

Abstract
Corneal infections result through interaction between microbes and host innate immune receptors. Damage to the cornea occurs as a result of microbial virulence factors and is often exacerbated by lack of a controlled host immune response; the latter contributing to bystander damage to corneal structure. Understanding mechanisms involved in host microbial interactions is critical to development of novel therapeutic targets, ultimate control of microbial pathogenesis, and restoration of tissue homeostasis. Studies on these interactions continue to provide exciting findings directly related to this ultimate goal.
1. Introduction
Host microbe interactions at the corneal surface are facilitated by epithelial defects or injuries that predispose the eye to pathogens. Among bacterial pathogens, Staphylococcus aureus (S. aureus) and Pseudomonas aeruginosa (P. aeruginosa), are most often encountered. Fungi, including both filamentous and yeast (e.g., Fusarium, and Candida species), also induce destructive corneal disease. However, the leading cause of corneal ulcers and blindness worldwide is viral induced and follows recurrent infection of the cornea by herpes simplex virus (HSV-1). This virus is most commonly transmitted through contact with HSV in herpes lesions, mucosal surfaces, genital secretions, or oral secretions. This review will focus on several of these pathogens, their virulence factors and how the host responds.
2. Staphylococcus aureus (S. aureus)
S. aureus is a leading cause of keratitis globally and is the most virulent of the Staphylococcus species [1]. It possesses many virulence factors which allow for host adhesion, cytolytic activity, and evading the innate immune system. The bacterium uses collagen and fibronectin to adhere to corneal epithelial cells, initiating keratitis. A rabbit model, in which contact lenses soaked in S. aureus strains containing the collagen-binding adhesin (Cna+) or its isogenic mutant lacking the adhesin (Cna−), were placed on the injured cornea and outcome showed that Cna significantly contributed to bacterial adherence and corneal colonization [2]. The proteome of S. aureus isolates from keratitis [3] was analyzed and four adhesin virulence factors identified: fibronectin binding protein A, staphopain, glyceraldehyde-3-phosphate dehydrogenase 2 and extracellular adhesin proteins. When compared with a less virulent lab strain, the clinical isolate produced more potentially important virulence factors than the less virulent lab strain, accounting for ability of the clinical S. aureus isolate to cause more severe keratitis [4]. Fibronectin-binding proteins (FnBP) contribute to adherence in vitro (cultured human corneal epithelial cells, HCEC). Evidence suggests that FnBP promotes binding to the corneal epithelium, and ligation of α5β1 integrin (receptor) which allows actin polymerization, endocytic vesicle formation and pathogen invasion [3]. Recently, studies with HCEC showed that staphopain A, an extracellular protease, increased adhesion and cell invasion by increasing fibronectin binding; its inhibition decreased pathogenesis [5]. Further studies of staphopain A will be required to determine its potential as a target of treatment.
2.1. Toxins
The role of alpha (α)-toxin in corneal infection has been established by comparing virulence in animal models using α-toxin-deficient isogenic mutants with their wild-type parental strains [6]. α-toxin lyses erythrocytes and leukocytes, but not the predominant one, the neutrophil (PMN) [7]. The toxin binds to disintegrin and metalloproteinase 10 (ADAM10), resulting in binding to eukaryotic cells. Following binding, disruption of focal adhesions [8, 9], such as E-cadherin in epithelial cells, loss of epithelial barrier function and pore formation ensues [10]. The importance of other toxins: β-toxin, γ-toxin, and Panton–Valentine leukocidin, have been analyzed to a lesser degree and their roles in eye infections are less clear [11]. However, it has been shown that γ-toxin can cause corneal pathology and induce disease when injected intrastromally [12]. Other toxins such as the phenol-soluble modulins have yet to be examined in any animal models for their virulence in the eye [11].
Emergence of methicillin-resistant S. aureus (MRSA strains), resistant to the most commonly used fluoroquinolones necessitates development of new antimicrobials for bacterial keratitis. Besifloxacin, the first topical chlorofluoroquinolone was developed solely for ophthalmic use. It inhibits DNA gyrase and topoisomerase IV (ubiquitous in bacteria) and is essential for bacterial survival [13]. The drug has been shown effective in a rabbit model of keratitis and reduced MRSA colony-forming units more effectively than gatifloxacin or moxifloxacin [14]. A retrospective study showed that besifloxacin treatment of human cases of bacterial keratitis yielded no serious adverse effects and had low incidences of corneal scarring or neovascularization similar to that seen with moxifloxacin treatment [15]. The use of already existing agents to treat MRSA is also being studied. Lysostaphin is a zinc metalloproteinase extracted from S. simulans that lyses S. aureus by disrupting its peptidoglycan layer. It is effective in MRSA keratitis and endophthalmitis [16]. Las A protease (staphylolysin) targets the peptidoglycan of S. aureus and was comparable to vancomycin when treatment was begun early. It was more effective when treatment was begun in the late phase of infection, suggesting that Las A can lyse bacterial cell walls during the stationary growth phase [16]. Use of animal models of keratitis and endophthalmitis continue to provide insights into S. aureus virulence and host factors that are active in limiting these infections. It is hopeful that data from the animal studies will translate into potential human treatment.
3. Pseudomonas aeruginosa (P. aeruginosa)
P. aeruginosa, (a gram-negative bacterium), is widely dispersed in the environment [17] and causes acute and chronic infections in humans. P. aeruginosa can cause acute infection, including: keratitis, otitis externa, bacteremia, burn wound infections, and pneumonia associated with cystic fibrosis. It also can cause severe chronic infections [18]. The bacteria causes severe infections of the cornea, particularly in contact lens wearing patients [19] in industrialized countries, such as the USA, and it is one of the leading causes of corneal ulcers [20]. If untreated, or non-responsive to topical treatment, corneal perforation may occur, which necessitates a transplant; the annual cost to treat keratitis (approximately 175 million) is a financial, as well as a health burden [19,21].
3.1. Virulence factors and host response
P. aeruginosa expresses numerous virulence factors, including pili and non-pili adhesins, flagella, proteases, hemolysins, exoenzymes and exotoxins, that play a role in P. aeruginosa invasion and infection of the cornea. Antibacterial agents in tears and a stratified epithelium protect the cornea, but trauma, contact lens wear and other adverse events can compromise the tissue and allow attachment to host receptors, initiating disease. The glycocalyx prevents phagocytosis of invading P. aeruginosa by host cells [18]; while proteases (elastase and alkaline protease) and toxins (exotoxin A), compromise host defenses by degrading proteins (immunoglobulin A and surfactant protein D) [18]. Exotoxins, released by the type III secretion system (T3SS), which secretes ExoS, ExoT and ExoU, promote bacterial survival and spreading by inducing apoptosis of infiltrating PMN [22]. ExoU also has phospholipase activity and promotes stromal necrosis and inflammation [22]. Other studies have shown a novel virulence strategy in P. aeruginosa involving bleb niche formation (a niche within the plasma membrane of epithelial cells) in which bacteria survive, replicate and swim. Their intracellular survival involves ExoS ADP ribosyl activity [24] and implicates a connection between bleb niche formation and the known role of Exo-S mediated apoptosis and Rab GTPase activation [25].
P. aeruginosa lipopolysaccharide (LPS) and flagella bind to host cell surface receptors including TLR2, TLR4 and TLR5 [23]. Interactions initiate a signaling cascade in resident macrophages and corneal epithelial cells to produce inflammatory cytokines and chemokines, which promote PMN and other inflammatory cell recruitment to the cornea. PMN are essential for eradicating bacteria, but their persistence contributes to the severity of keratitis [20,23]. For instance, elevated expression of macrophage inflammatory protein-2 (MIP-2) and IL-1β, known chemoattractants for PMN [20], is consistent with increased disease severity, PMN infiltrate, persistence and bacterial load. This scenario is played out in P. aeruginosa infected susceptible C57BL/6 (cornea perforates) compared to resistant BALB/c mice (cornea heals) [20]. However, antibody depletion of PMN, the major inflammatory cell seen in keratitis, results in unhindered bacterial growth, and corneal destruction in both groups of mice (unpublished data, Fig.1), and in C57BL/6 mice, septicemia and death. In this regard, inhibition of IL-1β improves disease outcome in C57BL/6 mice by reducing, but not eliminating, PMN in the infected cornea. Besides PMN, CD4+ T cells and Langerhans cells, the latter especially in contact lens wear, migrate to the site of infection and contribute to the host immune response against P. aeruginosa [20]. It also was demonstrated that depleting CD4+ T cells in infected C57BL/6 mice prevented corneal perforation, while in controls, perforation occurred at 5–7 days after infection [20]. Suryawanshi et al. [26] added to this body of information and found that infection in C57BL/6 mice also induced Th17 cells (later in disease), which mediated corneal pathology, and that treatment with galectin-1 was efficacious. In addition to phagocytosis, PMN recruited to the corneal stroma produce reactive oxygen species (ROS) and matrix metalloproteinases (MMPs), including collagenases, to prevent microbial dissemination [23]. Specifically, recent confirmatory studies have shown that ROS and MMPs, including MMP13 [27] cause bystander damage to the cornea by disrupting the collagen matrix which causes corneal scarring, loss of corneal clarity and impaired visual acuity. Other studies [28] showed that HIF-1α, a transcription factor, is essential for effective PMN bacterial killing, antimicrobial peptide production and PMN apoptosis. PMN recruitment to the infected cornea were shown to limit biofilm production to prevent bacterial dissemination. During this process, PMN were found to move to the base of the forming biofilm where they underwent PMN extracellular trap formation (NETosis) in response to the bacterial T3SS. Further, blocking Psl exopolysaccharide and type III secretion, together with antibiotic treatment, broke the biofilm and reversed keratitis [29]. LasR (quorum sensing) defective mutants failed to induce robust NET formation compared to LAS-R sufficient strains [30], confirming the importance of this virulence factor in disease In the host, other factors, such as epithelium produced thymic stromal lymphopoietin also is protective in keratitis by targeting dendritic cells and in IL-23/IL-17 signaling [31]. Other groups using the C57BL/6 mouse model and previously documented approaches, including antibody neutralization and recombinant protein injection, showed downregulation of IL-24 reduced severity of keratitis. They also showed that recombinant IL-24 protein induced SOCS3, IL-24, IL-1β and IL-6 in primary human corneal epithelial cells [32]. This lab also reported that IL-1Ra and IL-36Ra have opposing effects and that IL-36 agonists could be used as an alternate therapeutic to IL-1β neutralization [33]; the latter confirming work originally shown before by Rudner et al [34]. In some of these studies, effects were measured at very early times (1 and 2 −16 h) after infection and although important mechanistically, it is difficult to understand how these studies will have patient applicability due to the narrow treatment window of these targets in the eye IL-10 is important in the resistance response of BALB/c mice, as recombinant IL-10 treatment rescued rapamycin treated mice that had decreased IL-10 levels, improving disease outcome [35]. It has also been shown that IL-33 promotes a Th2 type immune response in recombinant protein injected C57BL/6 mice and reduced inflammation by polarizing macrophage production of anti-inflammatory mediators in cornea [36]. Immunostaining showed that recombinant injection shifted macrophage polarization from NO synthase 2 to arginase production and that peritoneally elicited cells from C57BL/6 mice treated with LPS and recombinant IL-33 exhibited elevated ST2 levels and a shift from IL-12 to IL-10 (protective) production [36]. More recent work has shown that the NLRC4 vs NLRP3 inflammasome contributes to resistance through regulation of caspase-1, IL-1β and IL-18 in a CD11blow Ly6Glow cell population [37]. Others [38] found that NLRP12 decreased the severity of keratitis by reducing bacterial burden and inflammation through downregulating NFκB signaling and thereby cytokine production. Collectively, these studies provide much detail about disease pathogenesis, but treatment clinically remains elusive. In that regard, a silicone-gel contact lens model using the C57BL/6 mouse may prove useful, as studies revealed that without deliberate inoculation with P. aeruginosa, contact lens wear did not result in bacterial infection. Other immune cells in cornea were detected, however which may prime the cornea to respond to insult more quickly and vigorously than in the absence of a contact lens [39].P. aeruginosa has extreme ability to trigger antibiotic resistance mechanisms, making them difficult to eradicate even using current treatments [40]. For instance, inefficient porins in the P. aeruginosa outer membrane limit the rate of antibiotic penetration to the bacterial cell, resulting in efflux pump overexpression and enzymatic antibiotic modification[40,41]. In addition, P. aeruginosa acquires spontaneous genetic mutations based on the antibiotic therapy and the environment and transfers those mutated genetic elements horizontally between cells, further reducing its susceptibility to antibiotics [40]. Inappropriate use of antibiotics along with other risk factors cause a breakthrough resistance in P. aeruginosa, which results in emergence of multi-drug resistant (MDR), extensively drug resistant (EXD) and pan-drug resistant (PDR) P. aeruginosa strains, with no or minimum effective antibiotic therapy [40,42]. Alternate approaches that will enhance treatment are needed, as the number of new antibiotics in the pipe line are not keeping pace.
