

Abstract
Arrestin binding to active phosphorylated G protein-coupled receptors terminates G protein coupling and initiates another wave of signaling. Among the effectors that bind directly to receptor-associated arrestins are extracellular signal-regulated kinases 1/2 (ERK1/2), which promote cellular proliferation and survival. Arrestins may also engage ERK1/2 in isolation in a pre- or post-signaling complex that is likely in equilibrium with the full signal initiation complex. Molecular details of these binary complexes remain unknown. Here, we investigate the molecular mechanisms whereby arrestin-2 and arrestin-3 (a.k.a. β-arrestin1 and β-arrestin2, respectively) engage ERK1/2 in pairwise interactions. We find that purified arrestin-3 binds ERK2 more avidly than arrestin-2. A combination of biophysical techniques and peptide array analysis demonstrates that the molecular basis in this difference of binding strength is that the two non-visual arrestins bind ERK2 via different parts of the molecule. We propose a structural model of the ERK2- arrestin-3 complex in solution using size-exclusion chromatography coupled to small angle X-ray scattering (SEC-SAXS). This binary complex exhibits conformational heterogeneity. We speculate that this drives the equilibrium either toward the full signaling complex with receptor- bound arrestin at the membrane or toward full dissociation in the cytoplasm. As ERK1/2 regulates cell migration, proliferation, and survival, understanding complexes that relate to its activation could be exploited to control cell fate.
Keywords: arrestin, extracellular signal-regulated kinase 2, protein-protein interactions, protein scaffolds, small-angle X-ray scattering
Graphical Abstract
INTRODUCTION
Collectively, the two ubiquitous non-visual arrestins, arrestin-2 and arrestin-31, interact with at least 300 different proteins including more than 100 overlapping effectors and trafficking proteins (1). These interaction partners include clathrin and AP-2 (2,3), Src family kinases (4–6), leucine zipper kinases (7) and members of mitogen-activated protein kinase (MAPK) cascades (8–12). Among the best characterized arrestin effectors is extracellular signal-regulated kinase 1/2 (ERK1/2) (13), which is a MAPK. GPCR-dependent ERK1/2 activation is most commonly G protein dependent (14,15), but may also be arrestin-dependent (13,16,17) or arrestin-enhanced (18) depending upon the identity of the receptor-agonist pair and the arrestin isoform. ERK1/2 is also activated by other routes including upstream receptor tyrosine kinases (19). As the ERK1/2 cascade is implicated in cell migration, proliferation, and survival (20,21), modulation of ERK1/2 activation can potentially be used to control cell fate, warranting a deeper understanding of each activation pathway.
The two non-visual arrestins are ubiquitously expressed across many tissue types (22) and exhibit structural similarity (23,24). These two arrestins have significant functional overlap (25), which is not surprising given that they share 78% sequence identity (26,27). This raises the question of why vertebrates retained two non-visual arrestin subtypes for >400 million years of evolution (28). Considerable effort has been made to determine whether there are differences between the two arrestin isoforms. This includes studies that used siRNA knockdown of individual non-visual arrestins (29) and isoform-selective knockout mice (30,31).
Notably, sequence differences between arrestin-2 and arrestin-3 are more prevalent in the predicted effector engagement sites and the loops between structural elements. Recent work to probe whether these could underlie functional specialization showed that the absence of arrestin- 2-specific eight amino acids in an arrestin-3 loop likely allows the abundant cytoplasmic metabolite IP6 to induce the formation of arrestin-3, but not arrestin-2, trimer where each protomer is in receptor bound-like conformation (32). This ‘active’ conformation is believed to be required for the activation of many inducible signaling cascades.
Although some functional specialization of the arrestin isoforms has been established (3,33), ERK1/2 signaling appears to be facilitated by both isoforms (9,13,34–38). Interestingly, the role of arrestins in ERK1/2 activation is not consistent in various reports. Studies using distinct receptor-ligand combinations find that ERK1/2 activation can be arrestin-dependent (13,16,17), arrestin-independent (14,15), or arrestin-enhanced (18). While this has created much controversy in the field, the most parsimonious conclusion is that arrestin-dependent scaffolding and signal initiation depends on the receptor-ligand combination and the arrestin isoform, likely dictated by expression levels of these components in different cell types (29,39,40). However, more evidence is needed to support this idea as it is not clear how each of the two arrestin isoforms engage ERK1/2.
Here, we used the binary arrestin-ERK2 interaction to approach the issue of functional specialization of the non-visual arrestins. We analyzed the interaction sites between the arrestins and ERK2 employing a combination of biochemical, biophysical, and structural methods. These data show that arrestin-3 binds ERK2 more avidly and indicate that differences in the binding mode of ERK2 between the two isoforms constitute the structural basis of differential engagement. We also propose a model of the arrestin-3-ERK2 binary complex generated from SEC-SAXS data. Collectively, our data indicate that the two arrestins form distinct complexes with ERK2, suggesting different mechanisms of scaffolding ERK2 activation cascade by the two isoforms.
RESULTS
Purified arrestin-2 and −3 bind to purified inactive ERK2.
For initial screening of the arrestin-ERK2 interaction, we assessed whether MBP fusions of arrestin-2 and arrestin-3 directly bind purified, inactive ERK2 using an in vitro pull-down assay (Fig. 1, Supplementary Fig. S1). We immobilized MBP-fusions of arrestin-2/3 on amylose resin and used ERK2 as a prey protein. We detected statistically significant binding of ERK2 to both full-length arrestin-2 and arrestin-3. This suggests that ERK2 can interact with the non-visual arrestins in the absence of an activated receptor. In cells, we speculate that this binary interaction would form an intermediate toward formation or dissociation of a signal initiation complex that minimally contains the agonist-stimulated GPCR, the cellular membrane, an arrestin, the upstream kinases in the ERK2 activation cascade c-Raf1 and MEK1, and ERK2. Such a complex may also contain G protein and/or other trans-activating signaling proteins.
Figure 1. Purified nonvisual arrestins bind ERK2.
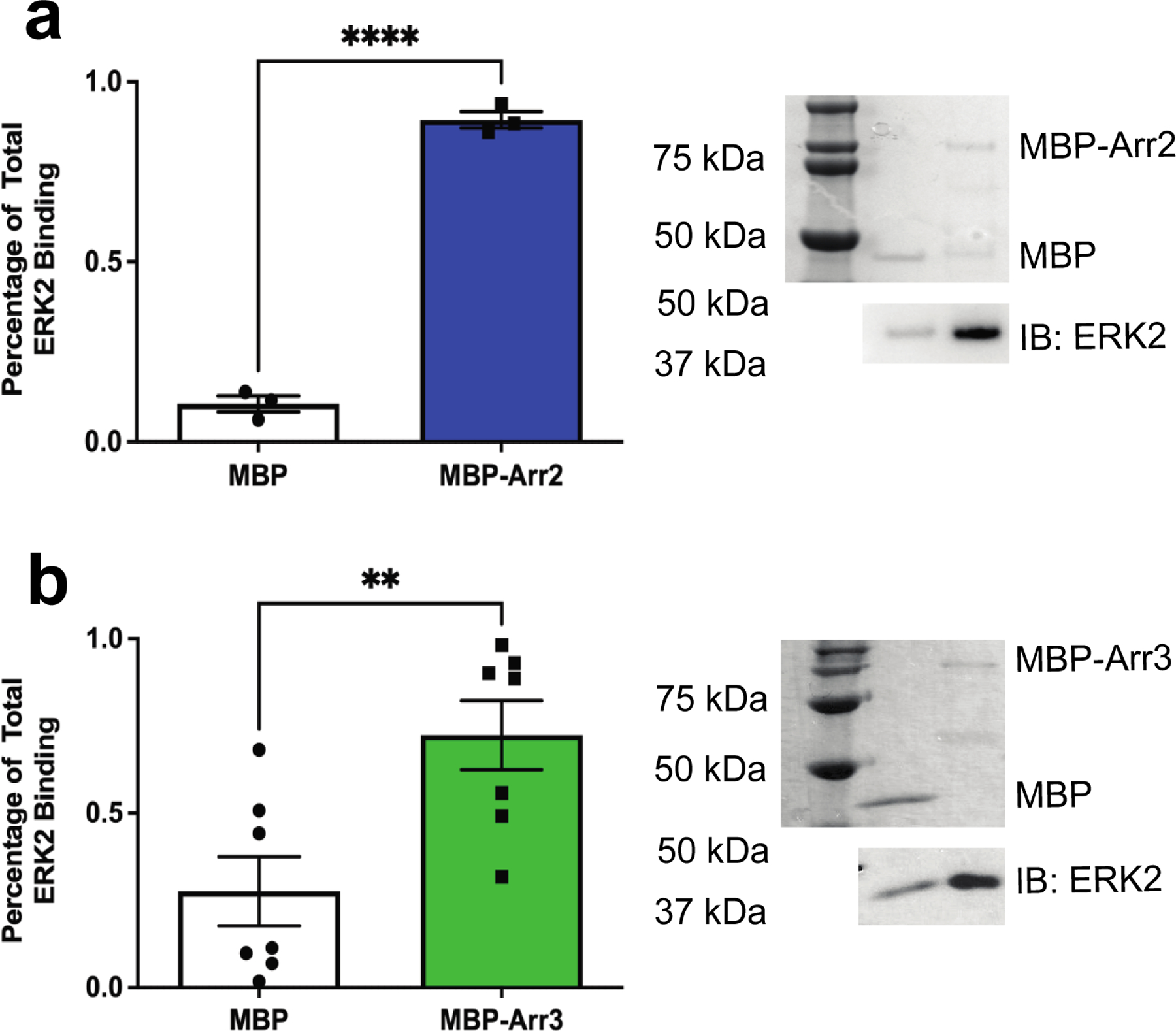
To determine whether the nonvisual arrestins directly interact with ERK2, we performed an in vitro pull-down assay for (a) MBP-arrestin-2 or (b) MBP-arrestin-3. Maltose binding protein (MBP) was used as an affinity tag on the arrestin proteins and MBP served as a negative control. We immobilized 5 μg of MBP or MBP-arrestin-2/3 (“bait” protein) on amylose resin before incubating with 5 μg purified rat ERK2 (“prey” protein). To test that a similar amount of each MBP-tagged protein was immobilized in the samples, we analyzed bound proteins using Coomassie staining after electrophoresis (top). Representative Coomassie stains are shown for both experimental setups and the corresponding lanes are labeled. To measure ERK2 bound to each isoform, we used western analysis (bottom) with anti-HA primary (#9102S, Cell Signaling Technology) and peroxidase-conjugated anti-rabbit secondary antibodies. ERK2 binding was quantified using Quantity One 1-D Analysis software (Bio-Rad Laboratories Inc., CA). Binding is reported as a percentage of total ERK2 and statistical analysis was performed using an unpaired Student’s t-test (N=3–7;**, p<0.05).
Arrestin-2/3-derived peptides that bind inactive ERK2.
To probe the interaction interface between the non-visual arrestins and ERK2 we used peptide array analysis, a method that allows parallel screening of hundreds of peptides (41). We screened peptides corresponding to the non-receptor-binding surface of arrestin-2/3 for ERK2 interaction. Complete lists of the screened peptides derived from arrestin-2 (Supplementary Table S1) and arrestin-3 (Supplementary Table S2) are shown in the supplement.
Over 100 non-visual arrestin peptides were assayed for interaction with ERK2 using far western analysis (Supplementary Fig. S2). Interaction intensity (high/low) was calculated as a percentage of total binding measured by densitometry. Surprisingly, the resulting heat maps for arrestin-2 and arrestin-3 revealed substantial differences. For arrestin-2, the peptides that interacted with ERK2 were derived from the first 35 amino acids of B1, the B4 region, and the B5 region (Fig. 2a). Mapping of these peptides onto the arrestin-2 crystal structure (PDB 1G4M (42)) revealed that if each of these elements interacts with ERK2 in the context of the folded protein, the ERK2 binding site would span both domains of arrestin-2 (Fig. 2b), in agreement with previous findings (35).
Figure 2. ERK2 binds differently to arrestin-2- and arrestin-3-derived peptides.
