

Abstract
The emerging of severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2) outbreak is associated with high morbidity and mortality rates globally. One of the most prominent characteristics of coronavirus disease-19 (COVID-19) is lymphopenia, which is in contrast to other viral infections. This controversy might be explained by the evaluation of impaired innate and adaptive immune responses, during the SARS-CoV-2 infection. During the innate immune response, poly-ADP-ribose polymerase hyperactivated due to virus entry and extensive DNA damage sequentially, leading to nicotinamide adenine dinucleotide (NAD)+ depletion, adenosine triphosphate depletion, and finally cell death. In contrast to the immune response against viral infections, cytotoxic T lymphocytes decline sharply in SARS-CoV-2 infection which might be due to infiltration and trapping in the lower respiratory tract. In addition, there are more factors proposed to involve in lymphopenia in COVID-19 infection such as the role of CD38, which functions as NADase and intensifies NAD depletion, which in turn affects NAD+–dependent Sirtuin proteins, as the regulators of cell death and viability. Lung tissue sequestration following cytokine storm supposed to be another reason for lymphopenia in COVID-19 patients. Protein 7a, as one of the virus-encoded proteins, induces apoptosis in various organ-derived cell lines. These mechanisms proposed to induce lymphopenia, although there are still more studies needed to clarify the underlying mechanisms for lymphopenia in COVID-19 patients.
Keywords: ADP-ribosyl cyclase 1, lymphopenia, NAD, SARS-CoV-2
Introduction
The coronavirus disease 19 (COVID-19), a novel infectious disease, caused by severe acute respiratory syndrome coronavirus 2 (SARS-CoV-2), which originated in Wuhan, China, has rapidly increased in pandemic scale with growing morbidity and mortality rate worldwide. In the past decades, two coronavirus family members were responsible for severe respiratory disease outbreaks which have been previously characterized as a major concern of public health; SARS and the Middle East respiratory syndrome (MERS) caused by SARS and MERS coronavirus, respectively.[1]
Coronaviruses spread broadly among humans leading to a spectrum of respiratory diseases ranging from flu-like symptoms or pneumonia to acute respiratory distress syndrome, which caused high mortality in human populations.[2]
Although the pathogenesis of SARS-CoV-2 is not yet fully understood, there are apparent similarities between SARS-CoV-2 and the original SARS-CoV. Extensive lung damage in COVID-19 patients seems to be associated with high initial viral load, increased number of monocytes, macrophage, and neutrophil infiltrated in the lungs, concomitant with elevated levels of serum pro-inflammatory cytokines and chemokines and a rapid decrease of peripheral T cell subsets.[3] Therefore, the clinical deterioration and tissue damage during SARS-CoV-2 infection may result from a combination of direct virus-induced cytopathic effects and maladjusted immune responses. As described earlier, one of the clinical features of SARS-CoV-2 infection is the reduction of peripheral T cell subsets, as a unique characteristic in COVID-19 patients during acute infection.[4] Recent data shed new light on the role of impaired immune response and eventual lymphopenia in SARS-CoV-2 pathogenesis.[5] Here, we provide a brief introduction to the immune response against SARS-CoV-2 and possible mechanisms underlying reduced peripheral T cell subsets in COVID-19 patients.
