

Abstract
Hydrogen cyanide (HCN) is a versatile synthon for generating carbon-carbon and carbon-heteroatom bonds. Unlike other one-carbon synthons (i.e., CO, CO2), HCN can function as a nucleophile (as in potassium cyanide, KCN) and an electrophile (as in cyanogen bromide, (CN)Br). The incorporation of the −CN motif into organic molecules generates nitriles, hydantoins and (thio)cyanates, which can be converted to carboxylic acids, aldehydes, amides and amines. Such versatile chemistry is particularly attractive in PET radiochemistry where diverse bioactive small molecules incorporating carbon-11 in different positions need to be produced. The first examples of making [11C]HCN for radiolabeling date back to the 1960s. During the ensuing decades, [11C]cyanide labeling was popular for producing biologically important molecules including 11C-labeled α-amino acids, sugars and neurotransmitters. [11C]cyanation is now reemerging in many PET centers due to its versatility for making novel tracers. Here, we summarize the chemistry of [11C]HCN, review the methods to make [11C]HCN past and present, describe methods for labeling different types of molecules with [11C]HCN, and provide an overview of the reactions available to convert nitriles into other functional groups. Finally, we discuss some of the challenges and opportunities in [11C]HCN labeling such as developing more robust methods to produce [11C]HCN and developing rapid and selective methods to convert nitriles into other functional groups in complex molecules.
Keywords: carbon-11, radiolabeling, [11C]HCN, radiopharmaceuticals, amino acids, sugars
Graphical Abstract
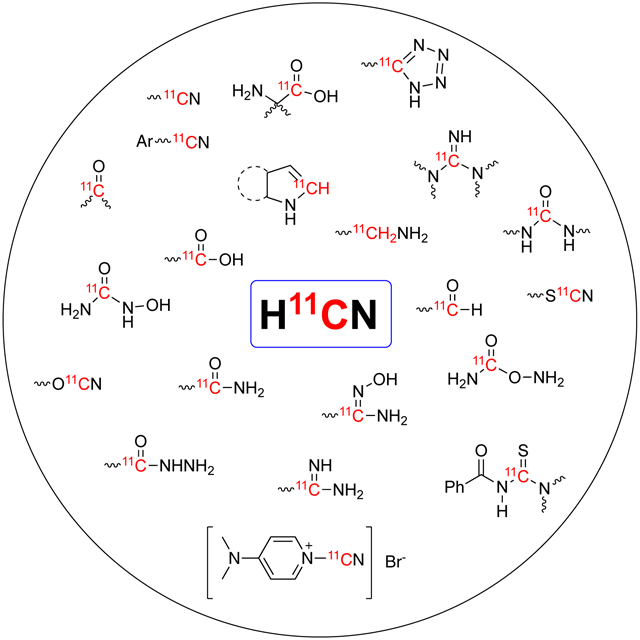
1. Introduction
Positron emission tomography (PET) is a molecular imaging technique that allows imaging the location of radiolabeled molecules inside living subjects. Unlike other molecular imaging techniques, PET can image deep inside the body as it relies in the detection of gamma rays generated by the annihilation of a positron emitted by the radiolabeled molecule and an electron of the surroundings. This feature, combined with its fully quantitative nature, makes PET a powerful tool for probing biological processes in live subjects, which can provide valuable information for disease diagnoses and drug development1–5. In order to reap the benefits of PET, radiotracers labeled with positron emitting isotopes are essential. In the case of small molecule radiotracers, the most common PET isotopes are fluorine-18 (t1/2 = 110 min)6–8 and carbon-11 (t1/2 = 20.3 min)9–11. While the longer half-life of fluorine-18 offers the opportunity to produce multiple doses from one batch production and distribute them to nearby hospitals, carbon-11 offers other advantages such as the opportunity to label molecules that do not contain fluorine and the opportunity to scan the same subject twice on the same day. Furthermore, the recent developments of Time-of-Flight (TOF) PET technology and the total-body PET scanner require shorter scan times and lower doses of radiotracer and are expected to make 11C-labeled radiotracers even more useful12–13,14.
Currently, the most common method for labeling small molecules with carbon-11 is carbon-11 methylation using [11C]methyl iodide or [11C]methyl triflate. However, 11C-methylation is mainly used for producing compounds bearing -Me, -OMe, -SMe, or -NRMe groups9, 11, 15–17. When introducing methyl is not possible or desirable, labeling with 11CO, 11CO2, H11CN and other 11C-synthons offer the opportunity to synthesize a variety of radiolabeled molecules (Fig. 1). Methods for labeling with 11CO and 11CO2 have been extensively reviewed recently9–11, 15, 17–28. However, H11CN labeling methods have only been covered succinctly in several more general reviews about 11C-labeling9, 11, 15, 17, 21, 26, 29 as well as a recent review on isotope cyanation chemistry focused on 14C, 13C and 11C cyanation reagents and applications30. Given the versatility of this synthon in PET radiochemistry and to facilitate and promote its use in radiopharmaceutical synthesis, here we present a comprehensive review of labeling with H11CN.
Fig. 1.
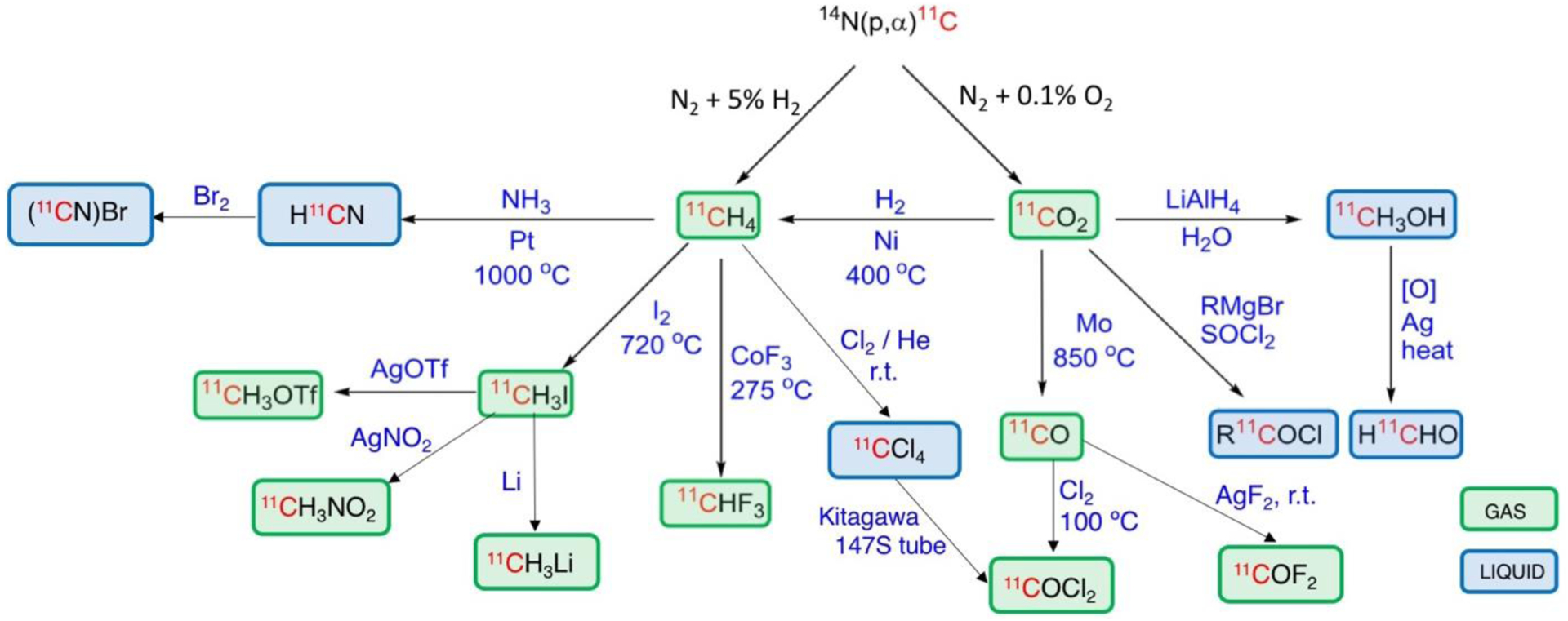
Carbon-11 reagents available for radiolabelin (original image courtesy of V. W. Pike and colleagues at NIMH, modified by the authors).
2. Properties of hydrogen cyanide
Hydrogen cyanide is a liquid at room temperature (bp. 25.6 °C) which, as discussed in the Section 3 (H11CN Production), makes it challenging to transfer within radiochemistry synthesizers. Hydrogen cyanide is slightly acidic (pKa 9.2) and partially ionizes in water to give cyanide ion (CN−). Cyanide can react with alkaline metals to generate salts (e.g,, NaCN, KCN, etc) and with transition metals to generate coordination compounds (e.g., Prussian blue, FeIII4[FeII(CN)6]3). Cyanide is a good nucleophile and can react with organic molecules containing electrophilic carbons to generate nitriles and cyanohydrins. Nitriles can be hydrolyzed to carboxamides (R(C=O)NH2) and carboxylic acids (RCOOH), reduced to primary amines (R2CH2NH2) and aldehydes (RCHO) as well as alkylated. Cyanohydrins are common precursors to amino acids. In addition, metal cyanides can be reacted with halogens such as bromine to generate cyanogen bromide (CNBr). Cyanogen bromide is a common reagent in organic syntheses that contains an electrophilic carbon and is prone to nucleophilic attack. All these properties make hydrogen cyanide an attractive synthon for radiolabeling.
3. H11CN production
Carbon-11 hydrogen cyanide (H11CN) was first reported by Dubrin and colleagues in 1964.31 H11CN was produced by bombarding nitrogen gas or nitrogen oxides (N2O, NO or NO2) with 11C ions generated through the neutron stripping of a 12C ion beam produced by a Heavy Ion Accelerator (Fig. 2A). These 11C ions reacted with gas molecules in the target chamber to produce a mixture of 11CO and 11CN• radical. Given the unstable nature of the 11CN• radical, hydrocarbons like ethylene were used to quench the radical which produced H11CN through hydrogen abstraction. In 1966, Ache and Wolf showed that H11CN was the main product of the proton bombardment,14N(p,α)11C reaction, of a gas mixture containing 89.5% N2 and 10.5% ethane (C2H6) which also occurred via 11CN• radical formation and hydrogen abstraction (Fig. 2B).32 In 1971, another two methods were described for the preparation of 11C cyanide.33 The first method involved the recoil synthesis, NaCN → Na11CN, via the 14N(p,α)11C and 12C(p,pn)11C reactions (Fig. 2C). The second method used the recoil synthesis, B2O3 → 11CO + 11CO2, via the 11B(p,n)11C, which after full oxidation to 11CO2, was converted to K11CN through the reaction: 11CO2 + K + NH3 → K11CN (Fig. 2D). These methods use non-standard liquid targets, suffer from low molar activity and require moisture and air sensitive reagents, which promoted the development a simple and reliable flow method for routine production of no-carrier-added, high-activity H11CN for radiopharmaceutical labeling by Christman and colleagues in 1973.34–35 This method consisted on reacting cyclotron-produced methane (11CH4) with ammonia gas (NH3) in a effluent stream over a platinum catalyst at 1,000 °C. A typical run produced 74 GBq (2 Ci) of no-carrier-added H11CN from a 45 min irradiation with a current of 30 μA of focused 25 MeV protons (Fig. 2E). In 1983, a method for producing H11CN using microwave discharge was reported by Niisawa et al (Fig. 2F).36 This method produces no-carrier added H11CN by the direct microwave discharge of cyclotron-produced 11CO2, with H2 and N2 gas mixture. This method typically yielded ca. 7.4 GBq (200mCi) of H11CN in ca. 20% radiochemical yield with molar activity of 7.33 GBq/μmol (199 mCi/μmol) within 20min of synthesis time.
Fig. 2.
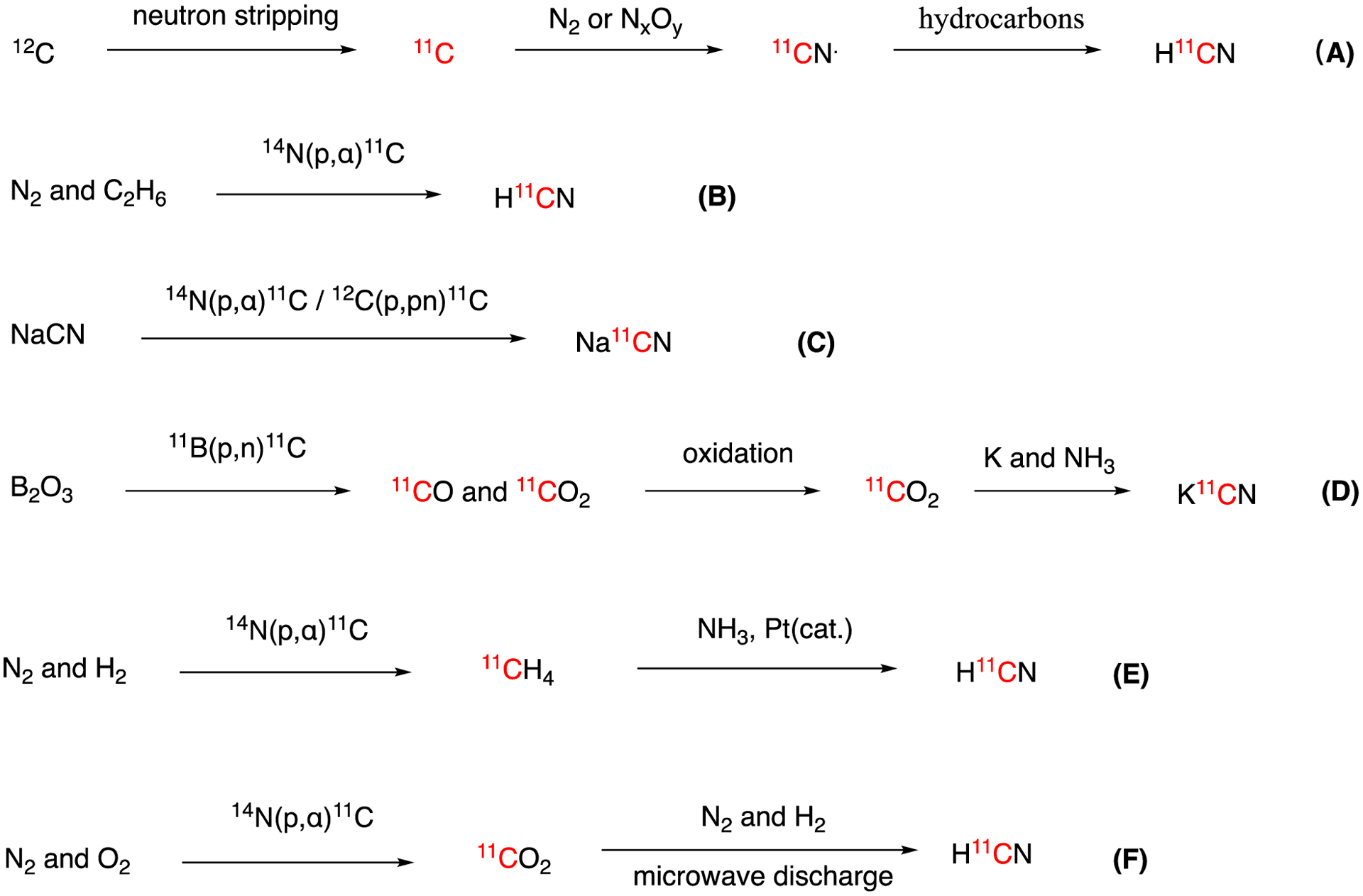
Summary of the H11CN/M11CN production methods.
Currently, the most common method to generate H11CN for radiopharmaceutical synthesis consists in the stepwise conversion of cyclotron-produced 11CO2 to 11CH4 and then to H11CN by reacting H2 gas over a nickel catalyst at 400 °C followed by reaction with NH3 gas over a platinum catalyst at 1000 °C as previously described (Fig. 2). This process is typically carried out in an automated flow reactor. While the chemistry is reproducible careful attention has to be paid to the flowrate and the dryness of NH3 in the effluent. Furthermore, when using these systems, it is imperative to use high purity anhydrous NH3 and an inline desiccant since moisture causes the formation NH411CN which tends adsorb to the walls of the delivery lines. Also, since HCN has a boiling point of 25.6 °C it can easily condense in the transfer lines making its transfer inefficient.
4. [11C]Cyanation of aliphatic compounds
4.1. Nucleophilic substitution reactions
Cyanide, CN−, is a very good nucleophile. The nucleophilic substitution reaction is the most direct method of introducing 11CN group into organic molecules to form [11C]nitriles. These [11C]nitriles can readily be generated by reaction of an alkyl electrophile with a cyanide salt (Na11CN, K11CN or NH411CN). These reactions typically proceed under mild conditions and give the excellent radiochemical yields.
4.1.1. Bisulfite, -SO3Na, as leaving group
The in situ reactions of aldehyde or ketone with sodium bisulfite, NaHSO3, give the corresponding bisulfite adduct. Although sodium bisulfite, -SO3Na, is not a commonly used leaving group, it is a good precursor for the synthesis of [11C]α-substituted carboxylic acids (e.g., [11C]α-amino acid and [11C]α-hydroxy acid). This reaction has been used to produce several biologically important molecules including [11C]dopamine (3) and [11C]norepinephrine (4) (Fig. 3).