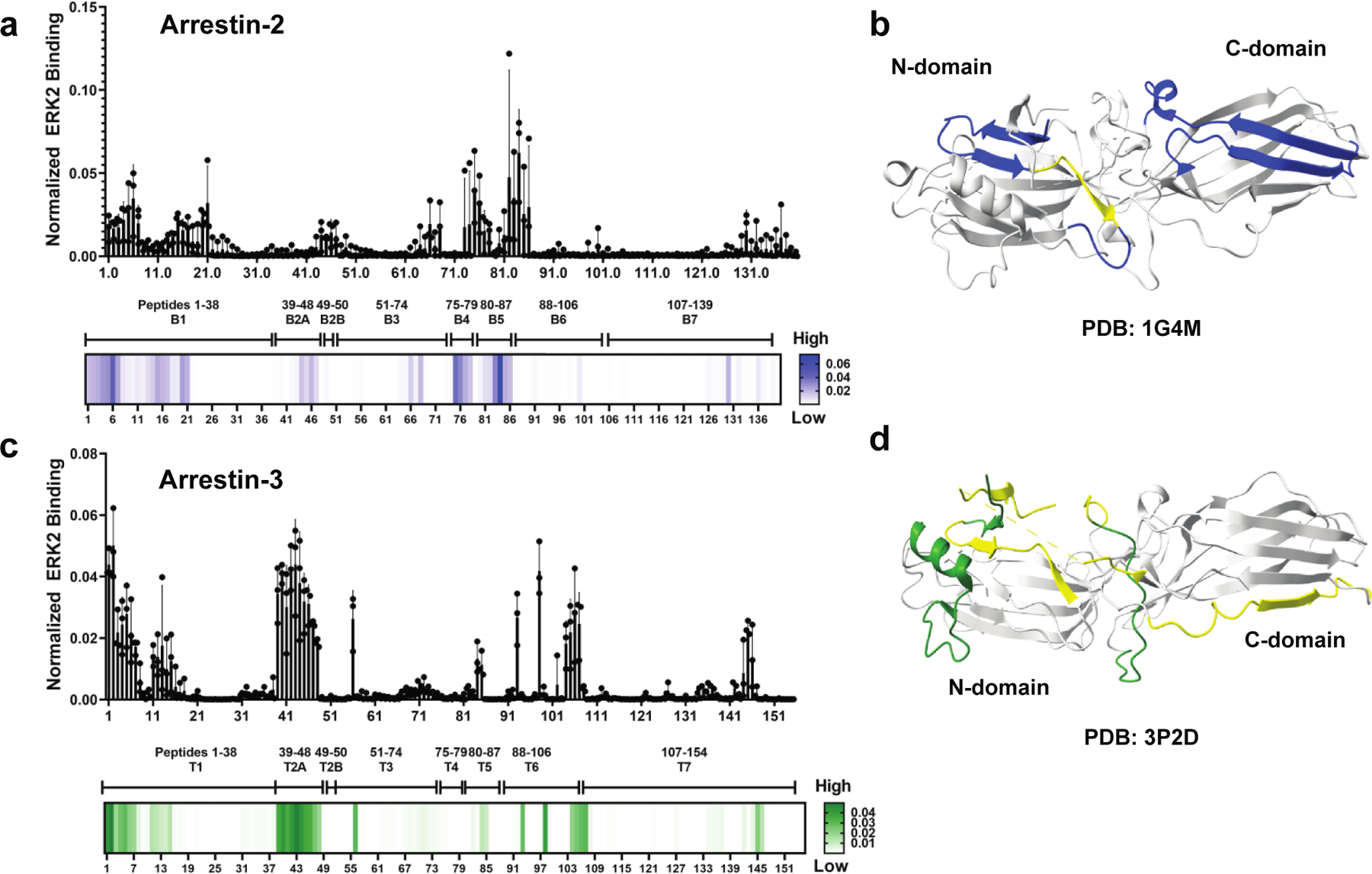
Peptides of the non-receptor-binding surface of arrestin-2 or arrestin-3 were synthesized as 15-mers using ResPep SL (Intavis Bioanalytical Instruments AG, Cologne, Germany) (1 amino acid shifts). Representative peptide arrays are shown in Supplementary Fig. S2. Quantification of ERK2 binding to the (a) arrestin-2- or (c) arrestin-3-derived peptides was performed using the peptide array analyzer on ImageJ (1) (n=3). A heat map using a single gradient to display ranges of binding was calculated using GraphPad Prism 8.0.2. ERK2 binding elements mapped on (b) arrestin-2 (PDB 1G4M (42)) and (d) arrestin-3 (PDB 3P2D (23)) crystal structures. Conditional formatting in Excel was used to detect peptides that exhibited moderate to strong binding to ERK2 (defined as >10% of the intensities). For arrestin-2, moderate binding is shown in yellow, strong binding is shown in blue. For arrestin-3, moderate binding is shown in yellow, strong binding is shown in forest green. The peptide with the highest binding for arrestin-2 was CKVYTLTPFLANNRE, while the peptide with the highest binding for arrestin-3 was the N-terminal, MGEKPGTRVFKKSSPN. Peptide residues are listed in Table S1 and Table S2.
In contrast to the arrestin-2-derived peptides, peptides corresponding to the T4 and T5 regions of arrestin-3 did not interact strongly with ERK2 (Fig. 2c). Instead, the highest intensity was observed for peptides containing 29 N-terminal amino acids (T1), peptides from T2A (aSwI (43)), and several fragments from the end of T6 (aSwIII (43)) to the beginning of T7 (peptides 98–107, residues T299-V362). As with arrestin-2, these sequences represent both domains of arrestin-3; however, most of the high-intensity regions corresponded to the N-domain (Fig. 2d). Collectively, these data show that the peptide sequences derived from homologous regions of the two non-visual arrestins interact with ERK2 differently.
Isoform differences in ERK2 binding in the context of folded proteins.
Peptide array analyses can yield false positives as many isolated peptides exhibit non-specific binding. Array analyses can also yield false negatives when binding is via a surface formed by non-contiguous regions of sequence. To validate the peptide array results in the context of folded proteins, we created arrestin-2/3 chimeras that swapped the binding regions predicted by the arrays and compared the binding of wild-type and chimeric arrestin-2 and arrestin-3 to ERK2 in cells (Fig. 3, Table S3).
Figure 3. Element-swapped chimeras of arrestin-2 and −3 affect binding affinity to ERK2.
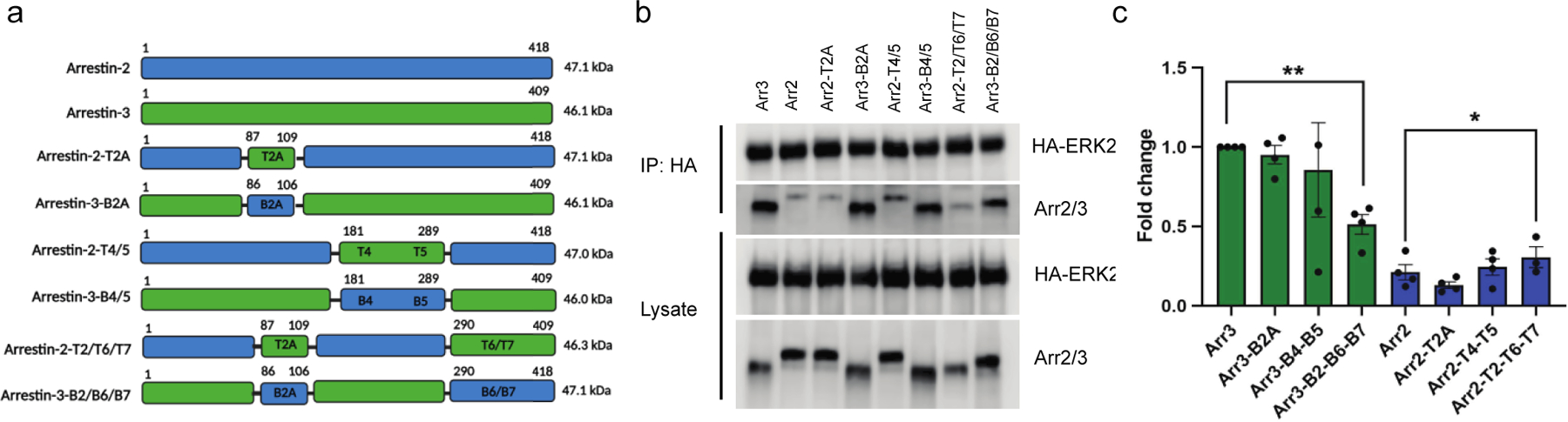
(a) Diagrammatic representation of various arrestin-2 and arrestin-3 chimeras with their anticipated molecular weights. Image made using Biorender.com (b) Coimmunoprecipitation detects the interactions of ERK2 and various arrestin-2/3 chimeras in a cellular context using the lysate of HEK293 arr2/3 KO cells co-expressing HA-ERK2 with wild-type and chimeric arrestins. A representative image from four independent experiments is shown. (c) Densitometry-based quantification of the immunoprecipitation. For the quantification, we used the signal of wild-type arrestin-3 as 100% ERK2 binding to show how binding compared between arrestin-2 and arrestin-3. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s multiple comparisons test (*, p<0.05).
We first compared binding of the wild-type arrestins by co-transfecting HEK293 arrestin-2 and −3 knockout cells with wild type arrestin-2 and arrestin-3 and HA-tagged ERK2. We evaluated binding via immunoprecipitation of the HA-ERK2 and blotting for arrestin. Intriguingly, arrestin-3 appears to interact with ERK2 much more avidly than arrestin-2 (Fig. 3), even though the latter interaction is better supported in the literature (44).
We then evaluated binding of the chimeras using the same experimental strategy. Based on the peptide arrays, we anticipated that arrestin-2-T2A and arrestin-2-T2/T6/T7 would bind ERK2 more strongly than wild-type arrestin-2, and this prediction was confirmed (Fig. 3). We also anticipated that introduction of the T4/T5 segment would decrease ERK2 binding to arrestin-2. We instead found that arrestin-2-T2A and arrestin-2-T4/T5 retained binding to ERK2 that was not statistically different from parental arrestin-2 (Fig. 3), potentially identifying the structurally equivalent T/B2A, T/B4, and T/B5 interactions as false positives in the array analysis.
For the arrestin-3 chimeras, the array data would predict that arrestin-3-B2A and arrestin- 3-B2/B6/B7 would have reduced binding to ERK2, and this was confirmed experimentally (Fig. 3). While peptide arrays predicted that arrestin-3-B2A and arrestin-3-B4/B5 would have increased binding, immunoprecipitation of ERK2 revealed no effect for the arrestin-3-B4/B5 chimeras, while the arrestin-3-B2/B6/B7 chimera demonstrated significant reduction in ERK2 interaction. This again supports the T/B4 and T/B5 interactions as false positives, but makes the role of T/B2A less clear.
Collectively, these data suggest that arrestin-3 binds to ERK2 more avidly than arrestin-2 and support a role for the B2/6/7 and T2/6/7 of the non-visual arrestins in the differential engagement of ERK2 in the binary complex. This finding indicates that ERK2 binding to both non-visual arrestins involves a folded surface, as was previously suggested (35) and also suggests that sequence differences in these regions likely underlie different affinities.
CW-EPR to identify ERK2-binding sites on arrestin-31−393 and arrestin-21–382.
Given the surprising findings that the two highly-homologous non-visual arrestins might engage ERK2 differently, we used an independent method to probe the interfaces involved in ERK2 binding by arrestin-31−393 and arrestin-21–382. We chose these versions of arrestins because they more readily transition into the receptor-bound-like conformation (45,46) and because data from several labs suggest that receptor-bound arrestins have higher affinity for ERK1/2 (13,34). Using cys-less versions of arrestin-2 and arrestin-3 that were previously shown to be functional (47) as base mutants, we introduced cysteines into identical locations and chemically modified these cysteines with a spin label. We then performed continuous wave electron paramagnetic resonance (CW EPR) spectroscopy in the absence or presence of a 2-fold molar excess of ERK2 (Fig. 4). The spectra obtained in the absence and presence of ERK2 were overlaid to reveal changes in spin label mobility induced by ERK2 because the mobility is expected to decrease when a surface label is located at or near the protein interaction interface.
Figure 4. Effects of ERK2 binding to arrestin-21−382 and arrestin-31−393 using CW EPR.
