

Abstract
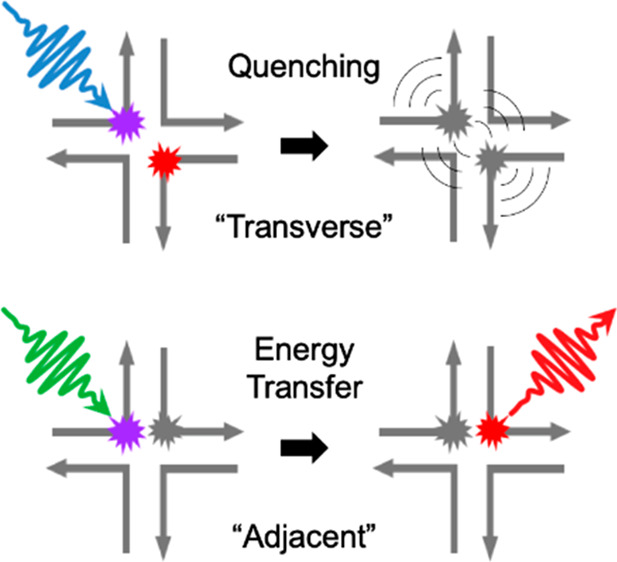
Molecular excitons, which propagate spatially via electronic energy transfer, are central to numerous applications including light harvesting, organic optoelectronics, and nanoscale computing; they may also benefit applications such as photothermal therapy and photoacoustic imaging through the local generation of heat via rapid excited-state quenching. Here we show how to tune between energy transfer and quenching for heterodimers of the same pair of cyanine dyes by altering their spatial configuration on a DNA template. We assemble “transverse” and “adjacent” heterodimers of Cy5 and Cy5.5 using DNA Holliday junctions. We find that the transverse heterodimers exhibit optical properties consistent with excitonically interacting dyes and fluorescence quenching, while the adjacent heterodimers exhibit optical properties consistent with nonexcitonically interacting dyes and disproportionately large Cy5.5 emission, suggestive of energy transfer between dyes. We use transient absorption spectroscopy to show that quenching in the transverse heterodimer occurs via rapid nonradiative decay to the ground state (∼31 ps) and that in the adjacent heterodimer rapid energy transfer from Cy5 to Cy5.5 (∼420 fs) is followed by Cy5.5 excited-state relaxation (∼700 ps). Accessing such drastically different photophysics, which may be tuned on demand for different target applications, highlights the utility of DNA as a template for dye aggregation.
Molecular (dye) aggregates are of interest in a number of fields including natural and artificial light harvesting,1−5 organic optoelectronics,6−8 and nanoscale computing.9−16 An essential element in each of these fields is the process of energy transfer. In natural light harvesting, for example, electronic energy is usually transported with high quantum efficiency from the antennae to the reaction centers, where charge separation occurs to drive the energy-storing chemical reaction. In organic optoelectronics, such as solar cells, efficient energy transfer to a donor:acceptor interface is needed for overall high light-to-electrical conversion efficiencies. In the case of nanoscale computing, efficient energy transfer is needed for efficient optical coupling to and from readouts in nanoscale computing materials.
An essential element of long-range, spatially directed energy transfer is that it is downhill, i.e., decreasing, in energy. In order for the process to be downhill in energy, at least two distinct transition energies are needed. One way in which to effect two distinct transition energies is to chemically modify dyes. Bringing two chemically distinct dyes together in close proximity forms a heteroaggregate. An advantage of heteroaggregates is that chemically distinct dyes are generally optically distinct—such selectivity can be leveraged in photophysical studies to access key and sometimes elusive mechanistic insight.
Dye aggregates also frequently undergo an aggregation-induced excited-state quenching process.17−30 In this case, a substantial amount of light (or electronic) energy is converted into heat. While this process would be detrimental to light harvesting, organic optoelectronics, or nanoscale computing, there are other fields that would benefit from highly efficient light-to-heat conversion, such as photothermal therapy (PTT) and photoacoustic imaging (PAI). In PTT, for example, cancer cells are targeted by selectively and locally exciting materials that efficiently convert light energy into heat and elevate the temperature in the vicinity of the cancer cells. Recently, polymer-encapsulated nanoparticles and nanomicelles of derivatives of heptamethine cyanine and boron dipyrromethene dyes have been demonstrated as candidate PTT and PAI agents.31,32
There are many different methods for aggregating dyes. Originally, dyes were aggregated spontaneously in concentrated solution. While this approach enabled numerous fundamental studies of dye aggregates,33−35 a disadvantage of spontaneous aggregation is the lack of control over the number of dyes present in the aggregates and dye packing within the aggregates. An alternative approach is to tether dyes to a conventional polymer, which provides control over the number of dyes that can be aggregated and the distance between them, though control over their relative orientation remains challenging.36 Nature has overcome these challenges by using proteins to template dye aggregates, resulting in sophisticated light harvesting assemblies.4,5,37 Similarly, an emerging approach to template dye aggregates is by using DNA.23−26,38−52 DNA templating is an appealing alternative due to its simpler assembly design rules, i.e., four nucleotides and Watson–Crick base pairing, which results in highly programmable assembly. Additionally, the configuration space of DNA templating is substantial. For example, there are over 1 million different configurations for an oligonucleotide sequence consisting of just 10 nucleotides. Bringing all of these factors together has led to the blossoming field of DNA nanotechnology, wherein arbitrary three-dimensional nanostructures have been designed and implemented.53−58
In this Letter, we show how to tune between two drastically different types of photophysics—unity quenching and unity energy transfer—in aggregates of the same pair of dyes. The pair of dyes at the focus of the study are the chemically distinct dyes Cy5 and Cy5.5 (Supporting Information, section S1). Cy5 and Cy5.5 are chosen for their optimal photophysical characteristics, namely, intense visible light absorption, bright fluorescence emission, and overlapping emission and absorption spectral bands.59,60 The dyes are covalently tethered to DNA oligonucleotides, which are then assembled into DNA Holliday junctions57,58 to form “transverse” and “adjacent” dimer aggregate configurations (Scheme 1). Details of the synthetic procedure for oligonucleotide labeling and assembly of DNA Holliday junctions are discussed in the Supporting Information (section S2).
Scheme 1. Schematic Representations of Cy5 and Cy5.5 Monomer and Heterodimer Constructs Templated Using DNA Holliday Junctions (HJs).
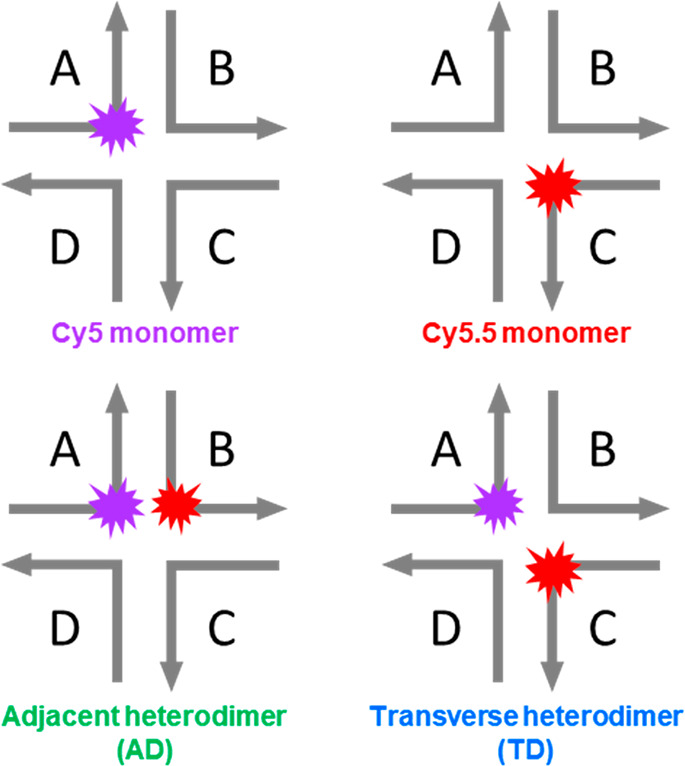
Each right-angle arrow represents a single oligonucleotide sequence consisting of 26 nucleotides, with 13 nucleotides on either side of the central dye. The actual HJ structure is believed to be somewhat planar with an estimated length × width of 10 nm × 10 nm based just on DNA extension. HJs are formed by adding stoichiometric amounts of each labeled and unlabeled A through D oligonucleotides in buffer and allowing them to hybridize in a polymerase chain reaction thermal cycler as described in the Supporting Information.
