanatos (hyperactivation of PARP1, increased mitochondrial apoptosis-inducible factor) (57); and mPTP-driven regulated necrosis (caspase-independent, p53-triggered mPTP opening results in oncotic rupture of mitochondria) (58, 59). Intervening against only one type of RCD is insufficient, as cells under stress will shunt to another form of cell death if able, and the inhibitor of one type can trigger another (52, 60, 61). Given the speed with which RCD pathways can be initiated (within minutes), timely intervention may extend tissue viability and prevent unnecessary cell death with resultant distant organ and immune dysfunction via DAMPs. Knowledge regarding the granular, tissue-specific proteomic and transcriptomic changes occurring in the setting of traumatic hemorrhagic shock is therefore needed to inform early treatment, reduce RCD, and prevent detrimental sequelae.
The Challenge of Traumatic Hemorrhage and Organ Failure
Ultimately, cellular dysfunction and death eventuate in damage to critical organs. In addition to organ dysfunction produced by ischemia, there is often the concomitant presence of tissue damage such as penetrating or blunt soft tissue injury, bone fracture, or traumatic brain injury. There is evidence that trauma affects cardiovascular and metabolic responses to hemorrhage and thus further exacerbates ischemic injury (62–64). First, traumatic injury and related nociceptive signals act to shunt blood away from metabolically active organs to relatively inactive tissues such as skeletal muscle after hemorrhage (62, 65). In fact, traumatic injury has been associated with decreased tolerance to hemorrhage, with nociception signaling as a possible mechanism (62, 66). Second, metabolic derangements, such as pyruvate and lactate accumulation and TCA cycle dysfunction, have been found in rats sustaining both trauma and hemorrhage (67). Pathophysiological changes induced by trauma not only further decrease tissue oxygen and energy supply but may also simultaneously increase the need for and increase systemic metabolic rate, thus synergistically exacerbating ischemia in metabolically active organs. Inflammatory responses following trauma can also increase and ATP depletion (68, 69). Furthermore, direct toxic effects of myoglobin, potassium, and other toxins released from injured muscle have been identified (70–72). Although these effects of traumatic injury are known, many animal models of hemorrhagic shock still do not combine hemorrhage with tissue damage. It should also be noted that, in addition to hemorrhage and traumatic injury effects, treatments such as prolonged tourniquet use on an extremity can also promote the release of such toxins and produce detrimental systemic effects upon tourniquet release.
Preserving organ function during prolonged care scenarios in warfighters with trauma and hemorrhage is therefore a critical goal for military research. Neurohumoral compensatory responses preferentially direct cardiac output to the brain and heart, which are critical organs requiring oxygen delivery, while “sacrificing” other organs. For example, although more resilient to ischemia than the brain and heart, the kidney is vulnerable to prolonged traumatic shock, and acute kidney injury (AKI) has long been a life-threatening complication to battlefield injuries. During World War II, mortality associated with AKI approached 100%; the development and application of dialysis techniques in the Korean War reduced mortality in patients with AKI to 60% (73, 74). Although there is a trend toward a reduction in overall rates of AKI due to less severe injury because of body armor wear and improved tactical field care in the current theaters, the overall mortality caused by AKI is still high, with any decrement in renal function significantly increasing morbidity and mortality (75, 76). Indeed, AKI occurred in current theaters despite the majority of evacuations occurring in <60 min (5); the frequency of AKI and dysfunction of other organs is expected to increase in delayed evacuation scenarios. Acute decreases in renal perfusion pressure due to hypotensive hemorrhage may overwhelm renal autoregulation and suppress glomerular filtration rate (i.e., prerenal injury), but this is reversible should renal perfusion be restored. However, prolonged ischemia may eventuate in acute tubular necrosis (intrarenal injury), in which tubules are damaged and autoregulation is lost (77). While AKI is one consequence of traumatic hemorrhage, dysfunction of other organs and the system as a whole (e.g., multiorgan failure) may also be a consequence of severe traumatic injury. Unfortunately, there is currently no preventive strategy for AKI and other sequelae of traumatic shock during delayed evacuation to a hospital setting. Current research efforts focus on preventing or delaying the cellular consequences of hemorrhage in an effort to protect vital organs for a prolonged period.
Another example of organ failure during hemorrhage is “blood failure.” Blood cells, circulating plasma components, and the vascular endothelium are in constant contact with one another, each contributing to the maintenance of homeostasis for the others. Accordingly, the blood and endothelium can be seen as a single organ system, as prone to failure during hemorrhage as any other (78). Taking this view, blood failure can be defined as “low tissue oxygen delivery, endotheliopathy, platelet dysfunction, and coagulopathy” (79). Hypoxia acts to disrupt metabolic function in endothelial cells just as in other cells, with the result that the endothelial barrier is compromised and becomes “leaky.” Importantly, normal endothelial regulation of blood flow at the microcirculatory level by various anticoagulant mechanisms is disrupted, the endothelial glycocalyx is degraded, and inflammatory cells are activated (79, 80).
In the blood failure concept, traumatic coagulopathy (i.e., the reduced ability to form clots) is a consequence of widespread cellular failure due to hypoxia, especially of platelets and the vascular endothelium (78). This failure of hemostasis is thought to be associated with both injury severity as well as poor outcomes after trauma (81); it is therefore of interest both for diagnostic potential and as a therapeutic target. Trauma patients can present with hyperfibrinolysis (enhanced destruction of formed clots), depletions in coagulation factors, as well as the activation of protein C, all of which contribute to coagulopathy and poor outcomes (82–84). Platelets become dysfunctional, further decreasing the ability to form clots. Therefore, the constellation of patient presentations and potential etiologies makes the coagulopathy of trauma very challenging to study and even more difficult to treat (85). Hypothermia and metabolic disturbances further amplify clotting dysfunction by decreasing platelet function and coagulation enzyme activity, leading to the well-known “lethal triad” of trauma: acidosis, hypothermia, and coagulopathy (86). Antifibrinolytic therapy with intravenous tranexamic acid (TXA; FIGURE 2) improves survival after trauma if given acutely (87) but worsens mortality if given >3 h after injury (88); TXA is now given routinely if administration is possible within 3 h of injury. In addition to its antifibrinolytic activation, TXA has beneficial effects on endothelial barrier function and reduces tissue edema (79) as well as other beneficial effects to include reducing inflammatory processes and protecting gut barrier function during ischemia-reperfusion injury (89–92).
Potential Treatment Strategies
Inherent in the discussion above is that there are a variety of physiological deficits in the hemorrhaging patient. However, the two most critical deficits are 1) the loss of blood itself leading to hemodynamic instability and 2) a decrease in oxygen delivery to vital organs. So the most effective treatment is fluid resuscitation to increase intravascular volume, thereby restoring blood pressure, and the optimal resuscitation fluid would also have the ability to carry oxygen. As discussed below, whole blood is currently considered the optimal resuscitative fluid as it replaces both volume and oxygen-carrying capacity. However, the logistics of blood delivery far-forward (e.g., the need for refrigeration, weight) may be daunting in future battlefield scenarios, and thus there is renewed interest in development of low-volume pharmacological adjuncts (“antishock drugs”) with the aim of altering the host response to the hemorrhagic insult to delay and/or prevent the onset of ischemic injury. For prehospital care, putative therapies with lower weight and size, a long shelf-life, and stability in variable environmental temperatures are desired. To this end, new pharmacological adjuncts are being considered that will further improve oxygen delivery and utilization, stabilize mitochondrial function, and/or prevent RCD. TABLE 2 provides a sample list of therapeutics currently under investigation, most already FDA approved and available for “off-label” use.
Table 2.
Pharmacological agents that have demonstrated beneficial effects after hemorrhage
| Drug | Mechanism of Action | Posthemorrhage Survival Time | Posthemorrhage Survival Proportion | Model Species | References |
|---|---|---|---|---|---|
| Tranexamic acid | Inhibits plasmin, suppresses DAMP release | + | + | Rat, human | (87, 139–142) |
| Polyethylene glycol-20k | Improves microcirculatory perfusion and | + | ? | Rat, pig | (113–115) |
| Trans-sodium crocetinate | Enhances oxygen diffusion into tissues | ? | + | Rat | (121) |
| Valproic acid | Histone deacetylase inhibitor; mitochondrial stabilization | + | + | Rat, pig | (143–149) |
| Cyclosporine A | Calcineurin inhibitor; mitochondrial stabilization | + | + | Rat | (150) |
| Centhaquine | α2β-Adrenergic receptors | + | + | Rat, pig, human | (107–110) |
| Naloxone, naltrexone | µ-, δ-Receptor antagonist | + | + | Dog, horse, primate, human | (151–156) |
| Melatonin | NF-κB inhibitor, antioxidant | + | + | Rat, pig | (135, 157, 158) |
| β-Hydroxy-butyrate | Ketone body to provide cellular fuel | + | + | Rat, pig | (135, 157, 159) |
| Minocycline/doxycycline | PARP/caspase/MMP inhibition | + | + | Mouse, rat | (160, 161) |
DAMP, damage-associated molecular patterns; , oxygen delivery; MMP, matrix metalloproteinase; NF-κB, nuclear factor kappa B; PARP, poly(ADP-ribose) polymerase.
