

Abstract
Background
Most patients with pancreatic cancer develop cachexia, which is characterized by progressive muscle loss. The mechanisms underlying muscle loss in cancer cachexia remain elusive. Pancreatic tumour organoids are 3D cell culture models that retain key characteristics of the parent tumour. We aimed to investigate the effect of pancreatic tumour organoid‐derived factors on processes that determine skeletal muscle mass, including the regulation of muscle protein turnover and myogenesis.
Methods
Conditioned medium (CM) was collected from human pancreatic cancer cell lines (PK‐45H, PANC‐1, PK‐1, and KLM‐1), pancreatic tumour organoid cultures from a severely cachectic (PANCO‐9a) and a non‐cachectic patient (PANCO‐12a), and a normal pancreas organoid culture. Differentiating C2C12 myoblasts and mature C2C12 myotubes were exposed to CM for 24 h or maintained in control medium. In myotubes, NF‐kB activation was monitored using a NF‐κB luciferase reporter construct, and mRNA expression of E3‐ubiquitin ligases and REDD1 was analysed by RT‐qPCR. C2C12 myoblast proliferation and differentiation were monitored by live cell imaging and myogenic markers and myosin heavy chain (MyHC) isoforms were assessed by RT‐qPCR.
Results
Whereas CM from PK‐1 and KLM‐1 cells significantly induced NF‐κB activation in C2C12 myotubes (PK‐1: 3.1‐fold, P < 0.001; KLM‐1: 2.1‐fold, P = 0.01), Atrogin‐1/MAFbx and MuRF1 mRNA were only minimally and inconsistently upregulated by the CM of pancreatic cancer cell lines. Similarly, E3‐ubiquitin ligases and REDD1 mRNA expression in myotubes were not altered by exposure to pancreatic tumour organoid CM. Compared with the control condition, CM from both PANCO‐9a and PANCO‐12a tumour organoids increased proliferation of myoblasts, which was accompanied by significant downregulation of the satellite cell marker paired‐box 7 (PAX7) (PANCO‐9a: −2.1‐fold, P < 0.001; PANCO‐12a: −2.0‐fold, P < 0.001) and myogenic factor 5 (MYF5) (PANCO‐9a: −2.1‐fold, P < 0.001; PANCO‐12a: −1.8‐fold, P < 0.001) after 48 h of differentiation. Live cell imaging revealed accelerated alignment and fusion of myoblasts exposed to CM from PANCO‐9a and PANCO‐12a, which was in line with significantly increased Myomaker mRNA expression levels (PANCO‐9a: 2.4‐fold, P = 0.001; PANCO‐12a: 2.2‐fold, P = 0.004). These morphological and transcriptional alterations were accompanied by increased expression of muscle differentiation markers such as MyHC‐IIB (PANCO‐9a: 2.5‐fold, P = 0.04; PANCO‐12a: 3.1‐fold, P = 0.006). Although the impact of organoid CM on myogenesis was not associated with the cachexia phenotype of the donor patients, it was specific for tumour organoids, as CM of control pancreas organoids did not modulate myogenic fusion.
Conclusions
These data show that pancreatic tumour organoid‐derived factors alter the kinetics of myogenesis, which may eventually contribute to impaired muscle mass maintenance in cancer cachexia.
Keywords: Cachexia, Skeletal muscle atrophy, Organoids, E3 ubiquitin ligases, Myogenesis
Introduction
Pancreatic cancer is a highly lethal malignancy that accounts for 4.5% of all cancer deaths worldwide. 1 Despite advances in our understanding of the complex tumour biology of pancreatic cancer and new treatment strategies, the prognosis of pancreatic cancer patients has only marginally improved. 2
A major contributor to the poor survival of pancreatic cancer patients is the cachexia syndrome, which is present in up to 80% of the patients at the time of diagnosis. 3 Cancer cachexia is a severe wasting disorder defined by involuntary loss of muscle mass, which cannot be fully reversed by conventional nutritional support and which leads to progressive functional impairment. 4 The pathophysiology of cancer cachexia remains to be elucidated, but it is well appreciated that it is driven by a complex interplay between tumour‐ and host‐derived factors that contributes to a negative protein and energy balance. 3 Because effective targeted therapies to treat cachexia are currently lacking, a better understanding of the cachexia‐inducing factors and mechanisms is crucial to prevent its onset and progression and to improve overall survival of pancreatic cancer patients.
Most of our current understanding of the cachexia‐inducing factors expressed and released by tumour cells is derived from in vitro and in vivo studies using established cancer cell lines. 5 , 6 , 7 , 8 , 9 , 10 In particular, studies with mouse C26‐colon carcinoma (C26) and Lewis lung carcinoma (LLC) cells have shown that tumour‐derived factors induce skeletal muscle atrophy by activation of NF‐κB and ubiquitin proteasome pathway (UPS)‐mediated proteolytic degradation of specific muscle proteins. 11 , 12 , 13 , 14 Moreover, impairments in myogenesis and its regulation have been implicated in the net loss of skeletal muscle in the context of cachexia. 15 , 16 , 17
Whereas these murine cell lines have contributed tremendously to our current understanding of cancer cachexia mechanisms, their suitability for modelling human cancer cachexia may not be optimal. 18 For example, C26 and LLC‐tumour bearing mice develop cachexia on a timescale of only a few weeks and their tumour volume typically represents more than 10% of the entire body mass, which is not the case in humans. 7 , 14 , 18 As such, human cell lines have been increasingly used to study cancer‐induced cachexia. 6 , 7 In particular, human pancreatic cancer cell lines like MiaPaCa‐2, Capan‐1, and Panc‐1 have been frequently applied because of the severity of cachexia in pancreatic cancer. 6 , 7 , 19 , 20 Unfortunately, the actual cachexia‐related clinical background of these cell lines is unknown. In addition, cell lines are routinely cultured on a stiff two‐dimensional (2D) plastic surface in the absence of physiological gradients of oxygen and nutrients and without functional cellular‐extracellular matrix interactions. 21 These comparatively artificial culture conditions may evoke important non‐physiological cellular adaptations that are associated with mutational and chromosomal instability. 22 As the majority of the cell lines used to study cancer cachexia mechanisms have been cultured in 2D for decades, currently available strains are likely to be genetically different compared with the originally isolated tumour cells, potentially affecting gene expression patterns. As a result, the established pancreatic cancer cell lines will likely have gained or lost cachexia‐inducing properties, contributing to inconsistent findings among studies. Taken together, these limitations of existing 2D cell lines highlight the need for better experimental models that contribute to our understanding of human cancer‐induced cachexia.
Recently, several aspects of cancer biology have been shown to be accurately modelled by three‐dimensional (3D) organoid cultures. 23 , 24 , 25 Pancreatic tumour organoids can be efficiently established by culturing primary epithelial tumour cells in basement membrane extract (BME) and a defined, tissue‐specific growth medium. These tumour cells self‐organize into 3D structures mimicking the architecture of the organ of origin, and have been shown to closely recapitulate pathophysiologically relevant aspects of pancreatic cancer both in vitro and in vivo. 23 Hence, we applied pancreatic tumour organoid cultures that were recently established in our lab 26 to investigate cachexia‐inducing properties of pancreatic cancer cells. We used tumour organoids from a severely cachectic and from a non‐cachectic patient to assess the direct effect of tumour‐derived factors on C2C12 skeletal muscle cells, focusing on muscle atrophy signalling and myogenesis. We hypothesized that tumour organoid‐derived CM induces myotube atrophy and impairs myogenesis.
Materials and methods
Human pancreatic organoid culture
Pancreatic tumour organoid cultures PANCO‐9a and PANCO‐12a were previously generated and characterized in our laboratory. 26 In short, ethical approval (METC 13‐4‐107) was obtained to pre‐operatively assess the cachexia status of pancreatic cancer patients undergoing surgical resection. A normal pancreas organoid culture (NP) was established at the Erasmus University Medical Center in Rotterdam (ethical approval obtained from the local Medical Ethics Committee, MEC‐2015‐085). Surgically removed pancreatic tumour tissue was used to generate pancreatic tumour organoids. This study has been performed in accordance with the ethical standards laid down in the 1964 Declaration of Helsinki and its later amendments.
