

Abstract
Parkinson’s disease (PD) is a clinically heterogeneous disorder with a multi-factorial pathology. Various molecular mechanisms are involved in the pathogenesis of PD, converging to oxidative stress and proteinopathy. The accumulation of reactive aldehydes (i.e., the dopamine metabolite DOPAL, lipid-peroxidation products, and advanced glycation end-products) has been reported in PD patients’ brains. Aldehydes easily react with primary amines such as lysine residues, which are involved in several regulatory processes in cells. Therefore, aldehyde adducts lead to severe consequences, including neuronal proteostasis, mitochondrial dysfunction, and cell death. In this review, we analyzed the scavenging role of amines toward toxic aldehydes in the brain. Interestingly, small molecules like metformin, rasagiline, hydralazine are already clinically available and used in the therapy for PD and other diseases. Hence, we propose to reevaluate this class of drugs as a disease-modifiers for PD, and we suggest that improved analysis of their pharmacology and bioavailability in the brain, together with a more precise patients stratification, should be considered before planning future clinical trials.
Keywords: Parkinson’s disease, aldehyde, scavenger, neuroprotection, patients stratification, biomarkers
1. INTRODUCTION
Parkinson’s disease (PD) is the second most common neurodegenerative disorder, and it currently affects about 1% of the population over 65 years old and more than 4-5% over 80 [1, 2]. According to the statistics provided by the Parkinson’s Foundation, nearly 10 million people are currently suffering from PD (https://www.parkinson.org/Understanding-Parkinsons/Statistics), with a higher prevalence in Europe, North America, and South America [3].
The diagnosis of PD is currently based on the presence of typical motor manifestations (tremor at rest, rigidity, bradykinesia, and postural instability) and underlined by the progressive loss of the dopaminergic neurons of the nigrostriatal circuits in the midbrain [4, 5]. Additional non-motor features are also present, and some, like olfactory dysfunction, cognitive impairment, psychiatric symptoms, rapid-eye- movement sleep disorder, may precede the motor manifestations by many years [6]. Along with the disease progression, further complications arise, causing significant disability that can hardly be managed by the available therapeutic approaches.
The considerable heterogeneity among patients in the age of onset, symptoms, and progression reflects the multisystemi and multi-factorial aspects of the pathogenesis of PD. There are diverse epidemiological variables like age, gender, and ethnicity, together with environmental as well as genetic variants, identified by genome-wide association studies (GWAS), which are considered risk factors [6, 7]. Most of the cases are classified as sporadic with an undefined etiology, but there are a fraction of monogenic forms (ranging from 5 to 10% among different world regions), with 19 disease-causing genes identified to date [8]. At the molecular level, in both genetic and idiopathic PD, several pathways are affected, that with few exceptions [9, 10] all converge to alpha-Synuclein (aSyn) aggregation and deposition in Lewy Bodies (LBs), lysosomal and mitochondrial dysfunction, oxidative stress, and neuroinflammation. According to the multiple-hit hypothesis for PD pathogenesis, many risk factors concomitantly affect neuronal homeostasis, where their synergistic action overcomes the homeostatic threshold and trigger the neurodegenerative process [11-13].
From a clinical perspective, such complexity in PD pathology still challenges the successful identification and development of disease-modifying interventions by specifically targeting the affected molecular pathways. The numerous currently available pharmacological and surgical interventions are mainly symptomatic and aim to replace the dopamine (DA) reservoir and action in the nigrostriatal pathway (DA precursor L-DOPA, DA agonists, inhibitors of DA catabolism).
In the present review, we discuss the potential benefits of the co-administration of antiparkinsonian agents with guanidine molecules to simultaneously target multiple processes. Small molecules like metformin, rasagiline, hydralazine, are already in clinical practice, and rasagiline has been extensively tested as a possible disease modifier in PD. Here, we point out their potential beneficial effect as scavengers of biogenic aldehydes whose aberrant accumulation and neurotoxicity have been described in PD [14-18]. Combined treatments with these drugs, together with an accurate evaluation of pharmacology and bioavailability in the brain and a precise patient stratification, might bring significant improvement in the definition of novel disease-modifying approaches for PD.
2. ALDEHYDES OF PATHOLOGICAL RELEVANCE IN PARKINSON’S DISEASE
Aldehydes are highly reactive molecules due to their electrophilic carbonyl groups. They undergo several reactions, such as nucleophilic additions that lead to the formation of alcohols, alkenes, diols, and cyanohydrins. Alternatively, the aldehyde moiety easily reacts with primary amines through a Schiff-base condensation producing imines. Hence, aldehydes have the intrinsic ability to irreversibly and covalently modify a variety of biomolecules in the cellular milieu, i.e., proteins (typically on lysines, cysteines, histidines), nucleic acid, amino-phospholipids, and other small molecules. Unlike free radicals, aldehydes are relatively stable, and they can diffuse far from the site where they are first generated. Thus, under physiological conditions, the rapid conversion of the aldehyde to less reactive moieties is rapidly ensured by dedicated enzymes (aldehyde dehydrogenases, aldehyde reductases, alcohol reductases) before any toxic down-stream cascade could be triggered by the aldehyde addition to relevant targets.
Interestingly, several studies reported that the alteration of specific cellular pathways in PD and other co-existing pathological conditions leads to the accumulation of neurotoxic aldehydes in the brain. As listed in (Fig. 1), alterations in the catabolic pathway of monoaminergic neurotransmitters (catecholamines and indoleamines) cause the accumulation of aldehydic intermediates as 3,4-dihydroxyphenylacetaldehyde (DOPAL) from DA, 3,4-dihydrophenylglycolaldehyde (DOPEGAL) from norepinephrine (NE), and epinephrine and 5-hydroxyondole-3-acetaldehyde (5-HIAL) from serotonin (5-HT). Also, as a consequence of the increased oxidative stress, lipid peroxidation generates sub-products like 4-hydroxy-2-nonenal (4-HNE), 4-oxo-2-nonenal (4-ONE), malondialdehyde (MDA), and acrolein. Moreover, the hyperglycaemia associated with PD patients with diabetic conditions causes increased glucose metabolism and consequently increased methylglyoxal (MGO) production.
Fig. (1).

Aldehydes of pathological relevance in Parkinson’s disease. Among them, aldehydic intermediates derived from the catabolism of dopamine, norepinephrine, and serotonin (green); lipid peroxidation products (blue); glycating agent derived by glucose metabolism in diabetic condition (red). (A higher resolution / colour version of this figure is available in the electronic copy of the article).
2.1. Altered Monoamines Catabolic Pathway
Although the dopaminergic neurons of the Substantia Nigra pars compacta (SNpc) are among the most affected in PD, neurodegeneration progressively pervades other neighbouring regions, leading to extensive loss of norepinephrinergic neurons of the locus coeruleus (LC), the epinephrinergic neurons of the rostral ventral lateral medulla (RVLM) and the serotoninergic neurons of the dorsal raphe nucleus (DRN). The overall monoaminergic neuron loss correlates with the entire spectrum of PD symptoms, from motor dysfunction to sleep disorders, cognitive deficits, and altered emotional behaviours.
Of note, all these neuron subtypes share similar metabolic pathways of their monoaminergic neurotransmitters. However, several studies in PD patients’ brains as well as PD experimental models pointed out an imbalance in monoamine homeostasis at multiple levels, identifying both genetic and environmental factors.
Under physiological conditions, DA and 5-HT are loaded in the synaptic vesicles as they are synthesized, whereas NE is produced in the lumen of the vesicles as the direct conversion of DA by the dopamine-β-hydroxylase enzyme. The small fraction of molecules that leaks from the vesicles rapidly undergoes catabolic conversion, starting with the oxidative deamination by monoamine oxidase (MAO) enzymes on the outer mitochondrial membrane to generate the aldehydic intermediates DOPAL, DOPEGAL, and 5-HIAL.
Conversely, post mortem studies on isolated synaptic vesicles from PD patients’ brains revealed a decreased activity of about 90% of the vesicular monoamine transporter type 2 (VMAT2), which is responsible for vesicles loading with dopamine and serotonin [19, 20], thus resulting in increasing concentration of cytosolic monoamines. Coherently, in vivo mouse models with reduced expression and activity of VMAT2 by genetic knockdown or inhibition by the anti-hypertensive drug reserpine resemble many features of the pathophysiology of PD and the age-related degeneration of both SNpc and LC [21-24]. Interestingly, from the genetic point of view, some polymorphisms in the Vmat2 gene have been associated with PD [25] and Infantile-Parkinsonism Dystonia 2, an infantile-onset movement disorder with severe parkinsonism, mood disturbance, and autonomic instability due to DA and 5-HT dyshomeostasis [26].
Once accumulated in the cytosol, DA and NE are oxidized by MAO-A (in the SNpc, LC, and RVLM neurons) [27], while 5-HT is reported to be metabolized by MAO-B in the DRN serotoninergic neurons [28]. Despite both MAO-A and MAO-B isoforms are expressed in SNpc dopaminergic neurons, MAO-B is abundantly expressed in striatal astrocytes [27, 29]. Once released in the synaptic cleft, a consistent fraction of DA is rapidly re-uptaken by the dopamine transporter DAT of astrocytes, where it is rapidly catabolized by MAO-B. Of note, an increased expression and activity of MAOs have been detected along with aging and neuroinflammation [30-33]. Also, a correlation between PD cases and some variants of the Mao-B gene, encoding for a hyperactive enzyme, has been described [34-36]. Hence, the upregulation of this first catabolic step results in sustained aldehydes production, together with the other by-product hydrogen peroxide, which likely exacerbates oxidative stress.
Increasing levels of DOPAL, 5-HIAL, and DOPEGAL have been found in PD patients’ brains, where they were not promptly detoxified by the aldehyde dehydrogenases (ALDHs) into their less reactive carboxylic derivatives 3,4-dihydroxyphenylacetic acid (DOPAC) and 5-hydroxyindoleacetic acid (5-HIAA) and by the aldehyde reductases (ARs) into the ethanol form 3,4-dihydroxyphenylglycol (DOPEG) [14, 17, 37].
ALDH1A1 and ALDH2 are the most expressed enzymes responsible for dopamine catabolism in the dopaminergic neurons of SNpc. Of note, studies on post-mortem PD patients’ SNpc revealed a significantly decreased expression and activity of ALDH1A1, assessing both at transcriptional and proteomic levels [38-41]. Consistently, the mouse model double knock-out for Aldh1a1 and Aldh2 shows an age-dependent loss of midbrain dopaminergic neurons, impaired motor performance, and increased striatal levels of DOPAL, 5-HIAL, and 4-HNE [42]. Although none of the Aldh genes have been reported in GWAS on PD [7], some variants and haplotypes have been associated with an increased risk to develop PD in Asian and American populations [43-46]. Additionally, ALDH2*2 mutant enzyme with no enzymatic activity, has been associated with cognitive impairments and sleep disturbances in PD patients [47, 48].
Environmental factors seem to play an important role in the accumulation of biogenic aldehydes. Many chemical compounds are known to be strong inhibitors of the ALDH enzymes [49]. Among them, drugs like the anti-alcohol abuse Disulfiram and Daidzin have shown to increased DOPAL and 5-HIAL concentration [18, 50, 51]. Importantly, heavy-dose consumption of Disulfiram by alcohol abusers has been associated with parkinsonism and enhanced toxicity in basal ganglia, thus corroborating the link among ALDH inhibition, degeneration of nigrostriatal axis, and PD [52]. Also, exposure to pesticides like Benomyl and Dieldrin (both irreversible inhibitors of ALDH2) are associated with an increased risk of developing PD in epidemiological studies [53, 54]. Finally, the ALDHs/ARs-mediated reactions rely on NADP+/NADPH as a cofactor. Hence, any kind of insult that would affect the mitochondrial physiology (i.e., complex I inhibition by rotenone exposure) and homeostatic level of the NADP+/NADPH reservoir is likely to undermine the DOPAL, DOPEGAL, and 5-HT detoxification [55].
2.1.1. Dopal, Dopegal, and 5-HIAL Neurotoxicity
Physiological intermediates of catecholamines catabolism, DOPAL, and DOPEGAL have been described as an endogenous neurotoxin; this was first hypothesized by Blashko in 1952 and then further experimentally substantiated by Burke and others in the late 90s [14, 56]. DOPAL and DOPEGAL concentrations were estimated to be around 2-3µM in the SNpc and 1.4µM in the LC, respectively. However, concentrations higher than physiological (>6μM) have been described as a threshold to elicit cytotoxic effects in various cell lines [57].
The main DOPAL and DOPEGAL-associated toxicity described in both in vitro and in vivo models refer to mitochondrial dysfunction by triggering the mitochondrial permeability transition pore [58], caspase, and apoptosis activation by calcium-mediated processes. Also, the ability to produce radical oxygen species (ROS) leading to oxidative stress and glutathione depletion has been described due to the tendency of the catechol group to self-oxidise to quinones. More recently, the ability of DOPAL to decrease cell viability, trigger apoptosis, and mitochondrial dysfunction has been assessed in an astrocytic cellular model [59].
A unique case is the synergy between the catechol and the aldehyde moieties of DOPAL, which is thought to enhance its reactivity towards proteins, targeting the thiol group of cysteines and the amino group of lysines [60-62]. Indeed, several studies highlight the ability of DOPAL to covalently modify a broad spectrum of targets triggering detrimental consequences for neuronal homeostases, such as enzyme inhibition (i.e., tyrosine hydroxylase, TH) by modification of functional residues in the active site [63], protein cross-linking and aggregation [60, 64].
In this frame, the DOPAL-induced oligomerization of aSyn has been widely investigated, being aSyn aggregates majorly involved in PD pathogenesis. Indeed, the covalent modification of aSyn lysines (15 lysines out of 140 amino acids) by DOPAL triggers aSyn aggregation, leading to synapse dysfunction and neuronal vulnerability, as demonstrated in various in vitro and in vivo models [65-68]. Along the same line, DOPEGAL modification of Tau lysines was showed to trigger Tau aggregation in a cellular model and after injected in the LC of a mouse model [69]. Besides, both DOPAL and DOPEGAL had been demonstrated to activate the asparagine endopeptidase (AEP) in the SNpc and LC, respectively [69, 70]. AEP-mediated cleavage of aSyn at N103 and Tau at N368 triggered protein aggregation [69, 71], leading to aSyn and Tau pathology, progressive neurodegeneration, motor impairment, and cognitive dysfunction. Although DOPEGAL-induced Tau aggregation in the LC was primarily associated with Alzheimer’s disease (AD), the alterations of the catecholamine metabolic pathway observed in PD might lead to similar DOPEGAL-dependent neurotoxicity in PD as well.
Finally, while 5-HIAL neurotoxicity was poorly investigated in PD models, the ability of 5-HIAL to covalently modify and trigger aSyn oligomerization was assessed [72].
2.2. Oxidative Stress and Lipid Peroxidation
Among the consequences of oxidative stress, the unregulated oxidation of polyunsaturated fatty acids (PUFAs) by free radicals represents a major issue in PD. The increasing levels of hydroxyl and superoxide radicals trigger the lipid peroxidation of PUFAs (i.e., arachidonic and linoleic acid) with further production of several reactive intermediates like 4-HNE, 4-ONE, MDA, and acrolein [73]. Being PUFAs the major component of membranous structures in all cells, the toxic action of lipid peroxidation products may lack specificity for neurons that are more affected in PD. Moreover, neurons in the SNpc are known to be particularly susceptible to oxidative stress due to the considerable energy demand that challenges the mitochondrial integrity, the lower antioxidant defences, and the catechol tendency to autoxidation [74]. Hence, the generation of lipid peroxidation products and the associated neurotoxicity are expected to be exacerbated in catecholaminergic neurons in PD.
Indeed, several studies on post-mortem PD patients’ brains pointed out the accumulation of 4-HNE-protein adducts in the nigrostriatal neurons and LBs [16, 75]. Similarly, rat models of PD injected with 6-OHDA displayed significantly increased MDA concentration and acrolein-bound proteins, specifically in the striatum [76, 77]. Also, the 4-HNE and MDA content in plasma and cerebrospinal fluid of PD patients was found to be significantly higher compared to healthy subjects, thus hinting at their quantification as potential biomarkers for PD [16, 78-80].
As for the monoamine-derived aldehydes, lipid peroxidation products are usually metabolized by ALDHs-mediated oxidation, reducted by ADHs, or conjugated with glutathione (GSH) [81]. However, in PD, ALDHs frequently display reduced expression and activity as described above, thus hindering the rapid 4-HNE and MDA detoxification. Coherently, a double knock-out mouse model lacking ALDH1A1 and glutathione peroxidase-1 (GPX1) displayed an age-related decrease in motor performance and accumulation of 4-HNE protein adducts [82]. Besides, concentrations of both 4-HNE and MDA up to 100µM were reported to strongly bind the catalytic site of ALDH1A1 and ALDH2 and to inhibit their activity both in vitro and in cellular models [64, 83, 84]. Hence, the aldehyde detoxification capacity of neurons is seriously compromised, leading to increasing levels of DOPAL and other biogenic aldehydes in a loop that exacerbates toxicity.
2.2.1. Neurotoxicity of Lipid Peroxidation Products
While the physiological concentration of lipid peroxidation products like 4-HNE varies between 0.1-3µM, it has been reported that under pathological conditions, it increases to 0.01-5mM [85]. As for other aldehydes, lipid peroxidation products have a very broad spectrum of reactivity, generating advanced lipoxidation end-products (ALEs) by covalent modification of proteins and relative structural and functional changes, protein aggregation, enzyme inhibition, and subcellular protein localization; DNA lipoxidative damage with potential mutagenic and carcinogenic effects and activation of signalling pathways (i.e., inflammatory and anti-oxidant responses); modification of amino-phospholipids and hindered membrane integrity [86]. Specifically, in the SNpc, 4-HNE has been shown to affect the dopaminergic pathway at multiple levels by modifying VMAT2 and preventing the neurotransmitter sorting into the vesicles by binding to the dopamine transporter (DAT), thus altering DA re-uptake and binding to the DA receptors D1 and D5 [80]. In addition, general modification of proteins by 4-HNE is associated with the accumulation of ubiquitinated proteins and impairment of the ubiquitin-proteasome system, activation of the apoptotic cascade, and further neurodegeneration [87, 88].