Resuscitation: Restoration of Fluid Volume and Blood Pressure
At the start of the recent conflicts, the standard for resuscitation of hemorrhaging trauma patients was prehospital infusion of crystalloid (e.g., lactated Ringer’s, saline) or colloid (e.g., Hextend) solutions, followed by administration of blood products upon hospital arrival. However, it became evident that prehospital infusion of these fluids produced detrimental effects, including hemodilution, endothelial dysfunction, and coagulopathy (93). By 2006, Army physicians recognized the superiority of fresh whole blood transfusion to conventional blood component therapy (94, 95), as whole blood both increases intravascular volume and provides hemoglobin for oxygen delivery (FIGURE 2). This was not new, as whole blood transfusions were used for treatment of military casualties in both World Wars, the Korean War, and the Vietnam War (93, 96). Transfusion of whole blood at or close to the point of injury, when available, has become the preferred prehospital resuscitation fluid for military forces, with Special Operations forces carrying both whole blood on combat missions and equipment to facilitate a “walking blood bank” (97). Because of the superiority of whole blood as a prehospital resuscitation fluid (98), the military experience has now been transferred to prehospital civilian care, with whole blood being carried by first responders in some United States cities (99).
In future conflicts, however, the need for blood during mass casualty scenarios will outstrip the supply of blood and the resources to collect blood from others, which is particularly problematic if donors also must continue the fight. One potential solution for trauma patients has been the development of artificial blood substitutes, including hemoglobin-based oxygen carriers; none are yet FDA approved. Another potential solution is to provide plasma, which contains clotting factors and is protective of endothelial function (FIGURE 2). Two recent civilian trials have been performed using thawed plasma in addition to standard of care during prehospital care, with somewhat conflicting results (100, 101); in one study, prehospital administration of plasma during helicopter transport resulted in decreased 30-day mortality (100), whereas in the other there was no survival benefit to the administration of plasma during ground ambulance transport (101). However, post hoc analysis of the combined data set demonstrated improved survival when transport times were >20 min (102). Although use of thawed plasma presents logistical challenges, freeze-dried (lyophilized) plasma has efficacy and physiological responses equivalent to thawed plasma (103). Again, this is not new, as freeze-dried plasma was used by the United States military during World War II but was halted after hepatitis outbreaks. Freeze-dried plasma products are currently in use by French, German, Norwegian, and Israeli forces; the United States military has had access to the French product since 2018 under the FDA’s expanded access program. Currently, the United States military and the Department of Health and Human Services are funding developmental efforts for freeze-dried plasma (104).
Another approach to maintain blood pressure is to pharmacologically produce vasoconstriction. Although general vasoconstriction with norepinephrine is counterproductive, recent studies suggest that selective venoconstriction may be helpful. Centhaquine, a promising resuscitative agent, acts by stimulating alpha2β-adrenergic receptors in the veins to cause constriction, which results in an increase in venous return, cardiac output, and mean arterial pressure in animal models of hemorrhage (FIGURE 2; Refs. 105–107). It has also been found to prevent AKI and restore renal blood flow in a rat hemorrhage model (108). Centhaquine has completed phase III clinical trials in hypovolemic shock patients and has been demonstrated to increase pulse pressure, reduce blood lactate levels, and improve base deficit associated with a reduction in multiple organ dysfunction and 28-day all-cause mortality (109, 110).
Oxygen Delivery Enhancement
Enhancing oxygen-carrying capability has long been considered a therapeutic strategy. However, although tissue perfusion is markedly impaired after hemorrhage, the decrease in arterial Po2 is usually minor (111). In fact, reduction in tissue microcirculatory perfusion is a major contributor to impaired tissue oxygen supply (111, 112). Therefore, compounds able to improve microcirculatory perfusion, such as polyethylene glycol-20k (FIGURE 2), may be more effective than oxygen carriers in maintaining organ oxygenation. Polyethylene glycol-20k molecules can enter the interstitial space and cause the movement of isotonic fluid from interstitial and intracellular spaces into the capillaries by osmotic forces (113–115), Polyethylene glycol-20k thus establishes effective gradients across cellular/interstitial/vascular spaces, prevents cell edema, and facilitates water movement from cell to capillary to produce volume expansion in the circulation and improve organ perfusion during hemorrhage (114, 115).
Facilitating oxygen diffusion into tissues is another potential strategy to maintain tissue oxygenation under ischemic conditions. The rate of oxygen diffusion from red blood cells into surrounding tissues is determined by the pressure gradient of oxygen and the resistance to oxygen transport, with plasma providing a great deal of this resistance (116, 117). Trans-sodium crocetinate is an oxygen diffusion-enhancing compound that increases hydrogen bonding among water molecules, thus enhancing the rate and distance of oxygen diffusion into tissues (FIGURE 2; Refs. 118, 119). Trans-sodium crocetinate has been used in clinical trials to enhance oxygen diffusion into hypoxic cancer cells (120). Previous studies have explored its therapeutic potential for treating hemorrhagic shock (121, 122).
Mitochondrial Stabilization
As discussed above, ROS generation is detrimental during ischemia, but ROS are also generated on reperfusion and reoxygenation of tissues. ROS both degrade mitochondrial function and serve as critical components of the cellular response to hypoxia; mitochondrial ROS may be required to stabilize the master-regulatory transcription factor hypoxia-inducible factor 1 alpha (HIF-1α) (123, 124). Interestingly, newly identified site-specific mitochondrial complex I (125) and III (126) inhibitors have shown that ROS generated at different sites in the electron transport chain have different impacts on cellular responses to hypoxia and reoxygenation (127). ROS from the matrix-facing quinone site on complex I appear to be destructive, whereas ROS from the intermembrane space-facing quinone site of complex III are critically required for stabilizing HIF-1α (127–129). The prevention of ROS generation at complex I without preventing ROS generation at complex III, such as suppressors of site 1Q electron leak (S1QEL; FIGURE 3), could therefore represent an appealing strategy for preserving mitochondrial function without preventing critical signaling mechanisms (127). Additionally, the preservation of cardiolipin in its native (i.e., multiply unsaturated) conformation with Szeto–Schiller peptides represents a promising approach to mitochondrial stabilization during stress (FIGURE 3; Ref. 130).
Prevention of RCD
Casualties suffering from traumatic shock have mere minutes before they begin to circulate DAMPs and exhibit early steps of RCD pathways (131); accordingly, favorable pharmacokinetics and route of administration become critical as the target tissues may have the most compromised circulatory supply. Ideally, such a therapeutic could be given at point of injury as an adjunct to resuscitation, delaying or preventing the onset of RCD, resultant DAMP release, and subsequent end organ injury. If targeting RCD, a combination of agents will likely be required, as preventing one RCD pathway typically leads to shunting toward another (51). Pharmacological agents proposed to ameliorate RCD include valproic acid (FIGURE 4), which helps to reduce apoptosis and is the subject of a current clinical trial to reduce AKI in liver transplant patients (https://clinicaltrials.gov/ct2/show/NCT04531592). Likewise, cyclosporine A (FIGURE 4) acts to stabilize mitochondria and prevent RCD after ischemia (132, 133). Although valproic acid and cyclosporine A seem to work for many types of apoptosis, it remains to be seen whether these potential treatments are a good option for reversing hemorrhagic shock, as these therapeutics rely on many time- and energy-consuming processes such as cellular entry, receptor binding, transcription, and protein translation and synthesis. Under the ischemic conditions following hemorrhage, there may not be enough cellular energy to allow these treatments to work.
The ideal of a rapidly induced hibernation state in humans has been explored, for obvious reasons; i.e., hibernating organisms survive with a greatly reduced metabolic rate, then rebound without loss of tissue or function (134–136). Unfortunately, the process of entering a proper hibernating/torpor state generally takes place over weeks, but perhaps could be compressed to days, or even hours; agents successful in rapidly inducing torpor states with small animals, however, have generally failed in larger-animal studies (137). Although the hemorrhaging casualty has only minutes before catastrophic changes begin to occur at the cellular level, possible clues may be taken from how hibernating organisms manage extreme metabolic deprivation. The Defense Advanced Research Projects Agency (DARPA) initiated the Biostasis program in an attempt to extend the Golden Hour by developing novel pharmacological approaches to slow metabolic processes, thereby resulting in a metabolic arrest that might protect functional capacity until definitive medical care is available (138).
Conclusions
Although the nature of war may change, the physiology underlying traumatic hemorrhage does not. However, the potential return to large-scale combat operations presents renewed challenges to a military medical system with historically low case fatality rates in recent wars; these may, in fact, represent an aberration in the history of warfare. Potential solutions to face these challenges must be based both on a greater understanding of the underlying physiology of hemorrhage and on the logistical realities of providing care in these scenarios. The relatively new understanding of the blood-endothelium as an organ that fails in hemorrhagic shock provides new opportunities for potential intervention. However, the rapid time course of RCD activation must also be taken into account when contemplating interventions. Undoubtedly, there is no one “silver bullet,” but ultimate resuscitation strategies will require combinations of oxygen-delivering solutions to increase blood volume and adjuncts to make the host tissue more tolerant to ischemia.
