

Abstract
Multiple drugs to treat traumatic brain injury (TBI) have failed clinical trials. Most drugs lose efficacy as the time interval increases between injury and treatment onset. Insufficient therapeutic time window is a major reason underlying failure in clinical trials. Few drugs have been developed with therapeutic time windows sufficiently long enough to treat TBI because little is known about which brain functions can be targeted if therapy is delayed hours to days after injury. We identified multiple injury parameters that are improved by first initiating treatment with the drug combination minocycline (MINO) plus N-acetylcysteine (NAC) at 72 hours after injury (MN72) in a mouse closed head injury (CHI) experimental TBI model. CHI produces spatial memory deficits resulting in impaired performance on Barnes maze, hippocampal neuronal loss, and bilateral damage to hippocampal neurons, dendrites, spines and synapses. neuMN72 treatment restores Barnes maze acquisition and retention, protects against hippocampal neuronal loss, limits damage to dendrites, spines and synapses, and accelerates recovery of microtubule associated protein 2 (MAP2) expression, a key protein in maintaining proper dendritic architecture and synapse density. These data show that in addition to the structural integrity of the dendritic arbor, spine and synapse density can be successfully targeted with drugs first dosed days after injury. Retention of substantial drug efficacy even when first dosed 72 hours after injury makes MINO plus NAC a promising candidate to treat clinical TBI.
Keywords: Diffuse brain injury, Therapeutic time window, Hippocampus, Spatial learning and memory, Dendritic morphology, Dendritic spines, Neuroprotection
Introduction
Traumatic brain injury (TBI) is a major cause of death and disability world-wide (Faul and Coronado, 2015). Despite this critical need, decades of research have not produced FDA-approved pharmacological treatments for TBI (Diaz-Arrastia et al., 2014). TBI is an acute biomechanical event that produces a primary injury immediately upon impact and initiates a progressive secondary injury that develops over time (Blennow et al., 2012). The position of the hippocampus within the medial temporal lobe makes it particularly vulnerable to biomechanical injury (Bigler, 2013). The risk of long term deficits increases as neural tissue is progressively damaged beyond the initial impact site (Bigler, 2013; Stocchetti and Zanier, 2016). As a result, TBI is best treated as early as possible. The majority of TBI’s, 70-80% of total incidence, are diagnosed as mild (Leibson et al., 2011). Despite the importance of early treatment, patients with a mild or moderate TBI often delay seeking treatment due to perceived symptom resolution, accessibility, or cost (Mohamadpour et al., 2019).
Most preclinical studies dose drugs within hours after injury, which does not consider the importance of treatment gap, the time between injury onset and treatment initiation (Tanielian, 2008; Demakis and Rimland, 2010). Treatment gap suggests that most TBIs will need to be treated with drugs with sufficiently long therapeutic time windows that target injury processes that develop over time (Diaz-Arrastia et al., 2014). Therapeutic time window in TBI is complex, as drug targets arise and disappear with differing kinetics (Mohamadpour et al., 2019). Despite the heterogeneity of TBI pathophysiology, most pharmaceutical interventions target a single injury mechanism (Xiong et al., 2013). Additionally, most preclinical studies have focused on treating injury in the hemisphere ipsilateral to the impact site. This focus may arise through the common use of unilateral open head experimental TBI models that directly injure the brain via a craniotomy (Failla and Wagner, 2015; Osier and Dixon, 2016). Neuronal loss and inflammation in open head models such as the controlled cortical impact model skew damage to regions proximal to the injury site. As a result, injury in regions distal to the impact site have been understudied (Hall et al., 2005; Tran et al., 2006). The closed head injury (CHI) model used in this study directly strikes an intact, unrestricted mouse skull with a piston. CHI produces a rapid acceleration-deceleration head movement resulting in a diffuse injury in both hemispheres (Johnson et al., 2015; White et al., 2017).
This study utilizes the diffuse brain injury produced by CHI to determine if the drug combination minocycline (MINO) plus N-acetylcysteine (NAC) retains efficacy when first dosed three days post-injury, and whether the therapeutic time window of MINO plus NAC differs in brain regions proximal or distal to the injury site. Both MINO and NAC have shown promise individually in preclinical experimental TBI models when first dosed within hours after injury (Xiong et al., 1999; Sanchez Mejia et al., 2001; Yi and Hazell, 2005; Hicdonmez et al., 2006; Bye et al., 2007; Chen et al., 2008; Abdel Baki et al., 2010; Homsi et al., 2010; Haber et al., 2017; Sangobowale et al., 2018). Although individual drugs alone showed reduced efficacy when first administered 24 hours after injury, MINO plus NAC synergized to lengthen the therapeutic time window in multiple outcomes when first dosed at 12 or 24 hours after CHI (Sangobowale et al., 2018). These data suggest that the drug combination of MINO plus NAC not only has higher efficacy, but also has a longer therapeutic time window than the individual drugs alone (Mohamadpour et al., 2019). MINO plus NAC restored acquisition of two hippocampal-dependent behavior tasks, active place avoidance and Barnes maze, and retained MAP2 and myelin levels in the ipsilateral hippocampus when dosed 12 hours after injury (Sangobowale et al., 2018). Unilateral hippocampal damage alone is not sufficient to induce spatial memory impairment. Lesion and inactivation studies demonstrate that spatial memory deficits need bilateral hippocampal destruction or inactivation (Smith et al., 1994; Poe et al., 2000; Tran et al., 2006). Injured mice first dosed 24 hours after injury (MN24) did not acquire active place avoidance, which requires two functional hippocampi, but did acquire Barnes maze, which only requires one functional hippocampus (Sunyer et al., 2007; Sangobowale et al., 2018). MN24 did not show histological improvement to the ipsilateral hippocampus, suggesting that the drug combination acted on the contralateral hippocampus to restore acquisition of Barnes maze.
Proper synapse formation and synaptic integration requires maintenance of the size, shape and complexity of dendritic arbors (Scott and Luo, 2001; Spruston, 2008). Dendrites, spines and synapses have high injury susceptibility since they are damaged by experimental TBI both ipsilateral and contralateral to the injury site (Scheff et al., 2005; Atkins, 2011; Gao and Chen, 2011; Gao et al., 2011; Campbell et al., 2012; Winston et al., 2013; Casella et al., 2014). Surviving neurons with damaged dendrites and fewer functional synapses results in disrupted brain circuitry. This study shows that damage to dendrites, spines and synapses is reduced by treatment with MINO plus NAC first dosed 3 days after CHI. Many of the therapeutic effects were seen in the contralateral hemisphere, suggesting regions distal to the initial injury can be targeted by drugs first dosed days after TBI.
Methods
Closed head injury model and drug treatment
All experiments were performed using male C57/BL6 mice (16 to 18 weeks old, 26-28gr). Mice were randomly assigned to control and experimental groups. Sham-CHI or CHI was administered as previously published (Grin’kina et al., 2016). Sham-injured mice received identical anesthesia treatment without the impact procedure, therefore the sham ipsilateral and contralateral hemispheres were combined. Immediately after injury, recovery of righting reflex was evaluated as a measure of acute neurological function and injury severity. Injured mice were randomly assigned to drug or saline treatment groups. At 72 hours after CHI or sham-CHI, mice received an intraperitoneal injection (i.p.) of MINO (22.5 mg/kg) plus NAC (75 mg/kg) in physiological saline or saline alone. Each group received two additional i.p. injections at 4 and 5 days post-injury. Sham-CHI mice only received saline treatment. All assessments done at 3 days after sham-CHI or CHI were on mice that did not receive drug or saline treatments. MINO plus NAC was prepared as previously described (Sangobowale et al., 2018). All drugs were from Sigma (St. Louis, MO). This study was carried out in strict accordance with the National Institutes of Health Guide for the Care and Use of Laboratory Animals. All studies were approved by the Institutional Animal Care and Use Committee of the State University of New York-Downstate Medical Center (protocol #18-10558). Experimenters were blinded to injury or treatment status since all mice were assigned a code number upon arrival at SUNY-Downstate that was used during all determinations.
Barnes Maze
Behavioral assessment on Barnes maze (n =13-14/group) was performed as described by Sangobowale et al. (Sangobowale et al., 2018). Testing began seven days after sham-CHI or CHI, after transient motor deficits in injured mice return to sham-CHI levels (Grin’kina et al., 2016). For four consecutive days, each mouse received four 3-minute spatial acquisition trials with a 15-minute intertrial interval. On the 5th day, mice received a 90-second probe trial with the escape box removed. Debut video software tracked the mouse position at 10 frames per second using a webcam (Tecknet C016 720p HD) located 1.5 m above the maze. Video records were analyzed using AnyMaze software (Stoelting) by an observer blind to the experimental treatment. The average of the four daily trials was calculated to produce the primary acquisition measure of latency to find the escape box. Latency, the number of erroneous holes searched, path length and speed were measured across the training trials. The probe trial was analyzed by time spent and number of holes searched in the target quadrant. Following behavioral assessment, animals were randomly assigned to histological analyses.
Golgi-Cox staining and analysis
Fourteen days after sham-CHI or CHI, mice were anesthetized with urethane (0.1 mL 40%) and whole brains were stained as described in the FD Rapid Golgi Stain kit (FD Neurotechnologies, Columbia, MD, USA). Coronal sections (180μm) were prepared on a vibratome (Leica VT 100M), mounted and stained. Z-stacks of Golgi-stained dorsal hippocampus (from Bregma −1.3 to −2.7) were obtained with a Nikon DS-U3 camera on a Nikon Eclipse Ci-L microscope and processed with NIS-Elements D 4.40.00 software. Individual CA3 and CA1 pyramidal neurons were analyzed that had a fully impregnated dendritic tree that was minimally obscured by the dendrites of nearby neurons. CA1 and CA3 pyramidal cells meeting these criteria (10 cells per region) in each group (n=5 animals/group) were reconstructed into 3-dimensional traces using Neurolucida 360 software (Version 11.03, MBF Bioscience). NeuroExplorer software (MBF Bioscience) measured the reconstructed neurons for surface area and volume, branch morphology (total number, length, surface area and volume), and node number. Basal and apical dendritic trees were analyzed separately. Built-in convex hull analysis (NeuroExplorer, MBF Bioscience) measured neuronal volume and dendritic field size. Built-in Sholl analysis measured dendritic intersection number at 10μm intervals from the neuronal soma.
Dendritic spine quantification and classification
Spines were quantified in CA1 apical dendrites proximal (<75 μm) and distal (>75μm) from the soma using NIS-Elements D 4.40.00 software. Secondary or tertiary dendritic branches were selected that did not overlap with other branches and were either parallel or at acute angles to the coronal section surface. Spine density per 10μm was assessed in at least 10 neurons and 50 branches per group (Table 7-1). Five distinct spine types (filopodia, stubby, thin, mushroom and branched) were manually sub-typed based on previously published methods (Arellano et al., 2007; Bloss et al., 2011; Risher et al., 2014; Afroz et al., 2016). Filopodia spines were defined as having a length of >2μm with no head; thin spines had a head/neck ration of >1:1 and a head diameter of <0.4μm; stubby spies had a length to width ratio of 1:1; mushroom spines had a head/neck ratio of >1:1 with a ≥0.4μm head width; and branched spine types contained 2 or more heads.
Immunohistochemistry
Mice (n=3-5/group) were deeply anesthetized with isoflurane (3–5%) in oxygen (0.8 L/min) and fixed transcardially with paraformaldehyde (4%, w/v). Brain were post-fixed overnight in paraformaldehyde (4%, w/v), embedded in paraffin, and parasagittal sections (10μm) prepared 0.8-2mm from the midline (HistoWiz, Brooklyn, NY). Sections were stained with antibodies against MAP2 (Abcam, catalog #ab32454; antibody registry ID: AB 776174; rabbit polyclonal 1:500), NeuN (Abcam, catalog #ab104225; antibody registry ID: 10711153; rabbit polyclonal 1:500), synaptophysin (LifeSpanBioSciences, catalog #LS-C203763; antibody registry ID: AB 2864290; rabbit polyclonal, 1:500), and PSD-95 (Abcam, catalog #ab2723; antibody registry ID: 303248; mouse monoclonal, 1:500) with the appropriate fluorescent secondary antibodies. Nuclei were stained with 4′,6-diamidino-2-phenylindole. The specificity of each antibody binding was tested by staining adjacent regions with only primary or secondary antibodies. Sections were imaged using an Olympus Fluoview FV100 laser scanning confocal microscope using a 60x oil immersion objective and Leica Application Suite 1.8.2. Image J software measured MAP2 immunoreactivity in CA3 and CA1. Changes in MAP2 protein expression were examined separately in apical and basal dendritic trees.