The thick arrows highlight the altered expression and/or activity of key enzymes due to ageing (magenta), genetic variants (yellow), and exposure to xenobiotics (green).
Importantly, also lipid peroxidation products are known to covalently modify aSyn and trigger its aggregation in various in vitro, cellular, and in vivo models. This has been described for acrolein [76], 4-HNE, and 4-ONE [89-94]. Of note, 4-HNE-induced aSyn aggregates display a curved proto-fibrillar morphology with β-sheet structures. Although small amounts of 4-HNE have been reported to trigger aSyn oligomerization, the principal modification site by 4-HNE is the sole histidine residue on aSyn sequence (H50), suggesting a different reactivity and aggregation kinetics compared to DOPAL modification, which occurs at several aSyn lysines.
2.3. Hyperglycemia and Type 2 Diabetes Mellitus
Type 2 Diabetes Mellitus (T2DM) is characterized by chronic hyperglycaemia. It affects approximately 9% of the global population over 40 years old, with increased incidence in Asian populations [95]. Under pathological conditions, the increased glucose concentration in the bloodstream and tissues promotes the formation of the glycolytic sub-product MGO and the accumulation of the advanced glycating end-products (AGEs) leading to diabetic nephropathy, retinopathy, and neuropathy. Of note, the correlation between T2DM and PD is gaining attention, as patients with T2DM have an estimated 40% increased risk to develop PD [96-98].
During glucose metabolism, MGO is produced by the spontaneous degradation of glyceraldehyde-3-phosphate and dihydroxyacetone-phosphate. MGO is then primarily metabolized to D-lactate by a two-steps reaction involving the Glyoxase 1 (Glo-1) and Glyoxase 2 (Glo-2) enzymes and GSH molecules [99]. However, the observed age-dependent decreased Glo-1 expression and depletion of GSH reservoir correlate with the progressive accumulation of MGO and AGEs [100-102].
In PD, the local accumulation of MGO may be further amplified by the high energy demand of the dopaminergic neurons of SNpc, which leads to an enhanced rate of glycolysis and mismatched glyoxalase system capacity [103]. Additionally, aberrant levels of the toxic aldehydes DOPAL and 4-HNE have been reported to modify and inhibit the glyceraldehyde-3-phosphate dehydrogenase (GAPDH) covalently, thus synergistically promoting the glyceraldehyde-3-phosphate conversion to MGO [104, 105].
In addition to the Glo-1/2 pathway, a detoxifying mechanism implies a direct conversion of MGO to pyruvate by the ALDHs [106]. It follows that the reported impaired ALDH efficiency in PD hinders this compensatory mechanism from buffering MGO. Indeed, a positive correlation between diabetic complications and ALDH2 polymorphisms has been reported, particularly in Asian populations [107, 108]. Furthermore, MGO itself has been demonstrated to inhibit ALDHs at concentrations associated with pathology [106].
2.3.1. Methylglyoxal Neurotoxicity
MGO physiological concentration in the plasma is about 150nM. However, in T2DM, it is known to increase up to 2-6 fold [109]. MGO aldehyde moiety is highly reactive towards cysteines, arginines, and lysines of proteins in the blood and the extra-cellular matrix, leading to AGEs accumulation. Also, MGO was showed to diffuse in cells and tissue, permeating the plasma membrane [110], where it is known to alter the cellular proteome, trigger mutagenic and apoptotic processes by nucleotide glycation (mainly deoxyguanosine), and affect membrane bilayer integrity by lipid glycation [99].
Increased amounts of AGEs in the brains of patients with synucleinopathies has been widely recognized, specifically in SNpc and LC of PD subjects [15, 16]. Once again, aSyn appears to be the preferential target of MGO covalent modification. Indeed, the MGO-induced aSyn aggregation and altered clearance were proved in cellular systems as well as in vivo mouse and fly models, together with the impairment of the autophagy-lysosomal pathway and general neurotoxic outcomes [111].
In (Fig. 2), the potential pathological mechanisms that generate increasing concentrations of biogenic aldehydes in the degenerating neurons in PD are represented, together with the mechanisms of aldehyde-associated neurotoxicity.
Fig. (2).

Cellular pathways lead to (a) biogenic aldehydes build-up and (b) aldehyde-associated neurotoxicity in neurons of SNpc, LC, and DRN in PD. (A higher resolution / colour version of this figure is available in the electronic copy of the article).
3. CLINICAL AND PRE-CLINICAL DRUGS WITH ALDEHYDE SCAVENGING PROPERTY AS THERAPEUTIC STRATEGY IN PARKINSON’S DISEASE
Based on the evidence reported above and on the complex network of intersecting pathways described (Fig. 2), what emerges is the need for effective and specific interventions to counteract the heavy burden of reactive aldehydes in the brain. Besides targeting the single steps in the metabolic processes, the use of small molecules as scavengers to remove the excess of biogenic aldehydes appears of interest.
Here, we will focus on drugs that have been investigated for the treatment of PD and other diseases but have also been found, both at the clinical and pre-clinical stage, to possess secondary aldehyde scavenging properties (Table 1). Among them, the anti-glycating agents, such as metformin, aminoguanidine, and pyridoxamine, were investigated and used as therapeutic strategies in diabetes and related conditions. Drugs like hydralazine, ambroxol, and rasagiline that are used in the therapy for hypertensive disorders, bronchopulmonary diseases, and idiopathic PD, respectively, showed secondary aldehyde scavenging activity through the primary amino group in their chemical structure. Similarly, the food supplements thiamine and carnosine were reported to possess aldehyde scavenging properties.
Table 1.
Chemical structures of clinical and pre-clinical drugs and food supplements with aldehyde scavenging property.
| Drug | Chemical Structure |
|---|---|
| Anti-glycating agents | |
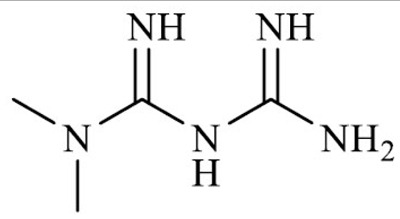
| Metformin (N, N-dimethylbiguanidine) |
|
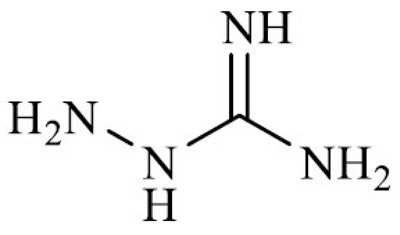
| Aminoguanidine (2-aminoguanidine) |
|
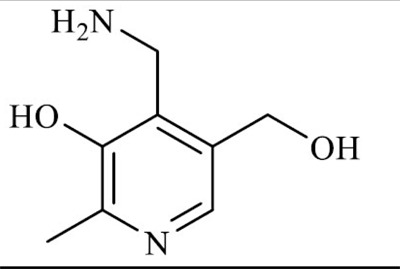
| Pyridoxamine (4-(amidomethyl)-5-(hydroxymethyl)-2-methylpyridin-3-ol) |
|
| Drugs with secondary aldehyde scavenging activity | |
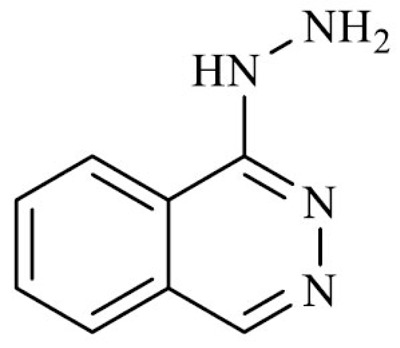
| Hydralazine (phthalazine-1-ylhydrazine) |
|
| Ambroxol (4-[(2-amino-3,5-dibromophenyl) methylamino] cyclohean-1-ol) |
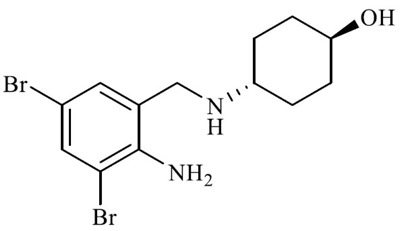
|
| Rasagiline ((1R)-N-prop-2-ynyl-2,3. dihydro-1H-inden-1-amine) |
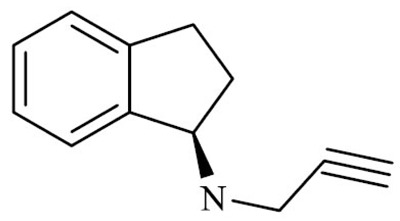
|
| Aminoidan (1-indanamine) |
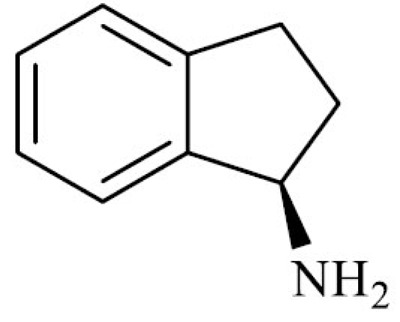
|
| Food supplements with aldehyde scavenging properties | |
| Thiamine (2-[3-[(4-amino-2-methylpyrimidin-5-yl) methyl]-4-methyl-1,3-thiazol-3-ium-5-yl] ethanol) |
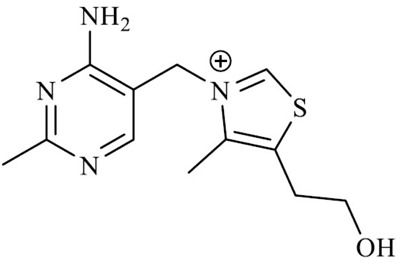
|
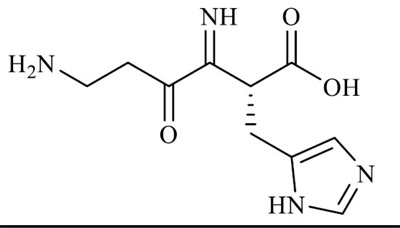
| Carnosine ((2S) -2-(3-aminopropanoylamino) -3-(1H-imidazol-5-yl) propanoic acid) |
|
The rationale is based on the capacity of primary amines (i.e., aminoguanidine, pyridoxamine, ambroxol, aminoidan, thiamine, carnosine) and hydrazine groups (i.e., metformin, hydralazine) to covalently react with the aldehyde moiety and form unreactive and hydrophilic reaction products (with high clearance). Several chemical reactions and multistep processes are usually involved, mainly through Schiff base formation with the generation of immine (Fig. 3a) and hydrazine (Fig. 3b) derivatives, following Michael addiction, condensation, or substitution.
Fig. (3).

Schiff-base reactions between the aldehyde and the scavenging groups in the proposed drugs. (a) The chemical reaction between the aldehyde and the primary amine generates an immine. (b) The chemical reaction between the aldehyde and the hydrazide produces hydrazine.
Drugs of this kind are of great interest, being that the pharmacodynamics, pharmacokinetics, and safety profile (see Table 2 for details) have already been assessed, and these could be included, as combination therapy, in regimens of PD patients.
Table 2.
Pharmacokinetics and safety data of clinical and pre-clinical drugs with aldehyde scavenging property.
| Drug | Dosage* and Administration | Bioavailability | BBB Passage | Metabolism | Half-Life | Elimination Route | Potential Side-Effects | FDA Approval | Refs. |
|---|---|---|---|---|---|---|---|---|---|
| Metformin | 500-1700 mg/dayin oral form | 40–60% | Yes | None,no hepatic metabolism | About 5 hours | Urine (90%), unchanged | GI irritation, lactic acidosis, hypoglycemia, decreased vitamin B12 absorption | In 1995, for T2DM therapy | [113, 114, 116, 118, 183] |
| Aminoguanidine | 300-600 mg/dayin oral form | 5%(rapid renal elimination) | Yes | Metabolized in the liver in 2-acetylaminoguanidine (4%) | About 4 hours | Urine, mostly unchanged | iNOS inhibition, altered immunological responses, vascular and respiratory deficits | Withdrawn in Phase III clinical trial (1999) | [126, 127, 131, 184, 185] |
| Pyridoxamine | 100-600 mg/dayin oral form | 65% | Yes | Partially metabolized to active forms pyridoxal phosphate and pyridoxamine phosphate | 1.5 hours(in rats) | Urine as 4-pyridoxic acid | Well tolerated | In Phase III clinical trial in T2DM patients | [134, 186-189] |
| Hydralazine | 20-40 mgby IV injection emergently; 40-200 mg/day in oral form | 16–35%Depending on patient’s acetylator phenotype | Yes | Significantly metabolized in the liver by acetylation;Phthalazine and pyruvic acid hydrazone (inactive metabolite) are main metabolites | 3-7 hours, depending on patient’s acetylator phenotype | Urine (65-90%); faeces (<10%); both unchanged and as metabolites | Headache, palpitations, GI irritation, vomiting, lupus-like syndrome, hypotension | In 1997, to treat hypertensive disorders | [146, 149, 190, 191] |
| Ambroxol | 525 - 1260 mg/day in oral form (PD clinical trials doses) | About 80% | Yes | Metabolized in the liver to di-bromoanthranilic acid and glucuronide forms | About 10 hours | Urine (85%) mostly as metabolites | Skin rashes, GI complaints, low risk of anaphylaxis | In 1979, used for respiratory diseases; in Phase II clinical trial for PD | [160, 165, 168, 169, 192, 193] |
| Rasagiline | 0.5-1 mg/dayin oral form; 1,25 mg/day by transdermal patch | About 36% | Yes | Hepatic, 1-aminoindan (major metabolite), 3-hydroxy-N-propargyl-1-aminoindan,and 3-hydroxy-1-aminoindan | 1.5–3.5 hours | Primarily via urine (62%) and secondarily via feces (7%), only 1% were excreted unchanged | Headache, dizziness, insomnia | In 2006, for PD therapy | [194-196] |
| Thiamine | 5-300 mg/dayby injection or in oral form | 3.7-5.3% | Yes | Acid metabolites (2-methyl-4-amino-5-pyrimidine carboxylic acid, 4-methyl-thiazole-5-acetic acid, and thiamine acetic acid | 3-5 hours | Urine, but applies only for the unphosphorylated and the acidic metabolite forms | Well tolerated, some allergic reactions and skin irritation | Approved as food supplement | [137, 175, 176, 181, 197-200] |
| Carnosine | 50-2000 mg/dayin oral form | Low, instable due to serum carnosinase | Yes | Entry in the blood stream mostly unchanged, serum carnosinase hydrolyzes it into β-alanine and L-histidine | Specific half-life of carnosine has not been established in humans | Urine, mostly as β-alanine and L-histidine, hydrolyzed also in the kidney | No side effects reported | No | [201-203] |
*The reported dosages were noted by clinical practice or by relevant clinical trials.
3.1. Anti-glycating Agents
3.1.1. Metformin
Metformin hydrochloride (1,1-dimethylbiguanide) is a biguanidine molecule and anti-hyperglycemic agent derived from galegine, a natural product from the plant Galega officinalis.
Approved by FDA in 1995, metformin (known under the trade name Glucophage, among others) is still considered the first-line therapy for the treatment of T2DM [112]. Indeed, the most important benefits of metformin are associated with the regulation of glucose metabolism in diabetes-related conditions as it reduces hepatic gluconeogenesis, increases insulin sensitivity, and promotes peripheral glucose uptake [113].
At a molecular level, multiple mechanisms of action have been proposed, at times multifaceted and not fully clarified. The anti-hyperglycemic effect of metformin is supposed to be mediated by both AMP-activated protein kinase (AMPK)-dependent and AMPK-independent pathways. It is also known to affect both the mitochondrial and lysosomal physiology by inhibiting the complex I of the respiratory chain and the glycerophosphate dehydrogenase and by modulating the mTOR signalling pathway [114].
In addition to the modes of action mentioned above, metformin displays a therapeutic effect as an aldehyde scavenger through the guanidino group and the primary amine in its structure. Specifically, it has been demonstrated to act as an anti-glycating agent by lowering the levels of circulating dicarbonyls, covalently binding to MGO, and producing inactive imidazolinone compounds that are secreted in the urine [115, 116]. Hence, metformin not only actively regulates the glucose homeostasis in diabetic patients but also protects from associated complications by reducing systemic AGEs and MGO-protein adducts.
Interestingly, the therapeutic potential of metformin in age-related diseases is increasingly discussed. The rationale relies on the positive correlation reported between the incidence of diabetes and neurodegenerative diseases [96-98]. Secondly, metformin acts on molecular mechanisms that are usually involved in degenerative processes, i.e., signalling pathways related to glucose metabolism, oxidative stress, proteostasis, and inflammation [117]. These mechanisms are all relevant for PD, and coherently, cellular and animal models have been used to demonstrate the neuroprotective effects of metformin, as extensively reviewed in Paudel et al. [118]. Briefly, metformin has been shown to improve motor performance and ameliorate the cognitive functions of MPTP rodent models of PD, as well as reducing their dopaminergic neuron loss in the SNpc [119]. The latter seems to depend on the regulating effect of metformin on mitochondrial functions and on reducing the ROS production and oxidative stress. Furthermore, both the autophagy induction, which promotes the clearance of misfolded and aggregated proteins like aSyn, and the decreased phosphorylation at serine 129 (a pathological signature of aSyn), are also related to the metformin action on the mTOR-dependent pathway [120, 121].
Despite these promising indications from the pre-clinical phase, clinical studies of metformin administration to PD patients provided conflicting results. When used in a Taiwanese population cohort in combination with sulfonylurea, another anti-hyperglycaemic agent, metformin counteracted the increased risk of PD in T2DM patients treated with sulfonylurea alone (without providing robust evidence of a neuroprotective role of metformin per se) [122]. Conversely, other studies correlated the use of metformin with PD, dementia, and cognitive decline in T2DM patients in Taiwan and Australia [123, 124]. However, these discrepancies might derive from the differences in the study designs, populations involved, diagnostic criteria, dose, and duration of metformin treatments. More recently, a large longitudinal study (5528 patients in total, with a median follow-up of 5.2 years) on US elderly veterans (mean age 63.2±10.9 years) with T2DM demonstrated that long-term metformin administration (more than 4 years) was associated with significantly decreased risk (HR 0.19, 95% CI 0.12 to 0.31) of both PD and AD [125], opening again for future interests in unravelling the therapeutic relevance of metformin in neurodegenerative diseases.
