

Abstract
BCOR‐ITD tumours form an emerging family of aggressive entities with an internal tandem duplication (ITD) in the last exon of the BCOR gene. The family includes cerebral tumours, termed central nervous system BCOR‐ITD (CNS BCOR‐ITD), and sarcomatous types described in the kidney as clear cell sarcoma of the kidney (CCSK), in the endometrium as high‐grade endometrial stromal sarcoma, and in the bone and soft tissue as undifferentiated round cell sarcoma or primitive myxoid mesenchymal tumour of infancy. Based on a series of 33 retrospective cases, including 10 CNS BCOR‐ITD and 23 BCOR‐ITD sarcomas, we interrogated the homogeneity of the entity regarding clinical, radiological, and histopathological findings, and molecular signatures. Whole‐transcriptomic sequencing and DNA methylation profiling were used for unsupervised clustering. BCOR‐ITD tumours mostly affected young children with a median age at diagnosis of 2.1 years (range 0–62.4). Median overall survival was 3.9 years and progression‐free survival was 1.4 years. This dismal prognosis is shared among tumours in all locations except CCSK. Histopathological review revealed marked differences between CNS BCOR‐ITD and BCOR‐ITD sarcomas. These two groups were consistently segregated by unsupervised clustering of expression (n = 22) and DNA methylation (n = 21) data. Proximity between the two groups may result from common somatic changes within key pathways directly related to the novel activity of the ITD itself. Conversely, comparison of gene signatures with single‐cell RNA‐Seq atlases suggests that the distinction between BCOR‐ITD sarcomas and CNS BCOR‐ITD may result from differences in cells of origin.
Keywords: CNS BCOR‐ITD, BCOR‐ITD sarcomas, CCSK, ESS, clustering, transcriptome, methylome
Introduction
BCOR‐internal tandem duplication (ITD) tumours are a heterogeneous group of neoplasms comprising sarcomas and neuroepithelial tumours of the brain. Sarcomas are described in the kidney as clear cell sarcomas of the kidney (CCSK) [1, 2, 3, 4, 5, 6, 7, 8], in the endometrium as high‐grade endometrial stromal sarcomas (HG‐ESS) [9, 10, 11], and in the bone [12, 13] and soft tissue as undifferentiated round cell sarcoma (URCS), but also including primitive myxoid mesenchymal tumour of infancy (PMMTI) [12, 14, 15]. In the central nervous system (CNS), tumours with BCOR‐ITD alteration were first identified by DNA methylation analyses and termed high‐grade neuroepithelial tumours BCOR (HGNET‐BCOR) [16, 17, 18, 19, 20, 21, 22, 23, 24]. They are now referred to as CNS BCOR‐ITD in the latest (2021) version of the WHO classification of brain tumours [25].
BCOR‐ITD tumours mostly affect infants and young children, except for the endometrial tumours, which occur exclusively in adult patients. The radiological and histopathological features of BCOR‐ITD tumours are not sufficiently specific to ascertain the diagnosis. Definitive diagnosis is achieved by molecular analysis through RNA‐Seq and DNA methylation studies or direct sequencing of the characteristic in‐frame partial duplication of BCOR exon 15 by RT‐qPCR.
The diversity of anatomical locations questions whether a single molecular alteration, i.e. BCOR‐ITD, drives similar oncogenic pathways throughout the various tumour entities. CNS BCOR‐ITD tumours were analysed through their methylation profiles, which proved to be specific [17, 22, 26]. BCOR‐ITD sarcomas were also found to localise within the group of BCOR‐rearranged tumours based on their transcriptomic profile [27]. Recently, hierarchical clustering based on the methylome of 1,077 prototypical sarcomas also identified the BCOR‐rearranged sarcomas as a coherent group [28].
To our knowledge, the molecular profiles of BCOR‐ITD sarcomas and CNS BCOR‐ITD have not been compared to date. Here, we report a retrospective study including BCOR‐ITD tumours from all described locations, with emphasis on the clinical, therapeutic, radiological, and histopathological aspects. We describe the features shared among all locations as well as the key differences, in particular the prognosis, histopathology, and immunohistochemical profiles. From this diversity emerges two distinct molecular signatures as revealed by unsupervised analysis of expression and methylation profiles: cranial versus extracranial BCOR‐ITD tumours.
Materials and methods
Study population
The population under study was defined as all patients with a BCOR‐ITD tumour diagnosed in France over the period 2010–2020, including retrospective diagnoses on archival material. Within this population, our cohort comprised all cases of BCOR‐ITD identified by RNA‐Seq analysis, as performed routinely for diagnostic purposes. This analysis was performed either at Institut Curie (Paris, France) or Centre Léon Bérard (Lyon, France), which were the sole centres to provide physicians with diagnostic RNA‐Seq. All BCOR‐ITD cases identified either by RT‐PCR or droplet‐digital PCR in other centres were not included because expression data were not available. Thirty‐three cases were identified: 7 CCSK, 4 HG‐ESS, 10 CNS BCOR‐ITD, 10 URCS, and 2 bone tumours. Five cases of URCS were reported previously in the study by Watson et al, although without clinical, radiological, and histopathological details [27]. Another case of URCS (P16) and one case of CNS BCOR‐ITD (P23) were also previously reported (in Refs [12] and [16], respectively).
This study was approved by the Institutional Clinical Research Board of Institut Curie, and complied with the reference methodology MR‐004 from the Commision Nationale de l'Informatique et des Libertés (CNIL). All fastq files have been deposited in the European Genome‐phenome Archive (URL pending).
Clinical review
The 33 patients were cared for within 17 medical centres in France and 1 centre abroad (Padova Hospital, Italy). Data collection in all 18 centres followed ethical rules approved by the Clinical Research Board of Institut Curie. Medical record could be collected for 31 cases (10 CNS BCOR‐ITD, 7 CCSK, 4 HG‐ESS, 8 URCS, 2 bone tumours).
Radiological review
Radiological images at diagnosis were available for 23 cases (10 CNS BCOR‐ITD, 5 CCSK, 2 HG‐ESS, 5 URCS, 1 bone tumour). Depending on the location, radiological review was based on centralised analysis of ultrasound, computed tomography (CT) scans, and/or magnetic resonance imaging (MRI). Images were qualitatively analysed using the viewer of the picture archiving and communicating system (PACS) of Institut Curie (Carestream‐Philips v 12.1.6).
Histopathological review and immunostaining
Central pathology review was performed conjointly by a neuropathologist (AT‐E) and a pathologist expert in paediatric sarcomas (AG) on 21 samples (9 CNS BCOR‐ITD, 4 CCSK, 2 HG‐ESS, 6 URCS). Of note, two bone tumours could not be evaluated histologically. Unstained 3‐μm‐thick slides of formalin‐fixed paraffin‐embedded (FFPE) tissues were obtained and submitted for immunostaining. Conditions of use of all primary antibodies are described in the Supplementary materials and methods. External positive and negative controls were used for all antibodies and stains.
Paired‐end RNA sequencing
Paired‐end RNA sequencing was performed on 22 frozen samples in Institut Curie (Paris, France) and 11 FFPE samples in Centre Léon Bérard (Lyon, France). Frozen and FFPE samples correspond to distinct cases: samples analysed in Paris and Lyon were obtained from different tumours. Details are given in supplementary material, Figure S1.
Identification of the BCOR‐ITD sequence
For frozen samples, gene expression values were extracted after alignment on Hg19 (GRCh37) genome annotation with SALMON (v0.13.1). For fusion gene discovery, sequencing reads were injected into various available tools: Defuse (v0.6.2), FusionCatcher (v1.0), STARFusion (v2.5), Arriba, and FusionMap integrated in Oshell (v10.0.1.50). Only fusion transcripts supported by at least two tools with at least two split reads were considered. Search for expressed mutations was performed after alignment on Hg19 genome (GRCh37) with STAR, using HaplotypeCaller and Mutect2 (GATK4). The BCOR‐ITD cases were identified by hierarchical clustering, using the R package Cluster v2.0.3 using the Pearson correlation distance and the Ward clustering method. Each sample, represented by its Salmon expression matrix, was compared with a panel of well‐characterised tumours including BCOR‐rearranged and BCOR‐ITD tumours. A sample was labelled ‘BCOR‐ITD’ if it segregates within the BCOR‐ITD family and is negative after the fusion search. The result was then confirmed by RT‐qPCR: total RNA was transcribed as cDNA (Multiscribe Reverse Transcriptase with random hexamers; Life Technologies, Carlsbad, CA, USA) and BCOR exon 15 was amplified with primers 5′‐3′ ACCATTGCAGACGGCAGAAT and 3′‐5′ ATGACACATATGCACAAGGATTAAC.