For maintaining cultures, organoids were resuspended in ice‐cold BME (Geltrex LDEV‐Free Reduced Growth Factor Basement Membrane Matrix, Gibco, Cat. No. 1413202) and three approximately 15 μL droplets of Geltrex‐cell suspension were allowed to solidify per well of a 24‐wells culture plate (Eppendorf,) at 37°C for 30 min. When the droplets were solidified, 500 μL of medium was added to each well. 26 The plate was transferred to a humidified 37°C/5% CO2 incubator and medium was changed every 2–3 days. The organoids were passaged every 7–10 days. Organoids were mechanically sheared, resuspended in ice‐cold BME, and re‐plated as described earlier.
Collection of pancreatic organoid conditioned medium
Conditioned medium (CM) was collected from both pancreatic tumour organoids (PANCO‐9a, PANCO‐12a) and a normal pancreas organoid culture (NP). One day before passaging, organoid growth medium was replaced by basal culture medium consisting of DMEM/F12 supplemented with 1% (v/v) HEPES and 1% (v/v) antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin, GIBCO). Additional wells containing empty Geltrex‐droplets overlaid with basal culture medium were included for the collection of control medium. After 24 h, CM was collected and centrifuged at 350×g for 10 min at 4°C. The supernatant was centrifuged for another 20 min at 2000×g at 4°C and the resulting CM cleared from cellular debris was aliquoted and stored at −80°C.
Culturing of 2D pancreatic cancer cell lines and collection of conditioned medium
Human pancreatic cancer cell lines PK‐45H, PANC‐1, 27 PK‐1, 28 and KLM‐1 29 (obtained from RIKEN BioResource Center) were cultured in high glucose (4.5 g/L) Dulbecco's Modified Eagle's Medium (DMEM) supplemented with 10% (v/v) foetal bovine serum (FBS) (Greiner Bio‐one, cat. no. 758093) and 1% (v/v) antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin, GIBCO). All cell lines were maintained at 37 °C, 5% CO2 in a humidified incubator.
Medium used for the production of CM consisted of DMEM (10% v/v) diluted in Hank's Balanced Salt solution (HBSS) (GIBCO) supplemented with NaHCO3 (Sigma‐Aldrich, Saint Louis, MO), 10% (v/v) FBS, and 1% (v/v) antibiotics. The medium was conditioned for 48 h using cells at approximately 60–80% confluency. CM was collected and stored at −80 °C.
C2C12 skeletal muscle cell culture
Murine C2C12 skeletal myoblasts (obtained from the American Type Culture Collection, #CRL1772) were cultured in growth medium (GM), composed of low glucose (1 g/L) DMEM supplemented with 10% (v/v) FBS and 1% (v/v) antibiotics (100 units/mL penicillin and 100 μg/mL streptomycin, GIBCO). The cells were maintained at 37°C, 5% CO2 in a humidified incubator.
Myotubes were generated by plating C2C12 myoblasts at a density of 1.0 × 104 cells per square centimetre on BD Matrigel‐coated (Matrigel® Matrix Basement Membrane–Growth factor reduced, Corning) (1:50 in low glucose DMEM) cell culture plates (Eppendorf); myoblasts were cultured in GM for 48 h before induction of differentiation. To induce differentiation, GM was replaced by differentiation medium (DM) consisting of DMEM supplemented with 1% heat‐inactivated FBS (30 min at 56°C) and 0.5% (v/v) antibiotics (50 units/mL penicillin and 50 μg/mL streptomycin, GIBCO). The medium was refreshed every 48 h. At indicated time points, C2C12 skeletal myoblasts or myotubes were exposed to 50% v/v CM diluted in differentiation medium.
NF‐κB‐luciferase activity
At Day 5 of differentiation, C2C12 myotubes with a luciferase construct containing three tandem NF‐κB luciferase responsive elements 30 were incubated for 4 h with CM. For the assessment of NF‐κB activation, myotubes were harvested in 1× luciferase buffer (5× Reporter Lysis Buffer, Promega) on ice and cell lysates were stored at −80°C. Luciferase activity was measured according to the manufacturer's protocol (Promega) using a luminometer (Berthold Technologies). Luciferase activity was corrected for total protein by using the BCA Protein assay according to the manufacturer's protocol (Pierce® BCA Protein Assay Kit, Thermo Scientific, Rockford, IL, USA).
Quantitative real‐time PCR
Total RNA was extracted from cell cultures using TRI Reagent (Sigma, St. Louis, MO) according to the manufacturer's instructions. RNA was reconstituted in RNase free water and stored at −80°C. The RNA concentration was measured using a DeNovix DS‐11 spectrophotometer and 375 ng RNA were reversed transcribed using the SensiFast cDNA Synthesis Kit according to the manufacturer's instructions (Bioline GmbH, Germany).
To quantify mRNA expression levels, quantitative real‐time PCR (qRT‐PCR) analysis was performed on the LightCycler480 (Roche) using a three‐step PCR programme followed by melt curve analysis. cDNA was amplified with the SensiMix SYBR Hi‐Rox Kit (Bioline, cat. No. QT605‐05). Specific primer pairs for each gene were ordered from Sigma as listed in Table S1. Relative gene expression levels were derived from the LinRegPCR (Version 2016.1) method 31 and normalized to the geometric average of three reference genes, cyclophilin A (CYPA), β‐2‐microglobulin (β2M), and 60S acidic ribosomal protein P0 (RPLP0).
IncuCyte™ NucLight rapid red proliferation assay
C2C12 myoblasts were plated on Matrigel‐coated 96‐wells cell culture plates (Eppendorf) at a density of 1 × 104 per square centimetre. The cells were cultured for 48 h in GM before the induction of differentiation. The IncuCyte NucLight Rapid Red Reagent (Essen Bioscience, Cat no. 4717) was diluted 1:250 in DM containing 2% (v/v) HI‐FBS. Subsequently, DM containing NucLight Rapid Red was diluted with either control (DMEM/F12) (50% v/v) or CM samples (50% v/v) resulting in a final NucLight Rapid Red dilution of 1:500 and 1% HI‐FBS in all conditions. Insulin growth factor 1 (IGF‐1) (Sigma), a well‐known anabolic stimulus, was included as a positive control and was added at the induction of differentiation. The differentiation of C2C12 myoblasts into myotubes was monitored through an IncuCyte® S3 Live‐Cell analysis system (Sartorius). Phase‐contrast images and red‐fluorescence images were captured every 2 h using the 10× objective. The integrated red object count metric tool was used to quantify the number of nuclei.
Statistics
Data are representative of three independent experiments performed in triplicate and are expressed as mean ± SEM. Raw data were analysed using IBM SPSS 25 for Microsoft Windows®. Statistical analyses were performed using the independent sample T‐test to compare differences between two groups. In case of more than two groups, the One‐way ANOVA test was used followed by Tukey's post‐hoc testing. A P value of P < 0.05 was considered statistically significant.