Figure 1.
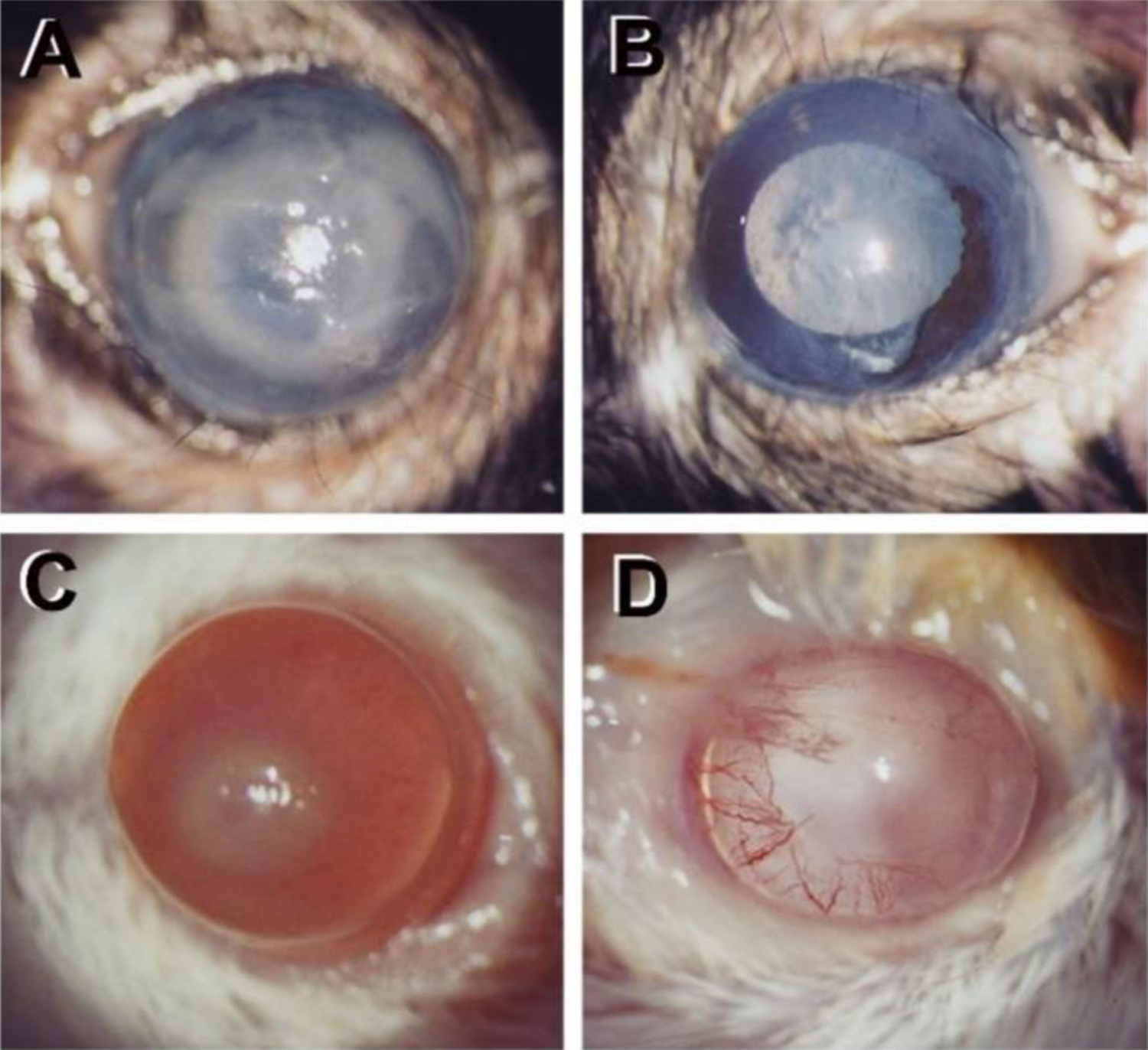
P. aeruginosa infected mice at 1 day after infection and depletion of PMN with anti-Ly-6G/GR-1 antibody (Ab). (A) Control C57BL/6 mouse cornea shows light opacity over entire cornea. (B) Ab treated C567BL/6 mouse cornea is clear (lack infiltrating PMN). (C) Control BALB/c mouse cornea with opacity over the pupil. (D) Ab treated BALB/c mouse cornea is clear (vessel ingrowth from the periphery).
3.2. Antimicrobial peptides (AMPs) and proteins
β-defensins are natural peptides produced by corneal and conjunctival epithelial cells. These peptides are cationic and interact with bacterial membranes, composed of negatively charged LPS. Such interaction enhances bacterial cell membrane permeability and cell lysis [43]. Using siRNA knockdown of mouse beta defensins (mBD) mBD2 and mBD3 in C57BL/6 mice, it was reported that both mBD2 and 3 enhanced resistance to disease [44]. Studies also revealed that mBD2 and mBD3 function together to regulate the expression of pro- and anti-inflammatory mediators, PMN infiltrate and bacterial load in the infected cornea. AMPs also have a range of non-microbicidal functions including signaling, immunomodulation, and influence vascularization and wound healing, all important for resolution of ocular surface disease in humans [45], Vasoactive intestinal peptide (VIP) is an anti-inflammatory neuropeptide with multiple functions that was shown to balance pro- and anti-inflammatory cytokines in the P. aeruginosa infected cornea and protect C57BL/6 mice against corneal perforation [46]. Li and coworkers [47] reported that OH-CATH30, a natural peptide in snakes, is protective alone or as adjunctive therapy with levofloxacin, and reduced the expression of pro-inflammatory cytokines (TNF-α, IL-1β), leukocyte infiltration, bacterial load and improved disease. Despite demonstrating various protective effects of AMPs and proteins, there are challenges to clinical use. These small peptides and proteins are effective in their native α-helical or β-sheet confirmation. However, extraction or synthetic conditions, as well as enrichment, (to facilitate delivery to the cell). may lead to loss of a native confirmation which would retard the activity of these molecules. In addition, the ability of these molecules to penetrate the cornea, the residence time and the stability in the ocular environment in the presence of a wide variety of proteases and other inhibitors, are other issues of concern, all of which slow progress.
High mobility group box-1 (HMGB1), a small 215 amino acid protein is an alarmin, belonging to the family of danger associated molecular patterns (DAMPs), which amplify inflammatory reactions. It is a ubiquitous molecule seen in the normal cornea and is released extracellularly after infection (Fig. 2 A,B). HMGB1 is an extracellular mediator of pro-inflammatory responses, and strategies to inhibit the molecule in various diseases are being tested. Silencing HMGB1, using small interfering RNA (siHMGB1), significantly improved disease outcome of pseudomonas keratitis in susceptible C57BL/6 mice [48]. HMGB1 knockdown downregulated pro-inflammatory cytokine expression, while upregulating anti-inflammatory cytokines. Treatment also downregulated IL-1β and MIP-2, reducing the PMN infiltrate. Since knockdown of HMGB1 would not be desirable clinically, pursuing use of other molecules such as glycyrrhizin (GLY), a natural anti-inflammatory and antiviral triterpene, to reduce HMGB1, appear promising and sensible. Experiments in mice using invasive (clinical isolate) and cytotoxic (ATCC strain 19660) pseudomonas strains, revealed that GLY treatment reduced HMGB1 levels, bacterial plate count, and inflammatory consequences and enhanced antibiotic therapy [49]. GLY was also effective against three ocular clinical isolates, reducing viability, biofilm formation and adherence [50]. It was also effective against multi-drug resistant isolates (systemic and ocular), of P. aeruginosa, even when treatment was delayed to 18h after infection. GLY is antimicrobial, and was found able to alter bacterial virulence factors (efflux pump activity), viability and increased the effects of ciprofloxacin to reduce plate count, and MPO activity [41]. The challenge for all of these approaches will be to bring successful candidates from animal models to the clinic for human testing/clinical trials.
Figure 2.
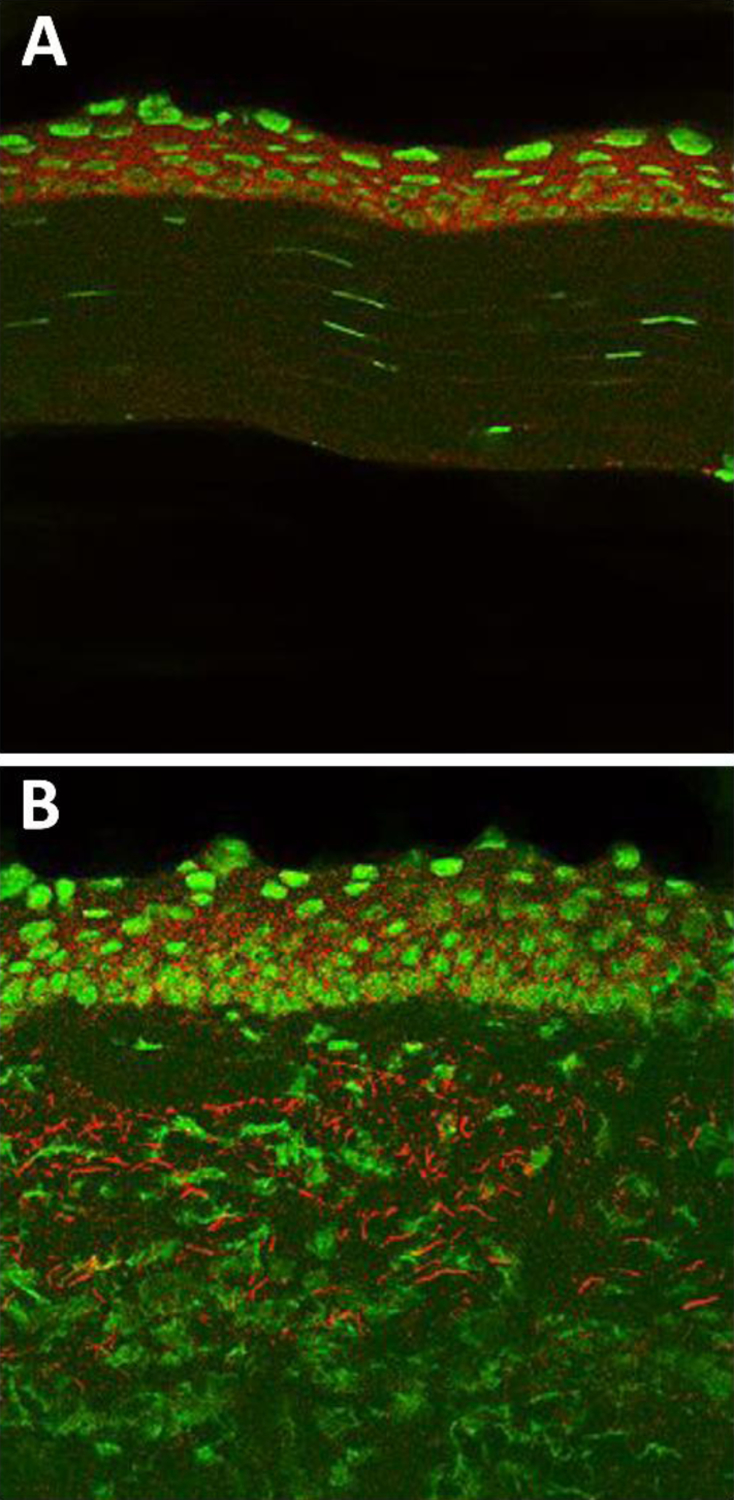
Normal and 5 day post infection C57BL/6 mouse cornea stained with an anti-HMGB1 Ab. (A) HMGB1 epithelial staining (red) in normal, uninfected cornea. (B) HMGB1 staining distributed throughout the epithelium and stroma after 5 days of infection. Nuclear marker=Sytox Green.
