

Abstract
Background
Skeletal muscle atrophy is a common clinical manifestation of various neurotrauma and neurological diseases. In addition to the treatment of primary neuropathies, it is a clinical condition that should be investigated. FoxO3 activation is an indispensable mechanism in denervation‐induced muscle atrophy; however, upstream factors that control FoxO3 expression and activity have not been fully elucidated. N6‐methyladenosine (m6A) methylation is a novel mode of epitranscriptional gene regulation that affects several cellular processes. However, the biological significance of m6A modification in FoxO3‐dependent atrophy is unknown.
Methods
We performed gain‐of‐function and loss‐of‐function experiments and used denervation‐induced muscle atrophy mouse model to evaluate the effects of m6A modification on muscle mass control and FoxO3 activation. m6A‐sequencing and mass spectrometry analyses were used to establish whether histone deacetylase 4 (HDAC4) is a mediator of m6A demethylase ALKBH5 regulation of FoxO3. A series of cellular and molecular biological experiments (western blot, immunoprecipitation, half‐life assay, m6A‐MeRIP‐qPCR, and luciferase reporter assays among others) were performed to investigate regulatory relationships among ALKBH5, HDAC4, and FoxO3.
Results
In skeletal muscles, denervation was associated with a 20.7–31.9% decrease in m6A levels (P < 0.01) and a 35.6–115.2% increase in demethylase ALKBH5 protein levels (P < 0.05). Overexpressed ALKBH5 reduced m6A levels, activated FoxO3 signalling, and induced excess loss in muscle wet weight (−10.3% for innervation and −11.4% for denervation, P < 0.05) as well as a decrease in myofibre cross‐sectional areas (−35.8% for innervation and −33.3% for denervation, P < 0.05) during innervation and denervation. Specific deletion of Alkbh5 in the skeletal muscles prevented FoxO3 activation and protected mice from denervation‐induced muscle atrophy, as evidenced by increased muscle mass (+16.0%, P < 0.05), size (+50.0%, P < 0.05) and MyHC expression (+32.6%, P < 0.05). Mechanistically, HDAC4 was established to be a crucial central mediator for ALKBH5 in enhancing FoxO3 signalling in denervated muscles. ALKBH5 demethylates and stabilizes Hdac4 mRNA. HDAC4 interacts with and deacetylates FoxO3, resulting in a significant increase in FoxO3 expression (+61.3–82.5%, P < 0.01) and activity (+51.6–122.0%, P < 0.001).
Conclusions
Our findings elucidate on the roles and mechanisms of ALKBH5‐mediated m6A demethylation in the control of muscle mass during denervation and activation of FoxO3 signalling by targeting HDAC4. These results suggest that ALKBH5 is a potential therapeutic target for neurogenic muscle atrophy.
Keywords: Muscle atrophy, Denervation, FoxO3, m6A modification, ALKBH5, HDAC4
Introduction
The skeletal muscle is the final executive organ of the human body for voluntary movement and postural support. Maintenance of its morphological structure and functional state depends on normal motor nerve innervation. However, under various pathological conditions, such as peripheral nerve injury, motor neuron and neuromuscular junction diseases, normal nerve signal transduction and neuronutritional supply of skeletal muscles are disturbed. Therefore, patients suffer from severe progressive muscle mass loss and weakness, known as denervation‐induced muscle atrophy. 1 Notably, nerve repair takes a long time. Primary neuropathy treatment does not significantly alleviate the problem as the muscle structure is seriously damaged due to long denervation periods. Therefore, the original shape and function of muscle fibres are not restored after they regain motor nerve signals. This implies that prevention of muscle atrophy is challenging in addition to treatment of primary neuropathy. Currently, due to the lack of a deep understanding of the regulatory mechanisms of skeletal muscle mass, there are no effective measures for preventing neurogenic muscle atrophy.
Overactivation of the ubiquitin‐proteasome proteolytic system is a major cellular process that results in denervation‐induced muscle atrophy, 2 , 3 and its activation is significantly correlated with upregulation of atrophy‐related genes (atrogenes), including Atrogin1, MuRF1, MUSA1, SMART. 4 , 5 Moreover, the transcription factor FoxO3, a member of Forkhead box (Fox) family, plays an important role in regulation of atrogene expression. FoxO3 binds promoter regions and activates atrogene transcription, resulting in the occurrence and progression of skeletal muscle atrophy induced by various pathological states. 5 , 6 , 7 , 8 However, as a human longevity gene, inhibition of FoxO3 activity increases the risk of age‐associated diseases. 9 Notably, development of inhibitors with the ability to target transcription factors is complex and challenging. 10 Therefore, studies should investigate upstream factors that regulate activation of FoxO3 signalling in denervation‐induced muscle atrophy to develop alternative therapies.
Post‐transcriptional modification of messenger RNA (mRNA) is a key mechanism in the control of gene expression levels. Among the more than 100 modifications in mRNA, N6‐methyladenosine (m6A) methylation is the most characterized and abundant post‐transcriptional modification in eukaryotic mRNA. In mammals, m6A is catalysed by RNA methyltransferase‐like 3 (METTL3) and methyltransferase‐like 14 (METTL14) (writers), is removed by demethylases fat mass and obesity‐associated protein (FTO) and alkB homologue 5 (ALKBH5) (erasers), and interacts with m6A‐binding proteins with YTH domain, such as YTHDF1/2/3, YTHDC1/2 (readers). 11 These three effectors regulate various cellular processes by dynamically modulating RNA splicing, translocation, stability, and translation efficiency. 12
In recent years, various studies have evaluated the biological functions of m6A methylation in mRNA. m6A is implicated in various physiological and pathological processes, including stem cell fate determination, 13 neural development, 14 cardiac hypertrophy, 15 tumorigenesis, and metastasis. 16 Dynamic m6A modification, mediated by METTL3 and FTO, plays an important role in promoting myogenic differentiation and muscle development. 17 , 18 Additionally, ALKBH5‐mediated m6A demethylation enhances FOXM1 expression in cancer cells, 19 , 20 suggesting that the Fox protein family is the likely potential target that is regulated by m6A. However, it has not been determined whether m6A is involved in the control of adult muscle mass, and whether it affects FoxO3‐dependent neurogenic muscle atrophy.
In this study, we found that ALKBH5‐meditaed m6A demethylation aggravates muscle mass loss during denervation and that HDAC4/FoxO3 signalling axis is an important pathways downstream of ALKBH5. Our findings provide a more promising enzyme treatment target for neurogenic muscle atrophy.
Materials and methods
An extended materials and methods section can be found online in the Supporting information.
Generation of muscle‐specific Alkbh5 knockout mice
Mice bearing the Alkbh5‐floxed allele (Alkbh5 fl/fl , Cyagen) were crossed with transgenic mice expressing Cre recombinase under the control of the Myl1 promoter (Myl1‐Cre; Stock No: 024713, The Jackson Laboratory) to generate muscle‐specific Alkbh5 knockout mice (Myl1‐Cre;Alkbh5 fl/fl ). Littermate Alkbh5 fl/fl mice were used as controls. Genotyping by tail DNA and PCR were performed at 4 weeks of age. The primers were shown in Table S1. Mice were housed under specific pathogen‐free conditions at 24 ± 2 °C with a 12:12 h light–dark cycle and ad libitum access to food and water. All experimental animal procedures strictly adhered to the National Institutes of Health Guide for the Care and Use of Laboratory Animals and approved by Jinan University Laboratory Animal Ethics Committee (IACUC Issue No. 20200318‐36).
m6A sequencing (m6A‐seq) and data analysis
m6A‐seq and data analysis were performed by Shanghai Jiayin Biotechnology Ltd. Briefly, total RNA from 2 week‐denervated gastrocnemius (GAS) muscle was extracted using the TRIzol reagent (Invitrogen), and fragmented into ∼200‐nucleotide‐long fragments. Approximately 5% of the fragmented RNA was used as the input RNA. Other RNA fragments were analysed by immunoprecipitation using anti‐m6A polyclonal antibodies. Sequencing was performed using an Illumina NovaSeq 6000 platform. m6A‐seq data were analysed as previously described. 21 m6A peaks with false discovery rate (FDR) < 0.05 were detected using transcriptome based peak caller MetPeak, and the top 5000 peaks were chosen for de novo motif analysis with MEME, which takes 200‐nt‐long peak summit‐centred sense sequences as input. Peak files of different groups were merged using the merge in bedtools toolset. Signals in two different groups on every merged m6A peaks was calculated using UCSC tools. m6A peaks were identified as differential peaks if they had m6A enrichment score fold change (FC) ≥ 2 and average m6A score greater than 6. Annotation of m6A peaks on the whole genome was obtained using ChIPseeker.