Analysis of neuronal and synaptic density
Hippocampal cell density was quantified in NeuN-stained sections using stereological methods in NIH ImageJ software (Wang et al., 2003; Sun et al., 2006). NeuN immunopositive cells (NeuN+) were manually counted within each region in three 10μm sections beginning 0.8mm from Bregma. NeuN+ cell number in each section was divided by the product of the area of counting frame and section thickness, averaged and expressed as number of neurons per mm3. Synaptic density was quantified by staining intensity and colocalization of fluorescently labeled synaptophysin and PSD-95 using the method of Chakroborty et al. (Chakroborty et al., 2019) performed with Metamorph software (Version 7.1, Molecular Devices, Sunnyvale, CA). A median filter was applied to background-subtracted red and green imaging channels to reduce noise, and images were set to inclusive threshold levels. Equally sized image boxes (100 μm x 100 μm) were placed over the CA1 stratum radiatum. Staining intensity was determined as the percentage of area containing puncta of synaptophysin or PSD-95, over a threshold intensity level. Colocalization was measured using the Measure Colocalization plugin and expressed as the percentage of overlap of red and green fluorescent puncta. For each image stack, staining intensity and colocalization was measured in 3 serial planes and the results were averaged.
Statistical Analysis
Two-way repeated measures (RM) ANOVA analyzed group differences in Barnes maze latency, errors, speed and path length on the factors of group and trial for each day assessed followed by pairwise comparisons using Tukey’s post-hoc test. Spearman rank correlation was used for righting reflex correlation analysis. A two-level mixed-effects model analyzed Sholl analysis on the factors of distance from the soma and treatment, followed by Tukey’s post hoc test. All remaining analyses used one-way ANOVA followed by pairwise comparisons using Tukey’s post hoc test. Statistical analysis was performed using Graphpad Prism software (La Jolla, CA) with significance set at 0.05. P-values in the text are from post-hoc comparisons unless noted otherwise as from ANOVA. This study used Barnes Maze latency as a primary outcome measure. Sample size was therefore determined using MN24 dosing from the study of Sangobowale to estimate an effect size of 0.9 that results in 13 mice per group and produces an actual power of 0.95 (Sangobowale, et al., 2018). Sample size was determined using G* power v. 3.1.9.6 software (Faul, F., Erdfelder, E., Buchner, A., Lang, A.-G. , Statistical power analyses using G*Power 3.1: Tests for correlation and regression analyses. Behav. Res. Meth. , 2009. 41: p. 1149-1160.) All values are reported as mean ± standard error of the mean.
Results
MN72 restores spatial navigation and memory deficits in Barnes maze after CHI
CHI produces a heterogenous injury due to directly striking the mouse’s head as well as allowing subsequent head movement during impact (Grin’kina et al., 2016). To reduce the variability in our injured group, an initial study was done comparing time to restore righting reflex after injury to Barnes maze acquisition (Figure 1-1). A significantly positive correlation was seen in mice with righting reflex over 450 seconds. Therefore, only mice that required more than 450 seconds to regain righting reflex were included in this study.
Beginning 7 days post-injury (PI), all groups were tested on Barnes maze. Latency had significant group effects on day and trial with a significant interaction of day and trial (day, F3,9 = 101.9, p < 0.0001; trial, F2,6 = 62.36, p < 0.0001; day x trial, F6,18 = 3.439, p < 0.05). Latency to find the escape hole significantly decreased from training days 1 to 4 in Sham-CHI and CHI-MN72 mice, but not CHI-Saline mice (Figure 1A). On Day 1, latency did not differ among the groups (p > 0.8). On Day 4, latency did not differ between Sham-CHI and CHI-MN72 mice and the latency of both groups was shorter than CHI-Saline mice (p < 0.005). Analysis of errors in Barnes maze testing yielded similar results (Table 1-1). Neither path length nor speed differed among the groups suggesting similar motor performance, therefore impaired navigation is the likely cause of differences in latency and errors (Table 1-1).
Figure 1. MN72 improves CHI-induced spatial navigation and memory deficits on Barnes Maze.
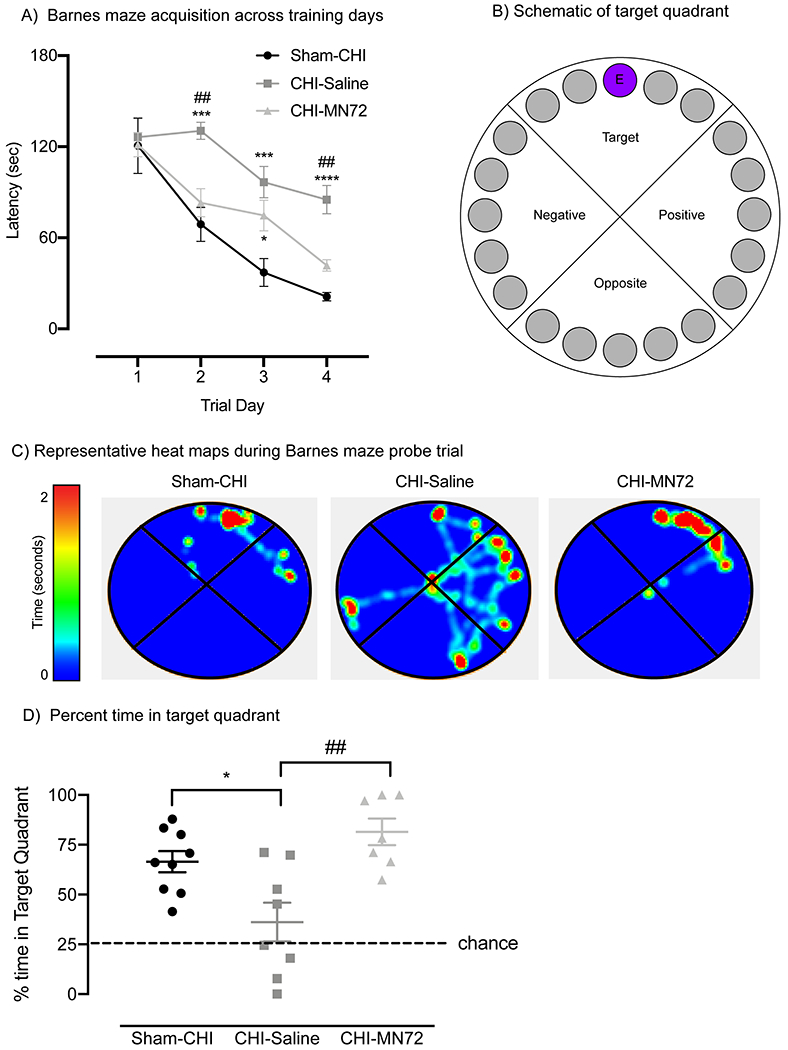
A, CHI causes deficits on Barnes Maze acquisition. Group differences in latency are indicated across training days. On day 1, all groups had similar latencies. On day 4, Sham-CHI and CHI-MN72 mice had significantly shorter latencies than CHI-Saline mice. B, Schematic of Barnes maze with a target quadrant containing an escape hole (E). C, Representative heat maps of probe trials. D, Probe trial performance. Sham-CHI and CHI-MN72 mice spent more time in the target quadrant than CHI-Saline mice. A hash tag (#) indicates a significant difference with CHI-MN72 group, and an asterisk (*) indicates a significant difference with the sham-CHI group; *p < 0.05, ***p<0.001, ****p<0.0001.
A probe trial assessed escape hole retention 24 hours after the last training trial (Figure 1B–D). The groups significantly differed in the percent time spent and holes searched in the target quadrant (% time, F2,21 = 9.35, p < 0.002; % holes, F2,21 = 13.24, p = 0.0002). Sham-CHI (p < 0.02) and CHI-MN72 mice (p < 0.002) spent significantly more time in the target quadrant than CHI-Saline mice (Figure 1D, Table 1-1). CHI-Saline mice searched significantly fewer holes than either Sham-CHI (p = 0.0002) or CHI-MN72 mice (p < 0.005; Table 1-1). Sham-CHI and CHI-MN72 mice did not differ in time spent or holes searched in the target quadrant (% time, p > 0.3; % holes, p > 0.7). These data strongly suggest that MN72 treatment restored the spatial navigation and memory deficits produced by CHI to uninjured levels.
MN72 prevents neuronal loss in contralateral CA3 and ipsilateral CA1
Hippocampal neuronal loss after experimental TBI correlates to severity of hippocampal-dependent deficits on behavior tasks (Smith et al., 1997; Atkins, 2011). Therefore NeuN+ cells in CA1 and CA3 stratum pyramidale at 14 days PI were quantified using stereological methods (Figure 2A) (Wang et al., 2003; Sun et al., 2006). NeuN+ cell density in CA3 and CA1 had a significant group effect (CA3, F4,17 = 5.2, p < 0.007; CA1, F4,15 = 5.7, p < 0.006). CA3 NeuN+ density decreased bilaterally in CHI-Saline mice as compared to Sham-CHI (ipsilateral, p < 0.005; contralateral, p < 0.05; Figure 2B). In contrast, NeuN+ density decreased only in the ipsilateral CA3 of CHI-MN72 mice (ipsilateral, p < 0.05; contralateral, p > 0.2). CA1 NeuN+ density decreased in the ipsilateral, but not contralateral, hippocampus of CHI-Saline mice as compared to Sham-CHI mice (ipsilateral, p < 0.02; contralateral, p > 0.1; Figure 2C). In contrast, CA1 NeuN+ density did not differ in either hippocampus between Sham-CHI or CHI-MN72 mice (ipsilateral, p > 0.1; contralateral, p > 0.9). These data suggest that CHI produced CA3 neuronal loss in both hippocampi and CA1 neuronal loss in the ipsilateral hippocampus by 14 days PI. MN72 prevented CA3 neuronal loss in the contralateral hippocampus and CA1 neuronal loss in the ipsilateral hippocampus.
Figure 2. MN72 maintains neuronal density in contralateral CA3 and ipsilateral CA1.
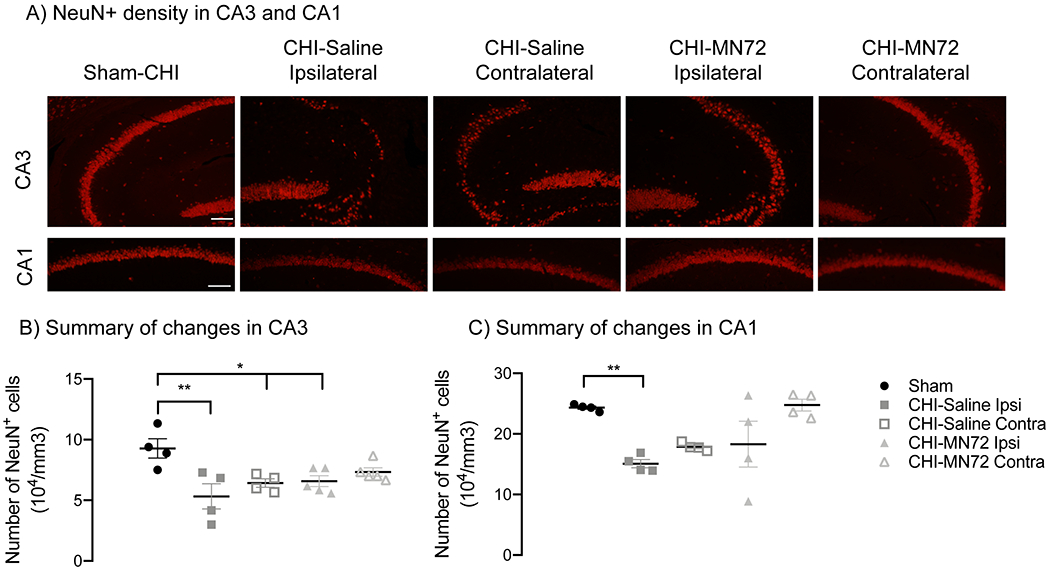
A, Representative photomicrographs of NeuN+ stained CA3 and CA1 at 14 days PI (scale bar, 100μm). B, Summary of CA3 NeuN+ density. Compared to Sham-CHI, CA3 NeuN+ density is bilaterally decreased in CHI-Saline mice, and decreased in the ipsilateral hippocampus in CHI-MN72 mice. C, Summary of CA1 NeuN+ density. Decreased ipsilateral CA1 NeuN+ density in CHI-Saline mice is prevented by MN72 treatment. An asterisk (*) represents significant differences with Sham-CHI, *p < 0.05, **p < 0.01.