Results
Tumour‐derived factors from established 2D pancreatic cancer cell lines do not consistently induce NF‐κB or atrophy signalling in mature C2C12 myotubes
Previous studies using conventional 2D colon and lung carcinoma lines have demonstrated that tumour‐derived factors may directly induce C2C12 skeletal muscle atrophy by activation of the ubiquitin proteasome pathway (UPS). Furthermore, proteolytic degradation of specific muscle proteins has been shown to be regulated via tumour factor‐promoted NF‐κB activation. 11 Given that pancreatic cancers generally have strong cachexia‐inducing abilities, we first investigated whether tumour‐derived factors from various established human 2D pancreatic cancer cell lines could initiate skeletal muscle atrophy signalling in vitro. Differentiated C2C12 myotubes were incubated with CM from four pancreatic cancer cell lines (PK‐45H, PANC‐1, PK‐1, and KLM‐1). First, we assessed the effect of CM on NF‐κB activation (Figure 1A). TNF‐α, a known inducer of NF‐κB signalling and here included as a positive control, significantly induced NF‐κB activity (5.0‐fold, P < 0.001) in C2C12 myotubes within 4 h. Whereas CM from PK‐1 (3.1‐fold, P < 0.001) and KLM‐1 (2.1‐fold, P = 0.01) also induced C2C12 NF‐κB activation, this was not observed after exposure to CM from PK‐45H and PANC‐1. Next, we assessed whether expression of Atrogin‐1/MAFbx and MuRF1, muscle‐specific E3‐ubiquitin ligases involved in muscle proteolysis, which have been reported to be upregulated under atrophy‐inducing conditions, 32 was affected by pancreatic cancer cell line‐derived factors (Figure 1B). Treatment with HBSS to mimic muscle atrophy as a consequence of nutrient deprivation caused significantly increased expression of Atrogin‐1/MAFbx (4.2‐fold, P < 0.001) and MuRF1 (3.6‐fold, P < 0.001), showing the responsiveness of the system. However, increased Atrogin‐1/MAFbx mRNA levels were only found after incubation with CM from PK‐1 (1.7‐fold, P = 0.015), and MuRF1 mRNA expression levels were only increased by CM from PK‐45H (2.0‐fold, P = 0.017). Collectively, these data show that whereas some human pancreatic cancer cell lines display certain skeletal muscle atrophy‐inducing properties, their effects are generally small and inconsistent. Moreover, cachexia‐related clinical data of the donor patients of these cell lines are not available, 27 , 28 , 29 questioning their application for modelling pancreatic cancer‐induced cachexia.
Figure 1.
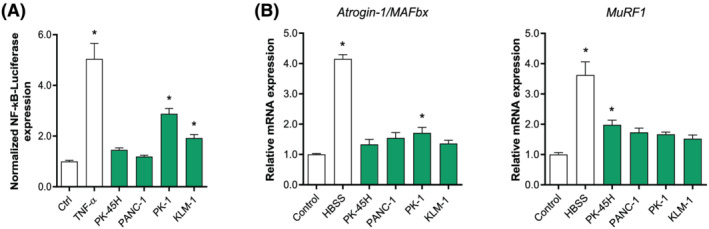
Effect of tumour‐derived factors from established 2D pancreatic cancer cell lines on C2C12 myotubes. Mature C2C12 myotubes were treated with CM from PK‐45H, PANC‐1, PK‐1, and KLM‐1. (A) After 4 h, NF‐κB luciferase activity luciferase was assessed. (B) mRNA expression of Atrogin‐1/MAFbx and MuRF1 were determined after 24 h. Data were normalized to CYPA, B2M, and RPLP0 reference genes and obtained from three independent experiments. Data are presented as mean ± SEM.
Human pancreatic tumour organoid‐derived factors do not induce skeletal muscle atrophy signalling in mature C2C12 myotubes
Given the limitations of human 2D pancreatic cancer cell lines and promising developments in organoid technology, we recently established an organoid biobank consisting of pancreatic tumour organoid cultures originating from patients whose cachexia phenotype was thoroughly assessed. 26 According to the international consensus definition of cancer cachexia, we selected an organoid culture from a cachectic (PANCO‐9a) and a non‐cachectic (PANCO‐12a) patient for the current study. These patients showed substantial differences concerning the percentage of weight loss over the past 6 months (PANCO‐9a: 13.4% vs. PANCO‐12a: 1.2%) and body composition parameters, including the L3‐skeletal muscle index (PANCO‐9a: 43.0 cm2/m2 vs. PANCO‐12a: 36.1 cm2/m2) and the L3‐visceral adipose tissue index (PANCO‐9a: 61.9 cm2/m2 vs. PANCO‐12a: 25.8 cm2/m2), which are representative for whole body muscle mass and fat mass, respectively. 26
We first investigated the potential of tumour organoid‐derived CM to induce NF‐κB activation in C2C12 myotubes. Mature C2C12 myotubes were exposed to pancreatic tumour organoid CM for 4 h (Figure 2A). Whereas TNF‐α administration caused a 6.9‐fold (P < 0.001) increase of NF‐κB activity, CM from PANCO‐12a resulted in a mere 1.8‐fold (P = 0.001) increase of NF‐κB activity, and PANCO‐9a‐derived CM did not induce any appreciable NF‐κB activation.
Figure 2.
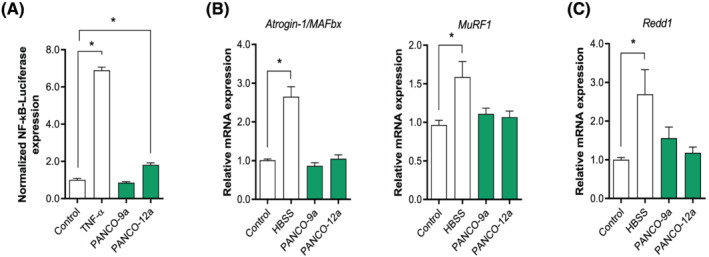
Pancreatic tumour organoid‐derived factors do not induce C2C12 muscle atrophy signalling. Mature C2C12 myotubes were treated with CM from PANCO‐9a and PANCO‐12a organoid cultures. (A) After 4 h, NF‐κB‐induced luciferase activity was assessed. (B) mRNA expression of Atrogin‐1/MAFbx, MuRF1 and (C) Redd1 was determined after 24 h. Data were normalized to CYPA, B2M, and RPLP0 reference genes and obtained from three independent experiments. Data are presented as mean ± SEM.
We next assessed whether tumour organoid‐derived CM induced upregulation of E3 ligases in mature C2C12 myotubes. Whereas HBSS resulted in significantly increased mRNA expression of Atrogin‐1/MAFbx (2.7‐fold, P = 0.004) and MuRF1 (1.6‐fold, P = 0.002), no increase of either E3‐ubiquitin ligase was observed in C2C12 myotubes incubated with CM from either tumour organoid culture (Figure 2B). In human primary myotubes incubated with CM from either tumour organoid culture, expression of these E3‐ubiquitin ligases was also unaltered, ruling out that interspecies differences accounted for the absence in activation of proteolysis signalling (Figure S1).
Because maintenance of skeletal muscle mass is coordinated by a balance between rates of protein synthesis and protein degradation, we also assessed the expression levels of REDD1, a repressor of protein synthesis signalling, which is elevated in skeletal muscle during various atrophic conditions. 33 , 34 As expected, REDD1 levels were significantly elevated after culturing C2C12 myotubes in HBSS (2.7‐fold, P = 0.011) (Figure 2C). In contrast, incubation with tumour organoid‐derived CM did not affect REDD1 expression (Figure 2C).
Human pancreatic tumour organoid‐derived factors promote myoblast fusion during differentiation
As myogenesis is an important process in the maintenance of muscle mass, we next explored the impact of tumour organoid factors on myogenesis. Impaired myogenesis and muscle regeneration have previously been observed in multiple muscle pathologies, including cancer‐associated cachexia. 15 , 16 , 35 , 36 , 37 When C2C12 myoblasts were exposed to tumour organoid‐derived CM during differentiation, pronounced disturbances in the morphology of the myotubes were observed after completion of the differentiation process (Figure 3A). The presence of either tumour organoid CM caused a highly heterogeneous appearance of both very large myotubes and detached myotubes. These pronounced morphological disturbances were not observed in the myotubes that were formed in the presence of either CM from a normal pancreas organoid culture or unconditioned control medium. To examine the impact of tumour organoid CM on myotube morphology in more detail, we monitored myotube formation using live cell imaging (Videos [Link], [Link], [Link], [Link]). Interestingly, compared with both control conditions, CM from PANCO‐9a and PANCO‐12a markedly accelerated the alignment of C2C12 myoblasts followed by strongly enhanced fusion into myotubes already after 48 h. Strikingly, detachment of large myotubes was frequently observed in the presence of tumour organoid‐derived CM, and occurred even prior to completion of myotube formation in control conditions (Figure S2). These observations prompted us to further investigate at which level myogenesis was affected by the pancreatic tumour organoid‐derived factors.