Acknowledgments
The opinions or assertions contained in this article are those of the authors and do not reflect the official policy or position of the U.S. Army Medical Department, Department of the Army, Department of Defense, or the U.S. Government.
This work was supported by the U.S. Army Combat Casualty Care Research Program, U.S. Army Medical Research and Development Command.
No conflicts of interest, financial or otherwise, are declared by the authors.
C.H.-L., I.L.H., E.R., L.X., and K.L.R. conceived and designed research; prepared figures; drafted manuscript; edited and revised manuscript; and approved final version of manuscript.
References
- 1.Centers for Disease Control and Prevention. Key Injury and Violence Data. https://www.cdc.go/injury/wisqars/overview/key_data.html. [2021 Jul 7].
- 2.Eastridge BJ, Mabry RL, Seguin P, Cantrell J, Tops T, Uribe P, Mallett O, Zubko T, Oetjen-Gerdes L, Rasmussen TE, Butler FK, Kotwal RS, Holcomb JB, Wade C, Champion H, Lawnick M, Moores L, Blackbourne LH. Death on the battlefield (2001-2011): implications for the future of combat casualty care. J Trauma Acute Care Surg 73: S431–S437, 2012. doi: 10.1097/TA.0b013e3182755dcc. [DOI] [PubMed] [Google Scholar]
- 3.Mazuchowski EL, Kotwal RS, Janak JC, Howard JT, Harcke HT, Montgomery HR, Butler FK, Holcomb JB, Eastridge BJ, Gurney JM, Shackelford SA. Mortality review of US Special Operations Command battle-injured fatalities. J Trauma Acute Care Surg 88: 686–695, 2020. doi: 10.1097/TA.0000000000002610. [DOI] [PubMed] [Google Scholar]
- 4.Kotwal RS, Howard JT, Orman JA, Tarpey BW, Bailey JA, Champion HR, Mabry RL, Holcomb JB, Gross KR. The effect of a golden hour policy on the morbidity and mortality of combat casualties. JAMA Surg 151: 15–24, 2016. doi: 10.1001/jamasurg.2015.3104. [DOI] [PubMed] [Google Scholar]
- 5.Howard JT, Kotwal RS, Santos-Lazada AR, Martin MJ, Stockinger ZT. Reexamination of a battlefield trauma golden hour policy. J Trauma Acute Care Surg 84: 11–18, 2018. doi: 10.1097/TA.0000000000001727. [DOI] [PubMed] [Google Scholar]
- 6.Kellerman AL, Elster E. (Editors). Out of the Crucible: How the US Military Transformed Combat Casualty Care in Iraq and Afghanistan. Fort Sam Houston, TX: Office of the Surgeon General, Borden Institute, 2017. [Google Scholar]
- 7.Howard JT, Kotwal RS, Stern CA, Janak JC, Mazuchowski EL, Butler FK, Stockinger ZT, Holcomb BR, Bono RC, Smith DJ. Use of combat casualty care data to assess the us military trauma system during the Afghanistan and Iraq conflicts, 2001-2017. JAMA Surg 154: 600–608, 2019. doi: 10.1001/jamasurg.2019.0151. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Davis MR, Rasmussen TE. Winds of change in military medicine and combat casualty care. J Trauma Acute Care Surg 87: S1–S2, 2019. doi: 10.1097/TA.0000000000002349. [DOI] [PubMed] [Google Scholar]
- 9.American College of Surgeons Committee on Trauma. Advanced Trauma Life Support (ATLS) Student Course Manual. Chicago, IL: American College of Surgeons, 2012. [Google Scholar]
- 10.Evans RG, Ventura S, Dampney RA, Ludbrook J. Neural mechanisms in the cardiovascular responses to acute central hypovolaemia. Clin Exp Pharmacol Physiol 28: 479–487, 2001. doi: 10.1046/j.1440-1681.2001.03473.x. [DOI] [PubMed] [Google Scholar]
- 11.Schadt JC, Ludbrook J. Hemodynamic and neurohumoral responses to acute hypovolemia in conscious mammals. Am J Physiol Heart Circ Physiol 260: H305–H318, 1991. doi: 10.1152/ajpheart.1991.260.2.H305. [DOI] [PubMed] [Google Scholar]
- 12.Sakai H, Hara H, Tsai AG, Tsuchida E, Johnson PC, Intaglietta M. Changes in resistance vessels during hemorrhagic shock and resuscitation in conscious hamster model. Am J Physiol Heart Circ Physiol 276: H563–H571, 1999. doi: 10.1152/ajpheart.1999.276.2.H563. [DOI] [PubMed] [Google Scholar]
- 13.Darlington DN, Shinsako J, Dallman MF. Responses of ACTH, epinephrine, norepinephrine, and cardiovascular system to hemorrhage. Am J Physiol Heart Circ Physiol 251: H612–H618, 1986. doi: 10.1152/ajpheart.1986.251.3.H612. [DOI] [PubMed] [Google Scholar]
- 14.Fejes-Tóth G, Brinck-Johnsen T, Naray-Fejes-Tóth A. Cardiovascular and hormonal response to hemorrhage in conscious rats. Am J Physiol Heart Circ Physiol 254: H947–H953, 1988. doi: 10.1152/ajpheart.1988.254.5.H947. [DOI] [PubMed] [Google Scholar]
- 15.Morris M, Kapoor V, Chalmers J. Plasma neuropeptide Y concentration is increased after hemorrhage in conscious rats: relative contributions of sympathetic nerves and the adrenal medulla. J Cardiovasc Pharmacol 9: 541–545, 1987. doi: 10.1097/00005344-198705000-00006. [DOI] [PubMed] [Google Scholar]
- 16.Rajani RR, Ball CG, Feliciano DV, Vercruysse GA. Vasopressin in hemorrhagic shock: review article. Am Surg 75: 1207–1212, 2009. doi: 10.1177/000313480907501212. [DOI] [PubMed] [Google Scholar]
- 17.Fabre LF Jr, Farmer RW, Davis HW, McBee G, Farrell G. Biphasic stimulation of aldosterone secretion during hemorrhage in dogs. Circ Res 24: 893–900, 1969. doi: 10.1161/01.res.24.6.893. [DOI] [PubMed] [Google Scholar]
- 18.Wade CE, Hannon JP, Bossone CA, Hunt MM, Loveday JA, Coppes RI Jr, Gildengorin V. Neuroendocrine responses to hypertonic saline/dextran resuscitation following hemorrhage. Circ Shock 35: 37–43, 1991. [PubMed] [Google Scholar]
- 19.Strittmatter RR, Schadt JC. Sex differences in the respiratory response to hemorrhage in the conscious, New Zealand white rabbit. Am J Physiol Regul Integr Comp Physiol 292: R1963–R1969, 2007. doi: 10.1152/ajpregu.00494.2006. [DOI] [PubMed] [Google Scholar]
- 20.Himeno Y, Ikebuchi M, Maeda A, Noma A, Amano A. Mechanisms underlying the volume regulation of interstitial fluid by capillaries: a simulation study. Integr Med Res 5: 11–21, 2016. doi: 10.1016/j.imr.2015.12.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Xiang L, Lu S, Mittwede PN, Clemmer JS, Husband GW, Hester RL. beta2-Adrenoreceptor blockade improves early posttrauma hyperglycemia and pulmonary injury in obese rats. Am J Physiol Heart Circ Physiol 307: H621–H627, 2014. doi: 10.1152/ajpheart.00208.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Silveira SA, Haibara AS, Coimbra CC. Hyperglycemic response to hemorrhage is modulated by baroreceptors unloading but not by peripheral chemoreceptors activation. Auton Neurosci 123: 36–43, 2005. doi: 10.1016/j.autneu.2005.08.007. [DOI] [PubMed] [Google Scholar]
- 23.Xiang L, Mittwede PN, Clemmer JS. Glucose homeostasis and cardiovascular alterations in diabetes. Compr Physiol 5: 1815–1839, 2015. doi: 10.1002/cphy.c150001. [DOI] [PubMed] [Google Scholar]
- 24.Burgos-Moron E, Abad-Jimenez Z, Maranon AM, Iannantuoni F, Escribano-Lopez I, Lopez-Domenech S, Salom C, Jover A, Mora V, Roldan I, Sola E, Rocha M, Victor VM. Relationship between oxidative stress, ER stress, and inflammation in type 2 diabetes: the battle continues. J Clin Med 8: 1385, 2019. doi: 10.3390/jcm8091385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Clyne AM. Endothelial response to glucose: dysfunction, metabolism, and transport. Biochem Soc Trans 49: 313–325, 2021. doi: 10.1042/BST20200611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Marik PE, Bellomo R. Stress hyperglycemia: an essential survival response!. Crit Care Med 41: e93–e94, 2013. doi: 10.1097/CCM.0b013e318283d124. [DOI] [PubMed] [Google Scholar]
- 27.Cooke WH, Ryan KL, Convertino VA. Lower body negative pressure as a model to study progression to acute hemorrhagic shock in humans. J Appl Physiol (1985) 96: 1249–1261, 2004. doi: 10.1152/japplphysiol.01155.2003. [DOI] [PubMed] [Google Scholar]
