

Abstract
Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) is a serine/threonine kinase that belongs to the DYRK family of proteins, a subgroup of the evolutionarily conserved CMGC protein kinase superfamily. Due to its localization on chromosome 21, the biological significance of DYRK1A was initially characterized in the pathogenesis of Down syndrome (DS) and related neurodegenerative diseases. However, increasing evidence has demonstrated a prominent role in cancer through its ability to regulate biologic processes including cell cycle progression, DNA damage repair, transcription, ubiquitination, tyrosine kinase activity, and cancer stem cell maintenance. DYRK1A has been identified as both an oncogene and tumor suppressor in different models, underscoring the importance of cellular context in its function. Here, we review mechanistic contributions of DYRK1A to cancer biology and its role as a potential therapeutic target.
Keywords: DYRK1A, quiescence, cell cycle, cell proliferation, tumor progression
Introduction
Dual-specificity tyrosine phosphorylation-regulated kinase 1A (DYRK1A) is a highly conserved kinase encoded on chromosome 21 in the Down syndrome critical region (DSCR). The DSCR contains a set of genes on the long arm of chromosome 21 (21q22.13–22.2) (1) that are associated with the Down syndrome (DS) phenotype (2). The Drosophila homolog of DYRK1A, minibrain (mnb), was identified as a contributor to postembryonic neurogenesis (3). In this report, the authors identified mnb as a novel serine/threonine protein kinase with significant homology to eukaryotic kinases that are known regulate cell growth and division; as such, they hypothesized that altered neuroblast proliferation observed in mnb kinase family mutants was due to a similar involvement in cell cycle regulation. Subsequent studies in mice demonstrated that complete loss of Dyrk1a is embryonically lethal, while Dyrk1a haploinsufficiency leads to intellectual disability, microcephaly, and growth defects, underscoring its critical role in neurologic development (4, 5). DYRK1A overexpression has also been implicated in neurodevelopmental delays, cognitive deficits, and motor impairment (6). These studies suggest that the impact of DYRK1A on various pathologies may occur in a dose-dependent fashion, whereby both under- and overexpression can drive disease. Cancer as a disease entity exemplifies this dynamic nature of DYRK1A and the heterogeneity of its function within different, and occasionally the same, cell types. In this review, we will discuss DYRK1A’s structure, function, regulation, and role in cancer.
Domain Composition of DYRK1A
The DYRK family of kinases is evolutionarily conserved and divided into two categories: class I (DYRK1A and DYRK1B) and class II (DYRK2, DYRK3, and DYRK4) (7) (Figure 1). A key feature of DYRK family members is their ability to autophosphorylate tyrosine residues, rendering them catalytically active to phosphorylate other substrates on serine and threonine residues, hence the nomenclature of a dual-specificity kinase (8, 9). Class I DYRK proteins were historically defined by their putative nuclear localization signals (NLS), which precede the catalytic domain (10). They both contain a C-terminal PEST domain (11) and a WD-repeat domain, which binds the scaffolding protein DDB1 and CUL4 Associated Factor 7 (DCAF7) (12). This binding motif is critical for regulating nuclear localization of DYRK1A and maintaining protein levels (13). However, the presence of a NLS is in fact not unique to class I DYRKs, as both DYRK2 and DYRK4 have been shown to have NLS in subsequent loss-of-function studies (14, 15). Class II DYRKs also contain a NAPA domain that mediates autophosphorylation, although they lack the PEST domain (16).
Figure 1: Domain composition of the DYRK proteins.
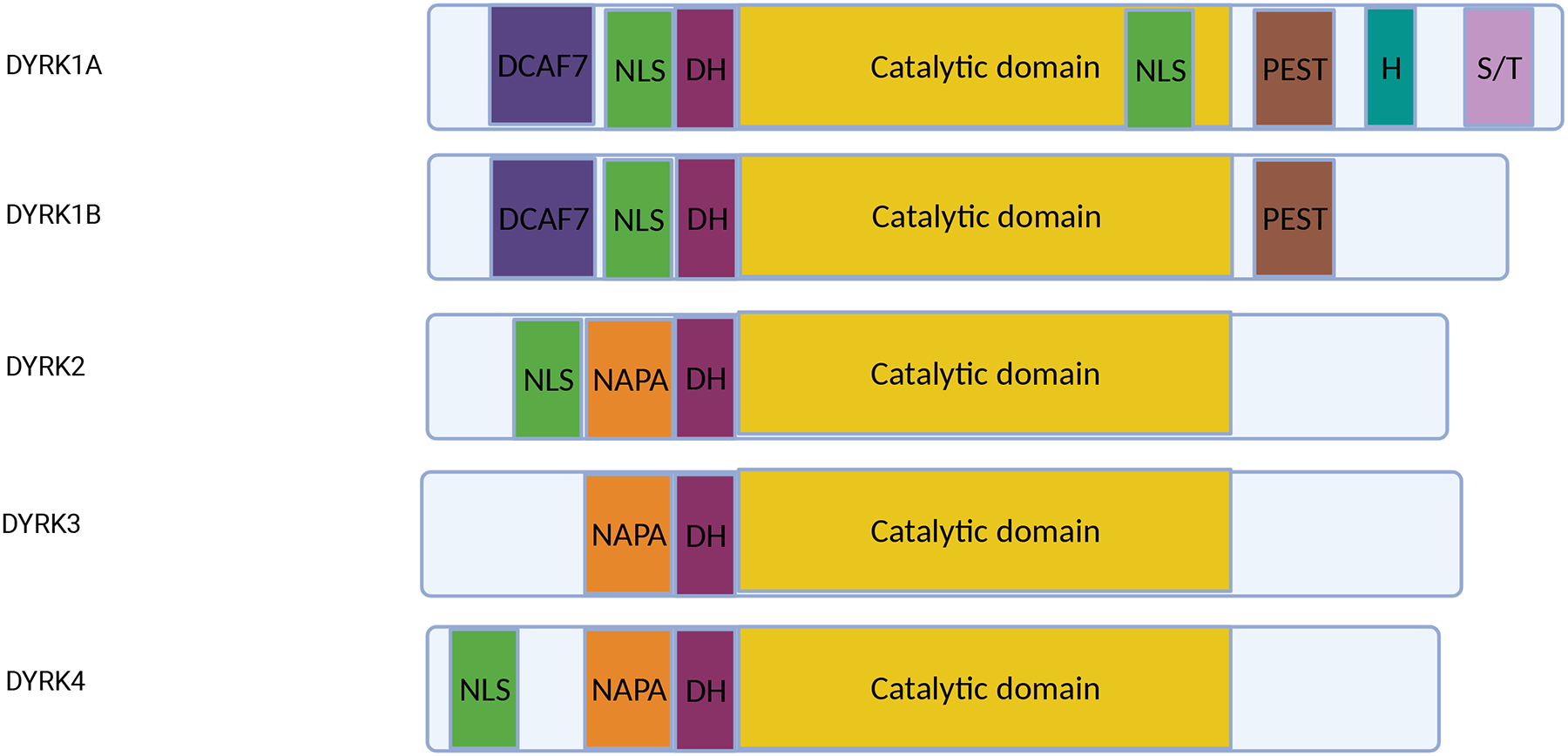
DYRK1 contains a DCAF7 binding domain, a nuclear localization site (NLS) at the N-terminus, a DYRK homology box (DH), and a proline, glutamate, serine, threonine (PEST) region. DYRK1A contains a second NLS within the catalytic domain, a histidine repeat (H), and serine/threonine (S/T) repeats near the C-terminus. DYRK2, DYRK3, and DYRK4 contain a NAPA domain, DH, and catalytic domain. DYRK2 and DYRK4 also have an N-terminus NLS.
DYRK1A is composed of three major domains (9). First, the DYRK homology (DH) box is essential for stabilizing tertiary structure in the N-terminus of autophosphorylated DYRK1A (9). Second, the conserved catalytic domain is required for kinase activity. Autophosphorylation of the critical activation-loop residue tyrosine 321 (Tyr321) within this domain takes place during or shortly after translation (8, 9, 17). Mutations in Tyr321 dramatically reduce the catalytic activity and impacts the overall function of its orthologs (9, 18). Third, DYRK1A has a PEST domain that facilitates DYRK1A degradation (10). In addition, DYRK1A contains a N-terminus NLS and a second within the catalytic domain (10), a C-terminal histidine repeat that promotes protein localization into nuclear speckles (19), and a Ser/Thr rich sequence that seems to regulate catalytic activity (20).
Regulation of DYRK1A
Transcriptional Regulation
Several proteins have been identified as upstream regulators of DYRK1A expression. RE1-silencing transcription factor (REST), also known as neuron-restrictive silencer factor (NRSF), is a zinc finger protein that acts as a master repressor of neuronal genes in differentiated non-neuronal tissues (21). REST activates DYRK1A transcription by binding to a neuron-restrictive silencer element located in the DYRK1A promoter region (22). In a negative feedback loop, DYRK1A can then phosphorylate REST, leading to its degradation (22). Using a transgenic mouse model of Dyrk1a overexpression, DYRK1A was found to interact with the SWI/SNF complex, which can bind REST, affecting its expression and dysregulating neuronal gene expression (23). However, this may not entirely be mediated by DYRK1A kinase activity. Separately, in embryonic stem cells and mice that model partial trisomy 21, REST was reported to be downregulated in a DYRK1A dose-dependent fashion (24). Consequently, the increase in DYRK1A expression and downregulation of REST was associated with a reduction in expression of pluripotency regulators and enhanced endoderm and mesoderm differentiation.
