

Abstract
Amprenavir (141W94) is extensively metabolized by P450 cytochromes, specifically, CYP3A4. Because hepatic insufficiency reduces P450-mediated metabolism, the concentrations in plasma of drugs metabolized through this pathway are often increased in subjects with liver disease. Following administration of a single, oral dose of 600 mg of amprenavir, pharmacokinetic parameters were determined for 10 subjects with severe cirrhosis, 10 subjects with moderate cirrhosis, and 10 healthy volunteers. Model-independent methods for determining the area under the plasma concentration-time curve (AUC) from time zero to infinity (AUC0–∞) showed an increase in amprenavir AUC0–∞ of 2.5-fold in the group with moderate cirrhosis and 4.5-fold in the group with severe cirrhosis compared with that in the control group of healthy volunteers (P < 0.05). AUC0–∞ was linearly related to the severity of liver disease, as assessed by the Child-Pugh score. Of the laboratory data used to calculate the Child-Pugh score, only the mean total bilirubin concentration showed a significant relationship with AUC0–∞. The relationship between the total bilirubin concentration and the AUC0–∞ of amprenavir was well characterized by a simple Emax model, suggesting that the total bilirubin concentration may be a useful parameter for predicting the amprenavir AUC in subjects with hepatic insufficiency. Finally, the sera of cirrhotic subjects showed significant decreases in the levels of α1-acid glycoprotein, the primary plasma binding protein for amprenavir. On the basis of the results of this study, for an exposure equivalent to a clinical dose of 1,200 mg twice daily in subjects without cirrhosis, subjects with Child-Pugh scores of 5 to 8 should receive a twice-daily 450-mg dose of amprenavir, and subjects with Child-Pugh scores of 9 to 15 should receive a twice-daily 300-mg dose of amprenavir.
Amprenavir (141W94) is a novel anti-human immunodeficiency virus (anti-HIV) agent recently approved for treatment of HIV infection. The mechanism of antiviral activity of amprenavir is inhibition of viral aspartic protease, with a Ki of 0.6 nM (7). Amprenavir is a potent inhibitor of HIV type 1 (HIV-1) replication in vitro, with 50% inhibitory concentrations of 0.084 and 0.080 mM for virus in human MT-4 cells and peripheral blood lymphocytes, respectively (15). Clinical studies have evaluated single doses of amprenavir over a range of 150 to 1,200 mg in HIV-1-infected adults and have shown that in the range evaluated, plasma amprenavir concentrations increased linearly with dose but the increases were slightly greater than dose proportional (13). Following administration of a single, oral dose of 1,200 mg of amprenavir, the mean plasma amprenavir concentration 12 h after drug administration was fourfold greater than the in vitro 50% inhibitory concentration for HIV in peripheral blood lymphocytes (13). The dose approved for treatment of HIV-1 infection in adults is 1,200 mg twice daily.
Liver dysfunction resulting from coinfection with hepatitis B or hepatitis C virus may be a complication of HIV infection (F. Moretti, R. Novati, G. Morsica, and A. Poli, Abstr. 12th Int. Conf. AIDS, p. 1124–1125, 1998). Like the rest of the currently licensed HIV-1 protease inhibitors used as antiviral drugs for HIV-1-infected subjects, amprenavir is extensively metabolized in the liver by cytochrome P450 enzymes, specifically, through the CYP3A4 pathway (7; J. Woolley, S. Studenberg, C. Boehlert, G. Bowers, A. Sinhabaru, and P. Adams, Abstr. 37th Intersci. Conf. Antimicrob. Agents Chemother., abstr. A-60, p. 12, 1997). Drug metabolism by cytochrome P450 enzymes has important clinical consequences because of the possibility of reduced drug metabolism and subsequent increased plasma drug concentrations in subjects with liver dysfunction (3). In addition, the potential for protease inhibitors to induce or inhibit specific P450 isozymes has clinical significance due to the possibility of drug-drug interactions in a patient population likely to require multiple concomitant medications. In vitro studies have shown that amprenavir inhibits CYP3A4 (Ki = 0.06 mM) at clinically achievable concentrations and CYP2C19 (Ki = 47 mM) at much higher levels of exposure but does not inhibit CYP1A2, CYP2C9, CYP2D6, or 2 CYP2E1 (Woolley et al., 37th ICAAC).
Like other HIV-1 protease inhibitors, amprenavir is bound to albumin and to α1-acid glycoprotein (AAG), both of which are synthesized in the liver. In vitro studies with the protease inhibitors indinavir, saquinavir, and ritonavir, as well as with investigational protease inhibitors A77003, A-80987, KN1-272, and CGP 61755, have shown that addition of AAG decreases in vitro drug activity (4, 5, 8, 10). In vivo, the situation is more complex as AAG is an acute-phase protein whose levels are changed in certain disease states (9; K. Stellrecht, G. L. Drusano, D. S. Stein, and J. A. Bilello, 3rd Conf. Retroviruses and Opportunistic Infections, abstract, p. 84, 1996). In addition, amprenavir and other protease inhibitors are hepatically cleared.