The Innate Immune Response
The SARS-CoV-2 spike protein includes a strong binding affinity for human angiotensin II receptor, which considered essential for host cell entry and subsequent viral progeny. During an acute respiratory tract infection, the innate immune response is that the first line of defense against the virus results in the initiation of a rapid immune response following virus–cell interaction.[6]
In conventional cell biology, ADP-ribosylation could be a common reversible posttranslational modification with proposed antiviral properties and impact on innate immunity. ADP-ribosylation is mediated by poly-ADP-ribose (ADPr) polymerase (PARP) gene family encoded proteins. Transfer of one or more ADPr groups from nicotinamide adenine dinucleotide (NAD+) to target protein is catalyzed within eukaryotic cells by members of the PARP, now called diphtheria toxin-like ADP-ribosyltransferases (ARTDs).[7] PARP-1 hub's role is sensing cellular metabolic stress, including oxidative stress, DNA repair, and pathogen infection, which result in activation and ultimately the determination of cell fate. The PARP activity is especially correlated with the regulation of the mammalian innate antiviral response.[8] Since the NAD+ level is critical for the regulation of energy metabolism and maintenance of redox homeostasis, PARP hyperactivation following extensive DNA damage upon viral infection results in rapid depletion of cellular NAD+ and reduced adenosine triphosphate (ATP) production and ultimately cell death.[9]
The Adaptive Immune Response
The adaptive immune response against viral infections began to develop exact and powerful protector immunity against viruses. The adaptive immune response to viral infections exerts through the effector function of cytotoxic T lymphocyte (CTL) response. CTLs are generated in response to intracellular invading pathogens, and they specifically recognize and kill virus-infected cells and/or release inhibitory antiviral-soluble factors. Thus, a pointy increase in CTL count is extremely expected in CTL patients with SARS-CoV-2 infection.[10]
However, unlike the traditional immune responses against viruses, in SARS-CoV-2 infection, a different response shaped which is accompanied by particular T cell lymphopenia, with a rapid decrease in both CD4 and CD8 T cell subsets. The latest report indicated the reduced number of CD4+ and CD8+ T cells within the peripheral blood of SARS-CoV-2-infected patients, similar to patients with SARS-CoV, with concomitant hyperactivated T cells shown by high proportions of CD4-restricted HLA-DR (3.47%) and CD38 (CD8, 39.4%) double-positive populations.[11]
The possible explanation of CTL reduction in COVID-19 patients could be an adverse outcome that needs further investigation. Infiltration of T cells and subsequent trapping in the lower respiratory tract, as well as immune cell death, could also participate in the underlying mechanisms of the acute decrease of peripheral T cell subsets detected in SARS-CoV-2 infection.
Here, the possible factors that could be involved in the general reduction of peripheral T lymphocytes during SARS-CoV-2 are further discussed in detail [Figure 1].
Figure 1.
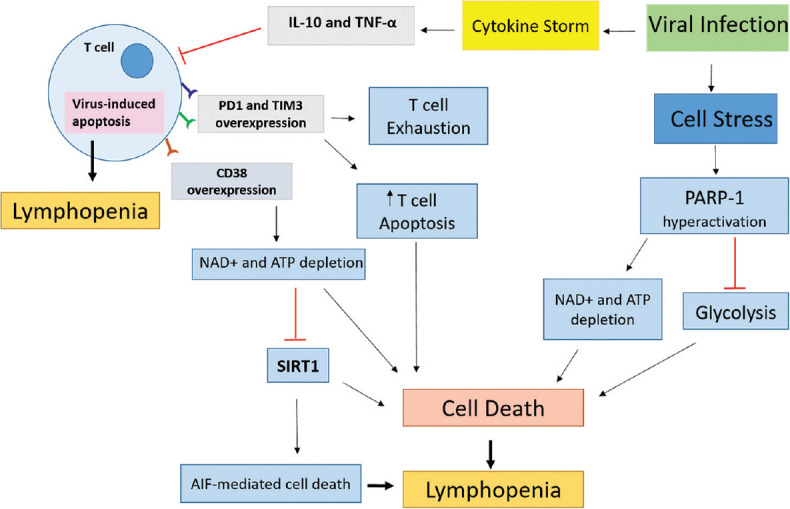
Possible mechanisms involved within the reduction of peripheral lymphocyte subsets in patients with severe acute respiratory syndrome coronavirus 2. The nicotinamide adenine dinucleotide + depletion subsequent to poly-ADP-ribose polymerase 1 hyperactivation and CD38 overexpression leads to cell death and T cell depletion. Further, the negative effect of cytokines on T cells and virus-induced apoptosis could also be involved in severe acute respiratory syndrome coronavirus 2-related lymphopenia
Nicotinamide adenine dinucleotide+ depletion
Expression of CD38 on immune system cells seems to play an important role in the context of host defense to infection. CD38 is a multifunctional enzyme that incorporates a key role in the degradation of NAD and involved in the control of cellular NAD+ levels. It has been postulated that CD38 is the major NADase in the cells and NAD+ levels significantly increased upon CD38 knockdown. Thus, it absolutely was hypothesized that NAD+ depletion upon CD38 overexpression on the T cells of COVID-19–infected patients is associated with disturbed metabolic regulation and cell viability. NAD+ is one among the energy currencies required for vital cellular processes mediating bio-energetic processes, metabolic homeostasis, response to damages, and immune reactions. Moreover, emerging evidence demonstrated that NAD+ is released during the early phase of inflammation and exerts the immunoregulatory role in vivo.[12,13]
As a part of the innate immune response, PARP hyperactivation, following to viral mediated-oxidative and/or nitrosative stress with CD38 overexpression during adaptive immunity consumes large amounts of NAD+ and lead to NAD+ depletion.