Liquid chromatography–tandem mass spectrometry for determination of m6A/A ratio
Approximately 500 ng of purified mRNA was digested with Nuclease P1 (1 U; Sigma) in 25 μL of reaction buffer (10 mM NaCl, 2.0 mM ZnCl2) containing 10 mM NH4OAc (pH 5.3) at 42°C for 2 h, followed by addition of NH4HCO3 (3 μL, 3 M) and alkaline phosphatase (1 μL, 1 U/μL; Sigma) and incubation at 37°C for 2 h. After neutralization using 1 μL HCl (3 M), samples were diluted to 50 μL and filtered (0.22 μm, Millipore). Then 5 μL of the solution was used as the injection volume for liquid chromatography–tandem mass spectrometry (LC–MS/MS). Nucleosides were separated by reverse‐phase ultra‐performance liquid chromatography on a C18 column (Phenomenex), followed by online mass spectrometry detection using a Thermo Fisher Q Exactive Focus Orbitrap UPLC mass spectrometer via the positive electrospray ionization mode. All nucleosides were quantified based on retention time and nucleoside‐to‐base ion mass transitions of 282 to 150 (m6A) and 268 to 136 (A). A standard curve was generated using pure nucleoside standards running in the same batch of samples. The ratio of m6A to A was calculated based on calibrated concentrations.
m6A dot blot assay
After being denatured by heating at 72°C for 5 min, mRNA in a volume of 1.5 μL was spotted on Hybond Nitrocellulose Membranes (Pall) and cross‐linked to the membrane by UV. One of the membranes was blocked using 5% non‐fat dry milk in TBST for 1 h at room temperature and inducted with rabbit anti‐m6A antibody (1:3000; Synaptic Systems) overnight at 4°C. Next day, the membrane was inducted with HRP‐conjugated goat anti‐rabbit IgG (1:5000) for 2 h at room temperature, then visualized on GNOME XRQ NPC (Syngene) using ECL chemiluminescence (Millipore). The other membrane was stained with 0.02% methylene blue in 0.3 M sodium acetate (pH 5.2) for 2 h as the loading control.
Statistical analysis
Statistical analyses were performed using the SPSS 13.0 software. Statistically significant differences between experimental groups were determined by a two‐tailed Student's t test or one‐way analysis of variance (ANOVA) followed by Bonferroni's multiple comparisons test. Data are presented as the mean ± SD or mean ± SEM (*P < 0.05; **P < 0.01; and ***P < 0.001).
Results
Suppressed m6A levels in denervated muscles were correlated with upregulation of ALKBH5
The LC–MS/MS and dot blot showed that m6A modification levels in muscles at different days after denervation were significantly low, compared with normal muscles (Figure 1A). Meanwhile, suppressed m6A levels correlated with increased demethylase ALKBH5 levels in denervated muscles, whereas changes in METTL3, METTL14, and FTO levels after denervation were not significant (Figure 1B–1E). Notably, upregulation of ALKBH5 was consistent with upregulation of FoxO3 on different days post‐denervation (Figure 1D and 1E). These findings show a strong association between ALKBH5‐mediated m6A demethylation with muscle wasting and FoxO3 expression, implying that ALKBH5 plays an important role in FoxO3‐dependent neurogenic muscle atrophy.
Figure 1.
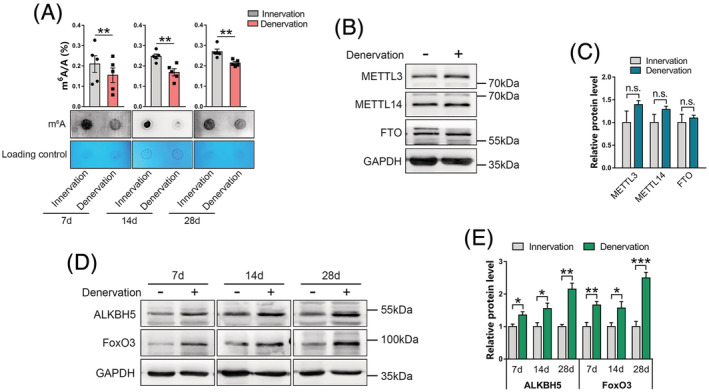
Decrease in m6A levels in denervated muscles is accompanied by upregulation of ALKBH5. (A) m6A mRNA levels in gastrocnemius (GAS) muscles on Days 7, 14, and 28 post‐denervation as determined using liquid chromatography–tandem mass spectrometry (LC–MS/MS) (up) and dot‐blot (down) (n = 5). (B and C) METTL3, METTL14, and FTO protein levels in innervated and denervated GAS muscles (n = 5). GAPDH was used as the internal reference. (D and E) expression and quantification of ALKBH5 and FoxO3 protein in GAS muscles at 7, 14, and 28 days post‐denervation (n = 5). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001, n.s., no significant by two‐tailed Student's t‐test.
ALKBH5 overexpression induces muscle mass loss and accelerates denervation‐induced muscle atrophy
To explore the potential role of ALKBH5 in muscle mass control and denervation‐induced muscle atrophy, ALKBH5 was overexpressed in tibialis anterior (TA) muscles through AAV‐ALKBH5 local injection. Infection with AAV‐ALKBH5 efficiently increased ALKBH5 expression at mRNA and protein levels in innervated and denervated TA muscles (Figure 2A–2C). Dot blot analysis showed that overexpressed ALKBH5 significantly suppressed the levels of m6A modifications (Figure 2D), thereby confirming the role of ALKBH5 as an m6A “eraser” of mRNA. Notably, overexpressed ALKBH5 significantly reduced muscle wet weight (AAV‐Control 59.59 ± 2.59 mg, AAV‐ALKBH5 53.47 ± 3.71 mg), myofibre CSA (AAV‐Control 4751.56 ± 568.24 μm2, AAV‐ALKBH5 3052.40 ± 252.78 μm2), and MyHC expression in innervated muscles (Figure 2B, 2C and 2E–2I). Furthermore, overexpressed ALKBH5 was associated with excessive loss in muscle mass (AAV‐Control 39.92 ± 4.20 mg, AAV‐ALKBH5 35.35 ± 2.23 mg) and size (AAV‐Control 3405.11 ± 487.64 μm2, AAV‐ALKBH5 2269.78 ± 289.79 μm2) in denervated muscles (Figure 2E–2H and 2J). Western blot and qPCR showed that overexpressed ALKBH5 not only promoted the levels of FoxO3 and its target genes (Atrogin1 and MuRF1) in TA muscles (Figure 2B, 2C, 2K and 2L) but also reduced the proportion of pFoxO3 (the phosphorylation status by which FoxO3 is exported from the nucleus and degraded in the cytoplasm 22 ) (Figure S1). These findings indicate that upregulation of ALKBH5 has the ability to drive normal muscle mass loss and aggravate denervation‐induced muscle atrophy by enhancing FoxO3 expression and activity.
Figure 2.
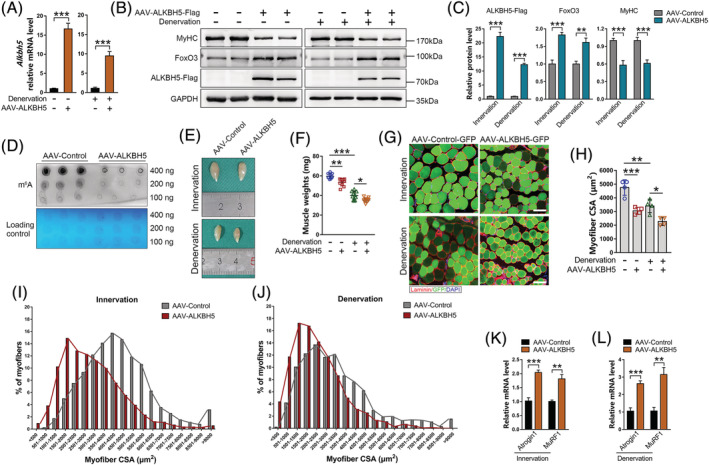
F ALKBH5 overexpression induces muscle mass loss and accelerates denervation‐induced muscle atrophy. (A) Alkbh5 mRNA levels after AAV‐ALKBH5 injection into tibialis anterior (TA) muscle (n = 4). (B and C) MyHC, FoxO3, and ALKBH5‐Flag protein levels in TA muscles after 6 weeks of AAV‐ALKBH5 infection and/or two weeks of denervation (n = 6). (D) mRNA dot blot analysis of m6A levels in AAV‐ALKBH5 infected TA muscle. (E and F) Representative images and TA muscle wet weight after six weeks of AAV‐ALKBH5 infection (n = 10). (G and H) Representative immunofluorescence images of TA muscles after AAV‐ALKBH5 injection labelled with laminin (red) and GFP (green), and quantification of myofibre cross‐sectional areas (CSAs) (n = 4). DAPI (blue) was used to label the nuclei. Scale bar = 100 μm. (I and J) Distribution of myofibre CSA from images in (G). (K and L) mRNA levels of Atrogin1 and MuRF1 in AAV‐ALKBH5 infected TA muscles with innervation or denervation (n = 4). Data are expressed as mean ± SEM (A, C, K, L) or mean ± SD (F, H). *P < 0.05, **P < 0.01, ***P < 0.001 by two‐tailed Student's t test (A, C, K, L) or one‐way ANOVA with Bonferroni's post‐hoc test (F, H).