The steady-state absorption spectra of the monomers and heterodimers in aqueous buffer solution are presented in Figure 1. The absorption spectrum of Cy5 monomer exhibits its most intense absorption feature at ∼650 nm, with two higher-lying bands at ∼610 and 560 nm. Because of its longer conjugation length, the absorption spectrum of Cy5.5 monomer is red-shifted relative to Cy5 monomer, exhibiting its most intense absorption feature at ∼695 nm. As with Cy5 monomer, the Cy5.5 monomer exhibits two higher-lying bands at ∼643 and 587 nm. The position of these higher-lying bands indicates that the electronic transition of both monomers is coupled to a vibrational mode with an energy of ∼1100 cm–1 (∼0.14 eV). In addition, the extinction coefficients of Cy5 and Cy5.5 are similar, ∼243000 and ∼244000 M–1 cm–1, respectively, in the vicinity of their respective absorption peak maxima (Supporting Information, section S5). Such large peak extinction coefficients are consistent with fully allowed electronic transitions (i.e., oscillator strength, f = 1) and maximal transition dipole moment (TDM) amplitudes.61 For example, ab initio quantum chemical calculations indicate the TDM amplitude of the S0 → S1 transition of Cy5 is ∼16 D.62,63
Figure 1.
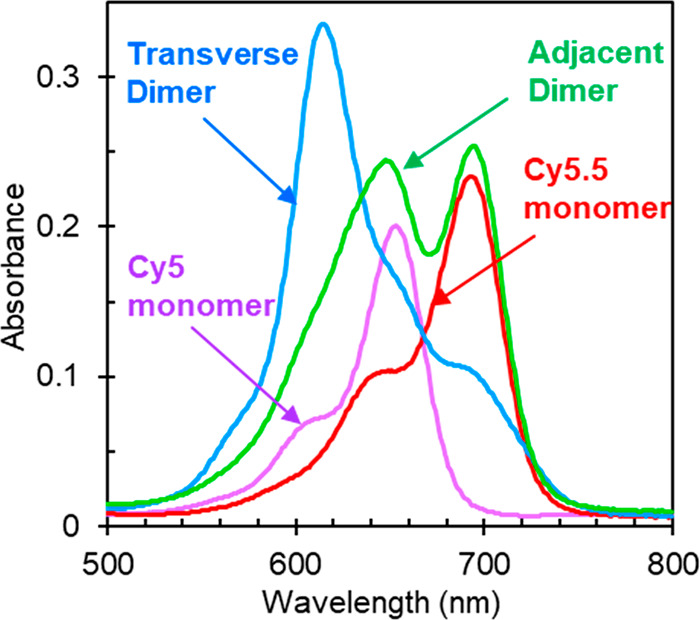
Steady-state absorption spectra of DNA-templated Cy5 and Cy5.5 monomers and heterodimers in 1× TAE buffer with 15 mM MgCl2 added and measured at room temperature. “Adjacent” and “transverse” describe the relative position of the dyes in the Holliday junction as shown in Scheme 1.
Figure 1 also displays the absorption spectrum of the transverse heterodimer solution, which exhibits considerable changes compared to the monomers. Specifically, the transverse heterodimer exhibits an intense, hypsochromically shifted (or blue-shifted) absorption band centered at ∼618 nm and suppression of absorption intensity at longer wavelengths. Additionally, the steady-state circular dichroism (CD) spectrum of the transverse heterodimer solution exhibits prominent positive- and negative-going features at 622 and 716 nm, respectively, whereas the corresponding spectra of the monomer solutions are featureless in the same spectral range (Supporting Information, section S6). These changes in the optical properties of the heterodimer compared with the monomers are consistent with relatively strong coupling and exciton delocalization. They also appear to be largely independent of the permutation of the DNA sequence used to template their aggregation (section S7), which suggests that while the DNA is essential to positioning the dyes precisely and bringing them into close proximity, the dyes themselves drive the aggregate packing. The interpretation that the dyes largely drive the aggregate packing is supported by the observation that the optical properties of Cy5 and Cy5.5 transverse homodimers exhibit similar trends (section S8), namely, an overall hypsochromic shift of the absorption spectrum compared with the respective monomer, indicating that the dyes in the homodimers and in the heterodimer adopt a face-to-face, or H-type, packing arrangement.64 Thus, we observe considerable spectral changes in the case of the transverse heterodimer, consistent with extensive coupling between Cy5 and Cy5.5 dyes, while the overall hypsochromic shift of the absorption spectrum indicates H-type packing between Cy5 and Cy5.5.
To gain additional insights into packing and coupling in the transverse dimer, we modeled its absorption and CD spectra. Specifically, we employed an approach based on Kühn–Renger–May (KRM) theory,65 which we have previously used to derive information about packing and coupling in dimer, trimer, and tetramer aggregates of Cy5,25,40,46 Square-660 (an indolenine-based squaraine),52 and SeTau-670 (an aniline-based squaraine rotaxane).38 Consistent with our expectations of H-aggregation, the modeling indicates that the “oblique” angle, α, between dyes (as the TDM is oriented along the long axis of the dye) is ∼22° (section S9). The α value of 22° for the transverse Cy5-Cy5.5 heterodimer is similar to values of ∼22°–27° reported for H-aggregate dimers of Square-660.52 Additionally, the modeling indicates that the dyes exhibit a center-to-center distance and coupling strength of ∼0.63 nm and ∼95 meV (∼770 cm–1), respectively. These values are intermediate between the values of ∼0.81 nm and ∼59 meV (590 cm–1) and ∼0.48 nm and ∼115 meV (930 cm–1) for Cy5 and Cy5.5 homodimers, respectively (section S9). Such coupling strengths are considerable, comparable to the largest value of ∼135 meV (∼1000 cm–1) reported for tetramer aggregates of Cy5,25 and likely arise from the large TDM of the constituent dyes along with their small center-to-center distance.64 Thus, the KRM modeling confirms that the dyes in the transverse heterodimer exhibit an H-type packing arrangement and are relatively strongly coupled with one another.