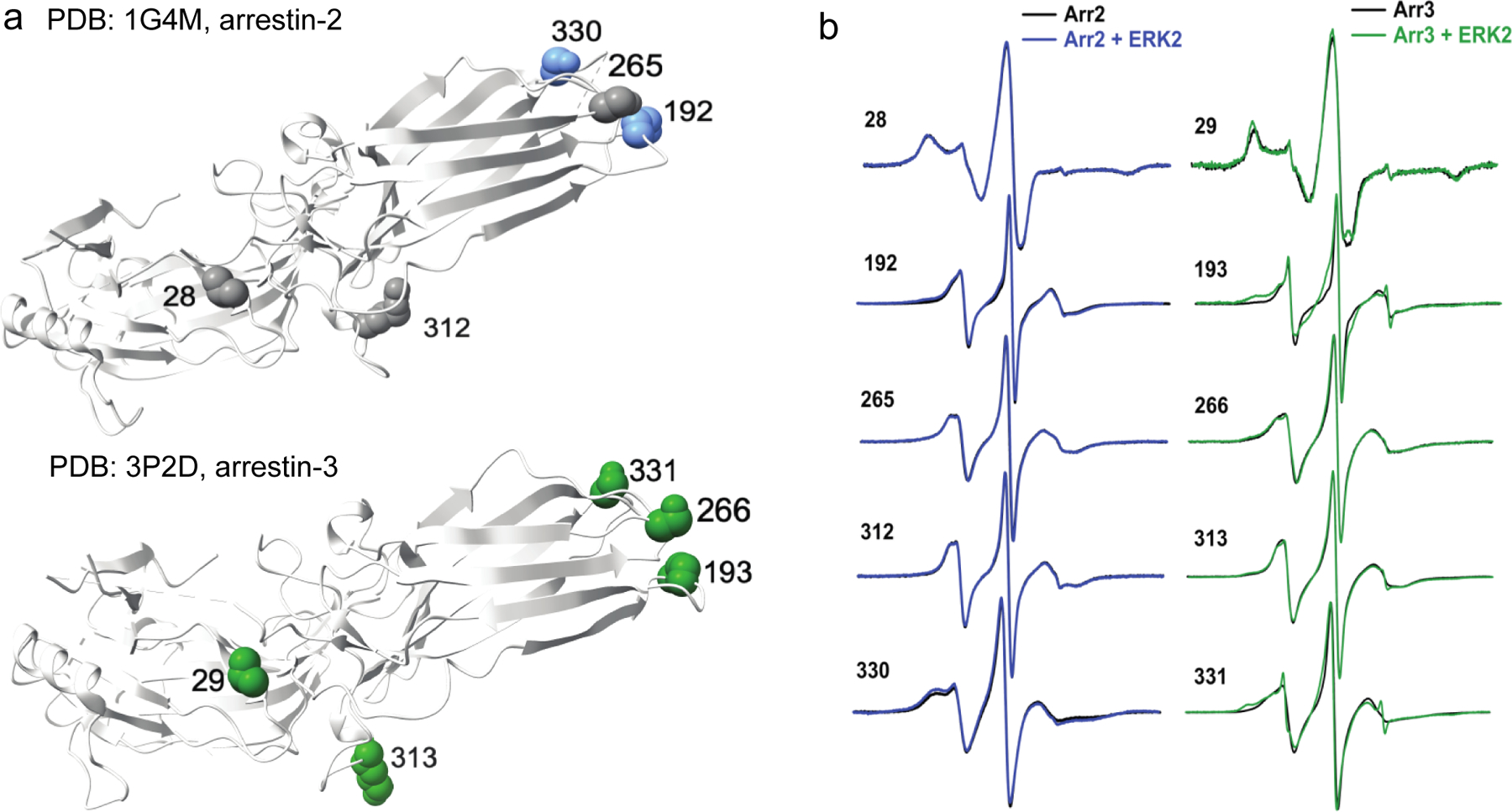
(a) Map of spectral changes upon mixing with ERK2 mapped on the crystal structure of arrestin-2 (PDB 1G4M (42)) or arrestin-3 (PDB 3P2D (23)). Dark green residues (arrestin-3) were found to have medium spectral line shape changes, while blue (arrestin-2) residues were found to have small spectral line shape changes, and residues colored gray had minimal or no changes in their EPR spectra. (b) Overlays of center line height-normalized X-band EPR spectra of 5 sites on arrestin-2 (50 μM) or arrestin-3 (50 μM) in the absence (black) and presence (blue or green, respectively) of unphosphorylated Cys-less ERK2 (100 μM). The complete set of overlays for arrestin-3 is shown in Supplementary Fig. S3.
ERK2-dependent spectral changes in arrestin-3 (Fig. 4a) were clustered on the distal loops of the C-terminal domain (involving M193 (aSwII (43)), S266, and S331), plus a site in the flexible linker region (K313 (aSwIII (43)), and V29 in the N-domain. V29, M193, and K313 agree with interaction sites identified using peptide array analysis (Fig. 2). These medium-to- small spectral changes upon ERK2 addition show that the label at these sites becomes less mobile, which suggests that they either are in or near arrestin elements that directly contact ERK2 or undergo conformational changes upon ERK2 binding. The 43R1, 88R1 (aSwI (43)), and 101R1 sites on arrestin-3 also exhibited small changes upon ERK2 binding. These residues clustered on the N-terminal domain and overlapped with high binding regions identified using peptide array analysis, notably the T2A region (Fig. 2). In contrast, a dozen other sites showed slight or no observable spectral changes, suggesting that these residues are not affected by the arrestin-3-ERK2 interaction (Supplementary Fig. S3). These residues were scattered across arrestin-3. The results generally agreed with little interaction detected by the peptide array analysis. The mobility changes observed by EPR indicate that arrestin-3 sites at the tip of the C-domain, flexible linker, and N-domain are affected by ERK2 binding, which is consistent with a cross-domain interaction. However, these sites were not clustered in such a way as to map a single ERK2 binding site on arrestin-3 definitively. This is not entirely unexpected, as previous studies of arrestin have shown that local conformational changes, such as alterations in the arrestin switch regions, can shift the arrestin equilibrium toward an active conformation (43).
In stark contrast, spectral changes in the same locations were minimal in arrestin-21–382. All homologous sites on arrestin-2 showed only small or no changes upon ERK2 binding (Fig. 4b), which is consistent with the stronger binding of arrestin-3 in the binary complex (Fig. 3) and engagement of different arrestin-2 and −3 elements by bound ERK2 (Fig. 2, Fig. 3). While this result is somewhat surprising, it might reflect the fact that we did not use activator for these studies and that arrestin-2 adopts the active conformation less frequently than arrestin-3 (23,48).
ERK2-derived peptides that bind arrestin-2/3.
We next reversed the peptide array analysis to define the “footprints” of the non-visual arrestins on ERK2. Here, ERK2-derived peptides were synthesized and tested for binding to purified versions of arrestins that lack the C-tail (arrestin-21−382 and arrestin-31−393). We screened over 300 peptides (Supplementary Table S4) that covered the entirety of the ERK2 protein using far western analysis (Supplementary Fig. S4).
Significant differences were observed in the ERK2-derived peptides that interacted with arrestin-21−382 versus arrestin-31–393. Arrestin-21−382 strongly bound just a few ERK2-derived peptides (Supplementary Fig. S5a,c). These include a small region that maps to the end of the N-lobe (peptides 83–90, residues I93-L114) and a segment that maps to the kinase domain (peptides 155–161, residues D165-Y185). Interestingly, the regions of the kinase domain include the TEY phosphorylation motif of the ERK2 activation loop, where dual phosphorylation on both the Thr and Tyr residues by MEK1 is required for full ERK2 activation (49). Models of MAPK activation predict that perturbation of this region could help to facilitate activation (50).
In contrast to arrestin-21−382, arrestin-31−393 bound strongly to numerous ERK2 peptides. These peptides are broadly distributed across the kinase structure and do not make a contiguous binding surface. This potentially suggests that either some of these are non-specific interactions, or that there is more than one mode of purified ERK2 engagement by isolated arrestin-3 in the absence of other components of the signal initiation complex (Supplementary Fig. S5b,c). The strongest interaction was detected with a peptide within the D recruitment site, which mediates ERK binding to upstream kinases. In the case of arrestin-31−393 we did not detect binding to the activation loop of ERK2. Thus, despite high homology between the two non-visual arrestins (26,27), these proteins engage distinct ERK2-derived peptides (Figs. 2 and 3).
Small-angle X-ray scattering identifies a heterogeneous binary complex between ERK2 and arrestin-31–393.
Recently, a model of the arrestin-2-ERK2 complex was reported based on data obtained by hydrogen/deuterium exchange-mass spectrometry, tryptophan-induced bimane fluorescence quenching, and NMR (44). As our data suggest that the arrestin-3-ERK2 binary complex is distinct from that of that of arrestin2-ERK2, we wished to develop a model of arrestin-3-ERK2 for comparison. Toward this goal, we generated fusion constructs of arrestin-21−382 and arrestin-31−393 with ERK2 (arrestin-2-ERK2 or arrestin-3-ERK2) joined with a 12 amino acid linker (GSLEVLFQGPGS). In conjunction with the inherent flexibility of the C-terminus of arrestin and N-terminus of ERK2, we reasoned that this linker would not interfere with the folding of either protein or restrict interaction modes in the binary complex. To test that these proteins folded, we assessed whether ERK2 in this construct can be phosphorylated upon isoproterenol-stimulation of β2-adrenergic receptor in HEK293 arrestin-2/3 knockout cells; this was reported to occur via a G protein-dependent pathway (15)(Fig. 5. Supplementary Figure S6). We found that ERK2 in the arrestin-2/3-ERK2 fusions was robustly phosphorylated even in the absence of receptor agonist, confirming correct folding.
Figure 5. The ERK2 in the arrestin-2-ERK2 and arrestin-3-ERK2 constructs undergo phosphorylation in cells.
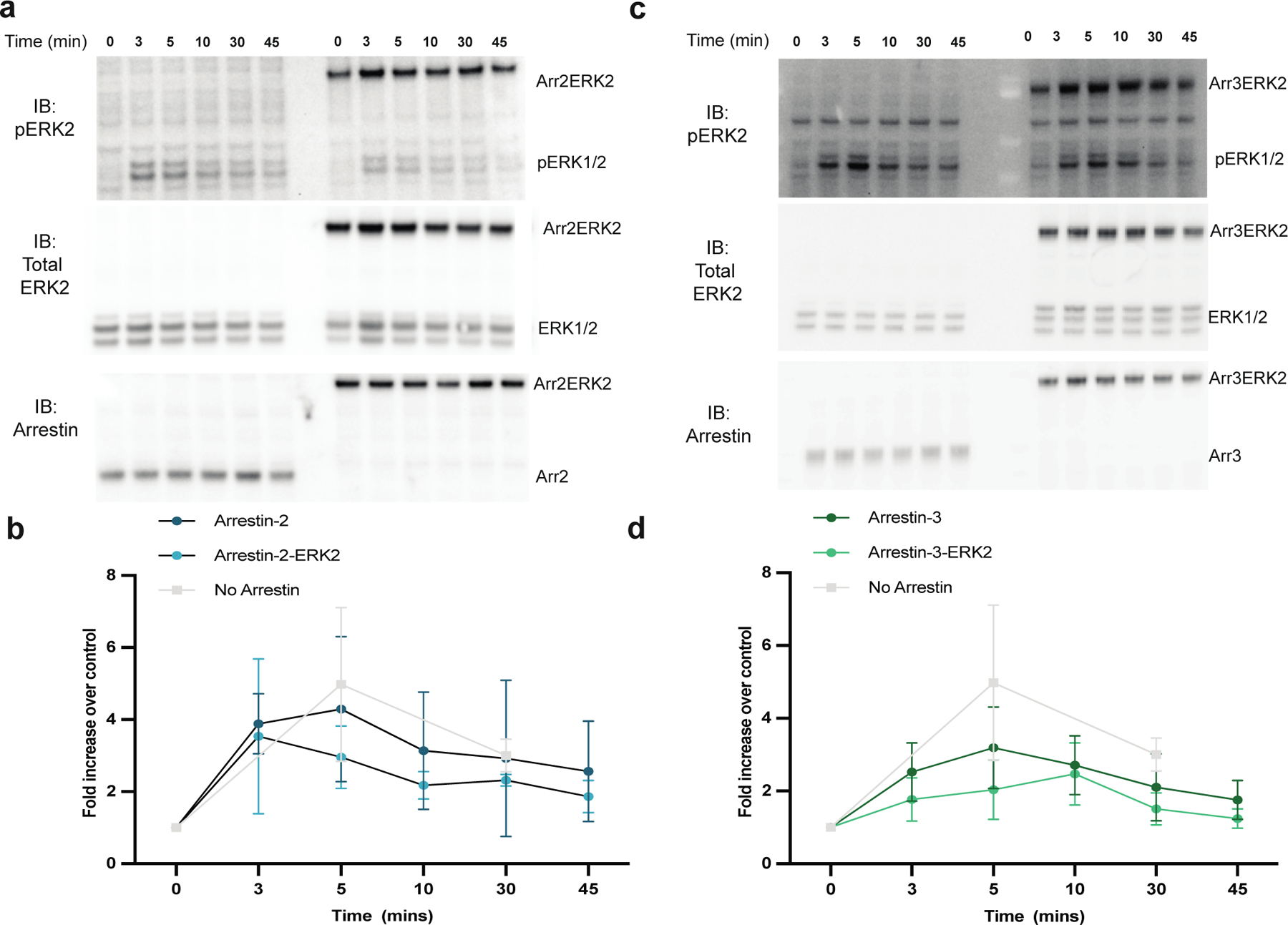
Knockout arrestin-2/3 HEK293 cells were transfected with the β2-adrenergic receptor (0.3 µg) and either free arrestin (arrestin-31−393 or arrestin-21−382) (0.1 µg) or an arrestin-ERK2 fusions (1.5 µg). Respective westerns of each transfection are shown for (a) arrestin-2 and (c) arrestin-3. After 48 hrs, the cells were incubated with 10 µM isoproterenol for indicated times before lysis with 400 µL SDS buffer. Western analysis was performed with antibodies against phosphorylated ERK1/2 (Cell Signaling Technology, #9101, 1:1,000), total ERK1/2 (Cell Signaling Technology, #4695, 1:1,000), or arrestin (F431, 1:5,000). (b,d) ImageJ software was used to measure band intensity of phosphorylated ERK2, which was normalized to total ERK1/2 and shown as fold change relative to the 0 min time point (N=3).