Conditional deletion of Alkbh5 in skeletal muscles did not cause developmental defects but abrogated denervation‐induced muscle atrophy
ALKBH5 overexpression was involved in initiation of muscle wasting; therefore, further experiments were performed to determine whether Alkbh5 deletion can increase muscle mass or prevent muscle atrophy. Alkbh5 fl/+ mice were bred with Myl1‐Cre transgenic mice to obtain muscle‐specific Alkbh5 knockout (Myl1‐Cre;Alkbh5 fl/fl ) mice, and their littermate control (Alkbh5 fl/fl ) mice (Figure 3A and 3B). qPCR and western blot revealed suppressed ALKBH5 mRNA and protein levels in skeletal muscles of Myl1‐Cre;Alkbh5 fl/fl mice (Figures 3C, 3D, and S2A), but not in other tissues (Figure S2B), which subsequently elevated m6A levels in denervated muscles (Figure 3E). Myl1‐Cre;Alkbh5 fl/fl mice were viable and did not show any morphological or growth abnormalities. Moreover, they had normal body weights (Figure S2C–S2E). Grip strength was comparable in the two genotypes (Figure S2F), and no histopathologic changes were observed in Alkbh5 knockout muscles (Figure S2G). In addition, wet weight and myofibre cross‐sectional area (CSA) of muscles in Alkbh5 knockout mice at 10 weeks of age were not significantly different, compared with those in littermate controls (Figure 3F–3J). This implies that deletion of Alkbh5 had no effect on normal muscle mass and function in mice. Further, the effects of Alkbh5 deficiency on denervation‐induced muscle atrophy were investigated. It was established that upon denervation, Alkbh5 knockout mice exhibited larger muscle weights (control 81.39 ± 8.34 mg, knockout 94.43 ± 8.65 mg) (Figure 3F and 3G) and myofibre CSA (control 1103.22 ± 262.42 μm2, knockout 1654.70 ± 376.75 μm2) (Figure 3H–3K), and higher MyHC expression levels (Figure 3L and 3M), compared with controls. Specifically, analysis of fibre types showed that Alkbh5 deficiency mainly inhibited type II fibres atrophy, but had no significant effect on type I fibres (Figure S3). This may be because the main myofibre in the GAS muscle was Type II or fast‐twitch fibres. 23 Western blot and qPCR showed that Alkbh5 knockout significantly suppressed FoxO3, Atrogin1 and MuRF1 levels in denervated GAS muscles, compared with the control group (Figure 3L–3N). These findings imply that muscle‐specific Alkbh5 knockout can prevent the activation of FoxO3 signalling and progression of muscle atrophy induced by denervation without causing muscle hypertrophy.
Figure 3.
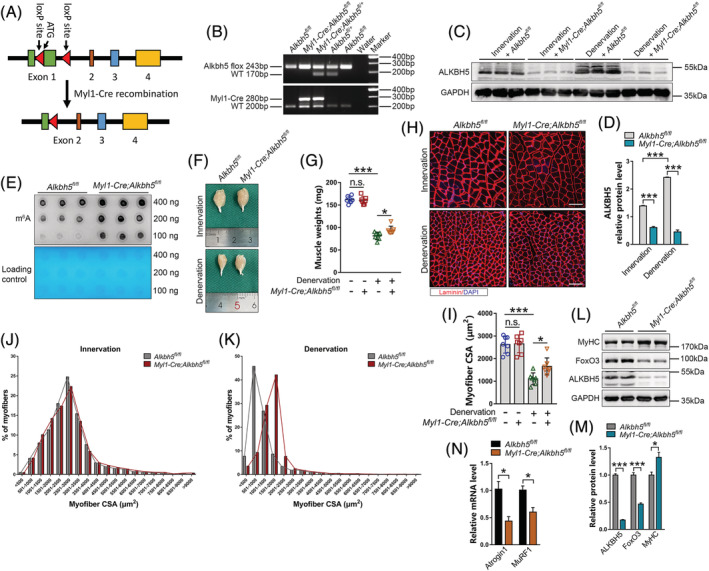
Conditional deletion of Alkbh5 in skeletal muscles causes no developmental defects, but abrogates muscle atrophy induced by denervation. (A) Schematic showing generation of muscle‐specific Alkbh5 knockout mice using the Cre‐LoxP recombination system. Exon 1 is deleted upon Myl1‐Cre‐mediated recombination. (B) PCR analysis with tail DNA from Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. (C–E) Western blot for ALKBH5 protein levels and dot blot for m6A abundance in Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl gastrocnemius (GAS) muscle with or without denervation (n = 3). (F and G) Representative image and GAS muscle wet weight in Alkbh5 wild‐type and knockout mice with or without denervation (n = 7). (H and I) Immunofluorescence staining and quantification of myofibre CSA of GAS muscle at 14 days post‐denervation in Alkbh5 wild‐type and knockout mice (n = 7). Red denotes laminin; blue denotes nuclei labelled with DAPI. Scale bar = 100 μm. (J and K) Distribution of myofibre CSA from images in (H). (L and M) MyHC, FoxO3, and ALKBH5 protein levels in Myl1‐Cre;Alkbh5 fl/fl GAS muscle after 14 days of denervation (n = 6). (N) qPCR showing reduction in Atrogin1 and MuRF1 mRNA levels at 14 days post‐denervation in muscles of Myl1‐Cre;Alkbh5 fl/fl mice (n = 4). Data are expressed as mean ± SEM (D, M, N) or mean ± SD (G, I). *P < 0.05, **P < 0.01, ***P < 0.001 by two‐tailed Student's t test (D, M, N) or one‐way ANOVA with Bonferroni's post‐hoc test (G, I).
Identification of m6A targets in denervation muscles
To establish the underlying mechanism of m6A modification in regulation of neurogenic muscle atrophy, m6A methylomes of skeletal muscles undergoing denervation were mapped by m6A sequencing (m6A‐seq). The ‘GGAC’ sequence motif was highly enriched within the m6A sites identified in both innervated and denervated muscles (Figure 4A), majority of the m6A peaks were located in 3′UTR and CDS regions, with a small subset of peaks located in 5′UTR and distal intergenic regions (Figure S4A and S4B). Guitar analysis revealed that m6A peaks were predominantly abundant in the vicinity of stop codons (Figure 4B). A total of 556 new m6A peaks and 779 disappearing peaks were identified in denervated muscles through m6A‐seq analysis, whereas the remaining 8209 peaks were unchanged in both innervated and denervated muscles (Figures 4C and S4C). For m6A‐regulated genes, m6A‐seq showed 441 new modifying genes and 621 genes loss of m6A modification in denervated muscles (Table S2), whereas the other 3890 genes were identified in both innervated and denervated muscles (Figure S4D). These results demonstrate that atrophic muscles unique peaks and genes had specific m6A targets.
Figure 4.
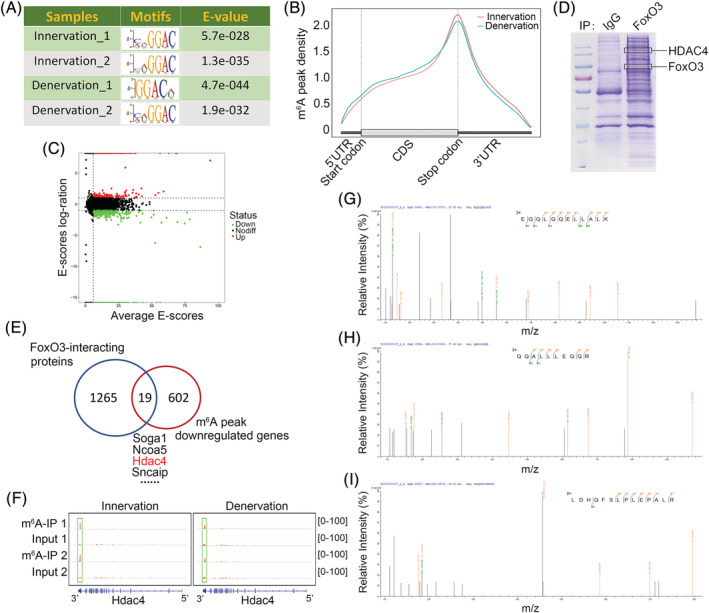
F Identification of m6A targets in muscles undergoing denervation. (A) Motif analysis using MEME program identified ‘GGAC’ as the m6A consensus motif in both the innervated and denervated muscles in m6A‐seq. (B) Density distribution of m6A peaks across the transcriptome of gastrocnemius (GAS) muscle with innervation or denervation. (C) Scatter plots showing the distribution of m6A peaks in the denervation group, compared with innervation group. (D) Coomassie staining of proteins co‐immunoprecipitated with FoxO3 antibodies in denervated muscles. The marked bands were identified by tandem mass spectrometry (MS/MS). (E) Venn diagram showing overlap between FoxO3‐interacting molecules and m6A‐downregulated genes in GAS muscles with denervation. (F) Integrative genomics viewer (IGV) tracks displaying m6A‐seq read distribution in Hdac4 transcript of innervated and denervated GAS muscles. (G–I) HDAC4 unique peptides (QQALLLEQQR; EQQLQQELLALK; LDHQFSLPLEPALR) identified by MS/MS analysis.