4. Fungal keratitis
Fungal keratitis is a common, severe eye infection, particularly common in tropical and subtropical areas of the world. Ocular trauma and contact with plant material, insects or other vehicles contaminated with fungal spores [51–53], are causative. In China and India, for example, agricultural work is the major risk factor; fungal keratitis increases during the harvest season, because of increased airborne soil and contaminated plant materials [51–55]. However, in industrialized countries use of contact lenses remains a key risk factor [56–58]. In either case, if the integrity of the corneal epithelium is breached due to trauma, fungi such as Aspergillus and Fusarium conidia or hyphae penetrate into the corneal stroma, and germinate causing ulceration, extreme pain, and visual impairment. Fungal keratitis patients are immunocompetent compared with systemically infected patients [59–61]. They are able to mount a vigorous immune response to Aspergillus and Fusarium hyphae characterized by infiltration of host innate immune cells and secretion of inflammatory, chemotactic, and regulatory cytokines. Other work has shown [62], in both mouse and human PMN, production of NADPH oxidase to control fungal growth. The antifungal activity depended on CD18, and not the β-glucan receptor dectin-1. Others, [63] studied the effects of calprotectin,(PMN source), and showed that zinc and manganese binding contribute to its anti-hyphal activity. However, calprotectin was not important in killing intracellular fungal conidia. It was also shown that dectin-1 dependent IL-6 production regulated expression of heme and siderophore binding proteins in infected mice and that human PMN synthesize lipocalin-1 that restricts fungal growth. Mice that were immunized with fungal conidia before infection had improved fungal killing and less stromal opacity versus unimmunized animals [62]. PMN, innate immune cells, that produced IL-17, as well as T cells producing IL-17, but not IFN-γ, contributed to improved fungal killing. IL-17 production in PMN from fungal keratitis patients, as well as uninfected people, were studied [64] and IL-17 levels were detected in all groups, but were significantly higher in PMN from keratitis patients. Toll-like receptors (TLR) and lectin-like-receptors also are important in the host innate immune response to fungi (e.g., Candida albicans (C. albicans) [65], leading to cytokine production. A main fungal recognition receptor, dectin-1 deficiency results in enhanced susceptibility to fungal infection in mice [66,67]. TLR2- and TLR4 amplification by dectin 1 leads to production of tumor necrosis factor (TNF) [68], and alone enhances IL-17, IL-6 and IL-10 [69]. In a clinical study, mutations in dectin-1 led to defective production of several cytokines after β-glucan challenge, but fungal phagocytosis and killing were unimpaired [70]. Analysis of fungal vs bacterial keratitis (human late stage) was done using comparative transcriptomics confirmed by RT-PCR [71]. The study revealed overlap in gene expression profiles, but showed about 50 unique genes to fungal disease that highlighted a corneal epithelial wound healing response. Those genes could provide new therapeutic targets and will require further investigation to determine their usefulness in treatment of fungal disease.
3.3. Microbiome
The gut microbiome is a complex ecosystem and plays a pivotal role in maintaining ocular health and affects immunity of the eye [72]. In mice, both genetics and environment play a role in its composition [73], which we have recently confirmed for two inbred strains (C57BL/6 and BALB/c), often used in eye studies. It is tempting to hypothesize that differences between the two, one susceptible and the other resistant to P. aeruginosa keratitis, may reflect the disparate gut microbiome (Fig. 3). This is an attractive hypothesis we believe, as bacteria that reside in the gut play a key role in the regulation of mucosal immunity, particularly in balancing Th17/Treg cells [72]. In fact, gut dysbiosis has been implicated in infectious keratitis [72]. A recent study in patients with bacterial keratitis showed dysbiosis in the gut bacterial and fungal communities when compared to healthy human subjects [74]. The study implicated that increased abundance of pro-inflammatory and pathogenic bacteria in bacterial keratitis subjects may contribute to the disease phenotype [74]. Another study, evaluating patients with fungal keratitis linked gut dysbiosis to disease phenotype. A decrease in abundance of beneficial bacteria and increase in abundance of pro-inflammatory and pathogenic bacteria was observed in these patients [75]. A study by Zaidi et al [76] evaluated the link between the gut microbiome and S. aureus infection in the eye. The results showed that the gut microbiota was important in immune cell maturation. Further, along with PMN recruitment mediated by ICAM-1, mature CD4+ T cells producing IL-17 and IL-22 were required for antibody-mediated adaptive immunity to S. aureus in the anterior eye compartment [76]. These studies indicate the presence of a gut-eye axis and provide a strong link between changes in the microbiota of the gut and eye infections. Future studies focused toward microbiome modulation can potentially lead to therapeutic interventions, both at the ocular surface and by modulating the gut microbiome.
Figure 3.
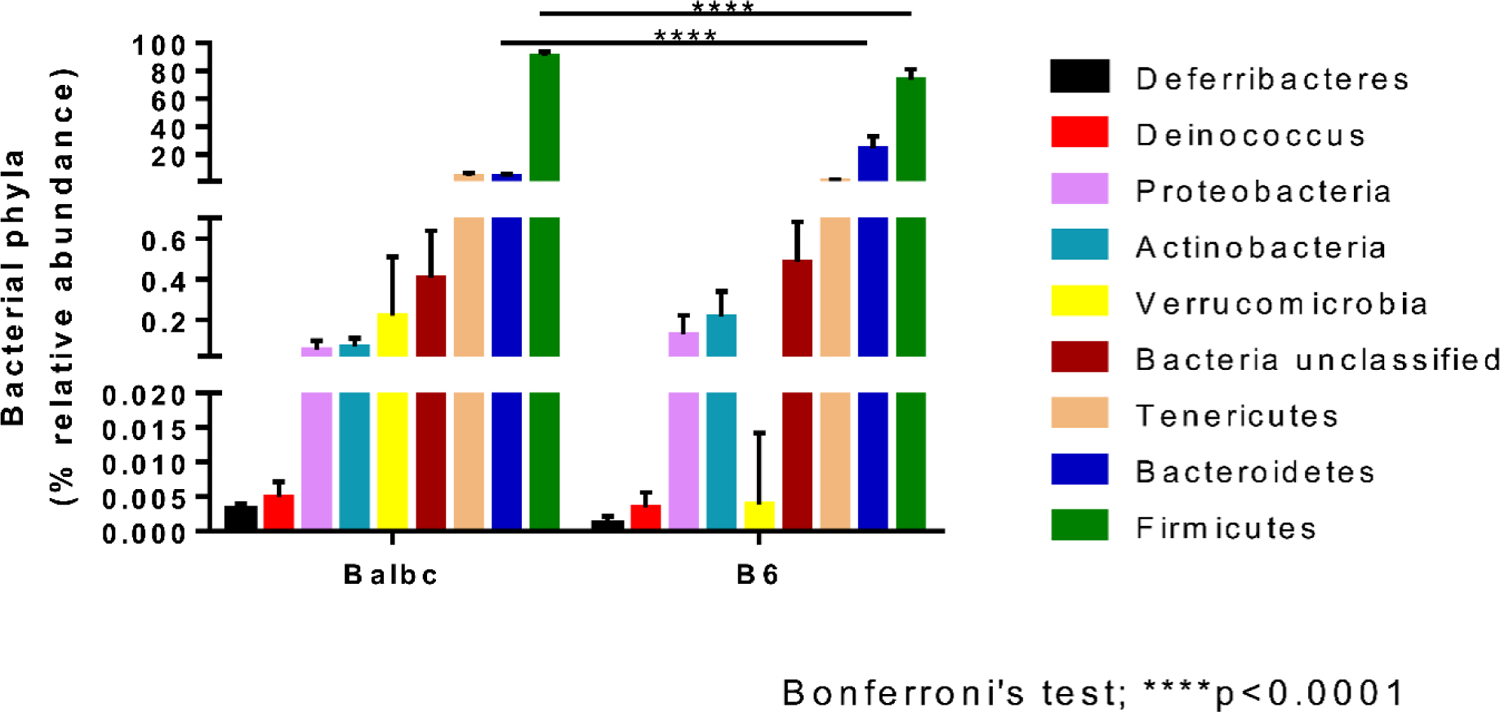
Normal BALB/c and C57BL/6 mice harbor a strain-specific gut bacteria phyla. Results are represented as % relative abundance and were analyzed with two-way ANOVA. Significant differences were present in Firmicutes and Bacteroidetes phyla. For Firmicutes, significantly higher (p<0.0001, Bonferroni’s test) levels were found in BALB/c compared to C57BL/6. For Bacteroidetes, BALB/c had significantly reduced (p<0.0001, Bonferroni’s test) levels when compared to C57BL/6.
Due to the lack of extensive studies, the role of the ocular microbiota in regulating ocular immunity is not well understood [72]. A classic study [77] however revealed that in mice an ocular commensal protected against corneal infection (Candida albicans and P. aeruginosa) by driving mucosal γδ T cells and IL-17. A few studies have investigated contact lens wear, as it is the leading cause of corneal ulcers, with P. aeruginosa, accounting for nearly 50% of all infections [72]. Increased levels of opportunistic pathogens and differences in the distribution of gram-negative bacteria were reported in the conjunctiva of contact lens vs non-lens wearers [72]. A pioneering study by Kugadas et al [78] provided the first insight in the role of ocular microbiota in regulating ocular immunity in P. aeruginosa-induced keratitis. This study revealed that in healthy mice, the ocular innate immune barrier was strengthened by the presence of ocular microbiota. A significant increase in the levels of immune effectors such as secretory IgA and complement proteins in the tear film were observed. Future studies dedicated to identifying the different species of ocular microbiota and microbiota-induced signaling that regulate PMN and bactericidal activities may be an avenue for development of new therapeutic interventions, including fecal transplant.
5. Herpes stromal keratitis (HSK)
Herpes simplex virus-1 (HSV-1) is a leading cause of corneal blindness worldwide [79] and reactivation of latent virus from trigeminal ganglion (TG) sensory neurons is a deleterious outcome. Upon reactivation, infectious virus and/or viral proteins travel through axons of the ophthalmic branch of the TG to penetrate the corneal epithelium through sensory nerve terminals [80]. In the epithelium, virus causes development of herpes epithelial keratitis (HEK) [81]. These lesions tend to be self-limiting, but heal more rapidly when treated. For example, topical ganciclovir (formulated as an ophthalmic gel), has low toxicity, and is well tolerated when treating acute herpetic keratitis [82]. Overall, HSV-1 can infect the corneal epithelium, and the stroma, with or without ulceration; it also can infect the corneal endothelium, manifesting as keratitis in each case [83]. Clinical manifestations of HSK include: stromal opacity, swelling, and neovascularization. Unfortunately, once corneal scarring has occurred, penetrating keratoplasty may be required and points to the need to develop new approaches to treatment [84–87]. Disruption of viral entry into cells, is amongst a number of novel means to treat this disease [88].