- 28.Guyenet PG, Stornetta RL, Bayliss DA. Central respiratory chemoreception. J Comp Neurol 518: 3883–3906, 2010. doi: 10.1002/cne.22435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Chaudry IH. Cellular mechanisms in shock and ischemia and their correction. Am J Physiol Regul Integr Comp Physiol 245: R117–R134, 1983. doi: 10.1152/ajpregu.1983.245.2.R117. [DOI] [PubMed] [Google Scholar]
- 30.Heckbert SR, Vedder NB, Hoffman W, Winn RK, Hudson LD, Jurkovich GJ, Copass MK, Harlan JM, Rice CL, Maier RV. Outcome after hemorrhagic shock in trauma patients. J Trauma 45: 545–549, 1998. doi: 10.1097/00005373-199809000-00022. [DOI] [PubMed] [Google Scholar]
- 31.Convertino VA, Koons NJ, Suresh MR. Physiology of human hemorrhage and compensation. Compr Physiol 11: 1531–1574, 2021. doi: 10.1002/cphy.c200016. [DOI] [PubMed] [Google Scholar]
- 32.Grover GJ, Atwal KS, Sleph PG, Wang FL, Monshizadegan H, Monticello T, Green DW. Excessive ATP hydrolysis in ischemic myocardium by mitochondrial F1F0-ATPase: effect of selective pharmacological inhibition of mitochondrial ATPase hydrolase activity. Am J Physiol Heart Circ Physiol 287: H1747–H1755, 2004. doi: 10.1152/ajpheart.01019.2003. [DOI] [PubMed] [Google Scholar]
- 33.Grover GJ, Malm J. Pharmacological profile of the selective mitochondrial F1F0 ATP hydrolase inhibitor BMS-199264 in myocardial ischemia. Cardiovasc Ther 26: 287–296, 2008. doi: 10.1111/j.1755-5922.2008.00065.x. [DOI] [PubMed] [Google Scholar]
- 34.Grover GJ, Marone PA, Koetzner L, Seto-Young D. Energetic signalling in the control of mitochondrial F1F0 ATP synthase activity in health and disease. Int J Biochem Cell Biol 40: 2698–2701, 2008. doi: 10.1016/j.biocel.2008.06.013. [DOI] [PubMed] [Google Scholar]
- 35.Rouslin W, Erickson JL, Solaro RJ. Effects of oligomycin and acidosis on rates of ATP depletion in ischemic heart muscle. Am J Physiol Heart Circ Physiol 250: H503–H508, 1986. doi: 10.1152/ajpheart.1986.250.3.H503. [DOI] [PubMed] [Google Scholar]
- 36.Rouslin W, Broge CW, Grupp IL. ATP depletion and mitochondrial functional loss during ischemia in slow and fast heart-rate hearts. Am J Physiol Heart Circ Physiol 259: H1759–H1766, 1990. doi: 10.1152/ajpheart.1990.259.6.H1759. [DOI] [PubMed] [Google Scholar]
- 37.Jennings RB, Reimer KA, Steenbergen C. Effect of inhibition of the mitochondrial ATPase on net myocardial ATP in total ischemia. J Mol Cell Cardiol 23: 1383–1395, 1991. doi: 10.1016/0022-2828(91)90185-o. [DOI] [PubMed] [Google Scholar]
- 38.Wengrowski AM, Kuzmiak-Glancy S, Jaimes R 3rd, Kay MW. NADH changes during hypoxia, ischemia, and increased work differ between isolated heart preparations. Am J Physiol Heart Circ Physiol 306: H529–H537, 2014. doi: 10.1152/ajpheart.00696.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Chouchani ET, Pell VR, Gaude E, Aksentijević D, Sundier SY, Robb EL, Logan A, Nadtochiy SM, Ord EN, Smith AC, Eyassu F, Shirley R, Hu CH, Dare AJ, James AM, Rogatti S, Hartley RC, Eaton S, Costa AS, Brookes PS, Davidson SM, Duchen MR, Saeb-Parsy K, Shattock MJ, Robinson AJ, Work LM, Frezza C, Krieg T, Murphy MP. Ischaemic accumulation of succinate controls reperfusion injury through mitochondrial ROS. Nature 515: 431–435, 2014. doi: 10.1038/nature13909. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Taegtmeyer H. Metabolic responses to cardiac hypoxia. Increased production of succinate by rabbit papillary muscles. Circ Res 43: 808–815, 1978. doi: 10.1161/01.res.43.5.808. [DOI] [PubMed] [Google Scholar]
- 41.Zhang J, Wang YT, Miller JH, Day MM, Munger JC, Brookes PS. Accumulation of succinate in cardiac ischemia primarily occurs via canonical Krebs cycle activity. Cell Rep 23: 2617–2628, 2018. doi: 10.1016/j.celrep.2018.04.104. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Robb EL, Hall AR, Prime TA, Eaton S, Szibor M, Viscomi C, James AM, Murphy MP. Control of mitochondrial superoxide production by reverse electron transport at complex I. J Biol Chem 293: 9869–9879, 2018. doi: 10.1074/jbc.RA118.003647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Chouchani ET, Pell VR, James AM, Work LM, Saeb-Parsy K, Frezza C, Krieg T, Murphy MP. A unifying mechanism for mitochondrial superoxide production during ischemia-reperfusion injury. Cell Metab 23: 254–263, 2016. doi: 10.1016/j.cmet.2015.12.009. [DOI] [PubMed] [Google Scholar]
- 44.Abramov AY, Scorziello A, Duchen MR. Three distinct mechanisms generate oxygen free radicals in neurons and contribute to cell death during anoxia and reoxygenation. J Neurosci 27: 1129–1138, 2007. doi: 10.1523/JNEUROSCI.4468-06.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Guo Y, Tan J, Miao Y, Sun Z, Zhang Q. Effects of microvesicles on cell apoptosis under hypoxia. Oxid Med Cell Longev 2019: 5972152, 2019., doi: 10.1155/2019/5972152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Arnould T, Michiels C, Alexandre I, Remacle J. Effect of hypoxia upon intracellular calcium concentration of human endothelial cells. J Cell Physiol 152: 215–221, 1992. doi: 10.1002/jcp.1041520127. [DOI] [PubMed] [Google Scholar]
- 47.Galimov ER. The role of p66shc in oxidative stress and apoptosis. Acta Naturae 2: 44–51, 2010. [PMC free article] [PubMed] [Google Scholar]
- 48.Battelli MG, Polito L, Bortolotti M, Bolognesi A. Xanthine oxidoreductase-derived reactive species: physiological and pathological effects. Oxid Med Cell Longev 2016: 3527579, 2016., doi: 10.1155/2016/3527579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Douzinas EE. Progressive hemorrhage: administer oxygen or early resuscitation? Intensive Care Med 35: 1664–1666, 2009. doi: 10.1007/s00134-009-1576-3. [DOI] [PubMed] [Google Scholar]
- 50.Hotchkiss RS, Strasser A, McDunn JE, Swanson PE. Cell death. N Engl J Med 361: 1570–1583, 2009. doi: 10.1056/NEJMra0901217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Galluzzi L, Bravo-San Pedro JM, Vitale I, Aaronson SA, Abrams JM, Adam D, et al. Essential versus accessory aspects of cell death: recommendations of the NCCD 2015. Cell Death Differ 22: 58–73, 2015. doi: 10.1038/cdd.2014.137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Galluzzi L, Kepp O, Krautwald S, Kroemer G, Linkermann A. Molecular mechanisms of regulated necrosis. Semin Cell Dev Biol 35: 24–32, 2014. doi: 10.1016/j.semcdb.2014.02.006. [DOI] [PubMed] [Google Scholar]
- 53.Kolb JP, Oguin TH 3rd, Oberst A, Martinez J. Programmed cell death and inflammation: winter is coming. Trends Immunol 38: 705–718, 2017. doi: 10.1016/j.it.2017.06.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Bergsbaken T, Fink SL, Cookson BT. Pyroptosis: host cell death and inflammation. Nat Rev Microbiol 7: 99–109, 2009. doi: 10.1038/nrmicro2070. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Jun W, Benjanuwattra J, Chattipakorn SC, Chattipakorn N. Necroptosis in renal ischemia/reperfusion injury: a major mode of cell death? Arch Biochem Biophys 689: 108433, 2020. doi: 10.1016/j.abb.2020.108433. [DOI] [PubMed] [Google Scholar]
- 56.Zhang X, Wu J, Liu Q, Li X, Li S, Chen J, Hong Z, Wu X, Zhao Y, Ren J. mtDNA-STING pathway promotes necroptosis-dependent enterocyte injury in intestinal ischemia reperfusion. Cell Death Dis 11: 1050, 2020. doi: 10.1038/s41419-020-03239-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Fatokun AA, Dawson VL, Dawson TM. Parthanatos: mitochondrial-linked mechanisms and therapeutic opportunities. Br J Pharmacol 171: 2000–2016, 2014. doi: 10.1111/bph.12416. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Baines CP. The mitochondrial permeability transition pore and ischemia-reperfusion injury. Basic Res Cardiol 104: 181–188, 2009. doi: 10.1007/s00395-009-0004-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Linkermann A, Bräsen JH, Darding M, Jin MK, Sanz AB, Heller JO, De Zen F, Weinlich R, Ortiz A, Walczak H, Weinberg JM, Green DR, Kunzendorf U, Krautwald S. Two independent pathways of regulated necrosis mediate ischemia-reperfusion injury. Proc Natl Acad Sci USA 110: 12024–12029, 2013. doi: 10.1073/pnas.1305538110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Harrison SA, Goodman Z, Jabbar A, Vemulapalli R, Younes ZH, Freilich B, Sheikh MY, Schattenberg JM, Kayali Z, Zivony A, Sheikh A, Garcia-Samaniego J, Satapathy SK, Therapondos G, Mena E, Schuppan D, Robinson J, Chan JL, Hagerty DT, Sanyal AJ. A randomized, placebo-controlled trial of emricasan in patients with NASH and F1-F3 fibrosis. J Hepatol 72: 816–827, 2020. doi: 10.1016/j.jhep.2019.11.024. [DOI] [PubMed] [Google Scholar]