Given that ALKBH5 promoted FoxO3 expression during denervation just at the protein level without affecting its mRNA (Figures 2B, 2C, and S5), we postulated that the increase in FoxO3 protein levels in denervated muscles may be caused by enhanced FoxO3 translation efficiency through ALKBH5‐mediated m6A demethylation. However, m6A‐seq analysis showed that FoxO3 is not one of the differential genes of m6A modification, implying that expressions of FoxO3 proteins in atrophic muscles are not directly regulated by m6A. Consistent with this finding, a previous study reported that FoxO3 translation is independent of m6A methylation. 24 Therefore, to determine how ALKBH5 regulates FoxO3 expression and muscle atrophy, we reviewed the literature and found that the function of FoxO3 protein in skeletal muscles is controlled by various post‐translational modifications, such as phosphorylation, acetylation, methylation and ubiquitination. 25 This implies that ALKBH5 may indirectly affect FoxO3 protein levels by modulating specific post‐translational modification molecule. To explore this postulate, co‐immunoprecipitation (Co‐IP) and tandem mass spectrometry analyses were performed. A total of 1284 FoxO3‐interacting proteins were identified in denervated muscles (Figure 4D and 4E; Table S3). These proteins were crossed with differential genes of m6A modification and 19 molecules overlapping between m6A‐downregulated genes and FoxO3‐interacting proteins in denervated muscles were obtained (Figure 4E; Table S4). Among the 19 identified molecules, histone deacetylase 4 (HDAC4), a critical driver of skeletal muscle atrophy, 26 , 27 exhibited highly enriched and specific m6A peaks near its stop codon and could interact with FoxO3 (Figure 4F–4I). These findings imply that HDAC4 may be the central mediator of ALKBH5‐mediated regulation of FoxO3 expression.
ALKBH5‐mediated m6A demethylation is required for Hdac4 mRNA stability
The mechanism underlying the regulation of HDAC4 by ALKBH5 in skeletal muscles was investigated. ALKBH5 is a demethylase that selectively binds m6A‐containing RNA to regulate RNA splicing and stability 28 , 29 ; therefore, expression levels of precursor (pre‐) and mature mRNA of Hdac4 were determined. Overexpressed ALKBH5 significantly increased the expression of mature Hdac4 (Figure 5A), whereas Alkbh5 knockout in denervated muscles significantly reduced mature Hdac4 mRNA levels, compared with the control group (Figure 5B). There were no significant differences in pre‐Hdac4 levels between control and ALKBH5‐modified muscles (Figure 5A and 5B), implying that Hdac4 mRNA splicing was not affected by ALKBH5. Notably, alterations in mature Hdac4 mRNA levels synchronously altered HDAC4 protein levels (Figure 5C and 5D). Then we determined whether ALKBH5 can affect the stability of Hdac4 mRNA by overexpressing ALKBH5 in vitro through infection of C2C12 cells with Adv‐ALKBH5, and used actinomycin D (Act‐D) to block transcription. The half‐life of mature Hdac4 mRNA, but not pre‐Hdac4 mRNA was found to be significantly shorter in control cells, compared with ALKBH5‐overexpressed cells (Figure 5E). These findings indicate that upregulated HDAC4 levels in denervated muscles can be attributed to ALKBH5‐induced intensification of the stability of mature Hdac4 mRNA.
Figure 5.
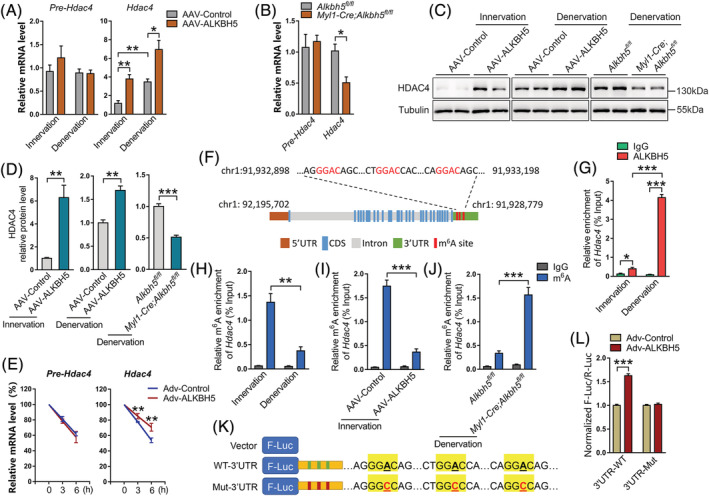
ALKBH5‐mediated m6A demethylation is required for Hdac4 mRNA stability. (A) Precursor and mature mRNA levels of Hdac4 in AAV‐ALKBH5 infected muscle with or without denervation (n = 4). (B) Levels of pre‐Hdac4 and mature Hdac4 in Alkbh5 knockout muscle with denervation (n = 4). (C and D) Protein expression of HDAC4 in ALKBH5 overexpressed and Alkbh5 knockout muscle with or without denervation (n = 4 or 6). (E) Half‐life of pre‐Hdac4 and mature Hdac4 mRNA at 0, 3, and 6 h after actinomycin D (5 μg/mL) treatment in C2C12 myotubes transduced with adenovirus expressing ALKBH5 (Adv‐ALKBH5) (n = 4). (F) Schematic representation of positions of m6A motifs within Hdac4 mRNA. (G) CLIP‐qPCR validation of ALKBH5 binding with 3′UTR of Hdac4 mRNA in both innervated and denervated gastrocnemius (GAS) muscles (n = 4). (H) MeRIP‐qPCR validation of m6A changes in Hdac4 mRNA in muscles during denervation (n = 4). (I and J) m6A‐MeRIP‐qPCR showed that ALKBH5 overexpression depleted m6A modification of Hdac4 mRNA (I), whereas m6A modification of Hdac4 mRNA was enriched after ALKBH5 knockdown (J) (n = 4). (K) Schematic presentation for the generation of wild‐type (WT) or three mutant (Mut; GGAC to GGCC) Hdac4 3′UTR firefly luciferase reporter. (L) Normalized F‐Luc/R‐Luc activity of the WT‐3′UTR and three Mut‐3′UTR reporter in Adv‐ALKBH5 infected C2C12 cells (n = 6). Data are expressed as mean ± SEM. *P < 0.05, **P < 0.01, ***P < 0.001 by two‐tailed Student's t test.
To establish the precise mechanisms through which ALKBH5 increases Hdac4 mRNA stability, m6A sites were searched on Hdac4 transcript based on m6A‐seq data. Three ‘GGAC’ motifs were identified near the stop codon in 3′UTR (Figure 5F), consistent with positions of peaks identified by m6A‐seq. Given that m6A‐seq showed that m6A peak of Hdac4 in 3′UTR significantly shrank with upregulation of ALKBH5 during denervation (Figures 1D, 1E and 4F), we conducted CLIP‐qPCR assays using anti‐ALKBH5 antibody in innervated and denervated muscles. We observed that ALKBH5 significantly enriched Hdac4 transcripts (Figure 5G), implying that ALKBH5 regulates HDAC4 at RNA level. Then, we performed m6A‐MeRIP‐qPCR using specific primers of these potential m6A sites to verify ALKBH5‐mediated m6A demethylation in Hdac4 mRNA. When compared with the IgG group, an obvious enrichment of Hdac4 mRNA was obtained by reaction with m6A‐specific antibody (Figure 5H). Notably, either denervation or ALKBH5 overexpression remarkably reduced m6A enrichment in the 3′UTR of Hdac4 mRNA (Figure 5H and 5I), whereas deletion of Alkbh5 caused an increase in m6A levels in these sites (Figure 5J).