5.1. Innate and adaptive immunity
Studies in mice have confirmed participation of the immune system in viral clearance and in development of HSK pathogenesis following experimental infection with HSV-1. These studies have shown that PMN are key inflammatory cells in control of viral load in HSV-1 infected corneas. In fact, these cells enhance the severity of HSK in the mouse model [56]. To test their role in disease, PMN were selectively depleted, and the data provided evidence that inflammatory monocytes, (not PMN), recruited by production of type I interferon by corneal epithelial cells were important in controlling viral load early after infection [86]. Type I interferon production in response to HSV-1 infection is regulated by an innate DNA sensor (IFI-16) [89]. Another study examined early depletion of resident corneal dendritic cells (DCs). Depleting these cells reduced infiltration of natural killer (NK) cells in infected corneas, suggesting their indirect role in controlling HSV-1 infection [90]. It is without question that innate immune cells are essential in controlling viral load in HSV-1 infected corneas. However, the importance of adaptive immunity, especially CD8+ T cells, in HSV-1 clearance from infected corneas has been demonstrated in a mouse model of primary ocular HSV-1 infection [89] and also will require further work to understand applicability to human disease. The latter is also true for studies of another virus, the Zika virus, a single-stranded RNA virus of the Flaviviridae family, which has recently been shown to infect and replicate in primary cultures of human corneal epithelial cells [91].
5.2. IL-17 and Foxp3+Treg
Studies using mouse models have established that CD4+ T cells are a principle effector cell for induction of HSK [87]. In this regard, data has shown that after experimental induction of ocular HSV-1 infection, only infiltrating corneal DCs took up viral antigens and trafficked to draining lymph nodes where they primed virus specific CD4+ T cells [92]. It also was shown that these primed cells migrate to the infected corneas where they orchestrate tissue damage. Recently, IL-17 producing CD4+ T cells were detected in inflamed corneas during the later stages of disease and in IL-17 knock out mice, significantly reduced ocular lesions were seen [93]. (γδ) T lymphocytes that produce IL-17 also were shown to infiltrate HSV-1 infected corneas early after infection, but whether IL-17 production by these cells during active viral infection is beneficial or detrimental is not clear. Nonetheless, IL-17 expression, which has been reported in HSK patients, is of importance, as these human data suggest IL-17 involvement in HSK pathogenesis [94]. Other studies examined the role of Foxp3 expressing regulatory CD4+ T cells in controlling HSK using mice. In vivo treatment with the fungal metabolite drug FTY720 was shown to increase the Foxp3+ Treg population, significantly diminishing HSV-1 induced ocular lesions [95]. However, discontinuation of the treatment resulted in relapsed inflammatory lesions. Similarly, in vivo expansion of Foxp3+ Tregs during the early stages of corneal HSV-1 infection, using IL-2/anti-IL-2 complex, was shown to reduce the development of HSK lesions but, increased Treg therapy was not effective upon priming and infiltration of pathogenic CD4+ T cells [96]. Thus, it appears from these data that increasing the Foxp3+ Treg population only would not sufficiently control HSK.
5.3. Hemangiogenesis and lymphangiogenesis
After HSV-1 infection, new blood vessels sprout from the corneal limbus and invade the avascular cornea. Vascular endothelial growth factor-A (VEGF-A), produced in HSV-1 infected corneas, is important for induction of hemangiogenesis in HSK [57]. Infection has been shown to reduce availability of soluble VEGF receptor-1 (sVEGFR-1), a molecule which inactivates VEGF [97]. Reduction in sVEGFR-1 is dependent upon matrix metalloproteinase 9 (MMP-9), produced by infiltrating PMN. Other angiogenic factors are also involved. Studies have shown that mice lacking Robo4, an endothelial receptor that counteracts VEGF downstream signaling, have increased angiogenesis vs control mice after ocular HSV-1 infection [98]. In contrast, microRNA 132 (miR-132) has been shown to enhance VEGF-A mediated angiogenesis [99]. HSV-1 infected corneas have recently been shown to have extensions of lymphatic vessels from the limbal region of the cornea. HSV-1 induced lymphangiogenesis is dependent on VEGF-A produced by virus infected corneal epithelial cells [100]. Besides VEGF-A, VEGF-C (released from infiltrating CD8+ T cells), promote lymphatic vessel growth into the central cornea [89]. VEGF-A/ VEGFR2 signaling also induces lymphangiogenesis in HSV-1 infected mouse corneas [51]. Inflammatory cytokines (TNF and IL-6) potentiate VEGF, promoting virus induced lymphangiogenesis in infected corneas [101]. Novel recent work has focused on metabolic changes in HSK lesions and showed the prevalence of glycolytic metabolism and the development of hypoxia in HSK lesions, information which may provide new targets for treatment development [102].
5.4. Corneal homeostasis
Alteration in homeostasis of resident cells in cornea may also contribute to initiation and or progression of HSK. A recent study used in vivo confocal microscopy, and demonstrated reduced corneal nerve density, branching of nerve trunks and loss of sensation in patients with HSK, in both the infected and uninfected eye [103]. Similarly, in a mouse model of HSK, reduced corneal innervation was detected in the corneal epithelium during the clinical period of disease [104]. Tarsorrhaphy (suturing eyelids), after ocular HSV-1 infection, prevented corneal desiccation and nerve loss, reducing HSK severity. The data suggest that inhibiting nerve retraction in the infected corneas could be important in controlling HSK severity. Corneal nerve terminals are closely associated with superficial corneal epithelial cells and thus, alteration in the corneal epithelium of HSV-1 infected eyes effect corneal innervation. In fact, in the superficial epithelium, increase in cell size, decrease in cell density, and squamous metaplasia and desquamating superficial corneal epithelial cells is reported in diseased eyes of patients with HSV keratitis, suggesting an altered corneal epithelial morphology correlated with changes in corneal innervation [105]. This suggests that further studies directed to the interaction of corneal nerves and immune cells may deliver new therapeutic targets with clinical potential.
6. microRNAs (miRNA)
Several pioneering studies have demonstrated that miRNAs play critical roles in the pathogenesis of infectious keratitis. These findings will be reviewed essentially focusing on how miRNAs modulate infectious keratitis through regulating host-microbe interactions.
6.1. miRNAs in P. aeruginosa keratitis
Yang et al. was first to report that miR-155 was induced by P. aeruginosa infection in macrophages in vitro and in mouse and human corneas in vivo[106]. Inactivation of miR-155 resulted in a reduced bacterial load, and decreased severity of experimental keratitis, suggesting that miR-155 enhanced bacterial burden and promoted corneal susceptibility to the bacteria. Macrophages of miR-155 knockout mice showed increased phagocytosis and intracellular bacterial killing; while over-expression of miR-155 inhibited them, and expression of induced nitric oxide synthase (iNOS) and the production of nitric oxide (NO)[106]. These effects are mediated by targeting Rheb, a gene known to interact with mammalian target of rapamycin (mTOR) and increase its activity[107]; inhibition of mTOR using rapamycin increased bacterial burden and reduced PMN bactericidal activity (Fig.4A)[108]. Data suggest that miR-155 could be a therapeutic target as knockdown increased phagocytosis and intracellular killing capacity by macrophages, cells that constitute a minority of the infiltrate after infection.
Figure 4.
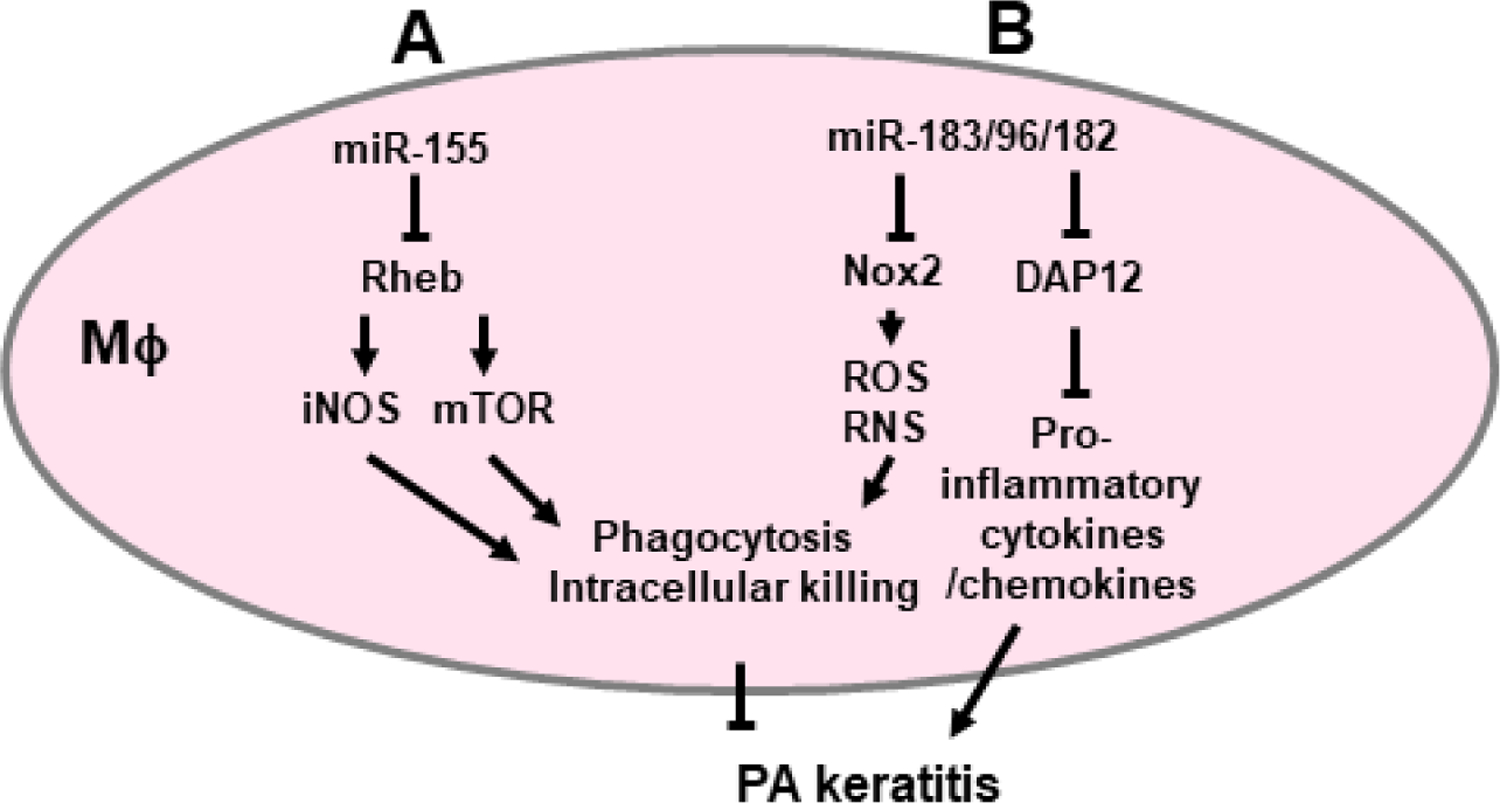
miR-155 and the miR-183/96/182 cluster modulate P. aeruginosa keratitis by regulating the function of macrophages (Mϕ). A. miR-155 targets the expression of Rheb, which enhances the production of iNOS and mTOR activity to promote phagocytosis and intracellular bacterial killing. B. miR-183/96/182 targets Nox2, important in ROS and RNS production, to promote phagocytosis/intracellular bacterial killing, and DAP12, which limits production of pro-inflammatory cytokines.