- 61.Fritsch M, Günther SD, Schwarzer R, Albert MC, Schorn F, Werthenbach JP, Schiffmann LM, Stair N, Stocks H, Seeger JM, Lamkanfi M, Krönke M, Pasparakis M, Kashkar H. Caspase-8 is the molecular switch for apoptosis, necroptosis and pyroptosis. Nature 575: 683–687, 2019. doi: 10.1038/s41586-019-1770-6. [DOI] [PubMed] [Google Scholar]
- 62.Kirkman E, Watts S. Haemodynamic changes in trauma. Br J Anaesth 113: 266–275, 2014. doi: 10.1093/bja/aeu232. [DOI] [PubMed] [Google Scholar]
- 63.Foëx BA. Systemic responses to trauma. Br Med Bull 55: 726–743, 1999. doi: 10.1258/0007142991902745. [DOI] [PubMed] [Google Scholar]
- 64.Xiang L, Calderon AS, Klemcke HG, Hudson IL, Hinojosa-Laborde C, Chung KK, Ryan KL. Extremity trauma exacerbates acute kidney injury following prolonged hemorrhagic hypotension. J Trauma Acute Care Surg 91: S113–S123, 2021. doi: 10.1097/TA.0000000000003311. [DOI] [PubMed] [Google Scholar]
- 65.Kirkman E, Zhang H, Spapen H, Little RA, Vincent JL. Effects of afferent neural stimulation on critical oxygen delivery: a hemodynamic explanation. Am J Physiol Regul Integr Comp Physiol 269: R1448–R1454, 1995. doi: 10.1152/ajpregu.1995.269.6.R1448. [DOI] [PubMed] [Google Scholar]
- 66.Parsons E, Phemuster D. Hemorrhage and “shock” in traumatized limbs. Surg Gynaecol Obstet 51: 196–207, 1930. [Google Scholar]
- 67.D’Alessandro A, Slaughter AL, Peltz ED, Moore EE, Silliman CC, Wither M, Nemkov T, Bacon AW, Fragoso M, Banerjee A, Hansen KC. Trauma/hemorrhagic shock instigates aberrant metabolic flux through glycolytic pathways, as revealed by preliminary 13C-glucose labeling metabolomics. J Transl Med 13: 253, 2015. doi: 10.1186/s12967-015-0612-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Taylor CT, Colgan SP. Hypoxia and gastrointestinal disease. J Mol Med (Berl) 85: 1295–1300, 2007. doi: 10.1007/s00109-007-0277-z. [DOI] [PubMed] [Google Scholar]
- 69.Kominsky DJ, Campbell EL, Colgan SP. Metabolic shifts in immunity and inflammation. J Immunol 184: 4062–4068, 2010. doi: 10.4049/jimmunol.0903002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Petejova N, Martinek A. Acute kidney injury due to rhabdomyolysis and renal replacement therapy: a critical review. Crit Care 18: 224, 2014. doi: 10.1186/cc13897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Nath KA. Heme oxygenase-1: a provenance for cytoprotective pathways in the kidney and other tissues. Kidney Int 70: 432–443, 2006. doi: 10.1038/sj.ki.5001565. [DOI] [PubMed] [Google Scholar]
- 72.Crepaldi C, Ocampo C, Ronco C. Myoglobin as a toxin. In: Critical Care Nephrology (2nd ed.), edited by Ronco C, Bellomo R, Kellum JA.. Philadelphia, PA: Saunders Elsevier, 2009, p. 1103–1109. [Google Scholar]
- 73.Teschan PE, Post RS, Smith LH Jr, Abernathy RS, Davis JH, Gray DM, Howard JM, Johnson KE, Klopp E, Mundy RL, O’Meara MP, Rush BF Jr.. Post-traumatic renal insufficiency in military casualties. I. Clinical characteristics. Am J Med 18: 172–186, 1955. doi: 10.1016/0002-9343(55)90233-3. [DOI] [PubMed] [Google Scholar]
- 74.Ronco C, Ricci Z, De Backer D, Kellum JA, Taccone FS, Joannidis M, Pickkers P, Cantaluppi V, Turani F, Saudan P, Bellomo R, Joannes-Boyau O, Antonelli M, Payen D, Prowle JR, Vincent JL. Renal replacement therapy in acute kidney injury: controversy and consensus. Crit Care 19: 146, 2015. doi: 10.1186/s13054-015-0850-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Stewart IJ, Sosnov JA, Howard JT, Chung KK. Acute kidney injury in critically injured combat veterans: a retrospective cohort study. Am J Kidney Dis 68: 564–570, 2016. doi: 10.1053/j.ajkd.2016.03.419. [DOI] [PubMed] [Google Scholar]
- 76.Heegard KD, Stewart IJ, Cap AP, Sosnov JA, Kwan HK, Glass KR, Morrow BD, Latack W, Henderson AT, Saenz KK, Siew ED, Ikizler TA, Chung KK. Early acute kidney injury in military casualties. J Trauma Acute Care Surg 78: 988–993, 2015. doi: 10.1097/TA.0000000000000607. [DOI] [PubMed] [Google Scholar]
- 77.Basile DP, Anderson MD, Sutton TA. Pathophysiology of acute kidney injury. Compr Physiol 2: 1303–1353, 2012. doi: 10.1002/cphy.c110041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Bjerkvig CK, Strandenes G, Eliassen HS, Spinella PC, Fosse TK, Cap AP, Ward KR. “Blood failure” time to view blood as an organ: how oxygen debt contributes to blood failure and its implications for remote damage control resuscitation. Transfusion 56, Suppl 2: S182–S189, 2016. doi: 10.1111/trf.13500. [DOI] [PubMed] [Google Scholar]
- 79.White NJ, Ward KR, Pati S, Strandenes G, Cap AP. Hemorrhagic blood failure: oxygen debt, coagulopathy, and endothelial damage. J Trauma Acute Care Surg 82: S41–S49, 2017. doi: 10.1097/TA.0000000000001436. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Torres Filho I. Hemorrhagic shock and the microvasculature. Compr Physiol 8: 61–101, 2017. doi: 10.1002/cphy.c170006. [DOI] [PubMed] [Google Scholar]
- 81.Tisherman SA, Schmicker RH, Brasel KJ, Bulger EM, Kerby JD, Minei JP, Powell JL, Reiff DA, Rizoli SB, Schreiber MA. Detailed description of all deaths in both the shock and traumatic brain injury hypertonic saline trials of the Resuscitation Outcomes Consortium. Ann Surg 261: 586–590, 2015. doi: 10.1097/SLA.0000000000000837. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Cohen MJ, Call M, Nelson M, Calfee CS, Esmon CT, Brohi K, Pittet JF. Critical role of activated protein C in early coagulopathy and later organ failure, infection and death in trauma patients. Ann Surg 255: 379–385, 2012. doi: 10.1097/SLA.0b013e318235d9e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Cohen MJ, Kutcher M, Redick B, Nelson M, Call M, Knudson MM, Schreiber MA, Bulger EM, Muskat P, Alarcon LH, Myers JG, Rahbar MH, Brasel KJ, Phelan HA, del Junco DJ, Fox EE, Wade CE, Holcomb JB, Cotton BA, Matijevic N; PROMMTT Study Group. Clinical and mechanistic drivers of acute traumatic coagulopathy. J Trauma Acute Care Surg 75: S40–S47, 2013. doi: 10.1097/TA.0b013e31828fa43d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Burggraf M, Payas A, Kauther MD, Schoeneberg C, Lendemans S. Evaluation of clotting factor activities early after severe multiple trauma and their correlation with coagulation tests and clinical data. World J Emerg Surg 10: 43, 2015. doi: 10.1186/s13017-015-0038-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Meledeo MA, Herzig MC, Bynum JA, Wu X, Ramasubramanian AK, Darlington DN, Reddoch KM, Cap AP. Acute traumatic coagulopathy: the elephant in a room of blind scientists. J Trauma Acute Care Surg 82: S33–S40, 2017. doi: 10.1097/TA.0000000000001431. [DOI] [PubMed] [Google Scholar]