MN72 maintains CA3 and CA1 neuron volume and dendritic field size
Damage to neuronal and dendritic architecture produced by experimental TBI is more extensive than neuronal loss (Gao and Chen, 2011; Gao et al., 2011). Therefore, multiple morphological and geometric characteristics were assessed in Golgi-Cox stained CA1 and CA3 neurons in both hemispheres (Table 3-1). Convex hull analysis assessed the volume of surviving CA1 and CA3 neurons (Figure 3A, 3D). CA3 neuron volume differed among groups (F4, 41 = 8.09, p < 0.0001; Figure 3B). Bilaterally, the volume of CA3 neurons from CHI-Saline mice was reduced compared to Sham-CHI (ipsilateral, p < 0.005; contralateral, p < 0.01). In contrast, only CA3 neurons from the ipsilateral hippocampus of CHI-MN72 mice had reduced volume (p < 0.007). CA3 neuron volume in the contralateral hippocampus of CHI-MN72 mice did not differ from Sham-CHI (p > 0.5) and was larger than CHI-Saline mice (p < 0.05). These data suggest that CHI bilaterally reduced CA3 neuron volume, which was prevented in the contralateral hemisphere by MN72 treatment.
Figure 3. MN72 prevents reductions in CA3 and CA1 neuronal volume and dendritic field size induced by CHI.
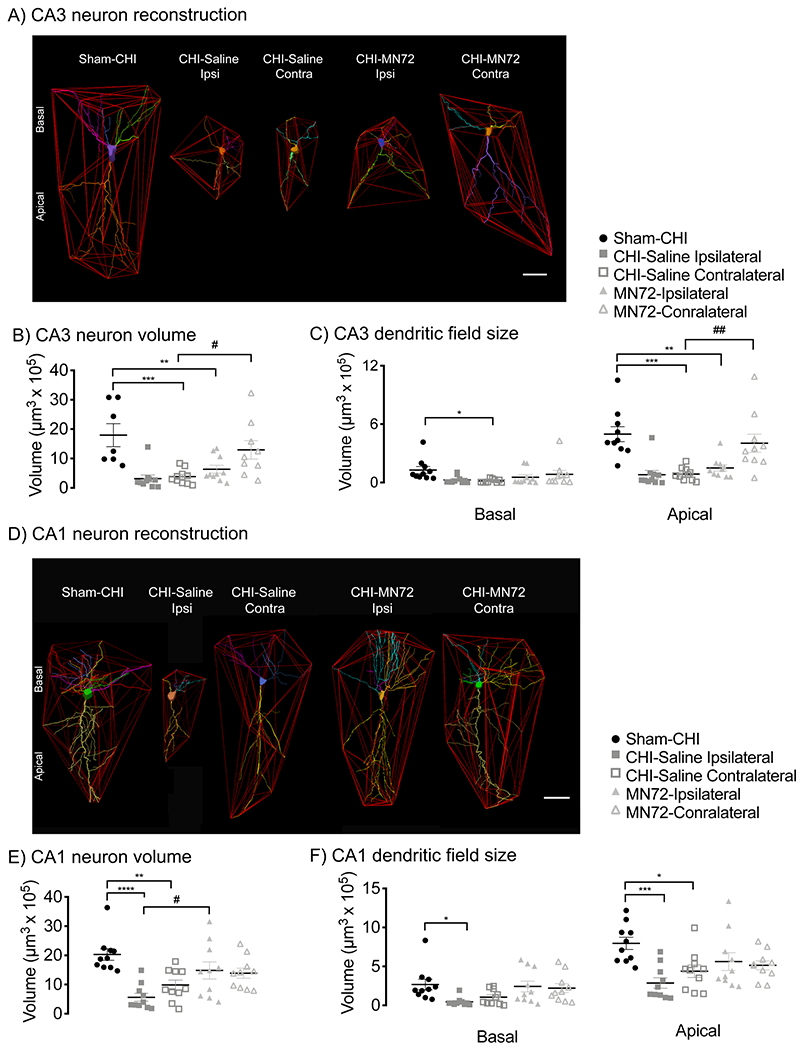
A, D, Polygons generated to assess CA3 (A) and CA1 (D) neuron volume and dendritic field size. B, CHI bilaterally reduced CA3 neuron volume which was prevented by MN72 in the contralateral hippocampus. C, CHI reduced contralateral CA3 basal dendritic field size and bilaterally reduced CA3 apical dendritic field size. MN72 prevented reduction of CA3 dendritic field size only in contralateral apical dendrites. E, CHI bilaterally reduced CA1 neuron volume which was prevented by MN72. F, MN72 bilaterally maintained CA1 basal and apical dendritic field sizes. An asterisk (*) represents significant difference with Sham-CHI, a hashtag (#) represents significant differences with CHI-MN72. * p < 0.05, ** p < 0.01, *** p < 0.001, **** p < 0.0001.
Convex hull analysis also measured basal and apical dendritic field size in CA3 and CA1 neurons. CA3 basal and apical dendritic field size significantly differed among groups (basal, F4, 45 = 2.79, p < 0.05; apical, F4, 45 = 10.32, p < 0.001; Figure 3C). In the ipsilateral hippocampus, CA3 basal dendritic field size was similar in CHI-Saline, CHI-MN72, and Sham-CHI mice. Ipsilateral apical dendritic field size was reduced in both CHI-Saline (p < 0.0002) and CHI-MN72 mice (p < 0.002). In the contralateral hippocampus, CA3 basal and apical dendritic field size in CHI-Saline mice was smaller than Sham-CHI mice (basal, p < 0.05; apical, p < 0.0005). In contrast, CA3 basal and apical dendritic field size was similar in CHI-MN72 and Sham-CHI mice (basal, p > 0.7; apical, p > 0.8). Apical dendritic field size was larger in CHI-MN72 and Sham-CHI mice than CHI-Saline mice (p < 0.005). These data suggest that, in CA3, CHI reduced basal dendritic field size in the contralateral hippocampus and reduced apical dendritic field size bilaterally. MN72 maintained CA3 apical dendritic field size in the contralateral hippocampus.
CA1 neuron volume also differed among groups (F4, 45 = 7.49, p = 0.0001; Figure 3E). Bilaterally, CA1 neurons from CHI-Saline mice had reduced volume compared to Sham-CHI mice (ipsilateral, p < 0.0001; contralateral, p < 0.005). In contrast, CA1 neurons from CHI-MN72 and Sham-CHI mice had similar volume (ipsilateral, p > 0.3; contralateral, p > 0.15). Ipsilateral CA1 neurons from CHI-MN72 mice had a larger volume than CHI-Saline mice (p < 0.02). These data suggest that MN72 bilaterally prevented the reduction of CA1 neuron volume produced by CHI.
CA1 basal and apical dendritic field size significantly differed among groups (basal, F4, 45 = 3.31, p <0.05; apical, F4, 45 = 5.17, p = 0.002; Figure 3F). In ipsilateral CA1, CHI-Saline mice had smaller basal and apical dendritic fields than Sham-CHI mice (basal, p < 0.05; apical, p < 0.01). In contralateral CA1, CHI-Saline mice had a similar basal but smaller apical dendritic field size (basal, p > 0.2; apical, p < 0.05). In contrast, CA1 basal and apical dendritic field size did not differ between CHI-MN72 or Sham-CHI mice in either hippocampus (basal, ipsilateral, p > 0.9, contralateral, p > 0.9; apical, ipsilateral, p > 0.2, contralateral, p > 0.1). These data suggest that CHI bilaterally reduced CA1 basal and apical dendritic field size, which was prevented by MN72.
MN72 maintains CA3 dendritic architecture in the contralateral hippocampus
The shape and complexity of the dendritic arbor are important determinants of neuronal input and synaptic integration and could underlie functional deficits (Ishizuka et al., 1995; Staff et al., 2000; Stuart and Spruston, 2015). Dendritic architecture was therefore evaluated by two complementary methods, quantifying nodes and Sholl analysis (Figures 4,5; Table 3-1). Node number indicates the complexity of the dendritic arbor; Sholl’s analysis provides additional spatial information on dendritic arborization that enhances node analysis.
Figure 4. MN72 maintains CA3 dendritic complexity and architecture in the contralateral hippocampus.
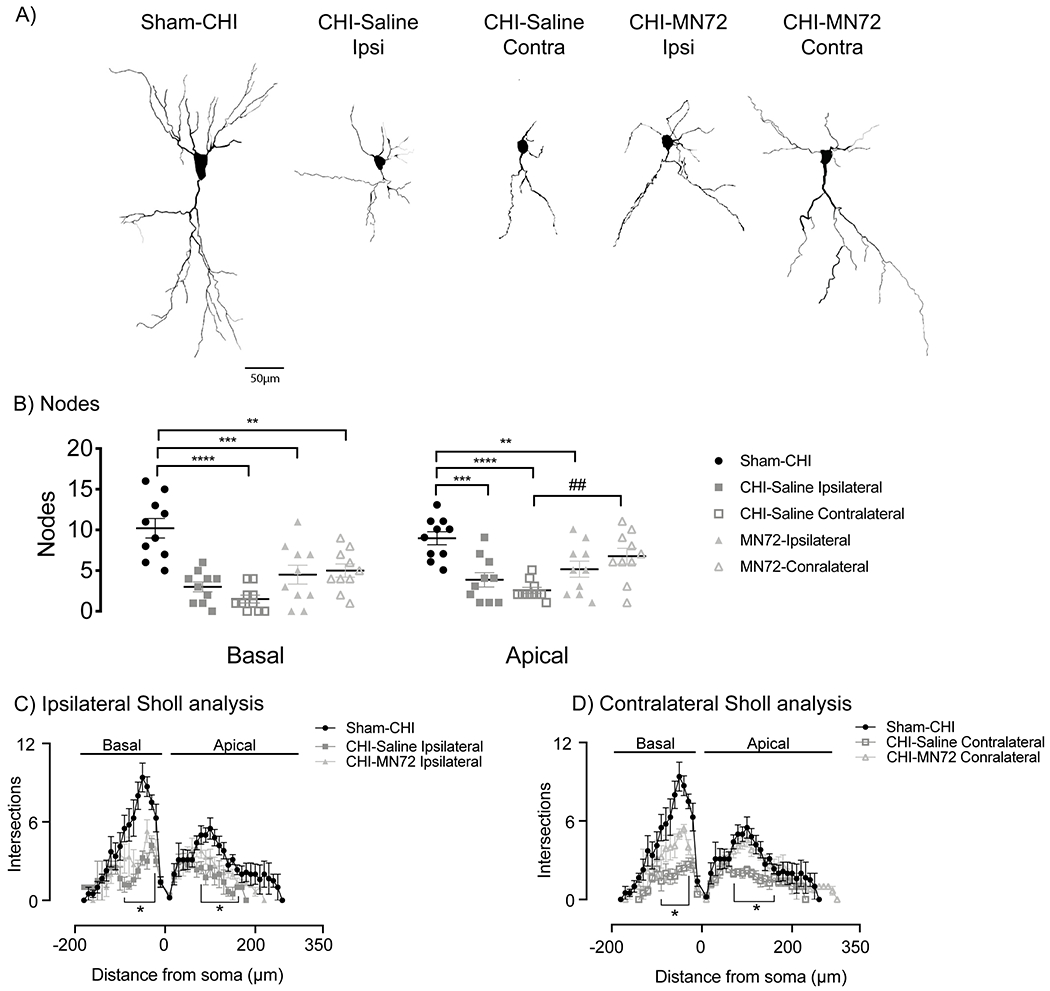
A Representative tracings of Golgi-Cox stained CA3 pyramidal neurons from Sham-CHI, CHI-Saline and CHI-MN72 mice. B, CHI bilaterally reduced node number in apical and basal dendrites compared to Sham-CHI. MN72 maintained node number in the contralateral CA3 apical dendrites. An asterisk (*) represents significant difference with Sham-CHI, a hashtag (#) represents significant differences with CHI-MN72. ** p < 0.01, *** p < 0.001, **** p < 0.0001. C-D, Sholl’s analysis. C, In the ipsilateral hippocampus, intersection number was reduced in CHI-Saline basal (30-90μm) and apical (80-160μm) dendrites compared to Sham-CHI. Intersection number was only lower in CHI-MN72 basal (30-60μm) dendrites. D, In the contralateral hippocampus, intersection number was reduced in CHI-Saline basal (30-70μm) and apical (70-160μm) dendrites compared to Sham-CHI. CHI-MN72 mice had fewer intersections in basal (30-50μm) dendrites than Sham-CHI mice, but more intersections (40-50μm) than CHI-Saline mice. Contralateral apical dendrites of Sham-CHI and CHI-MN72 mice had a similar number of intersections. An asterisk (*) indicates significant differences in intersection number.
Figure 5. MN72 maintains CA1 dendritic complexity and architecture in the ipsilateral and contralateral hippocampus.