Myocyte-specific enhance factor 2D (MEF2D) is a transcription factor initially determined to be essential for muscle differentiation in both embryogenesis and adult regeneration (25) but was also found to promote post-mitotic neuronal survival (26). Of note, MEF2D upregulates DYRK1A expression in glioblastoma cells through a MEF2D responsive element in the promoter region of DYRK1A (27). The same group demonstrated that DYRK1A phosphorylates MEF2D at Ser251 and that increased DYRK1A expression or activity is inversely correlated with MEF2D transcriptional in HEK293 and U87MG cells, which was phenocopied by expression of phosphomimetic and phosphodeficient Ser 251 alleles (28). This phosphorylation event results in dissociation of MEF2D from DNA and decreased transactivation, demonstrating another negative feedback mechanism of a transcriptional activator.
Additionally, a recent kinase-focused CRISPR screen in KMT2A-rearranged B-cell acute lymphoblastic leukemia (B-ALL) cells identified a dependency on DYRK1A (29). ChIP-Seq revealed that the KMT2A-AFF1 fusion complex directly binds the DYRK1A promoter and regulates its transcription. Notably, DYRK1A expression levels were lower in KMT2A-rearranged B-ALLs than in other B-ALL subtypes. The authors also demonstrated that chemical inhibition of DYRK1A suppressed the growth of B-ALL cells with this rearrangement.
While the exact interplay between DYRK1A and its upstream transcriptional regulators requires investigation in the context of cancer. Several of these regulators are involved in tissue specific differentiation and drive a negative feedback mechanism after activation of DYRK1A expression. These regulators also demonstrate conflicting oncogenic and tumor suppressor roles in a cell-dependent context; for example, REST promotes central nervous system tumors yet is reported to have anti-tumor properties in lung, breast, and colon cancer (21).
Regulation via Ubiquitin
Beyond transcriptional regulation, DYRK1A levels are also tightly controlled at the protein level through ubiquitination. SCFβTrCP, an E3 ubiquitin ligase that promotes neuronal development by targeting REST for degradation in embryonic and neural stem cells (30), has also been shown to ubiquitinate and promote degradation of DYRK1A in HEK293 cells (31). As expected, DYRK1A protein levels negatively correlate with SCFβTrCP throughout the cell cycle, contributing to increased DYRK1A levels in G0/G1 and decreased levels in S and G2/M. Similar to DYRK1A, SCFβTrCP has also been found to act in an oncogenic or tumor suppressor role in different cancers depending on its substrates (32), perhaps reflecting an antagonistic relationship between the two proteins, although this remains to be examined.
Another E3 ubiquitin ligase, TRAF2, which has been implicated in inflammation-mediated tumor growth (33), facilitates K63-linked ubiquitination of DYRK1A, promoting its translocation to a number of subcellular structures including membranous vesicles such as endosomes (34). Once localized to an endosome, DYRK1A can phosphorylate Sprouty2 at the Thr75 residue thereby negatively regulating endocytosis and recycling of EGFR thereby leading to its stabilization; this in turn promotes growth of glioma cell lines. Moreover, TRAF2 knockdown phenocopies EGFR degradation seen with DYRK1A knockdown in these cell lines. As such, loss of DYRK1A signaling by genetic and pharmacologic approaches slowed growth of glioma cell lines, which could not be rescued by TRAF2 overexpression.
A recent study reported that p53 activation leads to degradation of DYRK1A and subsequent downregulation of EGFR-ERK signaling, leading to cellular senescence in vitro and in vivo (35). It was also shown that MDM2, a p53 transcriptional target and ubiquitin ligase, directly binds DYRK1A and promotes its polyubiquitination. MDM2 can also bind p53 in a negative feedback loop; however, the use of Nutlin-3a, which selectively disrupts MDM2-p53 binding but not MDM2-DYRK1A binding, led to p53 activation and increased MDM2 expression, ultimately causing DYRK1A degradation. Moreover, a single study in embryonic neuronal cells proposed that DYRK1A phosphorylates p53 at Ser15 (36) to cause cell cycle arrest. While this does invoke the possibility of DYRK1A as a regulator of p53 signaling, further studies are needed to more precisely define the relationship between DYRK1A and p53 in different cellular contexts.
Contributions of DYRK1A to normal and malignant cell growth
DYRK1A is associated with a multitude of tumors (Table 1), where it acts by modifying proteins that play key roles in cellular processes such as cell cycle, DNA damage repair, pre-mRNA splicing, transcription, angiogenesis, and protein stability (Figure 2).
Table 1.
Summary of the role of DYRK1A in different cancers.
| Cancer | Role | Signaling Pathway | References |
|---|---|---|---|
| DS-AMKL | tumor-promoting | NFAT | (61) |
| ALL | tumor-promoting | FOXO1, STAT3 | (46) |
| AML | tumor-suppressing | c-Myc | (125) |
| Glioblastoma | both | REST, RNA Polymerase II, EGFR, ID2, cyclin B | (21, 53, 82, 85, 126) |
| Neuroblastoma | tumor-promoting | p27 and cyclin D1 | (41) |
| PDAC | tumor-promoting | c-MET | (80) |
| Ovarian | tumor-promoting | MuvB, DREAM | (39) |
| NSCLC | tumor-promoting | STAT3, EGFR, c-MET, Mcl-1 | (75, 127) |
| Bladder | tumor-promoting | FGF2 | (128) |
| Osteosarcoma | both | DREAM, SIRT1 | (38, 52) |
| Cervical | tumor-suppressing | RNF169, 53BP1 | (49) |
| HNSCC | tumor-promoting | FGF2, FOXO3A | (88, 129) |
| Epithelial cancer in individuals with DS | tumor-suppressing | NFAT | (59) |
DS-AMKL = Down syndrome-acute megakaryoblastic leukemia; PDAC = pancreatic ductal adenocarcinoma; NSCLC = non-small cell lung cancer; HNSCC = head and neck squamous cell carcinoma.
Figure 2: Known DYRK1A substrates.

Targets of DYRK1A include proteins involved in cell cycle regulation, DNA damage response, transcription and cell signaling regulation, angiogenesis, tyrosine kinase regulation, cancer stem cell (CSC) properties, and alternative splicing.
DYRK1A contributes to cell cycle regulation
The DREAM (dimerization partner, RB-like, E2F and multi-vulval class B) complex is a group of proteins that assemble in G0 to repress cell cycle dependent gene expression and cell cycle progression (37). In addition, a critical component of the multi-vulval class B (MuvB) subunit of the DREAM complex is LIN52, which was shown to be phosphorylated by DYRK1A at Ser28 (38). Inhibition of DYRK1A or expression of a phosphodeficient allele of LIN52 (Ser28Ala) disrupted the assembly of the DREAM complex and significantly reduced the ability of cells to enter quiescence. Moreover, overexpression of a kinase-inactive allele of DYRK1A (Lys188Arg) led to increased proliferation of U2OS cells, while overexpression of a wild-type allele reduced proliferative capacity by half. This interplay between DYRK1A and the DREAM complex may not be limited to quiescence; co-expression of oncogenic HRAS with either DYRK1A Lys188Arg or LIN52 Ser28Ala, both of which have dominant-negative activity, was also associated with reduced cellular senescence. Furthermore, DYRK1A-mediated DREAM complex assembly contributes to ovarian cancer dormancy as DYRK1A inhibition reduced spheroid viability and restored sensitivity to chemotherapy targeting actively proliferating cells, suggestive of cells exiting quiescence (39).
Cyclins are another set of critical DYRK1A substrates. For example, DYRK1A has been shown to prolong G1 by phosphorylating and degrading cyclin D1 (40), and sustained phosphorylation of cyclin D1 and p27Kip1 by DYRK1A in neuroblastoma and neural stem cells decreased proliferation and increased differentiation (41).
DYRK1A also orchestrates early lymphopoiesis through phosphorylation of cyclin D3 (42). Highly proliferative lymphoid precursor cells known as large pre-B cells and double-negative (DN) thymocytes normally enter quiescence to facilitate maturation into small pre-B cells and double-positive (DP) thymocytes, respectively. In the absence of DYRK1A, B and T cell maturation are halted at the large pre-B cell and DN thymocyte stages, diminishing the production of small pre-B cells, DP thymocytes, and more differentiated lymphocytes. In this context, DYRK1A promotes quiescence through phosphorylation of cyclin D3, at Thr283, which resides in a phosphodegron motif conserved across all D-type cyclins, thereby leading to its ubiquitination and proteasomal-mediated degradation. Consequently, loss or inhibition of DYRK1A impaired quiescence and maturation of large pre-B cells and DN thymocytes through de-repression of E2F-mediated gene transcription in a cell cycle-dependent manner (42). Though this mechanism was paralleled in both B and T cell lineages, curiously it was not seen in myeloid cells. Although DYRK1A-deficient lymphocyte precursors had impaired ability to enter quiescence, they had reduced proliferation compared to control cells and accumulated in G2/M, suggesting a concomitant late cell cycle defect.
DYRK1A has also been reported to phosphorylate the ubiquitin ligase CDC23, which mediates mitotic protein degradation, at Ser588 in the glioblastoma cell line U251 (43). In this model, DYRK1A inhibition decreased Ser588 phosphorylation, impairing APC complex assembly, thus preventing degradation of cyclin B and subsequently causing hyperactivation of CDK1. Indeed, DYRK1A inhibition promoted tumor growth and the fraction of Ki67-positive cells in this study.
Taken together, DYRK1A has diverse, non-redundant roles in the cell cycle by regulating the balance between cell cycle entry and quiescence, with substrates including LIN52, cyclin D1, cyclin D3, and CDC23. However, the ability of DYRK1A to promote or inhibit tumor growth and survival in these roles depends on the cellular context. Nevertheless, given the fundamental importance of cell cycle regulation in cancer biology, DYRK1A remains a promising target.