Figure 3.
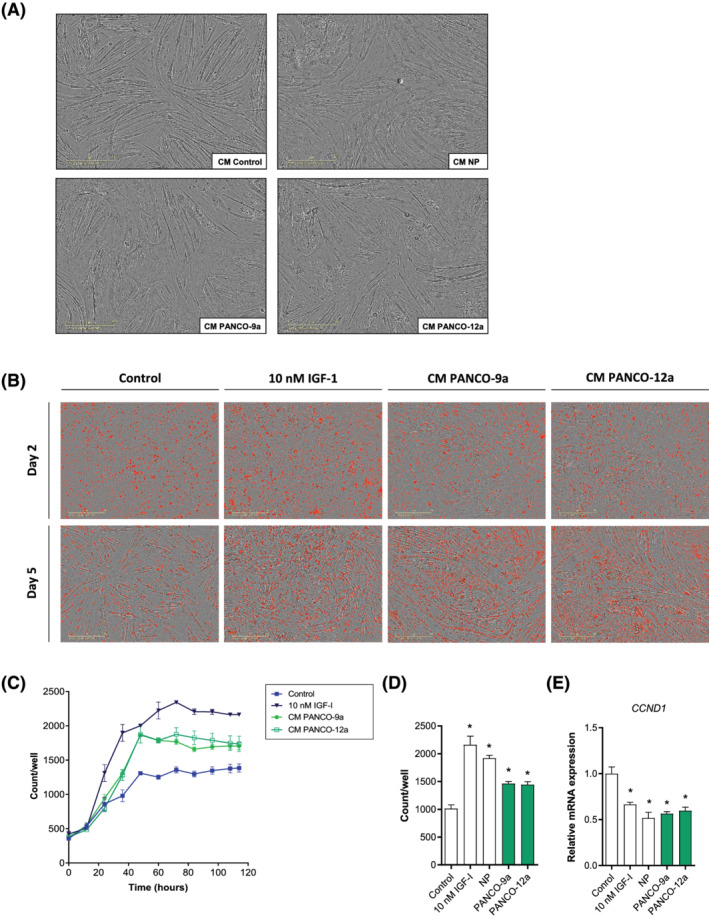
Tumour organoid‐derived CM transiently stimulates proliferation and increases fusion of myoblasts during differentiation. (A) Representative phase‐contrast images of C2C12 myoblasts differentiated for 5 days in DM control medium [DM, 50% (v/v) DMEM/F12] (control CM) or DM containing 50% (v/v) (tumour) organoid CM (NP; normal pancreas). Scale bar = 400 μm. (B) C2C12 myoblasts were differentiated in DM control medium [DM, 50% (v/v) DMEM/F12], DM containing 10 nM IGF‐1, or DM containing 50% (v/v) tumour organoid CM. NucLight rapid red was used to stain nuclei. Representative phase‐contrast images overlaid with red‐fluorescence images are presented. Scale bar = 400 μm. (C) The number of red stained nuclei on each individual image (12 h time interval) was quantified and plotted against time. (D) Bar graph showing the number of nuclei after 48 h of differentiation. Nuclei counts were obtained from three independent experiments. Data are presented as mean ± SEM. (E) mRNA expression of CCND1 was determined after 48 h. Data were normalized to CYPA, B2M, and RPLP0 reference genes and obtained from three independent experiments. Data are presented as mean ± SEM.
Transient increase of C2C12 myoblast proliferation by pancreatic tumour organoid‐derived factors
We first investigated myoblast proliferation during differentiation in the presence or absence of tumour organoid CM by live cell imaging of C2C12 cultures, using NucLight Red nuclear staining (Figure 3B). In all conditions, the number of cells increased throughout the first 48 h of differentiation, corresponding to the cell cycle exit which is required for terminal differentiation (Figure 3C–D). However, CM from both tumour organoid cultures significantly enhanced proliferation within the first 48 h in comparison with the unconditioned control medium (Figure 3C). The enhancement was intermediate to that observed after stimulation by IGF‐1, a highly potent inducer of myoblast proliferation and myogenesis (Figure 3D). CM from a normal pancreas organoid culture also stimulated myoblast proliferation (Figure 3D). As this was not followed by accelerated and increased myoblast fusion induced by PANCO‐9a and PANCO‐12a CM (Videos [Link], [Link], [Link], [Link]), these data suggest that enhanced myotube formation is unlikely to result from increased myoblast numbers but rather induced by tumour‐specific factors. Remarkably, transcript levels of cyclin D1 (CCND1), a gene with a cell cycle‐dependent expression pattern, was significantly decreased in C2C12 myoblasts differentiated with CM from PANCO‐9a (−1.8‐fold, P < 0.001) and PANCO‐12a (−1.7‐fold, P < 0.001) compared with control DM after 48 h (Figure 3E). This suggests that fewer cycling myoblasts remained, and that the proliferative effect of PANCO‐CM is transient in nature.
Increased expression of muscle‐specific genes by pancreatic tumour organoid‐derived factors
To investigate whether myogenic differentiation was affected by tumour organoid‐derived factors, we first validated the expression of key‐myogenic regulators, muscle specific genes, and genes involved in excitation‐contraction (EC)‐coupling in muscle (Figures 4A and S3), confirming the suitability of this system to monitor myogenesis in vitro. Next, C2C12 myoblasts were exposed to both CM from a normal pancreas organoid culture and tumour‐organoid CM from PANCO‐9a and PANCO‐12a during differentiation, and expression of transcription factor paired‐box 7 (Pax‐7), a satellite cell marker that has been postulated to be a critical mediator of asymmetric cell division of these muscle progenitor cells, 38 was monitored. After 48 h of differentiation, PAX7 was significantly downregulated (PANCO‐9a: −2.1‐fold, P < 0.001; and PANCO‐12a: −2.0‐fold, P < 0.001) compared with the control (Figure 4B). In addition, we observed significantly reduced mRNA levels of myogenic factor 5 (MYF5) (PANCO‐9a: −2.1‐fold, P < 0.001; and PANCO‐12a: −1.8‐fold, P = 0.01). After 48 h, mRNA expression levels of both myogenic differentiation 1 (MYOD) (PANCO‐9a: −1.4‐fold, P = 0.002; and PANCO‐12a: −1.4‐fold, P = 0.004) and myogenin (MYOG) (PANCO‐9a: −1.3‐fold, P = 0.01; and PANCO‐12a: −1.4‐fold, P = 0.006) were significantly lower after exposure to tumour organoid CM compared with CM from a normal pancreas organoid culture (Figure 4C). Furthermore, the transcript levels of muscle creatine kinase (MCK) were markedly elevated after exposure to tumour organoid CM (PANCO‐9a: 3.3‐fold, P = 0.005; and PANCO‐12a: 2.7‐fold, P = 0.032), indicating that myogenic differentiation was enhanced (Figure 4C). In line, mRNA levels of myomaker (MYMK) were significantly increased (PANCO‐9a: 2.4‐fold, P = 0.001; and PANCO‐12a: 2.2‐fold, P = 0.004) and corresponded to the strong increase in the number of nuclei residing inside myotubes (Figure 3C). Collectively, these data indicate that pancreatic tumour organoid‐derived factors accelerate the myogenic differentiation process.
Figure 4.