3.1.2. Aminoguanidine
Analogous to metformin in structure, aminoguanidine hydrochloride is a synthetic drug with nucleophilic hydrazine (-NHNH2) and a guanidine (-NHC(=NH)NH2) group. Although it was first synthesized more than a century ago, the translational potential of aminoguanidine emerged in the 90’ when it was proposed as an investigational drug for the treatment of diabetic nephropathy and retinopathy under the trade name of Pimagedine.
Because of its molecular structure, aminoguanidine is an effective scavenger of α,β-dicarbonyl glycating agents, both in vitro and in vivo experimental models. The reaction between aminoguanidine and MGO generates non-toxic compounds and prevents the formation of AGEs, whose accumulation is frequently associated with secondary complications in diabetic patients [126, 127]. In addition, besides the preferential reactivity towards MGO, aminoguanidine was also shown to act as scavengers of aldehydes derived from lipid peroxidation, preventing the amino acid and protein modification by 4-HNE and MDA in vitro experiments [128, 129].
Of note, the therapeutic potential of aminoguanidine as an anti-glycating drug has also been investigated in neurodegenerative diseases. For instance, aminoguanidine administration to an AD mouse model with and age-dependent inhibition of β-amyloid glycation significantly reduced the cognitive deficits, Tau hyperphosphorylation, and loss of synaptic elements in the Tg2576 mouse model of AD [130]. More recently, the treatment with aminoguanidine rescued the MGO-induced aggregation and impaired clearance of aSyn in cellular models, as well as the motor impairment in aSyn-overexpressing Drosophila, thus suggesting the aminoguanidine therapeutic potential in PD [111].
Unfortunately, when tested in the ACTION I and ACTION II clinical trials to prevent the development of nephropathy in T1DM and T2DM patients, some safety concerns and apparent lack of efficacy led to the decision to discontinue the aminoguanidine Phase III trial in ‘98-99 [131]. Indeed, aminoguanidine was also proved to inhibit the inducible nitric oxide synthase (iNOS) and diamine oxidase (DAO), potentially leading to aberrant immunological responses due to altered NO signalling pathway and severe vascular and respiratory deficits due to histamine accumulation in the blood, respectively [126]. Also, the molecule was well-absorbed but rapidly cleared by glomerular filtration, resulting in a bioavailability of only 5% [132]. Hence, a high dosage of aminoguanidine might be necessary, with the off- targets and side effects coming along.
On this ground, the clinical use of aminoguanidine as an aldehyde scavenger will first require the development of aminoguanidine derivatives to improve its bioavailability, as well as prodrug formulations to be activated in the regions of interest, thus improving the therapeutic odds of aminoguanidine in both T2DM and PD.
3.1.3. Pyridoxamine
Pyridoxamine, a naturally occurring metabolite and a structural analog of vitamin B6 (pyridoxine), has been developed as a broadly active inhibitor in the early and late stages of AGEs formation. It is currently under phase III clinical trial to determine its efficacy in reducing the rate of progression of nephropathy in T2DM patients [133, 134].
Interestingly, the molecule has multiple beneficial effects that are carried out by two functional groups, the hydroxyl and the aminomethyl group [135]. Among the effects is the inhibition of AGEs and ALEs formation by scavenging reactive carbonyl species [134, 136, 137], the sequestration of catalytic metal ions [138], the blockage of oxidative degradation of Amadori intermediates derived from glycated proteins [139], ROS and free radicals scavenging [140].
Although less effective than other molecules (i.e., hydralazine, aminoguanidine, and carnosine) as a scavenger of biogenic aldehydes, pyridoxamine was showed to react with 4-HNE, MGO and more efficiently with MDA by producing an N-propenal–pyridoxamine adduct [129], as already described in previous in vitro studies [141] and a mouse model of diabetes [142]. Also, the neuroprotective action of pyridoxamine was explored in the rotenone-exposed mouse model of PD, where it reduced neuronal cell death associated with microglial AGE-albumin, the most abundant AGE product in the human PD brain [143]. However, to date, limited investigation of pyridoxamine beneficial role in PD therapy is conducted, although a high (2.3-5.6 mg/day) dietary intake of vitamin B6 (pyridoxine) has been associated with a decreased risk of PD [144]. Conversely, a low intake of vitamin B6 is positively correlated with PD development [145]. Here, the neuroprotection by pyridoxamine can be related to its antioxidant properties and the regulation of homocysteine levels in the blood, but also its activity as aldehyde scavengers should be considered and further unraveled.
3.2. Drugs with Secondary Aldehyde Scavenging Activity
3.2.1. Hydralazine
Hydralazine hydrochloride (1-Phthalazinylhydrazine) is a hydrazine derivative, approved by the FDA under the trade name Apresoline (among others) for the treatment of essential hypertension and as an adjunct therapy to lower blood pressure in case of heart failure [146].
Hydralazine acts primarily as a vasodilator on smooth muscle cells of arterial vessels and arterioles, reducing peripheral resistance and lowering blood pressure. Although the molecular mechanisms of action are not completely understood yet, studies on animal models suggested that hydralazine might act by modulating the effect of purine-like compounds released from sympathetic nerve endings and by altering intracellular calcium release, thus interfering with smooth muscle cell calcium influx [147-149].
Since its discovery, additional studies have pointed out that hydralazine can act as a scavenger of biogenic aldehydes through its nucleophilic hydrazine moiety, which binds to free and protein-bound aldehydes to form stable hydrazones [129, 150, 151]. Its scavenging activity was shown both in cellular and animal models to be aimed mainly towards lipid peroxidation products, i.e., acrolein, MDA, and 4-HNE, exerting cytoprotective effects from free aldehydes and aldehyde-protein adducts [150, 152-154]. In addition, hydralazine was shown to be effective in reducing the formation of AGEs in plasma and kidney in a mouse model of T1DM [155].
Interestingly, the therapeutic potential of hydralazine for PD was studied in different toxin-based animal and cell models. Ambaw et al. demonstrated that hydralazine acted as an effective scavenger of acrolein generated by lipid peroxidation, in turn, induced by injection of 6-OHDA and rotenone in rat striatum. Furthermore, it also alleviated the aggregation of aSyn, neuronal cell death, and motor deficits [76]. Guo et al. documented that hydralazine confers protection in dopaminergic neurons in a MPTP model of PD, alleviating oxidative stress, and activating the NRF2 pathway [156, 157]. However, reported side effects of hydralazine should be considered in the case of a purported prolonged therapy in PD patients, like immunological reaction and lupus-like syndrome [158]. Also, hydralazine has been shown to act as a weak MAO inhibitor; hence it needs to be used carefully in patients concomitantly taking other MAO inhibitors to avoid a synergistic action and severe blood pressure decrease [159].
3.2.2. Ambroxol
Ambroxol hydrochloride (trans-4-((2-Amino-3,5-dibromobenzyl) amine) cyclohexanol) belongs to the class of organic compounds known as phenylethylamines. It is an active and synthetic derivative of vasicine, a quinazoline alkaloid extracted from the plant Adhatoda vasica.
In clinical practice, it is frequently used as an expectorant and mucolytic agent to treat bronchial asthma and chronic bronchitis [160]. Indeed, the main molecular mechanisms ascribed to ambroxol are related to mucokinetic properties, mucociliary activity, stimulation of surfactant production by type II pneumocytes, anti-inflammatory, and antioxidative actions, and the local anesthetic effect [160].
Besides, ambroxol exhibited protective action against free radical species and lipid peroxidation products, i.e., MDA [161-163]. Although the role of ambroxol as a direct scavenger of biogenic aldehydes has not been tested in vitro and in vivo yet, the data showing decreased lipid peroxidation and MDA levels are in line with this hypothesis. Action due to the presence of a primary amine in its structure allowed a Schiff-base reaction with reactive aldehydes.
Recently, ambroxol has gained importance as a potential therapy to slow down the neurodegeneration in PD and PD dementia (PDD), particularly in patients carrying mutations of lysosomal enzyme β-Glucocerebrosidase (GCase; gene name GBA1). First, the use of ambroxol in cell culture and animal models of PD resulted in reduced aSyn aggregation levels. Second, it acted as a pharmacological chaperone to increase the activity of GCase [164-167]. From this evidence and due to the excellent safety profile of this drug, two clinical trials were conducted (Clinical.gov: NCT02914366, NCT02941822) in PD patients with and without GBA1 mutations, confirming the ambroxol as a promising therapeutic approach for PD [168, 169].
3.2.3. Rasagiline
Rasagiline (N-propargyl-1(R)-aminoidan) is a selective and irreversible inhibitor of MAO-B and has been used in the therapy of PD since its EMA and FDA approval in 2005-2006.
The rationale is to elevate the dopamine levels by preventing its deamination by MAO in the first catabolic step, thus sustaining the activity of dopaminergic neurons and improving the motor symptoms.
Aside from the MAO activity inhibition, extensive literature now supports a more general neuroprotective action of rasagiline in several experimental models of PD [170]. The drug has been demonstrated to prevent neuron death through many molecular mechanisms, i.e., by acting as an antioxidant molecule, by inducing the gene expression of neurotrophic factors, antioxidant and anti-apoptotic enzymes, and by regulating mitochondrial physiology.
Moreover, rasagiline has been shown to interfere with aSyn aggregation through at least three mechanisms. First, rasagiline and its major metabolite R-1-aminoidan (produced by cytochrome P450-1A2 in the liver) were shown to interact with the N- and C-terminal regions of aSyn, forming a less-prone-to-aggregation loop structure [171]. Second, by blocking the formation of the monoamine-derived aldehydes DOPAL and 5-HIAL, rasagiline administration prevents the aldehyde-induced aSyn modification and aggregation [70, 71]. Finally, both rasagiline and R-1-aminoidan appeared to act as DOPAL scavenger through a Schiff-base reaction between the aldehyde and the amino group in the drugs’ structure, thus preventing aSyn aggregation and reducing DOPAL-induced toxicity in PC-12 cells [172].
Despite the many beneficial effects ascribed to rasagiline therapy in PD, the drug demonstrated disease modification at the dose of 1 mg but not at 2 mg, possibly because of inadequate study design and patients’ stratification [173, 174].
3.3. Food Supplements with Aldehyde Scavenging Properties
3.3.1. Thiamine
Thiamine hydrochloride, also known as vitamin B1, is an essential cofactor in several metabolic processes, such as glucose metabolism and cellular energy handling. This water-soluble vitamin is not endogenously produced in the human body; therefore, it must be taken with the diet. It is mostly found in leafy green vegetables, eggs, grain, and meats [175]. Hence, thiamine pharmaceutical supplementation is primarily used to treat and prevent its deficiency or related disorders, including Beriberi and Wernicke–Korsakoff syndromes.
In addition to its metabolic cofactor activity, thiamine was reported to exert a protective role against lipid peroxidation products in rat liver and kidney [176]. In vitro experiments also proved the thiamine pyrophosphate scavenging activity for AGEs [137], further confirmed in endothelial cellular models exposed to high glucose concentrations [177] and rodent models of diabetes [178]. Importantly, thiamine was shown to reduce circulating glucose levels when administered to hyperglycaemic patients [179]. Hence, these preliminary data and the presence of a primary amine in the molecular structure suggest a promising action of thiamine as an aldehyde scavenger, whose characterization should be extended to PD models.
In the last years, several studies pointed out a possible interplay between thiamine levels and PD [180, 181], where low plasma concentration of vitamin B1 was associated with mild cognitive decline in PD patients [182].
3.3.2. Carnosine
Carnosine is an endogenous histidyl dipeptide, consisting of a β-alanine bound to histidine. It is abundantly synthesized by the Carnosine Synthase 1 (CRNS1 or ATPGD1) in skeletal muscles and other tissues such as the brain, heart, spleen, and kidney [204], but it is also available as an oral drug and food supplement.
Endogenous carnosine displays a variety of defensive effects by exhibiting pH buffering capacity, metal chelating activity, and antioxidant properties. More interestingly, carnosine has been proved to act as an aldehyde scavenger, as demonstrated by several in vitro and in vivo studies [201, 202].
Carnosine has proven to prevent MGO protein modification and AGEs formation through different mechanisms, i.e., by a trans-glycating effect and by removing the glucosyl-adduct from proteins [205, 206]; directly quenching reactive carbonyl species (RCS) [129]; by inhibiting the metal- catalyzed oxidative conversion of glycated proteins to AGEs [207]. Also, carnosine is known to inhibit ALEs formation by reacting with α,β-unsaturated aldehydes, and forming covalent adducts that are excreted in the urine [208, 209]. Chemically, the detoxification occurs by a multistep reaction where both the amino group of β-alanine and the imidazole ring act synergistically to form the stable hemiacetal derivative [129]. For example, its quenching activity towards 4-HNE was showed both in vitro [129] and in vivo [210], demonstrating that dietary supplementation of carnosine could attenuate the development of dyslipidemia, hypertension, and carbonyl stress in rodent models of obesity and atherosclerosis [209, 211].
Of note, Nelson et al. recently demonstrated that DOPAL and DOPEGAL could be scavenged by carnosine, demonstrating the therapeutic potential of carnosine in diseases associated with catecholamine-related toxicity [212].
Interestingly, carnosine pre-treatment appeared to prevent neurodegeneration and apoptosis activation in 6-OHDA-injected rats [213]; to exert antioxidative and anti-inflammatory protection in the striatum of MPTP-treated mice [214]; to restore the normal antioxidant activity and physiological DA and 5-HT monoamine metabolism in MPTP-exposed rats [215]. Also, the group of Genter provided evidence that daily intranasal administration of carnosine had a positive impact on mitochondrial function and a protective role in preventing the progression of PD symptoms, motor deficits, and aSyn aggregation in the Thy-1 aSyn genetic mouse model [216, 217].
To date, one clinical trial investigated the efficacy of carnosine (1.5 g/day) in PD patients in combination with L- Dopa therapy [218], where a significantly increased efficacy of the L-DOPA treatment and improvements in clinical symptomatology were reported. Also, initiating in 2017, another PD clinical trial aimed to investigate the beneficial effects of exercise training and carnosine dietary supplementation on cognitive, motor, and metabolic functions have been reported (NCT03330470).
4. ALDEHYDE SCAVENGING THERAPY IN PARKINSON’S DISEASE: GENERAL CONSIDERATIONS
Since PD has multi-systemic and multifactorial pathology, the idea of simultaneously targeting multiple affected processes could be a promising therapeutic strategy [219]. On this basis, the previously mentioned drugs support this concept since, in addition to their aldehyde scavenging activity, they display multiple modes of action, targeting various molecular mechanisms associated with PD.
Molecules like metformin and ambroxol would scavenge toxic aldehydes in the brain and also activate protein clearance and lysosomal activity to avoid the accumulation of protein aggregates. Hydralazine, metformin, and aminoguanidine could prevent the burden of AGEs and ALEs derived by diabetic conditions or oxidative stress generated by long-term use of L-DOPA and other antiparkinsonian medications. Rasagiline would decrease DA catabolism in dopaminergic neurons and buffer any toxic aldehyde in the cellular environment. Food supplements like thiamine, carnosine, and pyridoxamine would respond to the metabolic imbalance in certain PD patients as well as scavenge excessive biogenic aldehydes. Hence, the administration of aldehyde scavengers in conjunction with the approved symptomatic therapies might assist in developing effective strategies, possibly towards disease-modifying actions.
These approaches would indeed require a careful evaluation of the clinical outcomes following long-term use. Dose and duration adjustment, the accurate analysis of potential side effects, and the metabolic fate of the aldehyde-scavenger reaction products should be examined in future experimental research. Also, while planning new clinical trials, two important factors should not be underestimated: the timing for starting the treatment and the selection of defined patient cohorts for whom the aldehyde scavenging therapy would prove most beneficial.
Usually, once the patient’s clinical picture presents a definite PD diagnosis, the neurodegeneration process would have already irreversibly expanded, making efforts at targeting pathogenic mechanisms unlikely to produce any radical changes to the course of the disease [220]. Thus, intervention in the early prodromal phases appears critical to achieving substantial disease modification. Furthermore, identifying small subgroups with a homogeneous pathophysiological basis would be necessary to accurately assess the positive outcomes from the proposed therapeutic approach [219, 220]. Therefore, it is necessary to define precise criteria for patients stratification to identify those subjects in the early stages of PD who have a higher vulnerability to excessive biogenic aldehyde yield.
According to the literature reviewed in this article, a promising strategy would comprise analysing genetic variations that were described to affect the activity of enzymes involved in the monoamines metabolism and the detoxification of aldehydes. Of note, the single nucleotide polymorphisms (SNPs) listed in Table (3) have also been associated, in epidemiological studies, with an increased risk to develop PD.
Table 3.
List of SNPs in genes encoding enzymes involved in monoamines metabolism and aldehyde degradation that have been associated with increased risk to develop PD.
| Gene | Genetic Variations |
| Vmat2 | rs363371 and rs363324 [25]; rs1392638187 [26] |
| Mao-b | rs1137070 and rs1799836 [36]; rs1799836 [35] |
| Aldh1a1 | rs3764435 [46] |
| Aldh2 | Haplotype rs737280, rs968529, rs16941667, rs16941669, rs9971942 [43] haplotype rs4767944, rs441, rs671 [44]; rs671 [45, 47, 48, 222]. |
As a first step toward patients stratification, the analysis of these variants can be introduced in the genetic panels currently used for PD, thus facilitating the early identification of patients at risk for aldehyde-related pathology. Here, the first candidates for genetic screening of these SNPs could be those who display early signs of prodromal symptoms of PD, like REM Behaviour Disturbances (RBD). Indeed, dysfunction in the serotoninergic system contributes heavily to both motor and non-motor symptoms of PD [221], and the accumulation of serotonin-derived aldehyde supposedly contributes to sleep disturbances. For instance, a recent study involving 83 idiopathic PD patients in Taiwan highlighted the increased difficulty in sleep maintenance in patients carrying the A allele in the rs671 SNPs of the Aldh2 gene, which encodes for an enzyme with significantly reduced enzyme activity [48]. It is worth mentioning that the rs671 genetic variant is highly frequent in Asian populations, where the genetic interchange has been limited compared to those from
Europe and America. Moreover, to our knowledge, the work by Lin et al. is the first study associated with sleep disturbances with impaired aldehyde metabolism. Further comprehensive investigations on RBD patients would accordingly be required, assessing larger cohorts reflective of populations with diverse genetic backgrounds. This approach should enhance the genetic stratification of patients to test the efficacy of an aldehyde scavenging-based therapy.