DYRK1A contributes to the DNA damage response
Recent studies have shed light on the role of DYRK1A in DNA damage response and apoptosis. Activated Forkhead box proteins (FOXO) proteins affect the cell cycle and proliferation in colon cancer, glioblastoma, osteosarcoma, acute myeloid leukemia, and head and neck squamous cell carcinoma (HNSCC) (44). As a transcription factor, FOXO1 plays a critical role in activating genes related to cell proliferation and apoptosis. During the G2/M phase, FOXO1 acts as a DNA damage sensor and slows cell cycle progression to accommodate DNA repair or trigger apoptosis (45). DYRK1A phosphorylates FOXO1 at Ser329 in humans (orthologous to Ser326 in mice) and promotes its nuclear export and degradation (46, 47). When DYRK1A activity is ablated in normal pre-B cells, this increases the expression of FOXO1 transcriptional targets such as GADD45A, CCNG2, and BCL2L11 to cause a G2/M halt in response to cell cycle dysregulation and DNA damage accumulation without substantially increasing apoptosis; however, loss of both FOXO1 and DYRK1A activity preferentially kills B-ALL cells through increased sensitivity to replicative stress (46). Moreover, DYRK1A inhibition was found to sensitize leukemic cells to conventional chemotherapies that induce genotoxic stress.
Quantitative mass spectrometry revealed interactions of DYRK1A with numerous proteins involved in DNA damage repair, including RNF169, an E3 ubiquitin ligase that is an essential component of the cellular response to DNA double-stranded breaks (DSB) (48–51). DYRK1A was found to be recruited to sites of DNA damage through this interaction with RNF169. Moreover, knockdown of DYRK1A conferred increased sensitivity to ionizing radiation in colony formation assays (48). DYRK1A has also been linked to the DNA damage response via its phosphorylation of Sirtuin 1 (SIRT1) at Thr522, resulting in deacetylation of p53 in U2OS cells (52). Thus, there is accumulating evidence for the role of DYRK1A in regulating DNA damage through several distinct substrates.
DYRK1A regulates transcription and cell signaling
DYRK1A has been reported to regulate transcription through kinase dependent and independent interactions with RNA polymerase II (RNAPII) (53). ChIP-Seq data in T98G and HeLa cells revealed that DYRK1A is recruited to the promoters of ribosomal biogenesis and translation regulation and that its biding sites are enriched for the palindromic TCTCGCGAGA sequence. This study also revealed that DYRK1A phosphorylates the carboxy-terminal domain (CTD) of RNAPII at Ser2 and Ser5. A reduction in phosphorylation of these two residues by DYRK1A knockdown was found to impair the ability of RNAPII to associate with promoters. More recently, Lu et al demonstrated that DYRK1A contains a histidine-rich domain (HRD), which allows it to form phase-separated liquid droplets in vitro and in cells, thereby promoting highly efficient hyperphosphorylation of the RNAPII CTD (54). Deletion of the HRD reduced both CTD phosphorylation and co-immunoprecipitation of RNAPII, though did not affect DCAF7 co-immunoprecipitation. However, another group reported that DCAF7 promotes DYRK1A-RNAPII interaction and is essential for myogenic differentiation and expression of key myogenic genes, including MYH2, CAV3, and MYOG (55), suggesting that there may be multiple mechanisms by which DYRK1A localizes to RNAPII in subcellular compartments.
DYRK1A also regulates transcription factors that control cell signaling. Nuclear factor of activated T-cells (NFAT) is phosphorylated by DYRK1A and subsequently exported from the nucleus, preventing transactivation (56–58). Impaired NFAT nuclear export leads to a more invasive phenotype of the breast cancer cell line 4T1 (59) through upregulation of the metalloproteinase ADAMTS1 (60). Increased levels of DYRK1A in DS also contributes to the development of acute megakaryoblastic leukemia (AMKL) through inhibition of NFAT signaling (61).
The transcription factor Signal Transducer and Activator of Transcription 3 (STAT3) is another DYRK1A substrate that regulates tumor proliferation. Constitutive activation of STAT3 has been reported in many cancers, including hematologic malignancies and solid tumors (62, 63), and correlates with a poor prognosis (64). Canonically, STAT3 is activated by JAK-mediated phosphorylation at Tyr705, resulting in its dimerization and translocation to the nucleus to activate transcription (65, 66). DYRK1A phosphorylates STAT3 at Ser727 (67, 68), a residue conserved in both humans and mice, and several studies show that phosphorylation of STAT3 at Ser727 is critical for STAT3 activity, including non-canonical mitochondrial pathways (69–71). Aberrant STAT3 activation due to hyperactivation of upstream tyrosine kinases, and overexpression of stimulatory receptor-ligand interactions promote tumor progression (72, 73). In a mouse model of DS, DYRK1A overexpression enhances STAT3 activity and promotes astrogliogenesis (74). In non-small cell lung cancer (NSCLC), DYRK1A inhibition decreases STAT3 activity and decreases NSCLC proliferation due to impaired EGFR/MET signaling (75). Recently, phosphorylation of STAT3 Ser727 by DYRK1A has been linked to survival of B-ALL tumor cells through maintenance of canonical Tyr705 signaling and by reducing cellular stress induced by reactive oxidative species (ROS) (46).
In a recent study, Li et. al examined the activity of B-cell activating factor (BAFF) in autoimmunity and B-ALL (76). They report that BAFF promotes non-canonical NF-kB signaling in a DYRK1A-dependent manner. Specifically, DYRK1A phosphorylates TRAF3, a protein involved in ubiquitin mediated degradation of a noncanonical NF-kB inducing kinase, at Ser29 and facilitates B-cell tumor development.
DYRK1A in angiogenesis
In 2009, Ryeom and colleagues proposed that DYRK1A and DSCR1, which contribute to calcium homeostasis, control angiogenesis, providing a potential explanation for the decreased incidence of solid tumors in people with DS (77). Another study found that inhibition or silencing of DYRK1A in primary endothelial cells led to decreased intracellular Ca2+ influx in response to VEGF and reduced downstream NFAT activation (78). Such modulation of Ca2+/NFAT signaling by DYRK1A was discovered to be mediated through VEGF receptor 2 (VEGFR2) stability. Moreover, Dyrk1a haploinsufficient mice showed defects in developmental retinal vascularization, providing additional evidence that DYRK1A influences the angiogenic response (78). Finally, the kinase activity of DYRK1A is required for vascular formation in zebrafish via regulation of calcium signaling (79).
DYRK1A regulates tyrosine kinases involved in tumor growth
Major tyrosine kinase substrates of DYRK1A that drive tumor growth include c-MET and EGFR (75, 80). DYRK1A is upregulated in both pancreatic ductal adenocarcinoma (PDAC) and non-small cell lung cancer (NSCLC), where it has been associated with tumor growth and maintenance by modulating the activity of several downstream pathways. For example, DYRK1A expression is correlated with c-MET levels in the PANC-1 pancreatic cells, and DYRK1 knockdown led to reduced proliferation, suggesting that it may be a therapeutic target (80). In EGFR wild-type NSCLC cells, DYRK1A knockdown also decreased proliferation (75).
Like c-MET, EGFR degradation is also inhibited by DYRK1A(34). During adult neural progenitor cell division, DYRK1A inhibits EGFR degradation via phosphorylation of Thr75 on Sprouty2, a regulator of receptor tyrosine kinase turnover (81). In glioblastomas, increased DYRK1A expression correlated with increased EGFR levels, and inhibition of DYRK1A impaired self-renewal capacity in EGFR dependent glioblastoma cells (82). Conversely, in NSCLC, inhibition of DYRK1A reduced the levels of EGFR and MET, and consequently sensitized cells to AZD9291, an EGFR tyrosine kinase inhibitor (75). Of note, EGFR signaling is also required for Kras oncogene driven carcinogenesis in PDAC (83). Further studies have shown that EGFR cooperates and activates the AKT and STAT3 signaling pathways that together promote Kras driven oncogenic signals (83). As DYRK1A increases EGFR stability, these observations suggest an important tumor-inducing role of this pathway in PDAC as well.
DYRK1A regulates cancer stem cell-like properties
Recent data have shown that DYRK1A regulates the cancer stem cell population (84). Lee and colleagues discovered that DYRK1A-mediated phosphorylation of inhibitor of DNA binding 2 (ID2) at Thr27 blocks the ID2-VHL interaction and leads to HIF2α stabilization and cancer cell stemness in glioma (85). Downregulation of DYRK1A was found to increase HIF2α, suggesting that DYRK1A can act as a tumor suppressor in this setting. Similarly, DYRK1A induced cancer stem cell (CSC) differentiation by downregulating CDK5-SOX2 in the glioma line U251 (86). Conversely, DYRK1A inhibition in gliomas limited self-renewal capacity through decreased EGFR stability (82). DYRK1A may also induce stemness through REST (87), suggesting it can also function as an oncogene in gliomas. These data highlight DYRK1A’s context and tumor-dependent action within cancers of the same origin. Beyond gliomas, DYRK1A can induce FGF2 to repress differentiation and promoted CSC self-renewal capacity in oropharyngeal squamous cell carcinoma (OSCC) Notably, DYRK1A inhibition abrogates CSC maintenance, increases sensitivity to chemotherapy, and suppresses migration of OSCC (88).