Fig. 3.
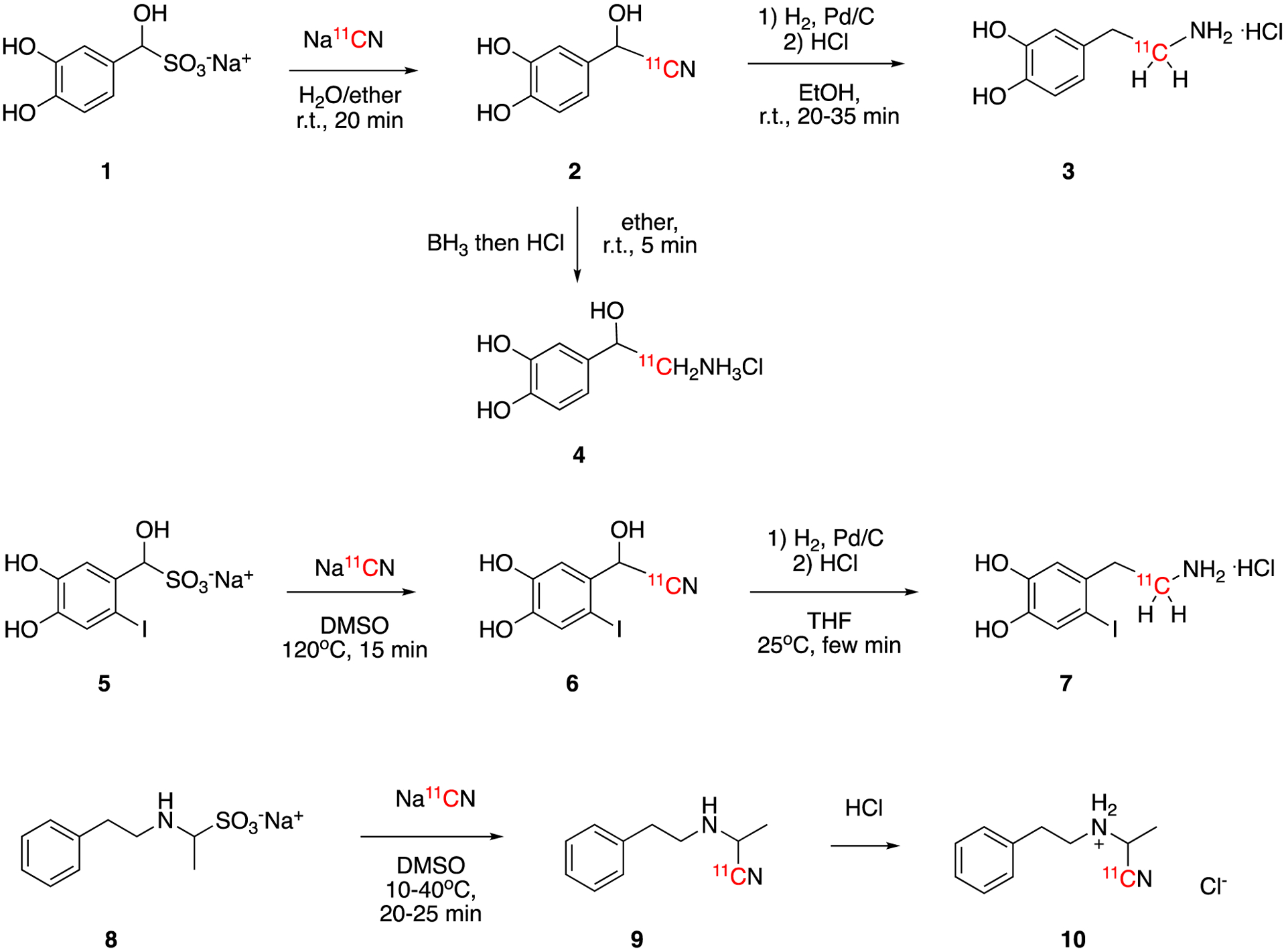
Synthesis of [11C]dopamine hydrochloride (3), [11C]norepinephrine hydrochloride (4), [11C]6-iododopamine hydrochloride (7), and [11C]2-N-benzylaminobutanenitrile hydrochloride (10).
Dopamine is a hormone and a neurotransmitter that plays important roles in the brain and body. [11C]dopamine hydrochloride (3) was first reported by Chrisman et al at 197037. In this first report, precursor 1 was prepared by the reaction of sodium bisulfite with 3,4-dihydroxybenzaldehyde in water and used without isolation. The addition of carrier-added Na11CN generated intermediate 2. This was then reacted with hydrogen gas over palladium catalyst and hydrochloric acid to generate [11C]dopamine hydrochloride (3). The molar activity was 0.23 MBq/μmol (0.0062 mCi/μmol) calculated to end of bombardment (EoB).
Since the molar activity of 3 prepared from carrier-added Na11CN was very low and limited its biological application, the same research group developed the synthesis [11C]dopamine hydrochloride (3) from no-carrier-added H11CN later38. Using no-carrier added Na11CN, 3 was prepared with high molar activity of 383 GBq/μmol (10.3 Ci/μmol). One year later, Fowler et al reported the preparation of [11C]norepinephrine hydrochloride (4) in 2.5% decay-corrected radiochemical yield using a similar no-carrier-added method.39
The iodo- derivative of dopamine, [11C]6-iododopamine hydrochloride (7), was also prepared using similar no-carrier-added method. In this case, precursor 5 was prepared by the in situ reaction of 6-iodo-3,4-dihydroxybenzaldehyde with sodium bisulfite. Further substitution of -SO3Na by 11CN− yielded intermediate 6, which was then reduced to produce [11C]6-iododopamine hydrochloride (7) in 11% TLC-based radiochemical yield.40 This represents a good example of a selective reduction to remove the hydroxyl group while preserving the nitrile group.
[11C]2-N-Phenethylaminoalkanenitriles were synthesized using a similar method.41 The corresponding in situ prepared aldehyde-sodium bisulfite addition adducts was reacted with phenethylamine and carrier added Na11CN to give [11C]2-N-phenethylaminoalkanenitrile (9). Further acidification gave the corresponding hydrochloride salt (10), which was used for biodistribution studies. The synthesis times were 51–66 min and 0.22–1.30GBq (6–35 mCi) of products were obtained.
In 1987, Iwata et al reported the use of sodium bisulfite precursors to produce several amino acids labeled at the carboxylic carbon (Fig. 4). Specifically, they reported the synthesis of no-carrier-added phenylalanine, valine, isoleucine and others via [11C]aminonitriles intermediates.42 The precursors for labeling, amino sulfites (12a-12i), were prepared by the reaction between ammonium hydroxide with the corresponding aldehyde or ketone bisulfite adducts (11a-11i). The following [11C]cyanation and nitrile-hydrolysis lead to the corresponding racemic [11C]amino acids (14a-14i) in good radiochemical yields. As such, this chemistry makes the synthesis and purification of [11C]amino acids simple and suitable for automation. In 1990, Takahashi et al reported the synthesis of DL-[11C]alanine from H11CN using this method43. Starting from sodium 1-hydroxyethanesulfite, DL-[11C]alanine was obtained in 75% decay-corrected radiochemical yield within 40 min.
Fig. 4.
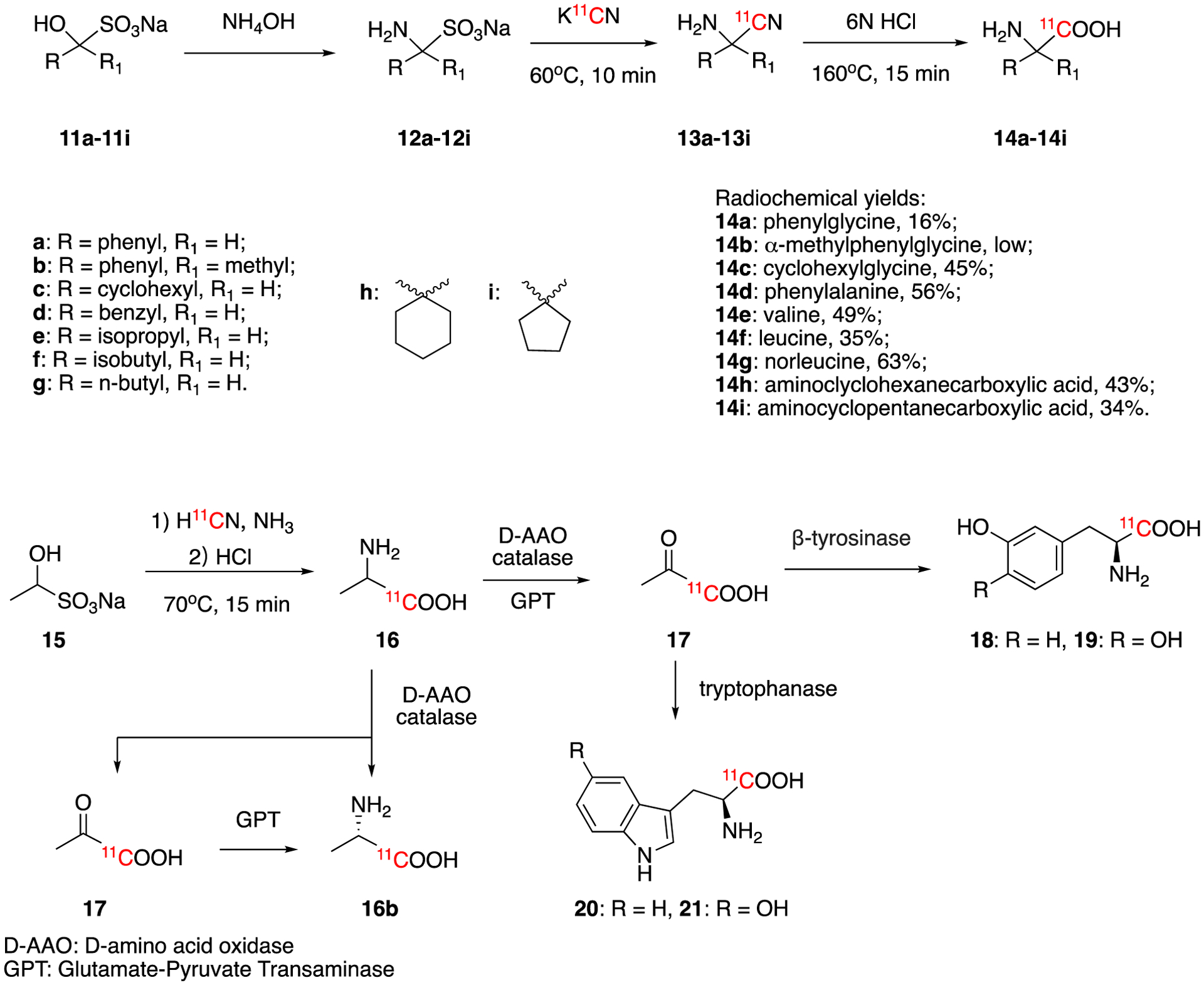
Synthesis of [11C]α-amino acids (14a-14i), L-[11C]alanine(16b), L-[11C]tyrosine (18), L-[11C]DOPA (19), L-[11C]tryptophan (20) and [11C]5-hyroxy-L-tryptophan (21).
In 1995, a new on-column synthesis system was developed for the synthesis of 1-aminocyclopentane-1-[11C]carboxylic acid ([11C]ACPC, 14i) from H11CN using the same chemistry.44 This on-column method greatly simplifies the synthesis procedure and enables the automation for routine synthesis of PET tracers. This compound, also known as cycloleucine, is a metabolically stable amino acid used for oncologic imaging.45 The preparation has been successfully accommodated to routine production of [11C]ACPC. The on-column synthesis method could generate [11C]ACPC in over 60% decay-corrected radiochemical yield in 40 min and >98% of radiochemical purity.
Although this chemistry produces a mixture of the D and L amino acids, Bjurling and colleagues were able to prepare optically pure L-amino acids by combining this method with enzymatic reactions. Specifically, they could convert racemic [1-11C]alanine (16) to [1-11C]pyruvate (17) with D-amino acid oxidase (D-AAO) and further convert 17 to optically pure L-[11C]alanine (16b) with glutamic-pyruvic transaminase (GPT)46. In addition, they could also use 17 to generate L-[11C]tyrosine (18) and L-[11C]3,4-dihydroxyphenylalanine (19, L-[11C]DOPA) with β-tyrosinase as well as L-[11C]tryptophan (20) and [11C]5-hyroxy-L-tryptophan (21) with tryptophanase. These products were generated in 45–60% decay-corrected radiochemical yield, >99% of enantiomeric purity and molar activity 0.41–2.00 GBq/μmol (0.011–0.054 Ci/μmol) in 45 – 50 min synthesis time.
In the same year, the same group also reported the synthesis of L-1-[11C]lactic acid (22) from the corresponding racemic 1-[11C]alanine (16) using similar enzyme catalyzed reactions (Fig 5).47 Racemic 1-[11C]alanine (16) was converted to 1-[11C]pyruvic acid (17) by D-amino acid oxidase (D-AAO)/catalase and glutamic-pyruvic transaminase (GPT). Finally, 17 was reduced to L-[1-11C]lactic acid (22) by L-lactic dehydrogenase (L-LDH). The total synthesis time was ca. 40 min and the decay-corrected radiochemical yields were ca. 70%. In 2006, Drandarov et al reported a nonenzymatic automated synthesis method of D- and L-[1-11C]lactic acid (22) from the same precursor (15).48 In this case, direct conversion from 16 to 22 under acidic conditions produced the two [11C]lactic acid enantiomers which were separated by chiral ligand-exchange chromatography. This fully automated radiosynthesis resulted in >80% decay-corrected radiochemical yield in 45min of synthesis time. The products were obtained in >99% radiochemical, chemical and enantiomeric purity. The molar activity at the end of the synthesis was ca. 370.30 GBq/μmol (10.8 Ci/μmol).
Fig. 5.
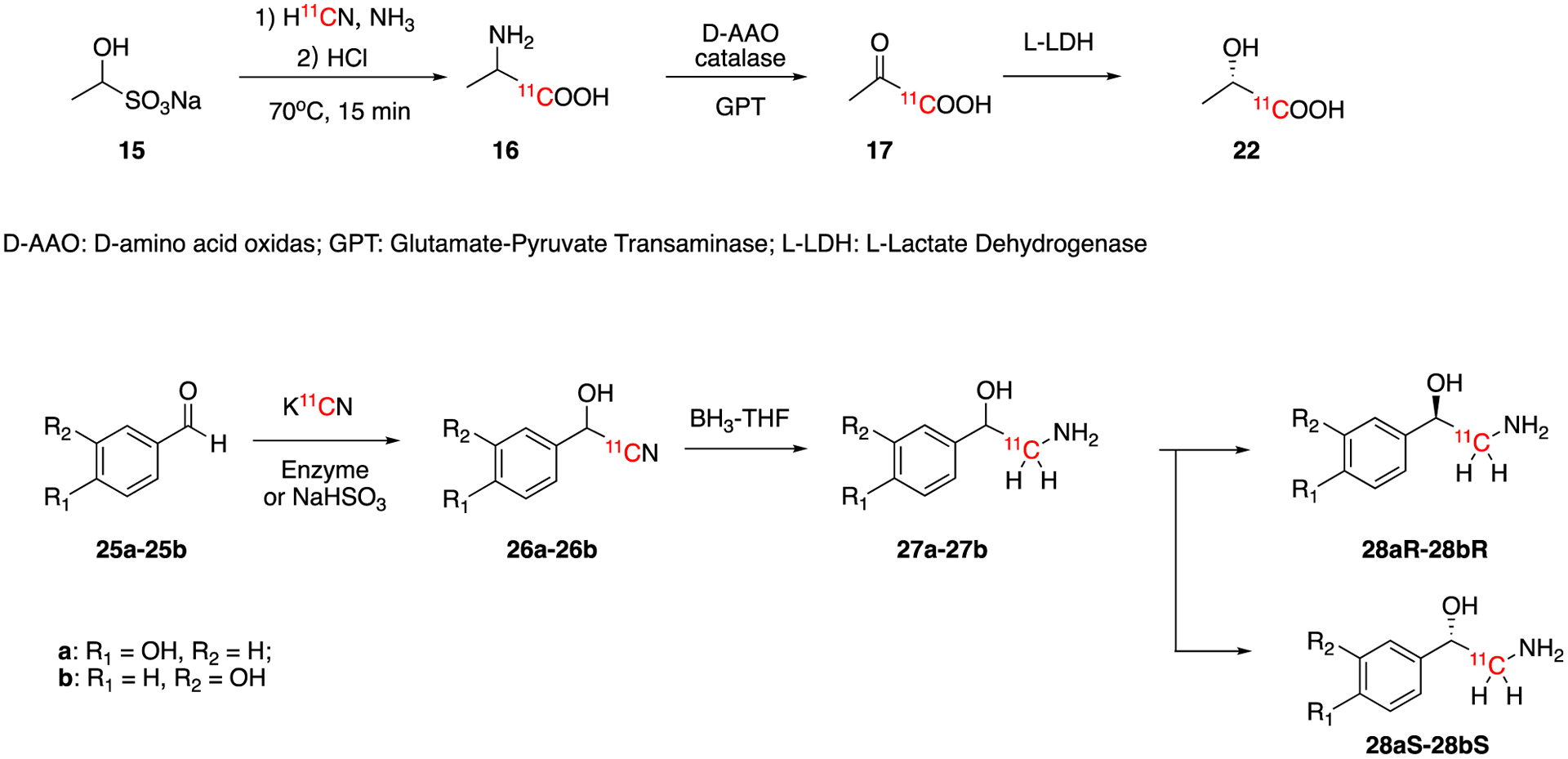
Synthesis of L-[1-11C]lactic acid (22), p-[11C]octopamine (28aR and 28aS), and m-[11C]octopamine (28bR and 28bS).