We purified the arrestin-3-ERK2 fusion protein (Supplementary Fig. S7), collected fractions 12–18, and performed SEC-SAXS analysis (Supplementary Table S6), which identified four components (Supplementary Fig. S8). Of these, two components contained the arrestin-3-ERK2 fusion protein (components 2 and 3, colored brown and green, respectively). The other two components (1 and 4) occurred before and after the peak of interest; these were identified as contaminants and excluded (Supplementary Fig. S8, Fig. 6a). The scattering profiles corresponding to the arrestin-3-ERK2 components appeared monodisperse based on Guinier analysis (Fig. 6b). The radius of gyration (Rg) and scattering intensity at zero angle (I(0)) were estimated for both components from the Guinier analysis (Fig. 6b) and P(r) functions (Supplementary Fig. S8, Supplementary Table S5). In both cases the P(r) function showed an extended tail (Supplementary Fig. S9), which could be characteristic of flexibility or of larger contaminants in solution and gave a maximum dimension of 160 Å and 140 Å for components 2 and 3 respectively.
Figure 6. Models of arrestin-3-ERK2 binary complexes.
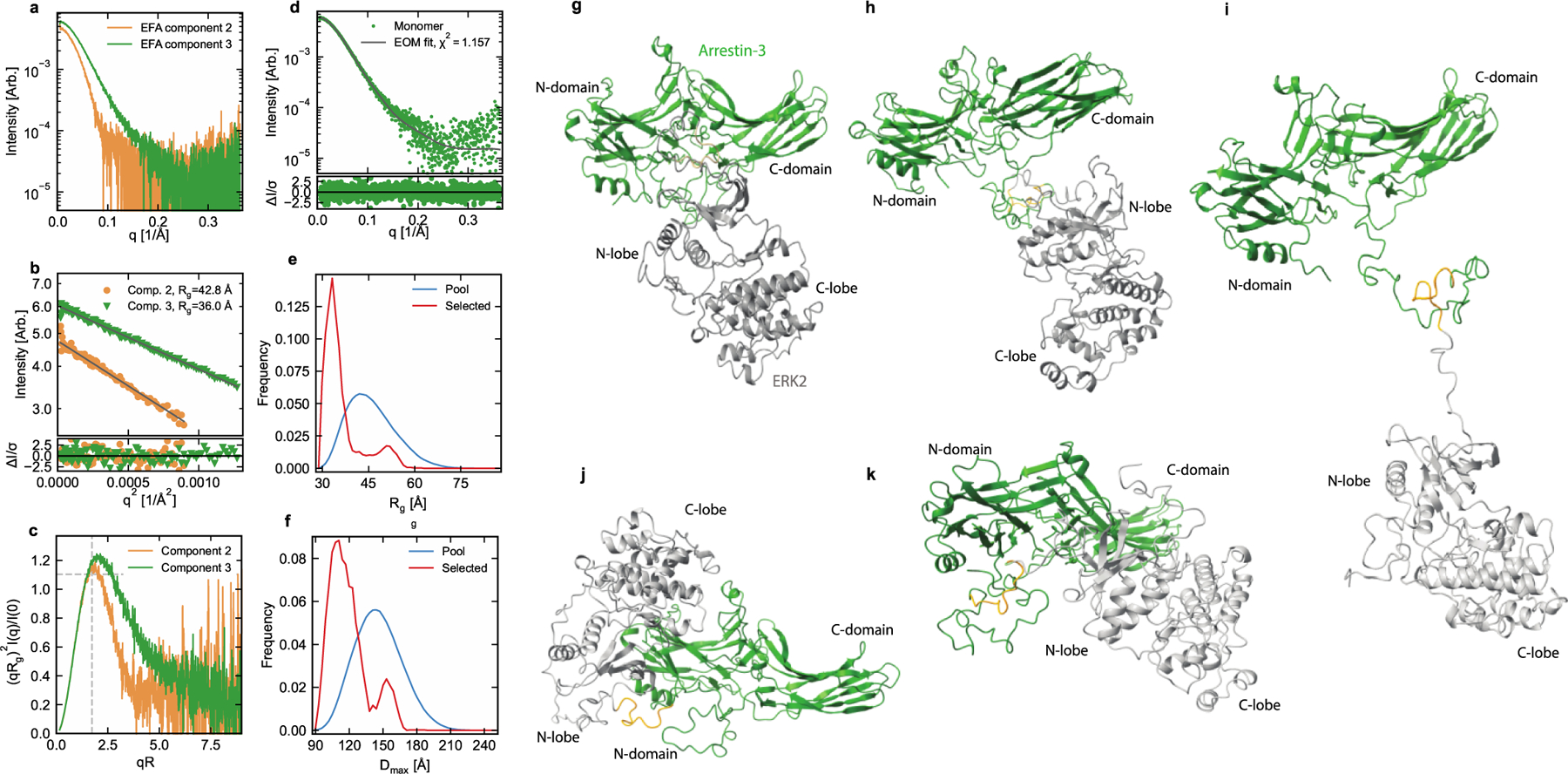
(a) Plot of the scattering profiles of the two components. Note that the overall scale factor from evolving factor analysis (EFA) is arbitrary and does not represent the two components’ relative concentration and molecular weights. (b) Guinier fit (black lines) to qRg=1.3 for both profiles (top panel) and normalized fit residuals (bottom panel). The normalized residuals show no systematic deviations, indicating high-quality Guinier fits. (c) Dimensionless Kratky plot for both profiles. The dashed gray lines indicate peak position and height for a compact globular object. The peak position and height shift to higher values and the extended tailing to larger qRg for component 3 indicates an extended and/or flexible macromolecule. (d) Monomer (component 3) scattering profile and ensemble optimization (EOM) fit (top panel) and the normalized fit residuals (bottom panel). Only very small systematic deviations are seen in the residual, indicating a high-quality fit. (e,f) The distribution of values in the full randomly generated structure pool (blue) and the EOM selected sub-ensemble of structures (red) for Rg, and Dmax, and (e, f), respectively. (g-k) EOM provided five structures representative of the full ensemble of structures in solution. For EOM, the high-resolution active structure of arrestin-3 (PDB 5VT1 (43)) was used with residues 1–7 and 350–393 removed for flexibility. The ERK2 structure (PDB 1ERK (87)) with residues 1–18 and 352–358 removed was used.
Molecular weight analysis determined by both methods calculated 147 kDa and 159 kDa for the larger component 2 (Fig. 6c) and 88 kDa and 75 kDa for the smaller component 3 (Fig. 6c). Dimensionless Kratky plots showed that the component 2 was essentially globular and compact (Fig. 6c). Component 3 had an extended Kratky plot, thus while most of the protein is compact, it likely contains additional extended and/or flexible states (Fig. 6c). However, analysis of the larger molecular weight species (component 2) revealed inconsistencies that resulted in poor quality bead model reconstructions. As a result, we could not be confident that this consisted of a single monomeric species. In contrast, component 3 (Fig. 6d) agreed well with the expected monomeric MW of 88 kDa (Fig. 6e,f). Therefore, component 3 was analyzed further.
Ensemble optimization analysis (EOM) on component 3 (Fig. 6d) tested both the active and basal conformation of arrestin-3 on this binary complex and resulted in five reconstructions representative of the full ensemble of arrestin-3-ERK2 binary structures in solution (Fig. 6g-k). Importantly, these models rely on quite distinct protein-protein contacts. This heterogeneity explains why some of results from arrays, chimeras, and EPR might initially seem contradictory. For example, the model in Fig. 6g appears to involve the gate loop of arrestin, which was identified as a contact in peptide array analysis (Fig 2). In contrast, the model in Fig. 6h primarily involves the C-domain with contacts near T6, which was identified as a contact through peptide array analysis and chimeragenesis (Figs 2 and 3). A full list of contacts, both from our and previously published results, and how these support each EOM model is shown in Fig. 7 and listed in Table 7. Each of these could represent pathway intermediates in the assembly or disassembly of the larger signaling complex.
Figure 7. Overview of ERK2 interaction on arrestin-2 and arrestin-3 as determined by orthogonal methods.
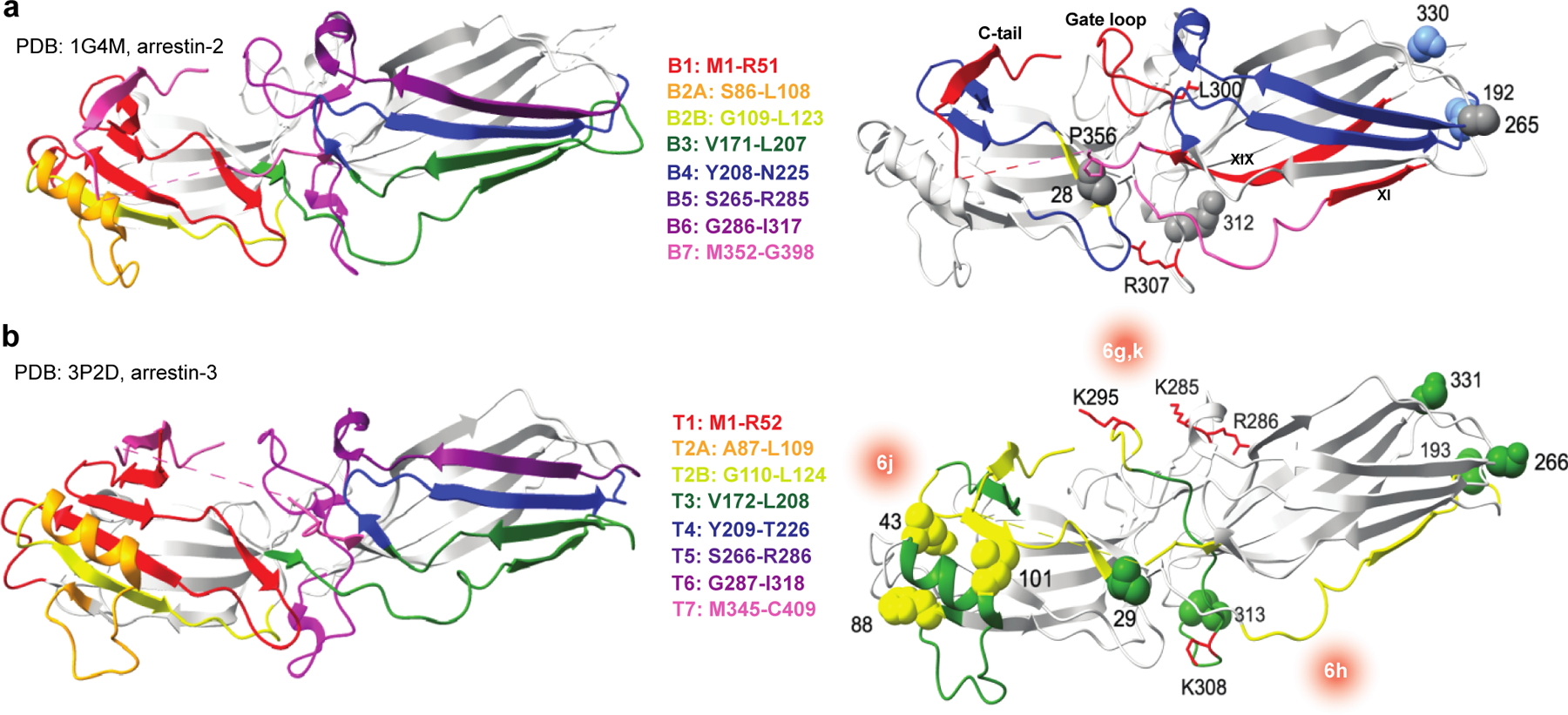
Residues corresponding to the non-receptor binding interface are shown for (a) arrestin-2 (PDB 1G4M (42)) and (b) arrestin-3 (PDB 3P2D (23)). Data from the peptide array analysis, CW-EPR, and SEC-SAXS for each nonvisual arrestin are shown on respective crystal structures. Blue (arrestin-2) or green (arrestin-3) represent regions of high interaction, yellow indicates intermediate interaction, and gray represents small to no interaction as measured by peptide array analysis and CW-EPR. Interfaces for ERK2 on arrestin-3 as determined by SEC-SAXS are represented by red gradients with the corresponding EOM model highlighted in the center using nomenclature from Fig. 6. Additional interaction sites as determined by other groups are highlighted in red on both structures. Full description of these interactions is in the supplemental Table S7.