Recently, it has been shown that the conserved, paralogous miRNA cluster, miR-183/96/182 cluster (miR-183/96/182), modulates the corneal response to P. aeruginosa infection through its regulation of pathogenesis at multiple levels. The miR-183/96/182 was originally identified as a sensory organ-specific miRNA cluster [109] required for normal development and function of all major sensory domains[110]. Inactivation of the cluster in mice resulted in decreased corneal nerve density in the subbasal plexus and the expression of nociceptor transient receptor potential vanilloid 1 (TRPV1) and multiple pro-inflammatory neuropeptides, including Tac1 [the precursor gene for substance P (sP)], calcitonin gene-related peptide (CGRP) and chemokine (C-X3-C motif) ligand 1 (Cx3cl1)[111]. miR-183/96/182 knockout mice showed a significantly decreased corneal inflammatory response to P. aeruginosa infection and reduced severity of disease[111]. Furthermore, the cluster was also expressed on innate immune cells, including macrophages and PMN[111]. Inactivation of miR-183/96/182 resulted in increased phagocytosis and intracellular bacterial killing capacity of both groups of cells (Fig.4A)[111]. miR-183/96/182 targets Nox2, a key enzyme required for generation of superoxide (O2−) and other microbicidal products [112]; inactivation of miR-183/96/182 resulted in increased Nox2 and enhanced production of reactive nitrogen (RNS) and ROS in macrophages. In addition, miR-183/96/182 promotes the production of pro-inflammatory cytokines in macrophages by targeting DAP12[112], consistent with the observation of an overall decreased level of pro-inflammatory cytokines in the cornea in response to P. aeruginosa infection (Fig.4B)[111]. Data further demonstrated that, in addition to its regulation on circulating macrophages and PMN, miR-183/96/182 has direct regulatory effects on corneal resident macrophages [113].
miR-183/96/182 is not only expressed in innate immune cells, but also it is one of the highest induced miRNAs during helper T (Th)-17 cell differentiation, and promotes Th17 cell pathogenicity by negatively regulating the expression of transcription factor Foxo1, a negative regulator of IL-1R1 [114]. Inactivation of miR-183/96/182 in mice resulted in decreased pathogenicity of Th17 cells and a reduced production of pro-inflammatory cytokines, leading to decreased severity of experimental autoimmune encephalomyelitis [114]. Since IL-17 activity and Th17 have been shown to promote PMN infiltration and the severity of P. aeruginosa keratitis[115], miR-183/96/182 may also by regulation of Th17, effect P. aeruginosa keratitis.
6.2. miRNAs in herpes simplex virus (HSV)-1 keratitis
At least 18 pre-miRNAs and 27 mature miRNAs have been identified in the HSV-1 genome based on the most updated miRbase (Release 22.1. October 2018). These miRNAs are actively involved in the control of productive infection/latency/reactivation[116]. During latency, viral gene expression is restricted to select non-coding RNAs, including the latency-associated transcript (LAT), which is critical to the HSV-1 latency-reactivation cycle through its anti-apoptotic and immune evasion properties, and viral miRNAs[116,117]. At least ten LAT-associated miRNAs (miRs H1–4, 6–8, H14, 15, 27) have been identified in and near the LAT locus of the HSV-1 genome[118].
6.3. HSV miRNAs modulate viral virulence
LAT-derived miR-H2 is in antisense to infected cell protein (ICP)0 gene, an immediate early gene which promotes viral replication, trans-activates lytic gene expression and facilitates reactivation from latency. It targets and regulates ICP0 expression at protein levels[118]. Disruption of miR-H2 without altering the amino acid sequence of the ICP0 gene resulted in increased production of ICP0 in host cells, and enhanced neurovirulence in mice after ocular infection with HSV-1 and more rapid reactivation[119,120]. Simultaneous inactivation of LAT transcript and miR-H2 resulted in a more robust reaction[120]. These data suggest that miR-H2 suppresses HSV-1 neurovirulence and reactivation and promotes latency through targeting ICP0. miR-H6, a HSV-1 miRNA derived from the promoter region of the LAT gene[118], showed sequence complementarity to ICP4, a transcription factor required for the expression of most HSV-1 genes during productive infection[120]; it also repressed ICP4 protein expression, an essential immediately-early lytic viral gene [118], suggesting a potential role in promoting or maintaining latency. Consistently, in human corneal epithelial cells, miR-H6 inhibited HSV-1 replication[121], but animal modeling is needed to confirm this.
6.4. HSV miRNAs regulate host genes
In addition to its direct regulation of ICP4 and latency, miR-H6 of HSV-1 modulates the expression of IL-6 in human corneal epithelial cells [121]. Overexpression of miR-H6 in these cells significantly reduced HSV-1-induced upregulation of IL-6[121]. These data are derived from an in vitro study and the direct target of miR-H6 mediating this effect is unidentified. However, since IL-6 is known to promote an inflammatory response, reactivation [122] and neovascularization during HSV-1 infection[123], it suggests that miR-H6 has a potential to inhibit the above effects of HSV-1[121]. Further study on miR-H6’s regulation on HSV-1 induced IL-6 in the cornea in vivo is warranted.
6.5. HSV-1 can regulate host miRNA expression
During acute HSV-1 infection, miR-183/96/182 is selectively upregulated in rat primary superior cervical ganglion neurons and human primary fibroblasts within 2–3 hours after HSV-1 infection. It is increased up to 30–40 fold by 1 day post-infection, gradually dropping over the next 6 days[124,125]. Lutz et al. further demonstrated that the immediate early protein ICP0, an E3 ubiquitin ligase, is required and sufficient to induce the expression of miR-183/96/182[124]. The induction of miR-183/96/182 by ICP0 is mediated by members of the transcriptional repressor ZEB family, ZEB1 and ZEB2. ZEB1/ZEB2 suppresses miR-183/96/182 transcription; ICP0 promotes the degradation ZEB1 (/ZEB2) proteasome through its E3 ubiquitin ligase activity and therefore releases the inhibition on miR-183/96/182 expression by ZEB1 (/ZEB2)[124]. Once miR-183/96/182 is induced by ICP0 in acute infection, its expression may be maintained at a lower but significant expression level over the latency phase because of a negative feedback regulation of miR-183/96/182 on ZEB1/ZEB2[126–128], when ICP0 expression is downregulated during latency. Although target genes of miR-183/96/182 and its exact functions during HSV-1 infection have not been identified, its upregulation in acute infection and modest expression during latency suggest a role in various phases of disease. In addition, HSV-1 is not the only herpesvirus to induce the expression of miR-183/96/182. It is among the most strongly upregulated miRNAs during the productive infection cycle of human cytomegalovirus, a β-herpesvirus[129]. The γ-herpesvirus Epstein-Barr virus (EBV) selectively downregulates miR-183/96/182 during type II and III latency[130]. These suggest that miR-183/96/182 may be important, with a conserved role in the pathogenesis of all three major herpesvirus subfamilies.
6.6. Host miRNA can regulate HSV-1 gene expression
A host neuron-specific miRNA, miR-138, targets and represses the expression of ICP0 and other lytic genes of HSV-1. A mutant HSV-1 (M138) with disrupted miR-138 target sites in ICP0 mRNA exhibited increased expression of ICP0 and other lytic proteins in infected neuronal cells in culture[131]. Consistently, in vivo, after corneal inoculation, M138-infected mice showed higher expression of ICP0 and other lytic transcripts in the TG during latency establishment, and exhibited increased mortality and encephalitis symptoms[131]. After full establishment of latency, an increased fraction of TG neurons expressed lytic transcripts in M138-infected mice. These data suggest that host miR-138 represses HSV-1 lytic gene expression and promotes host survival and viral latency[131]. As most of the studies on latency/reactivation are limited to in vitro or ex vivo level, further in vivo studies are needed to test the roles of miRNAs in these processes.
6.7. Host miRNAs regulates immune response to HSV-1
miR-155 is one of the first miRNAs shown to play a role in HSK through its regulation of the host immune system. It is significantly upregulated in the cornea at 2, 7 and 15 days after HSV-1 infection in the mouse[132]. miR-155 knockout mice showed decreased angiogenesis and reduced severity of HSK, and fewer infiltrating CD4+ T cells and Th1 and Th17 responses in both the infected cornea and lymphoid organs, including draining lymph nodes and the spleen[132]. miR-155 targets Ship1 and IFN-γRα, which are important regulators of IFN-γ expression and Th1 differentiation [133,134]. Inactivation of miR-155 resulted in significantly increased expression of both Ship1 and IFN-γRα in activated CD4+ T cells of draining lymph nodes[132], promoting Th1 cell development and a decrease in HSK severity in miR-155 knockout vs wildtype mice[132]. In vivo silencing of miR-155 by conjunctival injection of antigomir-155 nanoparticles at 1 day after infection and at an early clinical stage (5 days after infection) reduced infiltration of CD4+ T cells, PMN, pro-inflammatory cytokine production, and decreased the severity of HSK[132].
Corneal neovascularization allows immune/inflammatory cells to gain access to the cornea contributing to the pathogenesis of HSK[135,136]. Controlling neovascularization is critical to treatment of HSK[136]. miR-132 is known to activate the endothelium and facilitate pathological angiogenesis by targeting p120RasGAP- a negative regulator of angiogenic Ras activity[137]. Mulik et al showed that HSV-1 infection upregulates IL-17 production, which induces VEGF expression in the cornea; VEGF further induces the expression of miR-132, which inhibits p120RasGAP expression and contributes to neovascularization in the cornea in response to HSV-1 infection [138]. Knocking down VEGF by VEGF trap (soluble VEGF receptor) or inactivation of IL-17A receptor in mice prevented the upregulation of miR-132 in HSV-1 infected cornea, suggesting miR-132 mediates VEGF and IL-17A-induced neovascularization[138].