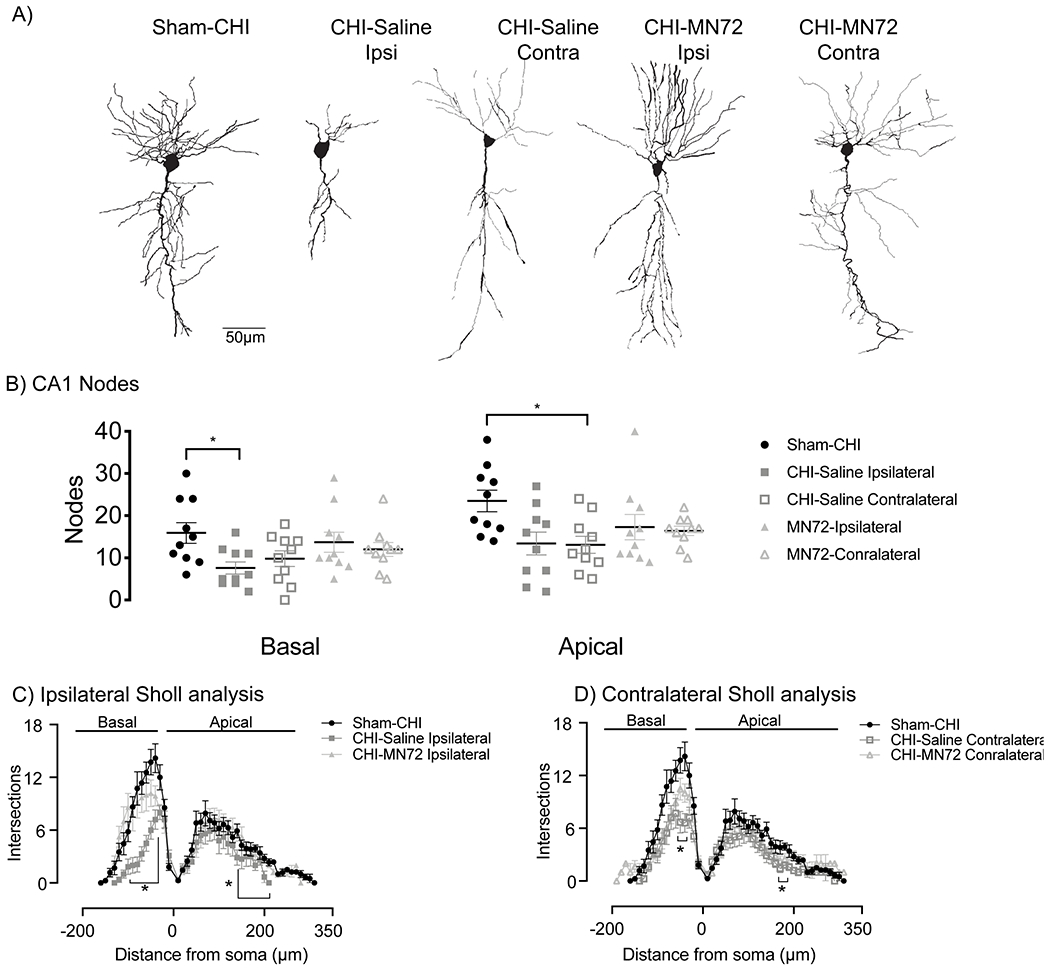
A Representative tracings of Golgi-Cox stained CA1 pyramidal neurons from Sham-CHI, CHI-Saline and CHI-MN72 mice. B, CHI reduced node number in ipsilateral basal dendrites and in ipsilateral and contralateral apical dendrites. MN72 bilaterally maintained node number in both apical and basal dendrites. An asterisk (*) represents significant difference with Sham-CHI. * p < 0.05. C-D, Sholl’s analysis. C, In the ipsilateral hippocampus compared to Sham-CHI, intersection number was reduced in CHI-Saline basal (40-100μm) and apical (140-220μm) dendrites. Sham-CHI and CHI-MN72 mice had similar intersection number in ipsilateral basal and apical dendrites. D, In the contralateral hippocampus compared to Sham-CHI, intersection number was reduced in CHI-Saline basal (40-50μm) and apical (170-180μm) dendrites. Sham-CHI and CHI-MN72 mice had similar intersection number in contralateral basal and apical dendrites. An asterisk (*) indicates significant differences in intersection number.
Node number in CA3 basal and apical dendrites significantly differed among groups (basal, F4, 45 = 13.4, p < 0.0001; apical, F4, 45 = 9.01, p < 0.0001; Figure 4B). Bilaterally, apical and basal dendrites in CHI-Saline mice had fewer nodes than Sham-CHI mice (basal, ipsilateral, p < 0.0001, contralateral, p < 0.0001; apical, ipsilateral, p = 0.007, contralateral, p < 0.0001). In the ipsilateral hippocampus, apical and basal dendrites in CHI-MN72 mice had fewer nodes than Sham-CHI mice (basal, p < 0.002; apical, p < 0.05). In the contralateral hippocampus, CHI-MN72 mice had fewer nodes in basal, but not apical dendrites (basal, p < 0.002; apical, p > 0.3). Contralateral CA3 apical dendrites in CHI-MN72 mice had more nodes than CHI-Saline mice (p < 0.008).
Sholl’s analysis investigated dendritic architecture as a function of distance from the soma by assessing the number of dendritic crossing intersections with concentric circles radiating from the soma (Figures 4C,D). Treatment and distance had a significant group effect on intersection number (basal, treatment, F4, 45 = 12.79, p <0.0001, distance, F3.2, 145.2 = 58.17, p < 0.0001, treatment x distance, F68, 765 = 4.12, p < 0.0001; apical, treatment, F4, 45 = 14.52, p <0.0001, distance, F3.9, 174.9 = 51.66, p < 0.0001, treatment x distance, F116, 1305 = 2.79, p < 0.0001; Figures 4C, D). CHI bilaterally reduced intersection number across the CA3 basal and apical dendritic arbor compared to Sham-CHI (p < 0.05). MN72 treatment increased intersection number at discrete locations in the contralateral basal dendritic arbor (p <0.01; Figure 4D). Despite this improvement from CHI-Saline mice, CA3 basal dendrites in CHI-MN72 mice had fewer intersections than Sham-CHI mice near the soma in both hippocampi (p < 0.05). In contrast, in both ipsilateral and contralateral CA3 apical dendrites, intersection number did not differ between Sham-CHI and CHI-MN72 mice (p > 0.1). MN72 treatment also increased intersection number at discrete locations in the contralateral apical dendritic arbor compared to CHI-Saline mice (p < 0.05). These data suggest that CHI bilaterally altered branch distribution and reduced the complexity of CA3 apical and basal dendritic architecture. MN72 bilaterally maintained dendritic architecture with a greater therapeutic effect in contralateral CA3 dendrites.
MN72 maintains CA1 dendritic architecture in the ipsilateral and contralateral hippocampus
Total node number in CA1 basal and apical dendrites significantly differed among groups (basal, F4, 45 = 2.64, p < 0.05; apical, F4, 45 = 3.14, p < 0.05; Figure 5B). In ipsilateral CA1 dendrites, CHI-Saline mice had fewer nodes compared to Sham-CHI (basal, p < 0.05; apical, p < 0.05). In contralateral CA1, CHI-Saline mice had fewer nodes in apical, but not basal dendrites (basal, p > 0.2; apical, p < 0.05). In contrast, node number in MN72-treated mice did not differ with Sham-CHI mice in either hippocampus (basal, ipsilateral, p > 0.9, contralateral, p > 0.6; apical, ipsilateral, p > 0.3, contralateral, p > 0.2).
In Sholl analysis, treatment and distance had a significant group effect on intersection number in CA1 dendrites (basal, treatment, F4, 47 = 6.89, p = 0.0002, distance, F2.973, 139.7 = 120.6, p < 0.0001, treatment x distance, F72, 846 = 3.93, p < 0.0001; apical, treatment, F4, 48 = 3.76, p = 0.01, distance, F3.7, 171.6 = 75.67, p < 0.0001; treatment x distance, F120, 1380 = 1.18, p < 0.09; Figure 5C,D). CHI bilaterally reduced intersections across CA1 apical and basal dendrites compared to Sham-CHI (p < 0.05). MN72 treatment increased intersection number across the CA1 apical and basal dendritic arbor compared to CHI-Saline mice (p < 0.05). In both hippocampi, intersection number did not differ between Sham-CHI and CHI-MN72 mice (basal, p > 0.08; apical, p > 0.1). These data suggest that CHI bilaterally altered CA1 dendritic branch distribution and reduced complexity with more pronounced injury in the ipsilateral hippocampus. MN72 bilaterally maintained CA1 dendritic architecture.
MN72 either prevents MAP2 loss or accelerates MAP2 recovery in CA3 dendrites
MAP2 is a major dendritic cytoskeletal protein; loss of MAP2 expression accompanies dendritic damage in multiple TBI models (Taft et al., 1992; Posmantur et al., 1996; Grin’kina et al., 2016; Sangobowale et al., 2018). Lowered MAP2 expression induces dendritic instability and subsequent loss; both instability and loss can be restored by maintaining MAP2 expression. MN72 treatment preserves dendritic architecture (Figures 4, 5). We therefore examined a time course of changes in hippocampal MAP2 expression at 3, 7 and 14 days PI. CA3 pyramidal neurons in Sham-CHI mice had prominent MAP2 dendritic labeling and dendrites that projected orthogonally from CA3 stratum pyramidale into stratum lucidum and stratum radiatum (Figure 6A). In contrast, pyramidal neurons from CHI-Saline mice had diminished dendritic labeling and disorganized, non-linear dendrites. Contralateral CA3 pyramidal neurons from CHI-MN72 mice had restored dendritic labeling and linear organization.
Figure 6. MN72 either prevents MAP2 loss or accelerates MAP2 recovery.
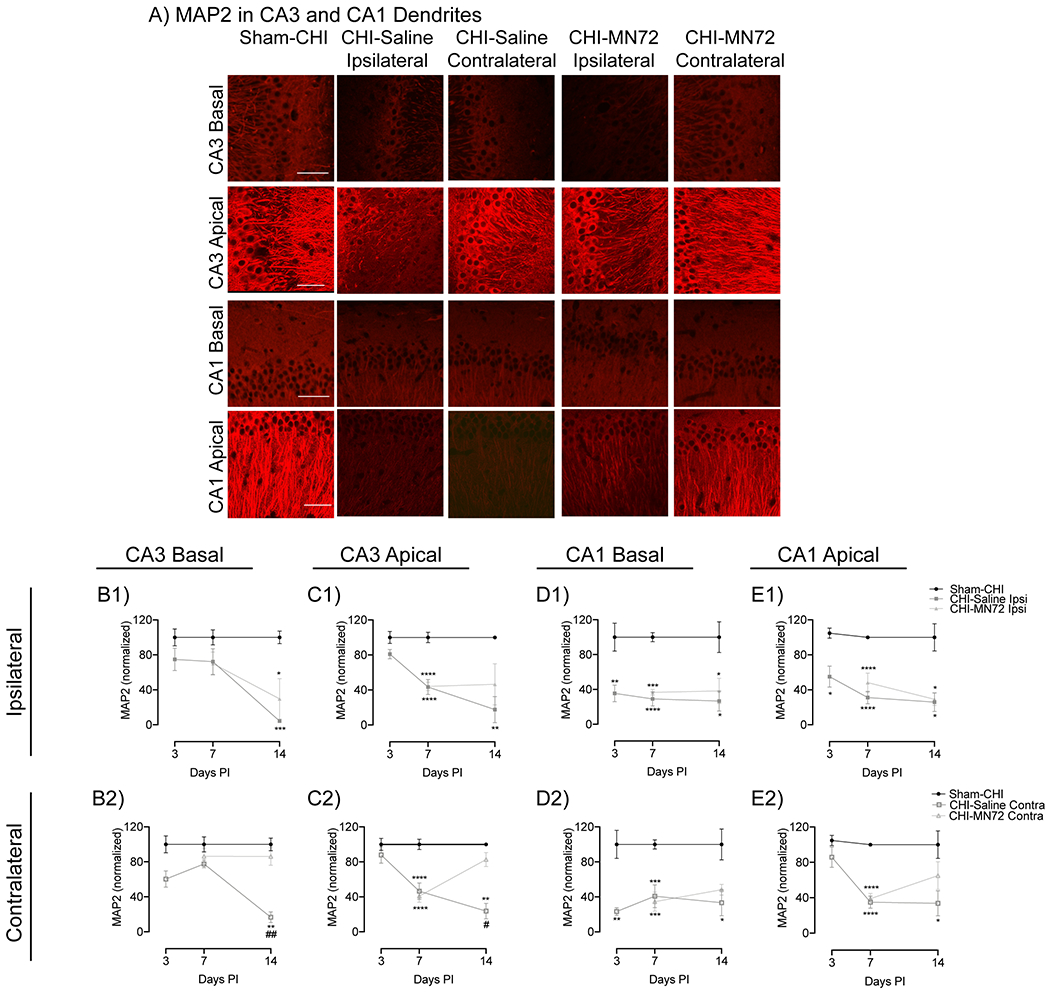
A, Representative photomicrographs of MAP2 immunoreactivity in CA3 and CA1 apical and basal dendrites at 14 days PI. Scale bar, 50μm. Summary of changes in MAP2 expression in the ipsilateral (B1, C1, D1, E1) or contralateral (B2, C2, D2, E2) hippocampus. In all regions, MAP2 expression was unchanged in Sham-CHI mice. B1, In ipsilateral CA3 basal dendrites, MAP2 loss occurs between days 7-14 in CHI-Saline and CHI-MN72 mice. B2, In contralateral CA3 basal dendrites, MAP2 loss occurs between days 7-14 in CHI-Saline mice. MN72 treatment prevents MAP2 loss. C1, In ipsilateral CA3 apical dendrites, MAP2 loss occurs between days 3-7 in CHI-Saline and CHI-MN72 mice, and persists through days 7-14 in CHI-Saline mice. C2, In contralateral CA3 apical dendrites, MAP2 loss occurs between days 3-7 in CHI-Saline and CHI-MN72 mice, and persists through days 7-14 in CHI-Saline mice. In contrast, MAP2 expression increases between days 7-14 in CHI-MN72 mice. D1, In ipsilateral CA1 basal dendrites MAP2 loss occurs by day 3 and persists through day 14 in CHI-Saline and CHI-MN72 mice. D2, In contralateral CA1 basal dendrites, MAP2 loss occurs by day 3 in CHI-Saline mice and persists through day 14. In CHI-MN72 mice, MAP2 loss increases by day 14. E1, In ipsilateral CA1 apical dendrites, MAP2 loss by day 3 persists through day 14 in CHI-Saline and CHI-MN72 mice. E2, In contralateral CA1 apical dendrites, MAP2 loss occurs by day 7 in CHI-Saline and CHI-MN72 mice. MAP2 loss persists through day 14 in CHI-Saline mice. In contrast MAP2 is restored by day 14 in CHI-MN72 mice. An asterisk (*) represents a significant difference with Sham-CHI, a hashtag (#) represents significant difference with CHI-MN72. *p < 0.05, **p< 0.01. ***p < 0.001, ****p < 0.0001.