Alignment of the ensembles on the structure of rhodopsin in complex with arrestin-1 (PDB 4ZWJ(51)) or neurotensin receptor 1 in complex with arrestin-2 (PDB 6UP7(52)) showed that the models in Fig. 6g, 6h, and 6i could bind to active receptor. In contrast, there are potential structural clashes between receptor and the arrestin-3-ERK2 ensembles shown in Fig. 6j and Fig. 6k (Supplementary Fig. S11). Thus, these particular modes of interaction may be transient states during the process of forming and disassembling an inducible signal initiation complex before arrestin binding to the receptor.
DISCUSSION
High-resolution structural information on arrestin-effector complexes is currently restricted to the terminal domain of clathrin bound either to full-length arrestin-2 (PDB 3GC3 (53)) or to an arrestin-3 peptide (PDB 1C9L and 1C9I (54)). The lack of structural data is striking considering the biological importance of arrestin-effector complexes in cellular signaling (55) and considering that both non-visual arrestins directly interact with over 100 shared effectors, plus numerous effectors specific for each isoform (56). One potential hurdle to high-resolution structural information is that many of these interactions are anticipated to occur within the context of a complex that contains the cellular membrane, an agonist-stimulated receptor, arrestin, and the effector(s), but may also contain G protein (57,58). In addition, arrestins are inherently flexible and this larger complex is expected to be in equilibrium with smaller transient complexes.
Instead, low-resolution and hybrid approaches have given hints to how arrestin engages effectors. Beginning with the arrestin conformation, previous work on the arrestin-ERK interaction focused on ERK2 engagement by arrestin-2, where purified proteins showed a direct interaction between arrestin-2 and ERK2 (34,59). A subset of these experiments focused on the arrestin conformation that binds ERK2. Here, in-cell assays showed that arrestin-2 preferentially engages ERK2 following recruitment to a stimulated receptor (13,34,60). This was supported by work with purified proteins, which showed enhanced binding of ERK2 to pre-activated arrestin-2 (arrestin- 21−384) (44). Collectively, these data support a model where an active receptor-bound arrestin conformation interacts with ERK2 in a biologically relevant fashion.
The previous mapping of the arrestin-2 interaction with ERK2 used various techniques and resulted in several models of MAPK signaling module organization on arrestin (4,35,61). Each of these is generally consistent with our data for the arrestin-2-ERK2 interaction. For example, in-cell assays of separated arrestin-2/3 domains (4,35,62), suggest that ERK2 binds both domains. This is consistent with our peptide array analysis showing that peptides corresponding to both the N- and C-domain of arrestin-2 and −3 engage purified ERK2. Reported computational modeling to dock multiple cascade partners suggested that arrestin can simultaneously scaffold multiple kinases of the ERK2 activation cascade (38).
The most extensive past work on the arrestin-2-ERK2 interaction combined HDX-MS, Trp-induced bimane fluorescence quenching, and NMR spectroscopy to suggest a model for arrestin-2 scaffolding ERK1/2, MEK1, and Raf1 (44). That model proposes that the arrestin-2 interaction with ERK2 involves the arrestin gate loop (residues D290-N299) and interdomain loop 2. This is consistent with our peptide array data, which indicates a strong interaction of the gate loop of arrestin-2 with ERK2 (Fig. 2a, peptides within B5). It should be noted, however, that our work and those in the past used binary arrestin-ERK2 complexes. These might differ from the interaction mode when receptor-arrestin complex scaffolds the full ERK2 activation cascade.
In contrast to the interaction between arrestin-2 and ERK2, the interaction between arrestin-3 and ERK2 is less well studied and in some cases seemed inconsistent with the data on arrestin-2 and ERK2. For arrestin-3, past work suggests that ERK1/2 can bind both the N- and C-domains of arrestin-3 (35), and identifies C-domain residues 271–295 of arrestin-3 (comprises part of T5 and T6) as the main binding site (63). ERK1/2 activation was also uniquely dependent on residues Lys285, Arg286, and Lys295 of arrestin-3 (35). Finally, docking studies suggest that ERK2 binds to arrestin-3 such that phosphorylated residues T202 and Y204 of ERK activation loop are near the active site of upstream kinase MEK1 (38). Our data also support these as part of the arrestin-3-ERK2 binding site.
Importantly, our data combined with those in the literature suggest that arrestin-3 engages ERK2 more readily and quite differently from arrestin-2 (Fig. 3). A trivial interpretation of this finding is that arrestin-3 does adopt the active conformation more readily (23,48). If a much greater percentage of arrestin-3 adopts the active conformation in the purified system, and this conformation binds more strongly, this would give an apparent higher affinity for the population. However, we also show that arrestin-2 and arrestin-3 use different regions of sequence to engage ERK2, as supported by peptide array analysis (Fig. 2) chimeragenesis (Fig. 3), CW-EPR (Fig. 4), and SEC-SAXS (Fig. 6). Considering their high homology (26,27), distinct binding modes for the two arrestin isoforms with ERK2 may seem surprising. However, available evidence supports the idea that the two non-visual arrestins are functionally different (32,35,43,64,65). The binding plasticity likely extends to all arrestin interactions, so it is conceivable that the highly similar arrestin isoforms might interact with the same effector differently. In fact, different binding modes of arrestin-2 and arrestin-3 to the same effector can reconcile past experimental evidence on the two arrestin isoforms that seemed inconsistent.
Interestingly, our data also indicate that arrestin-3 can bind ERK2 in more than one way in the binary complex. Three of these binary models (Fig 6g-i) appear to be consistent with how arrestin might simultaneously engage other components of an inducible signal initiation complex, that includes the receptor, membrane, MEK1, c-Raf1, and potentially G protein (57,58). Arrestins are naturally flexible proteins, and the binding partners that weren’t present in our experiments may also shift the conformational equilibrium of the binary complex in order to form a more homogeneous pose in the signal initiation complex.
As the two non-visual arrestins do not appear to be functionally redundant, this could influence biological outcomes. One potential impact of the stronger association of arrestin-3 with ERK2 is that cellular differences in the expression of arrestin-2 and arrestin-3 could allow for biological signals to be directed to different signaling cascades. Our study suggests how components of the ERK2 activation cascade could influence the arrestin-3 conformation and how the inducible signal initiation complex is formed. The binary complex is flexible, and interactions with additional components could preclude a subset of possible structural states of arrestin and increase the homogeneity. As a result, the insights from these data also provide a first step toward stabilizing the arrestin-containing signal initiation complexes for future biochemical and structural studies.
MATERIALS AND METHODS
Protein Purification.
Pre-activated, truncated forms of bovine arrestin-21−382 (42) and −31−393 (23), Cys-less arrestin-2 and −3, rat NpT7–5 His6-ERK2 (66), and MBP-fusion proteins were purified, as described previously (67–69). The arrestin-3-ERK2-His10 construct was made by Mutagenex Inc. (Suwanee, GA). In brief, the construct had the following structure in a pTrc vector: bovine arrestin-21−382 or −31−393 cDNA, linker with TEV cleavage site (SGLEVLFQGPGS), rat ERK2 cDNA, thrombin cleavage site (GSLVPRGSGS), and 10X His-tag. The purification was performed in three steps: HisTrap, Q/SP, and size-exclusion chromatography. The initial HisTrap purification was performed using a 15–300 mM imidazole gradient in running buffer (25 mM Tris-HCl pH 7.5, 500 mM NaCl, 10% glycerol, 1 mM TCEP, and 100 µM PMSF). The fractions were analyzed by SDS-PAGE and those enriched in fusion protein were loaded onto a Q/SP column (GE Healthcare, Chicago, IL) at approximately 50 mM NaCl (dilution was performed during loading in 25 mM Tris-HCl pH 7.5, 10% glycerol, and 1 mM TCEP). The fusion protein was eluted with a gradient of 25–800 mM NaCl. Finally, size exclusion chromatography of the pooled peak fractions was used to exchange arrestin-3-ERK2-His10 into its storage buffer (20 mM MOPS pH 7.5, 150 mM NaCl, 1 mM TCEP, and 5% glycerol).
MBP Pull-down.
N-terminal MBP-fusions of bovine arrestin-2 or −3 were purified as previously described (68,70). Arrestins were subcloned into a pMal-p2T vector with a 10 amino acid linker, TLVPRGSPGF, between MBP and arrestin-2 or −3. Purified MBP was used as a negative control in all pull-down experiments and non-specific binding was subtracted from the total observed binding for each construct. MBP-fusion proteins (10 μg in 50 μL of 50 mM HEPES-Na, pH 7.3/150 mM NaCl) were immobilized on amylose resin (25 μL, 50% slurry, New England Biolabs, Ipswich, MA) for 1 h at 4 °C with slight rotation. Prey protein (His6-ERK2, 10 μg in 50 μL of 20 mM Tris/150 mM NaCl) was added to the immobilized MBP constructs and incubated with gentle rotation for 2 h at 4 °C. Samples were transferred to centrifuge filters (Durapore®-PVDF-0.65 μm) and washed three times with ice-cold 50 mM HEPES-Na, pH 7.3, 150 mM NaCl. Proteins were eluted with 100 µL 50 mM maltose in wash buffer and concentrated by methanol precipitation. The pellets were air dried, dissolved in SDS sample buffer (30 μL Laemmli, 2x) and analyzed by SDS-PAGE and western blotting, as described (68) (Total-ERK antibody, #9102S, Cell Signaling Technology, Danvers, MA). Statistical analysis was performed using paired Student’s t-test.
Peptide Array Synthesis.
The ResPep SL peptide synthesizer was used with conventional SPOT synthesis protocols using 15 residue long peptides, as previously described (71). Both a blank spot and a 15-mer glycine peptide were used as negative controls, and non-specific binding to these controls was not statistically different. Peptides were assembled by the ResPep SL on solid support via a peptide amide linker using Fmoc-chemistry. The sequences of the peptides were derived from bovine arrestin-2, bovine arrestin-3, or rat ERK2. After synthesis, the amino-protecting group was removed using piperidine (20% (v/v) solution in dimethylformamide) followed by extensive washing. The membranes were then submerged in a solution of 95% trifluoroacetic acid (TFA) and 3% triisopropylsilane for 1 h with agitation. Following removal of the TFA solution, the membranes were washed in dichloromethane four times 10 min each, followed by four 10 min washes in dimethylformamide, and two 2 min washes in ethanol. The membranes were then dried in the hood and stored at 4°C.
Peptide Membrane Blots.
Dried membranes were soaked in 100% ethanol for 5 min and then rehydrated by washing twice in water for 5 min. The membranes were then incubated in blocking solution (5% non-fat dry milk in Tris-buffered saline with 0.1% Tween 20 (TBS-T)) for 1 h. The membranes were washed five times for 5 min before overnight incubation at 4°C with the prey protein (0.5 µM) in binding buffer (20 mM MOPS pH 7.5, 150 mM NaCl, 2 mM TCEP). The following morning, the membranes were rinsed five times for 5 min in TBS-T. Membranes were incubated with the appropriate primary antibody for 1 h at room temperature (#9102S, p44/42 MAPK ERK1/2, Cell Signaling Technology; anti-arrestin rabbit polyclonal F431 antibody (72)), then washed five times for 5 min in TBS-T. The corresponding HRP-conjugated secondary antibody (1:10,000 dilution) was prepared in TBS-T, incubated with the membrane for 1 h, followed by two 5 min washes in TBS-T at RT. The spots were visualized using the SuperSignal West Pico Stable Peroxide Solution kit (Thermo Fisher Scientific, Waltham, MA). Images were obtained using the Bio-Rad Gel Doc Imager and analyzed by densitometry using ImageJ protein array analyzer. For ResPep analysis, the signal in individual dots was measured as a percentage of total binding density detected on the membrane. This allowed for comparison across all peptide experiments. Statistical analysis was performed using one-way ANOVA followed by Dunnett’s post-hoc test using GraphPad Prism.