Octopamines are neurotransmitters from invertebrate animals used clinically as adrenergic drugs. The synthesis of p- and m-[11C]octopamine hydrochloride (27a and 27b) were reported in 1990 (Fig. 5).49 3- and 4-hydroxybenzaldehyde (25a and 25b) were reacted with sodium bisulfite and [11C]HCN to generate 26a and 26b. Alternatively, 26a and 26b were also produced by enzymatic conversion using mandelonitrile lyase. 26a and 26b were reduced using borane-THF to give final products (27a and 27b). The enantiomeric pure chiral isomer of p- and m-octopamine were separated using chiral column chromatography. This complex procedure which required ether extraction and drying under a stream of argon of the intermediate yielded the p- and m-octopamine in 1.2% and 2.3% non-decay-corrected isolated yields in ca. 40–60 min.
Enantiomerically pure [11C]tyrosine (Tyr, 32) and phenylalanine (Phe, 14d) were prepared in 2003 by Studenov et al using nonenzymatic conversions (Fig. 6).50 Starting from sodium bisulfite adducts of the aldehydes (29 and 11d), the corresponding 11C-labeled amino acids were produced by reacting with ammonia and 11CN−, followed by basic hydrolysis. The solid-phase extraction and chiral HPLC allowed individual enantiomers to be separated in 12–16% decay-corrected radiochemical yields within 40 – 45 min of total synthesis time. Enantiomeric excess of each isomer was >98% with the molar activity was 74–111 GBq/μmol (2–3 Ci/μmol).
Fig. 6.
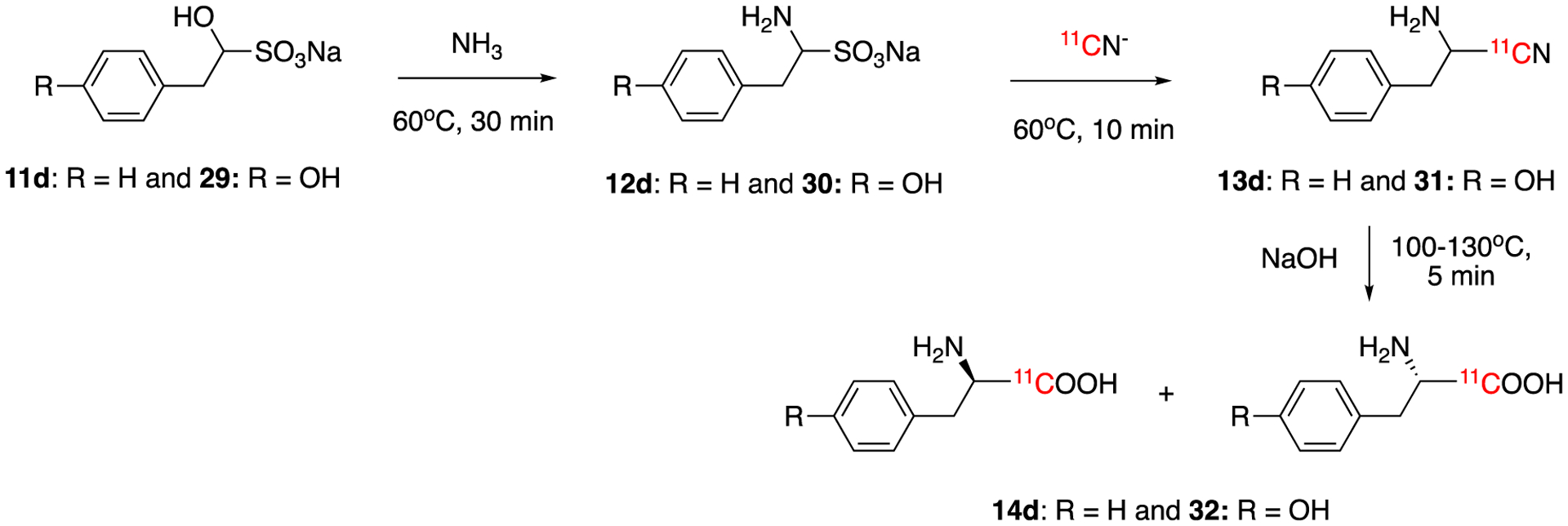
Synthesis of enantiomerically pure [11C]tyrosine (Tyr, 32) and phenylalanine (Phe, 14d).
Levetiracetam (LEV), (S)-α-ethyl-2-oxo-1-pyrrolidine acetamide, is a widely used epilepsy and central nervous system (CNS) disorder drug that binds to synaptic vesicle protein 2A (SV2A). [11C]levetiracetam 38 was reported in 2014 as PET tracer for SV2A expression (Fig. 7).51 The bisulfite adduct of propionaldehyde reacted with ammonia to give the precursor amino sulfonate 34, which further reacted with H11CN yielding 35 in aqueous solution. The intermediate 35 was converted to final product 38 by first reacting it with 4-chlorobutyryl chloride followed by hydrolysis or vice versa. Both approaches resulted in low radiochemical yield (~1.0%) of [11C]LEV within 1 hour of synthesis time. Given the low radiochemical yield, a modified multistep one-pot reaction based using H11CN addition to carbonyl addition reaction was later developed (see Section 4.2.2).
Fig. 7.
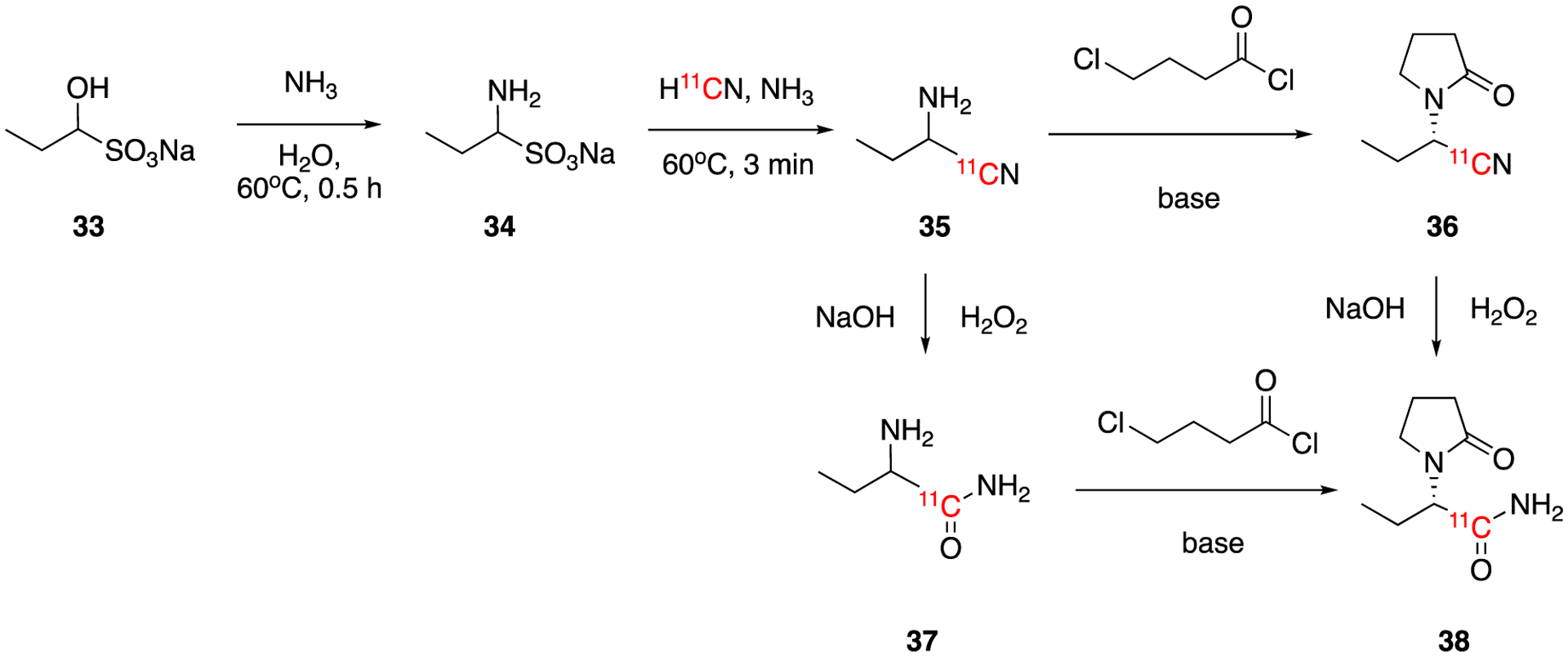
Synthesis of [11C]levetiracetam 38.
4.1.2. Triflate, -OTf, as leaving group
Triflate is an excellent leaving group for cyanation. However, its use in radiochemistry has been limited due the instability of many triflate precursors. Nevertheless, a triflate precursor was used to synthesize an important compound in radiochemistry: 2-deoxy-D-[1-11C]glucose (43).
It was the pioneer work by Sokoloff and colleagues using 2-deoxy-D-glucose labeled with carbon-14 the late 1960s and 70s that established the foundation for PET imaging. Following his autoradiography studies, Sokoloff sought to collaborate with radiochemists Alfted Wolf and Joanna Fowler at Brookhaven National Lab to develop derivatives of 2-deoxy-D-glucose that could be imaged noninvasively in humans. This led to the development of 2-deoxy-2-[18F]fluoro-D-glucose, [18F]FDG, the most important and widely used PET tracer today. Soon after the 11C analogue, 2-deoxy-D-[1-11C]glucose, was developed (Fig. 8).52–54. The reaction of precursor 40 with H11CN gave intermediate 2,3:4,5-di-O-isopropylidene-D-arabinononitrile (41). 2-Deoxy-D-[1-11C]glucose (42) was synthesized by the reduction-hydrolysis of intermediate 41. 2-Deoxy-D-[1-11C]glucose was obtained in 25–40% decay-corrected radiochemical yield with a synthesis time of 45–50 min according to different reports.55–56 Later in 1982, a remote semiautomated radiochemical synthesis system for 2-deoxy-D-[1-11C]glucose was reported.57 The system consists five flasks, two chromatography columns, a flow-through gamma detector, and a sterilization unit. A typical run could produce 0.74–1.30 GBq (20–35 mCi) of sterile, pyrogen-free 2-deoxy-D-[1-11C]glucose dose within a synthesis time of 50 min with radiochemical purity of greater than 98% and decay-corrected radiochemical yields of 25–30%.
Fig. 8.
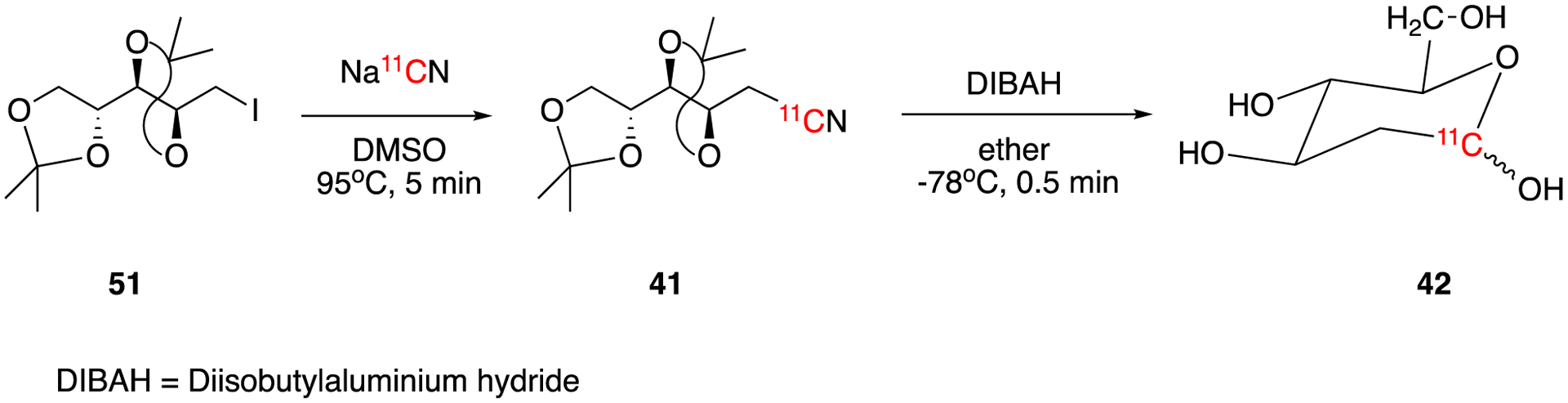
Synthesis of 2-deoxy-D-[1-11C]glucose (42).
4.1.3. Halides as leaving group
Halides, specifically iodide, bromide, and chloride, are among the most widely used leaving groups in organic chemistry. Many halide precursors are benchtop stable and widely available. Consequently, numerous 11CN− based nucleophilic substitution reactions have been developed. In the following paragraphs (figures 9–14), we review examples for making amino acids, sugars, aliphatic diamines, fatty acids and Pt(II) based anticancer drugs via 11C-cyanation of halide precursors.
Fig. 9.
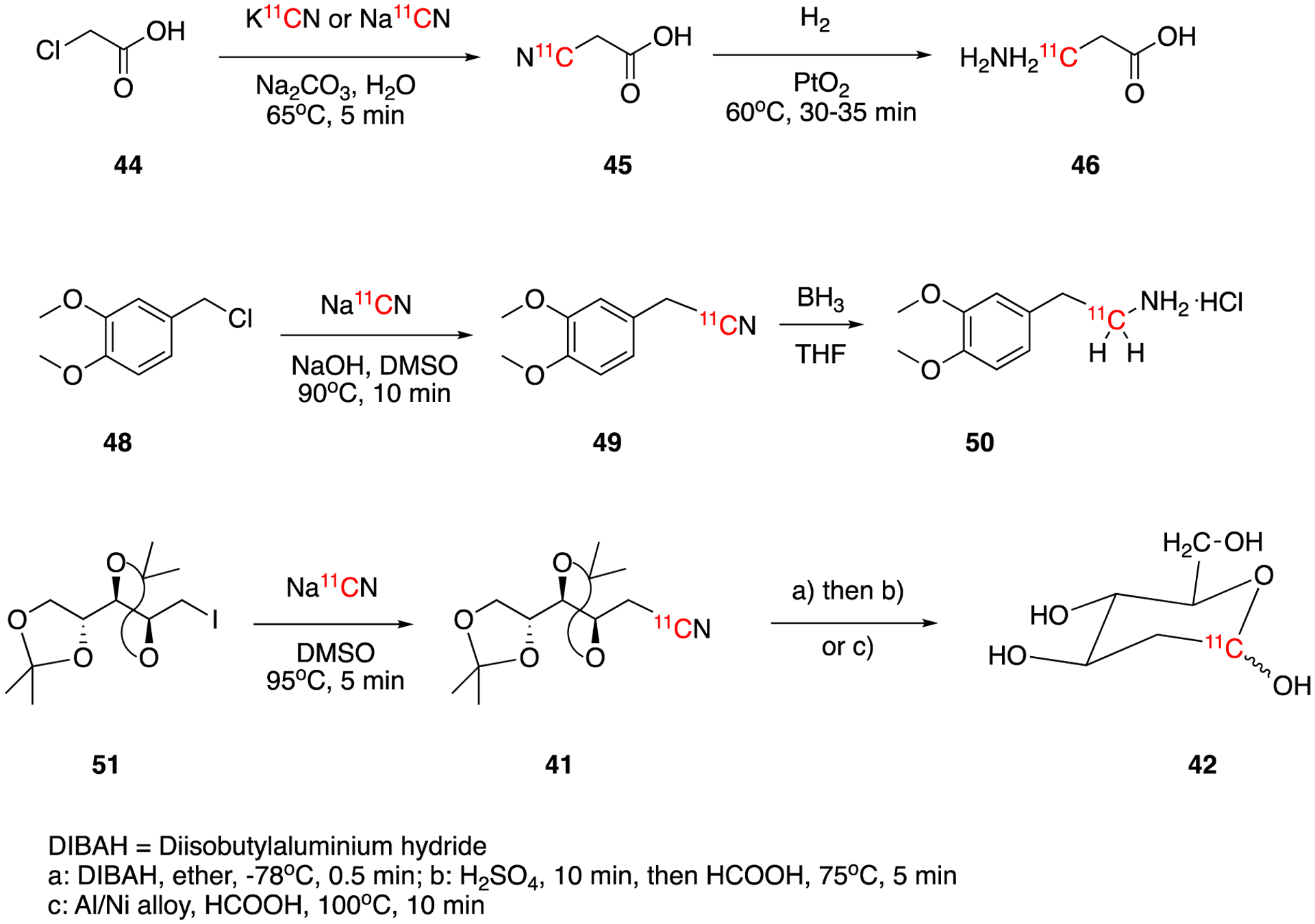
Synthesis of [3-11C]β-Alanine (46), [11C]3,4-dimethoxyphenethylamine (50), and 2-deoxy-D-[1-11C]glucose (42).