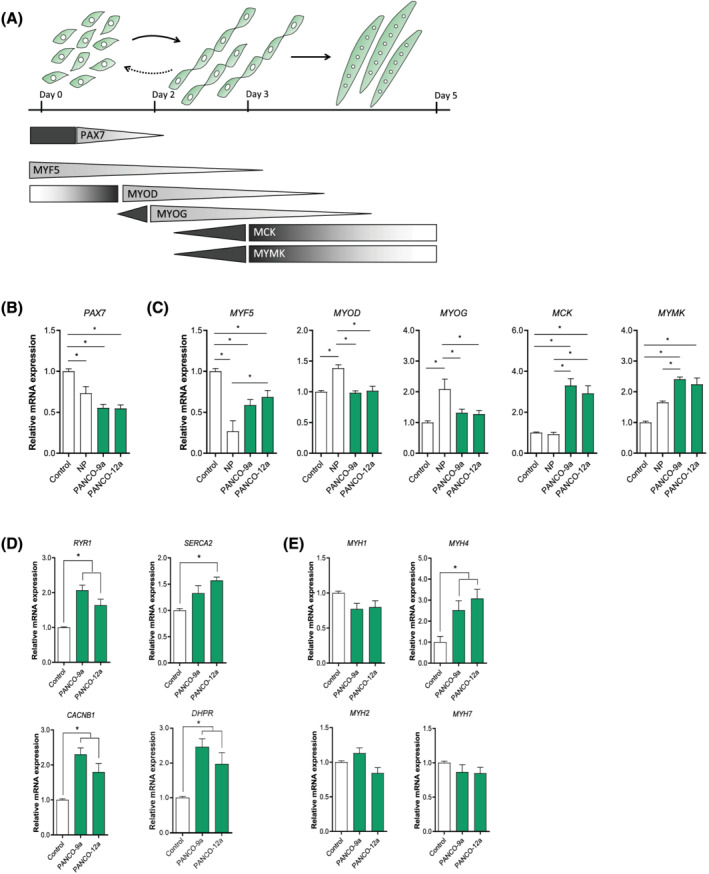
Enhanced myogenic differentiation induced by tumour organoid‐derived CM is accompanied by increased MyHC‐IIb expression and suppression of self‐renewal markers. (A) Graphical representation of the myogenic process. Myoblasts have the ability to proliferate, self‐renew, differentiate, and fuse into myotubes. During proliferation, myoblasts express the paired‐box 7 (PAX7) transcription factor. Upon myogenic commitment, a decrease in Pax7 expression results in cell cycle arrest, which is accompanied by increased expression of myogenic factor 5 (MYF5) and myoblast determination (MYOD) transcription factors. Myogenin (MYOG) is a transcription factor that is highly expressed during the fusion of myoblasts into myotubes. This protein results in the transcription of genes required for the fusion of myoblasts into myotubes, including myomaker (MYMK). mRNA expression of (B) satellite cell marker PAX7, (C) key‐myogenic regulators and muscle specific genes (MYF5, MYOD, MYOG, MCK, MYMK), and (D) Ca2+ flux regulating signalling channels (RYR1, SERCA2, CACNB1, DHPR) were determined after 48 h. (E) Expression of myosin heavy chain isoforms (MYH1, MYH2, MYH4, MYH7) was determined after 72 h. Data were normalized to CYPA, B2M, and RPLP0 reference genes and obtained from three independent experiments. Data are presented as mean ± SEM. CACNB1, calcium voltage‐gated channel auxiliary subunit beta1; DHPR, dihydropteridine reductase; MCK, muscle creatine kinase; MYH1, myosin heavy chain 1, MyHC‐IIx; MYH2, myosin heavy chain 2, MyHC‐IIa; MYH4, myosin heavy chain 4, MyHC‐IIb; MYH7, myosin heavy chain 7, MyHC‐I; RYR1, ryanodine receptor 1; SERCA2, sarcoplasmic/endoplasmic reticulum calcium ATPase 2.
Tumour organoid‐derived factors increase the expression of glycolytic fast‐twitch fibre MyHC‐IIB
Recent evidence indicates that disturbances of EC‐coupling may represent a common underlying phenomenon in the pathophysiology of many skeletal muscular disorders including cachexia and sarcopenia. 39 , 40 In particular, downregulation of Ca2+ flux‐regulating signalling channels impairs muscle function. However, in line with the enhanced myogenesis, we also observed significantly increased expression of Ca2+ flux regulating signalling channels (RYR1, SERCA2, DHPR, and CACNB1) in differentiating C2C12 myoblasts that were stimulated with CM from either tumour organoid (Figure 4D).
Finally, we investigated whether organoid‐derived CM affected the expression of genes encoding contractile proteins, focusing on myosin heavy chain (MyHC) isoforms. Compared with the control, mRNA expression levels of the glycolytic fast‐twitch myofibre MyHC‐IIB (MYH4) was significantly increased by CM from PANCO‐9a (2.5‐fold, P = 0.04) and PANCO‐12a (3.1‐fold, P = 0.006) after 72 h of differentiation (Figure 4E). Altogether, these data imply that pancreatic tumour organoid‐derived factors accelerate myogenesis, which is accompanied by a shift towards a myosin expression pattern that is frequently observed in cachexia.
Discussion
The loss of skeletal muscle is a clinically significant feature of cancer cachexia and is assumed to result from direct and/or indirect actions of tumour‐derived factors. At present, there is a lack of adequate pre‐clinical models that can be used to assess the direct effect of tumour‐derived factors of cachectic cancer patients on muscle wasting. In the current study, we first showed that the factors released by four different conventional human pancreatic cancer cell lines cultured in 2D had minimal and inconsistent effects on atrophy signalling pathways in skeletal muscle cells. Similarly, CM from pancreatic tumour organoids obtained from either a cachectic or non‐cachectic patient did not induce muscle atrophy signalling in mature C2C12 myotubes. Unexpectedly, however, pancreatic tumour organoid‐derived factors transiently increased proliferation of C2C12 myoblasts and accelerated their alignment and fusion. This enhanced myogenesis was accompanied by altered expression of factors governing muscle cell differentiation and a selective increase in MyHC‐IIB, which is preferentially expressed in atrophy‐sensitive glycolytic myofibres. Interestingly, these tumour organoid CM effects were not associated with the cachexia phenotype of the donor patients.
The loss of skeletal muscle tissue in cachexia has been suggested to result from tumour‐derived factors that activate the ubiquitin‐proteasome pathway to target major contractile skeletal muscle proteins for degradation. However, we found that the secretome of most conventionally cultured human pancreatic cancer cell lines only minimally affected the expression of the E3‐ubiquitin ligases Atrogin‐1/MAFbx and MuRF1 in mature C2C12 myotubes. Similar results were obtained in our pancreatic tumour organoid experiments. These data are not in line with previous in vitro studies that showed tumour‐induced muscle atrophy via activation of the ubiquitin proteasome pathway. For example, treatment of C2C12 mouse skeletal muscle myotubes with C26‐ and LLC‐derived factors has been reported to induce significant upregulation of the E3‐ubiquitin ligase Atrogin1/MAFbx, loss of myosin heavy chain proteins, and reduction of myofibre size. 41 , 42 However, in line with our results, Talbert et al. did not detect differential expression of these genes in muscle biopsies from cachectic versus non‐cachectic pancreatic cancer patients. 18 Similar observations were made in muscles from cachectic lung cancer patients: compared with healthy subjects, cachectic lung cancer patients showed no differences in E3 ubiquitin ligase expression despite reduced muscle mass, muscle fibre atrophy, and decreased quadriceps strength. 43 Furthermore, we did not observe noticeable morphological muscle cell atrophy after exposure to tumour cell‐derived or tumour organoid‐derived factors. Although this contrasts with findings of other studies, 8 , 41 , 42 our data are in line with a recent study of Guigni et al. in which the effect of murine and human lung tumour secreted factors on skeletal muscle was investigated. 44 Their report revealed that in fully matured C2C12 myotubes, myotube size and myosin heavy chain content were not affected by tumour‐derived factors. Altogether, these data do not support activation of the proteasome by E3‐ubiquitin ligases and subsequent myofibre atrophy as a direct effect of pancreas tumour‐derived factors. In line with these in vitro observations, we did not find evidence for the activation of the ubiquitin‐proteasome pathway (i.e. no upregulation of the E3 ubiquitin‐ligases Atrogin‐1/MAFbx and MuRF1) in the muscles of mice transplanted with PANCO‐9 and PANCO‐12 organoids, even though wet muscle weights generally tended to correlate with tumour weight (manuscript under review at JCSM*). Instead, other processes in muscle cells may be affected by tumour‐derived factors and contribute to the loss of muscle mass in human cancer‐induced cachexia.