Thereafter, to investigate the essential roles of m6A methylation in the Hdac4 3′UTR region, a luciferase reporter was generated with a wild‐type (WT) Hdac4–3′UTR sequence or mutant (Mut) counterpart whose putative m6A sites were mutated (GGAC to GGCC, Figure 5K). Co‐transfection of ALKBH5 obviously increased expression levels of WT‐3′UTR, but had no significant effects on the activity of Mut‐3′UTR in C2C12 cells (Figure 5L). These findings indicate that ALKBH5‐mediated m6A demethylation on 3′UTR is involved in m6A modification‐regulated Hdac4 expression.
Taken together, these findings show that ALKBH5 abrogates m6A methylation at the 3′UTR region of Hdac4 mRNA to inhibit m6A‐dependent mRNA degradation, which further increases the expression levels of HDAC4.
HDAC4 binds FoxO3 and enhances FoxO3 signalling via deacetylation
Mass spectrometry analysis showed that HDAC4 combines with FoxO3 in denervated muscles (Figure 4D, and 4G–4I). To further verify this finding, we investigated their interactions and colocalizations. Co‐IP experiments revealed that HDAC4 and FoxO3 physically interacted with each other in denervated muscles (Figure 6A). Confocal microscopy showed that HDAC4 was colocalized with FoxO3 in muscle nucleus (Figure 6B). HDAC4 is a deacetylase; therefore, the role of HDAC4 in deacetylating FoxO3 in denervation‐induced muscle atrophy was investigated. Upregulation of HDAC4 was correlated with a significant decrease in acetylated FoxO3 proteins immunoprecipitated with an antibody against acetylated‐lysine (Ace‐lys) after denervation, compared with the control group (Figure 6C). This finding implies that HDAC4 can remove acetylation modification on FoxO3. To verify that FoxO3 is a deacetylation substrate of HDAC4, Myc‐tagged HDAC4 and Flag‐tagged FoxO3 were expressed in HEK293T. Co‐IP experiments showed that exogenous HDAC4 strongly interacted with exogenous FoxO3, thereby reducing the acetylation levels of FoxO3 (Figure 6D).
Figure 6.
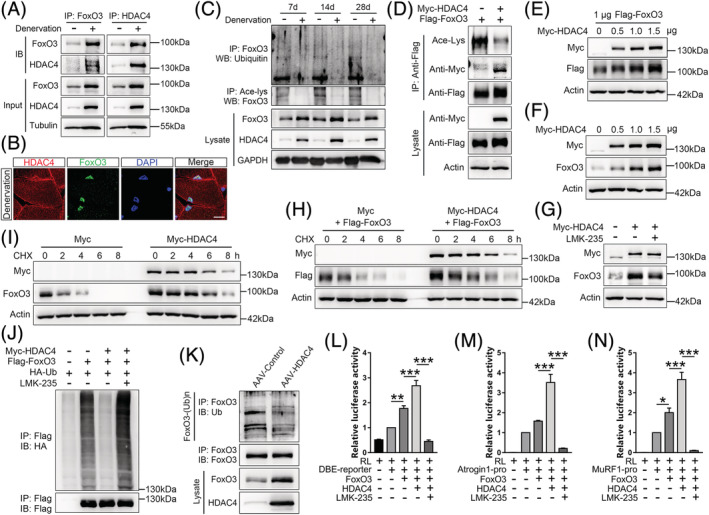
HDAC4 binds FoxO3 and enhances FoxO3 signalling by deacetylation. (A) HDAC4 endogenously interacts with FoxO3 in denervated muscles as shown by co‐immunoprecipitation (Co‐IP) with FoxO3 (left) or HDAC4 (right) antibody. (B) Confocal images showing colocalization between HDAC4 (red) and FoxO3 (green) in the nucleus (blue, DAPI‐labelled) of gastrocnemius (GAS) muscles after 14 days of denervation. Scale bar = 10 μm. (C) Co‐IP and immunoblot showing decrease in acetylation and ubiquitination levels of FoxO3 proteins in GAS muscles on Days 7, 14, and 28 post‐denervation. (D) Immunoblot analysis with anti‐Ace‐Lys, anti‐Myc, and anti‐Flag for proteins that co‐immunoprecipitated with Flag‐tagged FoxO3 from lysates of HEK293T cells that were transfected with plasmids encoding the proteins indicated in the above blots. (E and F) Increasing amounts of Myc‐HDAC4 expression plasmid were transfected into NIH/3T3 cells with or without Flag‐FoxO3 plasmid (1 μg). Exogenous (E) and endogenous (F) protein levels of FoxO3 were determined by immunoblotting with a specific antibody 24 h after transfection. (G) NIH/3T3 cells were transfected with Myc‐HDAC4 plasmid (1 μg), followed by LMK‐235 (2 μM) treatment for 12 h. Endogenous protein levels of FoxO3 were then determined. (H and I) Myc‐HDAC4 plasmid (1 μg) was transfected into NIH/3T3 cells together with or without Flag‐FoxO3 plasmid (1 μg), and cells were treated with cycloheximide (CHX) at 80 μg/mL for the indicated times. Half‐life of exogenous (H) and endogenous (I) FoxO3 proteins were determined by western blot. (J) HEK293T cells transfected with plasmids were treated with LMK‐235 (2 μM, 12 h) and proteasome inhibitor MG132 (20 μM, 8 h). Polyubiquitination of exogenous FoxO3 was detected by anti‐HA antibodies. (K) TA muscles were infected with AAV‐HDAC4 for 6 weeks. Endogenous FoxO3 was immunoprecipitated with anti‐FoxO3, and its polyubiquitination detected by anti‐Ubiquitin (Ub). (L–N) Relative luciferase activities of DBE (L), Atrogin1‐pro (M) or MuRF1‐pro (N) reporter in C2C12 cells that had been transfected with Myc‐HDAC4, Flag‐FoxO3 and RL reporter (n = 6 or 8). Data are expressed as mean ± SEM. **P < 0.01, ***P < 0.001 by one‐way ANOVA with Bonferroni's post‐hoc test.
Deacetylation is capable of increasing FoxO3 protein levels by protecting it against ubiquitination‐mediated degradation. 30 Therefore, we first determined whether HDAC4‐mediated deacetylation regulates FoxO3 protein levels. Western blot showed that ectopic expression of HDAC4 significantly increased the protein levels of exogenous FoxO3 in a dose‐dependent manner (Figure 6E). Meanwhile, HDAC4 promoted endogenous FoxO3 protein levels (Figure 6F). In contrast, promotion of FoxO3 protein levels by HDAC4 was blocked by treatment with LMK‐235, an inhibitor of HDAC4 (Figure 6G). Second, to ascertain the effects of HDAC4 on the stability of FoxO3 proteins, turnover rate of FoxO3 in the presence or absence of HDAC4 was determined using the protein synthesis inhibitor cycloheximide (CHX). Transfection of HDAC4 expression plasmid in NIH/3T3 cells remarkably prolonged the half‐life of both exogenous and endogenous FoxO3, compared with the empty vector (Figure 6H and 6I), suggesting that HDAC4 specifically stabilized FoxO3 in cells. Third, to confirm the role of HDAC4 in preventing FoxO3 ubiquitination degradation, Myc‐HDAC4, Flag‐FoxO3, and HA‐Ubiquitin expression vectors were transfected into HEK293T cells, followed by treatment with LMK‐235 or DMSO. FoxO3 ubiquitination was greatly suppressed by HDAC4 (lane 3), compared with the control vector (lane 2), while inhibition of HDAC4 by LMK‐235 elevated FoxO3 ubiquitylation levels (lane 4) (Figure 6J). Consistently, inhibitory effects of HDAC4 on FoxO3 ubiquitination degradation were observed in vivo (Figure 6K).
Given that deacetylation is necessary for activation of FoxO3, 31 we assessed the effects of HDAC4‐mediated deacetylation on FoxO3 transcriptional activities using a FoxO sensor that contained six FoxO DNA‐binding elements (DBE reporter) upstream of the luciferase gene. 32 Overexpressed HDAC4 elevated luciferase reporter activities, compared with the empty vector, whereas luciferase activities were significantly suppressed after LMK‐235 addition (Figure 6L). Similar findings were observed when the transfection of Atrogin1 or MuRF1 promoters controlled luciferase reporter (Figure 6M and 6N).
These findings demonstrate that HDAC4‐mediated deacetylation serves as a switch to control the ubiquitination levels of FoxO3, thereby modulating protein expressions and transcriptional activities of FoxO3 in skeletal muscles.