In contrast to the transverse heterodimer solution, the absorption spectrum of the adjacent heterodimer solution does not exhibit obvious signatures of exciton delocalization across the dyes (Figure 1). Rather, at first glance, the absorption spectrum of the adjacent heterodimer solution appears to resemble a linear combination of the absorption spectra of the constituent monomers. For example, the absorption spectrum of the adjacent heterodimer solution exhibits its two most intense absorption features in the vicinity of the most intense absorption features of the constituent monomers. If the absorption spectrum of the heterodimer is well-modeled by a linear combination of the constituent monomers, this would indicate negligible coupling between dyes in the dimer. On the other hand, if the absorption spectrum of the heterodimer is not well-modeled in this manner, this would indicate non-negligible coupling between dyes in the dimer and/or heterogeneity in the solution. We thus performed a linear combination analysis and found that the absorption spectrum of the adjacent heterodimer solution cannot be fully modeled as a linear combination of the monomer spectra (section S10). Specifically, while the sum of an equal fraction of Cy5 and Cy5.5 monomer absorption spectra can model most of the adjacent heterodimer solution spectrum, there is a non-negligible residual, constrained to only have positive values, with features peaking at ∼625 and 700 nm. Thus, either the adjacent dimer solution is homogeneous and there is non-negligible (i.e., intermediate or strong) excitonic coupling between dyes or the adjacent heterodimer solution exhibits structural heterogeneity, i.e., is a mixture of structures of excitonically interacting and nonexcitonically interacting dyes. To further investigate the optical properties of the adjacent dimer solution, we performed CD measurements. The CD spectrum of the adjacent dimer solution exhibits negative- and positive-going features at ∼625 and ∼700 nm, respectively (section S6). These results are consistent with the interpretation that the adjacent dimer solution is a heterogeneous mixture of structures of both excitonically interacting dyes and nonexcitonically interacting dyes. For example, if the solution were a homogeneous mixture of one structure of excitonically interacting dyes, we can expect, to first order, that the CD features should appear at the same peak positions corresponding to the absorption maxima of ∼648 and ∼694 nm. In contrast, the locations of the spectral features in the CD spectrum do not appear in these positions; rather, they match the peak locations associated with the residual in the linear combination analysis. Thus, the linear combination analysis and the CD measurements indicate that the adjacent heterodimer solution is heterogeneous and contains one subpopulation of structures consisting of nonexcitonically interacting dyes and another subpopulation of structures where the dyes exhibit non-negligible excitonic interactions.
To gain additional insight into the electronic structure and excited-state dynamics of the monomers and dimer aggregates, we performed steady-state fluorescence emission measurements. Fluorescence emission spectra are displayed in Figure 2. The fluorescence emission spectra of Cy5 and Cy5.5 monomer solutions are as expected; these spectra largely resemble mirror images of their corresponding absorption spectra (section S11). Additionally, we observe that the emission intensity of the series, with each spectrum scaled by dividing the intensity by the absorptance (i.e., 1 – transmittance) at the excitation wavelength at 615 nm, varies as ICy5 > ICy5.5 > IAD > ITD. These results indicate that Cy5 monomer is the brightest emitter, followed by Cy5.5 monomer and the heterodimer solutions.
Figure 2.
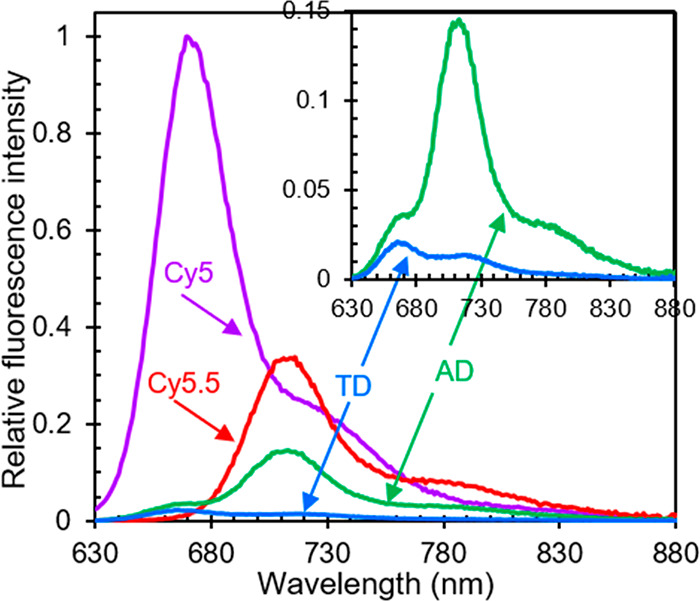
Relative fluorescence emission spectra of monomers and heterodimer solutions. The excitation wavelength for the measurements was 615 nm. The fluorescence emission intensity of each sample was scaled by dividing the intensity by the absorptance (i.e., 1 – transmittance) at the excitation wavelength. Each data set was subsequently scaled by the normalization factor for Cy5 emission. A close-up of the relative emission spectra of transverse and adjacent heterodimers is presented in the inset.
To better understand variations in the relative emission intensity of the monomer and heterodimer solutions, we performed fluorescence quantum yield (FQY) measurements. The FQY values for the Cy5 monomer, Cy5.5 monomer, transverse heterodimer, and adjacent heterodimer solutions were determined to be 0.45, 0.28, 0.010, and 0.11, respectively (Table 1). These results are consistent with the relative fluorescence emission intensities shown in Figure 2 while providing additional quantitative insight. For example, the FQY of the monomers are both large (i.e., both dyes are “bright” or highly emissive) and are generally quite similar, with the FQY of Cy5 monomer being ∼50% larger than that of Cy5.5 monomer. In contrast, the FQY of the transverse heterodimer solutions is suppressed considerably, which is consistent with excited-state quenching, i.e., the presence of a new, significant nonradiative pathway, concomitant with aggregation. On the other hand, the FQY of the adjacent heterodimer solution is intermediate between that of the transverse heterodimer solution and that of the monomer solutions.
Table 1. Fluorescence Quantum Yield of Monomer and Heterodimer Solutionsa.
| solution | fluorescence quantum yield (ΦF) |
|---|---|
| Cy5 monomer | 0.45 ± 0.06 |
| Cy5.5 monomer | 0.28 ± 0.04 |
| transverse heterodimer | 0.010 ± 0.002 |
| adjacent heterodimer | 0.11 ± 0.02 |
Excitation wavelength of 615 nm was used to measure the relative fluorescence quantum yield for all samples. Oxazine 720 and Rhodamine 800 were used as relative standards for all the samples. Uncertainty was determined from the error propagation of three measurements of all samples. For additional details, see Section S3.
The spectral profiles of the transverse and adjacent heterodimer solutions provide additional insight into the origin of the fluorescence emission (inset of Figure 2). Curiously, the fluorescence spectra do not resemble a mirror image of their corresponding absorption spectra. A linear combination analysis of the fluorescence emission spectra of the transverse and adjacent heterodimer solutions indicates that these data can be fairly well modeled by a linear combination of the monomer fluorescence emission spectra (section S12). Thus, the emission from the heterodimer solutions is attributed to emission from Cy5 and Cy5.5 monomers. To model the emission spectrum of transverse heterodimer solution, we needed an ∼3:1 weighting factor of Cy5 and Cy5.5. This is not surprising because one would anticipate 3:1 weighting factor of Cy5 and Cy5.5 to model the emission spectrum of transverse heterodimer solution (Figure 2) where the stoichiometric ratio of Cy5 and Cy5.5 dyes is 1:1. Surprisingly, a disproportionately large weighting of 1:4 of Cy5:Cy5.5 was required to model the adjacent heterodimer emission spectrum. Given that Cy5 and Cy5.5 are also present in this solution in a 1:1 stoichiometric ratio, this is ∼12× higher compared to the expected 3:1 Cy5:Cy5.5 weighting factor. This observation suggests that energy transfer may be taking place from Cy5 to Cy5.5 in the adjacent heterodimer.