A similar approach was applied to FFPE samples, as detailed in the Supplementary materials and methods.
Methylome analysis
DNA was extracted and converted by bisulphite treatment for 21 samples from both frozen (n = 15) and FFPE (n = 6) samples, without overlap between the two sample types. DNA methylation profiling was performed as described [29] at the Integragen Core Facility (Paris, France) with Illumina HumanMethylation450 BeadChip array (450k array) (Illumina, San Diego, CA, USA).
Clustering studies
Two segregation methods were applied separately to the RNA‐Seq dataset and the methylome dataset.
The RNA‐Seq dataset was compiled from the 22 available frozen samples (8 CNS BCOR‐ITD, 6 CCSK, 7 URCS, 1 bone, no HG‐ESS). Paraffin samples were excluded as there is no guarantee that their expression matrices are comparable with frozen samples, given the differences in the methods employed to produce libraries. The DNA methylation dataset was compiled from 21 BCOR‐ITD samples (9 CNS BCOR‐ITD, 6 CCSK, 6 URCS). Control samples were sourced differently depending on the dataset. Tumours in the RNA‐Seq dataset were compared with 181 control sarcoma cases extracted by Watson et al [27], and 165 control brain tumours from an in‐house collection. Control cases for the methylome dataset were taken from the publicly available database provided by the German Cancer Research Center DKFZ and the Heidelberg University Hospital [28, 29]. They included 310 cerebral tumours and 212 sarcomas. Methods for data alignment, scaling, and segregation are detailed in Supplementary materials and methods.
Differential expression analysis on RNA‐Seq dataset
Reads were aligned with STAR 2.6.0a [30] to the human genome (gencodeV19). Gene expression values (FPKM = fragments per kilobase per million reads) were computed by Cufflinks v2.2.1 [31] on Ensembl Release 75 and further normalisation between samples was done using quantile normalisation (log2 (FPKM + 2), R/Bioconductor package limma [32]). Differential analysis was performed with the Welch's t‐test and the P value was adjusted using the Benjamini–Hochberg correction. For differential analysis, significance was given by p < 0.05 and |fold‐change| > 1.5. All control cases used in this analysis were obtained from in‐house clinical cohorts: medulloblastomas (21 samples), ependymomas (10 samples), and Ewing sarcomas (31 samples). The list of significantly different genes was further analysed using the ToppGene suite, in particular ToppFun (Cincinnati Children's Hospital Medical Center).
BCOR‐ITD signatures from single‐cell RNA‐Seq atlases
Differential expression analysis (see previous section) between 8 CNS BCOR‐ITD and 15 BCOR‐ITD sarcomas using two different fold‐change thresholds (1.5 and 2.5) led to 356 and 117 differentially expressed genes (DEGs) enriched in CNS BCOR‐ITD and 129 and 54 DEG enriched in BCOR‐ITD sarcomas, respectively. These gene sets were compared with two human single‐cell/single‐nuclei RNA‐Seq atlases, namely Cao et al and Han et al [33, 34], taken from the https://descartes.brotmanbaty.org website (‘sampled data’) and from the Gene Expression Omnibus website under GEO accession number GSE134355, respectively. They contain over 70 and 100 carefully annotated cell identities, respectively, extracted from adult, foetal, cord, and induced pluripotent stem (iPS) tissue. Counts in both atlases were natural log‐normalised with the NormalizeData function in Seurat 4.0.3 with default options and signatures for all gene sets were calculated on the transformed data using Seurat's in‐built AddModuleScore method, which accounts for background expression. Signature intensities can thus be assimilated to an average natural log‐fold change with the background expression (i.e. a signature intensity of 0.4 can be considered as a fold‐change of about e0.4 = 1.5. Dot plots were built such that dot colour represents the average signature intensity for a given cell type and dot size the number of cells with a signature intensity strictly higher than 0 (brought to the [0, 1] range for each row).
GSEA on RNA‐Seq dataset
The GSEA (Gene Set Enrichment Analysis) software was used as described originally in [35] after downloading from https://www.gsea.msigdb.org/gsea. Gene sets representing canonical pathways were selected in the Molecular Signature Database (MSigDB). The list is provided in Supplementary materials and methods.
Statistical analysis
Kaplan–Meier analyses were performed to estimate the overall survival (OS) and progression‐free survival (PFS) of patients from different subgroups. The survival rates were calculated from the date of treatment start and the date of last follow‐up or event (relapse or death). Survival data are provided as percentages with 95% confidence intervals.
Results
Clinical features, therapeutic data, and survival analyses
Thirty‐three cases of BCOR‐ITD tumours were identified and distributed as follows: 10 URCS, 10 CNS BCOR‐ITD, 4 HG‐ESS, 7 CCSK, and 2 bone tumours (supplementary material, Figure S1). Clinical and therapeutic data were collected for 31 cases. The demographic features as well as the disease presentation at diagnosis are shown in Table 1 and, patient by patient, in supplementary material, Table S1. The disease was mainly localised at presentation, with the exception of HG‐ESS. The median age at diagnosis was 2.1 years (range 0–62.4). Adult cases were found exclusively in the HG‐ESS and bone groups. The overall sex ratio was balanced, 1:1.1 (M:F), without considering the four female cases of HG‐ESS. The differences in sex ratio within each group were not significant. Overall, these findings show that demographic features are homogeneous among all locations, except the adult cases of endometrium and the bone sites.
Table 1.
Demographic features
| Tumour type | n | Gender | Age at presentation, years (minimum; maximum) |
|---|---|---|---|
| URCS | 8 |
M: 6 F: 2 |
0.7 (0.1; 19.7) |
| Bone | 2 |
M: 2 F: 0 |
48.5 (48.3; 48.7) |
| CCSK | 7 |
M: 2 F: 5 |
2.0 (0.5; 18.9) |
| CNS BCOR‐ITD | 10 |
M: 3 F: 7 |
1.8 (1.2; 7.6) |
| HG‐ESS | 4 |
M: 0 F: 4 |
51.1 (25.8; 62.4) |
| Total | 31 |
M: 13 F: 18 |
2.1 (0; 62.4) |
The cohort was divided into the five reported histopathological types of BCOR‐ITD tumour. The corresponding number of cases with the available clinical data, the distribution of genders, and the age at presentation are shown.
F, females; M, males.
The initial therapeutic strategy was mainly multimodal including most often the first‐line surgery and adjuvant chemotherapy. Eighteen cases underwent complementary radiotherapy. Details are shown in supplementary material, Table S2. The median follow‐up was 50.3 months (range 1–200), with a median OS of 47 months (95% CI 38–not applicable [NA]) (Figure 1A,C) and median PFS of 17 months (95% CI 12–NA) (Figure 1B,D). Our data suggest that this overall dismal prognosis did not affect the CCSK group: OS and PFS were significantly higher in this subgroup (Figure 1E,F).
Figure 1.
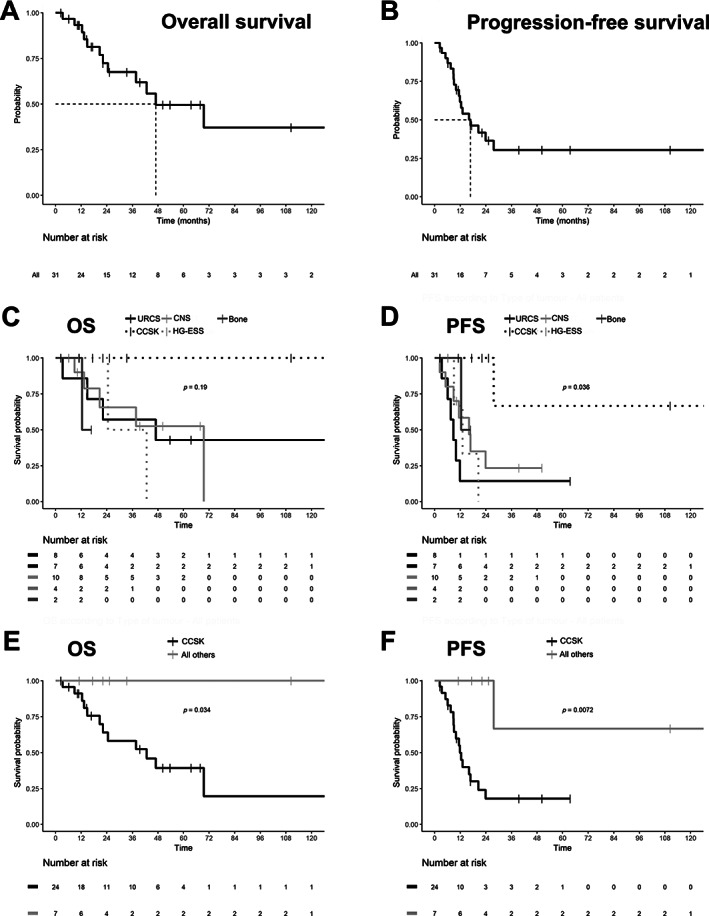
Prognosis of BCOR‐ITD tumours. Kaplan–Meier plots showing OS and PFS of the whole BCOR‐ITD cohort (A, B) and the five histological subgroups (C, D). The two metrics are also shown for the CCSK subgroup in comparison with all other groups (E, F). Survival was calculated as the delay between the date of first treatment and the date of decease or last medical visit. Log‐rank test P values are shown in C–F. NS, not significant; p, P value.