Immunoprecipitation with DSP cross-linking.
HEK293 arrestin-2/3 knockout cells were co-transfected in a 6-well plate with HA-tagged ERK2 and arrestin-2/3 or indicated chimeras (Fig. 3). 48 h post-transfection, before lysis the cells were treated with 1 mM cross-linking reagent DSP (Dithiobis(succinimidyl-propionate)) (#A35393, Pierce) for 30 min and the reaction was quenched by adding Tris-HCl, pH 7.5 (final concentration 20 mM) for 15 min at room temperature. Cells were lysed with 500 μL lysis buffer (50 mM Tris pH 7.5, 250 mM NaCl, 10% glycerol, 0.5% NP-40, 20 mM NaF, 1 mM Na orthovanadate, 2 mM benzamidine, and 1 mM phenylmethylsulfonyl fluoride (PMSF)) for 15 min on ice. After centrifugation to remove cell debris, the supernatant was precleared with 20 μL protein G-agarose (#sc-2002, Santa Cruz) for 30 min at 4 °C. Then the supernatant was incubated with anti-HA rat antibody (ROCHE, clone 3F10) for 2 h followed by the 20 μL of protein G-agarose beads for 2 h with slow agitation at 4 °C. The mixtures were transferred into centrifugal filters (# UFC30DV00, Millipore) and washed three times with 500 μL of ice-cold lysis buffer by centrifugation at 2000 x g for 30 sec. The proteins were eluted with 50 μL of 2 x Laemmli sample buffer, and analyzed by western blotting with rabbit polyclonal anti-arrestin (F431)(72) and anti-HA (#3724, Cell Signaling Technology) primary antibodies, followed by appropriate HRP-conjugated secondary antibodies and SuperSignal West Pico Stable Peroxide Solutions following manufacturer’s instructions. The protein bands were visualized and quantified using LI-COR C-DiGit Blot Scanner and Image Studio Lite software (LI-COR Biosciences U.S.).
Recombinant pcDNA3-arrestin-3-ERK2 construction.
To make the arrestin-3-ERK2-His10 construct the cDNAs encoding bovine arrestin-31−393 and full-length rat ERK2 (66) (based on cDNA from Mutagenex Inc., Suwanee, GA). were subcloned into a pcDNA3 vector between BamHI and HindIII sites. All restriction enzymes were from New England Biolabs (Ipswich, MA). The final construct consisted of arrestin-31−393 followed by a PreScission protease site (GSLEVLFQGPGS), full length ERK2, thrombin cleavage site (GSLVPRGSGS), and a His10 tag.
Continuous-wave EPR.
Purified arrestin-2 and arrestin-3 single cysteine mutants were spin labeled with 10-fold molar excess 1-oxyl-2,2,5,5-tetramethyl-Δ3-pyrroline-3-methyl methanethiosulfonate spin label (MTSL, Toronto Research Chemicals) to form the R1 side chain, whereupon free spin label was removed by extensive dialysis against 50 mM MOPS, 100 mM NaCl, pH7.0 buffer. CW EPR spectra were collected over 100 G under non-saturating power at room temperature in glass capillaries using a Bruker X-band E500 spectrometer and a SHQ cavity at the National Biomedical EPR Center (Medical College of Wisconsin, Milwaukee, WI). Samples contained 50 μM spin labeled arrestin-2 or arrestin-3 in the presence and absence of 100 μM unphosphorylated cysteine-free ERK2.
ERK2 phosphorylation assay in HEK293 arrestin-2/3 knockout cells.
HEK293 arrestin-2/3 knockout cells (72)(a generous gift from Dr. Asuka Inoue, Graduate School of Pharmaceutical Sciences, Tohoku University) were maintained in DMEM containing 10% FBS (Invitrogen, Waltham, MA), penicillin, and streptomycin at 37°C and 5% CO2. Cells were transfected at 90–100% confluency in 6-well plates with arrestin constructs (0–0.25 μg) or arrestin-ERK2-His10 constructs (0–1.5 µg) and 0.3 µg of β2-adrenergic receptor at a 1:2.5 ratio of DNA:Lipofectamine 2000 (Invitrogen, Waltham, MA), according to manufacturer’s instructions. All constructs were in pcDNA3. At 48 hr post-transfection, the cells were incubated with 10 µM agonist isoproterenol (Sigma-Aldrich, St. Louis, MO) for indicated times (0–45 mins). Cells were then lysed with 400 μL RIPA buffer and centrifuged at 15,000xg for 15 min to pellet debris. The supernatant was mixed 1:1 with 2x SDS sample buffer (Sigma-Aldrich, St. Louis, MO), subjected to 10% SDS-PAGE, and the proteins were transferred to PVDF membrane (Millipore, Bedford, MA). Membranes were blocked by 1% non-fat dry milk in TBS-T and incubated with respective primary antibodies: anti-totalERK2 (#9102S, Cell Signaling Technology, Danvers, MA), anti-phospho-ERK2 (#9101S, Cell Signaling Technology, Danvers, MA), and anti-arrestin (rabbit polyclonal F431 antibody (72)). After washing, membranes were incubated with the appropriate HRP-conjugated secondary antibodies (Jackson ImmunoResearch Laboratories, Inc., West Grove, PA). Bands were detected using the Bio-Rad Gel Doc Imager with enhanced chemiluminescence (ECL, Pierce Biotechnology, Waltham, MA) and quantification was done using ImageJ software (73).
Small-angle X-ray Scattering (SAXS).
SAXS was performed at BioCAT (beamline 18ID at the Advanced Photon Source, Chicago) with in-line size exclusion chromatography (SEC-SAXS) to separate samples from aggregates and other contaminants thus ensuring optimal sample quality. The protein sample was loaded onto a Superdex 200 Increase 10/300 GL column, run at 0.7 ml/min by an AKTA Pure FPLC (GE Healthcar, Chicago, IL) and the eluate after it passed through the UV monitor was flown through the SAXS flow cell. The flow cell consisted of a 1.0 mm ID quartz capillary with ~20 µm walls. A coflowing buffer sheath separates samples from the capillary walls, helping prevent radiation damage (74). Scattering intensity was recorded using a Pilatus3 X 1M (Dectris) detector placed 3.64 m from the sample giving us access to a q-range of 0.0046 Å−1 to 0.36 Å-1. 0.5 s exposures were acquired every 2 seconds during elution and data was reduced to 1D profiles using BioXTAS RAW 1.6.0 (75). RAW automatically determined an appropriate buffer region preceding the peak, which was then averaged and subtracted from every frame in the data set.
Overlapping peaks within the SEC-SAXS elution profile were deconvolved into the constituent components using evolving factor analysis (EFA) (76) (Supplementary Fig. S8) with default settings as implemented in RAW (75). The validity of the overall deconvolution was assessed based on the mean error weighted χ2 for the whole deconvolution range and the deconvolved component concentration profiles. Individual components were assessed based on the quality of the scattering profile and SAXS derived MW of the scattering profile compared to expected values. Guinier analysis was carried out in RAW, as was molecular weight analysis, which was carried out using both the adjusted Porod volume and volume of correlation methods (77,78). Initial flexibility analysis was carried out using a dimensionless Kratky plot (79) and showed that component 2 was significantly globular, while component 3 was either extended or flexible. Further analysis, described below, was carried out using programs from the ATSAS suite (versions 2.8.4 and 3.0.3) (80).
P(r) curves were created using GNOM (81) (Supplementary Fig. S9). Bead modeling was initially carried out for components 2 and 3 using 15 reconstructions from DAMMIF in slow mode, averaged by DAMAVER, and a final structure was refined in DAMMIN (82–84). AMBIMETER (85) was used to assess the ambiguity of reconstructions. For component 2 the ambiguity score of the reconstructions was 0.699, indicating potentially unique reconstructions. Of the individual models, 14 of 15 were included in the average, which gave a normalized spatial discrepancy (NSD) of 1.011 ± 0.077. Clustering analysis found four distinct clusters in the models. Individual χ2 for each model was ~2.4 (range: 2.357–2.366). The final refined model had an Rg of 43.05 Å, Dmax of 159.2 Å and an estimated MW of 236.5 kDa. The first two values are in good agreement with other results for the data, while the estimated molecular weight is ~1.5x that of other analysis methods. Given the relatively high NSD, the number of clusters, and χ2, as well as the disagreement in MW estimates, the bead model reconstructions seem to have failed for this system.
For component 3, the ambiguity score of the reconstructions was 1.602, indicating possibly ambiguous reconstructions. All 15 were included in the average of the individual models, which gave a NSD of 1.075 ± 0.052. Clustering analysis found two distinct clusters in the models. Individual χ2 for each model was ~1.2 (range: 1.205–1.207). The final refined model had an Rg of 37.2 Å, Dmax of 148.2 Å and an estimated MW of 99.4 kDa, in good agreement with other results for the data. The high NSD, ambiguity score, and existence of distinct clusters indicate that the bead model results for this sample are not useable. However, the individual reconstructions seem valid based on the χ2 and size metrics, which indicates that the spread of solutions shown by the ambiguity, NSD, and clustering likely represents many possible states in solution, again indicating a flexible system.
Because the arrestin-3-ERK2 fusion contains a flexible linker between the two proteins and our results showed flexibility in the SAXS data for component 3, Ensemble Optimization Method (EOM) was used to analyze the flexibility of component 3. EOM is a technique that starts by generating a large pool of random flexible structures. The pool was generated by fixing the structure of the known high-resolution domains while letting the linker and N- and C-terminus of the construct be flexible. EOM then selected an ensemble of curves from the pool that matched the SAXS data set using a genetic algorithm (Fig. 6g-k, Supplementary Fig. S10). It is important to note that EOM cannot yield precise structural solutions for a system, but rather provides statistics that inform on the full set of solutions (86).
Both the active and basal forms for the arrestin-3 domain were used for EOM. The high-resolution structure used for EOM with the basal form was PDB 3P2D with residues 1–6 and 351–393 removed as they represent likely flexible N- and C-terminus regions. For the active form, PDB 5TV1 was used, with residues 1–7 and 350–393 removed for the same reason. Additionally, residues 308–312 are missing from the 5TV1 structure and were assumed to be flexible. The ERK2 domain was assumed to be folded, the high-resolution structure used for EOM was PDB 1ERK with residues 1–18 and 352–358 truncated as they represent likely flexible N- and C-terminus regions. The active and basal structures of arrestin-3 EOM were used to generate 50,000 possible profiles for the base pool using default settings, and the genetic algorithm was run 10 times using default settings. The histogram average Rg and Dmax for the selected ensemble (pool) were 37.44 Å (4 Å) and 127.83 Å (146.67 Å), respectively. This supports the visual data that the selected ensemble is significantly more compact than the random pool. The Rflex value is represented as a % from 0 (rigid) to 100 (completely flexible), and for the selected ensemble (pool) was 66.19% (83.17%). The Rflex value compared to the pool shows less flexibility, which typically gives Rσ<1, in this case the Rσ value was 0.82. All of this supports the conclusion that the system is adopting very compact structures in solution and is very similar to the results using the basal state structure.
Supplementary Material
HIGHLIGHTS.
Non-visual arrestins act as scaffolds for signaling cascades, including ERK1/2
Free arrestins form a binary pre-complex with ERK1/2
Purified arrestin-2 and −3 bind purified ERK2 differently in binary complexes
The ERK2 interaction with arrestin can be modulated using arrestin-2/3 chimeras
Small-angle X-ray scattering suggests flexibility of the arrestin-3-ERK2 complex
Acknowledgements:
The authors are grateful to Dr. Asuka Inoue for the generous gift of the arrestin-2/3 knockout HEK293 cells. We also thank Drs. Tamer Kaoud, Andrea Piserchio, and Ranajeet Ghose for sharing their knowledge of the ERK2 interaction with non-visual arrestins.