In addition to genetic screening, the development and assessment of specific biomarkers of biogenic aldehyde build-up would reinforce the criteria for identifying the best candidates. The updated list of cerebrospinal fluid (CSF) and blood biomarkers includes read-outs of well-established pathological elements, i.e., the presence of aggregated proteins like aSyn, Tau, and amyloid-beta, the activity of lysosomal enzymes, and neurofilament light chain levels [223]. However, other studies investigated the diagnostic potential of elevated aldehyde concentrations in biological fluids. For instance, in a study conducted in West Bengal in 2009, a significantly increased MDA concentration in the plasma of PD patients was estimated compared to the healthy subjects (7.48 ± 1.55 nmol/ml vs. 5.10 ± 1.26 nmol/ml) [78]. More recently, the analysis of various biomarkers of immuno-inflammation and ROS in PD patients from a Brazilian cohort revealed the quantification of MDA in the peripheral blood as the most sensitive and specific biomarker for PD. It accordingly described increasing MDA levels in early PD (201.9 ± 18.9 µM/mg of proteins) and late PD (214.1 ± 29.7 µM/mg of proteins) subjects compared to healthy controls (168.1 ± 18.3 µM/mg of proteins) [79].
Furthermore, there have been investigations on the read-outs of an altered dopamine metabolic pathway. In a study, the analysis of dopamine and its metabolites in the CSF of patients with PD and parkinsonian multiple system atrophy (MSA-P) demonstrated a correlation between the pathology and the decreased concentration of DOPAC (1.60 ± 0.12 pmol/ml in healthy control vs. 0.94 ± 0.09 pmol/ml in PD patients and 0.89 ± 0.08 pmol/ml in MSA patients) [224]. Together with a decreased DOPAC/DA ratio and increased 5-S-Cysteinyl-dopamine/DOPAC ratio, this data suggested elevated oxidative stress and impaired ALDH activity in the SNpc dopaminergic neurons of parkinsonian patients, potentially leading to the accumulation of toxic aldehydes. Coherently, another metabolomic study assessed a statistically significant increase in DOPAL levels (p-value 0.000177) in PD patients’ plasma compared to healthy subjects [225]. Finally, the analysis of plasma samples from PD patients from a Chinese cohort compared to healthy controls identified aSyn species specifically modified by dopamine and DOPAL on lysine residues that could only derive from diseased dopaminergic neurons in PD patients [226].
Although very promising, the plasma/CSF concentration of these biogenic aldehydes would require more comprehensive studies to confirm their robustness as a biomarker for PD. First, a large-scale collection of samples from various patient cohorts should be analysed to define thresholds that identify prospective or already established PD patients. Second, RBD patients and subjects with prodromal PD signs should be enrolled to perform longitudinal post-hoc studies and assess whether the measured aldehyde levels are consistent with the following diagnosis of PD. Accordingly, this approach may outline values to identify the heterogenous clinical phenotypes and varying disease progressions [227] to adjust the therapeutic approach further. Finally, as an additional level of complexity, the plasma and CFS aldehyde profiling ought to consider possible inconsistency due to concurrent use of vitamins and food supplements, prescribed medications (i.e., Antabuse drugs, anti-glycating agents, L-DOPA), and exposure to toxins and pesticides that are known to interfere with the aldehyde metabolic pathways.
Taken together, the implementation of genetic panels with variants on Vmat2, Mao-b, Aldh1a1, and Aldh2 genes and the definition of biomarkers of aldehydes and aldehyde-adducts in the CSF or peripheral blood would represent powerful diagnostic and prognostic tools. On those bases, introducing the aldehyde scavenging therapy in subjects at the earliest stages of PD may help to prevent the toxic accumulation of biogenic aldehydes and exacerbate concomitant pathological mechanisms, thus acting as neuroprotective agents and delaying the progression of the disease.
CONCLUSION
The development of successful therapeutic interventions to modify PD progression still presents a great challenge given the complexity of the pathology and the broad spectrum of clinical manifestations.
At a molecular level, several pathological mechanisms contribute to the progressive dopaminergic neuron loss, among which the excessive accumulation of biogenic aldehydes in the brain appears to play an important role. On this basis, introducing an aldehyde scavenging therapy combined with other antiparkinsonian medications would contribute to elaborating disease-modifying strategies for PD.
Accordingly, this work described a series of drugs that may prove useful because of their secondary aldehyde scavenging properties, assessed in pre-clinical studies. Ideally, it would eventually be possible to predict which aldehyde would be more prone to accumulate in a given patient and which scavenger would exploit the best neuroprotective action as a sort of personalized medicine.
Of course, a deep analysis of the pharmacology and bioavailability of the proposed drugs in the brain, as well as the potential side effects from associated long-term use, ought to be conducted. However, a drug repurposing operation for these molecules, some of which are already off-patent, would encounter several legal, regulatory, and economic barriers. Although repositioning an already approved medication should entail fewer risks, lower costs, and shorter timelines for approval, the incentives to assess a new dosage and toxicology according to the new target and to run new clinical trials are still probably insufficient [228-230].
Moreover, to unequivocally prove or disprove the efficacy of aldehyde scavenging therapy in PD, the design of clinical trials in selected groups based on precise criteria for patients’ stratification will be necessary. Accordingly, in the first instance, it is proposed that there be an upgrade of the genetic screening panels with variants on genes involved in aldehyde metabolic pathways. Second, subjects with prodromal symptoms of PD, i.e., RBD patients, should be tested for blood biomarkers of biogenic aldehyde accumulation. Finally, it would be critical to perform studies on a larger scale, enrolling subjects representative of diverse genetic backgrounds and clinical histories to match the broad spectrum of onset, symptoms, and pathology of PD.
ACKNOWLEDGEMENTS
Declared none.
CONSENT FOR PUBLICATION
Not applicable.
FUNDING
A.M. and L.B. receive research support from the Ministry of Education University and Research (MIUR) Grant ARS01_01081 and PRIN 2017LYTE9M. M.S. receives an unrestricted grant from Lundbeck. A.A. receives research support from Chiesi Pharmaceuticals, Lundbeck, Horizon 2020 – PD_Pal Grant 825785, MIUR Grant ARS01_01081 and Cariparo Foundation.
CONFLICT OF INTEREST
The authors declare no conflict of interest, financial or otherwise.
REFERENCES
- 1.Dawson T.M., Dawson V.L. Neuroprotective and neurorestorative strategies for Parkinson’s disease. Nat. Neurosci. 2002;5(Suppl.):1058–1061. doi: 10.1038/nn941. [DOI] [PubMed] [Google Scholar]
- 2.Tysnes O.B., Storstein A. Epidemiology of Parkinson’s disease. J. Neural Transm. (Vienna) 2017;124(8):901–905. doi: 10.1007/s00702-017-1686-y. [DOI] [PubMed] [Google Scholar]
- 3.Pringsheim T., Jette N., Frolkis A., Steeves T.D.L. The prevalence of Parkinson’s disease: A systematic review and meta-analysis. Mov. Disord. 2014;29(13):1583–1590. doi: 10.1002/mds.25945. [DOI] [PubMed] [Google Scholar]
- 4.Sulzer D., Surmeier D.J. Neuronal vulnerability, pathogenesis, and Parkinson’s disease. Mov. Disord. 2013;28(1):41–50. doi: 10.1002/mds.25095. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Zarow C., Lyness S.A., Mortimer J.A., Chui H.C. Neuronal loss is greater in the locus coeruleus than nucleus basalis and substantia nigra in Alzheimer and Parkinson diseases. Arch. Neurol. 2003;60(3):337–341. doi: 10.1001/archneur.60.3.337. [DOI] [PubMed] [Google Scholar]
- 6.Kalia L.V., Lang A.E. Parkinson’s disease. Lancet. 2015;386(9996):896–912. doi: 10.1016/S0140-6736(14)61393-3. [DOI] [PubMed] [Google Scholar]
- 7.Nalls M.A., Blauwendraat C., Vallerga C.L., Heilbron K., Bandres-Ciga S., Chang D., Tan M., Kia D.A., Noyce A.J., Xue A., Bras J., Young E., von Coelln R., Simón-Sánchez J., Schulte C., Sharma M., Krohn L., Pihlstrøm L., Siitonen A., Iwaki H., Leonard H., Faghri F., Gibbs J.R., Hernandez D.G., Scholz S.W., Botia J.A., Martinez M., Corvol J.C., Lesage S., Jankovic J., Shulman L.M., Sutherland M., Tienari P., Majamaa K., Toft M., Andreassen O.A., Bangale T., Brice A., Yang J., Gan-Or Z., Gasser T., Heutink P., Shulman J.M., Wood N.W., Hinds D.A., Hardy J.A., Morris H.R., Gratten J., Visscher P.M., Graham R.R., Singleton A.B., Adarmes-Gómez A.D., Aguilar M., Aitkulova A., Akhmetzhanov V., Alcalay R.N., Alvarez I., Alvarez V., Barrero F.J., Bergareche Yarza J.A., Bernal-Bernal I., Billingsley K., Blazquez M., Bonilla-Toribio M., Botía J.A., Boungiorno M.T., Brockmann K., Bubb V., Buiza-Rueda D., Cámara A., Carrillo F., Carrión-Claro M., Cerdan D., Chelban V., Clarimón J., Clarke C., Compta Y., Cookson M.R., Craig D.W., Danjou F., Diez-Fairen M., Dols-Icardo O., Duarte J., Duran R., Escamilla-Sevilla F., Escott-Price V., Ezquerra M., Feliz C., Fernández M., Fernández-Santiago R., Finkbeiner S., Foltynie T., Garcia C., García-Ruiz P., Gomez H.M.J., Gómez-Garre P., González M.M., Gonzalez-Aramburu I., Guelfi S., Guerreiro R., Hardy J., Hassin-Baer S., Hoenicka J., Holmans P., Houlden H., Infante J., Jesús S., Jimenez-Escrig A., Kaishybayeva G., Kaiyrzhanov R., Karimova A., Kinghorn K.J., Koks S., Kulisevsky J., Labrador-Espinosa M.A., Leonard H.L., Lewis P., Lopez-Sendon J.L., Lovering R., Lubbe S., Lungu C., Macias D., Manzoni C., Marín J., Marinus J., Marti M.J., Martínez Torres I., Martínez-Castrillo J.C., Mata M., Mencacci N.E., Méndez-del-Barrio C., Middlehurst B., Mínguez A., Mir P., Mok K.Y., Muñoz E., Narendra D., Ojo O.O., Okubadejo N.U., Pagola A.G., Pastor P., Perez E.F., Periñán-Tocino T., Pihlstrom L., Plun-Favreau H., Quinn J. 23andMe Research Team; System Genomics of Parkinson’s Disease Consortium; International Parkinson’s Disease Genomics Consortium. Identification of novel risk loci, causal insights, and heritable risk for Parkinson’s disease: a meta-analysis of genome-wide association studies. Lancet Neurol. 2019;18(12):1091–1102. doi: 10.1016/S1474-4422(19)30320-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Deng H., Wang P., Jankovic J. The genetics of Parkinson disease. Ageing Res. Rev. 2018;42:72–85. doi: 10.1016/j.arr.2017.12.007. [DOI] [PubMed] [Google Scholar]
- 9.Schneider S. A., Alcalay R. N. Neuropathology of genetic synucleinopathies with parkinsonism: review of the literature. Mov. Disord. 2017;32(11):1504–1523. doi: 10.1002/mds.27193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Johansen K.K., Torp S.H., Farrer M.J., Gustavsson E.K., Aasly J.O. Parkinson’s Disease with no lewy body pathology due to a homozygous exon deletion in Parkin. Case Rep. Neurol. Med. 2018;2018:6838965. doi: 10.1155/2018/6838965. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Carvey P.M., Punati A., Newman M.B. Progressive dopamine neuron loss in Parkinson’s disease: the multiple hit hypothesis. Cell Transplant. 2006;15(3):239–250. doi: 10.3727/000000006783981990. [DOI] [PubMed] [Google Scholar]
- 12.Sulzer D. Multiple hit hypotheses for dopamine neuron loss in Parkinson’s disease. Trends Neurosci. 2007;30(5):244–250. doi: 10.1016/j.tins.2007.03.009. [DOI] [PubMed] [Google Scholar]
- 13.George S., Brundin P. Solving the conundrum of insoluble protein aggregates. Lancet Neurol. 2017;16(4):258–259. doi: 10.1016/S1474-4422(17)30045-5. [DOI] [PubMed] [Google Scholar]
- 14.Burke W.J., Li S.W., Chung H.D., Ruggiero D.A., Kristal B.S., Johnson E.M., Lampe P., Kumar V.B., Franko M., Williams E.A., Zahm D.S. Neurotoxicity of MAO metabolites of catecholamine neurotransmitters: role in neurodegenerative diseases. Neurotoxicology. 2004;25(1-2):101–115. doi: 10.1016/S0161-813X(03)00090-1. [DOI] [PubMed] [Google Scholar]
- 15.Castellani R., Smith M.A., Richey P.L., Perry G. Glycoxidation and oxidative stress in Parkinson disease and diffuse Lewy body disease. Brain Res. 1996;737(1-2):195–200. doi: 10.1016/0006-8993(96)00729-9. [DOI] [PubMed] [Google Scholar]
- 16.Dalfó E., Portero-Otín M., Ayala V., Martínez A., Pamplona R., Ferrer I. Evidence of oxidative stress in the neocortex in incidental Lewy body disease. J. Neuropathol. Exp. Neurol. 2005;64(9):816–830. doi: 10.1097/01.jnen.0000179050.54522.5a. [DOI] [PubMed] [Google Scholar]
- 17.Goldstein D.S., Sullivan P., Holmes C., Kopin I.J., Basile M.J., Mash D.C. Catechols in post-mortem brain of patients with Parkinson disease. Eur. J. Neurol. 2011;18(5):703–710. doi: 10.1111/j.1468-1331.2010.03246.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rooke N., Li D.J., Li J., Keung W.M. The mitochondrial monoamine oxidase-aldehyde dehydrogenase pathway: A potential site of action of daidzin. J. Med. Chem. 2000;43(22):4169–4179. doi: 10.1021/jm990614i. [DOI] [PubMed] [Google Scholar]
- 19.Pifl C., Rajput A., Reither H., Blesa J., Cavada C., Obeso J.A., Rajput A.H., Hornykiewicz O. Is Parkinson’s disease a vesicular dopamine storage disorder? Evidence from a study in isolated synaptic vesicles of human and nonhuman primate striatum. J. Neurosci. 2014;34(24):8210–8218. doi: 10.1523/JNEUROSCI.5456-13.2014. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Goldstein D.S., Sullivan P., Holmes C., Miller G.W., Alter S., Strong R., Mash D.C., Kopin I.J., Sharabi Y. Determinants of buildup of the toxic dopamine metabolite DOPAL in Parkinson’s disease. J. Neurochem. 2013;126(5):591–603. doi: 10.1111/jnc.12345. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21.Caudle W.M., Richardson J.R., Wang M.Z., Taylor T.N., Guillot T.S., McCormack A.L., Colebrooke R.E., Di Monte D.A., Emson P.C., Miller G.W. Reduced vesicular storage of dopamine causes progressive nigrostriatal neurodegeneration. J. Neurosci. 2007;27(30):8138–8148. doi: 10.1523/JNEUROSCI.0319-07.2007. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Fernandes V.S., Santos J.R., Leão A.H.F.F., Medeiros A.M., Melo T.G., Izídio G.S., Cabral A., Ribeiro R.A., Abílio V.C., Ribeiro A.M., Silva R.H. Repeated treatment with a low dose of reserpine as a progressive model of Parkinson’s disease. Behav. Brain Res. 2012;231(1):154–163. doi: 10.1016/j.bbr.2012.03.008. [DOI] [PubMed] [Google Scholar]