Radiological features
Imaging results are summarised in supplementary material, Table S3 and detailed in supplementary material, Text S1, for 23 reviewed cases. All BCOR‐ITD tumours were well‐circumscribed, non‐infiltrative, heterogeneous masses of variable volumes (median volume 128 ml [range 6–918]). Using CT scan or MRI, the injection of a contrast medium brings out the density/signal moderately and heterogeneously in most cases (supplementary material, Figure S2). BCOR‐ITD sarcomas (URCS, CCSK, bone, HG‐ESS) differed from CNS BCOR‐ITD, the latter displaying more necrosis (9/10 versus 5/13), more calcification (1/10 versus 1/13), and more haemorrhage (8/10 versus 4/13).
Histopathological and immunohistochemical analyses
The histopathological findings were available for 21 cases (9 CNS BCOR‐ITD, 4 CCSK, 2 ESS, and 6 URCS) and are summarised in Table 2. Complete description is provided in supplementary material, Text S2. All cases presented as highly cellular tumours, well‐delimited from host tissue. The predominant histopathological pattern varied according to the subtype: ependymoma‐like or fascicular in CNS BCOR‐ITD (Figure 2A,E), fascicular or diffuse in URCS (Figure 2B), diffuse in HG‐ESS (Figure 2C), and alveolar in CCSK (Figure 2D). Of note, in our series, a myxoid background was encountered only in CNS BCOR‐ITD and not in sarcomas, excluding PMMTI in our series. Additional patterns were frequently identified, such as perivascular pseudorosettes in CNS BCOR‐ITD (Figure 2E), URCS (Figure 2F), and HG‐ESS (Figure 2G), and microcysts in CNS BCOR‐ITD (Figure 2I), HG‐ESS (Figure 2K), and CCSK (Figure 2L). Cytology comprised round‐to‐ovoid basophil or spindle cells in all sarcomas (Figure 2B,C), and spindle cells only in CNS BCOR‐ITD. Necrosis (Figure 2A,J), haemorrhage, and calcification were predominantly encountered in CNS BCOR‐ITD, consistent with the radiological findings. The level of cell proliferation was similar among the four categories. In line with the favourable prognosis of CCSK, the mitotic count was found to be minimal in this subgroup, and no high‐grade cytological or histological features were identified.
Table 2.
Main histopathological features of the BCOR‐ITD tumours
| Number of cases | Histological pattern | Cytology | Pseudorosettes | Microcystic background | Haemorrhage | Calcification | Necrosis | Mitotic count per 10 hpf | |
|---|---|---|---|---|---|---|---|---|---|
| CNS BCOR‐ITD | 9 |
EPN‐like (6/9) Fascicular (3/9) |
Spindle cells | 9/9 | 7/9 | 6/9 | 2/9 | Palisading, geographic and calcified (6/9) | 5–62 |
| URCS | 6 |
Fascicular Diffuse |
Round‐to‐ovoid, spindle cells | 1/6 | 0 | 4/6 | 0 | 0 | 1.7–13.7 |
| HG‐ESS | 2 | Diffuse | Spindle cells | 1/2 | 2/2 | 2/2 | 0 | 0 | 14–21 |
| CCSK | 4 |
Diffuse Alveolar |
Round‐to‐ovoid, spindle cells | 0 | 1/4 | 0 | 0 | 0 | 0–18 |
EPN, ependymoma; hpf, high‐power field.
Figure 2.
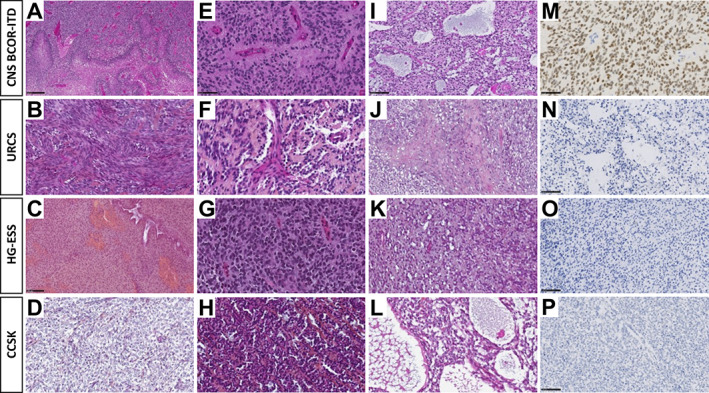
Histopathology of BCOR‐ITD tumours. Each line shows histopathological and immunohistochemical findings of one representative case of CNS BCOR‐ITD, URCS, ESS, and CCSK. A case of CNS BCOR‐ITD showing pseudo‐palisading necrosis (A, HPS, magnification, ×100), ependymal pseudorosettes (E, HPS, magnification, ×400), microcystic modifications (I, HPS, magnification, ×200), and diffuse Olig2 immunoexpression (M, magnification, ×400). A case of URCS showing a fascicular pattern composed of spindle cells (B, HPS, magnification, ×400), with some papillary structures and pseudorosettes (F, HPS, magnification, ×400), necrosis, and apoptotic bodies (J, HPS, magnification, ×200), without immunoexpression for Olig2 (N, magnification, ×200). A case of ESS showing spindle cells (C, HPS, magnification, ×200), some pseudorosettes (G, HPS, magnification, ×400), and microcysts (K, HPS, magnification, ×200), with no expression of Olig2 (O, magnification, ×200). A case of CCSK showing an alveolar pattern (D, HPS, magnification, ×400), with some pseudorosettes (H, HPS, magnification, ×400), and microcysts (L, HPS, magnification, ×400), but without immunostaining for Olig2 (P, magnification, ×200). HPS, haematoxylin phloxine saffron. Black scale bars represent 50 μm (E, M), 100 μm (I, N, C, O, P), and 250 μm (A).
The immunohistochemical results are summarised in supplementary material, Table S4 and detailed in supplementary material, Text S2.
CNS BCOR‐ITD cases exhibited non‐constant expression of the glial markers GFAP and Olig2 (Figure 2M), with varying degrees of distribution. These co‐stained with the neuronal markers neurofilament and, almost constantly, with NeuN. In contrast, no staining for glial or neuronal markers was found in BCOR‐ITD sarcomas (Figure 2N–P). There was shared staining among all BCOR‐ITD tumours for BCOR protein and four additional markers: SATB2, Cyclin D1, BCL2, and TLE1. These markers were tested for specificity by comparing the mean mRNA levels of each marker in BCOR‐ITD tumours versus selected families of sarcomas and brain tumours. The resulting boxplots show that BCOR, SATB2, and TLE1 tend to be overexpressed in the family of BCOR‐rearranged tumours, which includes BCOR‐ITD, BCOR‐CCNB3, and YWHAE‐NUTM2A/B members (supplementary material, Figure S3).
Among potential therapeutic targets, we identified EGFR as strongly and diffusely expressed in all CNS BCOR‐ITD cases and three cases of sarcoma. YAP1, as a surrogate of Shh signalling, was expressed diffusely in most cases. NTRK (pan‐Trk) was expressed only in CNS BCOR‐ITD. No significant nuclear accumulation of β‐catenin was observed in any case.