One of these processes is myogenesis, which involves activation and asymmetric cell division of satellite cells, followed by either repletion of this pool of quiescent muscle precursor cells, or by proliferation of daughter myoblasts and subsequent myogenic differentiation and fusion into myotubes. 45 Counterintuitively, pancreatic tumour‐derived factors provoked the development of both enlarged and disrupted myotubes after 5 days of differentiation. Live cell imaging‐based monitoring of morphological changes of C2C12 myoblasts during differentiation revealed that the disrupted myotube morphology resulted from early detachment of large myotubes which was followed by formation of new myotubes. This was preceded by accelerated alignment of C2C12 myoblasts and increased expression of muscle‐specific genes. Given that the myogenic process is driven by highly dynamic and transient changes in these muscle‐specific genes, 46 the mere evaluation of expression levels of these genes at a single point in time only cannot discriminate between an impaired myogenic programme or an accelerated myogenic programme. Our data therefore also underscore the importance of monitoring the morphological changes during the myogenic process by live‐cell imaging. Furthermore, the transient increase in myoblast proliferation in the presence of tumour‐derived factors that we observed is in line with previous reports on increases in the number of proliferating muscle precursor cells in atrophying muscle. 15 , 16 Similar observations have previously been made in cancer cachexia studies. For example, increased numbers of Pax‐7 expressing cells have been found in the muscle of tumour‐bearing animals and cachectic pancreatic cancer patients, which was interpreted as an aberrant proliferative response of muscle precursor cells. 14 , 36 While we found that myoblast proliferation initially increased in response to tumour‐derived factors, cyclin D1 expression was markedly reduced thereafter, in line with aberrant myoblast proliferative kinetics. In vivo, myoblast proliferation is subject to asymmetric cell division by activated muscle progenitor cells, governed by Pax‐7 expression. 45 Upon entering the myogenic programme, myoblasts committing to terminal differentiation lose Pax‐7 expression, whereas Pax‐7 expression is maintained by myoblasts that become quiescent to replete the satellite cell population. The reduced Pax‐7 expression in myoblast cultures in response to the tumour organoid‐derived factors may therefore reflect a depletion of the population of replication competent cells, referred to as ‘reserve cells’, which represent the in vitro equivalents of quiescent satellite cells. 47 In vivo, such a depletion of muscle precursor cells will interfere with long‐term myogenic potential, ultimately contributing to muscle mass loss. In support of this notion, cultured muscle precursor cells isolated from cachectic tumour‐bearing mice have been reported to differentiate more readily compared with those of control mice. 36 , 48 Taken together, these data imply that in response to tumour‐derived factors, muscle precursor cells are directed towards myogenic commitment at the expense of the quiescent satellite cell repopulation, which may contribute to the impaired muscle mass maintenance that is observed in cachexia.
Next to the altered myogenesis, we observed increased levels of the glycolytic fast‐twitch myofibre MyHC‐IIB (MYH4) in response to tumour organoid‐derived factors. This is consistent with the shift in muscle fibre type composition that has been reported in mice bearing C26 carcinomas: the type IIb myosin isoform comprised 19% of their total myosin fibre content, whereas it was not detected in control mice. 49 Because it has been suggested that type 2 muscle fibres are more sensitive to atrophy‐inducing stimuli, a tumour factor‐driven shift towards more fast twitch MyHC‐II expression may sensitize muscle fibres to additional catabolic triggers in cancer cachexia. 50 , 51 In line with this, wasting of muscle fibres in cancer cachexia is more pronounced in fast‐twitch type II‐containing muscles, such as tibialis anterior and gastrocnemius, compared with slow‐twitch type I muscles, such as the soleus. 52 , 53 This implies that pancreatic tumour organoid‐derived factors could promote the development of muscle fibres that are more prone to atrophy‐inducing stimuli.
Interestingly, the pronounced effects of tumour organoid‐derived factors on myogenesis and myosin heavy chain expression were not related to the cachexia status of the patients contributing the organoid cultures. However, despite the fact that patient PANCO‐9 was considered cachectic and patient PANCO‐12 was considered non‐cachectic based on the consensus definition, close evaluation of the actual cachexia‐related phenotype of these patients shows that they both had a skeletal muscle index below the sarcopenia cut‐off. 26 In addition, only CM from PANCO‐12a, originating from a non‐cachectic patient, caused mild NF‐κB activation in mature C2C12 myotubes. We have previously shown that PANCO‐12a organoids express relatively higher levels of interleukin 1 (IL‐1) and TNF‐α, known inducers of NF‐κB, which may explain the increased NF‐κB activity in myotubes after exposure to PANCO‐12a CM. 26
In conclusion, we here showed that pancreatic tumour organoid‐derived factors do not promote upregulation of skeletal muscle specific E3 ubiquitin ligases, but rather alter the kinetics of myogenesis. Given these data, more detailed analyses of the organoid secretome to identify the factors responsible are warranted. These studies will also help to distinguish direct and indirect effects of tumour‐derived factors on skeletal muscle (patho)physiology in relation to the cachexia status of the donor patient, thereby contributing to a better understanding of cachexia‐inducing mechanisms.
Conflict of interest
The authors have nothing to disclose.
Funding
R.D.W. Vaes is supported as a PhD candidate by the NUTRIM Graduate Programme.
Supporting information
Figure S1. Tumour‐derived factors from pancreatic tumour organoids do not induce muscle atrophy signalling in human primary myotubes. Human C25 myoblasts (HSM) were cultured as described previously (Kneppers et al. Skeletal Muscle (2018) 8:4). HSM cells were differentiated for 5 days in DM and subsequently mature HSM myotubes were treated for 24 h with 50% (v/v) organoid CM from PANCO‐9a and PANCO‐12a. Data were normalized to CYPA, B2M, and RPLP0 reference genes. Data are presented as mean ± SEM.
Figure S2. Tumour organoid‐derived CM induces morphological disturbances during the differentiation of myoblasts into myotubes. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) organoid CM (PANCO‐9a and PANCO‐12a). Sequential images (2 h time interval) show the detachment of large myotubes during differentiation. Scale bar = 400 μm.
Figure S3. Gene expression of myogenic markers during myogenesis. C2C12 myoblasts were differentiated for 3 days in DM and cell lysates were collected at 0 h, 48 h, and 72 h after differentiation was initiated. mRNA expression of A) PAX7, B) key‐myogenic regulators and muscle‐specific genes (MYF5, MYOD, MYOG, MYMK, MCK), C) genes involved in excitation‐contraction (EC)‐coupling in muscle (RYR1, SERCA2, DHPR, CACNB1), and D) myosin heavy chain isoforms (MYH1, MYH2, MYH4, MYH7) were determined. Data were normalized to CYPA, B2M, and RPLP0 reference genes and obtained from three independent experiments. Data are presented as mean ± SEM.
Video S1. Myogenic differentiation – Control. C2C12 myoblasts were differentiated for 5 days in DM control medium (DM, 50% (v/v) DMEM/F12). Scale bar = 400 μm.
Video S2. Myogenic differentiation – CM normal pancreas. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) CM from a normal pancreas organoid culture. Scale bar = 400 μm.
Video S3. Myogenic differentiation – CM PANCO‐9a. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) PANCO‐9a‐derived CM. Scale bar = 400 μm.