In order to investigate the effect of hepatic impairment on amprenavir pharmacokinetics, we undertook a phase I clinical trial (Glaxo Wellcome study PROB-1008) designed to compare the pharmacokinetics of amprenavir in healthy volunteers to that in subjects with moderate or severe cirrhosis. Because amprenavir is extensively metabolized in the liver and is approximately 90% bound to AAG, amprenavir pharmacokinetics might be significantly altered in subjects with liver disease, in whom both P450 enzyme activities and AAG concentrations might be reduced. For safety, the dose of amprenavir administered in this study was 600 mg, or one-half the 1,200-mg dose under clinical evaluation in phase III trials. The primary goal of this trial was to evaluate the differences in pharmacokinetics of amprenavir among the three groups of subjects and thereby to determine whether a dose reduction is necessary for subjects with cirrhosis.
MATERIALS AND METHODS
Study design.
This study was an open-label, single-period, single-dose, parallel-group, phase I study conducted at seven study centers in France during the period from March through November 1997. The study was designed to include 30 subjects, both male and female, divided equally among three groups. The three groups were composed of 10 subjects with severe cirrhosis, 10 subjects with moderate cirrhosis, and 10 healthy volunteers who were chosen as controls to match the subjects with moderate cirrhosis for gender, smoking status, weight, and age. Subjects with cirrhosis (moderate or severe) were enrolled at five study centers: Hôpital Avicenne, Bobigny; Hôpital Broussais, Paris; Hôpital Dupuytren, Limoges; Hôpital de Rangueil, Toulouse; and Hôtel-Dieu, Nantes. Healthy volunteers were enrolled at Therapharm Recherche in Boulogne and at ASTER in Paris. In accordance with French law, the study was registered with the French Ministry of Health and was approved by the authorized Ethics Review Board prior to initiation. Subjects provided written informed consent prior to enrollment in the study.
The study period consisted of a screening assessment, performed within 2 weeks prior to dosing, and a dosing period, during which subjects received study drug and were monitored clinically in the unit or hospital for 24 h (control subjects) or 96 h (cirrhotic subjects). Subjects were admitted to the unit or hospital the evening before the dosing day and remained there until the final study assessments were completed. Subjects fasted after midnight and received study medication at 8 a.m. the next morning; standard meals were served at 2, 6, and 12 h after dosing. Subjects were discharged after the final procedures were completed at 24 or 96 h, and no additional follow-up assessments were performed.
Subjects.
Ten subjects were enrolled in each of the three groups: healthy volunteers, subjects with moderate cirrhosis, and subjects with severe cirrhosis. All subjects were required to meet the following criteria for enrollment: agreed to consume no more than 4 units of alcohol per day until the end of the study (1 unit = 1/2 pint of beer, 1 glass of wine, or 1 measure of spirits); were capable of giving informed consent; and was affiliated with the French Social Security System for health care coverage. Healthy male and female volunteers were eligible for the study if they met the following criteria: were 18 to 65 years of age; had body weight (±10 kg), age (±5 years), gender, and smoking habits matched to those of subjects with moderate cirrhosis; and had good general health and were free from significant disease as determined by physical examination, medical history, and screening assessments. Healthy volunteers were ineligible if they were considered unfit by the investigator or if any of the following criteria were met: history of drug allergy or other allergy that contraindicated participation; blood donation within the previous month; current use of medication; current daily consumption of more than 4 units of alcohol; participation in an investigational drug study within the previous 3 months; positive antibody screen for HIV, hepatitis B virus, or hepatitis C virus; pregnant; breast-feeding female; or female of childbearing potential not using effective contraception.
Male and female subjects with moderate or severe cirrhosis were eligible for the study if they met the following criteria: were 18 to 65 years of age and were free of clinically significant organic or psychiatric disease that might affect amprenavir pharmacokinetics, other than the expected consequences of cirrhosis. Subjects with moderate cirrhosis were required to have biopsy-proven cirrhosis or a history of prior severe liver disease, as defined by this protocol. For subjects with moderate cirrhosis, the following laboratory values were required: albumin concentration, ≥28 g/liter; prothrombin activity, ≥55% of normal; and bilirubin concentration, ≤60 μmol/liter. Subjects with severe cirrhosis were required to have one of the following: a history of ascites, a history of hepatic encephalopathy, or esophageal varices (stage, ≥II). In addition, subjects with severe cirrhosis were required to have at least one of the following laboratory values: albumin concentration, <28 g/liter; prothrombin activity, <55% of normal; and bilirubin concentration, >60 μmol/liter. Cirrhotic subjects were ineligible if any of the following criteria were met: clinically unstable in the judgment of the investigator; participation in a clinical trial within the previous 3 months or blood donation within the previous 2 months; evidence of current active hepatitis; gastrointestinal malabsorption that might affect drug absorption; pregnant; breast-feeding female; female of childbearing potential not using effective contraception; evidence of drug abuse; currently active encephalopathy; creatinine clearance, <40 ml/min (6); or current use of an antacid drug or a drug that acts on intestinal motility (the drug had to be stopped on the day prior to amprenavir administration) or a drug known to be a P450 enzyme inducer or inhibitor (inducers had to be stopped 2 weeks prior to amprenavir administration, and inhibitors had to be stopped 1 week prior to amprenavir administration). Use of concomitant medications known to be metabolized by CYP3A4 was prohibited during the study.