DYRK1A and splicing
DYRK1A localizes to nuclear speckles and phosphorylates several key splicing factors including SRSF1, SRSF2, SRSF6, and SF3B1 (19, 89–93). Several studies have shown that DYRK1A modulates alternative splicing in neurobiology; for example, DYRK1A promotes alternative splicing of Tau through modulation of SRSF1 and SRSF2 (90, 94). New data from Abdel-Wahab and colleagues demonstrate that venetoclax-resistant AML cell lines have enhanced sensitivity to DYRK1A and CDC2-like kinase (CLK) inhibition, possibly through SR proteins (95). Further effort into elucidating DYRK1A’s impact in this process deserves special attention. (96).
Prospects for targeting DYRK1A
Given the multifaceted roles of DYRK1A in the phenotypes of DS, neurodegenerative diseases, and cancer, there has been significant interest in the development of potent and selective DYRK1A inhibitors (Table 2). In this section, we will review select DYRK1A-targeting therapies tested in cancer.
Table 2.
DYRK1A inhibitors described in cancer studies
| DYRK1A Inhibitors | Class of compound | Cancer/Disease tested in | Natural/Synthetic | References |
|---|---|---|---|---|
| harmine | β-carboline | In vitro: colon, gastric, pancreatic, lung, liver, breast ovarian, glioblastoma; In vivo: glioma, HNSCC | natural | (82, 129, 130) |
| L41 | Leucettine | In vitro: glioblastoma | natural | (43) |
| licocoumarone | flavonoid | In vitro: PDAC | natural | (131) |
| EGCG | Polyphenol | Clinical trials: colon cancer and prostate cancer | natural | (132, 133) |
| Lamerallins | Chromenoindole | In vitro: leukemia, prostate, melanoma, colon, ovarian, renal, glioma, breast, NSCLC | synthetic | (134, 135) |
| INDY | benzothiazol | In vitro: ovarian, glioblastoma | synthetic | (39, 82) |
| Meriolins | Pyrimidinylindol/azaindol | In vitro: glioma | synthetic | (136) |
| Meridianins | Pyrimidinylindol/azaindol | In vitro: breast, cervical, ovarian leukemia, HNSCC | synthetic | (137–139) |
| EHT 1610 | Thiazolo[5,4-f]quinazoline | In vitro/in vivo: B-ALL | synthetic | (46) |
| EHT 5372 | Thiazolo[5,4-f]quinazoline | In vitro: pancreatic cancer | synthetic | (108) |
| FC 162 | Thiazolo[5,4-f]quinazoline | In vitro: neuroblastoma | synthetic | (105) |
| pyrido[3,4-g]quinazoline derivatives | pyrido[3,4-g]quinazoline | In vitro: colon, breast, neuroblastoma, osteosarcoma | synthetic | (140) |
| AnnH-75 | β-carboline | In vitro: cervical cancer, neuroblastoma | synthetic | (141) |
| Compound 34 | pyrrolopyrimidine |
In vivo: glioblastoma
In vitro: osteosarcoma |
synthetic | (120) |
Natural compounds that inhibit DYRK1A
Several small molecules, including natural products, have been investigated for their ability to inhibit DYRK1A. For example, a clinical trial conducted in 2018 showed that epigallocatechine gallate (EGCG), a potent catechin found in green tea, improved visual recognition memory and working memory performance in patients with DS (97). While being a potent DYRK1A inhibitor in vitro, EGCG also binds and inhibits p38-regulated/activated protein kinase (PRAK)(98). EGCG has been tested both in vitro and in vivo in several cancers, including hepatocellular carcinoma, pancreatic cancer, prostate cancer, breast cancer, melanoma, head and neck cancer, and digestive cancers (99). In head and neck cancer, EGCG treatment was found to decrease cellular proliferation due to the suppression of the Notch pathway, while in triple negative breast cancer, EGCG induces apoptosis by scavenging ROS. However, EGCG has several disadvantages for in-vivo usage such as poor bioavailability and heterogeneous effects on signaling pathways.
Another widely used DYRK1A inhibitor for in vitro and in vivo studies has been a β-carboline alkaloid named harmine, an ATP-competitive inhibitor that was initially assayed for activity in Parkinson’s disease (100). Harmine has been tested in multiple cancer subtypes, including breast, pancreatic, HNSCC and ovarian cancer and has been shown to effectively reduce tumor progression in mice (101). Although harmine is a potent DYRK1A inhibitor, it also targets other DYRKs and monoamine oxidase (MAO-A), resulting in side effects that limit its therapeutic potential (102). To overcome this limitation, several derivatives of harmine are being synthesized that are more selective for DYRK1A (103, 104).
The natural compound inhibitor L41, a type of leucettine, has potent activity against DYRK1A(105) as well as CLK. L41 has been found to decrease memory impairments and neurotoxicity in mice treated with Aβ25–35 peptide that represents a non-transgenic model that mimics Alzheimer’s disease (AD)-like toxicity (106). Indeed, L41 displays effective activity (IC50=40nm) against U251 cells in vitro (43).
Race to develop selective inhibitors
Synthetic DYRK1A inhibitors Methyl 9-anilinothiazolo (5,4-f)quinazoline-2-carbimidates 1 and 2, commonly known as EHT 5372 and EHT 1610, have been shown to inhibit DYRK1A in neurologic disease (107) and in cancer. In PANC-1 tumor cells, EHT 5372 induced exit from quiescence and entry into the cell cycle while also increasing DNA damage and apoptosis (108). Exposure of murine pre-B cells to EHT 1610 recapitulated the phenotype seen upon Dyrk1a silencing, including loss of pre-B cell colony formation (42), and EHT 1610 also demonstrated anti-tumor activity in models of B-ALL (46). Moreover, treatment of KMT2A-rearranged B-ALL cell lines with EHT 1610 inhibited their proliferation similar to what was observed with genetic inhibition of DYRK1A using CRISPR (29). A more recent derivative of EHT 1610, FC162, displayed similar effects on B cell growth (105, 109).
CLK inhibitors target alternative splicing but demonstrate off-target effects on DYRK1A due to similarities in structure between their respective catalytic kinase domains (110, 111). CX-4945 (Silmitasertib), an orally bioavailable CLK inhibitor, exerts its effects through multiple survival pathways (112) and notably displays potent DYRK1A inhibition (IC50=6.8nm). Thus far, CX-4945 has demonstrated efficacy across multiple tumor cell lines, including lymphoid, myeloid, and gastric tumor cells (113–117). Preliminary data from a phase Ib/II trial using CX-4945 with gemcitabine and cisplatin for patients with locally advanced or metastatic cholangiocarcinoma revealed improved outcomes (118). Furthermore, clinical trials in medulloblastoma (NCT03904862), multiple myeloma (NCT01199718, NCT00891280), and basal cell carcinoma (NCT03897036) are underway.
Finally, a number of other selective inhibitors have been reported though structure-based discovery (119–123). From these advanced studies, it appears that we are close to identifying clinically viable compounds for multiple indications, including metabolic, neurologic, and oncologic disorders.
It should be highlighted that most DYRK1A inhibitors also target DYRK1B, which is upregulated in many cancers and is considered to be tumorigenic (124). Similar to DYRK1A, DYRK1B regulates cell proliferation, cell cycle and has been shown to regulate ROS levels in response to stress (108). Due to the similar function and upregulation of these two DYRKs in cancer, it is unclear whether the effect mediated by these inhibitors is due to reduced activity of DYRK1A or DYRK1B. Development of more selective compounds and genetic studies to target each homolog individually are needed to clearly establish the activities of these two genes to cancer.
Summary
DYRK1A is linked to many cellular processes, including proliferation, self-renewal, DNA damage, transcriptional regulation, apoptosis, ubiquitination, cancer stem cell maintenance, and alternative splicing. However, DYRK1A can promote or inhibit tumor growth based on cancer subtype and stage. Since DYRK1A is a potent regulator of quiescence, DYRK1A inhibition in cancer has the potential to trigger relapse of dormant cancer cells. To exploit this possibility, one strategic therapeutic option might be to administer a DYRK1A inhibitor in parallel with chemotherapy. In this light, combination chemotherapy could enable targeting dormant cancer cells by pushing them out of quiescence and into cell cycling; this has recently been demonstrated in pre-clinical studies of B-ALL (46). Additionally, recent studies have shown that DYRK1A can enhance sensitivity to radiation, and thus DYRK1A inhibition may offer a novel approach to radiosensitization (48, 49). Future studies must address ongoing issues with DYRK1A as a potential therapeutic target in cancer, including distinguishing off-target/on-target effects of inhibitors in vivo, characterizing the effects of DYRK1A inhibition in normal tissues, elucidating redundant and non-redundant roles of DYRK1A and DYRK1B in malignancy, and optimizing the pharmacokinetics of small molecule inhibitors. In this review, we summarized studies from various types of cancer that implicate DYRK1A in their pathogenesis and persistence. Additional studies are needed to validate many of the cell-specific findings; however, the emerging importance of DYRK1A in cancer biology warrants the ongoing development of novel, selective, and clinically efficacious inhibitors.
Acknowledgements
This review was supported in part by the NIH (R35 CA253096). The content is solely the responsibility of the authors and does not necessarily represent the official views of the NIH. Additional support was provided St. Jude/ALSAC. EH is supported by the American Society of Hematology’s Medical Student Physician-Scientist Award.
Footnotes
Conflict of Interest
The authors declare that they have no conflicts of interest.