The NAD+ depletion linked with a reduced glycolytic activity which can, in turn, affect ATP levels, since cells consume ATP for NAD+ replenishment. On the other hand, NAD+ depletion leads to increased production and release of pro-inflammatory cytokines, reactive oxygen species, and macrophage infiltration via Sirtuin-1 (SIRT1) inhibition.[14,15]
Proteins of the SIRT family (sirtuins) are NAD+–dependent histone deacetylases which govern the balance between cellular durability and death. SIRT proteins are thought to exert their function through the control of genomic stability, DNA repair, and transcriptional regulation. In addition to SIRT proteins, PARP operates convergent with SIRT proteins for maintenance of the balance that determines cell fate in response to stress.[16]
The functional cross-talk between SIRT proteins and PARP is suggested following the consumption of the common intracellular NAD+ pool by both of them. The previous results showed that overactivation of PARP subsequent to DNA damage leads to apoptosis-inducing factor-mediated cell death in the absence of SIRT1.[14,17,18]
Pro-inflammatory cytokines and chemokines
Patients infected with COVID-19 showed significantly increased levels of plasma pro-inflammatory cytokines including monocyte chemoattractant protein 1, macrophage inflammatory protein (MIP) 1α, MIP1β, interleukin (IL) 1-β, interleukin-1 receptor antagonist (IL-1RA), IL7, IL8, IL9, IL10, interferon gamma inducible protein-10 (IP-10), platelet-derived growth factor B, basic fibroblast growth factor 2, granulocyte colony-stimulating factor, granulocyte-macrophage colony-stimulating factor, interferon gamma γ, tumor necrosis factor α, and vascular endothelial growth factor.[3,19]
This cytokine profile indicates rapid recruitment and increased trafficking of the monocyte–macrophage lineage into the lung very early in the infection process. The immune cell infiltration into the lower respiratory tract, leading to uncontrolled immune responses with subsequent hyperinflammation and cytokine storm, results in organ failure, pulmonary tissue damage, and reduced lung capacity.[20] Thus, tissue sequestration might play a major role in the reduction of peripheral blood lymphocyte count of SARS-CoV-2 infected patients.
Recently, Diao et al. reviewed the quantity of T cells from data of 522 patients with laboratory-confirmed COVID-19. Their results showed that the number of total T cells, CD4+ T cells, and CD8+ T cells was significantly reduced in COVID-19 patients. T cell numbers were negatively correlated with patient survival and serum IL-6, IL-10, and TNF-α concentration. These data suggest that the decreased number of T cells seen in COVID-19 patients could also be the results of high serum concentration of TNF-α and IL-10, which negatively regulating T cell survival or proliferation. In addition, this study indicated that T cells from COVID-19 patients expressed considerably higher levels of the exhausted marker PD-1 and Tim-3, which can negatively affect the function of these cells.[19]
Protein 7a-induced apoptosis
Among the virus-encoded proteins, protein 7a, a structural protein specifically encoded by SARS-CoV-2, combines to mature virions and plays an essential role in the pathogenesis of SARS-CoV-2. This protein induces apoptosis, arrests the cell cycle, and promotes the production of pro-inflammatory cytokines. In addition, it has been specified that the 7a protein inhibits cellular gene expression.