To investigate the origin of the significant nonradiative decay observed in the transverse dimer solution, we performed transient visible (VIS) absorption measurements. The results are shown in Figure 3; surface plots are shown in the top row, selected transient spectra (at a time delay of 5 ps) are shown in the middle row, and selected kinetics (in the vicinity of the most intense ground-state bleach [GSB] feature) are shown in the bottom row. The selected transient spectra shown in the middle row indicate that all of the materials, i.e., Cy5 monomer, Cy5.5 monomer, and the transverse heterodimer, exhibit distinct spectral features. Specifically, Cy5 monomer exhibits an intense GSB and stimulated emission (SE) feature peaking at ∼660 nm along with two prominent excited-state absorption (ESA) bands at shorter wavelength peaking at ∼455 and ∼505 nm, while Cy5.5 monomer exhibits an intense GSB/SE feature peaking at ∼700 nm along with a prominent ESA band at shorter wavelength peaking at ∼545 nm. The locations of the most prominent GSB/SE features of the Cy5 and Cy5.5 monomers are consistent with the spectral positions of their lowest- and highest-energy absorption and emission bands, which are distinct for these two dyes (Figures 1 and 2). Additionally, we find that the ESA bands, associated with transitions taking place from the lowest electronic excited state, i.e., S1, are also spectrally distinct for Cy5 and Cy5.5 monomers. In the case of the transverse dimer solution, the most prominent GSB feature is observed peaking at ∼615 nm, which matches well with the most intense feature in the steady-state absorption spectrum, while a single ESA band is observed at shorter wavelength, peaking at ∼470 nm.
Figure 3.

Transient visible absorption of (A) Cy5 monomer, (B) Cy5.5 monomer, and (C) transverse heterodimer solutions pumped at 650, 690, and 615 nm, respectively, corresponding to each solution’s absorption maximum. Surface plots are on the top, selected spectra are (at 5 ps) in the middle, and selected kinetics are plotted at the bottom. In the surface plots, vertical lines are included at the probe wavelength corresponding to the selected kinetics at the bottom. Selected spectra in the middle row show spectrally distinct ground-state bleach, stimulated emission, and excited-state absorption features for all samples. In the selected kinetics plots corresponding to the ground-state bleach maxima, fits from a global target analysis (section S13) overlay the kinetics traces. The global target analysis derives lifetimes of 1.7 ns, 930 ps, and 31 ps for Cy5 monomer, Cy5.5 monomer, and transverse heterodimer, respectively.
The transient kinetics shown in the bottom row of Figure 3 are revealing with respect to the lifetimes of the Cy5 and Cy5.5 monomers and the transverse heterodimer. By performing a global target analysis of the transient absorption data, the Cy5 and Cy5.5 monomer lifetimes are determined to be 1.7 ns and 930 ps, respectively. Corresponding transient kinetics traces associated with the most intense GSB/SE feature in each solution are shown overlaid with the associated fit from the global target analysis in the bottom row of Figure 3. Such long lifetimes for the monomers are consistent with their high quantum yields (Table 1). An analysis of the radiative and nonradiative decay rates (section S14) indicates that Cy5 and Cy5.5 monomers have similar radiative decay rates (i.e., kr), which is consistent with their similar peak extinction coefficients (section S5). Additionally, the analysis indicates that Cy5.5 monomers have a factor of 2 larger nonradiative decay rate (i.e., knr), which we attribute to the addition of aryl groups at both ends of the optically active π-conjugated network (section S1).
In contrast to the monomers, the spectral features associated with the transverse dimer, including GSB and SE features, decay on a much more rapid time scale of ∼31 ps. These results are consistent with prior observations of excited-state quenching—that is, accelerated nonradiative decay directly to the ground state, i.e., internal conversion (see also section S15)—in DNA-templated homoaggregates.23−26 In addition, long-lived, negative-going signals are observed at ∼660 and 700 nm. To explore the origin of these long-lived signals, we performed pump wavelength-dependent transient VIS absorption measurements (section S16). The results show that when the transverse dimer solution is pumped at 650 nm, prominent negative-going features at ∼660 and ∼700 nm are observed, which are consistent with GSB/SE features of Cy5 and Cy5.5 monomer, respectively. Compared with pumping the solution at ∼615 nm, the intensity of these features has increased while the intensity of the negative-going feature at ∼615 nm associated with the relatively strongly coupled transverse dimer structure has decreased. With a pump wavelength of 690 nm, additional insights are gained. Specifically, the feature associated with transverse dimer is further reduced in intensity, the feature associated with GSB/SE of Cy5 monomer is no longer present, and the most prominent negative-going feature is the GSB/SE feature associated with Cy5.5 monomer. These results are consistent with the preferential excitation of Cy5.5 monomer at 690 nm, which is the primary absorber at that wavelength. More broadly, the results indicate that the transverse heterodimer solution is heterogeneous and includes a small subpopulation of monomers, likely as a result of either static or dynamic heterogeneity.25 Here, because of the optical selectivity of the constituent dye monomers, such heterogeneity—albeit not a major contributor for the 615 nm pumping experiment—is immediately apparent when pumping the transverse dimer solution at 650 and 690 nm.
We next turn to transient VIS absorption measurements of the adjacent heterodimer solution. Owing to the appreciable FQY (Table 1) and disproportionately large amount of Cy5.5 emission (Figure 2 and section S12) in the adjacent heterodimer solution, we are especially motivated to uncover dynamics, such as energy transfer from Cy5 to Cy5.5, that might explain these observations. As such, we performed the measurement pumping the solution at ∼650 nm, i.e., in the vicinity of the absorption maximum of Cy5 monomer. Surface plots of such measurements are shown in Figure 4A. Crucially, a positive-going feature at ∼455 nm consistent with one of the ESA bands of Cy5 monomer is observed at early time delays, which decays at longer time delays where the surface plot largely exhibits prominent positive- and negative-going bands at ∼540 and ∼700 nm consistent with Cy5.5 ESA and GSB/SE features, respectively. Thus, in contrast to the case of the transverse heterodimer where ∼31 ps time scale quenching was observed, the observations in the adjacent heterodimer solution are consistent with the rapid transfer of energy from Cy5 to Cy5.5 on a few hundred femtosecond time scale. Further confirmation of the presence of Cy5 features at early time and the decay into Cy5.5 features at longer time is evident in the transient spectra plotted in Figure 4B. Here, selected transient spectra of the adjacent heterodimer solution at the time origin of the measurement (i.e., at “0” ps) are plotted along with transient spectra of the monomer solutions acquired at a time delay of 2 ps. The comparison clearly shows spectral signatures of Cy5 present in the transient spectrum of the adjacent dimer solution at the time origin of the measurement (an alternative way of visualizing these observations is shown in section S17). Namely, features in the vicinity of ∼450, 505, and 660 nm are associated with ESA and GSB/SE bands of Cy5 monomer. Such features are no longer evident in the transient spectrum of the adjacent dimer solution at 2 ps time delay, which largely remains the same for the duration of the measurement and decays on a several hundred picoseconds time scale (section S18). Furthermore, even though the measurement was performed by pumping directly in the vicinity of where Cy5 monomer exhibits its most intense absorption and where the solution itself exhibits considerable absorption, there is no bleaching in this spectral region at 2 ps. Taken together, these results are consistent with the transfer of energy from Cy5 to Cy5.5 in the adjacent dimer on a rapid, few hundred femtoseconds time scale, followed by the decay of Cy5.5 on a several hundred picoseconds time scale.
Figure 4.

Transient visible absorption of the adjacent heterodimer solution pumped at 650 nm. (A) displays the surface plot and (B) displays selected spectra at time delays of 0 and 2 ps along with corresponding reference spectra of Cy5 and Cy5.5 monomer control solutions at a 2 ps time delay. Vertical dashed lines in (B) highlight the positions of distinct spectral features associated with Cy5 and Cy5.5 monomers. Specifically, vertical dashed lines are shown for ESA and GSB/SE features associated with Cy5 at ∼450, 506, and 660 nm (purple) and for ESA and GSB/SE features associated with Cy5.5 at ∼540 and 700 nm (red). At the time origin of the measurement, the adjacent dimer solution exhibits spectral features attributable to both Cy5 and Cy5.5; by ∼2 ps, the spectral features associated with Cy5 are absent, and only spectral features associated with Cy5.5 are observed.