Drug supply and administration.
Drug was supplied by Laboratoire Glaxo Wellcome Evreux. Individual 600-mg doses were provided in plastic bottles containing four soft gelatin capsules of 150 mg of amprenavir free base. On the morning of the dosing day, subjects ingested the four capsules with 200 ml of water.
Clinical procedures.
At the screening assessment the following procedures were performed: medical history and full physical examination; 12-lead electrocardiogram (ECG); blood collection for clinical chemistry (including AAG) and hematology (complete blood count and differential); urinalysis; urine screen for illicit drugs; screens for hepatitis B virus, hepatitis C virus, and HIV; thyroid function tests; and urine pregnancy test, if appropriate. Procedures and assessments performed at 30 min predosing, at 24 h postdosing, and at 96 h postdosing (cirrhotic subjects only) included clinical chemistry, hematology, ECG, and vital signs. Female subjects of childbearing potential were given a urine pregnancy test on the evening prior to dosing. Adverse events were monitored throughout the dosing period by means of subject interviews and physical examination at the end of the study period.
Sample collection.
Blood samples taken predosing and during the first 24 h postdosing were drawn through an intravenous cannula; other samples were taken by venipuncture. For amprenavir assays, 3-ml blood samples were drawn into EDTA-containing tubes; the plasma was separated by centrifugation and was stored at −20°C until analysis. For hematology and clinical chemistry, 2-ml blood samples were drawn into EDTA-containing tubes and lithium heparin-containing tubes, respectively. For thyroid function tests, 3-ml blood samples were drawn into EDTA-containing tubes. For determination of amprenavir levels, plasma samples were collected from all subjects at 0.5 h predosing (baseline) and at 0.25, 0.5, 0.75, 1, 1.5, 2, 2.5, 3, 4, 5, 6, 8, 10, 12, 15, and 24 h postdosing. An additional five plasma samples were collected from subjects with cirrhosis, at 34, 48, 58, 72, and 96 h postdosing.
Plasma assay conditions.
Amprenavir concentrations were determined by liquid chromatography-mass spectrometry in the Department of Bioanalysis, Glaxo Wellcome, Research Triangle Park, N.C. Aliquots of 200 μl of acetonitrile containing 0.5 ng of internal standard per μl were dispensed into a clean 96-well plate. Aliquots (100 μl) of plasma samples and appropriate standards or controls were added to the wells, and the plate was mixed by vortexing. Following centrifugation at 2,500 rpm for 2 min, the supernatants were transferred to a separate 96-well plate containing 100 μl of 0.1% formic acid. Samples were mixed by vortexing and were injected (10 to 40 μl) at 4-min intervals onto a Waters Symmetry C18 analytical column (Waters Inc., Milford, Mass.). For the first 2 min the samples were eluted to the mass spectrometer in mobile phase B (55% acetonitrile and 45% water; vol/vol) at a flow rate of 0.35 ml/min. After 2 min the pumps were switched to mobile phase A (55% acetonitrile and 45% water with 0.1% formic acid; vol/vol) at the same flow rate. An API-300 triple quadruple mass spectrometer (PE Sciex, Toronto, Ontario, Canada) operated in the positive-ion multiple-reaction-monitoring mode was used to detect amprenavir and the internal standard by atmospheric pressure chemical ionization tandem mass spectrometry.
Stock solutions of amprenavir for calibration and quality control standards were prepared by dissolving amprenavir in 100% methanol to yield a stock solution of 1 mg/ml. Calibration standards and quality controls were prepared by dilution of the amprenavir stock solution. The final concentrations of calibration standards ranged from 10 to 5,000 ng/ml for calibration standards; quality control concentrations were 35, 800, and 4,000 ng of amprenavir per ml. The calibration curve was linear from 10 to 5,000 ng/ml. Accuracy (expressed as percent bias) ranged from −5.9 to 2.9% for validation controls. Intra-assay precision, expressed as percent coefficient of variation (CV), ranged from 1.0 to 5.7%, and interassay precision ranged from negligible to 4.6%.
Data analyses.