HDAC4 reversed the regulatory effects of ALKBH5 on muscle mass
To confirm that ALKBH5‐induced muscle mass loss was mediated by HDAC4, first, we inhibited HDAC4 by intraperitoneal injection of LMK‐235 in AAV‐ALKBH5‐infected mice. LMK‐235 injection significantly elevated acetyl‐Histone H3 levels, indicating that HDAC4 activities were successfully suppressed (Figure 7F and 7G). In addition, improved muscle weight and myofibre CSA were observed after LMK‐235 injection in ALKBH5‐induced muscle atrophy model (Figure 7A–7E). Notably, western blot showed that inhibition of HDAC4 significantly upregulated MyHC while significantly downregulating FoxO3 levels (Figure 7F and 7G). Further, we transfected AAV‐HDAC4 or AAV‐Control into TA muscles of Myl1‐Cre;Alkbh5 fl/fl mice and established denervation‐induced muscle atrophy model. Overexpression of HDAC4 reversed the protective effects of Alkbh5 knockout on muscle wasting, as indicated by reduction of muscle weight and CSA, downregulation of MyHC, and increase in FoxO3 expression (Figure 7H–7N). These findings show that HDAC4 is a vital mediator and its suppression can partially rescue the effect of ALKBH5 in promoting skeletal muscle atrophy.
Figure 7.
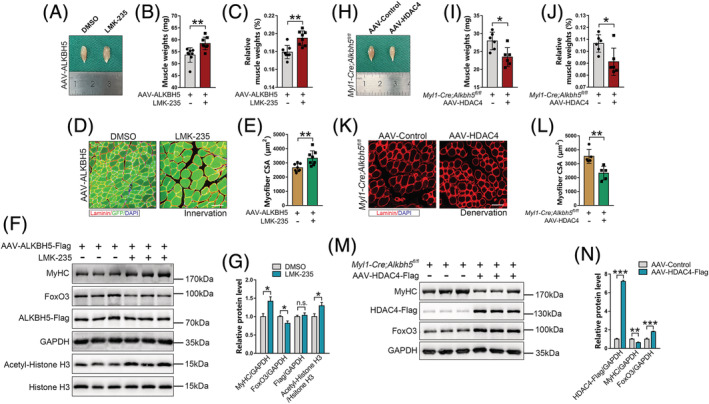
HDAC4 reversed the regulatory effects of ALKBH5 on muscle mass. (A–E) The wet weight (B), percentage of muscle wet weight relative to body weight (C), and myofibre cross‐sectional area (CSA) (E) of ALKBH5‐overexpressed TA muscles after 14 days of LMK‐235 injection (5 mg/kg/day) (n = 8). Scale bar = 100 μm. (F and G) Protein levels of MyHC, FoxO3, ALKBH5‐Flag, and acetyl‐Histone H3 in ALKBH5‐overexpressed TA muscle after 14 days of LMK‐235 injection (n = 5). GAPDH and Histone H3 were used as internal controls. (H–L) TA muscle wet weight (I), percentage of muscle wet weight relative to body weight (J), and myofibre CSA (L) of Alkbh5 knockout mice after 4 weeks of AAV‐HDAC4 infection, followed by 21 days of denervation (n = 5 or 6). Scale bar = 100 μm. (M and N) Protein levels of MyHC, FoxO3, and HDAC4‐Flag in Alkbh5 knockout TA muscles after 4 weeks of AAV‐HDAC4 infection, followed by 21 days denervation (n = 6). Data are expressed as mean ± SD (B, C, E, I, J, L) or mean ± SEM (G, N). *P < 0.05, **P < 0.01, ***P < 0.001 by two‐tailed Student's t test.
Discussion
Previous studies report that m6A modification in eukaryotic RNA regulates physiological homeostasis and development of diseases. 13 , 16 In skeletal muscles, a strong and intricate relationship between m6A modification and myogenesis has been documented. Dysregulation of m6A decreases MyoD expression in proliferative myoblasts, disrupts the specification and differentiation of myoblasts into myotubes, 17 suppresses mitochondrial biogenesis and energy production during muscle development. 18 We investigated the function of m6A on muscle phenotypes in adults. It was established that m6A regulates adult skeletal muscle mass and size, especially in denervation‐induced muscle atrophy. The m6A mRNA levels of skeletal muscles undergoing denervation significantly decreased, compared with control muscles. Moreover, expressions of m6A demethylase ALKBH5 was upregulated in denervated muscles. Overexpressed ALKBH5 reduced wet weight and fibre CSA in innervated and denervated muscles, whereas deletion of Alkbh5 protected mice against denervation‐induced muscle wasting with no effects on physical development and normal muscle morphologies. Even though studies have documented the biological and pathological roles of ALKBH5 in autophagy, 33 spermatogenesis, 29 ossification, 34 and cancer, 35 we, for the first time, proved the crucial function of ALKBH5 in controlling muscle mass in adults. Importantly, given the high feasibility and operability of developing drugs targeting enzyme protein, m6A demethylase ALKBH5 is a potential target for preventing neurogenic muscle atrophy.
FoxO3 is a transcription factor that is implicated in atrogene expression and muscle wasting in various pathological conditions, including denervation. 6 , 8 The findings of this study showed that ALKBH5 positively regulated the expressions of FoxO3 proteins during denervation, suggesting that FoxO3 may be a downstream molecule of ALKBH5. Then, m6A‐seq and FoxO3 co‐immunoprecipitation combined with mass spectrometry analyses were performed. The findings showed that FoxO3 is not a direct target for ALKBH5‐mediated m6A modification, however, it is indirectly regulated by the ‘bridge’ molecule HDAC4.
In this study, HDAC4 was identified as a novel substrate for ALKBH5 in denervated muscles. ALKBH5 improved mRNA stability of HDAC4 transcripts, leading to increased HDAC4 expressions through an m6A demethylation‐dependent mechanism. Regulation of mRNA stability is an important mechanism through which m6A modification affects gene expression. The diversity in cell type and state results in differences in effects of ALKBH5‐mediated m6A demethylation on mRNA stability. On the one hand, ALKBH5 demethylates and stabilizes PER1 mRNA and prevents pancreatic cancer progression. 36 ALKBH5 promotes cancer stem cell self‐renewal in acute myeloid leukaemia by abolishing m6A‐dependent TACC3 mRNA degradation. 37 On the other hand, ALKBH5 decreases the stability of CYR61 mRNA and inhibits trophoblast invasion at the maternal–foetal interface. 38 Overexpression of ALKBH5 can shorten the half‐life of LYPD1 mRNA and suppress the proliferation and metastasis of hepatocellular carcinoma. 39 In our study, we found that ALKBH5 enhances the stability of Hdac4 mRNA through m6A demethylation at the 3′UTR, which further increases its expression level in denervated muscles (Figure 8). Therefore, ALKBH5 is a novel mechanism for upregulating HDAC4 levels in denervation‐induced muscle atrophy.
Figure 8.
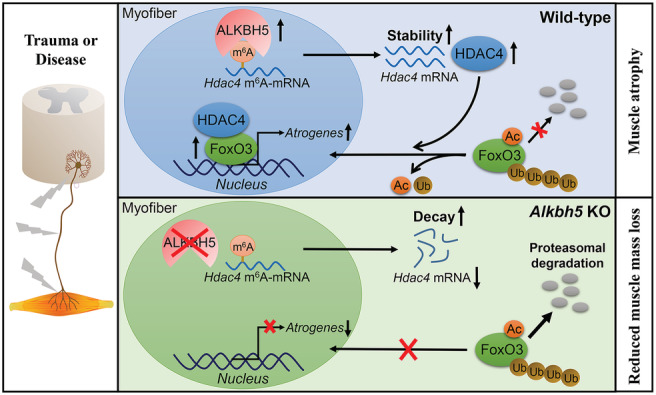
Schematic illustration of ALKBH5‐HDAC4‐FoxO3 axis promoting denervation‐induced muscle atrophy. In denervated muscles, ALKBH5‐mediated m6A demethylation stabilized and increased HDAC4 expression. HDAC4 interacted with FoxO3 and inhibited its degradation in a deacetylation‐dependent manner, ultimately leading to an increase in FoxO3 protein levels and transcriptional activities. Targeting ALKBH5 reduced HDAC4 levels, which in turn suppressed FoxO3 signalling activation and prevented denervation‐induced muscle atrophy.
Prevention of overactivation of FoxO3 signalling is essential to avoid muscle wasting. Actually, FoxO3 is regulated by many different post‐translational modifications, including acetylation/deacetylation and ubiquitination. 5 Acetyltransferase or deacetylase can negatively or positively modulate FoxO3 activities in skeletal muscles. For instance, p300/CBP‐mediated acetylation of FoxO3 suppresses its nuclear localization and makes it inactive in denervation‐induced muscle atrophy. 40 In addition, HDAC1‐mediated deacetylation activates FoxO3‐dependent atrophy during nutrient deprivation. 31 Although HDAC4 is involved in deacetylation and activation of FoxO3 in vascular endothelial cells, 41 the specific proteins mediating FoxO3 deacetylation in muscles undergoing denervation have not been identified. This study proves, for the first time, that HDAC4 directly deacetylates FoxO3 in skeletal muscles and is necessary for its upregulation and activation in response to denervation (Figure 8).