- 86.Moore EE, Moore HB, Kornblith LZ, Neal MD, Hoffman M, Mutch NJ, Schöchl H, Hunt BJ, Sauaia A. Trauma-induced coagulopathy. Nat Rev Dis Primers 7: 30, 2021. doi: 10.1038/s41572-021-00264-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Shakur H, Roberts I, Bautista R, Caballero J, Coats T, Dewan Y, El-Sayed H, Gogichaishvili T, Gupta S, Herrera J, Hunt B, Iribhogbe P, Izurieta M, Khamis H, Komolafe E, Marrero MA, Mejia-Mantilla J, Miranda J, Morales C, Olaomi O, Olldashi F, Perel P, Peto R, Ramana PV, Ravi RR, Yutthakasemsunt S; CRASH-2 trial collaborators. Effects of tranexamic acid on death, vascular occlusive events, and blood transfusion in trauma patients with significant haemorrhage (CRASH-2): a randomised, placebo-controlled trial. Lancet 376: 23–32, 2010. doi: 10.1016/S0140-6736(10)60835-5. [DOI] [PubMed] [Google Scholar]
- 88.CRASH-2 Collaborators, Roberts I, Shakur H, Afolabi A, Brohi K, Coats T, Dewan Y, Gando S, Guyatt G, Hunt BJ, Morales C, Perel P, Prieto-Merino D, Woolley T. The importance of early treatment with tranexamic acid in bleeding trauma patients: an exploratory analysis of the CRASH-2 randomised controlled trial. Lancet 377: 1096–1101, 2011. doi: 10.1016/S0140-6736(11)60278-X. [DOI] [PubMed] [Google Scholar]
- 89.Carter DW, Prudovsky I, Kacer D, Soul T, Kumpel C, Pyburn K, Palmeri M, Kramer R, Rappold J. Tranexamic acid suppresses the release of mitochondrial DAMPs and reduces lung inflammation in a murine burn model. J Trauma Acute Care Surg 86: 617–624, 2019. doi: 10.1097/TA.0000000000002177. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Diebel ME, Diebel LN, Liberati DM. Tranexamic acid and the gut barrier: Protection by inhibition of trypsin uptake and activation of downstream intestinal proteases. Am J Surg 213: 489–493, 2017. doi: 10.1016/j.amjsurg.2016.10.032. [DOI] [PubMed] [Google Scholar]
- 91.Diebel ME, Diebel LN, Manke CW, Liberati DM, Whittaker JR. Early tranexamic acid administration: a protective effect on gut barrier function following ischemia/reperfusion injury. J Trauma Acute Care Surg 79: 1015–1022, 2015. doi: 10.1097/TA.0000000000000703. [DOI] [PubMed] [Google Scholar]
- 92.Chu C, Yang C, Wang X, Xie T, Sun S, Liu B, Wang K, Duan Z, Ding W, Li J. Early intravenous administration of tranexamic acid ameliorates intestinal barrier injury induced by neutrophil extracellular traps in a rat model of trauma/hemorrhagic shock. Surgery 167: 340–351, 2020. doi: 10.1016/j.surg.2019.10.009. [DOI] [PubMed] [Google Scholar]
- 93.Cannon JW. Hemorrhagic shock. N Engl J Med 378: 1852–1853, 2018. doi: 10.1056/NEJMc1802361. [DOI] [PubMed] [Google Scholar]
- 94.Kauvar DS, Holcomb JB, Norris GC, Hess JR. Fresh whole blood transfusion: a controversial military practice. J Trauma 61: 181–184, 2006. doi: 10.1097/01.ta.0000222671.84335.64. [DOI] [PubMed] [Google Scholar]
- 95.Repine TB, Perkins JG, Kauvar DS, Blackborne L. The use of fresh whole blood in massive transfusion. J Trauma 60: S59–S69, 2006. doi: 10.1097/01.ta.0000219013.64168.b2. [DOI] [PubMed] [Google Scholar]
- 96.McCoy CC, Brenner M, Duchesne J, Roberts D, Ferrada P, Horer T, Kauvar D, Khan M, Kirkpatrick A, Ordonez C, Perreira B, Roberts D, Priouzram A, Cotton BA. Back to the future: whole blood resuscitation of the severely injured trauma patient. Shock 56: 9–15, 2021. doi: 10.1097/SHK.0000000000001685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Fisher AD, Miles EA, Broussard MA, Corley JB, Knight R, Remley MA, Cap AP, Gurney JM, Shackelford SA. Low titer group O whole blood resuscitation: military experience from the point of injury. J Trauma Acute Care Surg 89: 834–841, 2020. doi: 10.1097/TA.0000000000002863. [DOI] [PubMed] [Google Scholar]
- 98.Gurney J, Staudt A, Cap A, Shackelford S, Mann-Salinas E, Le T, Nessen S, Spinella P. Improved survival in critically injured combat casualties treated with fresh whole blood by forward surgical teams in Afghanistan. Transfusion 60, Suppl 3: S180–S188, 2020. doi: 10.1111/trf.15767. [DOI] [PubMed] [Google Scholar]
- 99.Zhu CS, Pokorny DM, Eastridge BJ, Nicholson SE, Epley E, Forcum J, Long T, Miramontes D, Schaefer R, Shiels M, Stewart RM, Stringfellow M, Summers R, Winckler CJ, Jenkins DH. Give the trauma patient what they bleed, when and where they need it: establishing a comprehensive regional system of resuscitation based on patient need utilizing cold-stored, low-titer O+ whole blood. Transfusion 59: 1429–1438, 2019. doi: 10.1111/trf.15264. [DOI] [PubMed] [Google Scholar]
- 100.Sperry JL, Guyette FX, Brown JB, Yazer MH, Triulzi DJ, Early-Young BJ, Adams PW, Daley BJ, Miller RS, Harbrecht BG, Claridge JA, Phelan HA, Witham WR, Putnam AT, Duane TM, Alarcon LH, Callaway CW, Zuckerbraun BS, Neal MD, Rosengart MR, Forsythe RM, Billiar TR, Yealy DM, Peitzman AB, Zenati MS; PAMPer Study Group. Prehospital plasma during air medical transport in trauma patients at risk for hemorrhagic shock. N Engl J Med 379: 315–326, 2018. doi: 10.1056/NEJMoa1802345. [DOI] [PubMed] [Google Scholar]
- 101.Moore HB, Moore EE, Chapman MP, McVaney K, Bryskiewicz G, Blechar R, Chin T, Burlew CC, Pieracci F, West FB, Fleming CD, Ghasabyan A, Chandler J, Silliman CC, Banerjee A, Sauaia A. Plasma-first resuscitation to treat haemorrhagic shock during emergency ground transportation in an urban area: a randomised trial. Lancet 392: 283–291, 2018. doi: 10.1016/S0140-6736(18)31553-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Pusateri AE, Moore EE, Moore HB, Le TD, Guyette FX, Chapman MP, Sauaia A, Ghasabyan A, Chandler J, McVaney K, Brown JB, Daley BJ, Miller RS, Harbrecht BG, Claridge JA, Phelan HA, Witham WR, Putnam AT, Sperry JL. Association of prehospital plasma transfusion with survival in trauma patients with hemorrhagic shock when transport times are longer than 20 minutes: a post hoc analysis of the PAMPer and COMBAT Clinical Trials. JAMA Surg 155: e195085, 2020. doi: 10.1001/jamasurg.2019.5085. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Mok G, Hoang R, Khan MW, Pannell D, Peng H, Tien H, Nathens A, Callum J, Karkouti K, Beckett A, da Luz LT. Freeze-dried plasma for major trauma—systematic review and meta-analysis. J Trauma Acute Care Surg 90: 589–602, 2021. doi: 10.1097/TA.0000000000003012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Pusateri AE, Butler FK, Shackelford SA, Sperry JL, Moore EE, Cap AP, Taylor AL, Homer MJ, Hoots WK, Weiskopf RB, Davis MR. The need for dried plasma—a national issue. Transfusion 59: 1587–1592, 2019. doi: 10.1111/trf.15261. [DOI] [PubMed] [Google Scholar]
- 105.Gulati A, Zhang Z, Murphy A, Lavhale MS. Efficacy of centhaquin as a small volume resuscitative agent in severely hemorrhaged rats. Am J Emerg Med 31: 1315–1321, 2013. doi: 10.1016/j.ajem.2013.05.032. [DOI] [PubMed] [Google Scholar]
- 106.Lavhale MS, Havalad S, Gulati A. Resuscitative effect of centhaquin after hemorrhagic shock in rats. J Surg Res 179: 115–124, 2013. doi: 10.1016/j.jss.2012.08.042. [DOI] [PubMed] [Google Scholar]
- 107.Papapanagiotou P, Xanthos T, Gulati A, Chalkias A, Papalois A, Kontouli Z, Alegakis A, Iacovidou N. Centhaquin improves survival in a swine model of hemorrhagic shock. J Surg Res 200: 227–235, 2016. doi: 10.1016/j.jss.2015.06.056. [DOI] [PubMed] [Google Scholar]
- 108.Ranjan AK, Zhang Z, Briyal S, Gulati A. Centhaquine restores renal blood flow and protects tissue damage after hemorrhagic shock and renal ischemia. Front Pharmacol 12: 616253, 2021. doi: 10.3389/fphar.2021.616253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Gulati A, Jain D, Agrawal N, Rahate P, Das S, Chowdhuri R, Dhibar D, Prabhu M, Haveri S, Agarwal R, Lavhale M. Clinical Phase II results of PMZ-2010 (centhaquin) as a resuscitative agent for hypovolemic shock. Crit Care Med 47: 12, 2019. doi: 10.1097/01.ccm.0000550815.69306.46. [DOI] [Google Scholar]