Pharmacokinetic calculations for determination of plasma amprenavir concentrations were performed for each subject by using WinNonlin (version 1.5; Scientific Consulting, Inc., Cary, N.C.). Model-independent methods were used to estimate the maximum concentration of amprenavir (Cmax), the time associated with Cmax (Tmax), the area under the concentration-time curve (AUC) from time zero to time t (AUC0–t), the half-life (t1/2), the apparent total clearance (CL/F), and the apparent volume of distribution during the elimination phase (VZ/F). The apparent terminal elimination rate constant (λZ) was estimated by log-linear regression of the terminal portions of the concentration-versus-time curves. For consistency with previously reported data, the AUC0–t was calculated by using the linear trapezoidal rule. AUC from time zero to infinity (AUC0–∞) was determined by extrapolation from AUC0–t with the addition of Clast/λZ, where Clast is the last measured concentration in plasma.
Analysis of variance (ANOVA) was used to compare pharmacokinetic parameters between the control group of healthy volunteers and each of the groups with liver disease. Prior to analysis the values for AUC0–t, AUC0–∞, Cmax, t1/2, CL/F, and VZ/F were loge transformed. Covariates selected to account for matching between groups included gender, age, weight, and smoking status. Geometric least-squares means were used to calculate the ratios of pharmacokinetic parameters in each liver disease group to those in the control group, along with 90% confidence intervals (CIs). Differences in pharmacokinetic parameters between two groups were considered statistically significant if the 90% CI did not include 1. The Wilcoxon rank sum test was used for comparison of the Tmax values between the control group and each liver disease group, and estimates of the median differences between groups were determined, along with 90% CIs.
The Student t test was used for comparison of AAG scores in each group. Because the normal range for AAG levels varied among the different laboratories used in the study, the following formula (14) was used to calculate a normalized AAG score for each subject: [Value − (H + L)/2]/(H − L), where Value is the mean of the screening and predosing AAG score, H is the upper limit of the normal range, and L is the lower limit of the normal range.
On the basis of laboratory evaluations and the medical histories of the subjects, liver disease in cirrhotic subjects was classified according to the Child-Pugh grading system (11); subjects in the control group (without liver disease) were assigned a Child-Pugh score of zero. Linear and nonlinear regression methods were used to assess the relationship between pharmacokinetic parameters and the Child-Pugh score. The relationship between log-transformed AUC0–∞ and Child-Pugh score was explored by an ANOVA model that included gender, Child-Pugh score, and the interaction between Child-Pugh score and gender, if significant. The relationship between log-transformed AUC0–∞ and each of the various laboratory values that contributed to the Child-Pugh score was examined by an ANOVA model which included gender and the log-transformed mean baseline values for albumin, prothrombin, and total bilirubin.
In order to evaluate the particular relationship between the AUC0–∞ and the mean total bilirubin concentrations (for screening and predosing values), a simple Emax model (equation 1) and a sigmoid Emax model (equation 2) were fit to the data by a nonlinear curve-fitting approach.
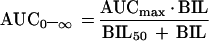 |
1 |
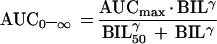 |
2 |
where AUCmax is the AUC0–∞ corresponding to the theoretical maximal effect of the mean bilirubin concentration; BIL50 is the mean bilirubin concentration at which 50% effect of AUCmax occurs; γ is an exponential parameter (shape parameter of the model); and BIL is the mean of the predosing and screening bilirubin values. Fitting was performed by the Gauss-Newton method by using WinNonlin (version 1.5; Scientific Consulting, Inc.). Analyses were conducted unweighted and weighted as 1/y, 1/y2, 1/ypredicted, and 1/y2predicted. Various goodness-of-fit measures were examined, including the CV of the estimated parameters, the planar 95% CI of the estimate, the Akaike Information Criterion (1), the coefficient of determination (r2), and various plots of weighted residual. Model weighting was compared by different empirical methods, and the effects on model fit were evaluated.
RESULTS
Subject enrollment and baseline characteristics.
Thirty subjects were enrolled in the study and completed the study. One of the subjects in the group with moderate cirrhosis was enrolled twice. After initial enrollment he was discontinued when it was discovered that he was taking disulfiram, a concomitant medication whose use was prohibited in this study. Treatment with disulfiram was interrupted for 2 weeks, and the subject was enrolled again and completed the study. Data from the subject's first enrollment are not included in the pharmacokinetic analysis.
Of the 30 evaluable subjects, 29 subjects were white and 1 subject was black. The 10 healthy subjects included 7 males and 3 females aged 41 to 60 years, the 10 subjects with moderate cirrhosis included 7 males and 3 females aged 37 to 64 years, and the 10 subjects with severe cirrhosis included 6 males and 4 females aged 33 to 64 years. Median heights and weights were 174.0 cm and 78.0 kg, 167.5 cm and 79.5 kg, and 170.0 cm and 58.5 kg for healthy subjects, subjects with moderate cirrhosis, and subjects with severe cirrhosis, respectively. The difference in weight between the group with severe cirrhosis and either of the other two groups is significant (P < 0.05). Three healthy subjects and two subjects from each of the cirrhosis groups had never used tobacco. Seven healthy subjects, seven subjects with moderate cirrhosis, and four subjects with severe cirrhosis were current smokers. One subject with moderate cirrhosis and four subjects with severe cirrhosis were former smokers. The median Child-Pugh score was 5.0 (range, 5 to 6) for the group with moderate cirrhosis and 9.0 (range, 5 to 12) for the group with severe cirrhosis. No subjects with a Child-Pugh score higher than 12 were enrolled in the study.