Transcriptome and methylome profiles identify BCOR‐ITD sarcomas and CNS BCOR‐ITD tumours as two close coherent subgroups
Unsupervised clustering was performed on RNA‐Seq data using 22 tumours from this series compared to 346 reference tumours. Visualisation after dimension reduction by the Uniform Manifold Approximation and Projection (UMAP) method delineated closely related but distinct clusters of BCOR‐ITD sarcomas and CNS BCOR‐ITD (Figure 3A). CNS BCOR‐ITD samples clustered with neither glial nor neuroectodermal tumours. Hierarchical clustering was applied to the same dataset leading to a higher resolution picture: while CNS BCOR‐ITD and BCOR‐ITD sarcomas were grouped within a single common clade of BCOR‐rearranged tumours (Figure 3B,C), they clearly formed two distinct subclusters. The same methods were applied to the DNA methylation data from 21 BCOR‐ITD cases compared with 528 reference tumours. Similar to transcriptomic data, CNS BCOR‐ITD and BCOR‐ITD sarcomas formed two distinct branches of the same clade (Figure 4).
Figure 3.
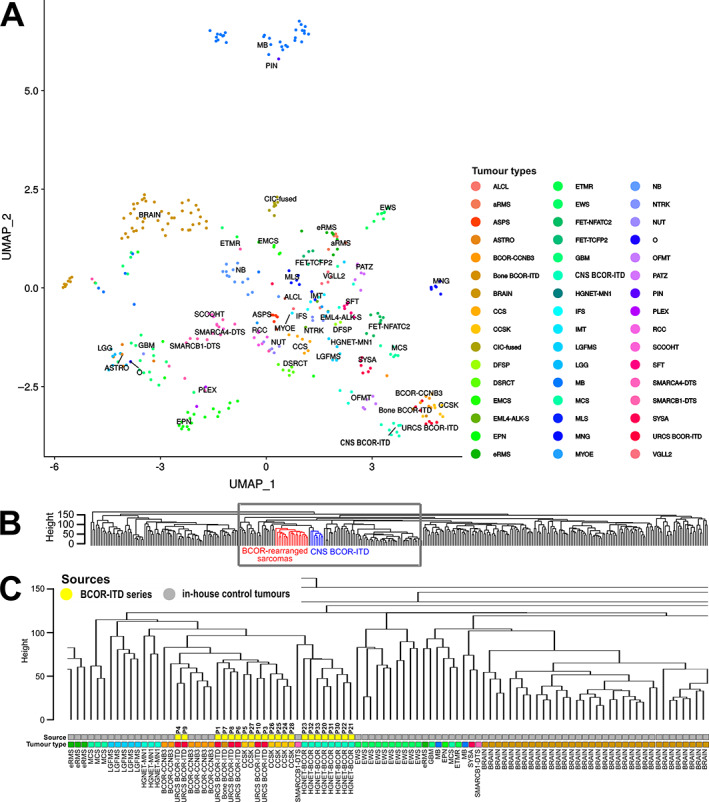
Unsupervised clustering using RNA‐Seq data. (A) Unsupervised clustering of gene expression levels of 22 BCOR‐ITD samples, with 346 reference tumours of known histology (previously published sarcomas from Watson et al [27] and unpublished brain tumours from in‐house clinical cohorts), using UMAP dimensionality reduction. (B) Dendrogram of hierarchical clustering using Euclidian distances and the complete linkage method applied to the same dataset as in (A). Locations of BCOR‐ITD sarcomas and CNS BCOR‐ITD tumours are depicted in red and blue, respectively. The grey square outlines the position of the zoom‐in shown in (C). (C) Zoom‐in on the dendrogram. The ‘Source’ bar refers to the control tumours in grey and the cases of the series in yellow, with the corresponding P numbers. The ‘Tumour type’ bar allocates a tumour type to each case, with the same colour code as in (A). Abbreviations: ALCL, anaplastic lymphoma; aRMS, alveolar rhabdomyosarcoma; ASPS, alveolar soft part sarcoma; ASTRO, astrocytoma; BRAIN, normal brain tissue from GTEX database; CCS, clear cell sarcoma of soft tissue; CCSK, clear cell sarcoma of the kidney; CIC‐fused, CIC‐fused sarcoma; DFSP, dermatofibrosarcoma protuberans; DSRCT, desmoplastic small round cell tumour; EMCS, extraskeletal myxoid chondrosarcoma; EML4‐ALK‐S, EML4‐ALK sarcoma; EPN, ependymoma; eRMS, embryonal rhabdomyosarcoma; ETMR, embryonal tumour with multi‐layered rosettes; EWS, Ewing sarcoma; FET‐NFATC2, FET‐NFATC2 sarcoma; FET‐TCFP2, FET‐TCFP2 sarcoma; GBM, glioblastoma; IFS, infantile fibrosarcoma; IMT, inflammatory myofibroblastic tumour; LGFMS, low‐grade fibromyxoid sarcoma; LGG, low‐grade astrocytoma and ganglioglioma; MB, medulloblastoma; MCS, chondrosarcoma; MLS, myxoid liposarcoma; MNG, meningioma; MYOE, myoepithelioma; NB, neuroblastoma; NTRK, NTRK fused sarcoma; NUT, NUT midline carcinoma; O, anaplastic oligodendroglioma; OFMT, ossifying fibromyxoid tumour; PATZ, EWSR1‐PATZ1 sarcoma; PIN, pineoblastoma; PLEX, choroid plexus carcinoma; RCC, renal cell carcinoma; SCCOHT, small cell carcinoma of the ovary hypercalcaemic type; SFT, solitary fibrous tumour; SMARCA4‐DTS, SMARCA4‐deficient thoracic sarcoma; SMARCB1‐DTS, SMARCB1‐deficient thoracic sarcoma; SYSA, synovial sarcoma; VGLL2, VGLL2‐fused sarcoma.
Figure 4.
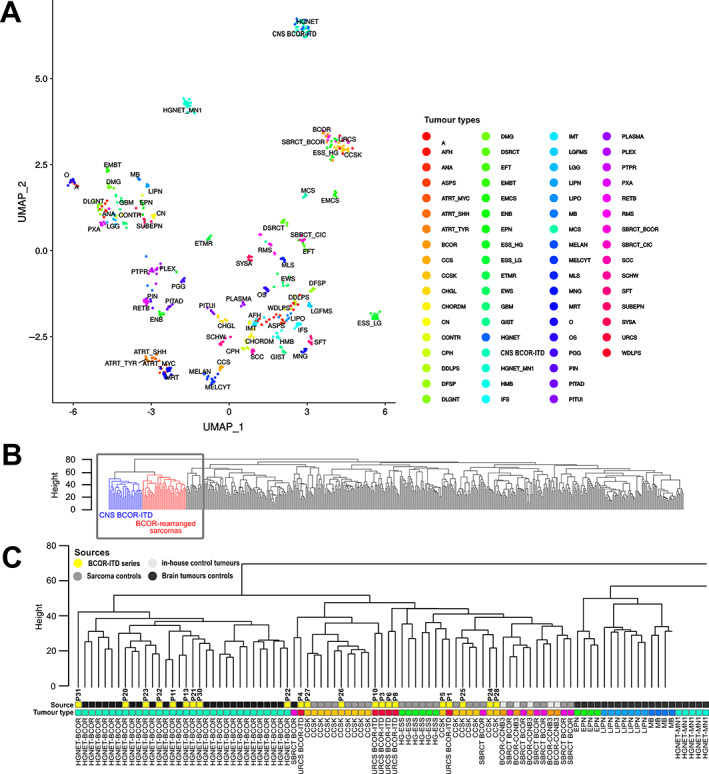
Unsupervised clustering of DNA methylation profiles. (A) UMAP dimension reduction of the 100 first principal components derived from the most variable methylation β values (SD > 0.2, n = 32,500 probes). Twenty‐one BCOR‐ITD samples, with 528 reference tumours (310 brain tumours from Capper et al [29], 212 sarcomas from Koelsche et a. [28], and 6 in‐house BCOR‐CCNB3 tumours). (B) Dendrogram of hierarchical clustering using Euclidian distances and the complete linkage method applied to the same dataset as in (A). Locations of BCOR‐ITD sarcomas and CNS BCOR‐ITD tumours are depicted in red and blue, respectively. The grey square outlines the position of the zoom‐in shown in (C). (C) Zoom‐in on the dendrogram. The ‘Source’ bar refers to the control tumours (light grey for sarcoma cases, dark grey for brain tumours) and the cases of the series in yellow, with the corresponding P numbers. The ‘Tumour type’ bar allocates a tumour type to each case, with the same colour code as in (A) and Figure 3. Abbreviations: A, diffuse astrocytoma IDH mutant; AFH, angiomatoid fibrous histiocytoma; ANA, anaplastic pilocytic astrocytoma; ASPS, alveolar soft part sarcoma; ATRT_MYC, atypical teratoid/rhabdoid tumour MYC subgroup; ATRT_SHH, atypical teratoid/rhabdoid tumour SHH subgroup; ATRT_TYR, atypical teratoid/rhabdoid tumour TYR subgroup; CCS, clear cell sarcoma of soft tissue; CCSK, clear cell sarcoma of the kidney; CHGL, chordoid glioma of the third ventricle; CHORDM, chordoma; CN, central neurocytoma; CPH, adamantinomatous craniopharyngioma; DDLPS, dedifferentiated liposarcoma; DFSP, dermatofibrosarcoma protuberans; DLGNT, diffuse leptomeningeal glioneuronal tumour; DMG, diffuse midline glioma H3K27M; DSRCT, desmoplastic small round cell tumour; EFT, CNS embryonal tumour NOS; EMCS, extraskeletal myxoid chondrosarcoma; EML4‐ALK‐S, EML4‐ALK sarcomas; ENB, esthesioneuroblastoma; EPN, ependymoma; ESS_HG, high‐grade endometrial stromal sarcoma; ESS_LG, low‐grade endometrial stromal sarcoma; ETMR, embryonal tumour with multi‐layered rosettes; EWS, Ewing sarcoma; GBM, glioblastoma; GIST, gastrointestinal stromal tumour; HMB, haemangioblastoma; IFS, infantile fibrosarcoma; IMT, inflammatory myofibroblastic tumour; LGFMS, low‐grade fibromyxoid sarcoma; LGG, low‐grade astrocytoma and ganglioglioma; LIPN, cerebellar liponeurocytoma; LIPO, lipoma; MB, medulloblastoma; MCS, chondrosarcoma; MELAN, malignant melanoma; MELCYT, melanocytoma; MLS, myxoid liposarcoma; MNG, meningioma; MRT, malignant rhabdoid tumour; O, anaplastic oligodendroglioma; OS, osteosarcoma; PGG, paraganglioma; PIN, pineoblastoma; PITAD, pituitary adenoma; PITUI, spindle cell oncocytoma; PLASMA, plasmacytoma; PLEX, choroid plexus carcinoma; PTPR, papillary tumour of the pineal region; PXA, pleiomorphic xanthoastrocytoma; RETB, retinoblastoma; RMS, rhabdomyosarcoma; SBRCT_BCOR, small blue round cell tumour with BCOR alteration; SBRCT_CIC, small blue round cell tumour with CIC alteration; SCC, squamous cell carcinoma; SCHW, melanocytic schwannoma; SFT, solitary fibrous tumour; SUBEPN, subependymoma; UMAP, Uniform Manifold Approximation and Projection