In addition, overexpression of protein 7a can induce apoptosis via a caspase-dependent pathway in cell lines derived from various organs, including lung, liver, and kidney. As a high copy number of virus has been detected in lymphocytes taken from SARS patients, it is possible that the lymphocytes are depleted as a result of virus-induced apoptosis. Based on these data, protein 7a expression could be considered as one of the mechanisms involved in lymphopenia.[21,22]
Conclusion
The innate and adaptive immune responses against SARS-CoV-2 and possible mechanisms involved in the reduction of peripheral lymphocyte subsets in patients with SARS-CoV-2 were discussed. Besides, we can infer from the above clues that NAD+ depletion, the direct effect of cytokines on T cells, and virus-induced apoptosis may be reasons leading to SARS-CoV-2–related lymphopenia. However, the precise mechanism underlying the acute lymphopenia during SARS-CoV-2 infection remains unclear, and further researches are vital for the identification of molecular mechanisms responsible for the pathogenesis of SARS-CoV-2 infection.
Financial support and sponsorship
Nil.
Conflicts of interest
There are no conflicts of interest.
References
- 1.Le TT, Andreadakis Z, Kumar A, Román RG, Tollefsen S, Saville M, et al. The COVID-19 vaccine development landscape. Nat Rev Drug Discov. 2020;19:305–6. doi: 10.1038/d41573-020-00073-5. [DOI] [PubMed] [Google Scholar]
- 2.Tian S, Hu N, Lou J, Chen K, Kang X, Xiang Z, et al. Characteristics of COVID-19 infection in Beijing. J Infect. 2020;80:401–6. doi: 10.1016/j.jinf.2020.02.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Li G, Fan Y, Lai Y, Han T, Li Z, Zhou P, et al. Coronavirus infections and immune responses. J Med Virol. 2020;92:424–32. doi: 10.1002/jmv.25685. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Cui W, Fan Y, Wu W, Zhang F, Wang JY, Ni AP. Expression of lymphocytes and lymphocyte subsets in patients with severe acute respiratory syndrome. Clin Infect Dis. 2003;37:857–9. doi: 10.1086/378587. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Li T, Qiu Z, Zhang L, Han Y, He W, Liu Z, et al. Significant changes of peripheral T lymphocyte subsets in patients with severe acute respiratory syndrome. J Infect Dis. 2004;189:648–51. doi: 10.1086/381535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Li X, Geng M, Peng Y, Meng L, Lu S. Molecular immune pathogenesis and diagnosis of COVID-19. J Pharm Anal. 2020;10:102–8. doi: 10.1016/j.jpha.2020.03.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Singatulina AS, Hamon L, Sukhanova MV, Desforges B, Joshi V, Bouhss A, et al. PARP-1 activation directs FUS to DNA damage sites to form PARG-reversible compartments enriched in damaged DNA. Cell Rep. 2019;27:1809–21.e5. doi: 10.1016/j.celrep.2019.04.031. [DOI] [PubMed] [Google Scholar]
- 8.Liu L, Su X, Quinn WJ, 3rd, Hui S, Krukenberg K, Frederick DW, et al. Quantitative analysis of NAD synthesis-breakdown fluxes. Cell Metab. 2018;27:1067–80.e5. doi: 10.1016/j.cmet.2018.03.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Hsieh CL, Hsieh SY, Huang HM, Lu SL, Omori H, Zheng PX, et al. Nicotinamide increases intracellular NAD+ content to enhance autophagy-mediated Group A streptococcal clearance in endothelial cells. Front Microbiol. 2020;11:117. doi: 10.3389/fmicb.2020.00117. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Cao X. COVID-19: Immunopathology and its implications for therapy. Nat Rev Immunol. 2020;20:269–70. doi: 10.1038/s41577-020-0308-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Xu Z, Shi L, Wang Y, Zhang J, Huang L, Zhang C, et al. Pathological findings of COVID-19 associated with acute respiratory distress syndrome. Lancet Respir Med. 2020;8:420–2. doi: 10.1016/S2213-2600(20)30076-X. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Chini CC, Tarragó MG, Chini EN. NAD and the aging process: Role in life, death and everything in between. Mol Cell Endocrinol. 2017;455:62–74. doi: 10.1016/j.mce.2016.11.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Chini EN, Chini CC, Espindola Netto JM, de Oliveira GC, van Schooten W. The pharmacology of CD38/NADase: An emerging target in cancer and diseases of aging. Trends Pharmacol Sci. 2018;39:424–36. doi: 10.1016/j.tips.2018.02.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cantó C, Sauve AA, Bai P. Crosstalk between poly (ADP-ribose) polymerase and sirtuin enzymes. Mol Aspects Med. 2013;34:1168–201. doi: 10.1016/j.mam.2013.01.004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Graeff R, Liu Q, Kriksunov IA, Kotaka M, Oppenheimer N, Hao Q, et al. Mechanism of cyclizing NAD to cyclic ADP-ribose by ADP-ribosyl cyclase and CD38. J Biol Chem. 2009;284:27629–36. doi: 10.1074/jbc.M109.030965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Aksoy P, Escande C, White TA, Thompson M, Soares S, Benech JC, et al. Regulation of SIRT 1 mediated NAD dependent deacetylation: A novel role for the multifunctional enzyme CD38. Biochem Biophys Res Commun. 2006;349:353–9. doi: 10.1016/j.bbrc.2006.08.066. [DOI] [PubMed] [Google Scholar]
- 17.Kolthur-Seetharam U, Dantzer F, McBurney MW, de Murcia G, Sassone-Corsi P. Control of AIF-mediated cell death by the functional interplay of SIRT1 and PARP-1 in response to DNA damage. Cell Cycle. 2006;5:873–7. doi: 10.4161/cc.5.8.2690. [DOI] [PubMed] [Google Scholar]
- 18.Jang KH, Hwang Y, Kim E. PARP1 impedes SIRT1-mediated autophagy during degeneration of the retinal pigment epithelium under oxidative stress. Mol Cells. 2020;43:632–44. doi: 10.14348/molcells.2020.0078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Diao B, Wang C, Tan Y, Chen X, Liu Y, Ning L, et al. Reduction and functional exhaustion of T cells in patients with coronavirus disease 2019 (COVID-19) Front Immunol. 2020;11:827. doi: 10.3389/fimmu.2020.00827. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.To KF, Lo AW. Exploring the pathogenesis of severe acute respiratory syndrome (SARS): The tissue distribution of the coronavirus (SARS-CoV) and its putative receptor, angiotensin-converting enzyme 2 (ACE2) J Pathol. 2004;203:740–3. doi: 10.1002/path.1597. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Jamwal S, Gautam A, Elsworth J, Kumar M, Chawla R, Kumar P. An updated insight into the molecular pathogenesis, secondary complications and potential therapeutics of COVID-19 pandemic. Life Sci. 2020;257:118105. doi: 10.1016/j.lfs.2020.118105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Kopecky-Bromberg SA, Martinez-Sobrido L, Palese P. 7a protein of severe acute respiratory syndrome coronavirus inhibits cellular protein synthesis and activates p38 mitogen-activated protein kinase. J Virol. 2006;80:785–93. doi: 10.1128/JVI.80.2.785-793.2006. [DOI] [PMC free article] [PubMed] [Google Scholar]