Video S4. Myogenic differentiation – CM PANCO‐12a. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) PANCO‐12a‐derived CM. Scale bar = 400 μm.
Table S1. qPCR mouse primer sequences
Acknowledgements
We would like to thank Bas Boonen and Marco Kelders for their excellent technical assistance. The authors certify that they comply with the ethical guidelines for authorship and publishing of the Journal of Cachexia, Sarcopenia, and Muscle. 54
Vaes R. D. W., van Dijk D. P. J., Farshadi E. A., Olde Damink S. W. M., Rensen S. S., and Langen R. C. (2022) Human pancreatic tumour organoid‐derived factors enhance myogenic differentiation, Journal of Cachexia, Sarcopenia and Muscle, 13, 1302–1313, 10.1002/jcsm.12917
Sander S. Rensen and Ramon C. Langen shared senior authorship.
References
- 1. Bray F, Ferlay J, Soerjomataram I, Siegel RL, Torre LA, Jemal A. Global cancer statistics 2018: GLOBOCAN estimates of incidence and mortality worldwide for 36 cancers in 185 countries. CA Cancer J Clin 2018;68:394–424. [DOI] [PubMed] [Google Scholar]
- 2. Kleeff J, Korc M, Apte M, La Vecchia C, Johnson CD, Biankin AV, et al. Pancreatic cancer. Nat Rev Dis Primers 2016;2:16022. [DOI] [PubMed] [Google Scholar]
- 3. Baracos VE, Martin L, Korc M, Guttridge DC, Fearon KCH. Cancer‐associated cachexia. Nat Rev Dis Primers 2018;4:17105. [DOI] [PubMed] [Google Scholar]
- 4. Fearon K, Strasser F, Anker SD, Bosaeus I, Bruera E, Fainsinger RL, et al. Definition and classification of cancer cachexia: an international consensus. Lancet Oncol 2011;12:489–495. [DOI] [PubMed] [Google Scholar]
- 5. Callaway CS, Delitto AE, Patel R, Nosacka RL, D'Lugos AC, Delitto D, et al. IL‐8 released from human pancreatic cancer and tumor‐associated stromal cells signals through a CXCR2‐ERK1/2 axis to induce muscle atrophy. Cancers (Basel) 2019;11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Delitto D, Judge SM, Delitto AE, Nosacka RL, Rocha FG, DiVita BB, et al. Human pancreatic cancer xenografts recapitulate key aspects of cancer cachexia. Oncotarget 2017;8:1177–1189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Henderson SE, Makhijani N, Mace TA. Pancreatic cancer‐induced cachexia and relevant mouse models. Pancreas 2018;47:937–945. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Jackman RW, Floro J, Yoshimine R, Zitin B, Eiampikul M, El‐Jack K, et al. Continuous release of tumor‐derived factors improves the modeling of cachexia in muscle cell culture. Front Physiol 2017;8:738. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Kandarian SC, Nosacka RL, Delitto AE, Judge AR, Judge SM, Ganey JD, et al. Tumour‐derived leukaemia inhibitory factor is a major driver of cancer cachexia and morbidity in C26 tumour‐bearing mice. J Cachexia Sarcopenia Muscle 2018;9:1109–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Zhang G, Liu Z, Ding H, Zhou Y, Doan HA, Sin KWT, et al. Tumor induces muscle wasting in mice through releasing extracellular Hsp70 and Hsp90. Nat Commun 2017;8:589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Wyke SM, Russell ST, Tisdale MJ. Induction of proteasome expression in skeletal muscle is attenuated by inhibitors of NF‐κB activation. Br J Cancer 2004;91:1742–1750. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Acharyya S, Butchbach ME, Sahenk Z, Wang H, Saji M, Carathers M, et al. Dystrophin glycoprotein complex dysfunction: a regulatory link between muscular dystrophy and cancer cachexia. Cancer Cell 2005;8:421–432. [DOI] [PubMed] [Google Scholar]
- 13. Cai D, Frantz JD, Tawa NE Jr, Melendez PA, Oh BC, Lidov HG, et al. IKKbeta/NF‐kappaB activation causes severe muscle wasting in mice. Cell 2004;119:285–298. [DOI] [PubMed] [Google Scholar]
- 14. Talbert EE, Metzger GA, He WA, Guttridge DC. Modeling human cancer cachexia in colon 26 tumor‐bearing adult mice. J Cachexia Sarcopenia Muscle 2014;5:321–328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Inaba S, Hinohara A, Tachibana M, Tsujikawa K, Fukada SI. Muscle regeneration is disrupted by cancer cachexia without loss of muscle stem cell potential. PLoS ONE 2018;13:e0205467. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Penna F, Costamagna D, Fanzani A, Bonelli G, Baccino FM, Costelli P. Muscle wasting and impaired myogenesis in tumor bearing mice are prevented by ERK inhibition. PLoS ONE 2010;5:e13604. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Wang G, Biswas AK, Ma W, Kandpal M, Coker C, Grandgenett PM, et al. Metastatic cancers promote cachexia through ZIP14 upregulation in skeletal muscle. Nat Med 2018;24:770–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Talbert EE, Cuitino MC, Ladner KJ, Rajasekerea PV, Siebert M, Shakya R, et al. Modeling human cancer‐induced cachexia. Cell Rep 2019;28:1612–22 e4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Shukla SK, Gebregiworgis T, Purohit V, Chaika NV, Gunda V, Radhakrishnan P, et al. Metabolic reprogramming induced by ketone bodies diminishes pancreatic cancer cachexia. Cancer Metab 2014;2:18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Togashi Y, Kogita A, Sakamoto H, Hayashi H, Terashima M, de Velasco MA, et al. Activin signal promotes cancer progression and is involved in cachexia in a subset of pancreatic cancer. Cancer Lett 2015;356:819–827. [DOI] [PubMed] [Google Scholar]
- 21. Baker BM, Chen CS. Deconstructing the third dimension: how 3D culture microenvironments alter cellular cues. J Cell Sci 2012;125:3015–3024. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Ben‐David U, Siranosian B, Ha G, Tang H, Oren Y, Hinohara K, et al. Genetic and transcriptional evolution alters cancer cell line drug response. Nature 2018;560:325–330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Boj SF, Hwang CI, Baker LA, Chio II, Engle DD, Corbo V, et al. Organoid models of human and mouse ductal pancreatic cancer. Cell 2015;160:324–338. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Clevers H. Modeling development and disease with organoids. Cell 2016;165:1586–1597. [DOI] [PubMed] [Google Scholar]
- 25. Lancaster MA, Huch M. Disease modelling in human organoids. Dis Model Mech 2019;12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. Vaes RDW, van Dijk DPJ, Welbers TTJ, Blok MJ, Aberle MR, Heij L, et al. Generation and initial characterization of novel tumour organoid models to study human pancreatic cancer‐induced cachexia. J Cachexia Sarcopenia Muscle 2020;11:1509–1524. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Lieber M, Mazzetta J, Nelson‐Rees W, Kaplan M, Todaro G. Establishment of a continuous tumor‐cell line (PANC‐1) from a human carcinoma of the exocrine pancreas. Int J Cancer 1975;15:741–747. [DOI] [PubMed] [Google Scholar]
- 28. Kobari M, Matsuno S, Sato T, Kan M, Tachibana T. Establishment of a human pancreatic cancer cell line and detection of pancreatic cancer associated antigen. Tohoku J Exp Med 1984;143:33–46. [DOI] [PubMed] [Google Scholar]
- 29. Kimura Y, Kobari M, Yusa T, Sunamura M, Kimura M, Shimamura H, et al. Establishment of an experimental liver metastasis model by intraportal injection of a newly derived human pancreatic cancer cell line (KLM‐1). Int J Pancreatol 1996;20:43–50. [DOI] [PubMed] [Google Scholar]
- 30. Langen RC, Schols AM, Kelders MC, Wouters EF, Janssen‐Heininger YM. Inflammatory cytokines inhibit myogenic differentiation through activation of nuclear factor‐κB. FASEB J 2001;15:1169–1180. [DOI] [PubMed] [Google Scholar]