7. Promising new therapeutic targets and future directions
Development of new therapeutic targets is the underlying goal of all studies focused on host-microbe interactions, and as antibiotic resistance remains a growing threat, the urgency for this work is heightened. A few emerging targets include reduction of infection and inflammation by decreasing bacterial load and HMGB1 levels using GLY, which has been shown efficacious in a mouse model of multi-drug resistant P. aeruginosa infection, as an example, or use of an anti-miR for miR-155 or knockdown of the miR-183/86/182 cluster. The role of the gut and eye microbiome [139,140] is another area of extreme interest, which provides important clues to bacterial, fungal and viral diseases; and more than likely, not only those involving microbial infections. Additional studies will be needed to test the clinical feasibility of any promising agent that works well in an animal model. How to achieve this goal is the key to future treatment successes.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Chang VS, Dhaliwal DK, Raju L, et al. Antibiotic resistance in the treatment of Staphylococcus aureus keratitis: a 20-year review. Cornea. 2015;34(6):698–703. doi: 10.1097/ICO.0000000000000431 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Rhem MN, Lech EM, Patti JM, et al. The collagen-binding adhesion is a virulence factor in Staphylococcus aureus keratitis. Infect Immun. 2000;68(6):3776–3779. doi: 10.1128/iai.68.6.3776-3779.2000 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Jett BD, Gilmore MS. Internalization of Staphylococcus aureus by human corneal epithelial cells: role of bacterial fibronectin-binding proteins and host cell factors. Infect Immun. 2002; 70(8):4697–4700. doi: 10.1128/iai.70.8.4697-4700.2002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Khan S, Cole N, Hume EB, et al. Identification of pathogenic factors potentially involved in Staphylococcus aureus keratitis using proteomics. Exp Eye Res. 2016;151:171–178. doi: 10.1016/j.exer.2016.08.016 [DOI] [PubMed] [Google Scholar]
- 5.Hume EB, Cole N, Khan S, Walsh BJ, Willcox MD. The role of staphopain a in Staphylococcus aureus keratitis. Exp Eye Res. 2020;193:107994. doi: 10.1016/j.exer.2020.107994 [DOI] [PubMed] [Google Scholar]
- 6.O’Callaghan RJ, Callegan MC, Moreau JM, et al. Specific roles of alpha-toxin and beta-toxin during Staphylococcus aureus corneal infection. Infect Immun. 1997;65(5):1571–1578. doi: 10.1128/IAI.65.5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Otto M Staphylococcus aureus toxins. Curr Opin Microbiol. 2014;17:32–37. doi: 10.1016/j.mib.2013.11.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Inoshima I, Inoshima N, Wilke GA, et al. A Staphylococcus aureus pore-forming toxin subverts the activity of ADAM10 to cause lethal infection in mice. Nat Med. 2011;17(10):1310–1314. doi: 10.1038/nm.2451 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wilke GA, Bubeck Wardenburg J. Role of a disintegrin and metalloprotease 10 in Staphylococcus aureus alpha-hemolysin-mediated cellular injury. Proc Natl Acad Sci U S A. 2010;107(30):13473–13478. doi: 10.1073/pnas.1001815107 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Nygaard TK, Pallister KB, DuMont AL, et al. Alpha-toxin induces programmed cell death of human T cells, B cells, and monocytes during USA300 infection. PLoS One. 2012;7(5):e36532. doi: 10.1371/journal.pone.0036532 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Astley R, Miller FC, Mursalin MH, et al. An eye on Staphylococcus aureus toxins: Roles in ocular damage and inflammation. Toxins (Basel). 2019;11(6):356. doi: 10.3390/toxins11060356 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Bierdeman MA, Torres AM, Caballero AR, et al. Reactions with antisera and pathological effects of Staphylococcus aureus gamma-toxin in the cornea. Curr Eye Res. 2017;42(8):1100–1107. doi: 10.1080/02713683.2017.1279636 [DOI] [PubMed] [Google Scholar]
- 13.Vuong C, Yeh AJ, Cheung GY, Otto M. Investigational drugs to treat methicillin-resistant Staphylococcus aureus. Expert Opin Investig Drugs. 2016;25(1):73–93. doi: 10.1517/13543784.2016.1109077 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Deschênes J, Blondeau J. Besifloxacin in the management of bacterial infections of the ocular surface. Can J Ophthalmol. 2015;50(3):184–191. doi: 10.1016/j.jcjo.2014.12.013 [DOI] [PubMed] [Google Scholar]
- 15.Schechter BA, Parekh JG, Trattler W. Besifloxacin ophthalmic suspension 0.6% in the treatment of bacterial keratitis: a retrospective safety surveillance study. J Ocul Pharmacol Ther. 2015;31(2):114–121. doi: 10.1089/jop.2014.0039 [DOI] [PubMed] [Google Scholar]
- 16.Mah FS, Davidson R, Holland EJ, et al. Current knowledge about and recommendations for ocular methicillin-resistant Staphylococcus aureus. J Cataract Refract Surg. 2014;40(11):1894–1908. doi: 10.1016/j.jcrs.2014.09.023 [DOI] [PubMed] [Google Scholar]
- 17.Willcox MD. Management and treatment of contact lens-related Pseudomonas keratitis. Clin Ophthalmol. 2012;6:919–924. doi: 10.2147/OPTH.S25168 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Hoge R; Pelzer A; Rosenau F, et al. Microbiology book series. Spain: weapons of a pathogen: proteases and their role in virulence of Pseudomonas aeruginosa; p. 383–395. [Google Scholar]
- 19.Eby AM, Hazlett LD. Pseudomonas keratitis, a review of where we’ve been and what lies ahead. J Microb Biochem Technol. 2015; 7:453–457. doi: 10.4172/1948-5948.1000254 [DOI] [Google Scholar]
- 20.Hazlett LD. Corneal response to Pseudomonas aeruginosa infection. Prog Retin Eye Res. 2004;23(1):1–30. doi: 10.1016/j.preteyeres.2003.10.002 [DOI] [PubMed] [Google Scholar]
- 21.Cope JR, Collier SA, Rao MM, et al. Contact lens wearer demographics and risk behaviors for contact lens-related eye infections--United States, 2014. MMWR Morb Mortal Wkly Rep. 2015;64(32):865–870. doi: 10.15585/mmwr.mm6432a2 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sun Y, Karmakar M, Taylor PR, Rietsch A, Pearlman E. ExoS and ExoT ADP ribosyltransferase activities mediate Pseudomonas aeruginosa keratitis by promoting neutrophil apoptosis and bacterial survival. J Immunol. 2012;188(4):1884–1895. doi: 10.4049/jimmunol.1102148 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Pearlman E, Sun Y, Roy S, et al. Host defense at the ocular surface. Int Rev Immunol. 2013;32(1):4–18. doi: 10.3109/08830185.2012.749400 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Angus AA, Evans DJ, Barbieri JT, Fleiszig SM. The ADP-ribosylation domain of Pseudomonas aeruginosa ExoS is required for membrane bleb niche formation and bacterial survival within epithelial cells. Infect Immun. 2010;78(11):4500–4510. doi: 10.1128/IAI.00417-10 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Ma X, Wang Q, Song F, et al. Corneal epithelial injury-induced norepinephrine promotes Pseudomonas aeruginosa keratitis. Exp Eye Res. 2020;195:108048. doi: 10.1016/j.exer.2020.108048 [DOI] [PubMed] [Google Scholar]
- 26.Suryawanshi A, Cao Z, Thitiprasert T, Zaidi TS, Panjwani N. Galectin-1-mediated suppression of Pseudomonas aeruginosa-induced corneal immunopathology. J Immunol. 2013;190(12):6397–6409. doi: 10.4049/jimmunol.1203501 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Gao N, Kumar A, Yu FS. matrix metalloproteinase-13 as a target for suppressing corneal ulceration caused by Pseudomonas aeruginosa infection. J Infect Dis. 2015;212(1):116–127. doi: 10.1093/infdis/jiv016 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Berger EA, McClellan SA, Vistisen KS, Hazlett LD. HIF-1α is essential for effective PMN bacterial killing, antimicrobial peptide production and apoptosis in Pseudomonas aeruginosa keratitis. PLoS Pathog. 2013;9(7):e1003457. doi: 10.1371/journal.ppat.1003457 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Thanabalasuriar A, Scott BNV, Peiseler M, et al. Neutrophil extracellular traps confine Pseudomonas aeruginosa ocular biofilms and restrict brain invasion. Cell Host Microbe. 2019;25(4):526–536.e4. doi: 10.1016/j.chom.2019.02.007 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Skopelja-Gardner S, Theprungsirikul J, Lewis KA, et al. Regulation of Pseudomonas aeruginosa-mediated neutrophil extracellular traps. Front Immunol. 2019;10:1670. doi: 10.3389/fimmu.2019.01670 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Cui X, Gao N, Me R, et al. TSLP protects corneas from Pseudomonas aeruginosa infection by regulating dendritic cells and IL-23-IL-17 pathway. Invest Ophthalmol Vis Sci. 2018;59(10):4228–4237. doi: 10.1167/iovs.18-24672 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Ross BX, Gao N, Cui X, et al. IL-24 promotes Pseudomonas aeruginosa keratitis in C57BL/6 mouse corneas. J Immunol. 2017;198(9):3536–3547. doi: 10.4049/jimmunol.1602087 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Gao N, Me R, Dai C, et al. Opposing effects of IL-1Ra and IL-36Ra on innate immune response to Pseudomonas aeruginosa Infection in C57BL/6 mouse corneas. J Immunol. 2018;201(2):688–699. doi: 10.4049/jimmunol.1800046 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Rudner XL, Kernacki KA, Barrett RP, Hazlett LD. Prolonged elevation of IL-1 in Pseudomonas aeruginosa ocular infection regulates macrophage-inflammatory protein-2 production, polymorphonuclear neutrophil persistence, and corneal perforation. J Immunol. 2000;164(12):6576–6582. doi: 10.4049/jimmunol.164.12.6576 [DOI] [PubMed] [Google Scholar]
- 35.Foldenauer ME, McClellan SA, Berger EA, Hazlett LD. Mammalian target of rapamycin regulates IL-10 and resistance to Pseudomonas aeruginosa corneal infection. J Immunol. 2013;190(11):5649–5658. doi: 10.4049/jimmunol.1203094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Hazlett LD, McClellan SA, Barrett RP, et al. IL-33 shifts macrophage polarization, promoting resistance against Pseudomonas aeruginosa keratitis. Invest Ophthalmol Vis Sci. 2010;51(3):1524–1532. doi: 10.1167/iovs.09-3983 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.McClellan SA, Jerome A, Suvas S, Hazlett LD. NLRC4 regulates caspase-1 and IL-1beta production in a CD11blowLy6Glow population of cells required for resistance to Pseudomonas aeruginosa keratitis. PLoS One. 2017;12(9):e0185718. doi: 10.1371/journal.pone.0185718 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Deng QC, Deng CT, Li WS, et al. NLRP12 promotes host resistance against Pseudomonas aeruginosa keratitis inflammatory responses through the negative regulation of NF-κB signaling. Eur Rev Med Pharmacol Sci. 2018;22(23):8063–8075. doi: 10.26355/eurrev_201812_16496 [DOI] [PubMed] [Google Scholar]
- 39.Metruccio MME, Wan SJ, Horneman H, et al. A novel murine model for contact lens wear reveals clandestine IL-1R dependent corneal parainflammation and susceptibility to microbial keratitis upon inoculation with Pseudomonas aeruginosa. Ocul Surf. 2019;17(1):119–133. doi: 10.1016/j.jtos.2018.11.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Breidenstein EB, de la Fuente-Núñez C, Hancock RE. Pseudomonas aeruginosa: all roads lead to resistance. Trends Microbiol. 2011;19(8):419–426. doi: 10.1016/j.tim.2011.04.005 [DOI] [PubMed] [Google Scholar]
- 41.Hazlett LD, Ekanayaka SA, McClellan SA, Francis R. Glycyrrhizin use for multi-drug resistant pseudomonas aeruginosa: in vitro and in vivo studies. Invest Ophthalmol Vis Sci. 2019;60(8):2978–2989. doi: 10.1167/iovs.19-27200 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Fernandes M, Vira D, Medikonda R, Kumar N. Extensively and pan-drug resistant Pseudomonas aeruginosa keratitis: clinical features, risk factors, and outcome. Graefes Arch Clin Exp Ophthalmol. 2016;254(2):315–322. doi: 10.1007/s00417-015-3208-7 [DOI] [PubMed] [Google Scholar]
- 43.Brandt CR. Peptide therapeutics for treating ocular surface infections. J Ocul Pharmacol Ther. 2014;30(9):691–699. doi: 10.1089/jop.2014.0089 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Wu M, McClellan SA, Barrett RP, et al. Beta-defensins 2 and 3 together promote resistance to Pseudomonas aeruginosa keratitis. J Immunol. 2009;183(12):8054–8060. doi: 10.4049/jimmunol.0902140 [DOI] [PubMed] [Google Scholar]