Fig. 14.
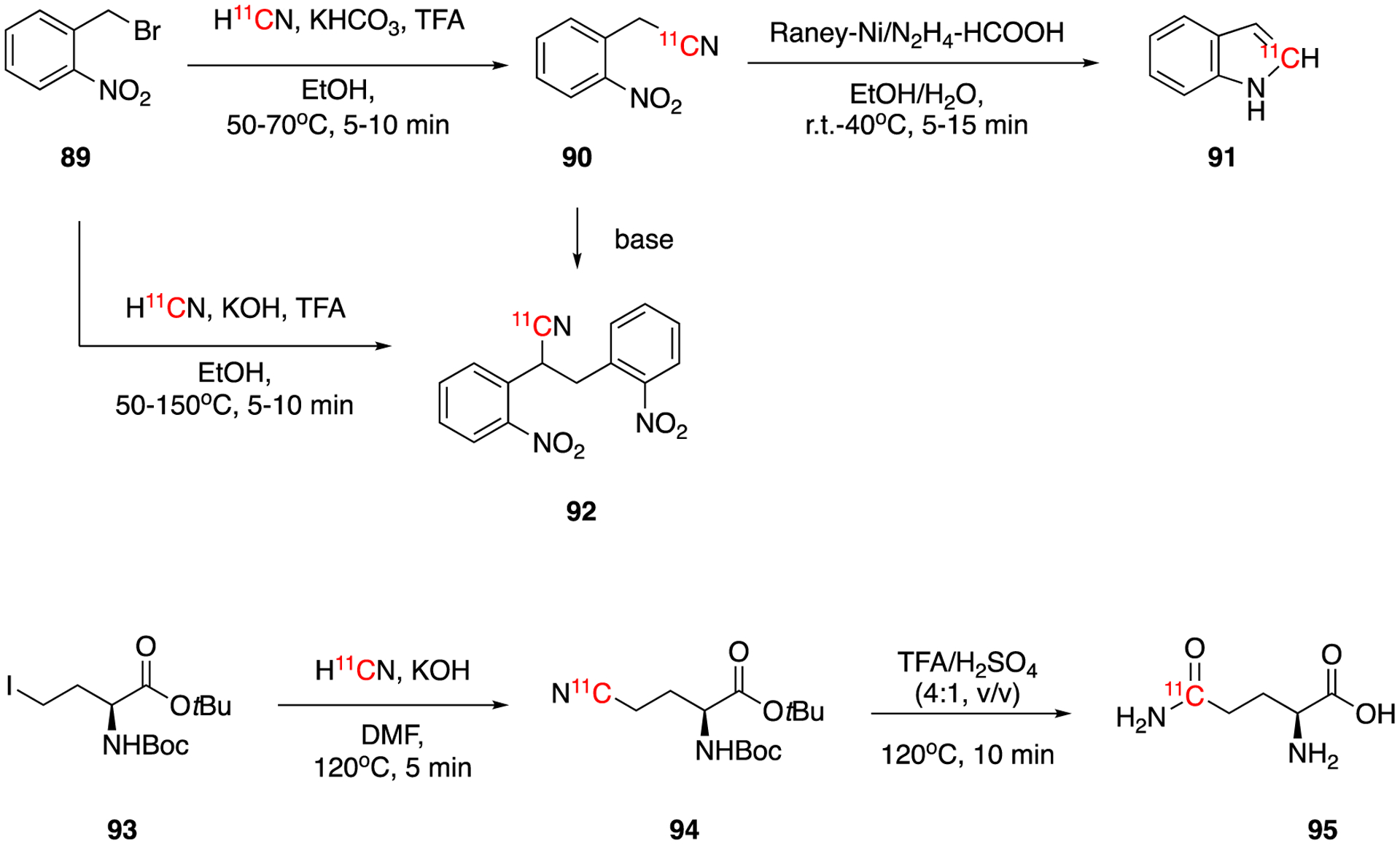
Synthesis of [2-11C]indole (91) and [5-11C]-glutamine (95).
[3-11C]β-Alanine (46) was first synthesized from the chloride precursor by Elias et al in 1972.58 2-Chloroacetic acid (44) was reacted with carrier-added Na11CN or K11CN yielding 2-isocyanoacetic acid (45) (Fig. 9). Subsequently, 45 was hydrogenated over PtO2 to generate [3-11C]β-alanine (46) in 35% chemical yield and 4.5% radiochemical yield.
In 1977, Mestelan et al reported the synthesis and biodistribution of [11C]3,4-dimethoxyphenethylamine (50).59 No-carrier-added H11CN was reacted with precursor 3,4-dimethoxybenzylchloride (48) in DMSO yielding intermediate 49, which was then reduced with borane to produce [11C]3,4-dimethoxyphenethylamine (50) in ca. 40% decay-corrected radiochemical yield.
In 1985, two research groups independently reported the optimized methods for 2-deoxy-D-[1-11C]glucose (42).60–61 Both papers describe the same new and bench stable labeling precursor, 1-deoxy-2,3:4,5-di-O-isopropylidine-1-iodo-D-arabitol (51). Stone-Elander et al used diethyl ether as solvent and diisobutylaluminium hydride (DIBAH) for the reduction of the intermediate (41).60 After the reduction, sulfuric acid and formic acid were used to hydrolyze the imine-aluminum complex and deprotect -OH groups. The decay-corrected radiochemical yield was ca. 20% the synthesis time around 50 min. van Haver et al purified the nitrile intermediate (41) by HPLC before reducing it with Raney nickel in formic acid61. The final product 2-deoxy-D-[1-11C]glucose (42) was isolated by ion exchange chromatography. The decay-corrected radiochemical yield was also around 20% and the synthesis time 50–55 min. The molar activity the final product (42) was 0.55–0.61 GBq/μmol (14.8–16.5 mCi/μmo) corresponding to a specific activity of 3.33–3.7 GBq/mg (90–100 mCi/mg).
In 1980, several 11C-labeled aliphatic diamines (54a-54c and 57a-57c) including putrescine (54a) and cadaverine (54b) were reported (Fig. 10).62 These diamines were synthesized from the reactions between the carrier-added Na11CN and the corresponding bromonitriles (52a-52c, n = 2, 3, and 4) or dibromoalkanes (55a-55c, n = 5, 6, and 7) followed by reduction with borane and acidification with hydrochloric acid. The decay-corrected radiochemical yields of these 11C-labeled aliphatic diamine were between 57% to 84%. Five years later, the synthesis of no-carrier-added [11C]putrescine was reported using a similar method.63 In this case, the decay-corrected radiochemical yield was 20% radiochemical yield and the synthesis time around 50–60 min. In 1989, the synthesis of L-[5-11C]ornithine was reported.64 The reaction of the bromo precursor 58 with K11CN gave intermediate 59, which was further reduced and deprotected yielding [5-11C]ornithine (6 in 25–40% decay-corrected radiochemical yield with the molar activity of >77.7 GBq/μmol (>2.1 Ci/μmol). The total synthesis and purification time was 50 min (EOB).
Fig. 10.
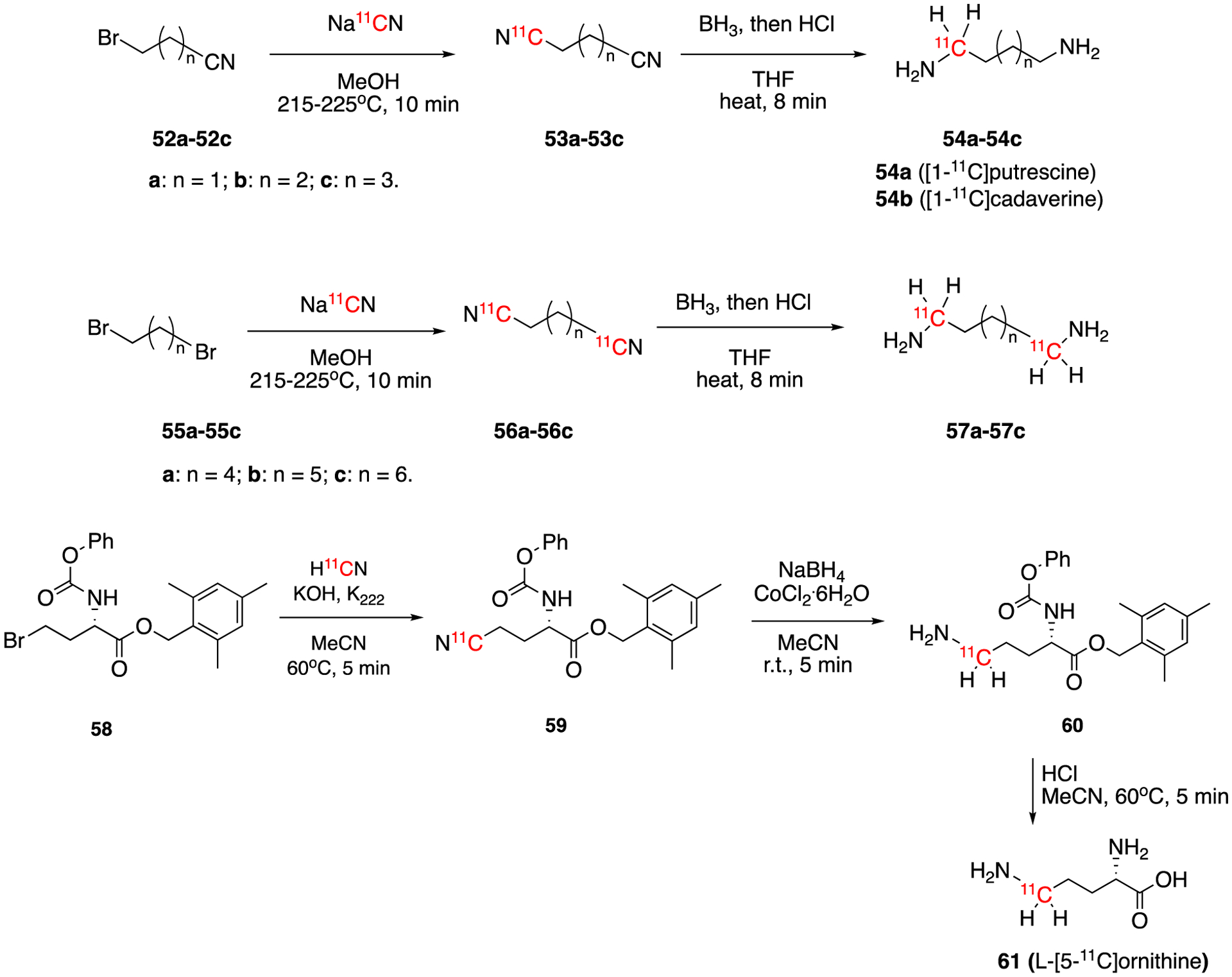
Synthesis of 11C labeled aliphatic diamines (54a-54c and 57a-57c) and [5-11C]ornithine (61).
The platinum(II) complex based anticancer drug, (ethylenediamine)(1-[11C]malonate)platinum(ll) ([11C]Ptenmal, [11C]EDMAL, or [11C]JM4O, 64), was also prepared using H11CN labeling.65 The labeling precursor bromoacetate (62) was reacted with H11CN to yield [11C]cyanoacetate (45), which was then converted to [11C]malonic acid (63) via hydrolysis. The resulting [11C]malonic acid was coordinated to Pt(II) diethylenediamine anion to give the desired final product 64 in 17–40% decay-corrected radiochemical yield within one hour after EoB. The molar activity was ca. 7.4 GBq/μmol (200 mCi/μmol).
Using a similar method, [1-11C]oxalic acid, 68 and [2-11C]2,3-dihydroxyquinoxaline, 69, were synthesized in 1993.66 Methyl chloroformate was reacted with no-carrier-added H11CN to give the intermediate [11C]methylcyanoformate, which was then converted to [1-11C]diethyl oxalate in ethanolic HCl solution. Subsequent hydrolysis of [11C]diethyl oxalate yielded [11C]oxalic acid (68). The time of synthesis was 6–7 min using combined microwave and thermal treatment. The radiochemical conversions of [11C]cyanide to [11C]diethyl oxalate and [11C]oxalic acid were ca. 80% and ca. 70%, respectively. The further reaction of phenylenediamine with [11C]oxalic acid gave [11C]-2,3-dihydroxyquinoxalin (69) in ca. 90% of radiochemical conversion.
Takahashi et al reported the synthesis of 11C-labeled multiple linear and branched fatty acid from H11CN in 1990 (Fig. 12).67 This method has several advantages over 11CO2 carboxylation via Grignard reaction such as not requiring absolutely anhydrous solvents, not being sensitive to ambient CO2 and being highly reproducible. The direct reaction of alkyl bromides (72a-72f) with K11CN in DMSO gave the corresponding [11C]alkyl nitriles intermediates (73a-73f). The hydrolysis of these intermediates with hydrochloric acid yielded the corresponding [11C]fatty acid (74a-74f). Four straight chain [1-11C]fatty acids (74a-74d) were synthesized in 70%-83% radiochemical yields and four branched chain ones (74e-74f) in 33%-42% decay-corrected radiochemical yields. The total synthesis times were 47–67 min from the EOB.
Fig. 12.
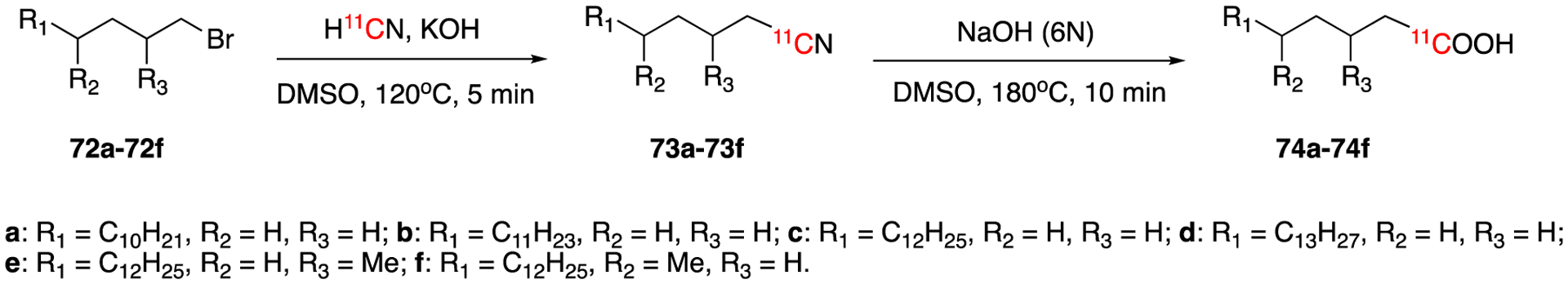
Synthesis of [1-11C]fatty acids (74a-74d).
The 11C-labeling of Busulfan, 1,4-bis(methanesulfonoxy)butane, a chemotherapy drug used for chronic myelocytic leukemia, was reported in 1991 (Fig. 13).68 The reaction of no-carrier-added NH411CN with 3-bromopropanol (75) followed by acid hydrolysis yielded the intermediate [1-11C]γ-butyrolactone, which was isolated using solid phase extraction and reduced to [1-11C]1,4-butanediol (78) with lithium aluminum hydride. Dimesylation of the 11C-labeled diol with methanesulfonyl chloride gave the final product [1-11C]busulfan (79) in 47% of decay-corrected isolated yield within a total synthesis and HPLC purification time of 65–75 min. Starting from no‐carrier‐added H11CN, several 11C labeled compounds, including ethyl [1‐11C]glycolate (82), [1‐11C]glycolic acid (83), [1‐11C]ethylene glycol (85), [2‐11C]2‐aminoethanol (84) were prepared by Thorell and colleages in 1994.69 The reaction of H11CN with chloromethyl pivalate generated the intermediate nitrile [11C]cyanomethyl pivalate (81), which was subsequently converted to 82-85 under the corresponding conditions shown in Fig. 13. The use of microwave reactions enabled the synthesis of the products in less than 3 min in ca. 90% conversions.
Fig. 13.