- 31. Ruijter JM, Ramakers C, Hoogaars WM, Karlen Y, Bakker O, van den Hoff MJ, et al. Amplification efficiency: linking baseline and bias in the analysis of quantitative PCR data. Nucleic Acids Res 2009;37:e45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Bodine SC, Baehr LM. Skeletal muscle atrophy and the E3 ubiquitin ligases MuRF1 and MAFbx/atrogin‐1. Am J Physiol Endocrinol Metab 2014;307:E469–E484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Britto FA, Cortade F, Belloum Y, Blaquiere M, Gallot YS, Docquier A, et al. Glucocorticoid‐dependent REDD1 expression reduces muscle metabolism to enable adaptation under energetic stress. BMC Biol 2018;16:65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Gordon BS, Williamson DL, Lang CH, Jefferson LS, Kimball SR. Nutrient‐induced stimulation of protein synthesis in mouse skeletal muscle is limited by the mTORC1 repressor REDD1. J Nutr 2015;145:708–713. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35. Arneson PC, Doles JD. Impaired muscle regeneration in cancer‐associated cachexia. Trends Cancer 2019;5:579–582. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. He WA, Berardi E, Cardillo VM, Acharyya S, Aulino P, Thomas‐Ahner J, et al. NF‐κB‐mediated Pax7 dysregulation in the muscle microenvironment promotes cancer cachexia. J Clin Invest 2013;123:4821–4835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Hogan KA, Cho DS, Arneson PC, Samani A, Palines P, Yang Y, et al. Tumor‐derived cytokines impair myogenesis and alter the skeletal muscle immune microenvironment. Cytokine 2018;107:9–17. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Kuang S, Kuroda K, Le Grand F, Rudnicki MA. Asymmetric self‐renewal and commitment of satellite stem cells in muscle. Cell 2007;129:999–1010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Agrawal A, Suryakumar G, Rathor R. Role of defective Ca2+ signaling in skeletal muscle weakness: Pharmacological implications. J Cell Commun Signal 2018;12:645–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Fontes‐Oliveira CC, Busquets S, Fuster G, Ametller E, Figueras M, Olivan M, et al. A differential pattern of gene expression in skeletal muscle of tumor‐bearing rats reveals dysregulation of excitation‐contraction coupling together with additional muscle alterations. Muscle Nerve 2014;49:233–248. [DOI] [PubMed] [Google Scholar]
- 41. Zhang G, Jin B, Li YP. C/EBPβ mediates tumour‐induced ubiquitin ligase atrogin1/MAFbx upregulation and muscle wasting. EMBO J 2011;30:4323–4335. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Zhang G, Liu Z, Ding H, Miao H, Garcia JM, Li YP. Toll‐like receptor 4 mediates Lewis lung carcinoma‐induced muscle wasting via coordinate activation of protein degradation pathways. Sci Rep 2017;7:2273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43. Op den Kamp CM, Langen RC, Snepvangers FJ, de Theije CC, Schellekens JM, Laugs F, et al. Nuclear transcription factor κB activation and protein turnover adaptations in skeletal muscle of patients with progressive stages of lung cancer cachexia. Am J Clin Nutr 2013;98:738–748. [DOI] [PubMed] [Google Scholar]
- 44. Guigni BA, van der Velden J, Kinsey CM, Carson JA, Toth MJ. Effects of conditioned media from murine lung cancer cells and human tumor cells on cultured myotubes. Am J Physiol Endocrinol Metab 2020;318:E22–E32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45. Motohashi N, Asakura A. Muscle satellite cell heterogeneity and self‐renewal. Front Cell Dev Biol 2014;2:1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Bentzinger CF, Wang YX, Rudnicki MA. Building muscle: molecular regulation of myogenesis. Cold Spring Harb Perspect Biol 2012;4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47. Yoshida N, Yoshida S, Koishi K, Masuda K, Nabeshima Y. Cell heterogeneity upon myogenic differentiation: down‐regulation of MyoD and Myf‐5 generates 'reserve cells'. J Cell Sci 1998;111:769–779. [DOI] [PubMed] [Google Scholar]
- 48. Talbert EE, Guttridge DC. Impaired regeneration: a role for the muscle microenvironment in cancer cachexia. Semin Cell Dev Biol 2016;54:82–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Diffee GM, Kalfas K, Al‐Majid S, McCarthy DO. Altered expression of skeletal muscle myosin isoforms in cancer cachexia. Am J Physiol Cell Physiol 2002;283:C1376–C1382. [DOI] [PubMed] [Google Scholar]
- 50. Matsakas A, Patel K. Skeletal muscle fibre plasticity in response to selected environmental and physiological stimuli. Histol Histopathol 2009;24:611–629. [DOI] [PubMed] [Google Scholar]
- 51. Schiaffino S, Reggiani C. Fiber types in mammalian skeletal muscles. Physiol Rev 2011;91:1447–1531. [DOI] [PubMed] [Google Scholar]
- 52. Acharyya S, Ladner KJ, Nelsen LL, Damrauer J, Reiser PJ, Swoap S, et al. Cancer cachexia is regulated by selective targeting of skeletal muscle gene products. J Clin Invest 2004;114:370–378. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Mendell JR, Engel WK. The fine structure of type II muscle fiber atrophy. Neurology 1971;21:358–365. [DOI] [PubMed] [Google Scholar]
- 54. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2021. J Cachexia Sarcopenia Muscle 2021;12:2259–2261. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Tumour‐derived factors from pancreatic tumour organoids do not induce muscle atrophy signalling in human primary myotubes. Human C25 myoblasts (HSM) were cultured as described previously (Kneppers et al. Skeletal Muscle (2018) 8:4). HSM cells were differentiated for 5 days in DM and subsequently mature HSM myotubes were treated for 24 h with 50% (v/v) organoid CM from PANCO‐9a and PANCO‐12a. Data were normalized to CYPA, B2M, and RPLP0 reference genes. Data are presented as mean ± SEM.
Figure S2. Tumour organoid‐derived CM induces morphological disturbances during the differentiation of myoblasts into myotubes. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) organoid CM (PANCO‐9a and PANCO‐12a). Sequential images (2 h time interval) show the detachment of large myotubes during differentiation. Scale bar = 400 μm.
Figure S3. Gene expression of myogenic markers during myogenesis. C2C12 myoblasts were differentiated for 3 days in DM and cell lysates were collected at 0 h, 48 h, and 72 h after differentiation was initiated. mRNA expression of A) PAX7, B) key‐myogenic regulators and muscle‐specific genes (MYF5, MYOD, MYOG, MYMK, MCK), C) genes involved in excitation‐contraction (EC)‐coupling in muscle (RYR1, SERCA2, DHPR, CACNB1), and D) myosin heavy chain isoforms (MYH1, MYH2, MYH4, MYH7) were determined. Data were normalized to CYPA, B2M, and RPLP0 reference genes and obtained from three independent experiments. Data are presented as mean ± SEM.
Video S1. Myogenic differentiation – Control. C2C12 myoblasts were differentiated for 5 days in DM control medium (DM, 50% (v/v) DMEM/F12). Scale bar = 400 μm.
Video S2. Myogenic differentiation – CM normal pancreas. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) CM from a normal pancreas organoid culture. Scale bar = 400 μm.
Video S3. Myogenic differentiation – CM PANCO‐9a. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) PANCO‐9a‐derived CM. Scale bar = 400 μm.
Video S4. Myogenic differentiation – CM PANCO‐12a. C2C12 myoblasts were differentiated for 5 days in DM containing 50% (v/v) PANCO‐12a‐derived CM. Scale bar = 400 μm.
Table S1. qPCR mouse primer sequences