The time course of MAP2 expression in CA3 basal dendrites had a significant group effect only at 14 days PI (Day 3, F2,8 = 3.07, p > 0.1; Day 7, F4,11 = 1.52, p > 0.2; Day 14, F4,11 = 13.82, p = 0.003, Figure 6B1, B2). MAP2 expression in CHI-Saline and CHI-MN72 mice was unchanged at 3 and 7 days PI as compared to Sham-CHI (Day 3, CHI-Saline, ipsilateral, p > 0.3, contralateral, p > 0.08; Day 7, CHI-Saline, ipsilateral, p > 0.3, contralateral, p > 0.5; CHI-MN72, ipsilateral, p > 0.2, contralateral, p > 0.8). At 14 days PI, MAP2 decreased bilaterally in CHI-Saline mice, and ipsilaterally in CHI-MN72 mice (CHI-Saline, ipsilateral, p = 0.001; contralateral, p < 0.002; CHI-MN72, ipsilateral, p < 0.02; contralateral, p > 0.9). Contralateral MAP2 in CHI-MN72 mice was significantly higher than in CHI-Saline mice (p < 0.01). These data suggest that MN72 prevented MAP2 loss in CA3 basal dendrites in the contralateral hippocampus.
In CA3 apical dendrites, MAP2 expression had significant group effects at 7 and 14 days PI (Day 3, F2,9 = 1.68, p > 0.2; Day 7, F4, 31 = 12.96, p < 0.001; Day 14, F4,11 = 7.59, p < 0.005; Figure 6C1, C2). At 7 days PI, MAP2 decreased bilaterally in CHI-Saline and CHI-MN72 mice as compared to Sham-CHI (p < 0.0001). At 14 days PI, bilateral MAP2 loss persisted in CHI-Saline mice but not in CHI-MN72 mice (CHI-Saline, ipsilateral, p < 0.01, contralateral, p < 0.01; CHI-MN72, ipsilateral, p > 0.09; contralateral, p > 0.8). Contralateral MAP2 in CHI-MN72 mice was significantly higher than CHI-Saline mice (p < 0.05). These data suggest that MN72 induced MAP2 recovery in contralateral CA3 apical dendrites, while preventing further MAP2 loss in ipsilateral CA3 apical dendrites.
MN72 accelerates MAP2 recovery in contralateral CA1 apical and basal dendrites
CA1 pyramidal neurons from Sham-CHI mice had prominent MAP2 labeling in long, linearly arranged dendrites (Figure 6A). In contrast, CHI-Saline mice had diminished MAP2 labeling in disorganized, non-linear dendrites. CHI-MN72 mice had increased MAP2 dendritic labeling and improved linear organization in the contralateral CA1 dendrites.
MAP2 expression in CA1 basal dendrites had significant group effects at 3, 7, and 14 days PI (Day 3, F2,8 = 15.66, p < 0.002; Day 7, F4, 20 = 13.52, p < 0.0001; Day 14, F4,20 = 4.090, p < 0.02, Figure 6D1, D2). At 3 days PI, MAP2 decreased bilaterally in CHI-Saline mice as compared to Sham-CHI (ipsilateral, p < 0.006, contralateral, p < 0.002). At 7 days PI, MAP2 decreased bilaterally in CHI-Saline and CHI-MN72 mice (CHI-Saline, ipsilateral, p < 0.0001, contralateral, p < 0.0004; CHI-MN72, ipsilateral, p < 0.0002, contralateral, p < 0.0003). MAP2 loss in CHI-Saline mice persisted bilaterally through 14 days PI (ipsilateral, p < 0.02, contralateral, p < 0.02). In contrast, MAP2 increased in the contralateral dendrites of CHI-MN72 mice (ipsilateral, p < 0.05, contralateral, p > 0.1).
In CA1 apical dendrites, MAP2 had significant group effects at 3, 7, and 14 days PI (Day 3, F2,10 = 5.79, p < 0.05; Day 7, F4,31 = 17.65, p < 0.0001; Day 14, F4,19 = 4.69, p < 0.009, Figure 6E1, E2). In Sham-CHI mice, MAP2 in CA1 apical dendrites was unchanged between days 3 and 14. At 3 days PI, ipsilateral MAP2 expression decreased in CHI-Saline mice as compared to Sham-CHI (ipsilateral, p < 0.02, contralateral, p > 0.4). At 7 days PI, MAP2 decreased bilaterally in both CHI-Saline and CHI-MN72 mice (p < 0.0001). Bilateral MAP2 loss in CHI-Saline mice persisted at 14 days PI (ipsilateral, p < 0.03, contralateral, p < 0.02). In contrast, at 14 days PI, CHI-MN72 mice had decreased MAP2 only in ipsilateral dendrites (ipsilateral, p < 0.05; contralateral, p > 0.4). These data suggest that MN72 accelerated MAP2 recovery in CA1 apical and basal dendrites in the contralateral hippocampus.
MN72 maintains total and mushroom spine density in CA1 apical dendrites
Experimental TBI reduces spine density in CA1 apical dendrites with a severity that varies with the distance from the soma (Winston et al., 2016). Spines receive different presynaptic inputs depending upon their location from the CA1 pyramidal cell soma. In CA1 apical dendrites, Schaffer collateral axons synapse on proximal dendrites and perforant path axons synapse on distal dendrites (Spruston, 2008). Loss of Schaffer collateral or perforant path synapses could lead to Barnes maze deficits, therefore both proximal and distal CA1 apical dendrites were examined for spine loss following CHI (Figure 7A, B). Total spine density on proximal and distal dendrites differed significantly among groups (proximal, F4,49 = 7.73, p < 0.0001; distal, F4, 50 = 5.54, p < 0.001; Figure 7 C, Table 7-1). CHI bilaterally lowered total spine density on both proximal and distal dendrites (proximal, ipsilateral, p < 0.002, contralateral, p < 0.008; distal, ipsilateral, p < 0.02, contralateral, p < 0.005). Total proximal and distal spine density in Sham-CHI and CHI-MN72 mice did not differ in either hippocampus (proximal, p > 0.8; distal, p > 0.4 ). In the ipsilateral hippocampus, CHI-MN72 mice had a higher proximal spine density than CHI-Saline mice (p < 0.005). In the contralateral hippocampus, CHI-MN72 mice had a higher distal spine density (p < 0.02).
Figure 7. MN72 maintains spine density in CA1 apical dendrites.
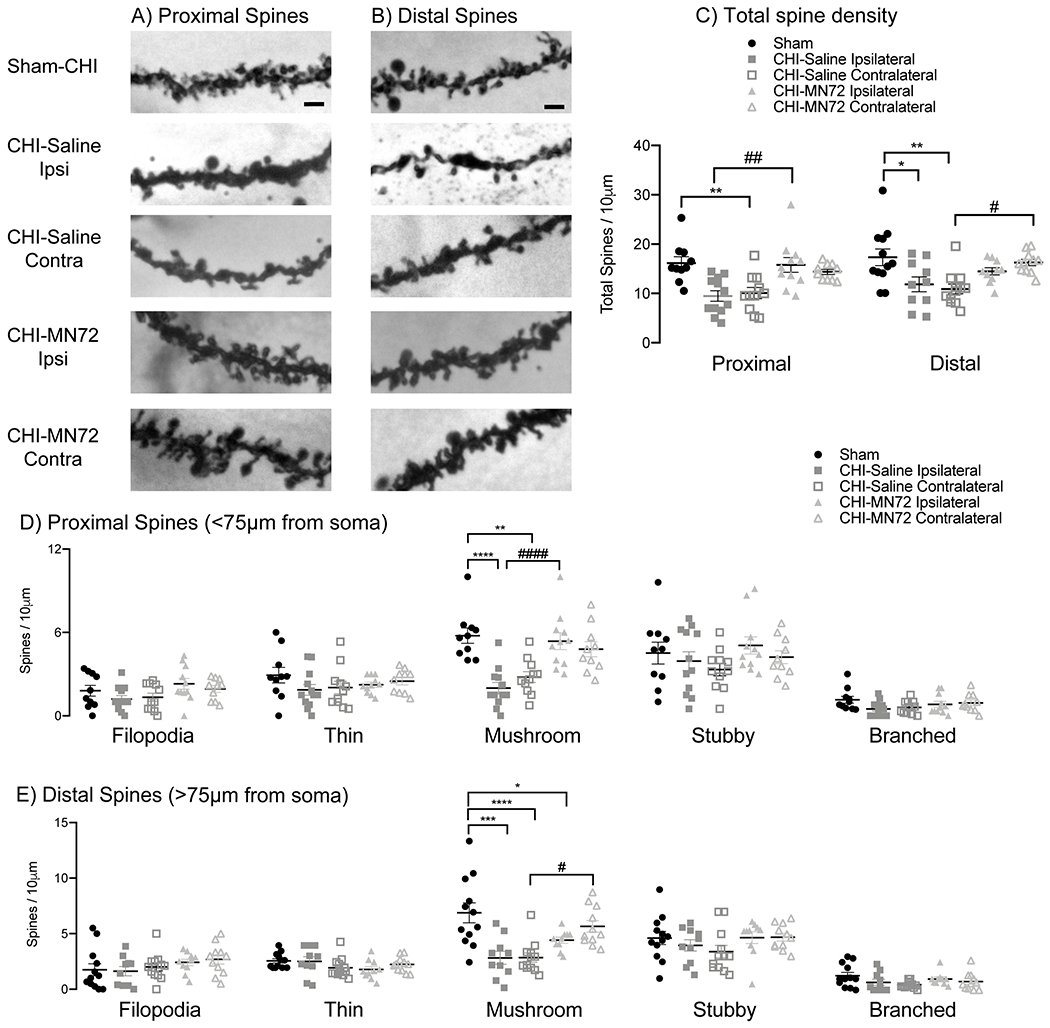
A-B, Representative images of proximal (A) and distal (B) Golgi-Cox stained CA1 apical dendrites. C, Summary of differences in total spine density. CHI bilaterally reduced total spine density in proximal and distal dendritic regions, which was maintained bilaterally by MN72. D, Individual spine type density in proximal dendrites. CHI bilaterally reduced proximal mushroom spine density. MN72 treatment maintained mushroom spine density. E, Individual spine type density in distal dendrites. CHI bilaterally reduced distal mushroom spine density. MN72 treatment maintained distal mushroom spine density in the contralateral hippocampus. An asterisk (*) represents significant a difference with Sham-CHI, a hashtag (#) represents a significant difference with CHI-MN72; *p < 0.05, ** p < 0.01, *** p < 0.001, ****p < 0.0001. Scale bar, 2μm.