Safety assessments.
Five subjects reported a total of five adverse events, of which three were considered possibly drug related: one episode of rhinitis that occurred 10 h postdosing resolved after 4 days and was experienced by a 43-year-old female in the group with moderate cirrhosis; one episode of moderate epigastric pain that occurred 10 h postdosing resolved after 16 h and was experienced by a 32-year-old female in the group with severe cirrhosis; and one episode of thrombocytopenia (platelet count, 146,000/mm3) was experienced at 96 h postdosing, was resolved at the end of the study but was clinically insignificant, and was experienced by a 59-year-old male with severe cirrhosis. The remaining two adverse events reported in the study were considered not drug related and consisted of one episode of diarrhea, experienced by a subject in the group with severe cirrhosis, and one episode of palpitations, experienced by a subject in the group with moderate cirrhosis. No adverse events were reported by the healthy subjects.
One healthy subject with a normal ECG both at screening and at predosing experienced clinically significant nonspecific ST-T changes in the ECG at 24 h. This event was not considered an adverse event and was unresolved at the end of the study. A second subject, in the group with moderate cirrhosis, had a normal ECG at the screening assessment but showed sinus bradycardia at the predosing ECG and at the 24-h-postdosing ECG. The ECG for this subject was normal at 96 h postdosing.
Laboratory abnormalities reported for the subjects with cirrhosis were consistent with abnormalities expected for that population. No new or unexpected adverse events were reported for any subjects in the study. No serious adverse events were reported.
Pharmacokinetic analysis.
Median plasma amprenavir profiles for each group are shown in Fig. 1. For all subjects, plasma amprenavir concentrations reached a peak within 0.25 to 3 h postdosing and declined in a biphasic pattern with a t1/2 value of approximately 5 to 8 h. Although blood samples for pharmacokinetic analysis continued to be obtained through 96 h for cirrhotic subjects, median concentrations of amprenavir were below the lower limit of quantification (10 ng/ml) at 34 and 48 h postdosing for subjects with moderate and severe cirrhosis, respectively. Median concentrations of amprenavir were detectable at 24 h postdosing (end of study) in the control group.
FIG. 1.

Median plasma amprenavir concentration profiles.
The pharmacokinetic parameters for the three groups are summarized in Table 1. As indicated by the higher AUC values and lower CL/F values for both groups of cirrhotic subjects compared to healthy subjects, the clearance of amprenavir was decreased in subjects with cirrhosis.
TABLE 1.
Summary of amprenavir pharmacokinetic parameters in healthy subjects and subjects with cirrhosis
| Pharmacokinetic parameters | Study group
|
||
|---|---|---|---|
| Healthy volunteers (n = 10) | Subjects with moderate cirrhosis (n = 10) | Subjects with severe cirrhosis (n = 10) | |
| AUC0–∞ (ng · h/ml) | |||
| Arithmetic mean (% CV) | 11,999 (37) | 25761 (57) | 38,656 (42) |
| GLS meana (95% CI) | 9,679 (6,965, 13,451) | 23,815 (17,280, 32,823) | 43,699 (31,786, 60,076) |
| Mean ratiob (90% CI) | NAc | 2.46 (1.76, 3.44)d | 4.51 (3.06, 6.67) |
| Cmax (ng/ml) | |||
| Arithmetic mean (% CV) | 4,901 (28) | 6,483 (35%) | 9,435 (28%) |
| GLS mean (95% CI) | 4,712 (3,436, 6,463) | 6,049 (4,471, 8,184) | 9240 (6,752, 12,645) |
| Mean ratio (90% CI) | NA | 1.28 (0.95, 1.74) | 1.96 (1.34, 2.87) |
| Tmax (h) | |||
| Arithmetic mean (% CV) | 0.98 (31) | 1.08 (38) | 1.08 (80) |
| Mediana (95% CI) | 0.88 (0.75, 1.25) | 1.00 (0.75, 1.50) | 0.90 (0.50, 1.78) |
| Median difference (90% CI) | NA | 0.00 (−0.25, 0.50) | −0.25 (−0.50, 0.50) |
| t1/2 (h) | |||
| Arithmetic mean (% CV) | 5.56 (25) | 7.81 (65) | 7.93 (50) |
| GLS mean (95% CI) | 4.78 (3.41, 6.70) | 6.04 (4.38, 8.34) | 6.35 (4.54, 8.87) |
| Mean ratio (90% CI) | NA | 1.26 (0.91, 1.75) | 1.33 (0.89, 1.99) |
| CL/F (ml/min) | |||
| Arithmetic mean (% CV) | 946 (37) | 564 (73) | 295 (35) |
| GLS mean (95% CI) | 1,033 (743, 1437) | 420 (305, 579) | 229 (166, 315) |
| Mean ratio (90% CI) | NA | 0.41 (0.29, 0.57)d | 0.22 (0.15, 0.33)d |
| VZ/F (liter) | |||
| Arithmetic mean (% CV) | 462 (49) | 458 (148) | 196 (56) |
| GLS mean (95% CI) | 389 (225, 674) | 255 (151, 431) | 124 (72, 214) |
| mean ratio (90% CI) | NA | 0.66 (0.39, 1.11) | 0.32 (0.16, 0.62)d |
GLS mean, geometric least-squares mean of log-transformed parameters.