Of cause, there are some weaknesses in this study to ameliorate by future works. First, in consideration of the easy transfection of HEK293T and NIH/3T3 cells, we used these two non‐muscle cells rather than myocyte/myotube models to explore the regulatory relationship between HDAC4 and FoxO3. Second, if we use HDAC4 constructs deficient in enzyme activity instead of chemical inhibitor, the results may be more convincing. These weaknesses are also the area of interest for our future investigation.
In summary, through analysis of regulatory relationships among ALKBH5, HDAC4, and FoxO3, we revealed that ALKBH5 demethylates and stabilizes Hdac4, leading to upregulation and activation of FoxO3, which accelerates denervation‐induced muscle wasting. Our findings elucidate on the pivotal regulatory role of m6A in muscle health and diseases and shed light on the development of new therapeutic strategies for neurogenic muscle atrophy.
Conflict of interest
The authors declare that they have no conflict of interest.
Supporting information
Figure S1. Western blot shows that the proportion of pFoxO3 (S253) decreased significantly when ALKBH5 was overexpressed in TA muscles (n = 4). Data are expressed as mean ± SEM. ***P < 0.001 by two‐tailed Student's t‐test.
Figure S2. Alkbh5 conditional knockout mice showed no morphological defects. (A) qPCR analysis of Alkbh5 mRNA in denervated GAS muscles from Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice (n = 5). (B) Western blot of ALKBH5 protein in muscles, skeletal, brains, heart, lungs, liver, and kidneys from Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. (C) Representative images of Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice at 12‐week old. (D and E) Growth curve of Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice from birth to 12‐week olds (n = 11–23 mice for each group). (F) Grip strength (normalized by body weight) was evaluated in Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice (n = 7). (G) H&E staining of lower limb muscle transverse section in 10‐week‐old Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. Scale bar = 100 μm for the up column; Scale bar = 20 μm for the down column. Data are expressed as mean ± SEM. ***P < 0.001, n.s., no significant by two‐tailed Student's t‐test.
Figure S3. Deletion of Alkbh5 prevents type II fibre atrophy of GAS muscles. (A) Representative photomicrographs of GAS muscle sections at 14 days post‐denervation in Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. Red denotes MyHC IIA, IIX and IIB; Green denotes laminin; Blue denotes nuclei labelled with DAPI. Scale bar = 100 μm. (B) Measurement of mean fibre CSA of type I (empty) or Type II (filled with red colour in A) fibres (n = 6). Data are expressed as mean ± SD. **P < 0.01, n.s., no significant by two‐tailed Student's t‐test.
Figure S4. Characterization of m6A‐regulated genes in muscles undergoing denervation. (A and B) Pie charts depicting the fractions of total m6A peaks in the indicated regions of mRNA transcripts from innervated (A) and denervated muscles (B). UTR: untranslated region. (C and D) Number of m6A peaks (C) and m6A modified genes (D) identified in m6A‐seq from innervated and denervated muscles.
Figure S5. qPCR shows that expressions of FoxO3 mRNA is not affected by ALKBH5 overexpression in both innervated and denervated muscles (n = 4). Data are expressed as mean ± SEM. n.s., no significant by two‐tailed Student's t‐test.
Data S1. Supporting Information
Table S1. PCR primers for genotyping.
Table S2. m6A‐regulated genes.
Table S3. FoxO3‐interacting proteins.
Table S4. Molecules overlapping between m6A‐downregulated genes and FoxO3‐interacting proteins.
Table S5. Mouse qPCR primers.
Acknowledgements
This work was supported by the National Natural Science Foundation of China (No. 32100915), Shenzhen Municipal Science and Technology Innovation Committee Project (SGLH20180625141602256, JCYJ20180305164544288, JSGG20180504170427135, JCYJ20190807145011340), and Guangdong Basic and Applied Basic Research Foundation (2019A1515110402). The authors of this manuscript certify that they comply with the ethical guidelines for authorship and publishing in the Journal of Cachexia, Sarcopenia and Muscle. 42
Liu Y., Zhou T., Wang Q., Fu R., Zhang Z., Chen N., Li Z., Gao G., Peng S., and Yang D. (2022) m6A demethylase ALKBH5 drives denervation‐induced muscle atrophy by targeting HDAC4 to activate FoxO3 signalling, Journal of Cachexia, Sarcopenia and Muscle, 13, 1210–1223, 10.1002/jcsm.12929
Contributor Information
Yuantong Liu, Email: liuytog@gmail.com.
Zhizhong Li, Email: lizhizhongjd@163.com.
Guoyong Gao, Email: guoyonggao@hotmail.com.
Songlin Peng, Email: dyffyy2@mail.sustech.edu.cn.
Dazhi Yang, Email: yangdazhi1111@163.com.
References
- 1. Ehmsen JT, Höke A. Cellular and molecular features of neurogenic skeletal muscle atrophy. Exp Neurol 2020;331:113379. [DOI] [PubMed] [Google Scholar]
- 2. Vainshtein A, Sandri M. Signaling pathways that control muscle mass. Int J Mol Sci 2020;21:4759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3. Liu H, Thompson LV. Skeletal muscle denervation investigations: selecting an experimental control wisely. Am J Physiol Cell Physiol 2019;316:C456–C461. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4. Furrer R, Handschin C. Muscle wasting diseases: novel targets and treatments. Annu Rev Pharmacol Toxicol 2019;59:315–339. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5. Sartori R, Romanello V, Sandri M. Mechanisms of muscle atrophy and hypertrophy: implications in health and disease. Nat Commun 2021;12:330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Milan G, Romanello V, Pescatore F, Armani A, Paik JH, Frasson L, et al. Regulation of autophagy and the ubiquitin‐proteasome system by the FoxO transcriptional network during muscle atrophy. Nat Commun 2015;6:6670. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7. Rom O, Reznick AZ. The role of E3 ubiquitin‐ligases MuRF‐1 and MAFbx in loss of skeletal muscle mass. Free Radic Biol Med 2016;98:218–230. [DOI] [PubMed] [Google Scholar]
- 8. O'Neill BT, Bhardwaj G, Penniman CM, Krumpoch MT, Suarez Beltran PA, Klaus K, et al. FoxO transcription factors are critical regulators of diabetes‐related muscle atrophy. Diabetes 2019;68:556–570. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9. Zhang W, Zhang S, Yan P, Ren J, Song M, Li J, et al. A single‐cell transcriptomic landscape of primate arterial aging. Nat Commun 2020;11:2202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Calissi G, Lam EW, Link W. Therapeutic strategies targeting FOXO transcription factors. Nat Rev Drug Discov 2021;20:21–38. [DOI] [PubMed] [Google Scholar]
- 11. Lan Q, Liu PY, Haase J, Bell JL, Hüttelmaier S, Liu T. The critical role of RNA m6A methylation in cancer. Cancer Res 2019;79:1285–1292. [DOI] [PubMed] [Google Scholar]
- 12. Shi H, Wei J, He C. Where, when, and how: context‐dependent functions of RNA methylation writers, readers, and erasers. Mol Cell 2019;74:640–650. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13. Zhang M, Zhai Y, Zhang S, Dai X, Li Z. Roles of N6‐methyladenosine (m6A) in stem cell fate decisions and early embryonic development in mammals. Front Cell Dev Biol 2020;8:782. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Wang CX, Cui GS, Liu X, Xu K, Wang M, Zhang XX, et al. METTL3‐mediated m6A modification is required for cerebellar development. PLoS Biol 2018;16:e2004880. 10.1371/journal.pbio.2004880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Dorn LE, Lasman L, Chen J, Xu X, Hund TJ, Medvedovic M, et al. The N(6)‐methyladenosine mRNA methylase METTL3 controls cardiac homeostasis and hypertrophy. Circulation 2019;139:533–545. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Wang T, Kong S, Tao M, Ju S. The potential role of RNA N6‐methyladenosine in cancer progression. Mol Cancer 2020;19:88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Kudou K, Komatsu T, Nogami J, Maehara K, Harada A, Saeki H, et al. The requirement of Mettl3‐promoted MyoD mRNA maintenance in proliferative myoblasts for skeletal muscle differentiation. Open Biol 2017;7:170119, 10.1098/rsob.170119 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Wang X, Huang N, Yang M, Wei D, Tai H, Han X, et al. FTO is required for myogenesis by positively regulating mTOR‐PGC‐1alpha pathway‐mediated mitochondria biogenesis. Cell Death Dis 2017;8:e2702, 10.1038/cddis.2017.122 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Zhang S, Zhao BS, Zhou A, Lin K, Zheng S, Lu Z, et al. m6A demethylase ALKBH5 maintains tumorigenicity of glioblastoma stem‐like cells by sustaining FOXM1 expression and cell proliferation program. Cancer Cell 2017;31:591–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Hao L, Yin J, Yang H, Li C, Zhu L, Liu L, et al. ALKBH5‐mediated m6A demethylation of FOXM1 mRNA promotes progression of uveal melanoma. Aging 2021;13:4045–4062. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Zeng Y, Wang S, Gao S, Soares F, Ahmed M, Guo H, et al. Refined RIP‐seq protocol for epitranscriptome analysis with low input materials. PLoS Biol 2018;16:e2006092, 10.1371/journal.pbio.2006092 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Liu Y, Li J, Shang Y, Guo Y, Li Z. CARM1 contributes to skeletal muscle wasting by mediating FoxO3 activity and promoting myofiber autophagy. Exp Cell Res 2019;374:198–209. [DOI] [PubMed] [Google Scholar]