Individual spine types were also assessed on proximal and distal dendrites. Mushroom spine density on proximal and distal dendrites differed among groups (proximal, F4,49 = 11.36, p <0.0001; distal, F4,50 = 9.05, p <0.0001; Figure 7D, E). CHI bilaterally decreased mushroom spine density on proximal and distal dendrites as compared to Sham-CHI (proximal, ipsilateral, p < 0.0001, contralateral, p < 0.002; distal, ipsilateral, p < 0.0005, contralateral, p < 0.0001). Bilaterally, mushroom spine density did not differ on proximal dendrites in Sham-CHI and CHI-MN72 mice (p > 0.6; Figure 7D). CHI-MN72 mice had more mushroom spines on proximal dendrites in the ipsilateral hippocampus than CHI-Saline mice (p < 0.001); increased mushroom spines in the contralateral hippocampus trended toward significance (p = 0.055). In contrast, Sham-CHI and CHI-MN72 mice had similar mushroom spine density on distal dendrites only in the contralateral hippocampus (ipsilateral, p < 0.05; contralateral, p > 0.5; Figure 7E). In the contralateral hippocampus, distal mushroom spine density was higher in CHI-MN72 mice than in CHI-Saline mice (p < 0.05). Spine types other than mushroom spines were unchanged among the groups (Table 7-1). These data suggest that CHI bilaterally decreased total hippocampal spine density on proximal and distal regions of CA1 apical dendrites. Spine loss was greater in the ipsilateral hippocampus. Additionally, CHI selectively lowered mushroom spine density. MN72 treatment limited the loss of mushroom spines as well as overall spine density.
MN72 bilaterally maintains CA1 synaptic density after CHI
Preservation of spine density is suggestive of synapse preservation, yet Golgi-Cox visualizes only post-synaptic structures. Therefore, synapse density was measured by co-localization of the presynaptic marker synaptophysin with the postsynaptic marker PSD-95 (Figure 8A). Synaptophysin staining density had a significant group effect (F4, 23 = 7.12, p < 0.001) with CHI-Saline mice having less staining in the ipsilateral hippocampus than Sham-CHI and CHI-MN72 mice (p < 0.005; Figure 8B1). In contrast, synaptophysin staining density did not differ in either hippocampus in CHI-MN72 and Sham-CHI mice (p > 0.9). PSD-95 staining density did not significantly differ among groups (F4, 23 = 2.03, p > 0.1; Figure 8B1). Synaptophysin and PSD-95 immunostaining revealed a punctate distribution consistent with colocalization of presynaptic boutons and the PSD. Colocalization of synaptophysin and PSD-95 had a significant group effect (F4, 23 = 4.71, p < 0.01, Figure 8B2). Bilaterally, CHI-Saline mice had significantly fewer colocalized puncta as compared to Sham-CHI (ipsilateral, p < 0.05; contralateral, p < 0.005). In contrast, CHI-MN72 and Sham-CHI mice had similar amounts of colocalized puncta (p > 0.1). These data suggest that CHI lowered ipsilateral presynaptic termini and bilaterally reduced synaptic density. MN72 prevented the loss of presynaptic termini and the reduction of synaptic density.
Figure 8. MN72 bilaterally maintains CA1 synaptic density after CHI.
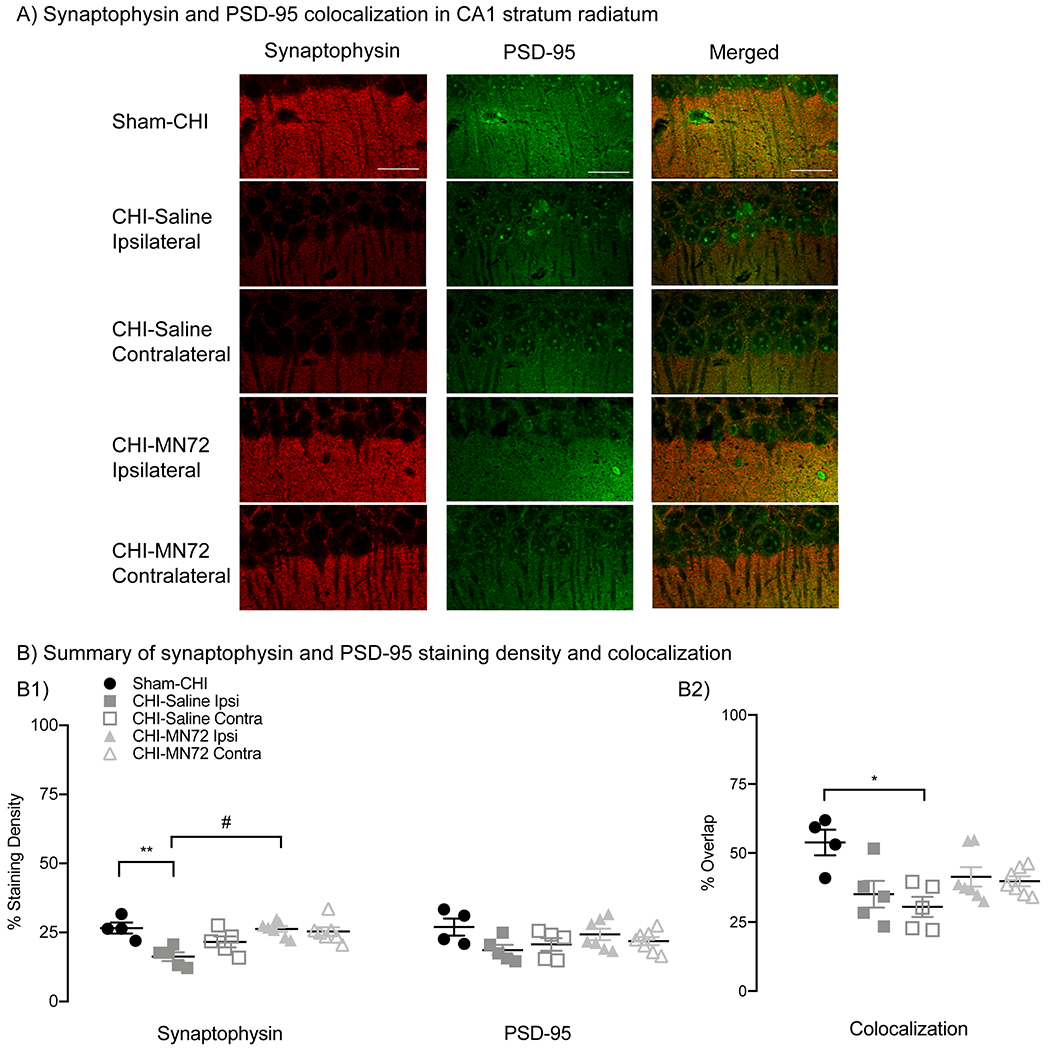
A, CA1 stratum radiatum stained for synaptophysin (red) and PSD-95 (green) from Sham-CHI, CHI-Saline and CHI-MN72 mice. B, Synaptophysin and PSD-95 density and colocalization. B1, Synaptophysin immunoreactivity in ipsilateral CA1 of CHI-Saline mice is lower than Sham-CHI and CHI-MN72 mice. PSD-95 immunoreactivity does not differ among the groups. B2, Synaptophysin and PSD-95 colocalization in CHI-Saline mice is decreased bilaterally compared to Sham-CHI mice; colocalization in CHI-MN72 and Sham-CHI mice does not differ. Scale bar, 50 μm, # represents significant difference with CHI-MN72, *p < 0.05, **p < 0.01, significant difference with Sham-CHI.
Discussion
TBI drug development has largely focused on acute neuroprotective interventions that prevent or reduce the early consequences of TBI (Atkins, 2011). A limitation of this approach is few drugs have clinically useful therapeutic time windows. Drug efficacy rapidly diminishes due to the speed and evolving complexity of TBI pathophysiology, therefore we need a better understanding of what aspects of TBI can be improved by delayed drug administration (Mohamadpour et al., 2019). We took advantage of the long therapeutic time window of the drug combination MINO plus NAC to identify elements of TBI pathophysiology that could be targeted with drugs first dosed 72 hours after injury. CHI impairs performance on Barnes maze, which can be acquired with only one functional hippocampus (Klur et al., 2009; Shinohara et al., 2012). A first dose of MINO plus NAC at 24 hours PI did not prevent damage to the ipsilateral hippocampus (Sangobowale et al., 2018). MN72 treatment improved acquisition and retention of Barnes maze (Figure 1). Therefore, we examined both the ipsilateral and contralateral hippocampi of CHI-injured mice treated with saline or MN72 to determine whether MN72 targeted the contralateral hippocampus.
Unilateral CHI produced extensive bilateral damage both proximal and distal to the impact site with greater neuronal loss in the ipsilateral than in the contralateral hippocampus (Figure 2). Unilateral hippocampal damage is not sufficient to induce spatial memory impairment; rather bilateral damage is needed to produce deficits (source #32 thesis). Thus, it is likely a combination of neuroprotective effects in both hemispheres that underlies the functional improvements produced by MN72 treatment. Despite CA1 being more proximal to the impact site, CA3 neurons were more vulnerable to mechanical injury due to greater tensile strains that extend over a larger area than in CA1 (Casella et al., 2014). CA1 neurons, in contrast, sustained dendritic damage without cell death. Consistent with these observations, CHI bilaterally reduced CA3 neuronal density, while neuronal loss was observed only in ipsilateral CA1 (Figure 2). CA3 cell loss deafferents CA1, leading to CA1 dendritic arbor damage (Casella et al., 2014). Loss of CA3 afferents may contribute to disrupted dendritic architecture and reduced spine density in CA1. Selective neuronal loss demonstrates that hippocampal subregions support specific memory functions. CA3 lesions suggest that CA3 maintains short-term spatial representations, which are critical to spatial memory formation and recall (Gilbert and Brushfield, 2009; Le Duigou et al., 2014; Cherubini and Miles, 2015). Selective CA1 neuronal loss correlates with spatial learning deficits after injury (Auer et al., 1989; Milani et al., 1998). Injury to both CA3 and CA1 likely contributes to the learning and memory deficits produced by CHI (Figure 1).
Two complementary analyses characterized neuronal morphology and dendritic architecture on surviving neurons (Table 3-1). Bilateral damage to dendrites, spines and synapses was more extensive than neuronal loss, suggesting neuronal loss underestimates hippocampal injury. Surviving ipsilateral neurons were smaller and sustained more dendritic damage than corresponding contralateral neurons (Figures 3–5). In contralateral CA1, dendritic degeneration and synapse loss occurred despite minimal cell death (Figures 5, 8). CHI bilaterally lowered CA1 spine and synaptic density (Figures 7,8; Table 7-1). Reduced synaptophysin expression suggested additional selective loss of ipsilateral presynaptic termini. Thus, assessment of drug efficacy by neuroprotection alone is insufficient due to widespread damage to surviving neurons. The broader characterization of brain damage following CHI in this study uncovered elements of TBI pathophysiology that can be targeted by drugs first given days after injury.
MN72 prevented neuronal loss, protected neuronal morphology and increased spine and synapse density (Figures 2–8). MN72 bilaterally maintained CA1 dendritic arborization and complexity, suggesting that CA1 dendritic architecture can be treated with delayed drug administration. In contrast, in CA3 MN72 therapeutic effects were limited to the contralateral hippocampus. Ipsilateral CA3 may have been more severely damaged, or the kinetics of pathophysiological events in CA3 differ or occur more rapidly than in CA1. Recent experiments suggest that MN12 bilaterally maintains CA3 neuronal density (Whitney and Bergold, unpublished result). The differing effects of MN12 and MN72 dosing suggests more rapid pathological processes in ipsilateral than contralateral CA3.