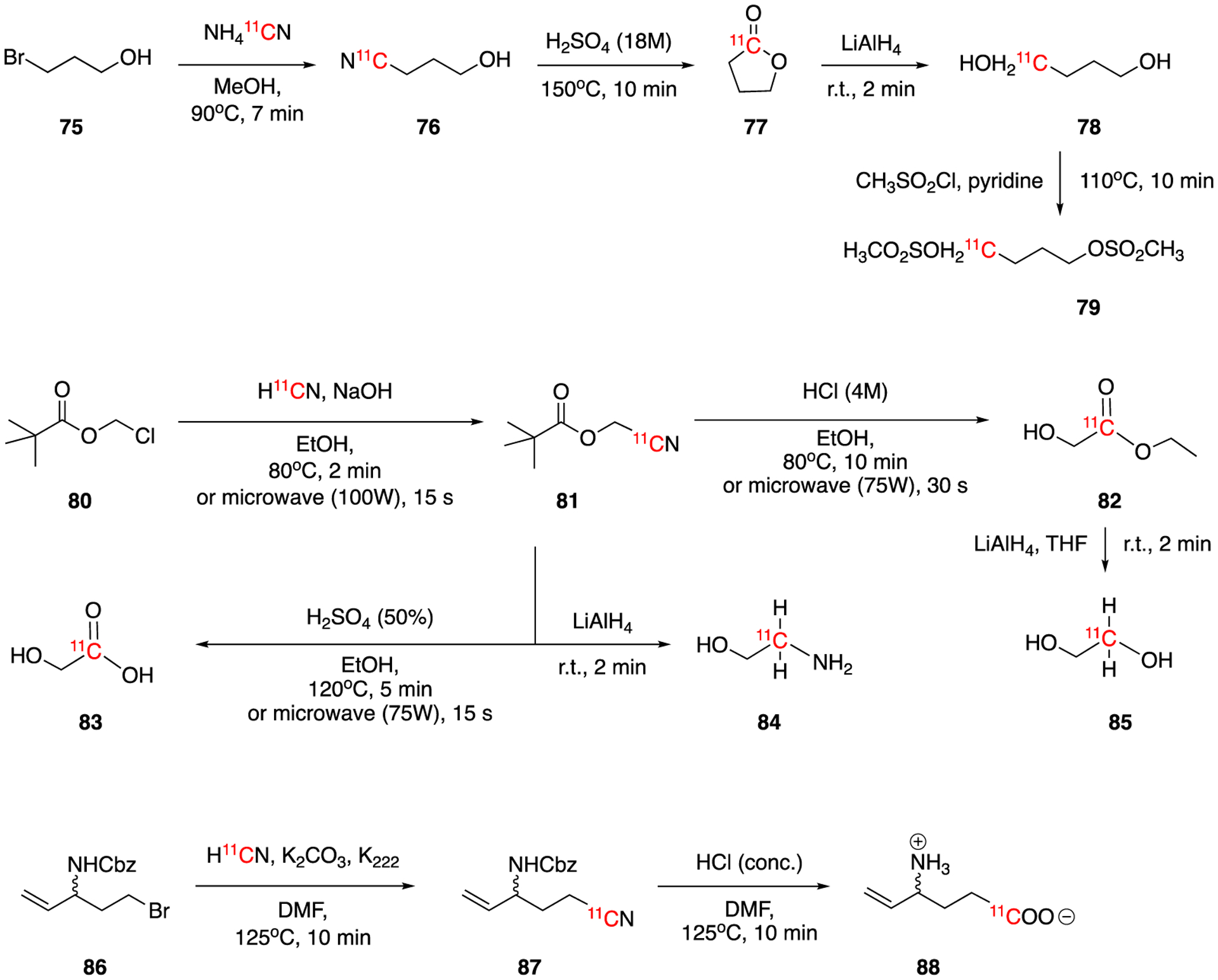
Synthesis of [1-11C]busulfan (79), ethyl [1‐11C]glycolate (82), [1‐11C]glycolic acid (83), [1‐11C]ethylene glycol (85), [2‐11C]2‐aminoethanol (84) and [1-11C]γ-vinyl-γ-aminobutyric acid (88).
The radiolabeling of γ-vinyl-γ-aminobutyric acid (GVG, Vigabatrin, 88) using H11CN was reported by Zhang et al in 2001.70 Vigabatrin inhibits the breakdown of GABA in the brain and it is used in the treatment of epilepsy and other psychiatric diseases. The precursor 5-bromo-3-(carbobenzyloxy)amino-1-pentene (86) was reacted with no-carrier-added H11CN, trapped with K2CO3 and K222, to generate intermediate 87. This compound was further hydrolysed to afford [1-11C]GVG (88) in 27 ± 9% (n=6, not optimized) of decay-corrected radiochemical yield. This one-pot, two-step radiosynthesis was completed with a synthesis time of 45min.
The synthesis of [11C]indole was first developed in 2015 using the nucleophilic cyanation method (Fig. 14).71 The 11C-cyanation of 2-nitrobenzyl bromide (89) with base-trapped H11CN gave 2-(2-nitrophenyl)-[1-11C]acetonitrile (90) as intermediate. The temperature and pH control of this reaction is crucial as high temperature or pH results in undesired side product 2,3-bis(2-nitrophenyl)-[1-11C]propanenitrile (92). The reductive cyclization of 90 catalyzed by Raney nickel yielded the desired product [2-11C]indole (91) in 21 ± 2.2% (n = 5, ranging from 18–24%) of decay-corrected radiochemical yield within 50–55 min of synthesis and HPLC purification time. The molar activity of [2-11C]indole was 176.12 ± 24.79 GBq/μmol (4.76 ± 0.67 Ci/μmol) (n = 5, ranging from 3.81–5.51 GBq/μmol).
11C-labeled L-glutamine was also prepared using a similar method.72 The iodination of L-homoserine gave the labeling precursor 93, which was then reacted with base-trapped H11CN to form intermediate 94. Hydrolysis of intermediate under acidic condition yielded the final product L-[5-11C]-glutamine (95) with a decay-corrected radiochemical yield of 63.8 ± 8.7 % (range from 51 to 74 %, n = 10), >90 % enantiomeric purity, and 7.03 ± 1.48 GBq/μmol (0.19 ± 0.04 Ci/μmol) of molar activity. The total synthesis and purification time was 40–50 min from the end of bombardment.
4.1.4. Carboxylate as leaving group
L-2,4-Diamino[4-11C]butyric acid is a natural nonproteinogenic amino acid that participates in amino acid metabolism and has been proposed as a potential tumor marker. L-2,4-Diamino[4-11C]butyric acid (DAB, 99a) was synthesized from H11CN using carboxylate as leaving group (Fig. 15).73 The enzyme (β-cyano-L-alanine synthase, BCAs) catalyzed reaction of carrier-added (0.1 μmol KCN) H11CN with O-acetyl-L-serine to give the intermediate β-[11C]cyano-L-alanine (97a). This was then reduced to L-2,4-diamino[4-11C]butyric acid (98% e.e.) in 30–40% of decay-corrected radiochemical yield, 96% of radiochemical purity and 32 min of synthesis time. The purified product was studied in vivo but it was less favorable for tumor imaging than other amino acids.
Fig. 15.
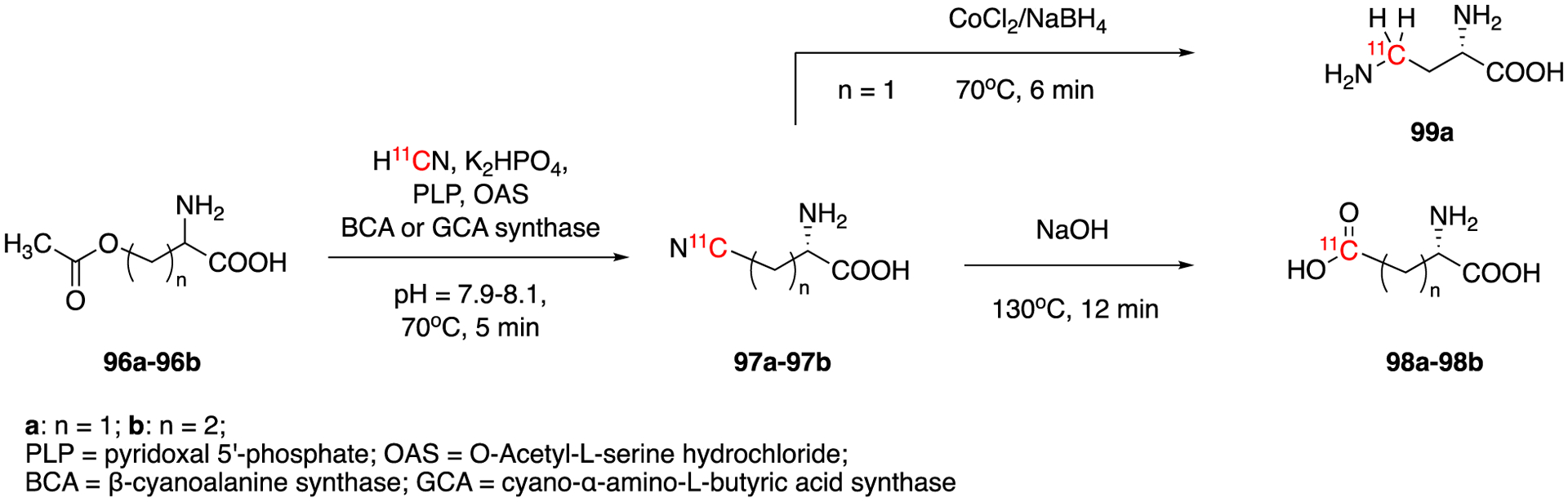
Synthesis of L-2,4-Diamino[4-11C]butyric acid (DAB, 99a), L-[4-11C]aspartate (98a) and L-[5-11C]glutamate (98b).
In 2001, the same group reported the preparation of L-[4-11C]aspartate (98a) and L-[5-11C]glutamate (98b) using a similar method.74 The intermediates, β-[11C]cyano-L-alanine (97a) and γ-[11C]cyano-α-amino-L-butyric acid (97b) were obtained by the reaction of O-acetyl-L-serine and O-acetyl-L-homoserine with carrier-added H11CN (0.1 μmol) using O-acetyl-L-serine sulfhydrylase and O-acetyl-L-homoserine sulfhydrylase as enzymes. The final products 98a and 98b were generated by alkaline hydrolysis of corresponding intermediates in 65% and 55% decay-corrected radiochemical yields. The molar activitiy was ca. 29.6 GBq/μmol (0.8mCi/μmol) and the enantiomeric purities were greater than 97.5%. The total reaction times were 50–55 min.
4.1.5. Amine as leaving group
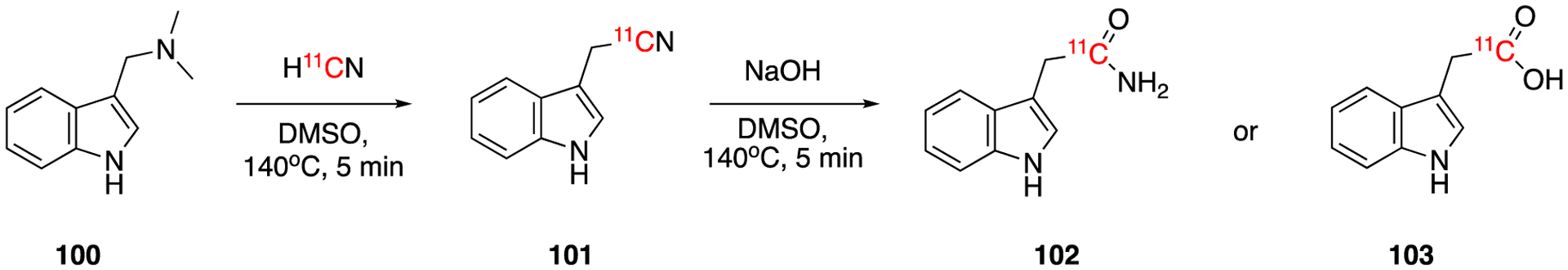
In 2011, 11C labeled 3-indolylacetic acid ([11C]IAA), the most common naturally occurring plant hormone, was synthesized by Reid et al using the reaction of H11CN with a precursor bearing a dimethyl amine leaving group (Fig. 16).75 The reaction of gramine (100) with H11CN gave the intermediate 3-indolyl[1-11C]acetonitrile (101). This intermediate was purified using HPLC and then hydrolysed with NaOH solution to give corresponding amide (102) side product and carboxylic acid (103). 3-indolyl[1-11C]acetic acid (103) was isolated in 28% of overall decay-corrected radiochemical yield after a second HPLC purification and 68 min of synthesis and purification time. The molar activity was 25.9 GBq/μmol (0.7 Ci/μmol). Three years later, the same group lead by Joanna Fowler reported an improved synthesis procedure of the same compound.76 This method utilized a streamlined semi-remote controlled production system which resulted in 61.0 ± 0.3 % (n = 10) decay-corrected radiochemical yield and 82.51 ± 36.26 GBq/μmol (2.23 ± 0.98 Ci/μmol) molar activity. The total synthesis time was 81.8 ± 3.0 minutes (n = 10). Finally, in 2015, they reported a one-pot synthesis procedure. To accomplish that they changed the solvent to tetraethylene glycol and both cyanation and hydrolysis reactions were performed without an intermediate purification step. The total synthesis time was reduced to 55 min. The decay-corrected radiochemical yield was 33 ± 9.5% of and the molar activity 47.36 ± 12.58 GBq/mmol (1.28 ± 0.34 Ci/mmol).77
Fig. 16.
Synthesis of 3-indolyl[1-11C]acetic acid (103).
4.2. Nucleophilic addition reaction
The nucleophile 11CN− can also attack electron-deficient centers of unsaturated compounds (e.g. C=O, electron-withdrawing group bearing C=C bond) leading to nucleophilic additions.
4.2.1. Bucherer-Strecker reaction
Bucherer-Strecker technique is a conventional method used for amino acid synthesis starting from cyanide. The reaction first generates the hydantoins, which are further hydrolyzed to generate the corresponding amino acids. Although amino acid synthesis by Bucherer-Strecker technique requires long reaction times, the reaction conditions can be optimized to shorten the reaction time making it suitable for 11C chemistry. (i.e., by increasing reaction temperature and pressure). 11C-labeled amino acids are of interest in oncologic imaging.
The Bucherer-Strecker technique was first used by Hayes et al to produce [11C]1-aminocyclopentanecarboxylic acid synthesis in 1976 (Fig. 17).78 In this report, 11C was produced by using a boron trioxide (B2O3) target as described in Section 3. H11CN was trapped by NaOH and then introduced into the reaction mixture containing clclopentanone, (NH4)2CO3, NH4Cl, and KCN. The reaction of carrier-added H11CN/KCN with cyclopentanone (105) generated the corresponding hydantoin intermediate which was subsequently hydrolyzed to [1-11C]1-aminocyclopentanecarboxylic acid (14i) (also known as cycloleucine). The decay-corrected radiochemical yield was ca. 40% with 1h synthesis time.
Fig. 17.
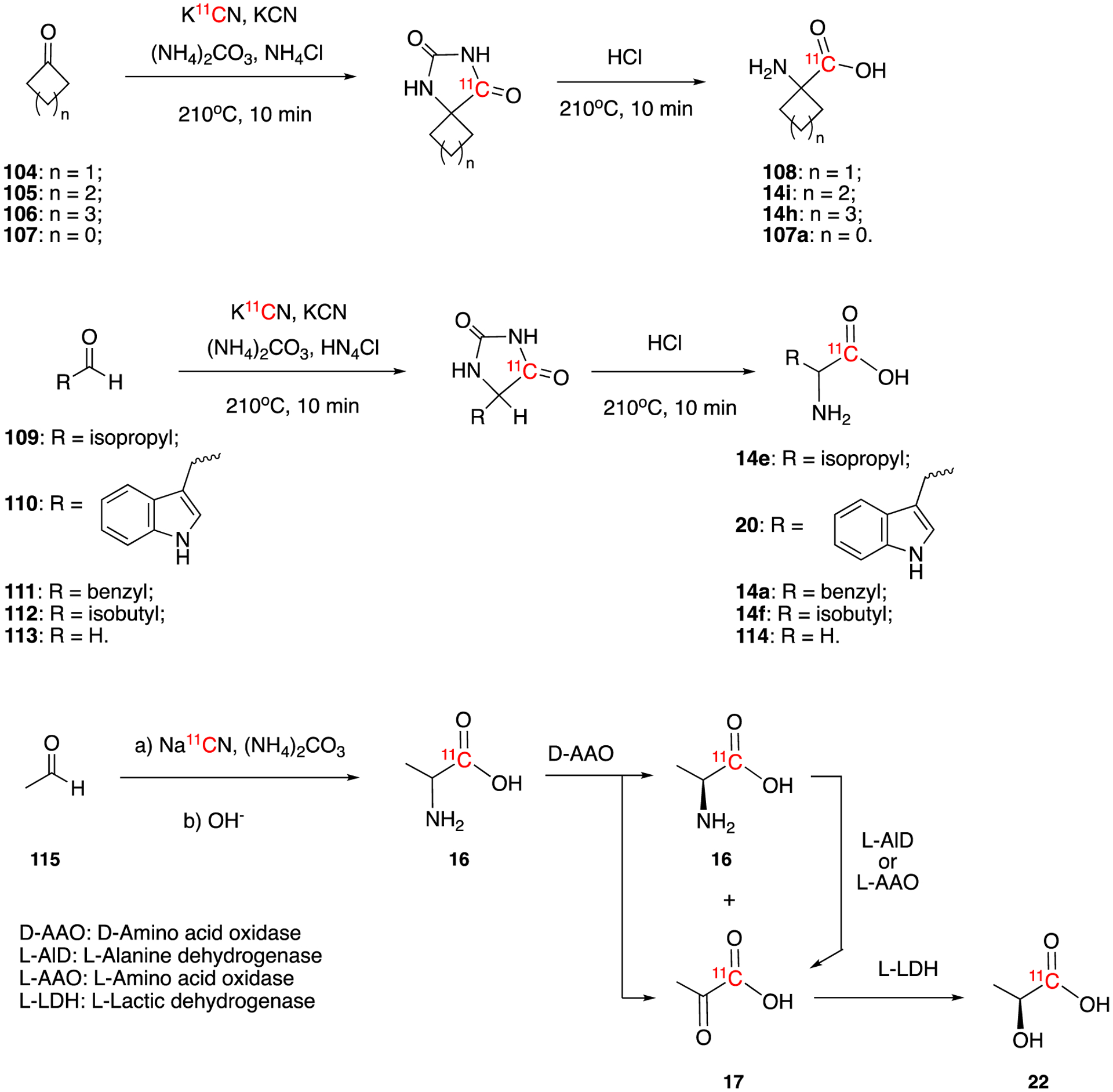
Synthesis of [11C]α-amino acid via Bucherer-Strecker reactions and the synthesis of L-[1-11C] lactic acid (22).