To gain further insight into the dynamics between Cy5 and Cy5.5 in the adjacent heterodimer, we performed transient near-infrared (NIR) absorption measurements. Transient NIR absorption measurements are particularly advantageous because GSB and SE bands are not present in this spectral range, thus providing an unobstructed view of ESA bands and simplifying interpretation of the excited-state electronic structure and dynamics.66−68 Furthermore, nonresonant signals, sometimes called “coherent artifacts”, are suppressed, which facilitates quantitative analyses of particularly rapid dynamics.66Figure 5A displays the transient NIR absorption surface plot for the adjacent dimer solution excited at 650 nm. A strong negative-going signal is observed below ∼950 nm, and strong positive-going features are observed at ∼1130 and 1360 nm. These features are even more obvious in the selected NIR absorption spectra displayed in Figure 5B. To determine the origin of these signals, we also performed transient NIR absorption measurements on Cy5 and Cy5.5 monomer control solutions (section S19). These results indicate that Cy5 and Cy5.5 exhibit prominent and distinct NIR ESA bands peaking at ∼1130 and 1360 nm, respectively. Additionally, Cy5.5 monomer exhibits larger relative negative-going intensity in the spectral region below ∼950 nm, which we assign to SE of Cy5.5 monomer based on its red-shifted fluorescence spectrum (Figure 2). Thus, we assign the features appearing at ∼1130 and 1360 nm in the adjacent dimer solution to Cy5 and Cy5.5 monomer, respectively, and attribute the negative-going signal below ∼950 nm as arising largely from Cy5.5 monomer.
Figure 5.

Transient near-infrared absorption of the adjacent heterodimer solution pumped at 650 nm. (A) displays the surface plot, (B) displays selected spectra at different time delays, and (C) displays selected kinetics traces at 1130 and 1359 nm. Purple and red lines in (A) at ∼1130 and 1360 nm, respectively, are centered at the peak of excited-state absorption bands associated with Cy5 and Cy5.5, respectively. The box outlined by a black, dashed line highlights the ESA band associated with Cy5 present only at early time delays. (B) shows the decay of Cy5 ESA band is associated with a simultaneous increase in the longest-wavelength Cy5.5 ESA band. (C) shows the kinetics of Cy5 and Cy5.5 ESA bands along with the fits from a global target analysis. A considerable portion of the early time kinetics (∼40%) are convolved with the pump pulse (see e.g. section S21), whose duration was measured to be ∼230 fs (see section S3).
As can be seen in both Figures 5A and 5B, the intensity of the NIR ESA band associated with Cy5 decreases over the course of a few hundred femtoseconds while the SE and ESA features associated with Cy5.5 increase in intensity. These results are consistent with the observations in the transient VIS absorption measurement (Figure 4), further confirming that energy is being transferred from Cy5 to Cy5.5 in the adjacent heterodimer. To determine the time scale of the energy transfer, we performed a global target analysis of the transient NIR absorption (section S20). Figure 5C displays the transient NIR kinetics associated with Cy5 and Cy5.5 ESA bands overlaid with the corresponding fits from the global target analysis. The results indicate that the transfer of energy between Cy5 and Cy5.5 in the adjacent heterodimer solution takes place on a rapid ∼420 ± 20 fs time scale and that Förster resonance energy transfer (FRET) likely mediates the process (sections S22–S24). A simple analysis based on kinetic competition using the Cy5 monomer rate of decay (i.e., ∼1/1700 ps–1) indicates such rapid energy transfer would result in an exceptionally large, ∼99.98% transfer efficiency. Such a large efficiency would be beneficial in any application, such as light harvesting or nanoscale computing, that utilizes this pair of dyes to transfer electronic excitation energy in a spatially directed manner.
We thus arrive at the following photophysical picture for the same pair of dyes assembled with the same DNA Holliday junction template but in distinct “adjacent” and “transverse” dimer configurations (Figure 6). In the case of the transverse dimer, rapid ∼30 ps relaxation directly to the ground state takes place, accelerated by an enhanced nonradiative decay process concomitant with aggregation. In the case of the adjacent dimer, the system does not rapidly relax to the ground state. Rather, electronic excitation energy is transferred from Cy5 to Cy5.5, via FRET, on a rapid ∼420 fs time scale, followed by the relaxation of Cy5.5 to the ground state on an ∼700 ps time scale. Additional insights into the exact nature of the dye packing may aid in better understanding the mechanism of the rapid ∼420 fs energy transfer process.
Figure 6.
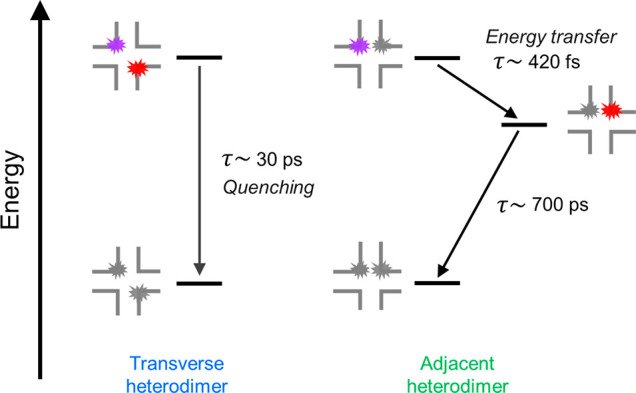
Photophysical diagram for the excited-state dynamics observed in the transverse and adjacent heterodimer structures, including schematic representations of these structures. The time scales for the kinetic steps identified in this work are indicated. We note that the transverse and adjacent heterodimer solutions exhibited signatures of structural heterogeneity (sections S10, S12, S13, S16, and S18). Thus, the diagrams shown here represent the subpopulation of dimers consisting of strongly excitonically (nonexcitonically) interacting dyes in the transverse (adjacent) heterodimer solution that undergo quenching (energy transfer).
In conclusion, we have shown that drastically different photophysics emerge for the same pair of dyes, Cy5 and Cy5.5, when templated by using the same DNA but arranged in different configurations. When the dyes are templated in a “transverse” configuration, the dyes primarily form a dimer that exhibits relatively strong coupling along with rapid and efficient quenching, which may benefit applications such as photothermal therapy. On the other hand, when the dyes are templated in an “adjacent” configuration, the dyes primarily form a dimer that exhibits negligible coupling and rapid ∼420 fs energy transfer from Cy5 to Cy5.5, which may benefit applications dependent on electronic energy transfer such as light harvesting, energy conversion, and nanoscale computing. The observation of such extremely opposite photophysics for the same pair of dyes simply oriented in different ways by using DNA is significant because it demonstrates that DNA templating may represent a useful platform to tune and optimize photophysics for target applications.
Acknowledgments
Research at Boise State was supported by the Department of the Navy, Office of Naval Research (ONR), via ONR Award No. N00014-19-1-2615. Research at the U.S. Naval Research Laboratory (NRL) was supported by NRL base funding, the NRL Nanosciences Institute, and ONR Award # N0001419WX01811. D.M. was supported by the National Institute of Biomedical Imaging and Bioengineering of the National Institutes of Health under Award No. K99EB030013. The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.jpclett.2c00017.
Figures S1–S21 and Tables S1–S14 (PDF)
The authors declare no competing financial interest.