REFERENCES
- 1.Song WJ, Sternberg LR, Kasten-Sportès C, Keuren ML, Chung SH, Slack AC, et al. Isolation of human and murine homologues of the Drosophila minibrain gene: human homologue maps to 21q22.2 in the Down syndrome “critical region”. Genomics. 1996;38(3):331–9. [DOI] [PubMed] [Google Scholar]
- 2.Delabar JM, Theophile D, Rahmani Z, Chettouh Z, Blouin JL, Prieur M, et al. Molecular mapping of twenty-four features of Down syndrome on chromosome 21. Eur J Hum Genet. 1993;1(2):114–24. [DOI] [PubMed] [Google Scholar]
- 3.Tejedor F, Zhu XR, Kaltenbach E, Ackermann A, Baumann A, Canal I, et al. minibrain: a new protein kinase family involved in postembryonic neurogenesis in Drosophila. Neuron. 1995;14(2):287–301. [DOI] [PubMed] [Google Scholar]
- 4.Altafaj X, Dierssen M, Baamonde C, Marti E, Visa J, Guimera J, et al. Neurodevelopmental delay, motor abnormalities and cognitive deficits in transgenic mice overexpressing Dyrk1A (minibrain), a murine model of Down’s syndrome. Hum Mol Genet. 2001;10(18):1915–23. [DOI] [PubMed] [Google Scholar]
- 5.Ji J, Lee H, Argiropoulos B, Dorrani N, Mann J, Martinez-Agosto JA, et al. DYRK1A haploinsufficiency causes a new recognizable syndrome with microcephaly, intellectual disability, speech impairment, and distinct facies. Eur J Hum Genet. 2015;23(11):1473–81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Feki A, Hibaoui Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain sciences. 2018;8(10):187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Becker W, Joost HG. Structural and functional characteristics of Dyrk, a novel subfamily of protein kinases with dual specificity. Prog Nucleic Acid Res Mol Biol. 1999;62:1–17. [DOI] [PubMed] [Google Scholar]
- 8.Himpel S, Panzer P, Eirmbter K, Czajkowska H, Sayed M, Packman LC, et al. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. Biochem J. 2001;359(Pt 3):497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Soundararajan M, Roos AK, Savitsky P, Filippakopoulos P, Kettenbach AN, Olsen JV, et al. Structures of Down syndrome kinases, DYRKs, reveal mechanisms of kinase activation and substrate recognition. Structure. 2013;21(6):986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Galceran J, de Graaf K, Tejedor FJ, Becker W. The MNB/DYRK1A protein kinase: genetic and biochemical properties. J Neural Transm Suppl. 2003(67):139–48. [DOI] [PubMed] [Google Scholar]
- 11.Soundararajan M, Roos Annette K, Savitsky P, Filippakopoulos P, Kettenbach Arminja N, Olsen Jesper V, et al. Structures of Down Syndrome Kinases, DYRKs, Reveal Mechanisms of Kinase Activation and Substrate Recognition. Structure. 2013;21(6):986–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Miyata Y, Nishida E. DYRK1A binds to an evolutionarily conserved WD40-repeat protein WDR68 and induces its nuclear translocation. Biochim Biophys Acta. 2011;1813(10):1728–39. [DOI] [PubMed] [Google Scholar]
- 13.Yousefelahiyeh M, Xu J, Alvarado E, Yu Y, Salven D, Nissen RM. DCAF7/WDR68 is required for normal levels of DYRK1A and DYRK1B. PLoS One. 2018;13(11):e0207779. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Taira N, Yamamoto H, Yamaguchi T, Miki Y, Yoshida K. ATM augments nuclear stabilization of DYRK2 by inhibiting MDM2 in the apoptotic response to DNA damage. The Journal of biological chemistry. 2010;285(7):4909–19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Papadopoulos C, Arato K, Lilienthal E, Zerweck J, Schutkowski M, Chatain N, et al. Splice variants of the dual specificity tyrosine phosphorylation-regulated kinase 4 (DYRK4) differ in their subcellular localization and catalytic activity. J Biol Chem. 2011;286(7):5494–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kinstrie R, Luebbering N, Miranda-Saavedra D, Sibbet G, Han J, Lochhead PA, et al. Characterization of a domain that transiently converts class 2 DYRKs into intramolecular tyrosine kinases. Sci Signal. 2010;3(111):ra16. [DOI] [PubMed] [Google Scholar]
- 17.Kentrup H, Becker W, Heukelbach J, Wilmes A, Schurmann A, Huppertz C, et al. Dyrk, a dual specificity protein kinase with unique structural features whose activity is dependent on tyrosine residues between subdomains VII and VIII. J Biol Chem. 1996;271(7):3488–95. [DOI] [PubMed] [Google Scholar]
- 18.Himpel S, Panzer P, Eirmbter K, Czajkowska H, Sayed M, Packman LC, et al. Identification of the autophosphorylation sites and characterization of their effects in the protein kinase DYRK1A. The Biochemical journal. 2001;359(Pt 3):497–505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Alvarez M, Estivill X, de la Luna S. DYRK1A accumulates in splicing speckles through a novel targeting signal and induces speckle disassembly. J Cell Sci. 2003;116(Pt 15):3099–107. [DOI] [PubMed] [Google Scholar]
- 20.Jin N, Yin X, Gu J, Zhang X, Shi J, Qian W, et al. Truncation and Activation of Dual Specificity Tyrosine Phosphorylation-regulated Kinase 1A by Calpain I: A MOLECULAR MECHANISM LINKED TO TAU PATHOLOGY IN ALZHEIMER DISEASE. The Journal of biological chemistry. 2015;290(24):15219–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Negrini S, Prada I, D’Alessandro R, Meldolesi J. REST: an oncogene or a tumor suppressor? Trends Cell Biol. 2013;23(6):289–95. [DOI] [PubMed] [Google Scholar]
- 22.Lu M, Zheng L, Han B, Wang L, Wang P, Liu H, et al. REST regulates DYRK1A transcription in a negative feedback loop. J Biol Chem. 2011;286(12):10755–63. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Lepagnol-Bestel A-M, Zvara A, Maussion G, Quignon F, Ngimbous B, Ramoz N, et al. DYRK1A interacts with the REST/NRSF-SWI/SNF chromatin remodelling complex to deregulate gene clusters involved in the neuronal phenotypic traits of Down syndrome. Human Molecular Genetics. 2009;18(8):1405–14. [DOI] [PubMed] [Google Scholar]
- 24.Canzonetta C, Mulligan C, Deutsch S, Ruf S, O’Doherty A, Lyle R, et al. DYRK1A-dosage imbalance perturbs NRSF/REST levels, deregulating pluripotency and embryonic stem cell fate in Down syndrome. Am J Hum Genet. 2008;83(3):388–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Liu N, Nelson BR, Bezprozvannaya S, Shelton JM, Richardson JA, Bassel-Duby R, et al. Requirement of MEF2A, C, and D for skeletal muscle regeneration. Proceedings of the National Academy of Sciences. 2014;111(11):4109–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Li M, Linseman DA, Allen MP, Meintzer MK, Wang X, Laessig T, et al. Myocyte enhancer factor 2A and 2D undergo phosphorylation and caspase-mediated degradation during apoptosis of rat cerebellar granule neurons. The Journal of neuroscience : the official journal of the Society for Neuroscience. 2001;21(17):6544–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wang P, Wang L, Chen L, Sun X. Dual-specificity tyrosine-phosphorylation regulated kinase 1A Gene Transcription is regulated by Myocyte Enhancer Factor 2D. Sci Rep. 2017;7(1):7240. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wang P, Zhao J, Sun X. DYRK1A phosphorylates MEF2D and decreases its transcriptional activity. J Cell Mol Med. 2021;25(13):6082–93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Hurtz C, Carroll MP, Tasian SK, Wertheim G, Bhansali RS, Lee SJ, et al. DYRK1A Is Required to Alleviate Replication Stress in KMT2A-Rearranged Acute Lymphoblastic Leukemia. Blood. 2020;136(Supplement 1):39–40. [Google Scholar]
- 30.Westbrook TF, Hu G, Ang XL, Mulligan P, Pavlova NN, Liang A, et al. SCFbeta-TRCP controls oncogenic transformation and neural differentiation through REST degradation. Nature. 2008;452(7185):370–4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Liu Q, Tang Y, Chen L, Liu N, Lang F, Liu H, et al. E3 Ligase SCFbetaTrCP-induced DYRK1A Protein Degradation Is Essential for Cell Cycle Progression in HEK293 Cells. J Biol Chem. 2016;291(51):26399–409. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Senft D, Qi J, Ronai ZA. Ubiquitin ligases in oncogenic transformation and cancer therapy. Nat Rev Cancer. 2018;18(2):69–88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Jin J, Xiao Y, Hu H, Zou Q, Li Y, Gao Y, et al. Proinflammatory TLR signalling is regulated by a TRAF2-dependent proteolysis mechanism in macrophages. Nature Communications. 2015;6(1):5930. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Zhang P, Zhang Z, Fu Y, Zhang Y, Washburn MP, Florens L, et al. K63-linked ubiquitination of DYRK1A by TRAF2 alleviates Sprouty 2-mediated degradation of EGFR. Cell Death Dis. 2021;12(6):608. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Xu X, Liu Q, Zhang C, Ren S, Xu L, Zhao Z, et al. Inhibition of DYRK1A-EGFR axis by p53-MDM2 cascade mediates the induction of cellular senescence. Cell Death Dis. 2019;10(4):282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Park J, Oh Y, Yoo L, Jung MS, Song WJ, Lee SH, et al. Dyrk1A phosphorylates p53 and inhibits proliferation of embryonic neuronal cells. J Biol Chem. 2010;285(41):31895–906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Iness AN, Litovchick L. MuvB: A Key to Cell Cycle Control in Ovarian Cancer. Front Oncol. 2018;8:223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Litovchick L, Florens LA, Swanson SK, Washburn MP, DeCaprio JA. DYRK1A protein kinase promotes quiescence and senescence through DREAM complex assembly. Genes Dev. 2011;25(8):801–13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.MacDonald J, Ramos-Valdes Y, Perampalam P, Litovchick L, DiMattia GE, Dick FA. A Systematic Analysis of Negative Growth Control Implicates the DREAM Complex in Cancer Cell Dormancy. Mol Cancer Res. 2017;15(4):371–81. [DOI] [PubMed] [Google Scholar]