Values are ratios of geometric least-squares means for the cirrhosis group to the geometric least-squares means for the control (healthy) group.
NA, not applicable.
P < 0.05 for each cirrhosis group versus control group.
The pharmacokinetic parameters for subjects with severe or moderate cirrhosis were compared to the pharmacokinetic parameters for healthy subjects (Table 1). There were statistically significant differences for AUC0–∞, AUC0–t, and CL/F for subjects with moderate cirrhosis compared to those for healthy volunteers and for Cmax, AUC0–∞, AUC0–t, CL/F, and VZ/F for subjects with severe cirrhosis compared to those for healthy volunteers. A comparison of amprenavir AUC0–∞ values among the three groups is shown in Fig. 2.
FIG. 2.

Relationship between amprenavir AUC0–∞ and Child-Pugh score. No subjects with a Child-Pugh score of >12 were enrolled; therefore, extrapolation of results to subjects with higher Child-Pugh scores should be made with caution.
The ANOVA model revealed that gender and the interaction of gender · group were significant for AUC0–∞, AUC0–t, and CL/F. Relative to male subjects, female subjects had higher AUC0–∞ and AUC0–t values, and the difference was more pronounced for the group with moderate cirrhosis than for the group with severe cirrhosis (data not shown). However, these differences resulted from the fact that the three female subjects in each group happened to have higher Child-Pugh scores than the male subjects. With the addition of Child-Pugh score to the model (see below), neither the gender nor the gender · group interaction terms were significant. No other covariates (weight, age, or smoking status) were significant influences on the pharmacokinetic parameters in the ANOVA model. The geometric least-squares mean ratio results presented in Table 1 for comparisons between each cirrhotic group versus healthy subjects were based on the model after adjustment for selection bias.
There was a significant relationship between AUC0–∞ and Child-Pugh score, as shown in Fig. 2, with AUC0–∞ increasing with increasing Child-Pugh score. Several models for the relationship between AUC0–∞ and Child-Pugh score were evaluated. Log transformation of AUC0–∞ and the use of curvilinear models resulted in marginally better statistical fits to the data than use of the linear regression analysis shown in Fig. 2 did, but the log transformation and curvilinear models predicted very narrow dosage-adjustment intervals for every 1 to 2 increments of the Child-Pugh score. Such fine resolution has little clinical relevance because of the composite nature and inherent variability in the Child-Pugh score. Additionally, body weight adjustments to AUC did not improve the fit; body weight is highly variable in subjects with ascites and does not predict lean body mass or the volume of distribution of highly lipophilic drugs like amprenavir.
On the basis of linear regression analysis, values of AUC0–∞ were predicted for every possible Child-Pugh score, and the ratio of the AUC0–∞ for healthy subjects to the AUC0–∞ estimated for each Child-Pugh score was used to estimate a dose of amprenavir which would be equivalent to the 1,200-mg dose proposed for subjects without liver disease. Results of these calculations are shown in Table 2. Since none of the enrolled subjects had a Child-Pugh score over 12, extrapolation of results to subjects with Child-Pugh scores over 12 should be made with caution, and higher concentrations of amprenavir may be observed in these subjects.
TABLE 2.
Estimated amprenavir AUC0–∞ for each Child-Pugh score and the proposed dosage required to achieve equivalence with the recommended 1,200-mg dosea
| Subject | Child-Pugh score | Estimated AUC0–∞ (ng · h/ml)b | AUC0–∞ ratio for healthy subjects:cirrhotic subjects | Recommended dose (mg) for equivalent exposure |
|---|---|---|---|---|
| Healthy subject | 0 | 9,790 | 1.00 | 1,200 |
| Hepatic failure, Child-Pugh score group A | 5 | 26,125 | 0.37 | 450 |
| 6 | 29,392 | 0.33 | 450 | |
| Hepatic failure, Child-Pugh score group B | 7 | 32,659 | 0.30 | 450 |
| 8 | 35,926 | 0.27 | 450 | |
| 9 | 39,193 | 0.25 | 300 | |
| Hepatic failure, Child-Pugh score group Cc | 10 | 42,460 | 0.23 | 300 |
| 11 | 45,727 | 0.21 | 300 | |
| 12 | 48,994 | 0.20 | 300 | |
| 13 | 52,261 | 0.19 | 300 | |
| 14 | 55,528 | 0.18 | 300 | |
| 15 | 58,795 | 0.17 | 300 |
Proposed doses were rounded up to the nearest 150-mg dose to account for the amprenavir capsule strength.