Overall, these data identify BCOR‐ITD as a homogeneous and reproducible molecular signature and define two related entities, intracranial and extracranial.
Differential analysis of expression data identifies biological differences between CNS BCOR‐ITD and BCOR‐ITD sarcomas and sheds light on their respective cell of origin
We aimed to define the molecular processes that drive the transcriptional traits underlying the differences between BCOR‐ITD sarcomas and CNS BCOR‐ITD.
The expression levels of 2,306 genes were found to be significantly different between CNS BCOR‐ITD and BCOR‐ITD sarcomas. In comparison, CNS BCOR‐ITD was more distant from medulloblastoma and ependymoma with 5,587 and 4,763 significantly different genes, respectively. Similarly, comparison between BCOR‐ITD sarcomas and bone or soft tissue Ewing sarcomas found 4,921 significant genes (supplementary material, Figure S4A–D).
The top 20 Gene Ontology (GO) Biological Processes enriched in CNS BCOR‐ITD in comparison with BCOR‐ITD sarcomas all refer to CNS development, neurogenesis, and glial cell differentiation (supplementary material, Table S5). Reciprocally, BCOR‐ITD sarcomas display an over‐representation of GO terms related to embryonic skeletal development and embryonic morphogenesis, in line with the 33 Hox genes found in the 100 top listed DEGs (supplementary material, Table S6).
DEGs were used as marker genes for CNS BCOR‐ITD and BCOR‐ITD sarcomas. Using publicly available single‐cell/single‐nuclei RNA‐Seq atlases, namely Cao et al (human foetal gene expression) and Han et al (adult, foetal, cord, and iPS tissue), we evaluated the enrichment of the BCOR‐ITD sarcoma signature and the CNS BCOR‐ITD signature in specific cell identities available in these atlases. The signature of CNS BCOR‐ITD sarcomas was found to be enriched in neurons, astrocytes, and oligodendrocytes (Figure 5 and supplementary material, Table S7A,C and Figure S5), while the signature of BCOR‐ITD sarcomas was enriched in stromal, endothelial, and mesangial cells of various organs (Figure 5 and supplementary material, Table S7B,D and Figure S5). This clear dichotomy suggests both types of tumours derive from distinct cells of origin.
Figure 5.
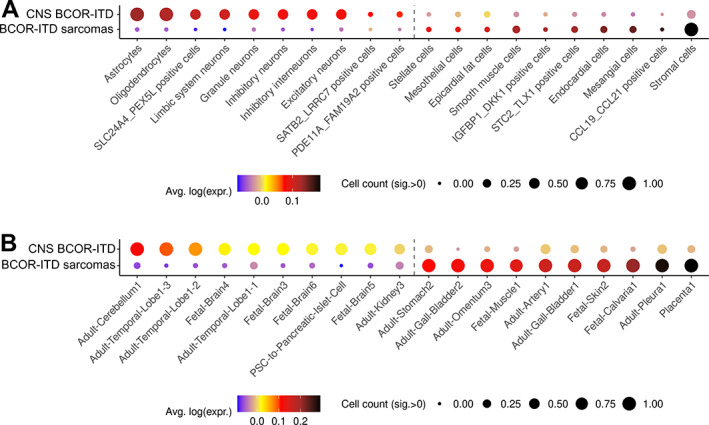
Dot plot summarising the distributions of CNS BCOR‐ITD and BCOR‐ITD sarcoma signatures over cell types independently analysed by single‐cell/single‐nuclei RNA‐Seq. The signatures were computed from the DEGs between CNS BCOR‐ITD and the BCOR‐ITD sarcoma with a minimal fold‐change of 2.5. Dot colour represents the average of each distribution and size represents the count of cells within a cell type with a signature intensity strictly higher than 0 brought to the [0, 1] range: a gene signature is highly represented in a cell type of the atlas when the dot gets bigger (several cells of one type express the signature) and tends towards black colour (several genes of the signature are expressed within a cell type). (A) Dot plot of the CNS BCOR‐ITD signature (upper line) and the BCOR‐ITD sarcomas signature (lower lines) over selected cell types of the Cao et al's atlas. Only significant cell types are shown. The complete list of this atlas' cell types is shown in supplementary material, Figure S5. The number of genes in common between the signatures and the reference database is as follows: CNS BCOR‐ITD n = 114; BCOR‐ITD sarcomas n = 43. (B) Dot plot of the CNS BCOR‐ITD signature (upper line) and the BCOR‐ITD sarcoma signature (lower line) over selected cell types of the Han et al's atlas. Only significant cell types are shown. The complete list of this atlas' cell types is shown in supplementary material, Figure S5. Number of genes in signatures is also in the reference database: CNS BCOR‐ITD n = 103; BCOR‐ITD sarcomas n = 38.
We then analysed the top listed DEGs to better characterise the molecular pathways governing CNS BCOR‐ITD and BCOR‐ITD sarcoma oncogenesis. Genes involved in targetable signalling pathways were scrutinised in particular. Compared with reference tumours of medulloblastoma and ependymoma types, CNS BCOR‐ITD displayed overexpression of NTRK2, NGFR, PDGFRA, and WNT7B (Table 3). Activation of the corresponding pathways could be confirmed by GSEA only for the PDGFRA pathway (CNS BCOR‐ITD versus medulloblastomas, Normalised Enriched Score [NES] 1.42, False Discovery Rate q‐value [FDRq] 0.085) (supplementary material, Figure S4E). In contrast, BCOR‐ITD sarcomas were better defined by overexpression of NTRK3, EGFR, and PDGFRA, in comparison with Ewing sarcomas (Table 4), as confirmed by the GSEA approach (NES 1.20, FDRq 0.244) (supplementary material, Figure S4F). Interestingly, two genes remained significantly differentially expressed when CNS BCOR‐ITD and BCOR‐ITD sarcomas were compared, NTRK2 and WNT7B, suggesting that these two may participate in the molecular subgrouping between the two entities.