Funding: This work was supported by American Heart Association 16PRE30180007 (NAP) and 18PRE34030017 (NAP), National Institutes of Health (NIH) 2T32GM008320–31 (IP), National Science Foundation Graduate Research Fellowship Program under Grant No. 1937963 (IP), NIH grants R35 GM122491 (VVG), R01 GM120569 (TMI), DA043680 (TMI/VVG), GM123252 (KND), a Vanderbilt Discovery Award (TMI/VVG), CPRIT RP140648 (KND), Welch F-1390 (KND), and Cornelius Vanderbilt Endowed Chair (VVG). Any opinions, findings, and conclusions or recommendations expressed in this material are those of the author(s) and do not necessarily reflect the views of the National Science Foundation. This work used facilities supported by NIH P41EB001980 (National Biomedical EPR Center) and P30EY008126 (Vanderbilt Core Grant in Vision Research) grants. This research used resources of the Advanced Photon Source, a U.S. Department of Energy (DOE) Office of Science User Facility operated for the DOE Office of Science by Argonne National Laboratory under Contract No. DE-AC02–06CH11357. This project was supported by grant 9 P41 GM103622 from the National Institute of General Medical Sciences of the National Institutes of Health. Use of the Pilatus 3 1M detector was provided by grant 1S10OD018090–01 from NIGMS.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
Declaration of Interest
The authors declare no conflict of interest.
Here we use the systematic names of arrestin proteins: arrestin-1 (historic names S-antigen, 48 kDa protein, visual or rod arrestin), arrestin-2 (β-arrestin or β-arrestin1), arrestin-3 (β-arrestin2 or hTHY-ARRX), and arrestin-4 (cone or X-arrestin; for unclear reasons its gene is called “arrestin 3” in the HUGO database).
CRediT author statement
Nicole Perry-Hauser: Conceptualization, Methodology, Investigation, Formal analysis, Writing, Visualization. Jesse Bennett Hopkins: Methodology, Software, Formal analysis, Data Curation. Ya Zhuo: Investigation, Formal analysis. Chen Zheng: Investigation, Formal analysis. Ivette Perez: Investigation, Formal analysis. Kathryn Schultz: Investigation, Formal analysis. Sergey Vishnivetskiy: Resources. Ali Kaya: Investigation, Resources. Pankaj Sharma: Methodology. Kevin Dalby: Resources, Writing – Review and Editing. Ka Young Chung: Writing – Review and Editing. Candice Klug: Methodology, Resources, Formal analysis, Writing – Review and Editing. Vsevolod Gurevich: Conceptualization, Writing, Supervision, Project management, Funding acquisition. TM Iverson: Conceptualization, Writing, Supervision, Project management, Funding acquisition
Declaration of interests
The authors declare that they have no known competing financial interests or personal relationships that could have appeared to influence the work reported in this paper.
Bibliography
- 1.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of beta-arrestin interactions revealed by proteomic analysis. Proc Natl Acad Sci USA 2007. Jul 17;104(29):12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Ferguson SS. Evolving concepts in G protein-coupled receptor endocytosis: the role in receptor desensitization and signaling. Pharmacol Rev 2001. Mar;53(1):1–24. [PubMed] [Google Scholar]
- 3.Goodman OB, Krupnick JG, Santini F, Gurevich VV, Penn RB, Gagnon AW, et al. Beta-arrestin acts as a clathrin adaptor in endocytosis of the beta2-adrenergic receptor. Nature 1996. Oct 3;383(6599):447–450. [DOI] [PubMed] [Google Scholar]
- 4.Imamura T, Huang J, Dalle S, Ugi S, Usui I, Luttrell LM, et al. beta-Arrestin-mediated recruitment of the Src family kinase Yes mediates endothelin-1-stimulated glucose transport. J Biol Chem 2001. Nov 23;276(47):43663–43667. [DOI] [PubMed] [Google Scholar]
- 5.Luttrell LM, Ferguson SS, Daaka Y, Miller WE, Maudsley S, Della Rocca GJ, et al. Beta-arrestin-dependent formation of beta2 adrenergic receptor-Src protein kinase complexes. Science 1999. Jan 29;283(5402):655–661. [DOI] [PubMed] [Google Scholar]
- 6.Perez I, Berndt S, Agarwal R, Castro MA, Vishnivetskiy SA, Smith JC, et al. A model for the signal initiation complex between arrestin-3 and the src family kinase Fgr. JMB 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Perry NA, Fialkowski KP, Kaoud TS, Kaya AI, Chen AL, Taliaferro JM, et al. Arrestin-3 interaction with maternal embryonic leucine-zipper kinase. Cell Signal 2019. Jul 25;63:109366. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Chavkin C, Schattauer SS, Levin JR. Arrestin-mediated activation of p38 MAPK: molecular mechanisms and behavioral consequences. Handb Exp Pharmacol 2014;219:281–292. [DOI] [PubMed] [Google Scholar]
- 9.DeFea KA. Beta-arrestins as regulators of signal termination and transduction: how do they determine what to scaffold? Cell Signal 2011. Apr;23(4):621–629. [DOI] [PubMed] [Google Scholar]
- 10.Good M, Tang G, Singleton J, Reményi A, Lim WA. The Ste5 scaffold directs mating signaling by catalytically unlocking the Fus3 MAP kinase for activation. Cell 2009. Mar 20;136(6):1085–1097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Guo C, Whitmarsh AJ. The beta-arrestin-2 scaffold protein promotes c-Jun N-terminal kinase-3 activation by binding to its nonconserved N terminus. J Biol Chem 2008. Jun 6;283(23):15903–15911. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Kook S, Zhan X, Kaoud TS, Dalby KN, Gurevich VV, Gurevich EV. Arrestin-3 binds c-Jun N-terminal kinase 1 (JNK1) and JNK2 and facilitates the activation of these ubiquitous JNK isoforms in cells via scaffolding. J Biol Chem 2013. Dec 27;288(52):37332–37342. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Luttrell LM, Roudabush FL, Choy EW, Miller WE, Field ME, Pierce KL, et al. Activation and targeting of extracellular signal-regulated kinases by beta-arrestin scaffolds. Proc Natl Acad Sci USA 2001. Feb 27;98(5):2449–2454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Grundmann M, Merten N, Malfacini D, Inoue A, Preis P, Simon K, et al. Lack of beta-arrestin signaling in the absence of active G proteins. Nat Commun 2018. Jan 23;9(1):341. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.O’Hayre M, Eichel K, Avino S, Zhao X, Steffen DJ, Feng X, et al. Genetic evidence that β-arrestins are dispensable for the initiation of β2-adrenergic receptor signaling to ERK. Sci Signal 2017. Jun 20;10(484). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Luttrell LM, Wang J, Plouffe B, Smith JS, Yamani L, Kaur S, et al. Manifold roles of β-arrestins in GPCR signaling elucidated with siRNA and CRISPR/Cas9. Sci Signal 2018. Sep 25;11(549). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Kaya AI, Perry NA, Gurevich VV, Iverson TM. Phosphorylation barcode-dependent signal bias of the dopamine D1 receptor. Proc Natl Acad Sci USA 2020. Jun 23;117(25):14139–14149. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Ahn S, Shenoy SK, Wei H, Lefkowitz RJ. Differential Kinetic and Spatial Patterns of β-Arrestin and G Protein-mediated ERK Activation by the Angiotensin II Receptor. J Bio Chem 2004;279(34):35518–35525. [DOI] [PubMed] [Google Scholar]
- 19.Luttrell LM. Location, location, location ‘: activation and targeting of MAP kinases by G protein-coupled receptors. J Mol Endocrinol 2003. Apr;30(2):117–126. [DOI] [PubMed] [Google Scholar]
- 20.Chambard J-C, Lefloch R, Pouysségur J, Lenormand P. ERK implication in cell cycle regulation. Biochim Biophys Acta 2007. Aug;1773(8):1299–1310. [DOI] [PubMed] [Google Scholar]
- 21.Zhang W, Liu HT. MAPK signal pathways in the regulation of cell proliferation in mammalian cells. Cell Res 2002. Mar;12(1):9–18. [DOI] [PubMed] [Google Scholar]
- 22.Wang P, Wu Y, Ge X, Ma L, Pei G. Subcellular Localization of β-Arrestins Is Determined by Their Intact N Domain and the Nuclear Export Signal at the C Terminus. J Bio Chem 2003;278(13):11648–11653. [DOI] [PubMed] [Google Scholar]
- 23.Zhan X, Gimenez LE, Gurevich VV, Spiller BW. Crystal structure of arrestin-3 reveals the basis of the difference in receptor binding between two non-visual subtypes. J Mol Biol 2011. Feb 25;406(3):467–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal structure of beta-arrestin at 1.9 A: possible mechanism of receptor binding and membrane Translocation. Structure 2001. Sep;9(9):869–880. [DOI] [PubMed] [Google Scholar]
- 25.Srivastava A, Gupta B, Gupta C, Shukla AK. Emerging Functional Divergence of β-Arrestin Isoforms in GPCR Function. Trends Endocrinol Metab 2015. Nov;26(11):628–642. [DOI] [PubMed] [Google Scholar]
- 26.Attramadal H, Arriza JL, Aoki C, Dawson TM, Codina J, Kwatra MM, et al. Beta-arrestin2, a novel member of the arrestin/beta-arrestin gene family. J Biol Chem 1992. Sep 5;267(25):17882–17890. [PubMed] [Google Scholar]
- 27.Sterne-Marr R, Gurevich VV, Goldsmith P, Bodine RC, Sanders C, Donoso LA, et al. Polypeptide variants of beta-arrestin and arrestin3. J Biol Chem 1993. Jul 25;268(21):15640–15648. [PubMed] [Google Scholar]
- 28.Indrischek H, Prohaska SJ, Gurevich VV, Gurevich EV, Stadler PF. Uncovering missing pieces: duplication and deletion history of arrestins in deuterostomes. BMC Evol Biol 2017. Jul 6;17(1):163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Ahn S, Nelson CD, Garrison TR, Miller WE, Lefkowitz RJ. Desensitization, internalization, and signaling functions of beta-arrestins demonstrated by RNA interference. Proc Natl Acad Sci USA 2003. Feb 18;100(4):1740–1744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Bohn LM, Lefkowitz RJ, Gainetdinov RR, Peppel K, Caron MG, Lin FT. Enhanced morphine analgesia in mice lacking beta-arrestin 2. Science 1999. Dec 24;286(5449):2495–2498. [DOI] [PubMed] [Google Scholar]
- 31.Conner DA, Mathier MA, Mortensen RM, Christe M, Vatner SF, Seidman CE, et al. beta-Arrestin1 knockout mice appear normal but demonstrate altered cardiac responses to beta-adrenergic stimulation. Circ Res 1997. Dec;81(6):1021–1026. [DOI] [PubMed] [Google Scholar]
- 32.Chen Q, Zhuo Y, Sharma P, Perez I, Francis DJ, Chakravarthy S, et al. An eight amino acid segment controls oligomerization and preferred conformation of the two non-visual arrestins. J Mol Biol 2020. Dec 30;166790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gong K, Li Z, Xu M, Du J, Lv Z, Zhang Y. A Novel Protein Kinase A-independent, Î2-Arrestin-1-dependent Signaling Pathway for p38 Mitogen-activated Protein Kinase Activation by Î22-Adrenergic Receptors. J Bio Chem 2008;283(43):29028–29036. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Coffa S, Breitman M, Hanson SM, Callaway K, Kook S, Dalby KN, et al. The effect of arrestin conformation on the recruitment of c-Raf1, MEK1, and ERK1/2 activation. PLoS One 2011. Dec 12;6(12):e28723. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Song X, Coffa S, Fu H, Gurevich VV. How does arrestin assemble MAPKs into a signaling complex? J Biol Chem 2009. Jan 2;284(1):685–695. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Jung S-R, Kushmerick C, Seo JB, Koh D-S, Hille B. Muscarinic receptor regulates extracellular signal regulated kinase by two modes of arrestin binding. Proc Natl Acad Sci USA 2017. Jul 11;114(28):E5579–E5588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Cassier E, Gallay N, Bourquard T, Claeysen S, Bockaert J, Crépieux P, et al. Phosphorylation of β-arrestin2 at Thr383 by MEK underlies β-arrestin-dependent activation of Erk1/2 by GPCRs. Elife 2017. Feb 7;6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Bourquard T, Landomiel F, Reiter E, Crépieux P, Ritchie DW, Azé J, et al. Unraveling the molecular architecture of a G protein-coupled receptor/β-arrestin/Erk module complex. Sci Rep 2015. Jun 1;5:10760. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Ahn S, Wei H, Garrison TR, Lefkowitz RJ. Reciprocal Regulation of Angiotensin Receptor-activated Extracellular Signal-regulated Kinases by Î2-Arrestins 1 and 2. J Bio Chem 2004;279(9):7807–7811. [DOI] [PubMed] [Google Scholar]