Supplementary Material
References
- Scholes G. D.; Rumbles G. Excitons in Nanoscale Systems. Nat. Mater. 2006, 5 (9), 683–696. 10.1038/nmat1710. [DOI] [PubMed] [Google Scholar]
- Gust D.; Moore T. A.; Moore A. L. Solar Fuels via Artificial Photosynthesis. Acc. Chem. Res. 2009, 42 (12), 1890–1898. 10.1021/ar900209b. [DOI] [PubMed] [Google Scholar]
- Wasielewski M. R. Self-Assembly Strategies for Integrating Light Harvesting and Charge Separation in Artificial Photosynthetic Systems. Acc. Chem. Res. 2009, 42 (12), 1910–1921. 10.1021/ar9001735. [DOI] [PubMed] [Google Scholar]
- Mirkovic T.; Ostroumov E. E.; Anna J. M.; van Grondelle R.; Govindjee; Scholes G. D. Light Absorption and Energy Transfer in the Antenna Complexes of Photosynthetic Organisms. Chem. Rev. 2017, 117 (2), 249–293. 10.1021/acs.chemrev.6b00002. [DOI] [PubMed] [Google Scholar]
- Brixner T.; Hildner R.; Köhler J.; Lambert C.; Würthner F. Exciton Transport in Molecular Aggregates - From Natural Antennas to Synthetic Chromophore Systems. Adv. Energy Mater. 2017, 7 (16), 1700236. 10.1002/aenm.201700236. [DOI] [Google Scholar]
- Brédas J.-L.; Norton J. E.; Cornil J.; Coropceanu V. Molecular Understanding of Organic Solar Cells: The Challenges. Acc. Chem. Res. 2009, 42 (11), 1691–1699. 10.1021/ar900099h. [DOI] [PubMed] [Google Scholar]
- Clarke T. M.; Durrant J. R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110 (11), 6736–6767. 10.1021/cr900271s. [DOI] [PubMed] [Google Scholar]
- Ostroverkhova O. Organic Optoelectronic Materials: Mechanisms and Applications. Chem. Rev. 2016, 116 (22), 13279–13412. 10.1021/acs.chemrev.6b00127. [DOI] [PubMed] [Google Scholar]
- Yurke B.; Kuang W. Passive Linear Nanoscale Optical and Molecular Electronics Device Synthesis from Nanoparticles. Phys. Rev. A 2010, 81 (3), 033814. 10.1103/PhysRevA.81.033814. [DOI] [Google Scholar]
- Graugnard E.; Kellis D. L.; Bui H.; Barnes S.; Kuang W.; Lee J.; Hughes W. L.; Knowlton W. B.; Yurke B. DNA-Controlled Excitonic Switches. Nano Lett. 2012, 12 (4), 2117–2122. 10.1021/nl3004336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- LaBoda C.; Duschl H.; Dwyer C. L. DNA-Enabled Integrated Molecular Systems for Computation and Sensing. Acc. Chem. Res. 2014, 47 (6), 1816–1824. 10.1021/ar500054u. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Davis P. H.; Lee J.; Kuang W.; Hughes W. L.; Graugnard E.; Yurke B.; Knowlton W. B. Excitonic AND Logic Gates on DNA Brick Nanobreadboards. ACS Photonics 2015, 2 (3), 398–404. 10.1021/ph500444d. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sawaya N. P. D.; Rappoport D.; Tabor D. P.; Aspuru-Guzik A. Excitonics: A Set of Gates for Molecular Exciton Processing and Signaling. ACS Nano 2018, 12 (7), 6410–6420. 10.1021/acsnano.8b00584. [DOI] [PubMed] [Google Scholar]
- Kellis D. L.; Sarter C.; Cannon B. L.; Davis P. H.; Graugnard E.; Lee J.; Pensack R. D.; Kolmar T.; Jäschke A.; Yurke B.; Knowlton W. B. An All-Optical Excitonic Switch Operated in the Liquid and Solid Phases. ACS Nano 2019, 13 (3), 2986–2994. 10.1021/acsnano.8b07504. [DOI] [PubMed] [Google Scholar]
- Buckhout-White S.; Brown C. W. III; Hastman D. A.; Ancona M. G.; Melinger J. S.; Goldman E. R.; Medintz I. L. Expanding Molecular Logic Capabilities in DNA-Scaffolded MultiFRET Triads. RSC Adv. 2016, 6 (100), 97587–97598. 10.1039/C6RA23079B. [DOI] [Google Scholar]
- Massey M.; Medintz I. L.; Ancona M. G.; Algar W. R. Time-Gated FRET and DNA-Based Photonic Molecular Logic Gates: AND, OR, NAND, and NOR. ACS Sensors 2017, 2 (8), 1205–1214. 10.1021/acssensors.7b00355. [DOI] [PubMed] [Google Scholar]
- Sundström V.; Gillbro T. Excited State Dynamics and Photophysics of Aggregated Dye Chromophores in Solution. J. Chem. Phys. 1985, 83 (6), 2733–2743. 10.1063/1.449275. [DOI] [Google Scholar]
- Kreller D. I.; Kamat P. V. Photochemistry of Sensitizing Dyes: Spectroscopic and Redox Properties of Cresyl Violet. J. Phys. Chem. 1991, 95 (11), 4406–4410. 10.1021/j100164a044. [DOI] [Google Scholar]
- Collini E.; Ferrante C.; Bozio R. Influence of Excitonic Interactions on the Transient Absorption and Two-Photon Absorption Spectra of Porphyrin J-Aggregates in the NIR Region. J. Phys. Chem. C 2007, 111 (50), 18636–18645. 10.1021/jp074974q. [DOI] [Google Scholar]
- Verma S.; Ghosh A.; Das A.; Ghosh H. N. Ultrafast Exciton Dynamics of J- and H-Aggregates of the Porphyrin-Catechol in Aqueous Solution. J. Phys. Chem. B 2010, 114 (25), 8327–8334. 10.1021/jp101643c. [DOI] [PubMed] [Google Scholar]
- Kakade S.; Ghosh R.; Palit D. K. Excited State Dynamics of Zinc–Phthalocyanine Nanoaggregates in Strong Hydrogen Bonding Solvents. J. Phys. Chem. C 2012, 116 (28), 15155–15166. 10.1021/jp304369r. [DOI] [Google Scholar]
- Dean J. C.; Oblinsky D. G.; Rafiq S.; Scholes G. D. Methylene Blue Exciton States Steer Nonradiative Relaxation: Ultrafast Spectroscopy of Methylene Blue Dimer. J. Phys. Chem. B 2016, 120 (3), 440–454. 10.1021/acs.jpcb.5b11847. [DOI] [PubMed] [Google Scholar]
- Cunningham P. D.; Kim Y. C.; Díaz S. A.; Buckhout-White S.; Mathur D.; Medintz I. L.; Melinger J. S. Optical Properties of Vibronically Coupled Cy3 Dimers on DNA Scaffolds. J. Phys. Chem. B 2018, 122 (19), 5020–5029. 10.1021/acs.jpcb.8b02134. [DOI] [PubMed] [Google Scholar]
- Huff J. S.; Davis P. H.; Christy A.; Kellis D. L.; Kandadai N.; Toa Z. S. D.; Scholes G. D.; Yurke B.; Knowlton W. B.; Pensack R. D. DNA-Templated Aggregates of Strongly Coupled Cyanine Dyes: Nonradiative Decay Governs Exciton Lifetimes. J. Phys. Chem. Lett. 2019, 10 (10), 2386–2392. 10.1021/acs.jpclett.9b00404. [DOI] [PubMed] [Google Scholar]