- 23.Taylor T. N., Alter S. P., Wang M., Goldstein D. S., Miller G. W. Reduced vesicular storage of catecholamines causes progressive degeneration in the Locus Ceruleus. Neuropharmacology. 2014;76(PART A):97–105. doi: 10.1016/j.neuropharm.2013.08.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Leão A.H.F.F., Sarmento-Silva A.J., Santos J.R., Ribeiro A.M., Silva R.H. Molecular, neurochemical, and behavioral hallmarks of reserpine as a model for parkinson’s disease: new perspectives to a long-standing model. Brain Pathol. 2015;25(4):377–390. doi: 10.1111/bpa.12253. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Brighina L., Riva C., Bertola F., Saracchi E., Fermi S., Goldwurm S., Ferrarese C. Analysis of vesicular monoamine transporter 2 polymorphisms in Parkinson’s disease. Neurobiol. Aging. 2013;34(6):1712.e9–1712.e13. doi: 10.1016/j.neurobiolaging.2012.12.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Rilstone J.J., Alkhater R.A., Minassian B.A. Brain dopamine-serotonin vesicular transport disease and its treatment. N. Engl. J. Med. 2013;368(6):543–550. doi: 10.1056/NEJMoa1207281. [DOI] [PubMed] [Google Scholar]
- 27.Graves S.M., Xie Z., Stout K.A., Zampese E., Burbulla L.F., Shih J.C., Kondapalli J., Patriarchi T., Tian L., Brichta L., Greengard P., Krainc D., Schumacker P.T., Surmeier D.J. Dopamine metabolism by a monoamine oxidase mitochondrial shuttle activates the electron transport chain. Nat. Neurosci. 2020;23(1):15–20. doi: 10.1038/s41593-019-0556-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Bortolato M., Chen K., Shih J.C. The degradation of serotonin: role of MAO. Handbook of Behavioral Neuroscience, 2010, 21, pp. 203-218. [Google Scholar]
- 29.Agid Y., Javoy F., Youdim M.B. Monoamine oxidase and aldehyde dehydrogenase activity in the striatum of rats after 6-hydroxydopamine lesion of the nigrostriatal pathway. Br. J. Pharmacol. 1973;48(1):175–178. doi: 10.1111/j.1476-5381.1973.tb08238.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Nicotra A., Pierucci F., Parvez H., Senatori O. Monoamine oxidase expression during development and aging. Neurotoxicology. 2004;25(1-2):155–165. doi: 10.1016/S0161-813X(03)00095-0. [DOI] [PubMed] [Google Scholar]
- 31.Damier P., Kastner A., Agid Y., Hirsch E.C. Does monoamine oxidase type B play a role in dopaminergic nerve cell death in Parkinson’s disease? Neurology. 1996;46(5):1262–1269. doi: 10.1212/WNL.46.5.1262. [DOI] [PubMed] [Google Scholar]
- 32.Chamoli M., Chinta S.J., Andersen J.K. An inducible MAO-B mouse model of Parkinson’s disease: A tool towards better understanding basic disease mechanisms and developing novel therapeutics. J. Neural Transm. (Vienna) 2018;125(11):1651–1658. doi: 10.1007/s00702-018-1887-z. [DOI] [PubMed] [Google Scholar]
- 33.Oreland L., Gottfries C.G. Brain and brain monoamine oxidase in aging and in dementia of Alzheimer’s type. Prog. Neuropsychopharmacol. Biol. Psychiatry. 1986;10(3-5):533–540. doi: 10.1016/0278-5846(86)90023-0. [DOI] [PubMed] [Google Scholar]
- 34.Kurth J.H., Kurth M.C., Poduslo S.E., Schwankhaus J.D. Association of a monoamine oxidase B allele with Parkinson’s disease. Ann. Neurol. 1993;33(4):368–372. doi: 10.1002/ana.410330406. [DOI] [PubMed] [Google Scholar]
- 35.Sampaio T.F., Dos Santos E.U.D., de Lima G.D.C., Dos Anjos R.S.G., da Silva R.C., Asano A.G.C., Asano N.M.J., Crovella S., de Souza P.R.E. MAO-B and COMT genetic variations associated with levodopa treatment response in patients with parkinson’s disease. J. Clin. Pharmacol. 2018;58(7):920–926. doi: 10.1002/jcph.1096. [DOI] [PubMed] [Google Scholar]
- 36.Sun Y-X., Wang X-H., Xu A-H., Zhao J-H. Functional polymorphisms of the MAO gene with Parkinson disease susceptibility: A meta-analysis. J. Neurol. Sci. 2014;345(1-2):97–105. doi: 10.1016/j.jns.2014.07.016. [DOI] [PubMed] [Google Scholar]
- 37.Brichta L., Greengard P. Molecular determinants of selective dopaminergic vulnerability in Parkinson’s disease: an update. Front. Neuroanat. 2014;8:152. doi: 10.3389/fnana.2014.00152. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Galter D., Buervenich S., Carmine A., Anvret M., Olson L. ALDH1 mRNA: presence in human dopamine neurons and decreases in substantia nigra in Parkinson’s disease and in the ventral tegmental area in schizophrenia. Neurobiol. Dis. 2003;14(3):637–647. doi: 10.1016/j.nbd.2003.09.001. [DOI] [PubMed] [Google Scholar]
- 39.Grünblatt E., Riederer P. Aldehyde dehydrogenase (ALDH) in Alzheimer’s and Parkinson’s disease. J. Neural Transm. (Vienna) 2016;123(2):83–90. doi: 10.1007/s00702-014-1320-1. [DOI] [PubMed] [Google Scholar]
- 40.Mandel S.A., Fishman T., Youdim M.B.H. Gene and protein signatures in sporadic Parkinson’s disease and a novel genetic model of PD. Parkinsonism Relat. Disord. 2007;13(Suppl. 3):S242–S247. doi: 10.1016/S1353-8020(08)70009-9. [DOI] [PubMed] [Google Scholar]
- 41.Werner C.J., Heyny-von Haussen R., Mall G., Wolf S. Proteome analysis of human substantia nigra in Parkinson’s disease. Proteome Sci. 2008;6(1):8. doi: 10.1186/1477-5956-6-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Wey M.C-Y., Fernandez E., Martinez P.A., Sullivan P., Goldstein D.S., Strong R. Neurodegeneration and motor dysfunction in mice lacking cytosolic and mitochondrial aldehyde dehydrogenases: implications for Parkinson’s disease. PLoS One. 2012;7(2):e31522. doi: 10.1371/journal.pone.0031522. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Fitzmaurice A.G., Rhodes S.L., Cockburn M., Ritz B., Bronstein J.M. Aldehyde dehydrogenase variation enhances effect of pesticides associated with Parkinson disease. Neurology. 2014;82(5):419–426. doi: 10.1212/WNL.0000000000000083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Zhang X., Ye Y-L., Wang Y-N., Liu F-F., Liu X-X., Hu B-L., Zou M., Zhu J-H. Aldehyde dehydrogenase 2 genetic variations may increase susceptibility to Parkinson’s disease in Han Chinese population. Neurobiol. Aging. 2015;36(9):2660.e9–2660.e13. doi: 10.1016/j.neurobiolaging.2015.06.001. [DOI] [PubMed] [Google Scholar]
- 45.Zhao C.C., Cai H.B., Wang H., Pan S.Y. Role of ADH2 and ALDH2 gene polymorphisms in the development of Parkinson’s disease in a Chinese population. Genet. Mol. Res. 2016;15(3) doi: 10.4238/gmr.15038606. [DOI] [PubMed] [Google Scholar]
- 46.Salas-Leal A.C., Sandoval-Carrillo A., Romero-Gutiérrez E., Castellanos-Juárez F.X., Méndez-Hernández E.M., La Llave-León O., Quiñones-Canales G., Arias-Carrión O., Salas-Pacheco J.M. rs3764435 Associated With Parkinson’s disease in mexican mestizos: case-control study reveals protective effects against disease development and cognitive impairment. Front. Neurol. 2019;10:1066. doi: 10.3389/fneur.2019.01066. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Yu R-L., Tan C-H., Lu Y-C., Wu R-M. Aldehyde dehydrogenase 2 is associated with cognitive functions in patients with Parkinson’s disease. Sci. Rep. 2016;6(1):30424. doi: 10.1038/srep30424. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Lin C.Y., Yu R.L., Wu R.M., Tan C.H. Effect of ALDH2 on sleep disturbances in patients with parkinson’s disease. Sci. Rep. 2019;9(1):18950. doi: 10.1038/s41598-019-55427-w. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Koppaka V., Thompson D.C., Chen Y., Ellermann M., Nicolaou K.C., Juvonen R.O., Petersen D., Deitrich R.A., Hurley T.D., Vasiliou V. Aldehyde dehydrogenase inhibitors: a comprehensive review of the pharmacology, mechanism of action, substrate specificity, and clinical application. Pharmacol. Rev. 2012;64(3):520–539. doi: 10.1124/pr.111.005538. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Helander A., Tottmar O. Effects of ethanol, acetaldehyde and disulfiram on the metabolism of biogenic aldehydes in isolated human blood cells and platelets. Biochem. Pharmacol. 1987;36(22):3981–3985. doi: 10.1016/0006-2952(87)90467-9. [DOI] [PubMed] [Google Scholar]
- 51.Legros H., Dingeval M-G., Janin F., Costentin J., Bonnet J-J. Toxicity of a treatment associating dopamine and disulfiram for catecholaminergic neuroblastoma SH-SY5Y cells: relationships with 3,4-dihydroxyphenylacetaldehyde formation. Neurotoxicology. 2004;25(3):365–375. doi: 10.1016/S0161-813X(03)00148-7. [DOI] [PubMed] [Google Scholar]
- 52.Laplane D., Attal N., Sauron B., De Billy A., Dubois B. Lesions of basal ganglia due to disulfiram neurotoxicity. J. Neurol. Neurosurg. Psychiatry. 1992;55(10):925–929. doi: 10.1136/jnnp.55.10.925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Corrigan F.M., Murray L., Wyatt C.L., Shore R.F. Diorthosubstituted polychlorinated biphenyls in caudate nucleus in Parkinson’s disease. Exp. Neurol. 1998;150(2):339–342. doi: 10.1006/exnr.1998.6776. [DOI] [PubMed] [Google Scholar]
- 54.Fitzmaurice A.G., Rhodes S.L., Lulla A., Murphy N.P., Lam H.A., O’Donnell K.C., Barnhill L., Casida J.E., Cockburn M., Sagasti A., Stahl M.C., Maidment N.T., Ritz B., Bronstein J.M. Aldehyde dehydrogenase inhibition as a pathogenic mechanism in Parkinson disease. Proc. Natl. Acad. Sci. USA. 2013;110(2):636–641. doi: 10.1073/pnas.1220399110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Lamensdorf I., Eisenhofer G., Harvey-White J., Hayakawa Y., Kirk K., Kopin I.J. Metabolic stress in PC12 cells induces the formation of the endogenous dopaminergic neurotoxin, 3,4-dihydroxyphenylacetaldehyde. J. Neurosci. Res. 2000;60(4):552–558. doi: 10.1002/(SICI)1097-4547(20000515)60:4<552::AID-JNR14>3.0.CO;2-U. [DOI] [PubMed] [Google Scholar]
- 56.Mattammal M.B., Haring J.H., Chung H.D., Raghu G., Strong R. An endogenous dopaminergic neurotoxin: implication for Parkinson’s disease. Neurodegeneration. 1995;4(3):271–281. doi: 10.1016/1055-8330(95)90016-0. [DOI] [PubMed] [Google Scholar]
- 57.Marchitti S.A., Deitrich R.A., Vasiliou V. Neurotoxicity and metabolism of the catecholamine-derived 3,4-dihydroxyphenylacetaldehyde and 3,4-dihydroxyphenylglycolaldehyde: the role of aldehyde dehydrogenase. Pharmacol. Rev. 2007;59(2):125–150. doi: 10.1124/pr.59.2.1. [DOI] [PubMed] [Google Scholar]
- 58.Kristal B.S., Conway A.D., Brown A.M., Jain J.C., Ulluci P.A., Li S.W., Burke W.J. Selective dopaminergic vulnerability: 3,4-dihydroxyphenylacetaldehyde targets mitochondria. Free Radic. Biol. Med. 2001;30(8):924–931. doi: 10.1016/S0891-5849(01)00484-1. [DOI] [PubMed] [Google Scholar]
- 59.Bagnoli E., Diviney T., FitzGerald U. Dysregulation of astrocytic mitochondrial function following exposure to a dopamine metabolite: implications for Parkinson’s Disease. Eur. J. Neurosci. 2020;53(9):2960–2972. doi: 10.1111/ejn.14764. [DOI] [PubMed] [Google Scholar]
- 60.Rees J.N., Florang V.R., Eckert L.L., Doorn J.A. Protein reactivity of 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite, is dependent on both the aldehyde and the catechol. Chem. Res. Toxicol. 2009;22(7):1256–1263. doi: 10.1021/tx9000557. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Anderson D.G., Florang V.R., Schamp J.H., Buettner G.R., Doorn J.A. Antioxidant-mediated modulation of protein reactivity for 3,4-Dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Chem. Res. Toxicol. 2016;29(7):1098–1107. doi: 10.1021/acs.chemrestox.5b00528. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Anderson D.G., Mariappan S.V.S., Buettner G.R., Doorn J.A. Oxidation of 3,4-dihydroxyphenylacetaldehyde, a toxic dopaminergic metabolite, to a semiquinone radical and an ortho-quinone. J. Biol. Chem. 2011;286(30):26978–26986. doi: 10.1074/jbc.M111.249532. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Mexas L.M., Florang V.R., Doorn J.A. Inhibition and covalent modification of tyrosine hydroxylase by 3,4-dihydroxyphenylacetaldehyde, a toxic dopamine metabolite. Neurotoxicology. 2011;32(4):471–477. doi: 10.1016/j.neuro.2011.03.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Rees J.N., Florang V.R., Anderson D.G., Doorn J.A. Lipid peroxidation products inhibit dopamine catabolism yielding aberrant levels of a reactive intermediate. Chem. Res. Toxicol. 2007;20(10):1536–1542. doi: 10.1021/tx700248y. [DOI] [PubMed] [Google Scholar]
- 65.Burke W.J., Kumar V.B., Pandey N., Panneton W.M., Gan Q., Franko M.W., O’Dell M., Li S.W., Pan Y., Chung H.D., Galvin J.E. Aggregation of alpha-synuclein by DOPAL, the monoamine oxidase metabolite of dopamine. Acta Neuropathol. 2008;115(2):193–203. doi: 10.1007/s00401-007-0303-9. [DOI] [PubMed] [Google Scholar]
- 66.Follmer C., Coelho-Cerqueira E., Yatabe-Franco D.Y., Araujo G.D.T., Pinheiro A.S., Domont G.B., Eliezer D. Oligomerization and membrane-binding properties of covalent adducts formed by the interaction of α-synuclein with the toxic dopamine metabolite 3,4-dihydroxyphenylacetaldehyde (dopal). J. Biol. Chem. 2015;290(46):27660–27679. doi: 10.1074/jbc.M115.686584. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Plotegher N., Berti G., Ferrari E., Tessari I., Zanetti M., Lunelli L., Greggio E., Bisaglia M., Veronesi M., Girotto S., Dalla S.M., Perego C., Casella L., Bubacco L. DOPAL derived alpha-synuclein oligomers impair synaptic vesicles physiological function. Sci. Rep. 2017;7(1):40699. doi: 10.1038/srep40699. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Liu G., Yu J., Ding J., Xie C., Sun L., Rudenko I., Zheng W., Sastry N., Luo J., Rudow G., Troncoso J.C., Cai H. Aldehyde dehydrogenase 1 defines and protects a nigrostriatal dopaminergic neuron subpopulation. J. Clin. Invest. 2014;124(7):3032–3046. doi: 10.1172/JCI72176. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Kang S.S., Liu X., Ahn E.H., Xiang J., Manfredsson F.P., Yang X., Luo H.R., Liles L.C., Weinshenker D., Ye K. Norepinephrine metabolite DOPEGAL activates AEP and pathological Tau aggregation in locus coeruleus. J. Clin. Invest. 2020;130(1):422–437. doi: 10.1172/JCI130513. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Kang S.S., Ahn E.H., Zhang Z., Liu X., Manfredsson F.P., Sandoval I.M., Dhakal S., Iuvone P.M., Cao X., Ye K. α-Synuclein stimulation of monoamine oxidase-B and legumain protease mediates the pathology of Parkinson’s disease. EMBO J. 2018;37(12):e201798878. doi: 10.15252/embj.201798878. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Zhang Z., Kang S.S., Liu X., Ahn E.H., Zhang Z., He L., Iuvone P.M., Duong D.M., Seyfried N.T., Benskey M.J., Manfredsson F.P., Jin L., Sun Y.E., Wang J.Z., Ye K. Asparagine endopeptidase cleaves α-synuclein and mediates pathologic activities in Parkinson’s disease. Nat. Struct. Mol. Biol. 2017;24(8):632–642. doi: 10.1038/nsmb.3433. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Jinsmaa Y., Cooney A., Sullivan P., Sharabi Y., Goldstein D.S. The serotonin aldehyde, 5-HIAL, oligomerizes alpha-synuclein. Neurosci. Lett. 2015;590:134–137. doi: 10.1016/j.neulet.2015.01.064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Fritz K. S., Petersen D. R. An overview of the chemistry and biology of reactive aldehydes. Free Rad. Biol. Med. 2013;59:85–91. doi: 10.1016/j.freeradbiomed.2012.06.025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Surmeier D.J., Obeso J.A., Halliday G.M. Selective neuronal vulnerability in Parkinson disease. Nat. Rev. Neurosci. 2017;18(2):101–113. doi: 10.1038/nrn.2016.178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Yoritaka A., Hattori N., Uchida K., Tanaka M., Stadtman E.R., Mizuno Y. Immunohistochemical detection of 4-hydroxynonenal protein adducts in Parkinson disease. Proc. Natl. Acad. Sci. USA. 1996;93(7):2696–2701. doi: 10.1073/pnas.93.7.2696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Ambaw A., Zheng L., Tambe M.A., Strathearn K.E., Acosta G., Hubers S.A., Liu F., Herr S.A., Tang J., Truong A., Walls E., Pond A., Rochet J.C., Shi R. Acrolein-mediated neuronal cell death and alpha-synuclein aggregation: Implications for Parkinson’s disease. Mol. Cell. Neurosci. 2018;88:70–82. doi: 10.1016/j.mcn.2018.01.006. [DOI] [PubMed] [Google Scholar]