- 45.Mohammed I, Said DG, Dua HS. Human antimicrobial peptides in ocular surface defense. Prog Retin Eye Res. 2017;61:1–22. doi: 10.1016/j.preteyeres.2017.03.004 [DOI] [PubMed] [Google Scholar]
- 46.Szliter EA, Lighvani S, Barrett RP, Hazlett LD. Vasoactive intestinal peptide balances pro- and anti-inflammatory cytokines in the Pseudomonas aeruginosa-infected cornea and protects against corneal perforation. J Immunol. 2007;178(2):1105–1114. doi: 10.4049/jimmunol.178.2.1105 [DOI] [PubMed] [Google Scholar]
- 47.Li SA, Liu J, Xiang Y et al. Therapeutic potential of the antimicrobial peptide OH-CATH30 for antibiotic-resistant Pseudomonas aeruginosa keratitis. Antimicrob Agents Chemother. 2014;58(6):3144–3150. doi: 10.1128/AAC.00095-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.McClellan S, Jiang X, Barrett R, Hazlett LD. High-mobility group box 1: a novel target for treatment of Pseudomonas aeruginosa keratitis. J Immunol. 2015;194(4):1776–1787. doi: 10.4049/jimmunol.1401684 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Ekanayaka SA, McClellan SA, Peng X, Barrett RP, Francis R, Hazlett LD. HMGB1 antagonist, Box A, reduces TLR4, RAGE, and inflammatory cytokines in the cornea of P. aeruginosa-infected mice [published online ahead of print, 2018 Nov 8]. J Ocul Pharmacol Ther. 2018;34(10):659–669. doi: 10.1089/jop.2018.0073 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Peng X, Ekanayaka SA, McClellan SA, et al. Characterization of three ocular clinical isolates of P. aeruginosa: viability, biofilm formation, adherence, infectivity, and effects of Glycyrrhizin. Pathogens. 2017;6(4):52. doi: 10.3390/pathogens6040052 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bharathi MJ, Ramakrishnan R, Meenakshi R, et al. Microbial keratitis in South India: influence of risk factors, climate, and geographical variation. Ophthalmic Epidemiol. 2007;14(2):61–69. doi: 10.1080/09286580601001347 [DOI] [PubMed] [Google Scholar]
- 52.Bharathi MJ, Ramakrishnan R, Meenakshi R, et al. Analysis of the risk factors predisposing to fungal, bacterial & Acanthamoeba keratitis in south India. Indian J Med Res. 2009;130(6):749–757. [PubMed] [Google Scholar]
- 53.Xie L, Zhong W, Shi W, Sun S. Spectrum of fungal keratitis in north China. Ophthalmology. 2006;113(11):1943–1948. doi: 10.1016/j.ophtha.2006.05.035 [DOI] [PubMed] [Google Scholar]
- 54.Wang L, Sun S, Jing Y, et al. Spectrum of fungal keratitis in central China. Clin Exp Ophthalmol. 2009; 37:763–771. [DOI] [PubMed] [Google Scholar]
- 55.Oliveira M, Ribeiro H, Delgado JL, Abreu I. The effects of meteorological factors on airborne fungal spore concentration in two areas differing in urbanisation level. Int J Biometeorol. 2009;53(1):61–73. doi: 10.1007/s00484-008-0191-2 [DOI] [PubMed] [Google Scholar]
- 56.Gaujoux T, Chatel MA, Chaumeil C, Laroche L, Borderie VM. Outbreak of contact lens-related Fusarium keratitis in France. Cornea. 2008;27(9):1018–1021. doi: 10.1097/ICO.0b013e318173144d [DOI] [PubMed] [Google Scholar]
- 57.Chang DC, Grant GB, O’Donnell K, et al. Multistate outbreak of Fusarium keratitis associated with use of a contact lens solution. JAMA. 2006;296(8):953–963. doi: 10.1001/jama.296.8.953 [DOI] [PubMed] [Google Scholar]
- 58.Khor WB, Aung T, Saw SM, et al. An outbreak of Fusarium keratitis associated with contact lens wear in Singapore. JAMA. 2006;295(24):2867–2873. doi: 10.1001/jama.295.24.2867 [DOI] [PubMed] [Google Scholar]
- 59.Siddiqui S, Anderson VL, Hilligoss DM, et al. Fulminant mulch pneumonitis: an emergency presentation of chronic granulomatous disease. Clin Infect Dis. 2007;45(6):673–681. doi: 10.1086/520985 [DOI] [PubMed] [Google Scholar]
- 60.Martire B, Rondelli R, Soresina A, et al. Clinical features, long-term follow-up and outcome of a large cohort of patients with Chronic Granulomatous Disease: an Italian multicenter study. Clin Immunol. 2008;126(2):155–164. doi: 10.1016/j.clim.2007.09.008 [DOI] [PubMed] [Google Scholar]
- 61.Gallien S, Fournier S, Porcher R, et al. Therapeutic outcome and prognostic factors of invasive aspergillosis in an infectious disease department: a review of 34 cases. Infection. 2008;36(6):533–538. doi: 10.1007/s15010-008-7375-x [DOI] [PubMed] [Google Scholar]
- 62.Leal SM Jr, Vareechon C, Cowden S, et al. Fungal antioxidant pathways promote survival against neutrophils during infection. J Clin Invest. 2012;122(7):2482–2498. doi: 10.1172/JCI63239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Clark HL, Jhingran A, Sun Y, et al. Zinc and manganese chelation by neutrophil s100a8/a9 (calprotectin) limits extracellular aspergillus fumigatus hyphal growth and corneal infection. J Immunol. 2016;196(1):336–344. doi: 10.4049/jimmunol.1502037 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Karthikeyan RS, Leal SM Jr, Prajna NV, et al. Expression of innate and adaptive immune mediators in human corneal tissue infected with Aspergillus or fusarium. J Infect Dis. 2011;204(6):942–950. doi: 10.1093/infdis/jir426 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Netea MG, Brown GD, Kullberg BJ, Gow NA. An integrated model of the recognition of Candida albicans by the innate immune system. Nat Rev Microbiol. 2008;6(1):67–78. doi: 10.1038/nrmicro1815 [DOI] [PubMed] [Google Scholar]
- 66.Taylor PR, Tsoni SV, Willment JA, et al. Dectin-1 is required for beta-glucan recognition and control of fungal infection. Nat Immunol. 2007;8(1):31–38. doi: 10.1038/ni1408 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Saijo S, Fujikado N, Furuta T, et al. Dectin-1 is required for host defense against Pneumocystis carinii but not against Candida albicans. Nat Immunol. 2007;8(1):39–46. doi: 10.1038/ni1425 [DOI] [PubMed] [Google Scholar]
- 68.Dennehy KM, Ferwerda G, Faro-Trindade I, et al. Syk kinase is required for collaborative cytokine production induced through Dectin-1 and Toll-like receptors. Eur J Immunol. 2008;38(2):500–506. doi: 10.1002/eji.200737741 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Gow NA, Netea MG, Munro CA, et al. Immune recognition of Candida albicans beta-glucan by dectin-1. J Infect Dis. 2007;196(10):1565–1571. doi: 10.1086/523110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Ferwerda B, Ferwerda G, Plantinga TS, et al. Human dectin-1 deficiency and mucocutaneous fungal infections. N Engl J Med. 2009;361(18):1760–1767. doi: 10.1056/NEJMoa0901053 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Chidambaram JD, Kannambath S, Srikanthi P, et al. Persistence of innate immune pathways in late stage human bacterial and fungal keratitis: results from a comparative transcriptome analysis. Front Cell Infect Microbiol. 2017;7:193. doi: 10.3389/fcimb.2017.00193 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Cavuoto KM, Banerjee S, Galor A. Relationship between the microbiome and ocular health. Ocul Surf. 2019;17(3):384–392. doi: 10.1016/j.jtos.2019.05.006 [DOI] [PubMed] [Google Scholar]
- 73.Korach-Rechtman H, Freilich S, Gerassy-Vainberg S, et al. Murine genetic background has a stronger impact on the composition of the gut microbiota than maternal inoculation or exposure to unlike exogenous microbiota. Appl Environ Microbiol. 2019;85(18):e00826–19. doi: 10.1128/AEM.00826-19 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Jayasudha R, Chakravarthy SK, Prashanthi GS, et al. Alterations in gut bacterial and fungal microbiomes are associated with bacterial Keratitis, an inflammatory disease of the human eye. J Biosci. 2018;43(5):835–856. [PubMed] [Google Scholar]
- 75.Kalyana Chakravarthy S, Jayasudha R, Ranjith K, et al. Correction: Alterations in the gut bacterial microbiome in fungal Keratitis patients. PLoS One. 2019;14(1):e0211757. Published 2019 Jan 30. doi: 10.1371/journal.pone.0211757 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Zaidi T, Zaidi T, Cywes-Bentley C, Lu R, Priebe GP, Pier GB. Microbiota-driven immune cellular maturation is essential for antibody-mediated adaptive immunity to Staphylococcus aureus infection in the eye. Infect Immun. 2014;82(8):3483–3491. doi: 10.1128/IAI.01951-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.St Leger AJ, Desai JV, Drummond RA, et al. An ocular commensal protects against corneal infection by driving an Interleukin-17 response from mucosal γδ t cells. Immunity. 2017;47(1):148–158.e5. doi: 10.1016/j.immuni.2017.06.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Kugadas A, Christiansen SH, Sankaranarayanan S, et al. Impact of microbiota on resistance to ocular Pseudomonas aeruginosa-induced keratitis. PLoS Pathog. 2016;12(9):e1005855. doi: 10.1371/journal.ppat.1005855 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Young RC, Hodge DO, Liesegang TJ, Baratz KH. Incidence, recurrence, and outcomes of herpes simplex virus eye disease in Olmsted County, Minnesota, 1976–2007: the effect of oral antiviral prophylaxis. Arch Ophthalmol. 2010;128(9):1178–1183. doi: 10.1001/archophthalmol.2010.187 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Shimeld C, Hill TJ, Blyth WA, Easty DL. Reactivation of latent infection and induction of recurrent herpetic eye disease in mice. J Gen Virol. 1990;71 (Pt 2):397–404. doi: 10.1099/0022-1317-71-2-397 [DOI] [PubMed] [Google Scholar]
- 81.Liesegang TJ. Classification of herpes simplex virus keratitis and anterior uveitis. Cornea. 1999;18(2):127–143. doi: 10.1097/00003226-199903000-00001 [DOI] [PubMed] [Google Scholar]
- 82.Colin J Ganciclovir ophthalmic gel, 0.15%: a valuable tool for treating ocular herpes. Clin Ophthalmol. 2007;1(4):441–453. [PMC free article] [PubMed] [Google Scholar]
- 83.Holland EJ, Schwartz GS. Classification of herpes simplex virus keratitis. Cornea. 1999;18(2):144–154. doi: 10.1097/00003226-199903000-00002 [DOI] [PubMed] [Google Scholar]
- 84.Wilhelmus KR, Gee L, Hauck WW, et al. Herpetic Eye Disease Study. A controlled trial of topical corticosteroids for herpes simplex stromal keratitis. Ophthalmology. 1994;101(12):1883–1896. doi: 10.1016/s0161-6420(94)31087-6 [DOI] [PubMed] [Google Scholar]
- 85.Jabs DA. Acyclovir for recurrent herpes simplex virus ocular disease. N Engl J Med. 1998;339(5):340–341. doi: 10.1056/NEJM199807303390510 [DOI] [PubMed] [Google Scholar]
- 86.Conrady CD, Zheng M, Mandal NA, et al. IFN-α-driven CCL2 production recruits inflammatory monocytes to infection site in mice. Mucosal Immunol. 2013;6(1):45–55. doi: 10.1038/mi.2012.46 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Doymaz MZ, Rouse BT. Herpetic stromal keratitis: an immunopathologic disease mediated by CD4+ T lymphocytes. Invest Ophthalmol Vis Sci. 1992; 33(7):2165–2173. [PubMed] [Google Scholar]
- 88.Lobo AM, Agelidis AM, Shukla D. Pathogenesis of herpes simplex keratitis: the host cell response and ocular surface sequelae to infection and inflammation. Ocul Surf. 2019;17(1):40–49. doi 10.1016/j.jtos.2018.10.002 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Conrady CD, Zheng M, Stone DU, Carr DJ. CD8+ T cells suppress viral replication in the cornea but contribute to VEGF-C-induced lymphatic vessel genesis. J Immunol. 2012;189(1):425–432. doi: 10.4049/jimmunol.1200063 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Frank GM, Buela KA, Maker DM, Harvey SA, Hendricks RL. Early responding dendritic cells direct the local NK response to control herpes simplex virus 1 infection within the cornea. J Immunol. 2012;188(3):1350–1359. doi: 10.4049/jimmunol.1101968 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Singh PK, Singh S, Farr D, Kumar A. Interferon-stimulated gene 15 (ISG15) restricts Zika virus replicationin primary human cornea; epithelial cells. Ocul Surf. 2019;17(3):551–559. doi 10.1016/j.jtos.2019.03.006 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Buela KA, Hendricks RL. Cornea-infiltrating and lymph node dendritic cells contribute to CD4+ T cell expansion after herpes simplex virus-1 ocular infection. J Immunol. 2015;194(1):379–387. doi: 10.4049/jimmunol.1402326 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Suryawanshi A, Veiga-Parga T, Rajasagi NK, et al. Role of IL-17 and Th17 cells in herpes simplex virus-induced corneal immunopathology. J Immunol. 2011;187(4):1919–1930. doi: 10.4049/jimmunol.1100736 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Maertzdorf J, Osterhaus AD, Verjans GM. IL-17 expression in human herpetic stromal keratitis: modulatory effects on chemokine production by corneal fibroblasts. J Immunol. 2002;169(10):5897–5903. doi: 10.4049/jimmunol.169.10.5897 [DOI] [PubMed] [Google Scholar]