- 40.Kim J, Ahn S, Rajagopal K, Lefkowitz RJ. Independent Î2-Arrestin2 and Gq/Protein Kinase Cζ Pathways for ERK Stimulated by Angiotensin Type 1A Receptors in Vascular Smooth Muscle Cells Converge on Transactivation of the Epidermal Growth Factor Receptor. J Bio Chem 2009;284(18):11953–11962. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Hilpert K, Winkler DFH, Hancock REW. Peptide arrays on cellulose support: SPOT synthesis, a time and cost efficient method for synthesis of large numbers of peptides in a parallel and addressable fashion. Nat Protoc 2007;2(6):1333–1349. [DOI] [PubMed] [Google Scholar]
- 42.Han M, Gurevich VV, Vishnivetskiy SA, Sigler PB, Schubert C. Crystal Structure of β-Arrestin at 1.9 Å. Structure 2001. Sep;9(9):869–880. [DOI] [PubMed] [Google Scholar]
- 43.Chen Q, Perry NA, Vishnivetskiy SA, Berndt S, Gilbert NC, Zhuo Y, et al. Structural basis of arrestin-3 activation and signaling. Nat Commun 2017. Nov 10;8(1):1427. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Qu C, Park JY, Woo Yun M, Yang F, He Q, Kim K, et al. Scaffolding mechanism ofarrestin-2 in the cRaf/MEK1/ERK signaling cascade. Proc Natl Acad Sci USA 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kovoor A, Celver J, Abdryashitov RI, Chavkin C, Gurevich VV. Targeted construction of phosphorylation-independent beta-arrestin mutants with constitutive activity in cells. J Biol Chem 1999. Mar 12;274(11):6831–6834. [DOI] [PubMed] [Google Scholar]
- 46.Celver J, Vishnivetskiy SA, Chavkin C, Gurevich VV. Conservation of the phosphate-sensitive elements in the arrestin family of proteins. J Biol Chem 2002. Mar 15;277(11):9043–9048. [DOI] [PubMed] [Google Scholar]
- 47.Zhuo Y, Vishnivetskiy SA, Zhan X, Gurevich VV, Klug CS. Identification of receptor binding-induced conformational changes in non-visual arrestins. J Biol Chem 2014. Jul 25;289(30):20991–21002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Sensoy O, Moreira IS, Morra G. Understanding the Differential Selectivity of Arrestins toward the Phosphorylation State of the Receptor. ACS Chem Neurosci 2016. Sep 21;7(9):1212–1224. [DOI] [PubMed] [Google Scholar]
- 49.McReynolds AC, Karra AS, Li Y, Lopez ED, Turjanski AG, Dioum E, et al. Phosphorylation or mutation of the ERK2 activation loop alters oligonucleotide binding. Biochemistry 2016. Mar 29;55(12):1909–1917. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Pegram LM, Liddle JC, Xiao Y, Hoh M, Rudolph J, Iverson DB, et al. Activation loop dynamics are controlled by conformation-selective inhibitors of ERK2. Proc Natl Acad Sci USA 2019. Jul 30;116(31):15463–15468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Kang Y, Zhou XE, Gao X, He Y, Liu W, Ishchenko A, et al. Crystal structure of rhodopsin bound to arrestin by femtosecond X-ray laser. Nature 2015. Jul 30;523(7562):561–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Huang W, Masureel M, Qu Q, Janetzko J, Inoue A, Kato HE, et al. Structure of the neurotensin receptor 1 in complex with β-arrestin 1. Nature 2020. Mar;579(7798):303–308. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Kang DS, Kern RC, Puthenveedu MA, von Zastrow M, Williams JC, Benovic JL. Structure of an arrestin2-clathrin complex reveals a novel clathrin binding domain that modulates receptor trafficking. J Biol Chem 2009. Oct 23;284(43):29860–29872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Ter Haar E, Harrison SC, Kirchhausen T. Peptide-in-groove interactions link target proteins to the beta-propeller of clathrin. Proc Natl Acad Sci USA 2000. Feb 1;97(3):1096–1100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Perry-Hauser NA, Zhan X, Iverson TM. Chapter 4. Arrestin scaffolding and activation of MAPK cascades. In: Gurevich VV, editor. Arrestins: Structure, Function, and Molecular Biology 2021. [Google Scholar]
- 56.Xiao K, McClatchy DB, Shukla AK, Zhao Y, Chen M, Shenoy SK, et al. Functional specialization of β-arrestin interactions revealed by proteomic analysis 2007;104(29):12011–12016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Thomsen ARB, Plouffe B, Cahill TJ, Shukla AK, Tarrasch JT, Dosey AM, et al. GPCR-G Protein-β-Arrestin Super-Complex Mediates Sustained G Protein Signaling. Cell 2016. Aug 11;166(4):907–919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Nguyen AH, Thomsen ARB, Cahill TJ, Huang R, Huang L-Y, Marcink T, et al. Structure of an endosomal signaling GPCR-G protein-β-arrestin megacomplex. Nat Struct Mol Biol 2019. Dec;26(12):1123–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Coffa S, Breitman M, Spiller BW, Gurevich VV. A single mutation in arrestin-2 prevents ERK1/2 activation by reducing c-Raf1 binding. Biochemistry 2011. Aug 16;50(32):6951–6958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Tohgo A, Pierce KL, Choy EW, Lefkowitz RJ, Luttrell LM. beta-Arrestin scaffolding of the ERK cascade enhances cytosolic ERK activity but inhibits ERK-mediated transcription following angiotensin AT1a receptor stimulation. J Biol Chem 2002. Mar 15;277(11):9429–9436. [DOI] [PubMed] [Google Scholar]
- 61.Meng D, Lynch MJ, Huston E, Beyermann M, Eichhorst J, Adams DR, et al. MEK1 binds directly to betaarrestin1, influencing both its phosphorylation by ERK and the timing of its isoprenaline-stimulated internalization. J Biol Chem 2009. Apr 24;284(17):11425–11435. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Eishingdrelo H, Sun W, Li H, Wang L, Eishingdrelo A, Dai S, et al. ERK and β-arrestin interaction: a converging point of signaling pathways for multiple types of cell surface receptors. J Biomol Screen 2015. Mar;20(3):341–349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Xu T-R, Baillie GS, Bhari N, Houslay TM, Pitt AM, Adams DR, et al. Mutations of beta-arrestin 2 that limit self-association also interfere with interactions with the beta2-adrenoceptor and the ERK1/2 MAPKs: implications for beta2-adrenoceptor signalling via the ERK1/2 MAPKs. Biochem J 2008. Jul 1;413(1):51–60. [DOI] [PubMed] [Google Scholar]
- 64.He Q-T, Xiao P, Huang S-M, Jia Y-L, Zhu Z-L, Lin J-Y, et al. Structural studies of phosphorylation-dependent interactions between the V2R receptor and arrestin-2. Nat Commun 2021. Apr 22;12(1):2396. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Miller WE, McDonald PH, Cai SF, Field ME, Davis RJ, Lefkowitz RJ. Identification of a Motif in the Carboxyl Terminus of β-Arrestin2 Responsible for Activation of JNK3. J Bio Chem 2001;276(30):27770–27777. [DOI] [PubMed] [Google Scholar]
- 66.Waas WF, Dalby KN. Transient protein-protein interactions and a random-ordered kinetic mechanism for the phosphorylation of a transcription factor by extracellular-regulated protein kinase 2. J Biol Chem 2002. Apr 12;277(15):12532–12540. [DOI] [PubMed] [Google Scholar]
- 67.Vishnivetskiy SA, Zhan X, Chen Q, Iverson TM, Gurevich VV. Arrestin expression in E. coli and purification. Curr Protoc Pharmacol 2014. Dec 1;67:Unit 2.11.1–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Zhan X, Perez A, Gimenez LE, Vishnivetskiy SA, Gurevich VV. Arrestin-3 binds the MAP kinase JNK3α2 via multiple sites on both domains. Cell Signal 2014. Apr;26(4):766–776. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Zhang F, Robbins DJ, Cobb MH, Goldsmith EJ. Crystallization and preliminary X-ray studies of extracellular signal-regulated kinase-2/MAP kinase with an incorporated His-tag. J Mol Biol 1993. Oct 5;233(3):550–552. [DOI] [PubMed] [Google Scholar]
- 70.Zhan X, Stoy H, Kaoud TS, Perry NA, Chen Q, Perez A, et al. Peptide mini-scaffold facilitates JNK3 activation in cells. Sci Rep 2016. Feb 12;6:21025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Yim YY, Betke K, Hamm H. Using peptide arrays created by the SPOT method for defining protein-protein interactions. Methods Mol Biol 2015;1278:307–320. [DOI] [PubMed] [Google Scholar]
- 72.Alvarez-Curto E, Inoue A, Jenkins L, Raihan SZ, Prihandoko R, Tobin AB, et al. Targeted Elimination of G Proteins and Arrestins Defines Their Specific Contributions to Both Intensity and Duration of G Protein-coupled Receptor Signaling. J Biol Chem 2016. Dec 30;291(53):27147–27159. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Schneider CA, Rasband WS, Eliceiri KW. NIH Image to ImageJ: 25 years of image analysis. Nat Methods 2012. Jul;9(7):671–675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kirby N, Cowieson N, Hawley AM, Mudie ST, McGillivray DJ, Kusel M, et al. Improved radiation dose efficiency in solution SAXS using a sheath flow sample environment. Acta Crystallogr D Struct Biol 2016. Dec 1;72(Pt 12):1254–1266. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Hopkins JB, Gillilan RE, Skou S. BioXTAS RAW: improvements to a free open-source program for small-angle X-ray scattering data reduction and analysis. J Appl Crystallogr 2017. Oct 1;50(Pt 5):1545–1553. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Meisburger SP, Taylor AB, Khan CA, Zhang S, Fitzpatrick PF, Ando N. Domain Movements upon Activation of Phenylalanine Hydroxylase Characterized by Crystallography and Chromatography-Coupled Small-Angle X-ray Scattering. J Am Chem Soc 2016. May 25;138(20):6506–6516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Piiadov V, Ares de Araújo E, Oliveira Neto M, Craievich AF, Polikarpov I. SAXSMoW 2.0: Online calculator of the molecular weight of proteins in dilute solution from experimental SAXS data measured on a relative scale. Protein Sci 2019;28(2):454–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rambo RP, Tainer JA. Accurate assessment of mass, models and resolution by small-angle scattering. Nature 2013. Apr 25;496(7446):477–481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Durand D, Vivès C, Cannella D, Pérez J, Pebay-Peyroula E, Vachette P, et al. NADPH oxidase activator p67(phox) behaves in solution as a multidomain protein with semi-flexible linkers. J Struct Biol 2010. Jan;169(1):45–53. [DOI] [PubMed] [Google Scholar]
- 80.Franke D, Petoukhov MV, Konarev PV, Panjkovich A, Tuukkanen A, Mertens HDT, et al. ATSAS 2.8: a comprehensive data analysis suite for small-angle scattering from macromolecular solutions. J Appl Crystallogr 2017. Aug 1;50(Pt 4):1212–1225. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Svergun DI. Determination of the regularization parameter in indirect-transform methods using perceptual criteria. J Appl Crystallogr 1992. Aug 1;25(4):495–503. [Google Scholar]
- 82.Svergun DI. Restoring low resolution structure of biological macromolecules from solution scattering using simulated annealing. Biophys J 1999. Jun;76(6):2879–2886. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Volkov VV, Svergun DI. Uniqueness of ab initio shape determination in small-angle scattering. J Appl Crystallogr 2003. Jun 1;36(3):860–864. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Franke D, Svergun DI. DAMMIF , a program for rapidab-initio shape determination in small-angle scattering. J Appl Crystallogr 2009. Apr 1;42(2):342–346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Petoukhov MV, Svergun DI. Ambiguity assessment of small-angle scattering curves from monodisperse systems. Acta Crystallogr Sect D, Biol Crystallogr 2015. May;71(Pt 5):1051–1058. [DOI] [PubMed] [Google Scholar]
- 86.Tria G, Mertens HDT, Kachala M, Svergun DI. Advanced ensemble modelling of flexible macromolecules using X-ray solution scattering. IUCrJ 2015. Mar 1;2(Pt 2):207–217. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Zhang F, Strand A, Robbins D, Cobb MH, Goldsmith EJ. Atomic structure of the MAP kinase ERK2 at 2.3 A resolution. Nature 1994. Feb 24;367(6465):704–711. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.