- 77.Romuk E.B., Szczurek W., Oleś M., Gabrysiak A., Skowron M., Nowak P., Birkner E. The evaluation of the changes in enzymatic antioxidant reserves and lipid peroxidation in chosen parts of the brain in an animal model of Parkinson disease. Adv. Clin. Exp. Med. 2017;26(6):953–959. doi: 10.17219/acem/63999. [DOI] [PubMed] [Google Scholar]
- 78.Sanyal J., Bandyopadhyay S.K., Banerjee T.K., Mukherjee S.C., Chakraborty D.P., Ray B.C., Rao V.R. Plasma levels of lipid peroxides in patients with Parkinson’s disease. Eur. Rev. Med. Pharmacol. Sci. 2009;13(2):129–132. [PubMed] [Google Scholar]
- 79.de Farias C.C., Maes M., Bonifácio K.L., Bortolasci C.C., de Souza Nogueira A., Brinholi F.F., Matsumoto A.K., do Nascimento M.A., de Melo L.B., Nixdorf S.L., Lavado E.L., Moreira E.G., Barbosa D.S. Highly specific changes in antioxidant levels and lipid peroxidation in Parkinson’s disease and its progression: Disease and staging biomarkers and new drug targets. Neurosci. Lett. 2016;617:66–71. doi: 10.1016/j.neulet.2016.02.011. [DOI] [PubMed] [Google Scholar]
- 80.Romano A., Serviddio G., Calcagnini S., Villani R., Giudetti A. M., Cassano T., Gaetani S. Linking lipid peroxidation and neuropsychiatric disorders: focus on 4-Hydroxy-2-Nonenal. Free Rad. Biol. Med. 2017;111:281–293. doi: 10.1016/j.freeradbiomed.2016.12.046. [DOI] [PubMed] [Google Scholar]
- 81.Vasiliou V., Pappa A., Estey T. Role of human aldehyde dehydrogenases in endobiotic and xenobiotic metabolism. Drug Metab. Rev. 2004;36(2):279–299. doi: 10.1081/DMR-120034001. [DOI] [PubMed] [Google Scholar]
- 82.Bai X., Wey M.C-Y., Martinez P.A., Shi C., Fernandez E., Strong R. Neurochemical and motor changes in mice with combined mutations linked to Parkinson’s disease. Pathobiol. Aging Age Relat. Dis. 2017;7(1):1267855. doi: 10.1080/20010001.2017.1267855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Florang V.R., Rees J.N., Brogden N.K., Anderson D.G., Hurley T.D., Doorn J.A. Inhibition of the oxidative metabolism of 3,4-dihydroxyphenylacetaldehyde, a reactive intermediate of dopamine metabolism, by 4-hydroxy-2-nonenal. Neurotoxicology. 2007;28(1):76–82. doi: 10.1016/j.neuro.2006.07.018. [DOI] [PubMed] [Google Scholar]
- 84.Jinsmaa Y., Florang V.R., Rees J.N., Anderson D.G., Strack S., Doorn J.A. Products of oxidative stress inhibit aldehyde oxidation and reduction pathways in dopamine catabolism yielding elevated levels of a reactive intermediate. Chem. Res. Toxicol. 2009;22(5):835–841. doi: 10.1021/tx800405v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 85.Uchida K. Progress in Lipid Research. Elsevier Ltd; 2003. 4-Hydroxy-2-Nonenal: A product and mediator of oxidative stress. ; pp. 318–343. [DOI] [PubMed] [Google Scholar]
- 86.Pamplona R. Chemico-Biological Interactions. Vol. 192. Elsevier; 2011. Advanced lipoxidation end-products. pp. 14–20. [DOI] [PubMed] [Google Scholar]
- 87.Hyun D.H., Lee M.H., Halliwell B., Jenner P. Proteasomal dysfunction induced by 4-hydroxy-2,3-trans-nonenal, an end-product of lipid peroxidation: a mechanism contributing to neurodegeneration? J. Neurochem. 2002;83(2):360–370. doi: 10.1046/j.1471-4159.2002.01125.x. [DOI] [PubMed] [Google Scholar]
- 88.Okada K., Wangpoengtrakul C., Osawa T., Toyokuni S., Tanaka K., Uchida K. 4-Hydroxy-2-nonenal-mediated impairment of intracellular proteolysis during oxidative stress. Identification of proteasomes as target molecules. J. Biol. Chem. 1999;274(34):23787–23793. doi: 10.1074/jbc.274.34.23787. [DOI] [PubMed] [Google Scholar]
- 89.Almandoz-Gil L., Welander H., Ihse E., Khoonsari P.E., Musunuri S., Lendel C., Sigvardson J., Karlsson M., Ingelsson M., Kultima K., Bergström J. Low molar excess of 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote oligomerization of alpha-synuclein through different pathways. Free Radic. Biol. Med. 2017;110:421–431. doi: 10.1016/j.freeradbiomed.2017.07.004. [DOI] [PubMed] [Google Scholar]
- 90.Cai Y., Lendel C., Österlund L., Kasrayan A., Lannfelt L., Ingelsson M., Nikolajeff F., Karlsson M., Bergström J. Changes in secondary structure of α-synuclein during oligomerization induced by reactive aldehydes. Biochem. Biophys. Res. Commun. 2015;464(1):336–341. doi: 10.1016/j.bbrc.2015.06.154. [DOI] [PubMed] [Google Scholar]
- 91.Näsström T., Fagerqvist T., Barbu M., Karlsson M., Nikolajeff F., Kasrayan A., Ekberg M., Lannfelt L., Ingelsson M., Bergström J. The lipid peroxidation products 4-oxo-2-nonenal and 4-hydroxy-2-nonenal promote the formation of α-synuclein oligomers with distinct biochemical, morphological, and functional properties. Free Radic. Biol. Med. 2011;50(3):428–437. doi: 10.1016/j.freeradbiomed.2010.11.027. [DOI] [PubMed] [Google Scholar]
- 92.Sardar S.M., Villamil G.A.M., Öllinger K., Hallbeck M., Civitelli L. Lipid vesicles affect the aggregation of 4-hydroxy-2-nonenal-modified α-synuclein oligomers. Biochim. Biophys. Acta Mol. Basis Dis. 2018;1864(9 Pt B):3060–3068. doi: 10.1016/j.bbadis.2018.06.020. [DOI] [PubMed] [Google Scholar]
- 93.Xiang W., Schlachetzki J.C.M., Helling S., Bussmann J.C., Berlinghof M., Schäffer T.E., Marcus K., Winkler J., Klucken J., Becker C.M. Oxidative stress-induced posttranslational modifications of alpha-synuclein: Specific modification of alpha-synuclein by 4-hydroxy-2-nonenal increases dopaminergic toxicity. Mol. Cell. Neurosci. 2013;54:71–83. doi: 10.1016/j.mcn.2013.01.004. [DOI] [PubMed] [Google Scholar]
- 94.Zhang S., Eitan E., Wu T.Y., Mattson M.P. Intercellular transfer of pathogenic α-synuclein by extracellular vesicles is induced by the lipid peroxidation product 4-hydroxynonenal. Neurobiol. Aging. 2018;61:52–65. doi: 10.1016/j.neurobiolaging.2017.09.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Zheng Y., Ley S. H., Hu F. B. Global aetiology and epidemiology of Type 2 diabetes mellitus and its complications. Nat. Rev. Endocrinol. 2018;14(2):88–98. doi: 10.1038/nrendo.2017.151. [DOI] [PubMed] [Google Scholar]
- 96.Xu Q., Park Y., Huang X., Hollenbeck A., Blair A., Schatzkin A., Chen H. Diabetes and risk of Parkinson’s disease. Diabetes Care. 2011;34(4):910–915. doi: 10.2337/dc10-1922. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Schernhammer E., Hansen J., Rugbjerg K., Wermuth L., Ritz B. Diabetes and the risk of developing Parkinson’s disease in Denmark. Diabetes Care. 2011;34(5):1102–1108. doi: 10.2337/dc10-1333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Yue X., Li H., Yan H., Zhang P., Chang L., Li T. Medicine. United States: Lippincott Williams and Wilkins; 2016. Risk of Parkinson Disease in diabetes mellitus: an updated meta-analysis of population-based cohort studies. ; p. e3549. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Xue M., Rabbani N., Thornalley P. J. Glyoxalase in ageing. Semin. Cell Develop. Biol. 2011;22(3):293–301. doi: 10.1016/j.semcdb.2011.02.013. [DOI] [PubMed] [Google Scholar]
- 100.Morcos M., Du X., Pfisterer F., Hutter H., Sayed A.A.R., Thornalley P., Ahmed N., Baynes J., Thorpe S., Kukudov G., Schlotterer A., Bozorgmehr F., El Baki R.A., Stern D., Moehrlen F., Ibrahim Y., Oikonomou D., Hamann A., Becker C., Zeier M., Schwenger V., Miftari N., Humpert P., Hammes H.P., Buechler M., Bierhaus A., Brownlee M., Nawroth P.P. Glyoxalase-1 prevents mitochondrial protein modification and enhances lifespan in Caenorhabditis elegans. Aging Cell. 2008;7(2):260–269. doi: 10.1111/j.1474-9726.2008.00371.x. [DOI] [PubMed] [Google Scholar]
- 101.Chen T.S., Richie J.P., Jr., Lang C.A. The effect of aging on glutathione and cysteine levels in different regions of the mouse brain. Proc. Soc. Exp. Biol. Med. 1989;190(4):399–402. doi: 10.3181/00379727-190-42879. [DOI] [PubMed] [Google Scholar]
- 102.Kuhla B., Boeck K., Lüth H.J., Schmidt A., Weigle B., Schmitz M., Ogunlade V., Münch G., Arendt T. Age-dependent changes of glyoxalase I expression in human brain. Neurobiol. Aging. 2006;27(6):815–822. doi: 10.1016/j.neurobiolaging.2005.04.006. [DOI] [PubMed] [Google Scholar]
- 103.Allaman I., Bélanger M., Magistretti P.J. Methylglyoxal, the dark side of glycolysis. Front. Neurosci. 2015;9:23. doi: 10.3389/fnins.2015.00023. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Vanle B.C., Florang V.R., Murry D.J., Aguirre A.L., Doorn J.A. Inactivation of glyceraldehyde-3-phosphate dehydrogenase by the dopamine metabolite, 3,4-dihydroxyphenylacetaldehyde. Biochem. Biophys. Res. Commun. 2017;492(2):275–281. doi: 10.1016/j.bbrc.2017.08.067. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Ishii T., Tatsuda E., Kumazawa S., Nakayama T., Uchida K. Molecular basis of enzyme inactivation by an endogenous electrophile 4-hydroxy-2-nonenal: identification of modification sites in glyceraldehyde-3-phosphate dehydrogenase. Biochemistry. 2003;42(12):3474–3480. doi: 10.1021/bi027172o. [DOI] [PubMed] [Google Scholar]
- 106.Izaguirre G., Kikonyogo A., Pietruszko R. Methylglyoxal as substrate and inhibitor of human aldehyde dehydrogenase: comparison of kinetic properties among the three isozymes. Comp. Biochem. Physiol. B Biochem. Mol. Biol. 1998;119(4):747–754. doi: 10.1016/S0305-0491(98)00051-0. [DOI] [PubMed] [Google Scholar]
- 107.Morita K., Saruwatari J., Miyagawa H., Uchiyashiki Y., Oniki K., Sakata M., Kajiwara A., Yoshida A., Jinnouchi H., Nakagawa K. Association between aldehyde dehydrogenase 2 polymorphisms and the incidence of diabetic retinopathy among Japanese subjects with type 2 diabetes mellitus. Cardiovasc. Diabetol. 2013;12(1):132. doi: 10.1186/1475-2840-12-132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Idewaki Y., Iwase M., Fujii H., Ohkuma T., Ide H., Kaizu S., Jodai T., Kikuchi Y., Hirano A., Nakamura U., Kubo M., Kitazono T. Association of genetically determined aldehyde dehydrogenase 2 activity with diabetic complications in relation to alcohol consumption in Japanese patients with Type 2 Diabetes mellitus: The fukuoka diabetes registry. PLoS One. 2015;10(11):e0143288. doi: 10.1371/journal.pone.0143288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 109.Lu J., Randell E., Han Y., Adeli K., Krahn J., Meng Q.H. Increased plasma methylglyoxal level, inflammation, and vascular endothelial dysfunction in diabetic nephropathy. Clin. Biochem. 2011;44(4):307–311. doi: 10.1016/j.clinbiochem.2010.11.004. [DOI] [PubMed] [Google Scholar]
- 110.Rabbani N., Thornalley P. J. Dicarbonyl stress in cell and tissue dysfunction contributing to ageing and disease. Biochem. Biophys. Res.Commun. 2015;458(2):221–6. doi: 10.1016/j.bbrc.2015.01.140. [DOI] [PubMed] [Google Scholar]
- 111.Vicente M.H., Szegő É.M., Oliveira L.M.A., Breda C., Darendelioglu E., de Oliveira R.M., Ferreira D.G., Gomes M.A., Rott R., Oliveira M., Munari F., Enguita F.J., Simões T., Rodrigues E.F., Heinrich M., Martins I.C., Zamolo I., Riess O., Cordeiro C., Ponces-Freire A., Lashuel H.A., Santos N.C., Lopes L.V., Xiang W., Jovin T.M., Penque D., Engelender S., Zweckstetter M., Klucken J., Giorgini F., Quintas A., Outeiro T.F. Glycation potentiates α-synuclein-associated neurodegeneration in synucleinopathies. Brain. 2017;140(5):1399–1419. doi: 10.1093/brain/awx056. [DOI] [PubMed] [Google Scholar]
- 112.Jia Y., Lao Y., Zhu H., Li N., Leung S. W. Is Metformin still the most efficacious first-line oral hypoglycaemic drug in treating Type 2 Diabetes? a network meta-analysis of randomized controlled trials. Obesity Rev. 2019:1–12. doi: 10.1111/obr.12753. [DOI] [PubMed] [Google Scholar]
- 113.Viollet B., Guigas B., Sanz Garcia N., Leclerc J., Foretz M., Andreelli F. Cellular and molecular mechanisms of metformin: an overview. Clinical science. Inserm. 2012;122(6):253–70. doi: 10.1042/CS20110386. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Rena G., Hardie D. G., Pearson E. R. The mechanisms of action of metformin. Diabetologia. 2017;60(9):1577–1585. doi: 10.1007/s00125-017-4342-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Beisswenger P.J., Howell S.K., Touchette A.D., Lal S., Szwergold B.S. Metformin reduces systemic methylglyoxal levels in type 2 diabetes. Diabetes. 1999;48(1):198–202. doi: 10.2337/diabetes.48.1.198. [DOI] [PubMed] [Google Scholar]
- 116.Kinsky O.R., Hargraves T.L., Anumol T., Jacobsen N.E., Dai J., Snyder S.A., Monks T.J., Lau S.S. Metformin scavenges methylglyoxal to form a novel imidazolinone metabolite in humans. Chem. Res. Toxicol. 2016;29(2):227–234. doi: 10.1021/acs.chemrestox.5b00497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 117.Rotermund C., Machetanz G., Fitzgerald J.C. The therapeutic potential of metformin in neurodegenerative diseases. Front. Endocrinol. (Lausanne) 2018;9:400. doi: 10.3389/fendo.2018.00400. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Paudel Y. N., Angelopoulou E., Piperi C., Shaikh M. F., Othman I. Emerging neuroprotective effect of metformin in Parkinson’s Disease: A molecular crosstalk. Pharmacol. Res. 2020;152:104593. doi: 10.1016/j.phrs.2019.104593. [DOI] [PubMed] [Google Scholar]
- 119.Lu M., Su C., Qiao C., Bian Y., Ding J., Hu G. Metformin prevents dopaminergic neuron death in MPTP/P-induced mouse model of Parkinson’s disease via autophagy and mitochondrial ROS Clearance. Int. J. Neuropsychopharmacol. 2016;19(9):1–11. doi: 10.1093/ijnp/pyw047. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Pérez-Revuelta B.I., Hettich M.M., Ciociaro A., Rotermund C., Kahle P.J., Krauss S., Di Monte D.A. Metformin lowers Ser-129 phosphorylated α-synuclein levels via mTOR-dependent protein phosphatase 2A activation. Cell Death Dis. 2014;5(5):e1209–e1209. doi: 10.1038/cddis.2014.175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Katila N., Bhurtel S., Shadfar S., Srivastav S., Neupane S., Ojha U., Jeong G.S., Choi D.Y. Metformin lowers α-synuclein phosphorylation and upregulates neurotrophic factor in the MPTP mouse model of Parkinson’s disease. Neuropharmacology. 2017;125:396–407. doi: 10.1016/j.neuropharm.2017.08.015. [DOI] [PubMed] [Google Scholar]
- 122.Wahlqvist M.L., Lee M-S., Hsu C-C., Chuang S-Y., Lee J-T., Tsai H-N. Metformin-inclusive sulfonylurea therapy reduces the risk of Parkinson’s disease occurring with Type 2 diabetes in a Taiwanese population cohort. Parkinsonism Relat. Disord. 2012;18(6):753–758. doi: 10.1016/j.parkreldis.2012.03.010. [DOI] [PubMed] [Google Scholar]
- 123.Kuan Y.C., Huang K.W., Lin C.L., Hu C.J., Kao C.H. Effects of metformin exposure on neurodegenerative diseases in elderly patients with type 2 diabetes mellitus. Prog. Neuropsychopharmacol. Biol. Psychiatry. 2017;79(Pt B):77–83. doi: 10.1016/j.pnpbp.2017.06.002. [DOI] [PubMed] [Google Scholar]
- 124.Moore E.M., Mander A.G., Ames D., Kotowicz M.A., Carne R.P., Brodaty H., Woodward M., Boundy K., Ellis K.A., Bush A.I., Faux N.G., Martins R., Szoeke C., Rowe C., Watters D.A. AIBL Investigators. Increased risk of cognitive impairment in patients with diabetes is associated with metformin. Diabetes Care. 2013;36(10):2981–2987. doi: 10.2337/dc13-0229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Shi Q., Liu S., Fonseca V.A., Thethi T.K., Shi L. Effect of metformin on neurodegenerative disease among elderly adult US veterans with type 2 diabetes mellitus. BMJ Open. 2019;9(7):e024954. doi: 10.1136/bmjopen-2018-024954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Nilsson B. O. Biological effects of aminoguanidine: An Update. Inflamm.Res. 1999;48(10):509–515. doi: 10.1007/s000110050495. [DOI] [PubMed] [Google Scholar]