Loss of the dendritic cytoskeletal protein MAP2 likely contributed to dendritic arbor damage. MAP2 degradation occurs rapidly following moderate TBI, and contributes to behavioral deficits (Taft et al., 1992). Previous studies demonstrated MAP2 loss in ipsilateral CA3 persisted for 1-month post-CHI (Grin’kina et al., 2016; Sangobowale et al., 2018). The additional analyses of hippocampal MAP2 expression in this study revealed regional differences in MAP2 loss and recovery. MAP2 loss occurred more rapidly in the ipsilateral than the contralateral hippocampus (Figure 6). In CA3, MAP2 loss was more rapid in basal dendrites, while in CA1 bilateral MAP2 loss occurred in both apical and basal dendrites within 3 days post-CHI (Figures 6). Similarly, MN72 therapeutic effects on MAP2 recovery differed by region. MN72 prevented MAP2 loss in contralateral CA3 basal dendrites (Figure 6B2), and accelerated or restored MAP2 levels in contralateral CA3 apical (Figure 6C2), CA1 basal (Figure 6D2), and CA1 apical dendrites (Figure 6E2). Decreased MAP2 degradation may result in MAP2 recovery since the injured neuron can reorganize and repair cytoskeletal structure (source #197 thesis). These results demonstrate distinct regional differences in the therapeutic window of MN72 for MAP2 expression. A recent study showed that L-cysteine prevented MAP2 loss in a weight drop TBI model, therefore NAC may underlie MN72 therapeutic effects on MAP2 expression (Ouyang et al., 2019).
In CA1 apical dendrites, Schaffer collaterals synapse on proximal branches, while perforant path axons make synapses on distal branches (Spruston, 2008). CHI bilaterally reduces dendritic complexity and spine density in both proximal and distal branches of apical dendrites (Figure 4, 5, 7). This likely disrupts dendritic integration of Schaffer collateral and perforant path inputs. MN72 bilaterally increased mushroom spine density on proximal branches. On distal dendrites, mushroom spine density increased only in the contralateral hippocampus. Mushroom spines are more stable than other spine types (Rodriguez et al., 2008). During memory formation, mushroom spine head size increases due to the transport of intracellular glutamate receptors that are essential for synaptic plasticity (Sebastian et al., 2013; Avila et al., 2017). Based on these properties, mushroom spines are hypothesized to be a physical substrate of long-term memories (Bourne and Harris, 2007). Restoration of spatial navigation and memory may arise, in part, by preserving dendritic geometry and mushroom spine density in the contralateral hippocampus. These data show that in addition to the structural integrity of the dendritic arbor, spine and synapse density can be successfully targeted with drugs first dosed days after injury.
The data in this study suggest that effective therapeutic interventions will target multiple injury mechanisms to reduce progressive secondary injury. Inflammation, mitochondrial damage and oxidative stress are secondary injuries that contribute to TBI pathogenesis and neuronal death (Blennow et al., 2012). Targeting multiple mechanisms likely results in a longer therapeutic window then targeting a single acute neuroprotective mechanism (Margulies et al., 2009). MINO plus NAC likely has multiple drug targets that underlie its many therapeutic actions. MINO plus NAC modulates inflammation (Chen et al., 2008; Garrido-Mesa et al., 2013; Haber et al., 2013; Eakin et al., 2014). MINO plus NAC may also reduce mitochondrial damage and oxidative stress. NAC restored mitochondrial dysfunction and brain glutathione levels after experimental TBI (Xiong et al., 1999). Neuroprotection on mitochondrial function may be related to the ability of NAC to directly scavenge free radicals and maintain mitochondrial calcium homeostasis (Eakin et al., 2014).
The CHI model used in this study reproduces many features of clinical TBI, however no TBI models recapitulate the complexity of human brain injury (Xiong et al., 2013). A limitation of most TBI models is the use of anesthesia during the injury procedure. Isoflurane, the anesthetic drug used in this study, may be neuroprotective, which could influence injury severity, pathology and recovery (Statler et al., 2006). In addition, this study only used male mice and therefore does not address sex as a biological variable. Sex differences in recovery outcomes and response to pharmacological therapies after injury are demonstrated both clinically and in TBI models (Wright et al., 2014; Failla and Wagner, 2015). This study assesses that effects of the drug combination, not the individual drugs. MINO or NAC were previously shown to have little potency when dosed as individual drugs at 24 hours post-injury (Sangobowale, et al., 2018). This study did not examine microglia even though that they participate in dendritic pruning and loss of spines and synapses (Hong S, Dissing-Olesen L, Stevens B. New insights on the role of microglia in synaptic pruning in health and disease. Curr Opin Neurobiol. 2016 36:128-34). Treatment with minocycline plus N-acetylcysteine at one hour post injury altered modulated microglial activation (Haber, 2018). The differential effects of MN72 on ipsilateral and contralateral dendrites, spines and synapses provides the impetus for future studies examining whether MN72 have differing effects on microglia in each hemisphere. Ipsilateral injury may result in contralateral synaptic and dendritic degeneration. This study, however, did not examine commissural pathways connecting the two hippocampi. Axons crossing the midline including corpus callosum and hippocampal commissures are particularly vulnerable in clinical TBI as well as experimental TBI models Victoria E. Johnson, William Stewart, Douglas H. Smith, Axonal pathology in traumatic brain injury, 2013 Exp. Neurol., 246: 35-43. The integrity of the hippocampal commissures has not been examined in the CHI model, but CHI produced substantial white matter damage in corpus callosum that was not repaired by MN24 treatment (Sangobowale, 2018). Thus, the therapeutic effects of MN72 on the contralateral hippocampus does not likely result from retention or repair of commissural axons. Work is ongoing in our laboratory focusing on these issues in TBI pathophysiology, outcomes and therapeutic efficacy.
A major finding of this study is that MN72 has a different therapeutic time window in discrete hippocampal regions. Both drugs readily enter the brain making it unlikely that pharmacokinetics is responsible for regional therapeutic effects (Diaz-Arrastia et al., 2014). It is more likely that the complexity of TBI pathophysiology underlies the regional effects of MN72. Discrete brain regions may express the same TBI pathophysiology with different kinetics. Alternatively, discrete brain regions may vary in TBI pathophysiology. Either of these possibilities help explain why the therapeutic time window differs among different brain regions. Most preclinical studies have focused on drug therapeutic action proximal to the injury site, which resulted in the development of drugs with short therapeutic windows (Mohamadpour et al., 2019). Increased understanding of TBI pathophysiology suggests that a therapeutic time window is likely not limited to the first few hours after injury. TBI progresses over time, therefore delayed treatment may benefit TBI patients who cannot be treated immediately after injury.
A key unanswered question is what constitutes a clinically relevant therapeutic window for TBI intervention. Enrollment data for recent Phase III clinical trials show that TBI patients at designated trauma centers can be treated within 4-7 hours after a moderate to severe TBI (Skolnick et al., 2014). The 4-7 hour therapeutic window is optimal for patients who are injured enough to seek immediate medical attention. Less specialized hospitals will take longer to initiate treatment, and patients with mild TBI often wait longer to seek treatment after injury (Alexander, 1995; Kushner, 1998). To treat the largest number of patients, therapeutic interventions will need to retain high efficacy with delayed dosing. The finding that distal injury can be targeted with drugs first given days after injury provides a new framework in developing and evaluating drugs with sufficiently favorable therapeutic windows to treat TBI. The findings of this study provide further impetus to study the protective and restorative mechanisms of MINO plus NAC.
Acknowledgements:
The authors thank Drs. Julie Parato, Sheryl Smith and Mr. Matthew Evrard, for their assistance with the Golgi-Cox studies. This work was supported by NIH grant NS108190-01 (to P.J.B.)
Abbreviations:
- CHI
closed head injury
- Contra
contralateral
- Ipsi
ipsilateral
- MAP2
microtubule associated protein 2
- MINO
Minocycline
- MN12
MINO plus NAC first dosed 12 hours after injury
- MN72
MINO plus NAC first dosed 72 hours after injury
- NAC
N-acetylcysteine
- TBI
Traumatic brain injury
Footnotes
Declarations of interest: none
References
- Abdel Baki SG, Schwab B, Haber M, Fenton AA, Bergold PJ (2010) Minocycline synergizes with N-acetylcysteine and improves cognition and memory following traumatic brain injury in rats. PLoS One 5:e12490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Afroz S, Parato J, Shen H, Smith SS (2016) Synaptic pruning in the female hippocampus is triggered at puberty by extrasynaptic GABAA receptors on dendritic spines. Elife 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Alexander MP (1995) Mild traumatic brain injury: pathophysiology, natural history, and clinical management. Neurology 45:1253–1260. [DOI] [PubMed] [Google Scholar]
- Arellano JI, Benavides-Piccione R, Defelipe J, Yuste R (2007) Ultrastructure of dendritic spines: correlation between synaptic and spine morphologies. Front Neurosci 1:131–143. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Atkins CM (2011) Decoding hippocampal signaling deficits after traumatic brain injury. Transl Stroke Res 2:546–555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Auer RN, Jensen ML, Whishaw IQ (1989) Neurobehavioral deficit due to ischemic brain damage limited to half of the CA1 sector of the hippocampus. J Neurosci 9:1641–1647. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Avila JA, Alliger AA, Carvajal B, Zanca RM, Serrano PA, Luine VN (2017) Estradiol rapidly increases GluA2-mushroom spines and decreases GluA2-filopodia spines in hippocampus CA1. Hippocampus 27:1224–1229. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bigler ED (2013) Traumatic brain injury, neuroimaging, and neurodegeneration. Front Hum Neurosci 7:395. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Blennow K, Hardy J, Zetterberg H (2012) The neuropathology and neurobiology of traumatic brain injury. Neuron 76:886–899. [DOI] [PubMed] [Google Scholar]
- Bloss EB, Janssen WG, Ohm DT, Yuk FJ, Wadsworth S, Saardi KM, McEwen BS, Morrison JH (2011) Evidence for reduced experience-dependent dendritic spine plasticity in the aging prefrontal cortex. J Neurosci 31:7831–7839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bourne J, Harris KM (2007) Do thin spines learn to be mushroom spines that remember? Curr Opin Neurobiol 17:381–386. [DOI] [PubMed] [Google Scholar]
- Bye N, Habgood MD, Callaway JK, Malakooti N, Potter A, Kossmann T, Morganti-Kossmann MC (2007) Transient neuroprotection by minocycline following traumatic brain injury is associated with attenuated microglial activation but no changes in cell apoptosis or neutrophil infiltration. Exp Neurol 204:220–233. [DOI] [PubMed] [Google Scholar]
- Campbell JN, Register D, Churn SB (2012) Traumatic brain injury causes an FK506-sensitive loss and an overgrowth of dendritic spines in rat forebrain. J Neurotrauma 29:201–217. [DOI] [PubMed] [Google Scholar]
- Casella EM, Thomas TC, Vanino DL, Fellows-Mayle W, Lifshitz J, Card JP, Adelson PD (2014) Traumatic brain injury alters long-term hippocampal neuron morphology in juvenile, but not immature, rats. Childs Nerv Syst 30:1333–1342. [DOI] [PubMed] [Google Scholar]
- Chakroborty S, Hill ES, Christian DT, Helfrich R, Riley S, Schneider C, Kapecki N, Mustaly-Kalimi S, Seiler FA, Peterson DA, West AR, Vertel BM, Frost WN, Stutzmann GE (2019) Reduced presynaptic vesicle stores mediate cellular and network plasticity defects in an early-stage mouse model of Alzheimer’s disease. Mol Neurodegener 14:7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen G, Shi J, Hu Z, Hang C (2008) Inhibitory effect on cerebral inflammatory response following traumatic brain injury in rats: a potential neuroprotective mechanism of N-acetylcysteine. Mediators Inflamm 2008:716458. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cherubini E, Miles R (2015) The CA3 region of the hippocampus: how is it? What is it for? How does it do it? Front Cell Neurosci 9:19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Demakis GJ, Rimland CA (2010) Untreated mild traumatic brain injury in a young adult population. Arch Clin Neuropsychol 25:191–196. [DOI] [PubMed] [Google Scholar]