- 40.Chen JY, Lin JR, Tsai FC, Meyer T. Dosage of Dyrk1a shifts cells within a p21-cyclin D1 signaling map to control the decision to enter the cell cycle. Mol Cell. 2013;52(1):87–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Soppa U, Schumacher J, Florencio Ortiz V, Pasqualon T, Tejedor FJ, Becker W. The Down syndrome-related protein kinase DYRK1A phosphorylates p27(Kip1) and Cyclin D1 and induces cell cycle exit and neuronal differentiation. Cell Cycle. 2014;13(13):2084–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Thompson BJ, Bhansali R, Diebold L, Cook DE, Stolzenburg L, Casagrande AS, et al. DYRK1A controls the transition from proliferation to quiescence during lymphoid development by destabilizing Cyclin D3. J Exp Med. 2015;212(6):953–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Recasens A, Humphrey SJ, Ellis M, Hoque M, Abbassi RH, Chen B, et al. Global phosphoproteomics reveals DYRK1A regulates CDK1 activity in glioblastoma cells. Cell Death Discovery. 2021;7(1):81. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Jiramongkol Y, Lam EWF. FOXO transcription factor family in cancer and metastasis. Cancer and Metastasis Reviews. 2020;39(3):681–709. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Furukawa-Hibi Y, Yoshida-Araki K, Ohta T, Ikeda K, Motoyama N. FOXO forkhead transcription factors induce G(2)-M checkpoint in response to oxidative stress. J Biol Chem. 2002;277(30):26729–32. [DOI] [PubMed] [Google Scholar]
- 46.Bhansali RS, Rammohan M, Lee P, Laurent AP, Wen Q, Suraneni P, et al. DYRK1A regulates B cell acute lymphoblastic leukemia through phosphorylation of FOXO1 and STAT3. The Journal of Clinical Investigation. 2021;131(1). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Woods YL, Rena G, Morrice N, Barthel A, Becker W, Guo S, et al. The kinase DYRK1A phosphorylates the transcription factor FKHR at Ser329 in vitro, a novel in vivo phosphorylation site. Biochem J. 2001;355(Pt 3):597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Roewenstrunk J, Di Vona C, Chen J, Borras E, Dong C, Arato K, et al. A comprehensive proteomics-based interaction screen that links DYRK1A to RNF169 and to the DNA damage response. Sci Rep. 2019;9(1):6014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Guard SE, Poss ZC, Ebmeier CC, Pagratis M, Simpson H, Taatjes DJ, et al. The nuclear interactome of DYRK1A reveals a functional role in DNA damage repair. Sci Rep. 2019;9(1):6539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Menon VR, Ananthapadmanabhan V, Swanson S, Saini S, Sesay F, Yakovlev V, et al. DYRK1A regulates the recruitment of 53BP1 to the sites of DNA damage in part through interaction with RNF169. Cell Cycle. 2019;18(5):531–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.An L, Dong C, Li J, Chen J, Yuan J, Huang J, et al. RNF169 limits 53BP1 deposition at DSBs to stimulate single-strand annealing repair. Proc Natl Acad Sci U S A. 2018;115(35):E8286–E95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Guo X, Williams JG, Schug TT, Li X. DYRK1A and DYRK3 promote cell survival through phosphorylation and activation of SIRT1. J Biol Chem. 2010;285(17):13223–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Di Vona C, Bezdan D, Islam AB, Salichs E, Lopez-Bigas N, Ossowski S, et al. Chromatin-wide profiling of DYRK1A reveals a role as a gene-specific RNA polymerase II CTD kinase. Mol Cell. 2015;57(3):506–20. [DOI] [PubMed] [Google Scholar]
- 54.Lu H, Yu D, Hansen AS, Ganguly S, Liu R, Heckert A, et al. Phase-separation mechanism for C-terminal hyperphosphorylation of RNA polymerase II. Nature. 2018;558(7709):318–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Yu D, Cattoglio C, Xue Y, Zhou Q. A complex between DYRK1A and DCAF7 phosphorylates the C-terminal domain of RNA polymerase II to promote myogenesis. Nucleic Acids Research. 2019;47(9):4462–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Leontovich AA, Jalalirad M, Salisbury JL, Mills L, Haddox C, Schroeder M, et al. NOTCH3 expression is linked to breast cancer seeding and distant metastasis. Breast Cancer Res. 2018;20(1):105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Macian F NFAT proteins: key regulators of T-cell development and function. Nat Rev Immunol. 2005;5(6):472–84. [DOI] [PubMed] [Google Scholar]
- 58.Arron JR, Winslow MM, Polleri A, Chang CP, Wu H, Gao X, et al. NFAT dysregulation by increased dosage of DSCR1 and DYRK1A on chromosome 21. Nature. 2006;441(7093):595–600. [DOI] [PubMed] [Google Scholar]
- 59.Jauliac S, Lopez-Rodriguez C, Shaw LM, Brown LF, Rao A, Toker A. The role of NFAT transcription factors in integrin-mediated carcinoma invasion. Nat Cell Biol. 2002;4(7):540–4. [DOI] [PubMed] [Google Scholar]
- 60.Quang CT, Leboucher S, Passaro D, Fuhrmann L, Nourieh M, Vincent-Salomon A, et al. The calcineurin/NFAT pathway is activated in diagnostic breast cancer cases and is essential to survival and metastasis of mammary cancer cells. Cell Death Dis. 2015;6(2):e1658. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Malinge S, Bliss-Moreau M, Kirsammer G, Diebold L, Chlon T, Gurbuxani S, et al. Increased dosage of the chromosome 21 ortholog Dyrk1a promotes megakaryoblastic leukemia in a murine model of Down syndrome. J Clin Invest. 2012;122(3):948–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Siveen KS, Sikka S, Surana R, Dai X, Zhang J, Kumar AP, et al. Targeting the STAT3 signaling pathway in cancer: role of synthetic and natural inhibitors. Biochim Biophys Acta. 2014;1845(2):136–54. [DOI] [PubMed] [Google Scholar]
- 63.Yin W, Cheepala S, Roberts JN, Syson-Chan K, DiGiovanni J, Clifford JL. Active Stat3 is required for survival of human squamous cell carcinoma cells in serum-free conditions. Mol Cancer. 2006;5:15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Wu P, Wu D, Zhao L, Huang L, Shen G, Huang J, et al. Prognostic role of STAT3 in solid tumors: a systematic review and meta-analysis. Oncotarget. 2016;7(15):19863–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Okada Y, Watanabe T, Shoji T, Taguchi K, Ogo N, Asai A. Visualization and quantification of dynamic STAT3 homodimerization in living cells using homoFluoppi. Sci Rep. 2018;8(1):2385. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Zhong Z, Wen Z, Darnell JE, Jr. Stat3: a STAT family member activated by tyrosine phosphorylation in response to epidermal growth factor and interleukin-6. Science. 1994;264(5155):95–8. [DOI] [PubMed] [Google Scholar]
- 67.Li D, Jackson RA, Yusoff P, Guy GR. Direct association of Sprouty-related protein with an EVH1 domain (SPRED) 1 or SPRED2 with DYRK1A modifies substrate/kinase interactions. The Journal of biological chemistry. 2010;285(46):35374–85. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Kurabayashi N, Nguyen MD, Sanada K. DYRK1A overexpression enhances STAT activity and astrogliogenesis in a Down syndrome mouse model. EMBO reports. 2015;16(11):1548–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Aznar S, Valeron PF, del Rincon SV, Perez LF, Perona R, Lacal JC. Simultaneous tyrosine and serine phosphorylation of STAT3 transcription factor is involved in Rho A GTPase oncogenic transformation. Mol Biol Cell. 2001;12(10):3282–94. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakaguchi M, Oka M, Iwasaki T, Fukami Y, Nishigori C. Role and regulation of STAT3 phosphorylation at Ser727 in melanocytes and melanoma cells. J Invest Dermatol. 2012;132(7):1877–85. [DOI] [PubMed] [Google Scholar]
- 71.Wen Z, Zhong Z, Darnell JE, Jr. Maximal activation of transcription by Stat1 and Stat3 requires both tyrosine and serine phosphorylation. Cell. 1995;82(2):241–50. [DOI] [PubMed] [Google Scholar]
- 72.Chan KS, Sano S, Kataoka K, Abel E, Carbajal S, Beltran L, et al. Forced expression of a constitutively active form of Stat3 in mouse epidermis enhances malignant progression of skin tumors induced by two-stage carcinogenesis. Oncogene. 2008;27(8):1087–94. [DOI] [PubMed] [Google Scholar]