- 110.Gulati A, Choudhuri R, Gupta A, Singh S, Ali SK, Sidhu GK, Haque PD, Rahate P, Bothra AR, Singh GP, Maheshwari S, Jeswani D, Haveri S, Agarwal A, Agrawal NR. A multicentric, randomized, controlled phase III study of centhaquine (Lyfaquin®) as a resuscitative agent in hypovolemic shock patients. Drugs 81: 1079–1100, 2021. doi: 10.1007/s40265-021-01547-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Matsen FA 3rd, Wyss CR, King RV, Simmons CW. Effect of acute hemorrhage on transcutaneous, subcutaneous, intramuscular, and arterial oxygen tensions. Pediatrics 65: 881–883, 1980. doi: 10.1542/peds.65.5.881. [DOI] [PubMed] [Google Scholar]
- 112.Wettstein R, Tsai AG, Erni D, Lukyanov AN, Torchilin VP, Intaglietta M. Improving microcirculation is more effective than substitution of red blood cells to correct metabolic disorder in experimental hemorrhagic shock. Shock 21: 235–240, 2004. doi: 10.1097/01.shk.0000114301.36496.ea. [DOI] [PubMed] [Google Scholar]
- 113.Parrish D, Plant V, Lindell SL, Limkemann A, Reichstetter H, Aboutanos M, Mangino MJ. New low-volume resuscitation solutions containing PEG-20k. J Trauma Acute Care Surg 79: 22–29, 2015. doi: 10.1097/TA.0000000000000682. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Plant V, Limkemann A, Liebrecht L, Blocher C, Ferrada P, Aboutanos M, Mangino MJ. Low-volume resuscitation using polyethylene glycol-20k in a preclinical porcine model of hemorrhagic shock. J Trauma Acute Care Surg 81: 1056–1062, 2016. doi: 10.1097/TA.0000000000001155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Wickramaratne N, Kenning K, Reichstetter H, Blocher C, Li R, Aboutanos M, Mangino MJ. Acute resuscitation with polyethylene glycol-20k: a thromboelastographic analysis. J Trauma Acute Care Surg 87: 322–330, 2019. doi: 10.1097/TA.0000000000002332. [DOI] [PubMed] [Google Scholar]
- 116.Holland RA, Shibata H, Scheid P, Piiper J. Kinetics of O2 uptake and release by red cells in stopped-flow apparatus: effects of unstirred layer. Respir Physiol 59: 71–91, 1985. doi: 10.1016/0034-5687(85)90020-9. [DOI] [PubMed] [Google Scholar]
- 117.Huxley VH, Kutchai H. The effect of the red cell membrane and a diffusion boundary layer on the rate of oxygen uptake by human erythrocytes. J Physiol 316: 75–83, 1981. doi: 10.1113/jphysiol.1981.sp013773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Laidig KE, Daggett V, Gainer JL. Altering diffusivity in biological solutions through modification of solution structure and dynamics. J Am Chem Soc 120: 9394–9395, 1998. doi: 10.1021/ja981656j. [DOI] [Google Scholar]
- 119.Gainer JL. Trans-Sodium Crocetinate, Methods of Making and Methods of USE THEREOF. US Patent US6060511A. May 9, 2000.
- 120.Gainer JL, Sheehan JP, Larner JM, Jones DR. Trans sodium crocetinate with temozolomide and radiation therapy for glioblastoma multiforme. J Neurosurg 126: 460–466, 2017. doi: 10.3171/2016.3.JNS152693. [DOI] [PubMed] [Google Scholar]
- 121.Singer M, Stidwill RP, Nathan A, Gainer JL. Intravenous crocetinate prolongs survival in a rat model of lethal hypoxemia. Crit Care Med 28: 1972–2000, 1968. [DOI] [PubMed] [Google Scholar]
- 122.Stennett AK, Murray RJ, Roy JW, Gainer JL. Trans-sodium crocetinate and hemorrhagic shock. Shock 28: 339–344, 2007. doi: 10.1097/shk.0b013e3180487b2d. [DOI] [PubMed] [Google Scholar]
- 123.Brunelle JK, Bell EL, Quesada NM, Vercauteren K, Tiranti V, Zeviani M, Scarpulla RC, Chandel NS. Oxygen sensing requires mitochondrial ROS but not oxidative phosphorylation. Cell Metab 1: 409–414, 2005. doi: 10.1016/j.cmet.2005.05.002. [DOI] [PubMed] [Google Scholar]
- 124.Chandel NS, Maltepe E, Goldwasser E, Mathieu CE, Simon MC, Schumacker PT. Mitochondrial reactive oxygen species trigger hypoxia-induced transcription. Proc Natl Acad Sci USA 95: 11715–11720, 1998. doi: 10.1073/pnas.95.20.11715. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Brand MD, Goncalves RL, Orr AL, Vargas L, Gerencser AA, Borch Jensen M, Wang YT, Melov S, Turk CN, Matzen JT, Dardov VJ, Petrassi HM, Meeusen SL, Perevoshchikova IV, Jasper H, Brookes PS, Ainscow EK. Suppressors of superoxide-H2O2 production at site IQ of mitochondrial complex I protect against stem cell hyperplasia and ischemia-reperfusion injury. Cell Metab 24: 582–592, 2016. doi: 10.1016/j.cmet.2016.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Orr AL, Vargas L, Turk CN, Baaten JE, Matzen JT, Dardov VJ, Attle SJ, Li J, Quackenbush DC, Goncalves RL, Perevoshchikova IV, Petrassi HM, Meeusen SL, Ainscow EK, Brand MD. Suppressors of superoxide production from mitochondrial complex III. Nat Chem Biol 11: 834–836, 2015. doi: 10.1038/nchembio.1910. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Watson MA, Wong HS, Brand MD. Use of S1QELs and S3QELs to link mitochondrial sites of superoxide and hydrogen peroxide generation to physiological and pathological outcomes. Biochem Soc Trans 47: 1461–1469, 2019. doi: 10.1042/BST20190305. [DOI] [PubMed] [Google Scholar]
- 128.Bell EL, Klimova TA, Eisenbart J, Moraes CT, Murphy MP, Budinger GR, Chandel NS. The Qo site of the mitochondrial complex III is required for the transduction of hypoxic signaling via reactive oxygen species production. J Cell Biol 177: 1029–1036, 2007. doi: 10.1083/jcb.200609074. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Guzy RD, Hoyos B, Robin E, Chen H, Liu L, Mansfield KD, Simon MC, Hammerling U, Schumacker PT. Mitochondrial complex III is required for hypoxia-induced ROS production and cellular oxygen sensing. Cell Metab 1: 401–408, 2005. doi: 10.1016/j.cmet.2005.05.001. [DOI] [PubMed] [Google Scholar]
- 130.Szeto HH. First-in-class cardiolipin-protective compound as a therapeutic agent to restore mitochondrial bioenergetics. Br J Pharmacol 171: 2029–2050, 2014. doi: 10.1111/bph.12461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hazeldine J, Naumann DN, Toman E, Davies D, Bishop JR, Su Z, Hampson P, Dinsdale RJ, Crombie N, Duggal NA, Harrison P, Belli A, Lord JM. Prehospital immune responses and development of multiple organ dysfunction syndrome following traumatic injury: a prospective cohort study. PLoS Med 14: e1002338, 2017. doi: 10.1371/journal.pmed.1002338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Hoyer AA, Klaeske K, Garnham J, Kiefer P, Salameh A, Witte K, Borger M, Dieterlen MT. Cyclosporine A-enhanced cardioplegia preserves mitochondrial basal respiration after ischemic arrest. Perfusion 2021: 026765912110257, 2021. doi: 10.1177/02676591211025746. [DOI] [PubMed] [Google Scholar]
- 133.Schnichels S, Schultheiss M, Klemm P, Blak M, Herrmann T, Melchinger M, Bartz-Schmidt KU, Löscher M, Zeck G, Spitzer MS, Hurst J. Cyclosporine A protects retinal explants against hypoxia. Int J Mol Sci 22: 10196, 2021. doi: 10.3390/ijms221910196. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Bellamy R, Safar P, Tisherman SA, Basford R, Bruttig SP, Capone A, Dubick MA, Ernster L, Hattler BG Jr, Hochachka P, Klain M, Kochanek PM, Kofke WA, Lancaster JR, McGowan FX Jr, Oeltgen PR, Severinghaus JW, Taylor MJ, Zar H. Suspended animation for delayed resuscitation. Crit Care Med 24: S24–S47, 1996. [PubMed] [Google Scholar]
- 135.Wolf A, Lusczek ER, Beilman GJ. Hibernation-Based approaches in the treatment of hemorrhagic shock. Shock 50: 14–23, 2018. doi: 10.1097/SHK.0000000000001094. [DOI] [PubMed] [Google Scholar]
- 136.Perez de Lara Rodriguez CE, Drewes LR, Andrews MT. Hibernation-based blood loss therapy increases survivability of lethal hemorrhagic shock in rats. J Comp Physiol B 187: 769–778, 2017. doi: 10.1007/s00360-017-1076-7. [DOI] [PubMed] [Google Scholar]
- 137.Dirkes MC, van Gulik TM, Heger M. The physiology of artificial hibernation. J Clin Transl Res 1: 78–93, 2015. [PMC free article] [PubMed] [Google Scholar]
- 138.Defense Advanced Research Projects Agency (DARPA). Slowing biological time to extend the Golden Hour for lifesaving treatment. https://www.darpa.mil/news-events/2018-03-01. [2021 Jun 25].