- 95.Sehrawat S, Rouse BT. Anti-inflammatory effects of FTY720 against viral-induced immunopathology: role of drug-induced conversion of T cells to become Foxp3+ regulators. J Immunol. 2008;180(11):7636–7647. doi: 10.4049/jimmunol.180.11.7636 [DOI] [PubMed] [Google Scholar]
- 96.Gaddipati S, Estrada K, Rao P, Jerome AD, Suvas S. IL-2/anti-IL-2 antibody complex treatment inhibits the development but not the progression of herpetic stromal keratitis. J Immunol. 2015;194(1):273–282. doi: 10.4049/jimmunol.1401285 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Suryawanshi A, Mulik S, Sharma S, Reddy PB, Sehrawat S, Rouse BT. Ocular neovascularization caused by herpes simplex virus type 1 infection results from breakdown of binding between vascular endothelial growth factor A and its soluble receptor. J Immunol. 2011;186(6):3653–3665. doi: 10.4049/jimmunol.1003239 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Gimenez F, Mulik S, Veiga-Parga T, Bhela S, Rouse BT. Robo 4 counteracts angiogenesis in herpetic stromal keratitis. PLoS One. 2015;10(12):e0141925. doi: 10.1371/journal.pone.0141925 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Mulik S, Xu J, Reddy PB, et al. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012;181(2):525–534. doi: 10.1016/j.ajpath.2012.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Wuest TR, Carr DJ. VEGF-A expression by HSV-1-infected cells drives corneal lymphangiogenesis. J Exp Med. 2010;207(1):101–115. doi: 10.1084/jem.20091385 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Bryant-Hudson KM, Gurung HR, Zheng M, Carr DJ. Tumor necrosis factor alpha and interleukin-6 facilitate corneal lymphangiogenesis in response to herpes simplex virus 1 infection. J Virol. 2014;88(24):14451–14457. doi: 10.1128/JVI.01841-14 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Rao P, Suvas S. Development of inflammatory hypoxia and prevalence of glycolytic metabolism in progressing herpes stromal keratitis lesions. J Immunol. 2019;202(2):514–526. doi: 10.4049/jimmunol.1800422 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Hamrah P, Cruzat A, Dastjerdi MH, et al. Corneal sensation and subbasal nerve alterations in patients with herpes simplex keratitis: an in vivo confocal microscopy study. Ophthalmology. 2010;117(10):1930–1936. doi: 10.1016/j.ophtha.2010.07.010 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Chucair-Elliott AJ, Zheng M, Carr DJ. Degeneration and regeneration of corneal nerves in response to HSV-1 infection. Invest Ophthalmol Vis Sci. 2015;56(2):1097–1107. Published 2015 Jan 13. doi: 10.1167/iovs.14-15596 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Hamrah P, Sahin A, Dastjerdi MH, et al. Cellular changes of the corneal epithelium and stroma in herpes simplex keratitis: an in vivo confocal microscopy study. Ophthalmology. 2012;119(9):1791–1797. doi: 10.1016/j.ophtha.2012.03.005 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Yang K, Wu M, Li M, et al. miR-155 suppresses bacterial clearance in Pseudomonas aeruginosa-induced keratitis by targeting Rheb. J Infect Dis. 2014;210(1):89–98. doi: 10.1093/infdis/jiu002 [DOI] [PubMed] [Google Scholar]
- 107.Garami A, Zwartkruis FJ, Nobukuni T, et al. Insulin activation of Rheb, a mediator of mTOR/S6K/4E-BP signaling, is inhibited by TSC1 and 2. Mol Cell. 2003;11(6):1457–1466. doi: 10.1016/s1097-2765(03)00220-x [DOI] [PubMed] [Google Scholar]
- 108.Foldenauer ME, McClellan SA, Berger EA, Hazlett LD. Mammalian target of rapamycin regulates IL-10 and resistance to Pseudomonas aeruginosa corneal infection. J Immunol. 2013;190(11):5649–5658. doi: 10.4049/jimmunol.1203094 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Xu S, Witmer PD, Lumayag S, Kovacs B, Valle D. MicroRNA (miRNA) transcriptome of mouse retina and identification of a sensory organ-specific miRNA cluster. J Biol Chem. 2007;282(34):25053–25066. doi: 10.1074/jbc.M700501200 [DOI] [PubMed] [Google Scholar]
- 110.Lumayag S, Haldin CE, Corbett NJ, et al. Inactivation of the microRNA-183/96/182 cluster results in syndromic retinal degeneration. Proc Natl Acad Sci U S A. 2013;110(6):E507–E516. doi: 10.1073/pnas.1212655110 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Muraleedharan CK, McClellan SA, Barrett RP, et al. Inactivation of the miR-183/96/182 cluster decreases the severity of Pseudomonas aeruginosa-induced keratitis. Invest Ophthalmol Vis Sci. 2016;57(4):1506–1517. doi: 10.1167/iovs.16-19134 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 112.Muraleedharan CK, McClellan SA, Ekanayaka SA, et al. The miR-183/96/182 cluster regulates macrophage functions in response to Pseudomonas aeruginosa. J Innate Immun. 2019;11(4):347–358. doi: 10.1159/000495472 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 113.Coku A, McClellan SA, Van Buren E, Back JB, et al. The miR-183/96/182 cluster regulates the functions of corneal resident macrophages. Immunohorizons 2020;4:729–44. doi: 10.4049/immunohorizons.2000091 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Ichiyama K, Gonzalez-Martin A, Kim BS, et al. The MicroRNA-183-96-182 Cluster Promotes T Helper 17 Cell Pathogenicity by Negatively Regulating Transcription Factor Foxo1 Expression. Immunity. 2016;44(6):1284–1298. doi: 10.1016/j.immuni.2016.05.015 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Zaidi TS, Zaidi T, Pier GB, Priebe GP. Topical neutralization of interleukin-17 during experimental Pseudomonas aeruginosa corneal infection promotes bacterial clearance and reduces pathology. Infect Immun. 2012;80(10):3706–3712. doi: 10.1128/IAI.00249-12 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 116.Cokarić Brdovčak M, Zubković A, Jurak I. Herpes Simplex Virus 1 deregulation of host MicroRNAs. Noncoding RNA. 2018;4(4):36. Published 2018 Nov 23. doi: 10.3390/ncrna4040036 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rock DL, Nesburn AB, Ghiasi H, et al. Detection of latency-related viral RNAs in trigeminal ganglia of rabbits latently infected with herpes simplex virus type 1. J Virol. 1987;61(12):3820–3826. doi: 10.1128/JVI.61.12.3820-3826.1987 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Umbach JL, Kramer MF, Jurak I, Karnowski HW, Coen DM, Cullen BR. MicroRNAs expressed by herpes simplex virus 1 during latent infection regulate viral mRNAs. Nature. 2008;454(7205):780–783. doi: 10.1038/nature07103 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Jiang X, Brown D, Osorio N, et al. A herpes simplex virus type 1 mutant disrupted for microRNA H2 with increased neurovirulence and rate of reactivation. J Neurovirol. 2015;21(2):199–209. doi: 10.1007/s13365-015-0319-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Jiang X, Brown D, Osorio N, Hsiang C, BenMohamed L, Wechsler SL. Increased neurovirulence and reactivation of the herpes simplex virus type 1 latency-associated transcript (LAT)-negative mutant dLAT2903 with a disrupted LAT miR-H2. J Neurovirol. 2016;22(1):38–49. doi: 10.1007/s13365-015-0362-y [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Duan F, Liao J, Huang Q, Nie Y, Wu K. HSV-1 miR-H6 inhibits HSV-1 replication and IL-6 expression in human corneal epithelial cells in vitro. Clin Dev Immunol. 2012;2012:192791. doi: 10.1155/2012/192791 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Kriesel JD, Gebhardt BM, Hill JM, et al. Anti-interleukin-6 antibodies inhibit herpes simplex virus reactivation. J Infect Dis. 1997;175(4):821–827. doi: 10.1086/513977 [DOI] [PubMed] [Google Scholar]
- 123.Biswas PS, Banerjee K, Kinchington PR, Rouse BT. Involvement of IL-6 in the paracrine production of VEGF in ocular HSV-1 infection. Exp Eye Res. 2006;82(1):46–54. doi: 10.1016/j.exer.2005.05.001 [DOI] [PubMed] [Google Scholar]
- 124.Lutz G, Jurak I, Kim ET, et al. Viral ubiquitin ligase stimulates selective host microrna expression by targeting zeb transcriptional repressors. Viruses. 2017;9(8):210. Published 2017 Aug 7. doi: 10.3390/v9080210 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Rutkowski AJ, Erhard F, L’Hernault A, et al. Widespread disruption of host transcription termination in HSV-1 infection. Nat Commun. 2015;6:7126. Published 2015 May 20. doi: 10.1038/ncomms8126 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Li XL, Hara T, Choi Y, et al. A p21-ZEB1 complex inhibits epithelial-mesenchymal transition through the microRNA 183-96-182 cluster. Mol Cell Biol. 2014;34(3):533–550. doi: 10.1128/MCB.01043-13 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Wellner U, Schubert J, Burk UC, et al. The EMT-activator ZEB1 promotes tumorigenicity by repressing stemness-inhibiting microRNAs. Nat Cell Biol. 2009;11(12):1487–1495. doi: 10.1038/ncb1998 [DOI] [PubMed] [Google Scholar]
- 128.Oba S, Mizutani T, Suzuki E, et al. A useful method of identifying of miRNAs which can down-regulate Zeb-2. BMC Res Notes. 2013;6:470. Published 2013 Nov 18. doi: 10.1186/1756-0500-6-470 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Stark TJ, Arnold JD, Spector DH, Yeo GW. High-resolution profiling and analysis of viral and host small RNAs during human cytomegalovirus infection. J Virol. 2012;86(1):226–235. doi: 10.1128/JVI.05903-11 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Oussaief L, Fendri A, Chane-Woon-Ming B, et al. Modulation of MicroRNA Cluster miR-183-96-182 expression by Epstein-Barr virus latent membrane protein 1. J Virol. 2015;89(23):12178–12188. doi: 10.1128/JVI.01757-15 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Pan D, Flores O, Umbach JL, et al. A neuron-specific host microRNA targets herpes simplex virus-1 ICP0 expression and promotes latency. Cell Host Microbe. 2014;15(4):446–456. doi: 10.1016/j.chom.2014.03.004 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 132.Bhela S, Mulik S, Gimenez F, et al. Role of miR-155 in the pathogenesis of herpetic stromal keratitis. Am J Pathol. 2015;185(4):1073–1084. doi: 10.1016/j.ajpath.2014.12.021 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 133.Huffaker TB, Hu R, Runtsch MC, et al. Epistasis between microRNAs 155 and 146a during T cell-mediated antitumor immunity. Cell Rep. 2012;2(6):1697–1709. doi: 10.1016/j.celrep.2012.10.025 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Banerjee A, Schambach F, DeJong CS, Hammond SM, Reiner SL. Micro-RNA-155 inhibits IFN-gamma signaling in CD4+ T cells. Eur J Immunol. 2010;40(1):225–231. doi: 10.1002/eji.200939381 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Biswas PS, Rouse BT. Early events in HSV keratitis--setting the stage for a blinding disease. Microbes Infect. 2005;7(4):799–810. doi: 10.1016/j.micinf.2005.03.003 [DOI] [PubMed] [Google Scholar]
- 136.Kim B, Tang Q, Biswas PS, et al. Inhibition of ocular angiogenesis by siRNA targeting vascular endothelial growth factor pathway genes: therapeutic strategy for herpetic stromal keratitis. Am J Pathol. 2004;165(6):2177–2185. doi: 10.1016/S0002-9440(10)63267-1 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Anand S, Majeti BK, Acevedo LM, et al. MicroRNA-132-mediated loss of p120RasGAP activates the endothelium to facilitate pathological angiogenesis. Nat Med. 2010;16(8):909–914. doi: 10.1038/nm.2186 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 138.Mulik S, Xu J, Reddy PB, et al. Role of miR-132 in angiogenesis after ocular infection with herpes simplex virus. Am J Pathol. 2012;181(2):525–534. doi: 10.1016/j.ajpath.2012.04.014 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Shimizu E, Ogawa Y, Saijo Y, et al. Commensal microflora in human conjunctiva; characteristics of microflora in the patients with chronic ocular graft-versus-host disease. Ocul Surf. 2019;17(2):265–271. 10.1016/j.jtos.2019.02.001 [DOI] [PubMed] [Google Scholar]
- 140.Ozkan J, Willcox M, Wemheuer B, et al. Biogeography of the human ocular microbiota. Ocul Surf. 2019;17(1):111–118. doi: 10.1016/j.jtos.2018.11.005 [DOI] [PubMed] [Google Scholar]