- Diaz-Arrastia R, Kochanek PM, Bergold P, Kenney K, Marx CE, Grimes CJ, Loh LT, Adam LT, Oskvig D, Curley KC, Salzer W (2014) Pharmacotherapy of traumatic brain injury: state of the science and the road forward: report of the Department of Defense Neurotrauma Pharmacology Workgroup. J Neurotrauma 31:135–158. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Eakin K, Baratz-Goldstein R, Pick CG, Zindel O, Balaban CD, Hoffer ME, Lockwood M, Miller J, Hoffer BJ (2014) Efficacy of N-acetyl cysteine in traumatic brain injury. PLoS One 9:e90617. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Failla MD, Wagner AK (2015) Models of Posttraumatic Brain Injury Neurorehabilitation. In: Brain Neurotrauma: Molecular, Neuropsychological, and Rehabilitation Aspects (Kobeissy FH, ed). Boca Raton (FL). [Google Scholar]
- Faul M, Coronado V (2015) Epidemiology of traumatic brain injury. Handb Clin Neurol 127:3–13. [DOI] [PubMed] [Google Scholar]
- Gao X, Chen J (2011) Mild traumatic brain injury results in extensive neuronal degeneration in the cerebral cortex. J Neuropathol Exp Neurol 70:183–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao X, Deng P, Xu ZC, Chen J (2011) Moderate traumatic brain injury causes acute dendritic and synaptic degeneration in the hippocampal dentate gyrus. PLoS One 6:e24566. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Garrido-Mesa N, Zarzuelo A, Galvez J (2013) Minocycline: far beyond an antibiotic. Br J Pharmacol 169:337–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gilbert PE, Brushfield AM (2009) The role of the CA3 hippocampal subregion in spatial memory: a process oriented behavioral assessment. Prog Neuropsychopharmacol Biol Psychiatry 33:774–781. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grin’kina NM, Li Y, Haber M, Sangobowale M, Nikulina E, Le’Pre C, El Sehamy AM, Dugue R, Ho JS, Bergold PJ (2016) Righting Reflex Predicts Long-Term Histological and Behavioral Outcomes in a Closed Head Model of Traumatic Brain Injury. PLoS One 11:e0161053. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Haber M, Abdel Baki SG, Grin’kina NM, Irizarry R, Ershova A, Orsi S, Grill RJ, Dash P, Bergold PJ (2013) Minocycline plus N-acetylcysteine synergize to modulate inflammation and prevent cognitive and memory deficits in a rat model of mild traumatic brain injury. Exp Neurol 249:169–177. [DOI] [PubMed] [Google Scholar]
- Haber M, James J, Kim J, Sangobowale M, Irizarry R, Ho J, Nikulina E, Grin’kina NM, Ramadani A, Hartman I, Bergold PJ (2018) Minocycline plus N-acteylcysteine induces remyelination, synergistically protects oligodendrocytes, and modifies neuroinflammation in a rat model of mild traumatic brain injury. J Cereb Blood Flow Metab. 38:1312–1326. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hall ED, Sullivan PG, Gibson TR, Pavel KM, Thompson BM, Scheff SW (2005) Spatial and temporal characteristics of neurodegeneration after controlled cortical impact in mice: more than a focal brain injury. J Neurotrauma 22:252–265. [DOI] [PubMed] [Google Scholar]
- Hicdonmez T, Kanter M, Tiryaki M, Parsak T, Cobanoglu S (2006) Neuroprotective effects of N-acetylcysteine on experimental closed head trauma in rats. Neurochem Res 31:473–481. [DOI] [PubMed] [Google Scholar]
- Homsi S, Piaggio T, Croci N, Noble F, Plotkine M, Marchand-Leroux C, Jafarian-Tehrani M (2010) Blockade of acute microglial activation by minocycline promotes neuroprotection and reduces locomotor hyperactivity after closed head injury in mice: a twelve-week follow-up study. J Neurotrauma 27:911–921. [DOI] [PubMed] [Google Scholar]
- Ishizuka N, Cowan WM, Amaral DG (1995) A quantitative analysis of the dendritic organization of pyramidal cells in the rat hippocampus. J Comp Neurol 362:17–45. [DOI] [PubMed] [Google Scholar]
- Johnson VE, Meaney DF, Cullen DK, Smith DH (2015) Animal models of traumatic brain injury. Handb Clin Neurol 127:115–128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Klur S, Muller C, Pereira de Vasconcelos A, Ballard T, Lopez J, Galani R, Certa U, Cassel JC (2009) Hippocampal-dependent spatial memory functions might be lateralized in rats: An approach combining gene expression profiling and reversible inactivation. Hippocampus 19:800–816. [DOI] [PubMed] [Google Scholar]
- Kushner D (1998) Mild traumatic brain injury: toward understanding manifestations and treatment. Arch Intern Med 158:1617–1624. [DOI] [PubMed] [Google Scholar]
- Le Duigou C, Simonnet J, Telenczuk MT, Fricker D, Miles R (2014) Recurrent synapses and circuits in the CA3 region of the hippocampus: an associative network. Front Cell Neurosci 7:262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Leibson CL, Brown AW, Ransom JE, Diehl NN, Perkins PK, Mandrekar J, Malec JF (2011) Incidence of traumatic brain injury across the full disease spectrum: a population-based medical record review study. Epidemiology 22:836–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Margulies S, Hicks R, Combination Therapies for Traumatic Brain Injury Workshop L (2009) Combination therapies for traumatic brain injury: prospective considerations. J Neurotrauma 26:925–939. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Milani H, Uemura UU, Oliveira RM, Lepri ER, Xavier GF (1998) Loss of CA1 cells following global ischaemia correlates with spatial deficits in the circular platform task. J Neurosci Methods 80:19–27. [DOI] [PubMed] [Google Scholar]
- Mohamadpour M, Whitney K, Bergold PJ (2019) The Importance of Therapeutic Time Window in the Treatment of Traumatic Brain Injury. Front Neurosci 13:07. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Osier ND, Dixon CE (2016) The Controlled Cortical Impact Model: Applications, Considerations for Researchers, and Future Directions. Front Neurol 7:134. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ouyang Z, Xu G, Jiang Y (2019) l-Cysteine augments microtubule-associated protein 2 levels and enhances antioxidant activity in rats following traumatic brain injury. 3 Biotech 9:280. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Poe GR, Teed RG, Insel N, White R, McNaughton BL, Barnes CA (2000) Partial hippocampal inactivation: effects on spatial memory performance in aged and young rats. Behav Neurosci 114:940–949. [DOI] [PubMed] [Google Scholar]
- Posmantur RM, Kampfl A, Taft WC, Bhattacharjee M, Dixon CE, Bao J, Hayes RL (1996) Diminished microtubule-associated protein 2 (MAP2) immunoreactivity following cortical impact brain injury. J Neurotrauma 13:125–137. [DOI] [PubMed] [Google Scholar]
- Risher WC, Ustunkaya T, Singh Alvarado J, Eroglu C (2014) Rapid Golgi analysis method for efficient and unbiased classification of dendritic spines. PLoS One 9:e107591. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez A, Ehlenberger DB, Dickstein DL, Hof PR, Wearne SL (2008) Automated three-dimensional detection and shape classification of dendritic spines from fluorescence microscopy images. PLoS One 3:e1997. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanchez Mejia RO, Ona VO, Li M, Friedlander RM (2001) Minocycline reduces traumatic brain injury-mediated caspase-1 activation, tissue damage, and neurological dysfunction. Neurosurgery 48:1393–1399; discussion 1399–1401. [DOI] [PubMed] [Google Scholar]
- Sangobowale M, Grin’kina NM, Whitney K, Nikulina E, St Laurent-Arriot K, Ho JS, Bazyan N, Bergold P (2018) Minocycline plus N-acetylcysteine reduce behavioral deficits and improve histology with a clinically useful time window. J Neurotrauma [DOI] [PubMed] [Google Scholar]
- Scheff SW, Price DA, Hicks RR, Baldwin SA, Robinson S, Brackney C (2005) Synaptogenesis in the hippocampal CA1 field following traumatic brain injury. J Neurotrauma 22:719–732. [DOI] [PubMed] [Google Scholar]
- Scott EK, Luo L (2001) How do dendrites take their shape? Nat Neurosci 4:359–365. [DOI] [PubMed] [Google Scholar]
- Sebastian V, Estil JB, Chen D, Schrott LM, Serrano PA (2013) Acute physiological stress promotes clustering of synaptic markers and alters spine morphology in the hippocampus. PLoS One 8:e79077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinohara Y, Hosoya A, Yamasaki N, Ahmed H, Hattori S, Eguchi M, Yamaguchi S, Miyakawa T, Hirase H, Shigemoto R (2012) Right-hemispheric dominance of spatial memory in split-brain mice. Hippocampus 22:117–121. [DOI] [PubMed] [Google Scholar]
- Skolnick BE, Maas AI, Narayan RK, van der Hoop RG, MacAllister T, Ward JD, Nelson NR, Stocchetti N, Investigators ST (2014) A clinical trial of progesterone for severe traumatic brain injury. N Engl J Med 371:2467–2476. [DOI] [PubMed] [Google Scholar]
- Smith DH, Lowenstein DH, Gennarelli TA, McIntosh TK (1994) Persistent memory dysfunction is associated with bilateral hippocampal damage following experimental brain injury. Neurosci Lett 168:151–154. [DOI] [PubMed] [Google Scholar]
- Smith DH, Chen XH, Pierce JE, Wolf JA, Trojanowski JQ, Graham DI, McIntosh TK (1997) Progressive atrophy and neuron death for one year following brain trauma in the rat. J Neurotrauma 14:715–727. [DOI] [PubMed] [Google Scholar]
- Spruston N (2008) Pyramidal neurons: dendritic structure and synaptic integration. Nat Rev Neurosci 9:206–221. [DOI] [PubMed] [Google Scholar]
- Staff NP, Jung HY, Thiagarajan T, Yao M, Spruston N (2000) Resting and active properties of pyramidal neurons in subiculum and CA1 of rat hippocampus. J Neurophysiol 84:2398–2408. [DOI] [PubMed] [Google Scholar]
- Statler KD, Alexander H, Vagni V, Dixon CE, Clark RS, Jenkins L, Kochanek PM (2006) Comparison of seven anesthetic agents on outcome after experimental traumatic brain injury in adult, male rats. J Neurotrauma 23:97–108. [DOI] [PubMed] [Google Scholar]
- Stocchetti N, Zanier ER (2016) Chronic impact of traumatic brain injury on outcome and quality of life: a narrative review. Crit Care 20:148. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stuart GJ, Spruston N (2015) Dendritic integration: 60 years of progress. Nat Neurosci 18:1713–1721. [DOI] [PubMed] [Google Scholar]
- Sun HS, Feng ZP, Miki T, Seino S, French RJ (2006) Enhanced neuronal damage after ischemic insults in mice lacking Kir6.2-containing ATP-sensitive K+ channels. J Neurophysiol 95:2590–2601. [DOI] [PubMed] [Google Scholar]
- Sunyer B, Patil S, Höger H, Lubec G (2007) Barnes maze, a useful task to assess spatial reference memory in the mice.
- Taft WC, Yang K, Dixon CE, Hayes RL (1992) Microtubule-associated protein 2 levels decrease in hippocampus following traumatic brain injury. J Neurotrauma 9:281–290. [DOI] [PubMed] [Google Scholar]
- Tanielian T, & Jaycox LH (2008) Invisible wounds of war: Psychological and cognitive injuries, their consequences, and services to assist recovery. Santa Monica, CA RAND Corporation. [Google Scholar]
- Tran LD, Lifshitz J, Witgen BM, Schwarzbach E, Cohen AS, Grady MS (2006) Response of the contralateral hippocampus to lateral fluid percussion brain injury. J Neurotrauma 23:1330–1342. [DOI] [PubMed] [Google Scholar]
- Wang J, Liu S, Fu Y, Wang JH, Lu Y (2003) Cdk5 activation induces hippocampal CA1 cell death by directly phosphorylating NMDA receptors. Nat Neurosci 6:1039–1047. [DOI] [PubMed] [Google Scholar]
- White ER, Pinar C, Bostrom CA, Meconi A, Christie BR (2017) Mild Traumatic Brain Injury Produces Long-Lasting Deficits in Synaptic Plasticity in the Female Juvenile Hippocampus. J Neurotrauma 34:1111–1123. [DOI] [PubMed] [Google Scholar]
- Winston CN, Chellappa D, Wilkins T, Barton DJ, Washington PM, Loane DJ, Zapple DN, Burns MP (2013) Controlled cortical impact results in an extensive loss of dendritic spines that is not mediated by injury-induced amyloid-beta accumulation. J Neurotrauma 30:1966–1972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winston CN, Noel A, Neustadtl A, Parsadanian M, Barton DJ, Chellappa D, Wilkins TE, Alikhani AD, Zapple DN, Villapol S, Planel E, Burns MP (2016) Dendritic Spine Loss and Chronic White Matter Inflammation in a Mouse Model of Highly Repetitive Head Trauma. Am J Pathol 186:552–567. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wright DW, Espinoza TR, Merck LH, Ratcliff JJ, Backster A, Stein DG (2014) Gender differences in neurological emergencies part II: a consensus summary and research agenda on traumatic brain injury. Acad Emerg Med 21:1414–1420. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong Y, Peterson PL, Lee CP (1999) Effect of N-acetylcysteine on mitochondrial function following traumatic brain injury in rats. J Neurotrauma 16:1067–1082. [DOI] [PubMed] [Google Scholar]
- Xiong Y, Mahmood A, Chopp M (2013) Animal models of traumatic brain injury. Nat Rev Neurosci 14:128–142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yi JH, Hazell AS (2005) N-acetylcysteine attenuates early induction of heme oxygenase-1 following traumatic brain injury. Brain Res 1033:13–19. [DOI] [PubMed] [Google Scholar]