Estimated for the 600-mg dose used in this study by the linear model y = 3,267x + 9,790. See Results for details.
No subjects with a Child-Pugh score of >12 were enrolled; therefore, extrapolation of results to subjects with higher Child-Pugh scores should be made with caution.
Once a relationship with AUC0–∞ and Child-Pugh score had been established, an ANOVA model was used to determine whether there was a correlation with AUC0–∞ and any of the individual laboratory parameters which are components of the Child-Pugh score. Of the relevant laboratory parameters examined (albumin, prothrombin, and bilirubin concentrations), only total bilirubin concentrations were significantly correlated with AUC0–∞ (P = 0.01). The simple Emax model best described the relationship between AUC0–∞ and bilirubin concentration on the basis of the Akaike Information Criterion and on the basis of the fact that the 95% CI for the value of γ in the sigmoid Emax model included 1. The final equation is plotted in Fig. 3, in which the amprenavir AUC0–∞ is plotted versus the mean baseline total bilirubin concentration. The percent CV of the two parameters AUCmax and BIL50 was less than 30%, and the coefficient of determination (r2) was 0.65 (P < 0.0001).
FIG. 3.

Plot of amprenavir AUC0–∞ versus mean baseline total bilirubin concentration.
Mean ± standard deviation AAG scores for subjects with moderate cirrhosis (−0.39 ± 0.20) and for those with severe cirrhosis (−0.62 ± 0.32) were significantly less than those for healthy subjects (−0.18 ± 0.14) (P < 0.05). When compared with healthy subjects, the mean AAG score was decreased by twofold for subjects with moderate cirrhosis and by fourfold for subjects with severe cirrhosis.
DISCUSSION
This study was designed to assess the impact of liver disease on amprenavir pharmacokinetics. Thirty subjects completed the study, including 10 subjects with moderate cirrhosis and 10 subjects with severe cirrhosis. No new or unexpected adverse events were attributed to amprenavir, and the majority of laboratory abnormalities that occurred in subjects with moderate or severe cirrhosis were consistent with those associated with serious liver disease. The pharmacokinetic profile was altered for subjects with cirrhosis, with higher AUC0–∞ values and two- to fourfold lower CL/F values for cirrhotic subjects relative to those for healthy subjects. Although serum AAG concentrations were reduced in cirrhotic subjects, there was no apparent increase in total clearance of amprenavir. Because amprenavir is extensively metabolized by CYP3A4, the findings are consistent with a significant reduction in CYP3A4 activity, porto-caval shunting, or both.
A linear relationship was observed between AUC0–∞ and the Child-Pugh score. Because there is also a linear relationship between the amprenavir dose administered and AUC0–∞ (13), we were able to construct a table that estimates the dose of amprenavir for subjects with a given Child-Pugh score required to obtain AUC levels comparable to those obtained with a 1,200-mg dose administered to a subject without liver disease. Mean baseline total bilirubin concentrations were significantly correlated with AUC values, suggesting that it may be possible to use bilirubin concentration to predict the initial dose of amprenavir appropriate for a patient with liver disease. This relationship suggests that there may be common transport mechanisms (e.g., p-glycoprotein) or metabolic pathways that involve both amprenavir and bilirubin.
As would be expected, the total clearance of amprenavir was reduced in subjects with hepatic impairment, consistent with a decrease in unbound (intrinsic) clearance from a loss of hepatic CYP3A4. Consistent with another clinical study of subjects with liver disease (Stellrecht et al., 3rd Conf. Retroviruses and Opportunistic Infections), there was also a decrease in the serum AAG concentrations. By itself, the decrease in AAG would result in a decrease in the total drug concentration in plasma and therefore an apparent increase in the total clearance. However, the opposite trend was observed. The percentage of unbound drug would vary inversely with the AAG concentration, although the absolute free drug concentrations would not be affected in the absence of a change in intrinsic clearance (12). These data therefore indicate that the decrease in intrinsic clearance outweighs the decrease in AAG concentrations with regard to its effect on the apparent clearance of total drug.
In summary, results from this study indicate that the dosing should be reduced in subjects with liver disease to obtain plasma amprenavir levels comparable to those achieved in healthy subjects given a 1,200-mg oral dose twice daily. For subjects with moderate cirrhosis and Child-Pugh scores of 5 to 8, the equivalent dose of amprenavir is estimated to be 450 mg twice daily. For subjects with severe cirrhosis and Child-Pugh scores of 9 to 15, the equivalent dose of amprenavir is estimated to be 300 mg twice daily.