Table 3.
Key DEGs
| Group A (light grey) versus group B (dark grey) | NTRK1 | NTRK2 | NTRK3 | NGFR | EGFR | PDGFRA | Wnt7B | |
|---|---|---|---|---|---|---|---|---|
| CNS BCOR‐ITD, n = 8 | BCOR‐ITD sarcomas, n = 15 | n/s | 28.0 | n/s | n/s | n/s | n/s | 15.8 |
| CNS BCOR‐ITD, n = 8 | Ependymomas, n = 10 | n/s | 5.7 | 7.3 | 24.9 | 4.0 | 22.2 | 14.0 |
| CNS BCOR‐ITD, n = 8 | Medulloblastomas, n = 21 | n/s | 33.0 | 1.8 | 26.4 | 9.4 | 13.1 | 39.1 |
| BCOR‐ITD sarcomas, n = 15 | Ewing sarcomas, n = 30 | −1.6 | 2.2 | 29.4 | 5.6 | 12.7 | 28.8 | 2.4 |
The fold‐change of expression of seven genes between tumours of group A (light grey) and tumours of group B (dark grey) is presented. The number of cases in each group is indicated. Fold‐change is shown in favour of the tumours of group A (light grey). Values shown in bold are considered of high significance.
Table 4.
Summary of differences and similarities between BCOR‐ITD sarcomas of all localisations
| Clinical data | Histology | Radiology | Molecular biology | ||||
|---|---|---|---|---|---|---|---|
| Age, median (range)) | Prognosis # deceased mean delay | Mitotic counts per 10 hpf (range) | Immunostaining | (features apply to all tumour types) | DEGs | ||
| BCOR‐ITD sarcomas | CCSK | 2.0 (0.5–18.9) | 0/7 | 0–18 |
BCOR SATB2, Cyclin D1, BCL2, TLE1 WT1‐neg, SMARCB1 + |
Heterogeneous masses Necrosis Haemorrhage Low‐to‐moderate contrast enhancement |
NTRK3 EGFR PDGFRa |
| HG‐ESS | 51.1 (25.8–62.4) |
2/4 36 months |
14–21 |
BCOR SATB2, Cyclin D1, BCL2, TLE1 Desmin‐neg, Caldesmon‐neg |
|||
| URCS | 0.7 (0.1–19.7) |
4/8 31 months |
1.7–13.7 |
BCOR SATB2, Cyclin D1, BCL2, TLE1 |
|||
| BCOR‐ITD CNS tumours | HGNET‐BCOR | 1.8 (1.2–7.6) |
6/10 28 months |
5–62 |
BCOR SATB2, Cyclin D1, BCL2, TLE1 NeuN GFAP+/−, Olig2 +/− |
NTRK2 Wnt7b PDGFRa |
|
hpf, high‐power field.
Discussion
We present here a comprehensive description of 33 BCOR‐ITD tumours, putting together sarcomas of all known locations and CNS BCOR‐ITD. Although this study is retrospective, with a relatively small number of cases in each subgroup, treated following disparate protocols, we explored the main similarities and differences, as summarised in Table 4. This analysis showed no clear clinical, radiological, or pathological specificity of BCOR‐ITD tumours, but a common transcriptomic signature. We speculate that a common tumorigenic scheme, driven by the ITD itself and applied to distinct cells of origin in different environments, leads to two distinctive lineages: BCOR‐ITD sarcomas and CNS BCOR‐ITD.
Similarities and differences of the clinical presentations
The family of BCOR‐ITD tumours offers a heterogeneous set of presentations, reflecting the variety of locations. The main features in common are the median age at diagnosis as previously reported in CCSK (2.0 years over 33 cases [1(p),2,3]), URCS (0.8 year over 28 cases [12]), and CNS BCOR‐ITD (3.5 years over 49 cases [16, 17, 18, 19, 20, 21, 22, 23, 24, 26, 36, 37, 38]); the local presentation; and the poor prognosis.
Two entities appear as clinical outliers. First, CCSK with BCOR‐ITD alteration display higher survival rates: among the seven cases in our series, six were alive at the end of the study and one was still on therapy. A remission was obtained in five cases after the first‐line treatment. No histological feature could easily explain this better prognosis. Interrogating the therapeutic strategy reveals that all CCSK cases underwent upfront extracapsular surgery with R0 outcomes. First‐line complete resections were also performed on most CNS BCOR‐ITD cases, but microscopic mid‐range dissemination of the disease remains possible as brain tumours are not encapsulated. In addition, the chemotherapy plan of CCSK includes platinum derivatives and anthracyclines, both being excluded from the fist‐line treatment of other localisations.
HG‐ESS also appears to be a separate entity as it occurs exclusively in adult uterine tissue, with a more severe presentation at diagnosis, either because the symptoms are detected later or the disease progresses faster. Again, there is no histopathological clue to decipher why this type of tumour presents differently.
We report two cases primarily localised in bones, which is a rare situation as compared with the related BCOR‐CCNB3 subgroup. Three previous cases were reported in adolescents [12, 13]. Our cases were older, in the fifth decade, and both presented with synchronous pulmonary metastases at diagnosis. As CCSK are known to evolve with very long‐term pulmonary and bone metastasis [39], it was tempting to document these bone/pulmonary tumours as metastases from an occult CCSK. We found no argument in the medical records arguing in favour of this hypothesis.
Cranial and extracranial BCOR‐ITD tumours form two distinct subgroups and question the cell of origin
Transcriptomic profile is becoming a major tool for the molecular diagnosis of paediatric and adult sarcomas. Classification of cerebral tumours routinely uses DNA methylation data. A recent report shows that methylome profiling also reliably defines clusters of sarcomas, coherently with histopathology and expression data [28]. We showed here that both techniques could be used to define the whole family and its subgroups.
CNS BCOR‐ITD and BCOR‐ITD sarcomas present close but distinct transcriptomic and DNA methylation signatures. This may result from a common oncogenic programme, acquired somatically and applied to cells of distinct types, within distinct environments. This was corroborated by immunohistopathological data that shows neuro‐glial markers in CNS BCOR‐ITD (GFAP, Olig2, NeuN, NF70) and not in BCOR‐ITD sarcomas. Conversely, the sharp segregation between the two groups may result from a ‘host–tissue effect’. Such bias cannot be formally excluded at this stage but is rather unlikely because the same methods of molecular analysis were applied to compare renal, soft tissue, and cerebral rhabdoid tumours, and did not document a host–tissue effect (compare malignant rhabdoid tumours and atypical teratoid rhabdoid tumours in Figure 4A).
We argue that the divergence between CNS BCOR‐ITD and BCOR‐ITD sarcomas comes from differences in their cell of origin. Accordingly, the distance between the two subgroups was higher in the UMAP based on methylation data compared to transcriptome data. Second, differential analyses of expression data revealed a gene signature for both subgroups, based on the DEG. The signature of CNS BCOR‐ITD was enriched in genes expressed in neuro‐glial cells while BCOR‐ITD sarcomas predominantly expressed embryonal/developmental genes. Furthermore, those gene signatures were compared to two single‐cell/single‐nuclei RNA‐Seq human atlases. Analysis of the top listed cell types extracted from the atlases coherently showed that the signatures of CNS BCOR‐ITD tumours and BCOR‐ITD sarcomas point towards distinct cell types. Altogether, these indirect arguments suggest that CNS BCOR‐ITD and BCOR‐ITD sarcomas differ in their cell of origin, with CNS BCOR‐ITD being derived from a primitive neuro‐glial progenitor cell, while BCOR‐ITD sarcomas derive as expected from mesenchymal progenitors. Hence, CNS BCOR‐ITD tumours may not be seen as sarcomas of the brain.
Expression data shed light on the activated pathways
As an attempt to elucidate the tumourigenesis of BCOR‐ITD tumours and identify the possible therapeutic targets, we studied the most significant DEGs and identified differential upregulation of NTRK2, WNT7B, and PDGFRa in CNS BCOR‐ITD cases (in comparison with medulloblastomas and BCOR‐ITD sarcomas) and NTRK3, EGFR, and PDGFRa in BCOR‐ITD sarcomas (in comparison with Ewing sarcomas).