- Huff J. S.; Turner D. B.; Mass O. A.; Patten L. K.; Wilson C. K.; Roy S. K.; Barclay M. S.; Yurke B.; Knowlton W. B.; Davis P. H.; Pensack R. D. Excited-State Lifetimes of DNA-Templated Cyanine Dimer, Trimer, and Tetramer Aggregates: The Role of Exciton Delocalization, Dye Separation, and DNA Heterogeneity. J. Phys. Chem. B 2021, 125 (36), 10240–10259. 10.1021/acs.jpcb.1c04517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mazuski R. J.; Díaz S. A.; Wood R. E.; Lloyd L. T.; Klein W. P.; Mathur D.; Melinger J. S.; Engel G. S.; Medintz I. L. Ultrafast Excitation Transfer in Cy5 DNA Photonic Wires Displays Dye Conjugation and Excitation Energy Dependency. J. Phys. Chem. Lett. 2020, 11 (10), 4163–4172. 10.1021/acs.jpclett.0c01020. [DOI] [PubMed] [Google Scholar]
- Martini I.; Hartland G. V.; Kamat P. V. Ultrafast Photophysical Investigation of Cresyl Violet Aggregates Adsorbed onto Nanometer-Sized Particles of SnO2 and SiO2. J. Phys. Chem. B 1997, 101 (24), 4826–4830. 10.1021/jp9702964. [DOI] [Google Scholar]
- Das S.; Kamat P. V. Can H-Aggregates Serve as Light-Harvesting Antennae? Triplet–Triplet Energy Transfer between Excited Aggregates and Monomer Thionine in Aersol-OT Solutions. J. Phys. Chem. B 1999, 103 (1), 209–215. 10.1021/jp983816j. [DOI] [Google Scholar]
- Nasr C.; Hotchandani S. Excited-State Behavior of Nile Blue H-Aggregates Bound to SiO2 and SnO2 Colloids. Chem. Mater. 2000, 12 (6), 1529–1535. 10.1021/cm0000752. [DOI] [Google Scholar]
- Kelbauskas L.; Bagdonas S.; Dietel W.; Rotomskis R. Excitation Relaxation and Structure of TPPS4 J-Aggregates. J. Lumin. 2003, 101 (4), 253–262. 10.1016/S0022-2313(02)00547-1. [DOI] [Google Scholar]
- Atchimnaidu S.; Perumal D.; Harikrishanan K. S.; Thelu H. V. P.; Varghese R. Phototheranostic DNA Micelles from the Self-Assembly of DNA-BODIPY Amphiphiles for the Thermal Ablation of Cancer Cells. Nanoscale 2020, 12 (22), 11858–11862. 10.1039/D0NR02622K. [DOI] [PubMed] [Google Scholar]
- St. Lorenz A.; Buabeng E. R.; Taratula O.; Taratula O.; Henary M. Near-Infrared Heptamethine Cyanine Dyes for Nanoparticle-Based Photoacoustic Imaging and Photothermal Therapy. J. Med. Chem. 2021, 64 (12), 8798–8805. 10.1021/acs.jmedchem.1c00771. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jelley E. E. Spectral Absorption and Fluorescence of Dyes in the Molecular State. Nature 1936, 138 (3502), 1009–1010. 10.1038/1381009a0. [DOI] [Google Scholar]
- Mastron J. N.; Roberts S. T.; McAnally R. E.; Thompson M. E.; Bradforth S. E. Aqueous Colloidal Acene Nanoparticles: A New Platform for Studying Singlet Fission. J. Phys. Chem. B 2013, 117 (49), 15519–15526. 10.1021/jp4057972. [DOI] [PubMed] [Google Scholar]
- Pensack R. D.; Tilley A. J.; Parkin S. R.; Lee T. S.; Payne M. M.; Gao D.; Jahnke A. A.; Oblinsky D. G.; Li P.-F.; Anthony J. E.; Seferos D. S.; Scholes G. D. Exciton Delocalization Drives Rapid Singlet Fission in Nanoparticles of Acene Derivatives. J. Am. Chem. Soc. 2015, 137 (21), 6790–6803. 10.1021/ja512668r. [DOI] [PubMed] [Google Scholar]
- Petkov B. K.; Gellen T. A.; Farfan C. A.; Carbery W. P.; Hetzler B. E.; Trauner D.; Li X.; Glover W. J.; Ulness D. J.; Turner D. B. Two-Dimensional Electronic Spectroscopy Reveals the Spectral Dynamics of Förster Resonance Energy Transfer. Chem. 2019, 5 (8), 2111–2125. 10.1016/j.chempr.2019.05.005. [DOI] [Google Scholar]
- van Amerongen H.; van Grondelle R.; Valkunas L.. Photosynthetic Excitons; World Scientific: 2000. [Google Scholar]
- Barclay M. S.; Roy S. K.; Huff J. S.; Mass O. A.; Turner D. B.; Wilson C. K.; Kellis D. L.; Terpetschnig E. A.; Lee J.; Davis P. H.; Yurke B.; Knowlton W. B.; Pensack R. D. Rotaxane Rings Promote Oblique Packing and Extended Lifetimes in DNA-Templated Molecular Dye Aggregates. Commun. Chem. 2021, 4 (1), 19. 10.1038/s42004-021-00456-8. [DOI] [PubMed] [Google Scholar]
- Banal J. L.; Kondo T.; Veneziano R.; Bathe M.; Schlau-Cohen G. S. Photophysics of J-Aggregate-Mediated Energy Transfer on DNA. J. Phys. Chem. Lett. 2017, 8 (23), 5827–5833. 10.1021/acs.jpclett.7b01898. [DOI] [PubMed] [Google Scholar]
- Cannon B. L.; Kellis D. L.; Patten L. K.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Coherent Exciton Delocalization in a Two-State DNA-Templated Dye Aggregate System. J. Phys. Chem. A 2017, 121 (37), 6905–6916. 10.1021/acs.jpca.7b04344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Markova L. I.; Malinovskii V. L.; Patsenker L. D.; Häner R. J- vs. H-Type Assembly: Pentamethine Cyanine (Cy5) as a near-IR Chiroptical Reporter. Chem. Commun. 2013, 49 (46), 5298. 10.1039/c3cc42103a. [DOI] [PubMed] [Google Scholar]
- Cunningham P. D.; Díaz S. A.; Yurke B.; Medintz I. L.; Melinger J. S. Delocalized Two-Exciton States in DNA Scaffolded Cyanine Dimers. J. Phys. Chem. B 2020, 124 (37), 8042–8049. 10.1021/acs.jpcb.0c06732. [DOI] [PubMed] [Google Scholar]
- Kringle L.; Sawaya N. P. D.; Widom J.; Adams C.; Raymer M. G.; Aspuru-Guzik A.; Marcus A. H. Temperature-Dependent Conformations of Exciton-Coupled Cy3 Dimers in Double-Stranded DNA. J. Chem. Phys. 2018, 148 (8), 085101. 10.1063/1.5020084. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Heussman D.; Kittell J.; Kringle L.; Tamimi A.; von Hippel P. H.; Marcus A. H. Measuring Local Conformations and Conformational Disorder of (Cy3) 2 Dimer Labeled DNA Fork Junctions Using Absorbance, Circular Dichroism and Two-Dimensional Fluorescence Spectroscopy. Faraday Discuss. 2019, 216, 211–235. 10.1039/C8FD00245B. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hart S. M.; Chen W. J.; Banal J. L.; Bricker W. P.; Dodin A.; Markova L.; Vyborna Y.; Willard A. P.; Häner R.; Bathe M.; Schlau-Cohen G. S. Engineering Couplings for Exciton Transport Using Synthetic DNA Scaffolds. Chem. 2021, 7 (3), 752–773. 10.1016/j.chempr.2020.12.020. [DOI] [Google Scholar]