Later in 1978, the same group optimized and expanded the Bucherer-Strecker technique to several 11C-labeled neutral and basic α-amino acids. Starting from different cyclic ketones (104-107) they were able to generate the corresponding cyclic amino acids (108, 14i, 14h, 107a). Using this method they also reported the synthesis of DL-valine (14e), and DL-tryptophan (20).79 All of these reactions required ca. 20 min for synthesis and an 25 min for purification. Decay-corrected radiochemical yields ranged from 10–70%. In 1985, Sambre et al reported the routine production of 1-[11C]aminocyclopentanecarboxylic acid (14i) for medical use.80 This system allowed them to carry out 30 preparations per year with 60±2% of decay-corrected radiochemical yield, which is significantly improved compared to the original report.
In 1981, Casey et al reported a method for the preparation of optically pure α-amino acid (D- and L-[11C]phenylalanine).81 The enatiomeric mixture of 11C-labeled DL-Phenylalanine (14a, 13.88 GBq (375 mCi)) was produced in 40 minutes and 65% decay-corrected radiochemical yield using the Bucherer-Strecker reaction. Subsequently, optically pure D-phenylalanine (yield 0.703 GBq (19mCi)) and L-phenylalanine (yield 1 GBq (27 mCi)) were separated by oxidative deamination using immobilized L- and D-amino acid oxidase in 35 min, respectively. Comparison of the uptake of the two amino acids in the pancreas (relative to liver) suggested that the natural L-amino acid accumulated over time whereas the other did not.
The synthesis of optically pure L-[11C]leucine was reported in 1983 by Barrio et al.82 Starting from the corresponding aldehyde 112, the enantiomeric mixture DL-[11C]leucine 14f was synthesized using a modified Bucherer-Strecker reaction. The L-isomer was obtained by using a D-amino acid oxidase/catalase enzyme complex immobilized on a Sepharose support with a decay-corrected radiochemical yield of 25%. The synthesis was fully automated and the production time was 30–40 min. As reviewed in Section 4.1.1, compound class 14, were also produced via cyanation of sodium bisulfite precursors with higher radiochemical yields.
[11C]α-aminoisobutyric acid ([11C]AIB, 107a) was also prepared by Bucherer-Strecker reaction.83 Since AIB is a non-metabolizable amino acid, it is a good candidate to study the amino acid transport in vivo without interference by radiolabeled metabolic products. Starting from acetone, ammonium carbonate, and carrier-added K11CN, [11C]AIB was prepared in 35 – 60 % decay-corrected radiochemical yields in 70–80 min of synthesis and purification time. The carrier-added synthesis gave the molar activity of 11.1 GBq/mmol (0.3 Ci/mmol).
Applying the same method and starting from formaldehyde, ammonium carbonate and H11CN, [11C]glycine (114) was prepared. The synthesis and chromatographic purification time was 30–35 min and the decay-corrected radiochemical yield was 35%. [11C]glycine was used together with [68Ga]EDTA and L-[methyl-11C]methionine for imaging anaplastic astrocytoma using PET.84
In 1984, L-[1-11C]lactic acid 22, was prepared by addition of 11CN− to acetaldehyde.85 This reaction produced racemic DL-[11C]alanine 16 which was then converted to [1-11C]pyruvic acid 17 and L-[1-11C]lactic acid 22 by enzymatic transformation as described in Section 4.1.1 (Fig. 5). The synthesis and purification time was 35–40 min and the decay-corrected radiochemical yields were 25% for L-[1-11C]lactic acid, 29% for [1-11C]pyruvic acid, and 20% for L-[1-11C]alanine). Finally, in 1991, Fissekis et al reported a remote-control apparatus for the routine synthesis of [11C]carboxy-labelled α-amino acids using modified Bucherer-Strecker method.86 The apparatus has been used for the synthesis of several α-amino acids including DL-valine, DL-leucine, DL-isoleucine, α-amino-isobutyric acid, and 1-amino-cyclopentane-1-carboxylic acids. The apparatus allows the sequential batch-production of two different ammo acids in average 1.48–2.59 GBq (40–70 mCi) with average molar activates of 92.5 GBq/mmol (2.5 Ci/mmol).
4.2.2. Addition to C=O
The nucleophilic additions of 11CN− to carbonyl compounds (mainly aldehydes) give cyanohydrins, which can be further converted to aldehydes (sugars), amines, and carboxyl acids with one carbon atom chain extended.
The Kiliani-Fischer synthesis is a widely-used method of extending the carbon atom chain of pentopyranoses. In this reaction, the addition of cyanide to a pentopyranose and subsequent hydrolysis yields the corresponding hexopyranose.
In 1985, the synthesis of [1‐11C]‐D‐glucose (118) and [1‐11C]‐D-mannose (119) from carrier added Na11CN was reported by Shiue and Wolf (Fig. 18).87 H11CN was trapped in NaCN/AcOH solution at pH = 8 to give carrier added Na11CN. This was further reacted with D‐arabinose (116) yielding intermediate [1‐11C]‐aldononitrile (117). [1‐11C]‐D‐glucose (118) and [1‐11C]‐D-mannose (119) were obtained by reducing 117 with Raney alloy in 30% formic acid. The ratio of [1‐11C]‐D‐glucose (118) and [1‐11C]‐D-mannose (119) was pH dependent and these two products could be separated by HPLC. The decay-corrected radiochemical yield of 118 was ca. 15% with a total synthesis and purification time of 70 min. Using a similar method, [1‐11C]‐D‐galactose (122) which was synthesized from D-lyxose (120) with a decay-corrected radiochemical yield of ca. 30% and a synthesis and purification time of 70 min.
Fig. 18.
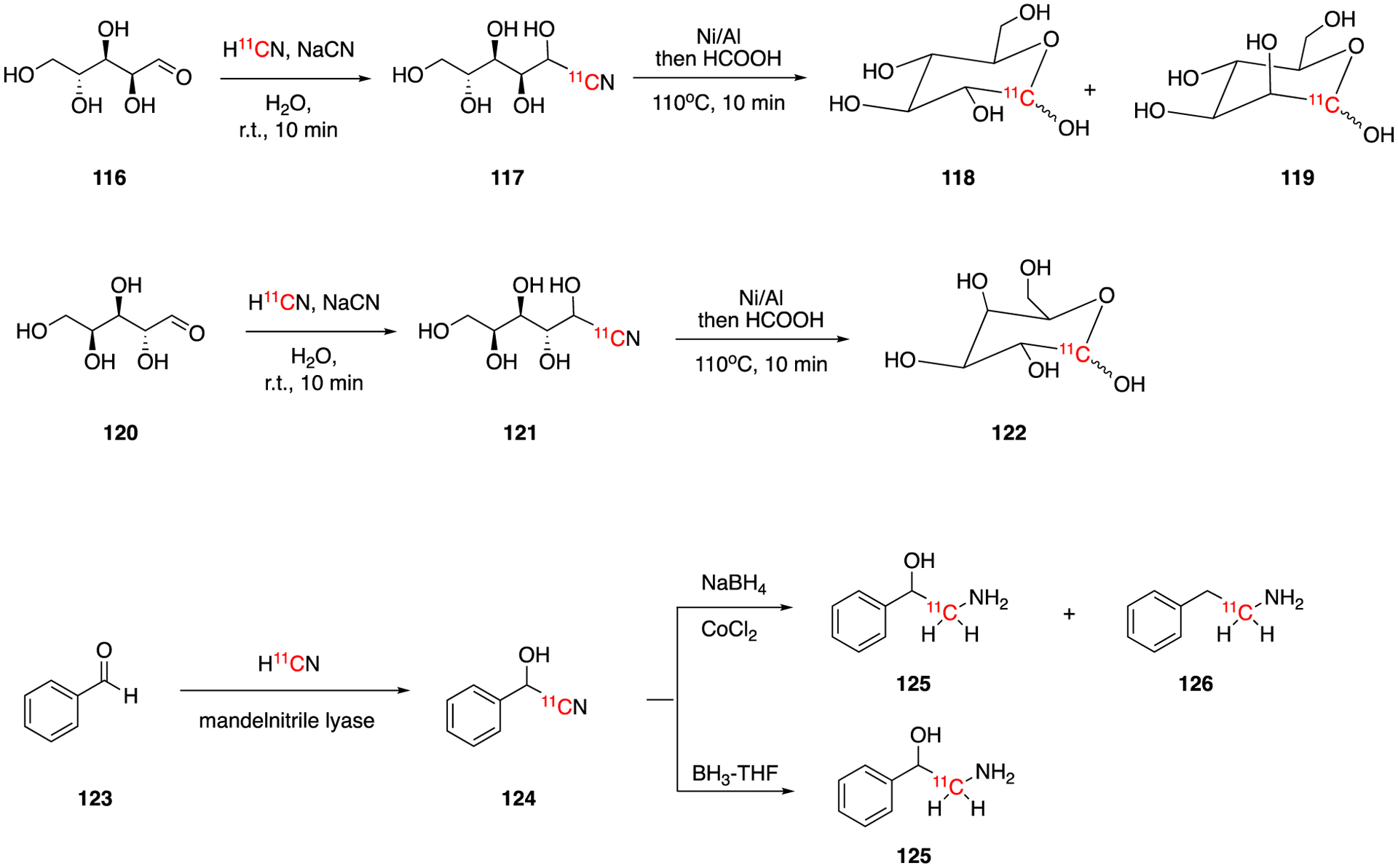
Synthesis of [1‐11C]‐D‐glucose (118), [1‐11C]‐D-mannose (119), [1‐11C]‐D‐galactose (122), and [11C]phenylethanolamine (125).
In 1988, [11C]phenylethanolamine (125) was prepared using no-carrier-added H11CN.88 The enzyme-catalyzed addition of H11CN to benzaldehyde gave the intermediate [11C]cyanohydrin (124), which was then reduced to produce [11C]phenylethanolamine (125) in 2–4% of radiochemical yields. The synthesis and purification took 50–60 min and the enantiomeric purities were dependent on the reducting reagent. NaBH4-CoCl2 reduction gave 60% e.e. while borane-THF reduction gave 80% e.e.
In 1982, D-[1-11C] glucopyranose (133) and D-[1-11C] galactopyranose (134) were also prepared using Kiliani-Fischer synthesis (Fig. 19).89 The reaction of D-arabinopyranose (127) / D-Lyxopyranose (128) with K11CN in the presence of base gave 131/132 via intermediates 129/130. 131/132 were further reduced by diborane yielding D-[1-11C] glucopyranose (133) and D-[1-11C] galactopyranose (134), which were purified by HPLC. The radiochemical yields of D-glucopyranose (133) and D-[1-11C] galactopyranose (134) were 17% and 50%, respectively. The total synthesis and purification time of 133/134 was ca. 60 min. In 1994, a fully automatic synthesis system of [1-11C]labelled aldoses, such as [1-11C]-D-glucose, and [1-11C]galactose, based on modified Kiliani-Fischer method was described.90 The system described had the ability of supplying reagents, performing the synthesis, purifying the 11C-labeled aldose, and preparing an injectable solution. In a typical run, 0.048 GBq (1.3 mCi, 11.9% of decay-corrected radiochemical yield) of [1-11C]-D-glucose was obtained in 49 min
Fig. 19.
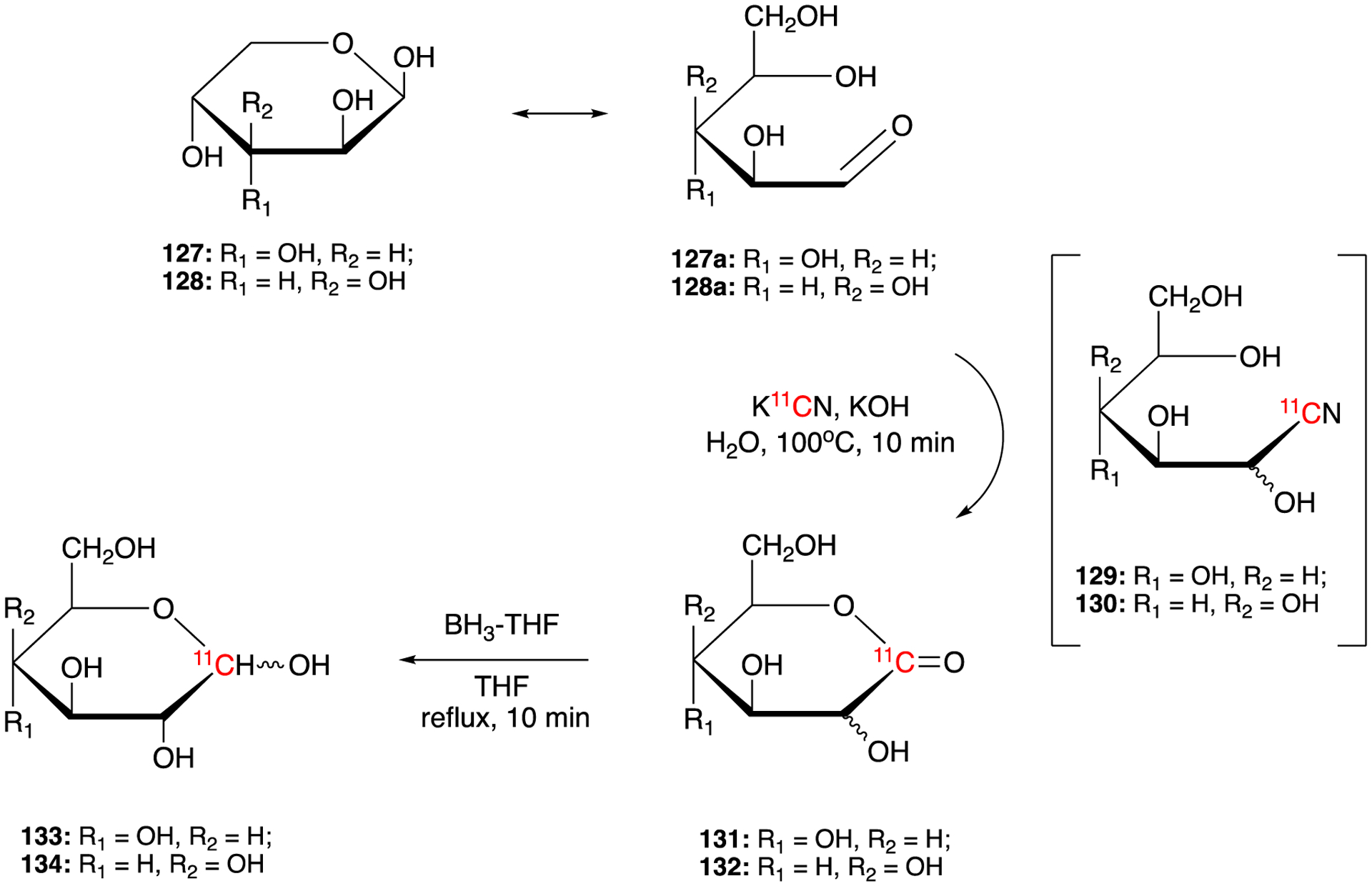
Synthesis of D-[1-11C] glucopyranose (133) and D-[1-11C] galactopyranose (134).
In Section 4.1.1, the radiochemical synthesis of [11C]levetiracetam (38) via nucleophilic substitution of bisulfite by [11C]cyanide was described (Fig. 20). Since this method gave low radiochemical yield an improved method of synthesis based on the nucleophilic addition of [11C]cyanide to carbonyl was developed. In this method, propionaldehyde was reacted with ammonia and H11CN to yield intermediate 35, which was then reacted with 4-chlorobutyryl chloride and followed by hydrolysis to give [11C]levetiracetam. After purification by chiral HPLC the decay-corrected radiochemical yield was 8.3 ± 1.6% (n = 8) in 50 ± 5.0 min, which is significantly better than the previous method (< 1% radiochemical yield).
Fig. 20.