- 23. Goto‐Inoue N, Morisasa M, Machida K, Furuichi Y, Fujii NL, Miura S, et al. Characterization of myofiber‐type‐specific molecules using mass spectrometry imaging. Rapid Commun Mass Spectrom 2019;33:185–192. [DOI] [PubMed] [Google Scholar]
- 24. Zhang Y, Wang X, Zhang X, Wang J, Ma Y, Zhang L, et al. RNA‐binding protein YTHDF3 suppresses interferon‐dependent antiviral responses by promoting FOXO3 translation. Proc Natl Acad Sci U S A 2019;116:976–981. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Tia N, Singh AK, Pandey P, Azad CS, Chaudhary P, Gambhir IS. Role of Forkhead Box O (FOXO) transcription factor in aging and diseases. Gene 2018;648:97–105. [DOI] [PubMed] [Google Scholar]
- 26. Moresi V, Williams AH, Meadows E, Flynn JM, Potthoff MJ, McAnally J, et al. Myogenin and class II HDACs control neurogenic muscle atrophy by inducing E3 ubiquitin ligases. Cell 2010;143:35–45. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Luo L, Martin SC, Parkington J, Cadena SM, Zhu J, Ibebunjo C, et al. HDAC4 controls muscle homeostasis through deacetylation of myosin heavy chain, PGC‐1α, and Hsc70. Cell Rep 2019;29:749–763. [DOI] [PubMed] [Google Scholar]
- 28. Zhang C, Samanta D, Lu H, Bullen JW, Zhang H, Chen I, et al. Hypoxia induces the breast cancer stem cell phenotype by HIF‐dependent and ALKBH5‐mediated m6A‐demethylation of NANOG mRNA. Proc Natl Acad Sci U S A 2016;113:E2047–E2056. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Tang C, Klukovich R, Peng H, Wang Z, Yu T, Zhang Y, et al. ALKBH5‐dependent m6A demethylation controls splicing and stability of long 3′‐UTR mRNAs in male germ cells. Proc Natl Acad Sci U S A 2018;115:E325–E333. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30. Tseng AH, Wu LH, Shieh SS, Wang DL. SIRT3 interactions with FOXO3 acetylation, phosphorylation and ubiquitinylation mediate endothelial cell responses to hypoxia. Biochem J 2014;464:157–168. [DOI] [PubMed] [Google Scholar]
- 31. Beharry AW, Sandesara PB, Roberts BM, Ferreira LF, Senf SM, Judge AR. HDAC1 activates FoxO and is both sufficient and required for skeletal muscle atrophy. J Cell Sci 2014;127:1441–1453. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Furuyama T, Kitayama K, Yamashita H, Mori N. Forkhead transcription factor FOXO1 (FKHR)‐dependent induction of PDK4 gene expression in skeletal muscle during energy deprivation. Biochem J 2003;375:365–371. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Song H, Feng X, Zhang H, Luo Y, Huang J, Lin M, et al. METTL3 and ALKBH5 oppositely regulate m6A modification of TFEB mRNA, which dictates the fate of hypoxia/reoxygenation‐treated cardiomyocytes. Autophagy 2019;15:1419–1437. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Wang HF, Kuang MJ, Han SJ, Wang AB, Qiu J, Wang F, et al. BMP2 modified by the m6A demethylation enzyme ALKBH5 in the ossification of the ligamentum flavum through the AKT signaling pathway. Calcif Tissue Int 2020;106:486–493. [DOI] [PubMed] [Google Scholar]
- 35. Wang J, Wang J, Gu Q, Ma Y, Yang Y, Zhu J, et al. The biological function of m6A demethylase ALKBH5 and its role in human disease. Cancer Cell Int 2020;20:347. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36. Guo X, Li K, Jiang W, Hu Y, Xiao W, Huang Y, et al. RNA demethylase ALKBH5 prevents pancreatic cancer progression by posttranscriptional activation of PER1 in an m6A‐YTHDF2‐dependent manner. Mol Cancer 2020;19:91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. Shen C, Sheng Y, Zhu AC, Robinson S, Jiang X, Dong L, et al. RNA demethylase ALKBH5 selectively promotes tumorigenesis and cancer stem cell self‐renewal in acute myeloid leukemia. Cell Stem Cell 2020;27:64–80. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Li XC, Jin F, Wang BY, Yin XJ, Hong W, Tian FJ. The m6A demethylase ALKBH5 controls trophoblast invasion at the maternal‐fetal interface by regulating the stability of CYR61 mRNA. Theranostics 2019;9:3853–3865. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Chen Y, Zhao Y, Chen J, Peng C, Zhang Y, Tong R, et al. ALKBH5 suppresses malignancy of hepatocellular carcinoma via m6A‐guided epigenetic inhibition of LYPD1. Mol Cancer 2020;19:123. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Bertaggia E, Coletto L, Sandri M. Posttranslational modifications control FoxO3 activity during denervation. Am J Physiol Cell Physiol 2012;302:C587–C596. [DOI] [PubMed] [Google Scholar]
- 41. Yang D, Xiao C, Long F, Su Z, Jia W, Qin M, et al. HDAC4 regulates vascular inflammation via activation of autophagy. Cardiovasc Res 2018;114:1016–1028. [DOI] [PubMed] [Google Scholar]
- 42. von Haehling S, Morley JE, Coats AJS, Anker SD. Ethical guidelines for publishing in the Journal of Cachexia, Sarcopenia and Muscle: update 2019. J Cachexia Sarcopenia Muscle 2019;10:1143–1145. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Figure S1. Western blot shows that the proportion of pFoxO3 (S253) decreased significantly when ALKBH5 was overexpressed in TA muscles (n = 4). Data are expressed as mean ± SEM. ***P < 0.001 by two‐tailed Student's t‐test.
Figure S2. Alkbh5 conditional knockout mice showed no morphological defects. (A) qPCR analysis of Alkbh5 mRNA in denervated GAS muscles from Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice (n = 5). (B) Western blot of ALKBH5 protein in muscles, skeletal, brains, heart, lungs, liver, and kidneys from Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. (C) Representative images of Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice at 12‐week old. (D and E) Growth curve of Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice from birth to 12‐week olds (n = 11–23 mice for each group). (F) Grip strength (normalized by body weight) was evaluated in Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice (n = 7). (G) H&E staining of lower limb muscle transverse section in 10‐week‐old Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. Scale bar = 100 μm for the up column; Scale bar = 20 μm for the down column. Data are expressed as mean ± SEM. ***P < 0.001, n.s., no significant by two‐tailed Student's t‐test.
Figure S3. Deletion of Alkbh5 prevents type II fibre atrophy of GAS muscles. (A) Representative photomicrographs of GAS muscle sections at 14 days post‐denervation in Alkbh5 fl/fl and Myl1‐Cre;Alkbh5 fl/fl mice. Red denotes MyHC IIA, IIX and IIB; Green denotes laminin; Blue denotes nuclei labelled with DAPI. Scale bar = 100 μm. (B) Measurement of mean fibre CSA of type I (empty) or Type II (filled with red colour in A) fibres (n = 6). Data are expressed as mean ± SD. **P < 0.01, n.s., no significant by two‐tailed Student's t‐test.
Figure S4. Characterization of m6A‐regulated genes in muscles undergoing denervation. (A and B) Pie charts depicting the fractions of total m6A peaks in the indicated regions of mRNA transcripts from innervated (A) and denervated muscles (B). UTR: untranslated region. (C and D) Number of m6A peaks (C) and m6A modified genes (D) identified in m6A‐seq from innervated and denervated muscles.
Figure S5. qPCR shows that expressions of FoxO3 mRNA is not affected by ALKBH5 overexpression in both innervated and denervated muscles (n = 4). Data are expressed as mean ± SEM. n.s., no significant by two‐tailed Student's t‐test.
Data S1. Supporting Information
Table S1. PCR primers for genotyping.
Table S2. m6A‐regulated genes.
Table S3. FoxO3‐interacting proteins.
Table S4. Molecules overlapping between m6A‐downregulated genes and FoxO3‐interacting proteins.
Table S5. Mouse qPCR primers.