- Cannon B. L.; Patten L. K.; Kellis D. L.; Davis P. H.; Lee J.; Graugnard E.; Yurke B.; Knowlton W. B. Large Davydov Splitting and Strong Fluorescence Suppression: An Investigation of Exciton Delocalization in DNA-Templated Holliday Junction Dye Aggregates. J. Phys. Chem. A 2018, 122 (8), 2086–2095. 10.1021/acs.jpca.7b12668. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seifert J. L.; Connor R. E.; Kushon S. A.; Wang M.; Armitage B. A. Spontaneous Assembly of Helical Cyanine Dye Aggregates on DNA Nanotemplates. J. Am. Chem. Soc. 1999, 121 (13), 2987–2995. 10.1021/ja984279j. [DOI] [Google Scholar]
- Wang M.; Silva G. L.; Armitage B. A. DNA-Templated Formation of a Helical Cyanine Dye J-Aggregate. J. Am. Chem. Soc. 2000, 122 (41), 9977–9986. 10.1021/ja002184n. [DOI] [Google Scholar]
- Garoff R. A.; Litzinger E. A.; Connor R. E.; Fishman I.; Armitage B. A. Helical Aggregation of Cyanine Dyes on DNA Templates: Effect of Dye Structure on Formation of Homo- and Heteroaggregates. Langmuir 2002, 18 (16), 6330–6337. 10.1021/la025742f. [DOI] [Google Scholar]
- Asanuma H.; Fujii T.; Kato T.; Kashida H. Coherent Interactions of Dyes Assembled on DNA. J. Photochem. Photobiol. C Photochem. Rev. 2012, 13 (2), 124–135. 10.1016/j.jphotochemrev.2012.04.002. [DOI] [Google Scholar]
- Melinger J. S.; Khachatrian A.; Ancona M. G.; Buckhout-White S.; Goldman E. R.; Spillmann C. M.; Medintz I. L.; Cunningham P. D. FRET from Multiple Pathways in Fluorophore-Labeled DNA. ACS Photonics 2016, 3 (4), 659–669. 10.1021/acsphotonics.6b00006. [DOI] [Google Scholar]
- Mass O. A.; Wilson C. K.; Roy S. K.; Barclay M. S.; Patten L. K.; Terpetschnig E. A.; Lee J.; Pensack R. D.; Yurke B.; Knowlton W. B. Exciton Delocalization in Indolenine Squaraine Aggregates Templated by DNA Holliday Junction Scaffolds. J. Phys. Chem. B 2020, 124 (43), 9636–9647. 10.1021/acs.jpcb.0c06480. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ong L. L.; Hanikel N.; Yaghi O. K.; Grun C.; Strauss M. T.; Bron P.; Lai-Kee-Him J.; Schueder F.; Wang B.; Wang P.; Kishi J. Y.; Myhrvold C.; Zhu A.; Jungmann R.; Bellot G.; Ke Y.; Yin P. Programmable Self-Assembly of Three-Dimensional Nanostructures from 10,000 Unique Components. Nature 2017, 552 (7683), 72–77. 10.1038/nature24648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hong F.; Zhang F.; Liu Y.; Yan H. DNA Origami: Scaffolds for Creating Higher Order Structures. Chem. Rev. 2017, 117 (20), 12584–12640. 10.1021/acs.chemrev.6b00825. [DOI] [PubMed] [Google Scholar]
- Wang D.; Yu L.; Huang C.-M.; Arya G.; Chang S.; Ke Y. Programmable Transformations of DNA Origami Made of Small Modular Dynamic Units. J. Am. Chem. Soc. 2021, 143 (5), 2256–2263. 10.1021/jacs.0c10576. [DOI] [PubMed] [Google Scholar]
- Rothemund P. W. K. Folding DNA to Create Nanoscale Shapes and Patterns. Nature 2006, 440 (7082), 297–302. 10.1038/nature04586. [DOI] [PubMed] [Google Scholar]
- Seeman N. C. Nucleic Acid Junctions and Lattices. J. Theor. Biol. 1982, 99 (2), 237–247. 10.1016/0022-5193(82)90002-9. [DOI] [PubMed] [Google Scholar]
- Kallenbach N. R.; Ma R.-I.; Seeman N. C. An Immobile Nucleic Acid Junction Constructed from Oligonucleotides. Nature 1983, 305 (5937), 829–831. 10.1038/305829a0. [DOI] [Google Scholar]
- Schobel U.; Egelhaaf H.-J.; Brecht A.; Oelkrug D.; Gauglitz G. New Donor–Acceptor Pair for Fluorescent Immunoassays by Energy Transfer. Bioconjugate Chem. 1999, 10 (6), 1107–1114. 10.1021/bc990073b. [DOI] [PubMed] [Google Scholar]
- Hohng S.; Joo C.; Ha T. Single-Molecule Three-Color FRET. Biophys. J. 2004, 87 (2), 1328. 10.1529/biophysj.104.043935. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Turro N. J.; Scaiano J. C.; Ramamurthy V.. Modern Molecular Photochemistry of Organic Molecules; University Science Books: Sausalito, CA, 2010. [Google Scholar]
- Spiegel J. D.; Fulle S.; Kleinschmidt M.; Gohlke H.; Marian C. M. Failure of the IDA in FRET Systems at Close Inter-Dye Distances Is Moderated by Frequent Low κ2 Values. J. Phys. Chem. B 2016, 120 (34), 8845–8862. 10.1021/acs.jpcb.6b05754. [DOI] [PubMed] [Google Scholar]
- Clarke T. M.; Durrant J. R. Charge Photogeneration in Organic Solar Cells. Chem. Rev. 2010, 110 (11), 6736–6767. 10.1021/cr900271s. [DOI] [PubMed] [Google Scholar]
- Kasha M.; Rawls H. R.; Ashraf El-Bayoumi M. The Exciton Model in Molecular Spectroscopy. Pure Appl. Chem. 1965, 11 (3–4), 371–392. 10.1351/pac196511030371. [DOI] [Google Scholar]
- Kühn O.; Renger T.; May V. Theory of Exciton-Vibrational Dynamics in Molecular Dimers. Chem. Phys. 1996, 204 (1), 99–114. 10.1016/0301-0104(95)00448-3. [DOI] [Google Scholar]
- Pensack R. D.; Ostroumov E. E.; Tilley A. J.; Mazza S.; Grieco C.; Thorley K. J.; Asbury J. B.; Seferos D. S.; Anthony J. E.; Scholes G. D. Observation of Two Triplet-Pair Intermediates in Singlet Exciton Fission. J. Phys. Chem. Lett. 2016, 7 (13), 2370–2375. 10.1021/acs.jpclett.6b00947. [DOI] [PubMed] [Google Scholar]
- Margulies E. A.; Miller C. E.; Wu Y.; Ma L.; Schatz G. C.; Young R. M.; Wasielewski M. R. Enabling Singlet Fission by Controlling Intramolecular Charge Transfer in π-Stacked Covalent Terrylenediimide Dimers. Nat. Chem. 2016, 8 (12), 1120–1125. 10.1038/nchem.2589. [DOI] [PubMed] [Google Scholar]
- Pensack R. D.; Tilley A. J.; Grieco C.; Purdum G. E.; Ostroumov E. E.; Granger D. B.; Oblinsky D. G.; Dean J. C.; Doucette G. S.; Asbury J. B.; Loo Y.-L.; Seferos D. S.; Anthony J. E.; Scholes G. D. Striking the Right Balance of Intermolecular Coupling for High-Efficiency Singlet Fission. Chem. Sci. 2018, 9 (29), 6240–6259. 10.1039/C8SC00293B. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.