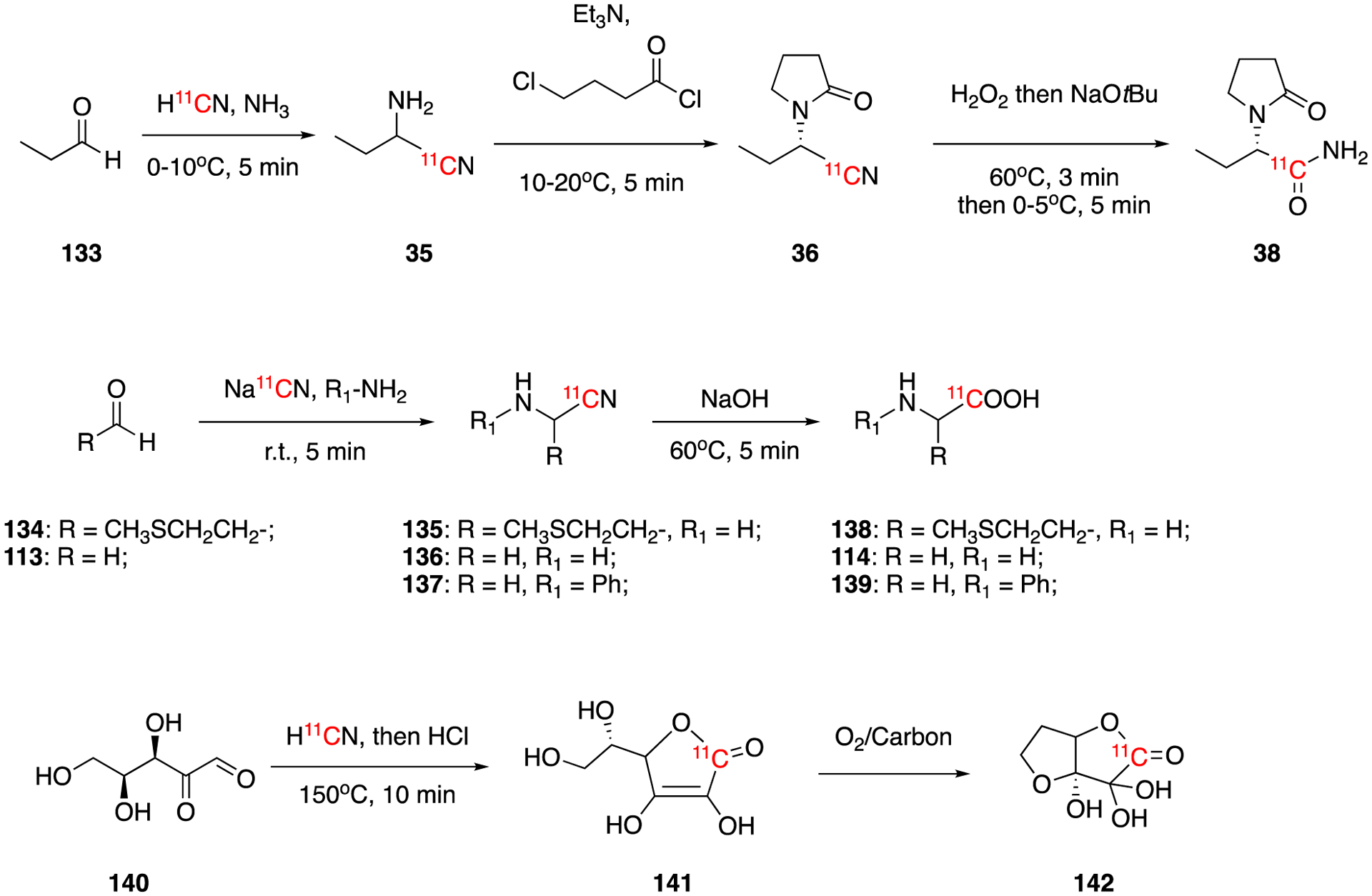
Synthesis of [11C]levetiracetam (38), [11C]methionine (138), [11C]glycine (114), [11C]-N-phenylglycine (139), [11C]ascorbic (141) and [11C]dehydroascorbic acid (142).
Nucleophilic addition of [11C]cyanide to carbonyl has also been applied in synthesis of 11C-labeled amino acids: [11C]methionine (138), [11C]glycine (114), and [11C]-N-phenylglycine (139).91 Using this method, the non decay-corrected radiochemical yields were 5%, 14%, and 2%, respectively. The molar activities were 46.62 GBq/μmol (1.26 Ci/μmol), 55.5 GBq/μmol (1.5 Ci/μmol), and 569.8 GBq/μmol (15.4 Ci/μmol), respectively. The protocol was fully automated using a commercially available radiochemistry synthesis module.
In 2016, the endogenous redox pair, [11C]ascorbic acid (vitamin C) and [11C]dehydroascorbic acid, were synthesized and used for sensing reactive oxygen species using PET.92 Aqueous solution of L-xylosone (140) was reacted with no-carrier-added or carrier-added H11CN/KCN to generate the addition cyanohydrin, which underwent cyclization upon reaction with HCl at 150°C to give the desired product [11C]ascorbic acid (141). [11C]ascorbic was completely oxidized to [11C]dehydroascorbic acid (142) in 10 min by bubbling O2 in the presence of activated charcoal. [11C]ascorbic (141) was produced in 14.3 ± 10.4% of decay-corrected radiochemical yield and 9.88 ± 5.48 GBq/μmol (267 ± 148 mCi/μmol) molar activity using no-carrier-added H11CN and in 29.7 ± 8.8 % to 45.4 ± 1.2 % of decay-corrected radiochemical yields and 0.148 ± 0.044 to 0.33 ± 0.137 GBq/μmol (4.0 ± 1.2 to 9.0 ± 3.7 mCi/μmol) of molar activity using carrier added H11CN.
4.2.3. Addition to C=C (Michael addition)
[11C]Cyanide can also react with the electron-withdrawing groups bearing α,β-unsaturated compounds via Michael addition reactions. These methods have been employed in the synthesis of [1-11C]putrescine (144) and γ-amino[4-11C]butyric acid (GABA, 146) (Fig. 21).
Fig. 21.
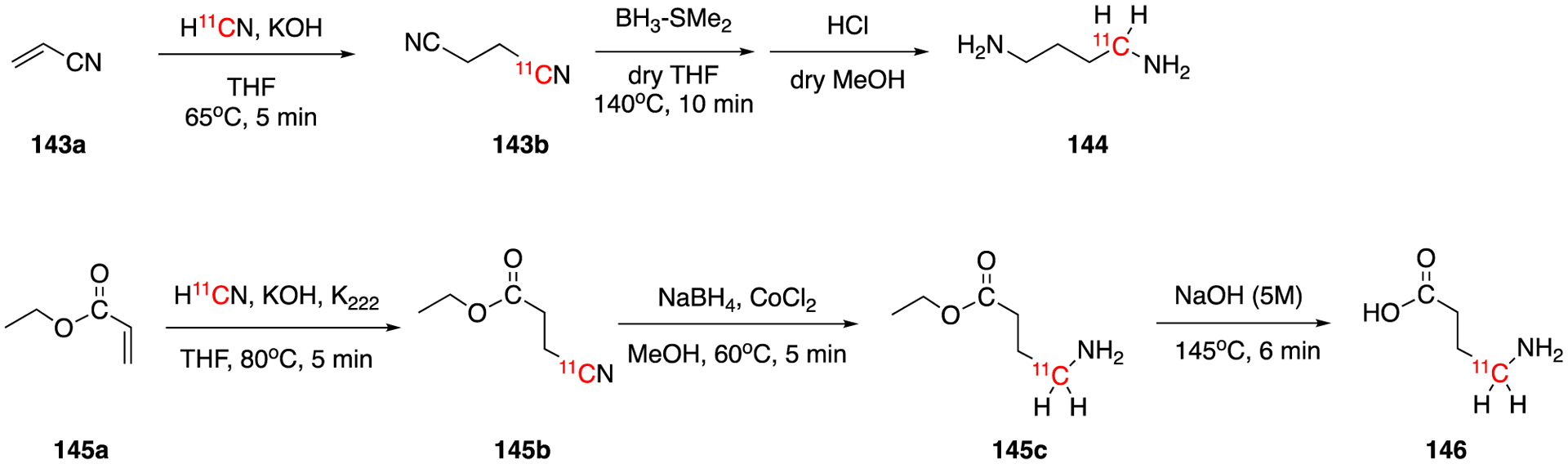
Synthesis of [1-11C]putrescine (144) and γ-amino[4-11C]butyric acid (GABA, 146).
In 1985, a synthetic method of no-carrier-added [1-11C]putrescine (144) using Michael addition was reported by McPherson et al.63 No-carrier-added H11CN was trapped in 1% KOH solution and reacted with acrylonitnile (143a) for 5 min at 65°C to give the intermediate succinonitrile (143b). This was then reduced to [1-11C]putrescine (144) using the same method reported previously (Section 4.1.3) in 20% radiochemical decay-corrected yield. The synthesis time was around 50 min.
In 1990, a silica gel based H11CN trap was developed by Somawardhana et al for the synthesis of base and moisture sensitive 11C-labeled compounds.93 The trap consisted on either coating silica gel particles directly with the substrate or adding KOH to the silica gel. The later version was employed for the synthesis of succinonitrile (143b) synthesis. After trapping the [11C]HCN in the silica-KOH, acrylonitrile was added to the column, the column was sealed and placed in a boiling water bath. Using this method the reaction time was reduced from 5 to 2 min and the decay-corrected radiochemical yield was increased to 73.9% to 84.7%. One year later, the same research group applied this method in the synthesis of [1-11C] putrescine.94 This method yielded [1-11C] putrescine in 53±4% decay-corrected radiochemical yield in 40 min of synthesis and purification time. The moisture-free semi-automated method allowed better radiochemical yields (53% vs. 20%), fewer side products and shorter synthesis time (40 vs. 50 min) compared to the previous report.63 It will be interesting to see whether this method can be applied to other reactions with [11C]HCN.
A one-pot synthesis of γ-amino[4-11C]butyric acid (GABA, 146) starting with H11CN based on Michael addition reaction was first reported by Antoni and Långström in 1989.95 No-carrier-added H11CN was first trapped in tetrahydrofuran/potassium hydroxide in the presence of Kryptofix 2.2.2. and then reacted with ethyl acylate to give intermediate 145b. This was further selectively reduced and hydrolyzed to yield [4-11C]GABA in 60–65% decay-corrected radiochemical yield. The total synthesis and purification time was ca. 40 min. Similar to previously described nucleophilic substitution methods, Michael addition can result in aliphatic amines although it may be less favorable for generating secondary and tertiary amines.
4.3. Ring opening reaction
The attack of the electrophilic center in aziridines by H11CN leads to ring-opening reaction, as reported by Gillings et al in 2001.96 In this example, the reaction of N-(tert-butoxycarbonyl)aziridine-2-isopropyl carboxylate (147) with H11CN gave intermediate DL-[4-11C]β-cyanoalanine (148). This was subsequently hydrolyzed or reduced-hydrolyzed to generate DL-[4-11C]asparagine (149a), DL-[4-11C]aspartic acid (149b), and DL-2,4-diamino[4-11C]butyric acid (149c) in 30–40% decay corrected radiochemical yield within 30 min. The racemization occurred during the ring-opening of racemic precursor 171 yield the racemic mixture products, which were resolved by chiral HPLC. It will be interesting to see if this ring opening reaction would work for other ring structures.
4.4. Oxidation reaction
The oxidation of H11CN gives the KO11CN, which can be further transferred to urea, (iso)hydroxyurea, and other related compounds. In 1978, [11C]hydroxyurea (150a), [11C]isohydroxyurea (150b) and [11C]urea (151) were synthesized from H11CN via [11C]cyanate (Fig. 23).97 H11CN was exchanged with KCN in aqueous solution giving carrier-added K11CN, which was then oxidized by KMnO4 and Cu(OH)2 yielding KO11CN in 37 min with 78.9% decay-corrected radiochemical yield. The further reaction of KO11CN with hydroxylamine-HCl gave the mixture of [11C]hydroxyurea (150a) and [11C]isohydroxyurea (150b) in 72% overall decay-corrected radiochemical yield and a total synthesis time of 124 min.
Fig. 23.
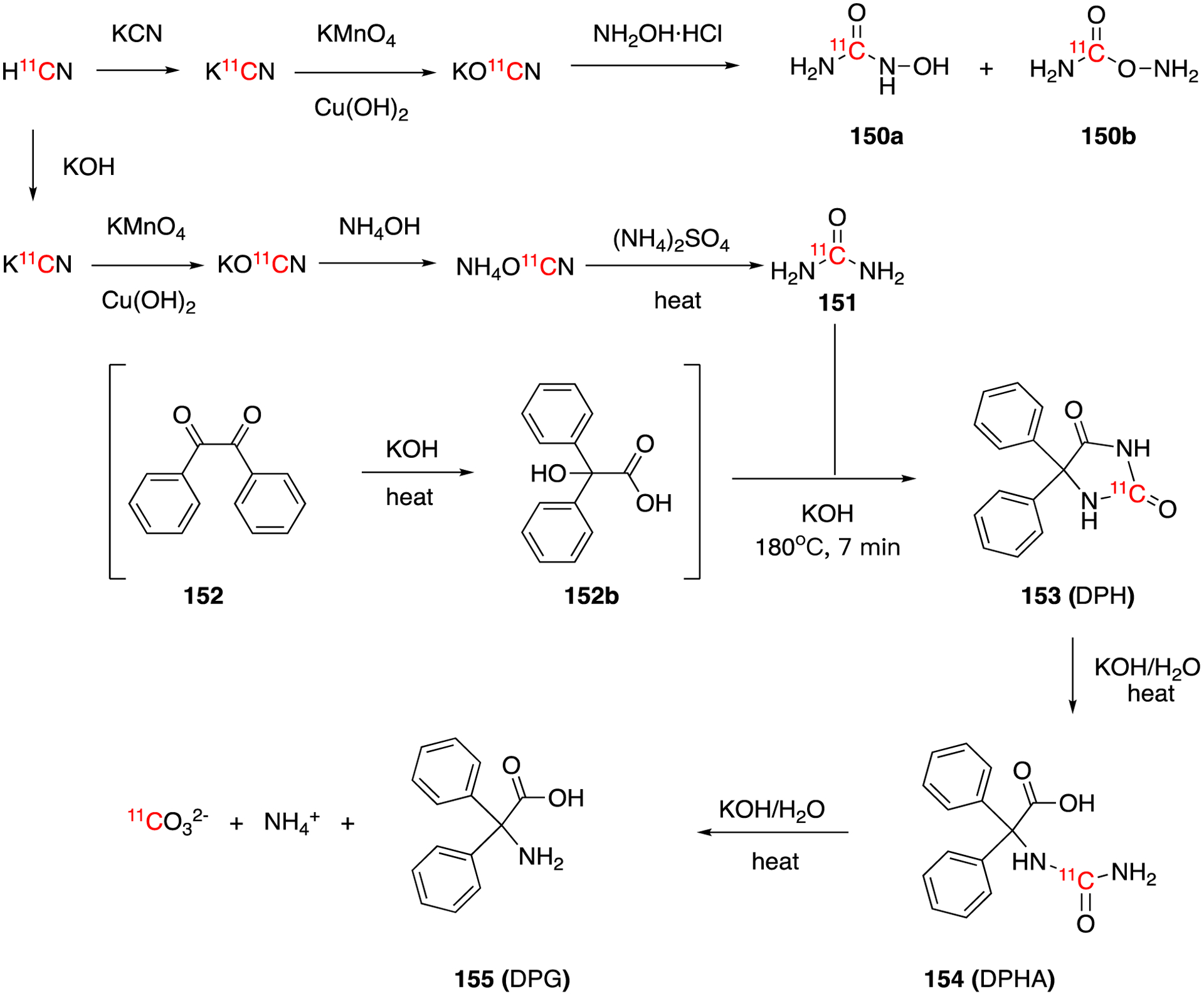
Synthesis of [11C]hydroxyurea (150a), [11C]isohydroxyurea (150b), [11C]urea (151), and [2-11C]5,5-diphenylhydantoin (DPH, 153).
Using a no-carrier added method, the synthesis of 11C-labeled urea (151) was reported by Emran et al in 1983 (Fig. 23).98 KO11CN, obtained through the oxidization of K11CN, was reacted with NH4OH to generate NH4O11CN intermediate. Heating of the intermediate yielded [11C]urea in 85 ± 5% decay-corrected radiochemical yield. The total synthesis time was less than 20 min. Two years later, the same research group reported several modifications which increased the decay-corrected radiochemical yield to 95 ± 2.5% and reduced the total synthesis time was reduced to 16±1 min. The molar activity of [11C]urea after the improvements was 129.5 ± 29.6 GBq/μmol (3.5 ± 0.8 Ci/μmol).99–101
5,5-diphenylhydantoin (DPH, phenytoin) is a widely prescribed anticonvulsant that works by blocking sodium channels in the brain. A 11C-labeled version of phenytoin (153) was synthesized by Emran et al via [11C]urea intermediate.102 [11C]urea was prepared from no-carrier added H11CN as previously described and reacted with a saturated alcoholic/aqueous benzil (1,2-diphenylethane-1,2-dione) KOH solution to generate [2-11C]-5,5-diphenylhydantoin (153). 153 was purified by preparative HPLC in an overall 30–35% decay-corrected radiochemical yield with high molar activity (118.4 ± 11.1 GBq/μmol; 3.2 ± 0.3 Ci/μmol) in 55–60 min. Prolonged reaction time resulted the dissociation of [11C]DPH to [11C]diphenylhydantoic acid ([11C]DPHA, 154), which could further undergo decarboxylation to generate diphenylglycine(DPG) and [11C]carbonate.