- 127.Thornalley P.J. Use of aminoguanidine (Pimagedine) to prevent the formation of advanced glycation endproducts. Arch. Biochem. Biophys. 2003;419(1):31–40. doi: 10.1016/j.abb.2003.08.013. [DOI] [PubMed] [Google Scholar]
- 128.Al-Abed Y., Bucala R. Efficient scavenging of fatty acid oxidation products by aminoguanidine. Chem. Res. Toxicol. 1997;10(8):875–879. doi: 10.1021/tx970035l. [DOI] [PubMed] [Google Scholar]
- 129.Colzani M., De Maddis D., Casali G., Carini M., Vistoli G., Aldini G. Reactivity, selectivity, and reaction mechanisms of aminoguanidine, hydralazine, pyridoxamine, and carnosine as sequestering agents of reactive carbonyl species: A Comparative Study. ChemMedChem. 2016;11(16):1778–1789. doi: 10.1002/cmdc.201500552. [DOI] [PubMed] [Google Scholar]
- 130.Li X.H., Du L.L., Cheng X.S., Jiang X., Zhang Y., Lv B.L., Liu R., Wang J.Z., Zhou X.W. Glycation exacerbates the neuronal toxicity of β-amyloid. Cell Death Dis. 2013;4(6):e673–e673. doi: 10.1038/cddis.2013.180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Freedman B.I., Wuerth J.P., Cartwright K., Bain R.P., Dippe S., Hershon K., Mooradian A.D., Spinowitz B.S. Design and baseline characteristics for the aminoguanidine clinical trial in overt type 2 diabetic nephropathy (ACTION II). Control. Clin. Trials. 1999;20(5):493–510. doi: 10.1016/S0197-2456(99)00024-0. [DOI] [PubMed] [Google Scholar]
- 132.Foote E.F., Look Z.M., Giles P., Keane W.F., Halstenson C.E. The pharmacokinetics of aminoguanidine in end-stage renal disease patients on hemodialysis. Am. J. Kidney Dis. 1995;25(3):420–425. doi: 10.1016/0272-6386(95)90103-5. [DOI] [PubMed] [Google Scholar]
- 133.Dwyer J.P., Greco B.A., Umanath K., Packham D., Fox J.W., Peterson R., Broome B.R., Greene L.E., Sika M., Lewis J.B. Pyridoxamine dihydrochloride in diabetic nephropathy (PIONEER-CSG-17): lessons learned from a pilot study. Nephron. 2015;129(1):22–28. doi: 10.1159/000369310. [DOI] [PubMed] [Google Scholar]
- 134.Voziyan P. A., Hudson B. G. Pyridoxamine as a multifunctional pharmaceutical: targeting pathogenic glycation and oxidative damage. Cell. Mol. Life Sci. 2005:1671–1681. doi: 10.1007/s00018-005-5082-7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Voziyan P.A., Hudson B.G. Pyridoxamine: The many virtues of a maillard reaction inhibitor. Ann. NY. Acad. Sci. 2005;1043:807–816. doi: 10.1196/annals.1333.093. [DOI] [PubMed] [Google Scholar]
- 136.Lee S.H., Matsunaga A., Oe T. Inhibition effect of pyridoxamine on lipid hydroperoxide-derived modifications to human serum albumin. PLoS One. 2018;13(4):e0196050. doi: 10.1371/journal.pone.0196050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Booth A.A., Khalifah R.G., Hudson B.G. Thiamine pyrophosphate and pyridoxamine inhibit the formation of antigenic advanced glycation end-products: comparison with aminoguanidine. Biochem. Biophys. Res. Commun. 1996;220(1):113–119. doi: 10.1006/bbrc.1996.0366. [DOI] [PubMed] [Google Scholar]
- 138.Adrover M., Vilanova B., Frau J., Muñoz F., Donoso J. The pyridoxamine action on Amadori compounds: A reexamination of its scavenging capacity and chelating effect. Bioorg. Med. Chem. 2008;16(10):5557–5569. doi: 10.1016/j.bmc.2008.04.002. [DOI] [PubMed] [Google Scholar]
- 139.Corzo-Martínez M., Moreno F.J., Olano A., Villamiel M. Role of pyridoxamine in the formation of the Amadori/Heyns compounds and aggregates during the glycation of β-lactoglobulin with galactose and tagatose. J. Agric. Food Chem. 2010;58(1):500–506. doi: 10.1021/jf902073t. [DOI] [PubMed] [Google Scholar]
- 140.Onorato J.M., Jenkins A.J., Thorpe S.R., Baynes J.W. Pyridoxamine, an inhibitor of advanced glycation reactions, also inhibits advanced lipoxidation reactions. Mechanism of action of pyridoxamine. J. Biol. Chem. 2000;275(28):21177–21184. doi: 10.1074/jbc.M003263200. [DOI] [PubMed] [Google Scholar]
- 141.Kang Z., Li H., Li G., Yin D. Reaction of pyridoxamine with malondialdehyde: mechanism of inhibition of formation of advanced lipoxidation end-products. Amino Acids. 2006;30(1):55–61. doi: 10.1007/s00726-005-0209-6. [DOI] [PubMed] [Google Scholar]
- 142.Murakoshi M., Tanimoto M., Gohda T., Hagiwara S., Ohara I., Toyoda H., Ishikawa Y., Horikoshi S., Tomino Y. Pleiotropic effect of pyridoxamine on diabetic complications via CD36 expression in KK-Ay/Ta mice. Diabetes Res. Clin. Pract. 2009;83(2):183–189. doi: 10.1016/j.diabres.2008.11.008. [DOI] [PubMed] [Google Scholar]
- 143.Bayarsaikhan E., Bayarsaikhan D., Lee J., Son M., Oh S., Moon J., Park H.J., Roshini A., Kim S.U., Song B.J., Jo S.M., Byun K., Lee B., Oh S. Microglial AGE-albumin is critical for neuronal death in Parkinson’s disease: a possible implication for theranostics. Int. J. Nanomed. 2016;10:281–292. doi: 10.2147/IJN.S95077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.de Lau L.M.L., Koudstaal P.J., Witteman J.C.M., Hofman A., Breteler M.M.B. Dietary folate, vitamin B12, and vitamin B6 and the risk of Parkinson disease. Neurology. 2006;67(2):315–318. doi: 10.1212/01.wnl.0000225050.57553.6d. [DOI] [PubMed] [Google Scholar]
- 145.Murakami K., Miyake Y., Sasaki S., Tanaka K., Fukushima W., Kiyohara C., Tsuboi Y., Yamada T., Oeda T., Miki T., Kawamura N., Sakae N., Fukuyama H., Hirota Y., Nagai M. Fukuoka Kinki Parkinson’s Disease Study Group. Dietary intake of folate, vitamin B6, vitamin B12 and riboflavin and risk of Parkinson’s disease: a case-control study in Japan. Br. J. Nutr. 2010;104(5):757–764. doi: 10.1017/S0007114510001005. [DOI] [PubMed] [Google Scholar]
- 146.McComb M. N., Chao J. Y., Ng T. M. H. Direct vasodilators and sympatholytic agents. J. Cardiovas. Pharmacol. Therap. 2016:3–19. doi: 10.1177/1074248415587969. [DOI] [PubMed] [Google Scholar]
- 147.Ellershaw D.C., Gurney A.M. Mechanisms of hydralazine induced vasodilation in rabbit aorta and pulmonary artery. Br. J. Pharmacol. 2001;134(3):621–631. doi: 10.1038/sj.bjp.0704302. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Bang L., Nielsen-Kudsk J.E., Gruhn N., Trautner S., Theilgaard S.A., Olesen S.P., Boesgaard S., Aldershvile J. Hydralazine-induced vasodilation involves opening of high conductance Ca2+-activated K+ channels. Eur. J. Pharmacol. 1998;361(1):43–49. doi: 10.1016/S0014-2999(98)00701-8. [DOI] [PubMed] [Google Scholar]
- 149.Jacobs M. Mechanism of action of hydralazine on vascular smooth muscle. Biochem. Pharmacol. 1984;33(18):2915–2919. doi: 10.1016/0006-2952(84)90216-8. [DOI] [PubMed] [Google Scholar]
- 150.Burcham P.C., Pyke S.M. Hydralazine inhibits rapid acrolein-induced protein oligomerization: Role of aldehyde scavenging and adduct trapping in cross-link blocking and cytoprotection. Mol. Pharmacol. 2006;69(3):1056–1065. doi: 10.1124/mol.105.018168. [DOI] [PubMed] [Google Scholar]
- 151.LoPachin R.M., Gavin T., Petersen D.R., Barber D.S. Molecular mechanisms of 4-hydroxy-2-nonenal and acrolein toxicity: Nucleophilic targets and adduct formation. Chem. Res. Toxicol. 2009;22(9):1499–1508. doi: 10.1021/tx900147g. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 152.Vindis C., Escargueil-Blanc I., Elbaz M., Marcheix B., Grazide M.H., Uchida K., Salvayre R., Nègre-Salvayre A. Desensitization of platelet-derived growth factor receptor-β by oxidized lipids in vascular cells and atherosclerotic lesions: prevention by aldehyde scavengers. Circ. Res. 2006;98(6):785–792. doi: 10.1161/01.RES.0000216288.93234.c3. [DOI] [PubMed] [Google Scholar]
- 153.Kaminskas L.M., Pyke S.M., Burcham P.C. Strong protein adduct trapping accompanies abolition of acrolein-mediated hepatotoxicity by hydralazine in mice. J. Pharmacol. Exp. Ther. 2004;310(3):1003–1010. doi: 10.1124/jpet.104.067330. [DOI] [PubMed] [Google Scholar]
- 154.Galvani S., Coatrieux C., Elbaz M., Grazide M.H., Thiers J.C., Parini A., Uchida K., Kamar N., Rostaing L., Baltas M., Salvayre R., Nègre-Salvayre A. Carbonyl scavenger and antiatherogenic effects of hydrazine derivatives. Free Radic. Biol. Med. 2008;45(10):1457–1467. doi: 10.1016/j.freeradbiomed.2008.08.026. [DOI] [PubMed] [Google Scholar]
- 155.Kesavan S.K., Bhat S., Golegaonkar S.B., Jagadeeshaprasad M.G., Deshmukh A.B., Patil H.S., Bhosale S.D., Shaikh M.L., Thulasiram H.V., Boppana R., Kulkarni M.J. Proteome wide reduction in AGE modification in streptozotocin induced diabetic mice by hydralazine mediated transglycation. Sci. Rep. 2013;3:2941. doi: 10.1038/srep02941. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 156.Guo X., Han C., Ma K., Xia Y., Wan F., Yin S., Kou L., Sun Y., Wu J., Hu J., Huang J., Xiong N., Wang T. Hydralazine protects nigrostriatal dopaminergic neurons from MPP+ and MPTP induced neurotoxicity: Roles of Nrf2-ARE signaling pathway. . Front. Neurol. 2019;10:271. doi: 10.3389/fneur.2019.00271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 157.Dehghan E., Zhang Y., Saremi B., Yadavali S., Hakimi A., Dehghani M., Goodarzi M., Tu X., Robertson S., Lin R., Chudhuri A., Mirzaei H. Hydralazine induces stress resistance and extends C. elegans lifespan by activating the NRF2/SKN-1 signalling pathway. Nat. Commun. 2017;8(1):2223. doi: 10.1038/s41467-017-02394-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 158.Arce C., Segura-Pacheco B., Perez-Cardenas E., Taja-Chayeb L., Candelaria M., Dueñnas-Gonzalez A. Hydralazine target: from blood vessels to the epigenome. J. Transl. Med. 2006;4:10. doi: 10.1186/1479-5876-4-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 159.Lyles G.A., Garcia-Rodriguez J., Callingham B.A. Inhibitory actions of hydralazine upon monoamine oxidizing enzymes in the rat. Biochem. Pharmacol. 1983;32(17):2515–2521. doi: 10.1016/0006-2952(83)90012-6. [DOI] [PubMed] [Google Scholar]
- 160.Malerba M., Ragnoli B. Ambroxol in the 21st Century: Pharmacological and Clinical Update. Expert Opin. Drug Metab. Toxicol. 2008:1119–1129. doi: 10.1517/17425255.4.8.1119. [DOI] [PubMed] [Google Scholar]
- 161.Felix K., Pairet M., Zimmermann R. The antioxidative activity of the mucoregulatory agents: Ambroxol, bromhexine and N-acetyl-L-cysteine. A pulse radiolysis study. Life Sci. 1996;59(14):1141–1147. doi: 10.1016/0024-3205(96)00431-6. [DOI] [PubMed] [Google Scholar]
- 162.Nowak D., Antczak A., Król M., Bialasiewicz P., Pietras T. Antioxidant properties of Ambroxol. Free Radic. Biol. Med. 1994;16(4):517–522. doi: 10.1016/0891-5849(94)90130-9. [DOI] [PubMed] [Google Scholar]
- 163.Nowak D., Pierscinski G., Drzewoski J. Ambroxol inhibits doxorubicin-induced lipid peroxidation in heart of mice. Free Radic. Biol. Med. 1995;19(5):659–663. doi: 10.1016/0891-5849(95)00028-V. [DOI] [PubMed] [Google Scholar]
- 164.Migdalska-Richards A., Daly L., Bezard E., Schapira A.H.V. Ambroxol effects in glucocerebrosidase and α-synuclein transgenic mice. Ann. Neurol. 2016;80(5):766–775. doi: 10.1002/ana.24790. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 165.Migdalska-Richards A., Ko W.K.D., Li Q., Bezard E., Schapira A.H.V. Oral ambroxol increases brain glucocerebrosidase activity in a nonhuman primate. Synapse. 2017;71(7):e21967. doi: 10.1002/syn.21967. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 166.Magalhaes J., Gegg M.E., Migdalska-Richards A., Schapira A.H. Effects of ambroxol on the autophagy-lysosome pathway and mitochondria in primary cortical neurons. Sci. Rep. 2018;8(1):1385. doi: 10.1038/s41598-018-19479-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 167.Mazzulli J.R., Zunke F., Tsunemi T., Toker N.J., Jeon S., Burbulla L.F., Patnaik S., Sidransky E., Marugan J.J., Sue C.M., Krainc D. Activation of β-glucocerebrosidase reduces pathological α-synuclein and restores lysosomal function in Parkinson’s patient midbrain neurons. J. Neurosci. 2016;36(29):7693–7706. doi: 10.1523/JNEUROSCI.0628-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 168.Mullin S., Smith L., Lee K., D’Souza G., Woodgate P., Elflein J., Hällqvist J., Toffoli M., Streeter A., Hosking J., Heywood W.E., Khengar R., Campbell P., Hehir J., Cable S., Mills K., Zetterberg H., Limousin P., Libri V., Foltynie T., Schapira A.H.V. Ambroxol for the treatment of patients with parkinson disease with and without glucocerebrosidase gene mutations: a nonrandomized, noncontrolled trial. JAMA Neurol. 2020;77(4):427–434. doi: 10.1001/jamaneurol.2019.4611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 169.Silveira C.R.A., MacKinley J., Coleman K., Li Z., Finger E., Bartha R., Morrow S.A., Wells J., Borrie M., Tirona R.G., Rupar C.A., Zou G., Hegele R.A., Mahuran D., MacDonald P., Jenkins M.E., Jog M., Pasternak S.H. Ambroxol as a novel disease-modifying treatment for Parkinson’s disease dementia: protocol for a single-centre, randomized, double-blind, placebo-controlled trial. BMC Neurol. 2019;19(1):20. doi: 10.1186/s12883-019-1252-3. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 170.Naoi M., Maruyama W., Shamoto-Nagai M. Rasagiline and selegiline modulate mitochondrial homeostasis, intervene apoptosis system and mitigate α-synuclein cytotoxicity in disease-modifying therapy for Parkinson’s disease. J. Neural Transm. (Vienna) 2020;127(2):131–147. doi: 10.1007/s00702-020-02150-w. [DOI] [PubMed] [Google Scholar]
- 171.Kakish J., Tavassoly O., Lee J.S. Rasagiline, a suicide inhibitor of monoamine oxidases, binds reversibly to α-synuclein. ACS Chem. Neurosci. 2015;6(2):347–355. doi: 10.1021/cn5002914. [DOI] [PubMed] [Google Scholar]
- 172.Kumar V.B., Hsu F-F., Lakshmi V.M., Gillespie K.N., Burke W.J. Aldehyde adducts inhibit 3,4-dihydroxyphenylacetaldehyde-induced α-synuclein aggregation and toxicity: Implication for Parkinson neuroprotective therapy. Eur. J. Pharmacol. 2019;845:65–73. doi: 10.1016/j.ejphar.2018.12.027. [DOI] [PubMed] [Google Scholar]
- 173.Ahlskog J.E., Uitti R.J. Neurology. Lippincott Williams and Wilkins; 2010. Rasagiline, Parkinson neuroprotection, and delayed-start trials: Still No Satisfaction? pp. 1143–1148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 174.Olanow C.W., Rascol O., Hauser R., Feigin P.D., Jankovic J., Lang A., Langston W., Melamed E., Poewe W., Stocchi F., Tolosa E. ADAGIO Study Investigators. A double-blind, delayed-start trial of rasagiline in Parkinson’s disease. N. Engl. J. Med. 2009;361(13):1268–1278. doi: 10.1056/NEJMoa0809335. [DOI] [PubMed] [Google Scholar]
- 175.Aleguas A. Critical Care Toxicology: Diagnosis and Management of the Critically Poisoned Patient. Springer International Publishing; 2017. Thiamine. pp. 2993–2999. [DOI] [Google Scholar]
- 176.Senapati S.K., Dey S., Dwivedi S.K., Patra R.C., Swarup D. Effect of thiamine hydrochloride on lead induced lipid peroxidation in rat liver and kidney. Vet. Hum. Toxicol. 2000;42(4):236–237. [PubMed] [Google Scholar]