Increased expression of NTRK3/TrkC receptors has already been reported in CCSK [40], BCOR‐ITD, and more generally in BCOR‐rearranged tumours [41]. Our data confirm this result and further argue that the whole NTRK3 pathway is active as GSEA shows its members to be significantly enriched in BCOR‐ITD sarcomas. Our study also shows that NTRK signalling is active in CNS BCOR‐ITD cases, given positive NTRK immunolabeling and upregulation of NTRK2 mRNA.
Results on Wnt7b in CNS BCOR‐ITD were corroborated neither by nuclear translocation of β‐catenin (as reported in Ref. [17]) nor by GSEA.
EGFR mRNA upregulation in BCOR‐ITD sarcomas and, to a lesser extent, in CNS BCOR‐ITD contrasts with the strong staining of EGFR protein in CNS BCOR‐ITD cases and in only a few BCOR‐ITD sarcomas. Despite these discrepancies, most probably related to technical artefacts, EGFR signalling appears to be an active pathway in the whole family, in line with the recent work showing strong positivity in nine cases of BCOR‐ITD URCS [42].
Concordant clues point towards NTRK, EGFR, and PDGFRa, but preclinical studies are required to evaluate the corresponding pathways and possibly document the role of the BCOR‐ITD protein in their activation.
Author contributions statement
YB, AT‐E, AG, LC, GS, FB, OD, GP and FD designed the study. YB, AT‐E, AG, LC, OD, GP and FD wrote the manuscript. YB and MC acquired and analysed the clinical data. LC reviewed all imaging data. AT‐E and AG reviewed all histopathological data, and performed immunohistochemistry. DG, DB, JV, SG, CQ, SW, FT, JM‐P and GP processed and analysed RNA‐Seq and methylome data. DO, NC, GS, FB, MS, CD, VM‐C, MB, FT, DP, MK, M‐CM, OD and FD recruited patients and provided samples and clinical information. All authors reviewed the manuscript.
Supporting information
Supplementary materials and methods
Text S1. Detailed imaging characteristics of BCOR‐ITD tumours
Text S2. Detailed histopathological and immunohistochemical analyses
Figure S1. Structure of the BCOR‐ITD cohort. For RNA‐Seq, the FFPE and frozen samples correspond to distinct tumours. There is no overlap between the two sample types
Figure S2. Examples of typical imaging findings in BCOR‐ITD sarcomas at diagnosis
Figure S3. Boxplots showing the mRNA levels of five markers in the BCOR‐ITD family in comparison with other families of sarcomas and brain tumours
Figure S4. Molecular divergence between CNS BCOR‐ITD and BCOR‐ITD sarcomas
Figure S5. Dot plot summarising loose (FC > 1.5) and stringent (FC > 2.5) CNS BCOR‐ITD and BCOR‐ITD sarcoma signature distributions over cell types independently analysed by sc/sn RNA‐Seq
Table S1. Tumour features at diagnosis, patient by patient.
Table S2. Treatment and prognosis, patient by patient
Table S3. Radiological features of BCOR‐ITD tumours
Table S4. Immunhistochemistry results
Table S5. Analysis of the DEGs between CNS BCOR‐ITD and BCOR‐ITD sarcomas
Table S6. Analysis of the DEGs between BCOR‐ITD sarcomas and CNS BCOR‐ITD
Table S7. List of the top 10 cell types where the gene signatures of CNS BCOR‐ITD and BCOR‐ITD sarcomas are enriched, based on two single‐cell/single‐nuclei RNA‐Seq atlases
Acknowledgements
We would like to thank the clinicians who shared medical reports: G Bisogno, R Alaggio, J Grill, P Berlanga, K Beccaria, S Puget, JC Gentet, C Coze, AS Defachelles, J Probert, P Schneider, S Le Gallic, S Ducassou, and M Toulmonde. We also thank the technicians who helped managing imaging transfer from one centre to the other: E Gasparollo, G Lemaire, S Kankonde, C Courio, F Becker, and K Legoupil.
This work was supported by the Institut National de la Santé et de la Recherche medical (INSERM), Institut Curie, the Institut National du Cancer (INCa), and the Direction Générale de l'offre de soins INCa‐DGOS_13219 (iMAPS project).
We are indebted to the following associations for providing essential support: L'Etoile de Martin, la Course de l'Espoir, M la vie avec Lisa, ADAM, Couleur Jade, Dans les pas du Géant, Courir pour Mathieu, Marabout de Ficelle, Les Bagouzamanon, Enfants et Santé, Les Amis de Claire, and Liv et Lumière.
No conflicts of interest were declared.
Contributor Information
Gaëlle Pierron, Email: gaelle.pierron@curie.fr.
François Doz, Email: francois.doz@curie.fr.
References
References 43 and 44 are cited only in the supplementary material.
- 1. Ueno‐Yokohata H, Okita H, Nakasato K, et al. Consistent in‐frame internal tandem duplications of BCOR characterize clear cell sarcoma of the kidney. Nat Genet 2015; 47: 861–863. [DOI] [PubMed] [Google Scholar]
- 2. Astolfi A, Melchionda F, Perotti D, et al. Whole transcriptome sequencing identifies BCOR internal tandem duplication as a common feature of clear cell sarcoma of the kidney. Oncotarget 2015; 6: 40934–40939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Roy A, Kumar V, Zorman B, et al. Recurrent internal tandem duplications of BCOR in clear cell sarcoma of the kidney. Nat Commun 2015; 6: 8891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Karlsson J, Valind A, Gisselsson D. BCOR internal tandem duplication and YWHAE‐NUTM2B/E fusion are mutually exclusive events in clear cell sarcoma of the kidney. Genes Chromosomes Cancer 2016; 55: 120–123. [DOI] [PubMed] [Google Scholar]
- 5. Kenny C, Bausenwein S, Lazaro A, et al. Mutually exclusive BCOR internal tandem duplications and YWHAE‐NUTM2 fusions in clear cell sarcoma of kidney: not the full story. J Pathol 2016; 238: 617–620. [DOI] [PubMed] [Google Scholar]
- 6. Gooskens SL, Gadd S, van den Heuvel‐Eibrink MM, et al. BCOR internal tandem duplications in clear cell sarcoma of the kidney. Genes Chromosomes Cancer 2016; 55: 549–550. [DOI] [PubMed] [Google Scholar]
- 7. Argani P, Perlman EJ, Breslow NE, et al. Clear cell sarcoma of the kidney: a review of 351 cases from the National Wilms Tumor Study Group Pathology Center. Am J Surg Pathol 2000; 24: 4. [DOI] [PubMed] [Google Scholar]
- 8. Argani P, Pawel B, Szabo S, et al. Diffuse strong BCOR immunoreactivity is a sensitive and specific marker for clear cell sarcoma of the kidney (CCSK) in pediatric renal neoplasia. Am J Surg Pathol 2018; 42: 1128–1131. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Mariño‐Enriquez A, Lauria A, Przybyl J, et al. BCOR internal tandem duplication in high‐grade uterine sarcomas. Am J Surg Pathol 2018; 42: 335–341. [DOI] [PubMed] [Google Scholar]
- 10. Brahmi M, Franceschi T, Treilleux I, et al. Molecular classification of endometrial stromal sarcomas using RNA sequencing defines nosological and prognostic subgroups with different natural history. Cancers 2020; 12: 2604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Micci F, Gorunova L, Agostini A, et al. Cytogenetic and molecular profile of endometrial stromal sarcoma. Genes Chromosomes Cancer 2016; 55: 834–846. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Antonescu CR, Kao Y‐C, Xu B, et al. Undifferentiated round cell sarcoma with BCOR internal tandem duplications (ITD) or YWHAE fusions: a clinicopathologic and molecular study. Mod Pathol 2020; 33: 1669–1677. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Malik F, Zreik RT, Hedges DJ, et al. Primary bone sarcoma with BCOR internal tandem duplication. Virchows Arch 2020; 476: 915–920. [DOI] [PubMed] [Google Scholar]
- 14. Kao Y‐C, Sung Y‐S, Zhang L, et al. Recurrent BCOR internal tandem duplication and YWHAE‐NUTM2B fusions in soft tissue undifferentiated round cell sarcoma of infancy: overlapping genetic features with clear cell sarcoma of kidney. Am J Surg Pathol 2016; 40: 1009–1020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Santiago T, Clay MR, Allen SJ, et al. Recurrent BCOR internal tandem duplication and BCOR or BCL6 expression distinguish primitive myxoid mesenchymal tumor of infancy from congenital infantile fibrosarcoma. Mod Pathol 2018; 31: 374. [DOI] [PubMed] [Google Scholar]