- 139.Roy M, Burggraf M, Lendemans S, de Groot H, Rohrig R. Tranexamic acid prolongs survival after controlled hemorrhage in rats. J Surg Res 208: 104–110, 2017. doi: 10.1016/j.jss.2016.09.023. [DOI] [PubMed] [Google Scholar]
- 140.Guyette FX, Brown JB, Zenati MS, Early-Young BJ, Adams PW, Eastridge BJ, Nirula R, Vercruysse GA, O’Keeffe T, Joseph B, Alarcon LH, Callaway CW, Zuckerbraun BS, Neal MD, Forsythe RM, Rosengart MR, Billiar TR, Yealy DM, Peitzman AB, Sperry JL; STAAMP Study Group. Tranexamic acid during prehospital transport in patients at risk for hemorrhage after injury: a double-blind, placebo-controlled, randomized clinical trial. JAMA Surg 156: 11–20, 2020. doi: 10.1001/jamasurg.2020.4350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 141.Morrison JJ, Dubose JJ, Rasmussen TE, Midwinter MJ. Military Application of Tranexamic Acid in Trauma Emergency Resuscitation (MATTERs) Study. Arch Surg 147: 113–119, 2012. doi: 10.1001/archsurg.2011.287. [DOI] [PubMed] [Google Scholar]
- 142.Wafaisade A, Lefering R, Bouillon B, Böhmer AB, Gäßler M, Ruppert M; TraumaRegister DGU. Prehospital administration of tranexamic acid in trauma patients. Crit Care 20: 143, 2016. doi: 10.1186/s13054-016-1322-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Russo R, Kemp M, Bhatti UF, Pai M, Wakam G, Biesterveld B, Alam HB. Life on the battlefield: valproic acid for combat applications. J Trauma Acute Care Surg 89: S69–S76, 2020. doi: 10.1097/TA.0000000000002721. [DOI] [PubMed] [Google Scholar]
- 144.Li Y, Liu B, Sailhamer EA, Yuan Z, Shults C, Velmahos GC, deMoya M, Shuja F, Butt MU, Alam HB. Cell protective mechanism of valproic acid in lethal hemorrhagic shock. Surgery 144: 217–224, 2008. doi: 10.1016/j.surg.2008.03.037. [DOI] [PubMed] [Google Scholar]
- 145.Hwabejire JO, Lu J, Liu B, Li Y, Halaweish I, Alam HB. Valproic acid for the treatment of hemorrhagic shock: a dose-optimization study. J Surg Res 186: 363–370, 2014. doi: 10.1016/j.jss.2013.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 146.Georgoff PE, Nikolian VC, Higgins G, Chtraklin K, Eidy H, Ghandour MH, Williams A, Athey B, Alam HB. Valproic acid induces prosurvival transcriptomic changes in swine subjected to traumatic injury and hemorrhagic shock. J Trauma Acute Care Surg 84: 642–649, 2018. doi: 10.1097/TA.0000000000001763. [DOI] [PubMed] [Google Scholar]
- 147.Biesterveld BE, Williams AM, Kemp MT, Wakam GK, Siddiqui AZ, O’Connell RL, Shamshad A, Chtraklin K, Bhatti UF, Li Y, Alam HB. Valproic acid decreases resuscitation requirements after hemorrhage in a prolonged damage-control resuscitation model. J Trauma Acute Care Surg 89: 752–760, 2020. doi: 10.1097/TA.0000000000002876. [DOI] [PubMed] [Google Scholar]
- 148.Biesterveld BE, Williams AM, Pai MP, Dennahy IS, Graham NJ, Chtraklin K, Siddiqui AZ, O’Connell RL, Bhatti UF, Liu B, Russo RM, Li Y, Alam HB. Dose optimization of valproic acid in a lethal model of traumatic brain injury, hemorrhage, and multiple trauma in swine. J Trauma Acute Care Surg 87: 1133–1139, 2019. doi: 10.1097/TA.0000000000002460. [DOI] [PubMed] [Google Scholar]
- 149.Williams AM, Bhatti UF, Biesterveld BE, Graham NJ, Chtraklin K, Zhou J, Dennahy IS, Kathawate RG, Vercruysse CA, Russo RM, Li Y, Alam HB. Valproic acid improves survival and decreases resuscitation requirements in a swine model of prolonged damage control resuscitation. J Trauma Acute Care Surg 87: 393–401, 2019. doi: 10.1097/TA.0000000000002281. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 150.Lei Y, Peng X, Liu L, Dong Z, Li T. Beneficial effect of cyclosporine A on traumatic hemorrhagic shock. J Surg Res 195: 529–540, 2015. doi: 10.1016/j.jss.2015.02.005. [DOI] [PubMed] [Google Scholar]
- 151.Gurll NJ, Reynolds DG, Holaday JW. Evidence for a role of endorphins in the cardiovascular pathophysiology of primate shock. Crit Care Med 16: 521–530, 1988. doi: 10.1097/00003246-198805000-00011. [DOI] [PubMed] [Google Scholar]
- 152.Gurll NJ, Reynolds DG, Vargish T, Lechner R. Naloxone without transfusion prolongs survival and enhances cardiovascular function in hypovolemic shock. J Pharmacol Exp Ther 220: 621–624, 1982. [PubMed] [Google Scholar]
- 153.Reynolds DG, Gurll NJ, Holaday JW, Lechner RB. The therapeutic efficacy of opiate antagonists in hemorrhagic shock. Resuscitation 18: 243–251, 1989. doi: 10.1016/0300-9572(89)90026-9. [DOI] [PubMed] [Google Scholar]
- 154.Tuggle DW, Horton JW. Effects of naloxone on splanchnic perfusion in hemorrhagic shock. J Trauma 29: 1341–1345, 1989. doi: 10.1097/00005373-198910000-00008. [DOI] [PubMed] [Google Scholar]
- 155.Weld JM, Kamerling SG, Combie JD, Nugent TE, Woods WE, Oeltgen P, Tobin T. The effects of naloxone on endotoxic and hemorrhagic shock in horses. Res Commun Chem Pathol Pharmacol 44: 227–238, 1984. [PubMed] [Google Scholar]
- 156.Boeuf B, Poirier V, Gauvin F, Guerguerian AM, Roy C, Farrell CA, Lacroix J. Naloxone for shock. Cochrane Database Syst Rev 4: CD004443, 2003. doi: 10.1002/14651858.CD004443. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Mulier KE, Lexcen DR, Luzcek E, Greenberg JJ, Beilman GJ. Treatment with beta-hydroxybutyrate and melatonin is associated with improved survival in a porcine model of hemorrhagic shock. Resuscitation 83: 253–258, 2012. doi: 10.1016/j.resuscitation.2011.08.003. [DOI] [PubMed] [Google Scholar]
- 158.Wolf A, Thakral S, Mulier KE, Suryanarayanan R, Beilman GJ. Evaluation of novel formulations of d-beta-hydroxybutyrate and melatonin in a rat model of hemorrhagic shock. Int J Pharm 548: 104–112, 2018. doi: 10.1016/j.ijpharm.2018.06.046. [DOI] [PubMed] [Google Scholar]
- 159.Klein AH, Wendroth SM, Drewes LR, Andrews MT. Small-volume d-beta-hydroxybutyrate solution infusion increases survivability of lethal hemorrhagic shock in rats. Shock 34: 565–572, 2010. doi: 10.1097/SHK.0b013e3181e15063. [DOI] [PubMed] [Google Scholar]
- 160.Chen CH, Tsai PS, Huang CJ. Minocycline ameliorates lung and liver dysfunction in a rodent model of hemorrhagic shock/resuscitation plus abdominal compartment syndrome. J Surg Res 180: 301–309, 2013. doi: 10.1016/j.jss.2012.04.036. [DOI] [PubMed] [Google Scholar]
- 161.Kholmukhamedov A, Czerny C, Hu J, Schwartz J, Zhong Z, Lemasters JJ. Minocycline and doxycycline, but not tetracycline, mitigate liver and kidney injury after hemorrhagic shock/resuscitation. Shock 42: 256–263, 2014. doi: 10.1097/SHK.0000000000000213. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 162.Blears EE, Carson JS, Finnerty CC, Ross E, Sommerhalder C, Herndon DN. Metabolism in surgical patients. In: Sabiston Textbook of Surgery, The Biological Basis of Modern Surgical Practice (21st ed.), edited by Townsend CM, Evers BM, Beauchamp RD, Mattox KL.. St. Louis, MO: Elsevier, 2021, p. 95–118. [Google Scholar]