- 73.Johnston PA, Grandis JR. STAT3 signaling: anticancer strategies and challenges. Mol Interv. 2011;11(1):18–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Kurabayashi N, Nguyen MD, Sanada K. DYRK1A overexpression enhances STAT activity and astrogliogenesis in a Down syndrome mouse model. EMBO Rep. 2015;16(11):1548–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Li YL, Ding K, Hu X, Wu LW, Zhou DM, Rao MJ, et al. DYRK1A inhibition suppresses STAT3/EGFR/Met signalling and sensitizes EGFR wild-type NSCLC cells to AZD9291. J Cell Mol Med. 2019;23(11):7427–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Li Y, Xie X, Jie Z, Zhu L, Yang J-Y, Ko C-J, et al. DYRK1a mediates BAFF-induced noncanonical NF-kB activation to promote autoimmunity and B cell leukemogenesis. Blood. 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Baek KH, Zaslavsky A, Lynch RC, Britt C, Okada Y, Siarey RJ, et al. Down’s syndrome suppression of tumour growth and the role of the calcineurin inhibitor DSCR1. Nature. 2009;459(7250):1126–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Rozen EJ, Roewenstrunk J, Barallobre MJ, Di Vona C, Jung C, Figueiredo AF, et al. DYRK1A Kinase Positively Regulates Angiogenic Responses in Endothelial Cells. Cell Rep. 2018;23(6):1867–78. [DOI] [PubMed] [Google Scholar]
- 79.Cho HJ, Lee JG, Kim JH, Kim SY, Huh YH, Kim HJ, et al. Vascular defects of DYRK1A knockouts are ameliorated by modulating calcium signaling in zebrafish. Dis Model Mech. 2019;12(5). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Luna J, Boni J, Cuatrecasas M, Bofill-De Ros X, Nunez-Manchon E, Gironella M, et al. DYRK1A modulates c-MET in pancreatic ductal adenocarcinoma to drive tumour growth. Gut. 2019;68(8):1465–76. [DOI] [PubMed] [Google Scholar]
- 81.Ferron SR, Pozo N, Laguna A, Aranda S, Porlan E, Moreno M, et al. Regulated segregation of kinase Dyrk1A during asymmetric neural stem cell division is critical for EGFR-mediated biased signaling. Cell Stem Cell. 2010;7(3):367–79. [DOI] [PubMed] [Google Scholar]
- 82.Pozo N, Zahonero C, Fernandez P, Linares JM, Ayuso A, Hagiwara M, et al. Inhibition of DYRK1A destabilizes EGFR and reduces EGFR-dependent glioblastoma growth. J Clin Invest. 2013;123(6):2475–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Navas C, Hernandez-Porras I, Schuhmacher AJ, Sibilia M, Guerra C, Barbacid M. EGF receptor signaling is essential for k-ras oncogene-driven pancreatic ductal adenocarcinoma. Cancer Cell. 2012;22(3):318–30. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Batlle E, Clevers H. Cancer stem cells revisited. Nature Medicine. 2017;23(10):1124–34. [DOI] [PubMed] [Google Scholar]
- 85.Lee SB, Frattini V, Bansal M, Castano AM, Sherman D, Hutchinson K, et al. An ID2-dependent mechanism for VHL inactivation in cancer. Nature. 2016;529(7585):172–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen B, McCuaig-Walton D, Tan S, Montgomery AP, Day BW, Kassiou M, et al. DYRK1A Negatively Regulates CDK5-SOX2 Pathway and Self-Renewal of Glioblastoma Stem Cells. International journal of molecular sciences. 2021;22(8):4011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Kamal MM, Sathyan P, Singh SK, Zinn PO, Marisetty AL, Liang S, et al. REST regulates oncogenic properties of glioblastoma stem cells. Stem cells (Dayton, Ohio). 2012;30(3):405–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Martin CE, Nguyen A, Kang MK, Kim RH, Park N-H, Shin K-H. DYRK1A is required for maintenance of cancer stemness, contributing to tumorigenic potential in oral/oropharyngeal squamous cell carcinoma. Experimental Cell Research. 2021;405(1):112656. [DOI] [PubMed] [Google Scholar]
- 89.Wegiel J, Kaczmarski W, Barua M, Kuchna I, Nowicki K, Wang KC, et al. Link between DYRK1A overexpression and several-fold enhancement of neurofibrillary degeneration with 3-repeat tau protein in Down syndrome. J Neuropathol Exp Neurol. 2011;70(1):36–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Qian W, Liang H, Shi J, Jin N, Grundke-Iqbal I, Iqbal K, et al. Regulation of the alternative splicing of tau exon 10 by SC35 and Dyrk1A. Nucleic Acids Res. 2011;39(14):6161–71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.de Graaf K, Czajkowska H, Rottmann S, Packman LC, Lilischkis R, Lüscher B, et al. The protein kinase DYRK1A phosphorylates the splicing factor SF3b1/SAP155 at Thr434, a novel in vivo phosphorylation site. BMC Biochemistry. 2006;7(1):7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Ding S, Shi J, Qian W, Iqbal K, Grundke-Iqbal I, Gong C-X, et al. Regulation of alternative splicing of tau exon 10 by 9G8 and Dyrk1A. Neurobiology of aging. 2012;33(7):1389–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Yin X, Jin N, Gu J, Shi J, Zhou J, Gong CX, et al. Dual-specificity tyrosine phosphorylation-regulated kinase 1A (Dyrk1A) modulates serine/arginine-rich protein 55 (SRp55)-promoted Tau exon 10 inclusion. J Biol Chem. 2012;287(36):30497–506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Shi J, Zhang T, Zhou C, Chohan MO, Gu X, Wegiel J, et al. Increased dosage of Dyrk1A alters alternative splicing factor (ASF)-regulated alternative splicing of tau in Down syndrome. The Journal of biological chemistry. 2008;283(42):28660–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Wang E, Bello Pineda JM, Bourcier J, Stahl M, Penson AV, Wakiro I, et al. Modulation of RNA Splicing Enhances Response to BCL2 Inhibition in Acute Myeloid Leukemia. Blood. 2021;138(Supplement 1):507-.34410352 [Google Scholar]
- 96.Zhang Y, Qian J, Gu C, Yang Y. Alternative splicing and cancer: a systematic review. Signal Transduction and Targeted Therapy. 2021;6(1):78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Feki A, Hibaoui Y. DYRK1A Protein, A Promising Therapeutic Target to Improve Cognitive Deficits in Down Syndrome. Brain Sci. 2018;8(10). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Lamoral-Theys D, Pottier L, Dufrasne F, Neve J, Dubois J, Kornienko A, et al. Natural polyphenols that display anticancer properties through inhibition of kinase activity. Curr Med Chem. 2010;17(9):812–25. [DOI] [PubMed] [Google Scholar]
- 99.Almatroodi SA, Almatroudi A, Khan AA, Alhumaydhi FA, Alsahli MA, Rahmani AH. Potential Therapeutic Targets of Epigallocatechin Gallate (EGCG), the Most Abundant Catechin in Green Tea, and Its Role in the Therapy of Various Types of Cancer. Molecules. 2020;25(14). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Djamshidian A, Bernschneider-Reif S, Poewe W, Lees AJ. Banisteriopsis caapi, a Forgotten Potential Therapy for Parkinson’s Disease? Mov Disord Clin Pract. 2016;3(1):19–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Zhang L, Li D, Yu S. Pharmacological effects of harmine and its derivatives: a review. Arch Pharm Res. 2020;43(12):1259–75. [DOI] [PubMed] [Google Scholar]
- 102.Kim H, Sablin SO, Ramsay RR. Inhibition of monoamine oxidase A by beta-carboline derivatives. Arch Biochem Biophys. 1997;337(1):137–42. [DOI] [PubMed] [Google Scholar]
- 103.Balint B, Weber C, Cruzalegui F, Burbridge M, Kotschy A. Structure-Based Design and Synthesis of Harmine Derivatives with Different Selectivity Profiles in Kinase versus Monoamine Oxidase Inhibition. ChemMedChem. 2017;12(12):932–9. [DOI] [PubMed] [Google Scholar]
- 104.Wurzlbauer A, Rüben K, Gürdal E, Chaikuad A, Knapp S, Sippl W, et al. How to Separate Kinase Inhibition from Undesired Monoamine Oxidase A Inhibition-The Development of the DYRK1A Inhibitor AnnH75 from the Alkaloid Harmine. Molecules. 2020;25(24). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Fruit C, Couly F, Bhansali R, Rammohan M, Lindberg MF, Crispino JD, et al. Biological Characterization of 8-Cyclopropyl-2-(pyridin-3-yl)thiazolo[5,4-f]quinazolin-9(8H)-one, a Promising Inhibitor of DYRK1A. Pharmaceuticals (Basel). 2019;12(4). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Naert G, Ferre V, Meunier J, Keller E, Malmstrom S, Givalois L, et al. Leucettine L41, a DYRK1A-preferential DYRKs/CLKs inhibitor, prevents memory impairments and neurotoxicity induced by oligomeric Abeta25–35 peptide administration in mice. Eur Neuropsychopharmacol. 2015;25(11):2170–82. [DOI] [PubMed] [Google Scholar]
- 107.Coutadeur S, Benyamine H, Delalonde L, de Oliveira C, Leblond B, Foucourt A, et al. A novel DYRK1A (dual specificity tyrosine phosphorylation-regulated kinase 1A) inhibitor for the treatment of Alzheimer’s disease: effect on Tau and amyloid pathologies in vitro. J Neurochem. 2015;133(3):440–51. [DOI] [PubMed] [Google Scholar]