ACKNOWLEDGMENTS
We gratefully acknowledge Cindy Rawls for performing the bioanalytical work and Barbara Rutledge and Belinda Ha for manuscript and writing assistance. We also gratefully acknowledge the subjects who participated in the study.
REFERENCES
- 1.Akaike H. A Bayesian analysis of the minimum AIC procedure. Ann Inst Statist Math. 1978;30:9–14. [Google Scholar]
- 2.Barry M, Gibbons S, Back D, Mulcahy F. Protease inhibitors in subjects with HIV disease: clinically important pharmacokinetic considerations. Clin Pharmacokinet. 1997;32:194–209. doi: 10.2165/00003088-199732030-00003. [DOI] [PubMed] [Google Scholar]
- 3.Benet L Z, Kroetz D L, Sheiner L B. Pharmacokinetics. The dynamics of dual absorption, distribution and elimination. In: Hardman J G, Linbird L E, Mobinoff P B, Ruddon R W, Gilman A G, editors. The pharmacological basis of therapeutics. New York, N.Y: McGraw-Hill Book Co.; 1996. p. 15. [Google Scholar]
- 4.Bilello J A, Bilello P A, Prichard M, Robins T, Drusano G L. Reduction of the in vitro activity of A77003, an inhibitor of human immunodeficiency virus protease, by human serum α1 acid glycoprotein. J Infect Dis. 1995;171:546–551. doi: 10.1093/infdis/171.3.546. [DOI] [PubMed] [Google Scholar]
- 5.Bilello J A, Bilello P A, Stellrecht K, Leonard J, Norbeck D W, Kempf D J, Robins T, Drusano G L. Human serum α1 acid glycoprotein reduces uptake, intracellular concentration, and antiviral activity of A-80987, an inhibitor of the human immunodeficiency virus type 1 protease. Antimicrob Agents Chemother. 1996;40:1491–1497. doi: 10.1128/aac.40.6.1491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Cockcroft D W, Gault M H. Prediction of creatinine clearance from serum creatinine. Nephron. 1976;16:31–41. doi: 10.1159/000180580. [DOI] [PubMed] [Google Scholar]
- 7.Decker C J, Laitinen L M, Bridson G W, Raybuck S A, Tung R D, Chaturvedi P R. Metabolism of amprenavir in liver microsomes: role of CYP3A4 inhibition for drug interactions. J Pharm Sci. 1998;87:803–807. doi: 10.1021/js980029p. [DOI] [PubMed] [Google Scholar]
- 8.Kageyama S, Anderson B D, Hoesterey B L, Hayashi H, Kiso Y, Flora K P, Mitsuya H. Protein binding of human immunodeficiency virus protease inhibitor KN1-272 and alteration of its in vitro antiretroviral activity in the presence of high concentrations of proteins. Antimicrob Agents Chemother. 1994;38:1107–1111. doi: 10.1128/aac.38.5.1107. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kremer J M H, Wilting J, Janssen L H M. Drug binding to human alpha-1 glycoprotein in health and disease. Pharmacol Rev. 1988;40:1–47. [PubMed] [Google Scholar]
- 10.Lazdins J K, Mestan J, Goutte G, Walker M R, Bold G, Capraro G, Klimkait T. In vitro effect of α1 acid glycoprotein on the anti-human immunodeficiency virus (HIV) activity of the protease inhibitor CGP 61755: a comparative study with other relevant HIV protease inhibitors. J Infect Dis. 1997;175:1063–1070. doi: 10.1086/520352. [DOI] [PubMed] [Google Scholar]
- 11.Pugh R N H, Murray-Lyon I M, Dawson J L, Pietroni M C, Williams R. Transection of the oesophagus for bleeding oesophageal varices. Br J Surg. 1973;60:646–649. doi: 10.1002/bjs.1800600817. [DOI] [PubMed] [Google Scholar]
- 12.Rolan P E. Plasma protein binding displacement interactions—why are they still regarded as clinically important. Br J Clin Pharmacol. 1994;37:125–128. doi: 10.1111/j.1365-2125.1994.tb04251.x. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Sadler B M, Hanson C D, Chittick G C, Symonds W T, Roskell N S. Safety and pharmacokinetics of amprenavir (141W94), an human immunodeficiency virus (HIV) type 1 protease inhibitor, following oral administration of single doses to HIV-infected adults. Antimicrob Agents Chemother. 1999;43:1686–1692. doi: 10.1128/aac.43.7.1686. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Smith A, Givens S V. Dealing with and defining abnormalities in laboratory data. Drug Infect J. 1993;27:771–778. [Google Scholar]
- 15.St. Clair M H, Millard J, Rooney J, Tisdale M, Parry N, Sadler B M, Blum M R, Painter G. In vitro antiviral activity of 141W94 (VX-478) in combination with other antiretroviral agents. Antivir Res. 1996;29:53–56. doi: 10.1016/0166-3542(95)00916-7. [DOI] [PubMed] [Google Scholar]