- 177.La Selva M., Beltramo E., Pagnozzi F., Bena E., Molinatti P.A., Molinatti G.M., Porta M. Thiamine corrects delayed replication and decreases production of lactate and advanced glycation end-products in bovine retinal and human umbilical vein endothelial cells cultured under high glucose conditions. Diabetologia. 1996;39(11):1263–1268. doi: 10.1007/s001250050568. [DOI] [PubMed] [Google Scholar]
- 178.Karachalias N., Babaei-Jadidi R., Rabbani N., Thornalley P.J. Increased protein damage in renal glomeruli, retina, nerve, plasma and urine and its prevention by thiamine and benfotiamine therapy in a rat model of diabetes. Diabetologia. 2010;53(7):1506–1516. doi: 10.1007/s00125-010-1722-z. [DOI] [PubMed] [Google Scholar]
- 179.Alaei S.F., Soares M.J., Zhao Y., Sherriff J. High-dose thiamine supplementation improves glucose tolerance in hyperglycemic individuals: A randomized, double-blind cross-over trial. Eur. J. Nutr. 2013;52(7):1821–1824. doi: 10.1007/s00394-013-0534-6. [DOI] [PubMed] [Google Scholar]
- 180.Lu’o’ng Kv., Nguyên L.T.H. Thiamine and Parkinson’s disease. J. Neurol. Sci. 2012;316(1-2):1–8. doi: 10.1016/j.jns.2012.02.008. [DOI] [PubMed] [Google Scholar]
- 181.Luong K.V.Q., Nguyễn L.T.H. The beneficial role of thiamine in Parkinson disease. CNS Neurosci. Ther. 2013;19(7):461–468. doi: 10.1111/cns.12078. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 182.Håglin L., Domellöf M., Bäckman L., Forsgren L. Low plasma thiamine and phosphate in male patients with Parkinson’s disease is associated with mild cognitive impairment. Clin. Nutr. ESPEN. 2020;37(0):93–99. doi: 10.1016/j.clnesp.2020.03.012. [DOI] [PubMed] [Google Scholar]
- 183.Graham G.G., Punt J., Arora M., Day R.O., Doogue M.P., Duong J.K., Furlong T.J., Greenfield J.R., Greenup L.C., Kirkpatrick C.M., Ray J.E., Timmins P., Williams K.M. Clinical pharmacokinetics of metformin. Clin. Pharmacokinet. 2011;50(2):81–98. doi: 10.2165/11534750-000000000-00000. [DOI] [PubMed] [Google Scholar]
- 184.Mahar Doan K.M., Lakhman S.S., Boje K.M. Blood-brain barrier transport studies of organic guanidino cations using an in situ brain perfusion technique. Brain Res. 2000;876(1-2):141–147. doi: 10.1016/S0006-8993(00)02643-3. [DOI] [PubMed] [Google Scholar]
- 185.Stuhlmiller D.F.E., Boje K.M.K. Characterization of L-arginine and aminoguanidine uptake into isolated rat choroid plexus: Differences in uptake mechanisms and inhibition by nitric oxide synthase inhibitors. J. Neurochem. 1995;65(1):68–74. doi: 10.1046/j.1471-4159.1995.65010068.x. [DOI] [PubMed] [Google Scholar]
- 186.Degenhardt T. Lippincott williams wilkins; 2002. Pharmacokinetics, Tolerability and Biologic Activity of Oral Pyridorin (TM), a novel age inhibitor, in patients with Type 1 diabetes and diabetic nephropathy. [Google Scholar]
- 187.Williams M.E., Bolton W.K., Khalifah R.G., Degenhardt T.P., Schotzinger R.J., McGill J.B. Effects of pyridoxamine in combined phase 2 studies of patients with type 1 and type 2 diabetes and overt nephropathy. Am. J. Nephrol. 2007;27(6):605–614. doi: 10.1159/000108104. [DOI] [PubMed] [Google Scholar]
- 188.Sakurai T., Asakura T., Mizuno A., Matsuda M. Absorption and metabolism of pyridoxamine in mice. I. Pyridoxal as the only form of transport in blood. J. Nutr. Sci. Vitaminol. (Tokyo) 1991;37(4):341–348. doi: 10.3177/jnsv.37.341. [DOI] [PubMed] [Google Scholar]
- 189.van der Ham M., Albersen M., de Koning T.J., Visser G., Middendorp A., Bosma M., Verhoeven-Duif N.M., de Sain-van der Velden M.G.M. Quantification of vitamin B6 vitamers in human cerebrospinal fluid by ultra performance liquid chromatography-tandem mass spectrometry. Anal. Chim. Acta. 2012;712:108–114. doi: 10.1016/j.aca.2011.11.018. [DOI] [PubMed] [Google Scholar]
- 190.Ludden T.M., McNay J.L., Jr., Shepherd A.M.M., Lin M.S. Clinical pharmacokinetics of hydralazine. Clin. Pharmacokinet. 1982;7(3):185–205. doi: 10.2165/00003088-198207030-00001. [DOI] [PubMed] [Google Scholar]
- 191.Carley D.W., Trbovic S.M., Bozanich A., Radulovacki M. Cardiopulmonary control in sleeping Sprague-Dawley rats treated with hydralazine. J. Appl. Physiol. 1997;83(6):1954–1961. doi: 10.1152/jappl.1997.83.6.1954. [DOI] [PubMed] [Google Scholar]
- 192.Jauch R., Bozler G., Hammer R., Koss F.W. Ambroxol, studies of biotransformation in man and determination in biological samples. Arzneimittel-Forschung/Drug Res. 1978;28(5):904–911. [PubMed] [Google Scholar]
- 193.Kim Y., Yum M., Heo S.H. Pharmacologic properties of high-dose ambroxol in four patients with Gaucher disease and myoclonic epilepsy. J. Med. Genetics. 2020;57:124–131. doi: 10.1136/jmedgenet-2019-106132. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 194.Zhou W., Lv C., Zhang Q., Zong S., Wang M. Pharmacokinetics, pharmacodynamics, and safety of rasagiline transdermal patch: a preliminary study in Healthy Chinese Subjects. Clin. Drug Investig. 2018;38(2):125–133. doi: 10.1007/s40261-017-0588-y. [DOI] [PubMed] [Google Scholar]
- 195.Lecht S., Haroutiunian S., Hoffman A., Lazarovici P. Therapeutics and Clinical Risk Management. Dove Press; 2007. Rasagiline - A Novel MAO B Inhibitor in Parkinson’s Disease Therapy. pp. 467–474. [PMC free article] [PubMed] [Google Scholar]
- 196.U. S. Food and Drug Administration. AZILECT® (Rasagiline Mesylate) Safely and Effectively. Highlights of prescribing information. 2014. https://Www.Accessdata.Fda.Gov/Drugsatfda_docs/Label/2014/021641s016s017lbl.Pdf
- 197.Smithline H.A., Donnino M., Greenblatt D.J. Pharmacokinetics of high-dose oral thiamine hydrochloride in healthy subjects. BMC Clin. Pharmacol. 2012;12:4. doi: 10.1186/1472-6904-12-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 198.Greenwood J., Pratt O.E. Comparison of the effects of some thiamine analogues upon thiamine transport across the blood-brain barrier of the rat. J. Physiol. 1985;369(1):79–91. doi: 10.1113/jphysiol.1985.sp015889. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 199.Tallaksen C.M.E., Sande A., Bøhmer T., Bell H., Karlsen J. Kinetics of thiamin and thiamin phosphate esters in human blood, plasma and urine after 50 mg intravenously or orally. Eur. J. Clin. Pharmacol. 1993;44(1):73–78. doi: 10.1007/BF00315284. [DOI] [PubMed] [Google Scholar]
- 200.Costantini A., Pala M.I., Grossi E., Mondonico S., Cardelli L.E., Jenner C., Proietti S., Colangeli M., Fancellu R. Long-term treatment with high-dose thiamine in parkinson disease: an open-label pilot study. J. Altern. Complement. Med. 2015;21(12):740–747. doi: 10.1089/acm.2014.0353. [DOI] [PubMed] [Google Scholar]
- 201.Boldyrev A. A., Aldini G., Derave W. Physiology and pathophysiology of carnosine. Physiol. Rev. 2013;93(45):1803–1845. doi: 10.1152/physrev.00039.2012. [DOI] [PubMed] [Google Scholar]
- 202.Hipkiss A.R. Carnosine and its possible roles in nutrition and health. Adv. Food Nutr. Res. 2009;57:87–154. doi: 10.1016/S1043-4526(09)57003-9. [DOI] [PubMed] [Google Scholar]
- 203.Gardner M.L., Illingworth K.M., Kelleher J., Wood D. Intestinal absorption of the intact peptide carnosine in man, and comparison with intestinal permeability to lactulose. J. Physiol. 1991;439(1):411–422. doi: 10.1113/jphysiol.1991.sp018673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 204.Drozak J., Veiga-da-Cunha M., Vertommen D., Stroobant V., Van Schaftingen E. Molecular identification of carnosine synthase as ATP-grasp domain-containing protein 1 (ATPGD1). J. Biol. Chem. 2010;285(13):9346–9356. doi: 10.1074/jbc.M109.095505. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 205.Seidler N.W., Yeargans G.S., Morgan T.G. Carnosine disaggregates glycated α-crystallin: An in vitro study. Arch. Biochem. Biophys. 2004;427(1):110–115. doi: 10.1016/j.abb.2004.04.024. [DOI] [PubMed] [Google Scholar]
- 206.Szwergold B.S. Carnosine and anserine act as effective transglycating agents in decomposition of aldose-derived Schiff bases. Biochem. Biophys. Res. Commun. 2005;336(1):36–41. doi: 10.1016/j.bbrc.2005.08.033. [DOI] [PubMed] [Google Scholar]
- 207.Nagai R., Murray D.B., Metz T.O., Baynes J.W. Chelation: A fundamental mechanism of action of AGE inhibitors, AGE breakers, and other inhibitors of diabetes complications. Diabetes. 2012;61(3):549–559. doi: 10.2337/db11-1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 208.Regazzoni L., de Courten B., Garzon D., Altomare A., Marinello C., Jakubova M., Vallova S., Krumpolec P., Carini M., Ukropec J., Ukropcova B., Aldini G. A carnosine intervention study in overweight human volunteers: bioavailability and reactive carbonyl species sequestering effect. Sci. Rep. 2016;6(1):27224. doi: 10.1038/srep27224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 209.Barski O.A., Xie Z., Baba S.P., Sithu S.D., Agarwal A., Cai J., Bhatnagar A., Srivastava S. Dietary carnosine prevents early atherosclerotic lesion formation in apolipoprotein E-null mice. Arterioscler. Thromb. Vasc. Biol. 2013;33(6):1162–1170. doi: 10.1161/ATVBAHA.112.300572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 210.Liu Y., Xu G., Sayre L.M. Carnosine inhibits (E)-4-hydroxy-2-nonenal-induced protein cross-linking: Structural characterization of carnosine-HNE adducts. Chem. Res. Toxicol. 2003;16(12):1589–1597. doi: 10.1021/tx034160a. [DOI] [PubMed] [Google Scholar]
- 211.Aldini G., Orioli M., Rossoni G., Savi F., Braidotti P., Vistoli G., Yeum K.J., Negrisoli G., Carini M. The carbonyl scavenger carnosine ameliorates dyslipidaemia and renal function in Zucker obese rats. J. Cell. Mol. Med. 2011;15(6):1339–1354. doi: 10.1111/j.1582-4934.2010.01101.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 212.Nelson M-A.M., Builta Z.J., Monroe T.B., Doorn J.A., Anderson E.J. Biochemical characterization of the catecholaldehyde reactivity of L-carnosine and its therapeutic potential in human myocardium. Amino Acids. 2019;51(1):97–102. doi: 10.1007/s00726-018-2647-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 213.Afshin-Majd S., Khalili M., Roghani M., Mehranmehr N., Baluchnejadmojarad T. Carnosine exerts neuroprotective effect against 6-hydroxydopamine toxicity in hemiparkinsonian rat. Mol. Neurobiol. 2015;51(3):1064–1070. doi: 10.1007/s12035-014-8771-0. [DOI] [PubMed] [Google Scholar]
- 214.Tsai S.J., Kuo W.W., Liu W.H., Yin M.C. Antioxidative and anti-inflammatory protection from carnosine in the striatum of MPTP-treated mice. J. Agric. Food Chem. 2010;58(21):11510–11516. doi: 10.1021/jf103258p. [DOI] [PubMed] [Google Scholar]
- 215.Kulikova O.I., Berezhnoy D.S., Stvolinsky S.L., Lopachev A.V., Orlova V.S., Fedorova T.N. Neuroprotective effect of the carnosine - α-lipoic acid nanomicellar complex in a model of early-stage Parkinson’s disease. Regul. Toxicol. Pharmacol. 2018;95:254–259. doi: 10.1016/j.yrtph.2018.03.025. [DOI] [PubMed] [Google Scholar]
- 216.Bermúdez M.L., Skelton M.R., Genter M.B. Intranasal carnosine attenuates transcriptomic alterations and improves mitochondrial function in the Thy1-aSyn mouse model of Parkinson’s disease. Mol. Genet. Metab. 2018;125(3):305–313. doi: 10.1016/j.ymgme.2018.08.002. [DOI] [PubMed] [Google Scholar]
- 217.Bermúdez M.L., Seroogy K.B., Genter M.B. Evaluation of carnosine intervention in the Thy1-aSyn mouse model of Parkinson’s Disease. Neuroscience. 2019;411:270–278. doi: 10.1016/j.neuroscience.2019.05.026. [DOI] [PubMed] [Google Scholar]
- 218.Boldyrev A., Fedorova T., Stepanova M., Dobrotvorskaya I., Kozlova E., Boldanova N., Bagyeva G., Ivanova-Smolenskaya I., Illarioshkin S. Carnosine [corrected] increases efficiency of DOPA therapy of Parkinson’s disease: A pilot study. Rejuvenation Res. 2008;11(4):821–827. doi: 10.1089/rej.2008.0716. [DOI] [PubMed] [Google Scholar]
- 219.Espay A.J., Kalia L.V., Gan-Or Z., Williams-Gray C.H., Bedard P.L., Rowe S.M., Morgante F., Fasano A., Stecher B., Kauffman M.A., Farrer M.J., Coffey C.S., Schwarzschild M.A., Sherer T., Postuma R.B., Strafella A.P., Singleton A.B., Barker R.A., Kieburtz K., Olanow C.W., Lozano A., Kordower J.H., Cedarbaum J.M., Brundin P., Standaert D.G., Lang A.E. Disease modification and biomarker development in Parkinson disease: Revision or reconstruction? Neurology. 2020;94(11):481–494. doi: 10.1212/WNL.0000000000009107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 220.Antonini A., Bravi D., Sandre M., Bubacco L. Immunization therapies for parkinson’s disease: state of the art and considerations for future clinical trials. Expert Opin. Investig. Drugs. 2020;29(7):685–695. doi: 10.1080/13543784.2020.1771693. [DOI] [PubMed] [Google Scholar]
- 221.Politis M., Niccolini F. Serotonin in Parkinson’s Disease. Behav. Brain Res. 2015;277:136–145. doi: 10.1016/j.bbr.2014.07.037. [DOI] [PubMed] [Google Scholar]
- 222.Perez R.G., Waymire J.C., Lin E., Liu J.J., Guo F., Zigmond M.J. A role for alpha-synuclein in the regulation of dopamine biosynthesis. J. Neurosci. 2002;22(8):3090–3099. doi: 10.1523/JNEUROSCI.22-08-03090.2002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 223.Parnetti L., Gaetani L., Eusebi P., Paciotti S., Hansson O., El-Agnaf O., Mollenhauer B., Blennow K., Calabresi P. CSF and blood biomarkers for Parkinson’s disease. Lancet Neurol. 2019;18(6):573–586. doi: 10.1016/S1474-4422(19)30024-9. [DOI] [PubMed] [Google Scholar]
- 224.Goldstein D.S., Holmes C., Sullivan P., Jinsmaa Y., Kopin I.J., Sharabi Y. Elevated cerebrospinal fluid ratios of cysteinyl-dopamine/3,4-dihydroxyphenylacetic acid in parkinsonian synucleinopathies. Parkinsonism Relat. Disord. 2016;31:79–86. doi: 10.1016/j.parkreldis.2016.07.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 225.Branco R., Ellsworth W., Niedzwiecki M., Butkovich L., Walker D., Huddleston D., Jones D., Miller G. Levodopa and dopamine dynamics in Parkinson’s disease metabolomics. BioRxiv. 2018;30:6266. [Google Scholar]
- 226.Zhao H., Huang S., Palanisamy S., Wang C., Rainer G., Zhang X. Alpha-synuclein dopaminylation presented in plasma of both healthy subjects and Parkinson’s disease patients. Proteomics Clin. Appl. 2020;14(5):e1900117. doi: 10.1002/prca.201900117. [DOI] [PubMed] [Google Scholar]
- 227.Kang J.H., Mollenhauer B., Coffey C.S., Toledo J.B., Weintraub D., Galasko D.R., Irwin D.J., Van Deerlin V., Chen-Plotkin A.S., Caspell-Garcia C., Waligórska T., Taylor P., Shah N., Pan S., Zero P., Frasier M., Marek K., Kieburtz K., Jennings D., Tanner C.M., Simuni T., Singleton A., Toga A.W., Chowdhury S., Trojanowski J.Q., Shaw L.M. Parkinson’s progression marker initiative. CSF biomarkers associated with disease heterogeneity in early Parkinson’s disease: The Parkinson’s progression markers Initiative study. Acta Neuropathol. 2016;131(6):935–949. doi: 10.1007/s00401-016-1552-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 228.Mucke H. A. M. Drug repositioning in the mirror of patenting: surveying and mining uncharted territory. Front. Pharmacol. 2017;8:927. doi: 10.3389/fphar.2017.00927. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 229.Breckenridge A., Jacob R. Overcoming the legal and regulatory barriers to drug repurposing. Nat. Rev. Drug Discov. 2018;18(1):1–2. doi: 10.1038/nrd.2018.92. [DOI] [PubMed] [Google Scholar]
- 230.Simsek M., Meijer B., van Bodegraven A. A., de Boer N. K. H., Mulder C.J.J. Finding hidden treasures in old drugs: The challenges and importance of licensing generics. Drug Discov. Today. 2014;23(1):17–21. doi: 10.1016/j.drudis.2017.08.008. [DOI] [PubMed] [Google Scholar]