- 16. Appay R, Macagno N, Padovani L, et al. HGNET‐BCOR tumors of the cerebellum: clinicopathologic and molecular characterization of 3 cases. Am J Surg Pathol 2017; 41: 1254–1260. [DOI] [PubMed] [Google Scholar]
- 17. Sturm D, Orr BA, Toprak UH, et al. New brain tumor entities emerge from molecular classification of CNS‐PNETs. Cell 2016; 164: 1060–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Haberler C, Reiniger L, Rajnai H, et al. Case of the month 1‐2019: CNS high‐grade neuroepithelial tumor with BCOR alteration. Clin Neuropathol 2019; 38: 4–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Ferris SP, Velazquez Vega J, Aboian M, et al. High‐grade neuroepithelial tumor with BCOR exon 15 internal tandem duplication—a comprehensive clinical, radiographic, pathologic, and genomic analysis. Brain Pathol 2020; 30: 46–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Yoshida Y, Nobusawa S, Nakata S, et al. CNS high‐grade neuroepithelial tumor with BCOR internal tandem duplication: a comparison with its counterparts in the kidney and soft tissue. Brain Pathol 2018; 28: 710–720. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Al‐Battashi A, Al Hajri Z, Perry A, et al. A cerebellar high‐grade neuroepithelial tumour with BCOR alteration in a five‐year‐old child: a case report. Sultan Qaboos Univ Med J 2019; 19: e153–e156. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Bremer J, Kottke R, Johann PD, et al. A single supratentorial high‐grade neuroepithelial tumor with two distinct BCOR mutations, exceptionally long complete remission and survival. Pediatr Blood Cancer 2020; 67: e28384. [DOI] [PubMed] [Google Scholar]
- 23. Kirkman MA, Pickles JC, Fairchild AR, et al. Early wound site seeding in a patient with central nervous system high‐grade neuroepithelial tumor with BCOR alteration. World Neurosurg 2018; 116: 279–284. [DOI] [PubMed] [Google Scholar]
- 24. Łastowska M, Trubicka J, Sobocińska A, et al. Molecular identification of CNS NB‐FOXR2, CNS EFT‐CIC, CNS HGNET‐MN1 and CNS HGNET‐BCOR pediatric brain tumors using tumor‐specific signature genes. Acta Neuropathol Commun 2020; 8: 105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Louis DN, Perry A, Wesseling P, et al. The 2021 WHO Classification of Tumors of the Central Nervous System: a summary. Neuro Oncol 2021; 23: 1231–1251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Reinhardt A, Stichel D, Schrimpf D, et al. Tumors diagnosed as cerebellar glioblastoma comprise distinct molecular entities. Acta Neuropathol Commun 2019; 7: 163. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Watson S, Perrin V, Guillemot D, et al. Transcriptomic definition of molecular subgroups of small round cell sarcomas: molecular classification of sarcoma subtypes. J Pathol 2018; 245: 29–40. [DOI] [PubMed] [Google Scholar]
- 28. Koelsche C, Schrimpf D, Stichel D, et al. Sarcoma classification by DNA methylation profiling. Nat Commun 2021; 12: 498. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Capper D, Jones DTW, Sill M, et al. DNA methylation‐based classification of central nervous system tumours. Nature 2018; 555: 469–474. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Dobin A, Davis CA, Schlesinger F, et al. STAR: ultrafast universal RNA‐seq aligner. Bioinformatics 2013; 29: 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31. Trapnell C, Williams BA, Pertea G, et al. Transcript assembly and quantification by RNA‐Seq reveals unannotated transcripts and isoform switching during cell differentiation. Nat Biotechnol 2010; 28: 511–515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Ritchie ME, Phipson B, Wu D, et al. limma powers differential expression analyses for RNA‐sequencing and microarray studies. Nucleic Acids Res 2015; 43: e47. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Cao J, O'Day DR, Pliner HA, et al. A human cell atlas of fetal gene expression. Science 2020; 370: eaba7721. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Han X, Zhou Z, Fei L, et al. Construction of a human cell landscape at single‐cell level. Nature 2020; 581: 303–309. [DOI] [PubMed] [Google Scholar]
- 35. Subramanian A, Tamayo P, Mootha VK, et al. Gene set enrichment analysis: a knowledge‐based approach for interpreting genome‐wide expression profiles. Proc Natl Acad Sci U S A 2005; 102: 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Paret C, Theruvath J, Russo A, et al. Activation of the basal cell carcinoma pathway in a patient with CNS HGNET‐BCOR diagnosis: consequences for personalized targeted therapy. Oncotarget 2016; 7: 83378–83391. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Rao S, Mitra S, Sugur H, et al. Central nervous system high grade neuroepithelial tumor with BCOR immunopositivity: is there a molecular heterogeneity? Brain Tumor Pathol 2021; 38: 41–49. [DOI] [PubMed] [Google Scholar]
- 38. Ishi Y, Shimizu A, Takakuwa E, et al. High‐grade neuroepithelial tumor with BCL6 corepressor‐alteration presenting pathological and radiological calcification: a case report. Pathol Int 2021; 71: 348–354. [DOI] [PubMed] [Google Scholar]
- 39. Aldera AP, Pillay K. Clear cell sarcoma of the kidney. Arch Pathol Lab Med 2020; 144: 119–123. [DOI] [PubMed] [Google Scholar]
- 40. Cutcliffe C, Kersey D, Huang C‐C, et al. Clear cell sarcoma of the kidney: up‐regulation of neural markers with activation of the sonic hedgehog and Akt pathways. Clin Cancer Res 2005; 11: 7986–7994. [DOI] [PubMed] [Google Scholar]
- 41. Kao Y‐C, Sung Y‐S, Argani P, et al. NTRK3 overexpression in undifferentiated sarcomas with YWHAE and BCOR genetic alterations. Mod Pathol 2020; 33: 1341–1349. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Salgado CM, Zin A, Garrido M, et al. Pediatric soft tissue tumors with BCOR ITD express EGFR but not OLIG2. Pediatr Dev Pathol 2020; 23: 424–430. [DOI] [PubMed] [Google Scholar]
- 43. Chen YA, Lemire M, Choufani S, et al. Discovery of cross‐reactive probes and polymorphic CpGs in the Illumina Infinium HumanMethylation450 microarray. Epigenetics 2013; 8: 203–209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cardoen L, Tauziède‐Espariat A, Dangouloff‐Ross V, et al. Imaging features with histopathologic correlation of CNS high‐grade neuroepithelial tumors with a BCOR internal tandem duplication. AJNR Am J Neuroradiol 2022; 43: 151–156. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary materials and methods
Text S1. Detailed imaging characteristics of BCOR‐ITD tumours
Text S2. Detailed histopathological and immunohistochemical analyses
Figure S1. Structure of the BCOR‐ITD cohort. For RNA‐Seq, the FFPE and frozen samples correspond to distinct tumours. There is no overlap between the two sample types
Figure S2. Examples of typical imaging findings in BCOR‐ITD sarcomas at diagnosis
Figure S3. Boxplots showing the mRNA levels of five markers in the BCOR‐ITD family in comparison with other families of sarcomas and brain tumours
Figure S4. Molecular divergence between CNS BCOR‐ITD and BCOR‐ITD sarcomas
Figure S5. Dot plot summarising loose (FC > 1.5) and stringent (FC > 2.5) CNS BCOR‐ITD and BCOR‐ITD sarcoma signature distributions over cell types independently analysed by sc/sn RNA‐Seq
Table S1. Tumour features at diagnosis, patient by patient.
Table S2. Treatment and prognosis, patient by patient
Table S3. Radiological features of BCOR‐ITD tumours
Table S4. Immunhistochemistry results
Table S5. Analysis of the DEGs between CNS BCOR‐ITD and BCOR‐ITD sarcomas
Table S6. Analysis of the DEGs between BCOR‐ITD sarcomas and CNS BCOR‐ITD
Table S7. List of the top 10 cell types where the gene signatures of CNS BCOR‐ITD and BCOR‐ITD sarcomas are enriched, based on two single‐cell/single‐nuclei RNA‐Seq atlases