- 108.Deng X, Friedman E. Mirk kinase inhibition blocks the in vivo growth of pancreatic cancer cells. Genes Cancer. 2014;5(9–10):337–47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Couly F, Harari M, Dubouilh-Benard C, Bailly L, Petit E, Diharce J, et al. Development of Kinase Inhibitors via Metal-Catalyzed C⁻H Arylation of 8-Alkyl-thiazolo[5,4-f]-quinazolin-9-ones Designed by Fragment-Growing Studies. Molecules. 2018;23(9). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Tazarki H, Zeinyeh W, Esvan YJ, Knapp S, Chatterjee D, Schröder M, et al. New pyrido[3,4-g]quinazoline derivatives as CLK1 and DYRK1A inhibitors: synthesis, biological evaluation and binding mode analysis. European Journal of Medicinal Chemistry. 2019;166:304–17. [DOI] [PubMed] [Google Scholar]
- 111.Coombs TC, Tanega C, Shen M, Wang JL, Auld DS, Gerritz SW, et al. Small-molecule pyrimidine inhibitors of the cdc2-like (Clk) and dual specificity tyrosine phosphorylation-regulated (Dyrk) kinases: Development of chemical probe ML315. Bioorganic & Medicinal Chemistry Letters. 2013;23(12):3654–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Siddiqui-Jain A, Drygin D, Streiner N, Chua P, Pierre F, O’Brien SE, et al. CX-4945, an Orally Bioavailable Selective Inhibitor of Protein Kinase CK2, Inhibits Prosurvival and Angiogenic Signaling and Exhibits Antitumor Efficacy. Cancer Research. 2010;70(24):10288–98. [DOI] [PubMed] [Google Scholar]
- 113.Quotti Tubi L, Gurrieri C, Brancalion A, Bonaldi L, Bertorelle R, Manni S, et al. Inhibition of protein kinase CK2 with the clinical-grade small ATP-competitive compound CX-4945 or by RNA interference unveils its role in acute myeloid leukemia cell survival, p53-dependent apoptosis and daunorubicin-induced cytotoxicity. J Hematol Oncol. 2013;6:78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Buontempo F, Orsini E, Martins LR, Antunes I, Lonetti A, Chiarini F, et al. Cytotoxic activity of the casein kinase 2 inhibitor CX-4945 against T-cell acute lymphoblastic leukemia: targeting the unfolded protein response signaling. Leukemia. 2014;28(3):543–53. [DOI] [PubMed] [Google Scholar]
- 115.Martins LR, Lúcio P, Melão A, Antunes I, Cardoso BA, Stansfield R, et al. Activity of the clinical-stage CK2-specific inhibitor CX-4945 against chronic lymphocytic leukemia. Leukemia. 2014;28(1):179–82. [DOI] [PubMed] [Google Scholar]
- 116.Prins RC, Burke RT, Tyner JW, Druker BJ, Loriaux MM, Spurgeon SE. CX-4945, a selective inhibitor of casein kinase-2 (CK2), exhibits anti-tumor activity in hematologic malignancies including enhanced activity in chronic lymphocytic leukemia when combined with fludarabine and inhibitors of the B-cell receptor pathway. Leukemia. 2013;27(10):2094–6. [DOI] [PubMed] [Google Scholar]
- 117.KIM HM, JEONG I, KIM HJ, KANG SK, KWON WS, KIM TS, et al. Casein Kinase 2 Inhibitor, CX-4945, as a Potential Targeted Anticancer Agent in Gastric Cancer. Anticancer Research. 2018;38(11):6171–80. [DOI] [PubMed] [Google Scholar]
- 118.Borad MJ, Bai L-Y, Chen M-H, Hubbard JM, Mody K, Rha SY, et al. Silmitasertib (CX-4945) in combination with gemcitabine and cisplatin as first-line treatment for patients with locally advanced or metastatic cholangiocarcinoma: A phase Ib/II study. Journal of Clinical Oncology. 2021;39(3_suppl):312-. [Google Scholar]
- 119.Weber C, Sipos M, Paczal A, Balint B, Kun V, Foloppe N, et al. Structure-Guided Discovery of Potent and Selective DYRK1A Inhibitors. J Med Chem. 2021;64(10):6745–64. [DOI] [PubMed] [Google Scholar]
- 120.Lee Walmsley D, Murray JB, Dokurno P, Massey AJ, Benwell K, Fiumana A, et al. Fragment-Derived Selective Inhibitors of Dual-Specificity Kinases DYRK1A and DYRK1B. Journal of Medicinal Chemistry. 2021;64(13):8971–91. [DOI] [PubMed] [Google Scholar]
- 121.Henderson SH, Sorrell F, Bennett J, Fedorov O, Hanley MT, Godoi PH, et al. Discovery and Characterization of Selective and Ligand-Efficient DYRK Inhibitors. J Med Chem. 2021;64(15):11709–28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Henderson SH, Sorrell F, Bennett J, Hanley MT, Robinson S, Hopkins Navratilova I, et al. Mining Public Domain Data to Develop Selective DYRK1A Inhibitors. ACS Medicinal Chemistry Letters. 2020;11(8):1620–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Yoon HR, Balupuri A, Choi K-E, Kang NS. Small Molecule Inhibitors of DYRK1A Identified by Computational and Experimental Approaches. International journal of molecular sciences. 2020;21(18):6826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Becker W. A wake-up call to quiescent cancer cells - potential use of DYRK1B inhibitors in cancer therapy. FEBS J. 2018;285(7):1203–11. [DOI] [PubMed] [Google Scholar]
- 125.Liu Q, Liu N, Zang S, Liu H, Wang P, Ji C, et al. Tumor suppressor DYRK1A effects on proliferation and chemoresistance of AML cells by downregulating c-Myc. PLoS One. 2014;9(6):e98853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Recasens A, Humphrey SJ, Ellis M, Hoque M, Abbassi RH, Chen B, et al. Global phosphoproteomics reveals DYRK1A regulates CDK1 activity in glioblastoma cells. Cell death discovery. 2021;7(1):81-. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Li Y, Zhou D, Xu S, Rao M, Zhang Z, Wu L, et al. DYRK1A suppression restrains Mcl-1 expression and sensitizes NSCLC cells to Bcl-2 inhibitors. Cancer Biol Med. 2020;17(2):387–400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Kottakis F, Polytarchou C, Foltopoulou P, Sanidas I, Kampranis SC, Tsichlis PN. FGF-2 regulates cell proliferation, migration, and angiogenesis through an NDY1/KDM2B-miR-101-EZH2 pathway. Mol Cell. 2011;43(2):285–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Radhakrishnan A, Nanjappa V, Raja R, Sathe G, Puttamallesh VN, Jain AP, et al. A dual specificity kinase, DYRK1A, as a potential therapeutic target for head and neck squamous cell carcinoma. Sci Rep. 2016;6:36132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Zhang L, Li D, Yu S. Pharmacological effects of harmine and its derivatives: a review. Archives of Pharmacal Research. 2020;43(12):1259–75. [DOI] [PubMed] [Google Scholar]
- 131.Zhao C, Wang D, Gao Z, Kan H, Qiu F, Chen L, et al. Licocoumarone induces BxPC-3 pancreatic adenocarcinoma cell death by inhibiting DYRK1A. Chem Biol Interact. 2020;316:108913. [DOI] [PubMed] [Google Scholar]
- 132.Shin CM, Lee DH, Seo AY, Lee HJ, Kim SB, Son WC, et al. Green tea extracts for the prevention of metachronous colorectal polyps among patients who underwent endoscopic removal of colorectal adenomas: A randomized clinical trial. Clin Nutr. 2018;37(2):452–8. [DOI] [PubMed] [Google Scholar]
- 133.Kumar NB, Dickinson SI, Schell MJ, Manley BJ, Poch MA, Pow-Sang J. Green tea extract for prevention of prostate cancer progression in patients on active surveillance. Oncotarget. 2018;9(102):37798–806. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Fan H, Peng J, Hamann MT, Hu J-F. Lamellarins and related pyrrole-derived alkaloids from marine organisms. Chemical reviews. 2008;108(1):264–87. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Quesada AR, García Grávalos MD, Fernández Puentes JL. Polyaromatic alkaloids from marine invertebrates as cytotoxic compounds and inhibitors of multidrug resistance caused by P-glycoprotein. Br J Cancer. 1996;74(5):677–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Jarry M, Lecointre C, Malleval C, Desrues L, Schouft MT, Lejoncour V, et al. Impact of meriolins, a new class of cyclin-dependent kinase inhibitors, on malignant glioma proliferation and neo-angiogenesis. Neuro Oncol. 2014;16(11):1484–98. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Park NS, Park YK, Ramalingam M, Yadav AK, Cho HR, Hong VS, et al. Meridianin C inhibits the growth of YD-10B human tongue cancer cells through macropinocytosis and the down-regulation of Dickkopf-related protein-3. J Cell Mol Med. 2018;22(12):5833–46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Rossignol E, Debiton E, Fabbro D, Moreau P, Prudhomme M, Anizon F. In-vitro antiproliferative activities and kinase inhibitory potencies of meridianin derivatives. Anticancer Drugs. 2008;19(8):789–92. [DOI] [PubMed] [Google Scholar]
- 139.Imperatore C, Aiello A, #039, Aniello F, Senese M, Menna M. Alkaloids from Marine Invertebrates as Important Leads for Anticancer Drugs Discovery and Development. Molecules. 2014;19(12):20391–423. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 140.Tazarki H, Zeinyeh W, Esvan YJ, Knapp S, Chatterjee D, Schroder M, et al. New pyrido[3,4-g]quinazoline derivatives as CLK1 and DYRK1A inhibitors: synthesis, biological evaluation and binding mode analysis. Eur J Med Chem. 2019;166:304–17. [DOI] [PubMed] [Google Scholar]
- 141.Rüben K, Wurzlbauer A, Walte A, Sippl W, Bracher F, Becker W. Selectivity Profiling and Biological Activity of Novel β-Carbolines as Potent and Selective DYRK1 Kinase Inhibitors. PLOS ONE. 2015;10(7):e0132453. [DOI] [PMC free article] [PubMed] [Google Scholar]