4.5. The reactions of [11C]cyanogen bromide (11CN+ reagent)
Through the reaction of H11CN with bromine, [11C]cyanogen bromide (11CN)Br can be made. This reaction converts the nucleophilic carbon center of cyanide, 11CN−, to an electrophilic carbon center cyanogen, 11CN+. Nucleophiles can attack the electrophilic carbon in [11C]cyanogen leading to very different chemical reactions compared with 11CN−.
In 1993, a new 11C-electrophilic labeling reagent, [11C]cyanogen bromide (11CN)Br, based on H11CN was developed by Westerburg et al (Fig. 24).103 H11CN was reacted with bromine in triethylenglycol dimethyl ether to give (11CN)Br in 70–80% decay-corrected radiochemical yield in 9–11 min. (11CN)Br (b.p. 61.5 °C) was separated from bromine (b.p. 137.8 °C) via distillation for further reactions. A typical run, which started from 20.35 GBq (550 mCi) of 11CO2, could yielded 10.14 GBq (274 mCi) of (11CN)Br. (11CN)Br was tested in the reaction with (thio)phenols, amines, pyridine, and disulfide compounds which yielded the corresponding [11C](thio)cyanates (156a-d), [11C]cyanamide (158-160), [1-11C]cyanopyridninium bromide (157), and [11C]thiocyanate (161) in 12–98% of radiochemical yields in 13–27 min from (11CN)Br production.
Fig. 24.
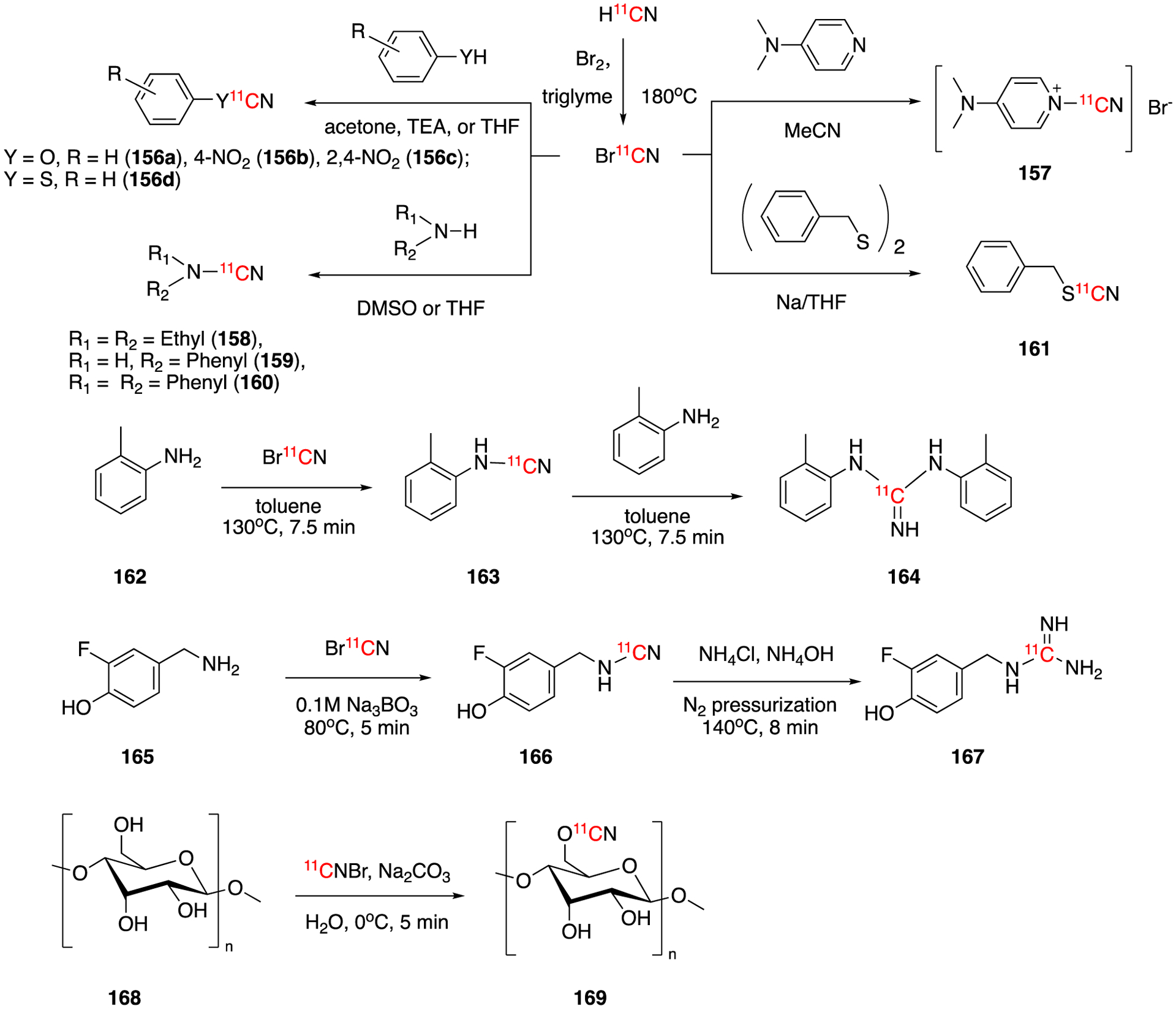
Radiochemistry of [11C]cyanogen bromide.
A second flow production system of (11CN)Br using solid pyridinium bromide perbromide instead of elemental bromine was developed in 1997.104 This simplified method produced (11CN)Br in 95% radiochemical yield (decay-corrected) within 3 min from the end of bombardment.
In 1994, Långström’s research group reported the synthesis of 1,3-di(2-tolyl)-[11C]guanidine (164) from o-toluidine and (11CN)Br.105 1,3-Di(2-tolyl)-[11C]guanidine was obtained in 87 % decay-corrected radiochemical yield within 31–33 min of synthesis time. The molar radioactivity of 164 was 118.4 GBq/μmol (3.2 Ci/μmol). In vitro autoradiographic studies showed specific binding of 164 on rat brain sections. However, in vivo PET imaging of Rhesus monkey suggested very low brain uptake, indicating that the 1,3-di(2-tolyl)-[11C]guanidine did not cross the blood-brain barrier. Very recently, Scott, Shao and co-workers reported the automated radiosynthesis of [11C]guanidines using (11CN)Br.106 The method was found to tolerate unprotected -OH and -NH groups and have broad substrate scope. Using this method they were able to prepare [11C]3F-PHPOG (167) which was found to have good cardiac uptake and heart retention in rabbits.
A similar method using (11CN)Br was reported for the synthesis [11C]polysaccharides in high molar radioactivity.107 The reaction of dextran and hyaluronan with (11CN)Br in basic solution gave the corresponding labeled products in 30–47% decay-corrected radiochemical yields. The synthesis times were 24–26 min and the molar activity activities were 4.44–114.7 GBq/μmol (0.12–3.1 Ci/μmol). The isolated fraction of [11C]hyaluronan (169) with 98% radiochemical purity was used for the biodistribution study in rats using PET imaging which suggested a rapid and displaceable uptake in liver.
4.6. Other reactions
H11CN can be directly reduced to [11C]methylamine (Fig. 25). This was accomplished by the hydrogenation-reduction of H11CN using Adams catalyst with H2 gas in diluted sulfuric acid solution. [11C]methylamine was synthesized in 50–70% decay-corrected radiochemical yield. The synthesis and purification time was 25 min.108
Fig. 25.
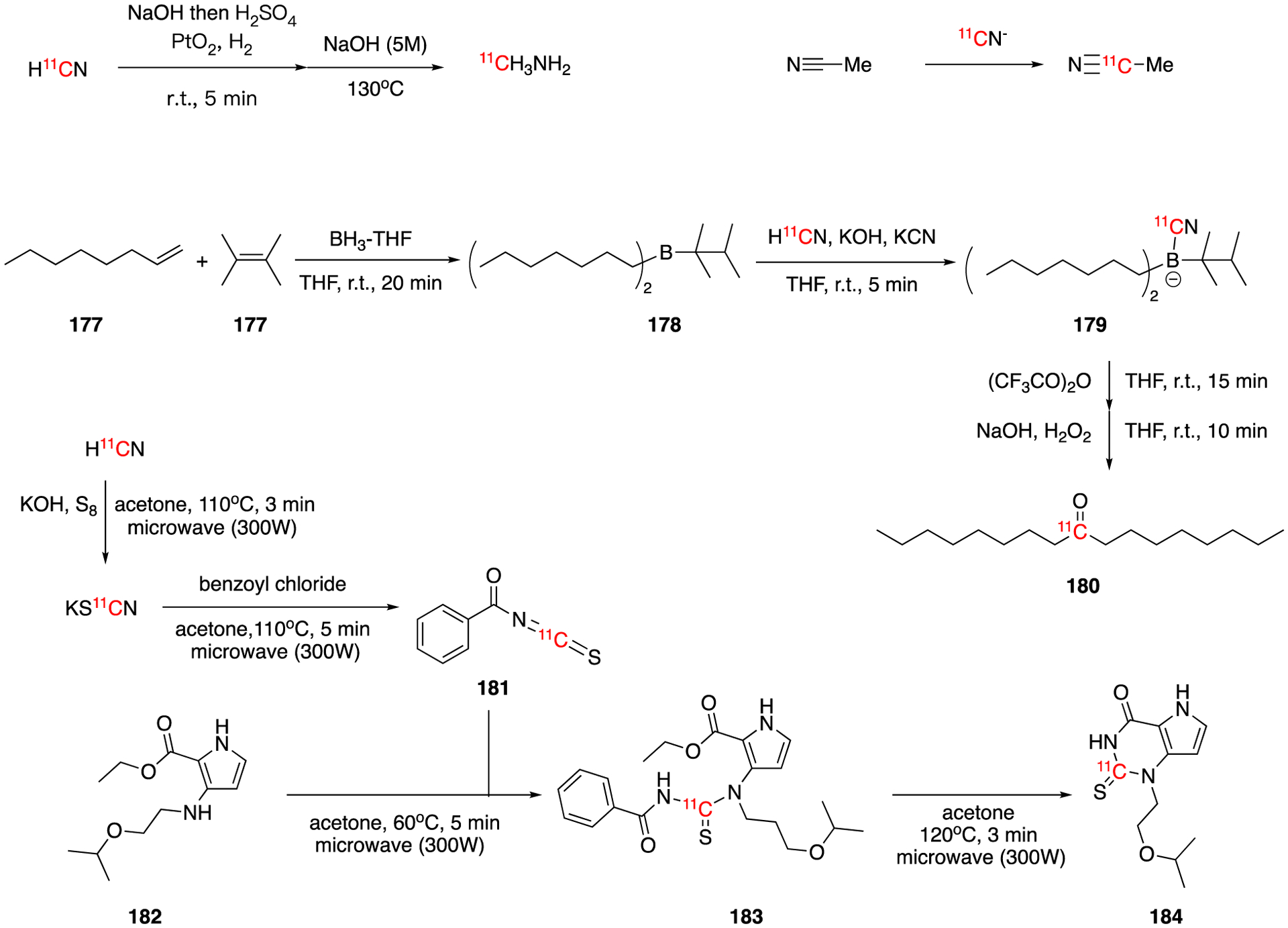
Synthesis of [11C]methylamine and 9-[11C]heptadecane-9-one (180) and cyanide exchange in acetonitrile.
In 1980, Jay et al reported the -CN exchange of the radiolabeled potassium cyanide with solvent acetonitrile in the present of crown ether (Fig. 25).109 Even though the exchange rate was slow this has implications in the choice of solvent for radiocyanations.
A method of synthesis 11C-labeled dialkyl ketones using H11CN was developed based on the reaction of H11CN with organoboranes by Kothari et al in 1986.110 The labeling precursor di-n-octylthexylborane (178) was synthesized by the reaction of 1-octene, tetramethylethylene, and BH3-THF. 178 was reacted with K11CN to give the adduct intermediate 179, which was further rearranged and oxidized to give 9-[11C]heptadecane-9-one (180) in in 50–70% overall decay-corrected radiochemical yield. The reaction and purification time was 55–60 min from the end of bombardment (EoB).
Similar to the oxidative reactions, the reaction of H11CN with S8 and KOH yields potassium [11C]thiocyanate, KS11CN, a heavier analogue of potassium cyanate, KO11CN. The myeloperoxidase inhibitor AZD3241 (184), a candidate drug for neurodegenerative brain disorders, can be synthesized via KS11CN labeling111. KS11CN was reacted with benzoyl chloride to generate benzoyl [11C]isothiocyanate (181), which was further converted to [11C]AZD3241 (184) via the reaction with ethyl 3-(2-isopropoxyethylamino)-1H-pyrrole-2-carboxylate and subsequent base-catalyzed cyclization. A typical synthesis gave 0.71 ± 0.29 GBq (19.19 ± 7.95 mCi) (mean ± SD, n = 7) of [11C]AZD3241 within 60 min with molar activity of 8.88 ± 4.07 GBq/μmol (0.24 ± 0.11 Ci/μmol) and 98% radiochemical purity. This compound was utilized for PET microdosing studies in cynomolgus monkeys.
5. [11C]cyanation of aromatic compounds
5.1. SNAr reaction with Cr(CO)3 activated arenes
The first example of an aromatic [11C]cyanation was first reported in a meeting abstract in 1986 by Balatoni, et. Al.112 with additional details published in the related paper by the same group in 1989.113 Nucleophilic displacement of arene chromium tricarbonyl complexes had previously been reported for [12C]cyanation conditions. The coordination to chromium helps withdraw the electron density from the ring making the nucleophilic displacement possible. This electron removal effect is similar to an aryl nitro group, commonly used in nucleophilic aromatic substitutions. Synthesizing an arene chromium tricarbonyl complex can be completed in milder conditions than the synthesis of aryl nitro derivatives in most cases.112
In 1986, one example of nucleophilic substitution by defluorination is reported (Fig. 26).112 No-carrier-added H11CN was bubbled into a solution of NaOH, dried, and then used under inert conditions to radiolabel η6-fluorobenzenetricarbonylchromium in DMSO at 150 °C for 5 min. A decay-corrected radiochemical yield of ~50% was obtained after HPLC purification.112 The authors noted that under these conditions, the decomplexation also occurs so an additional step is not needed. A control experiment using fluorobenzene produced no [11C]benzonitrile under the otherwise same conditions.112
Fig. 26.
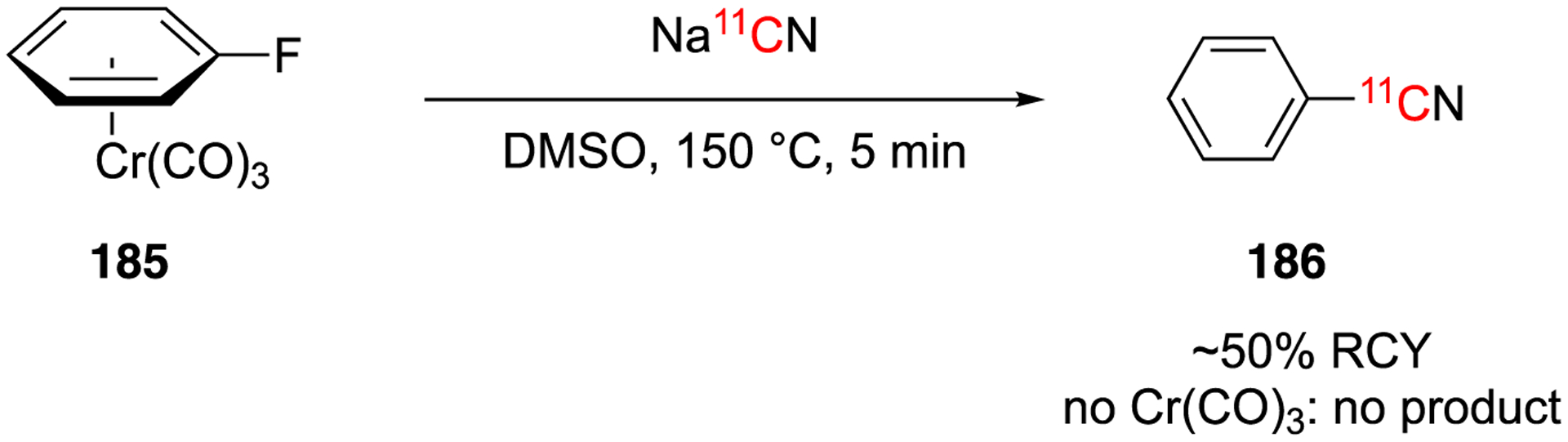
First example aromatic [11C]cyanation using arenechromium tricarbonyl complexes.
In 1989, the scope of the defluorination by nucleophilic displacement of arene chromium tricarbonyl complexes was expanded to four substrates (Fig. 27).113 Carrier-added Na11CN was used under inert conditions to radiolabel the aryl chromium tricarbonyl complexes in DMSO at 135 °C for 10 min. The radiochemical yields after HPLC purification ranged from 19–35%.113 Control experiments using the uncomplexed arenes provided no product under than same labeling conditions.
Fig. 27.
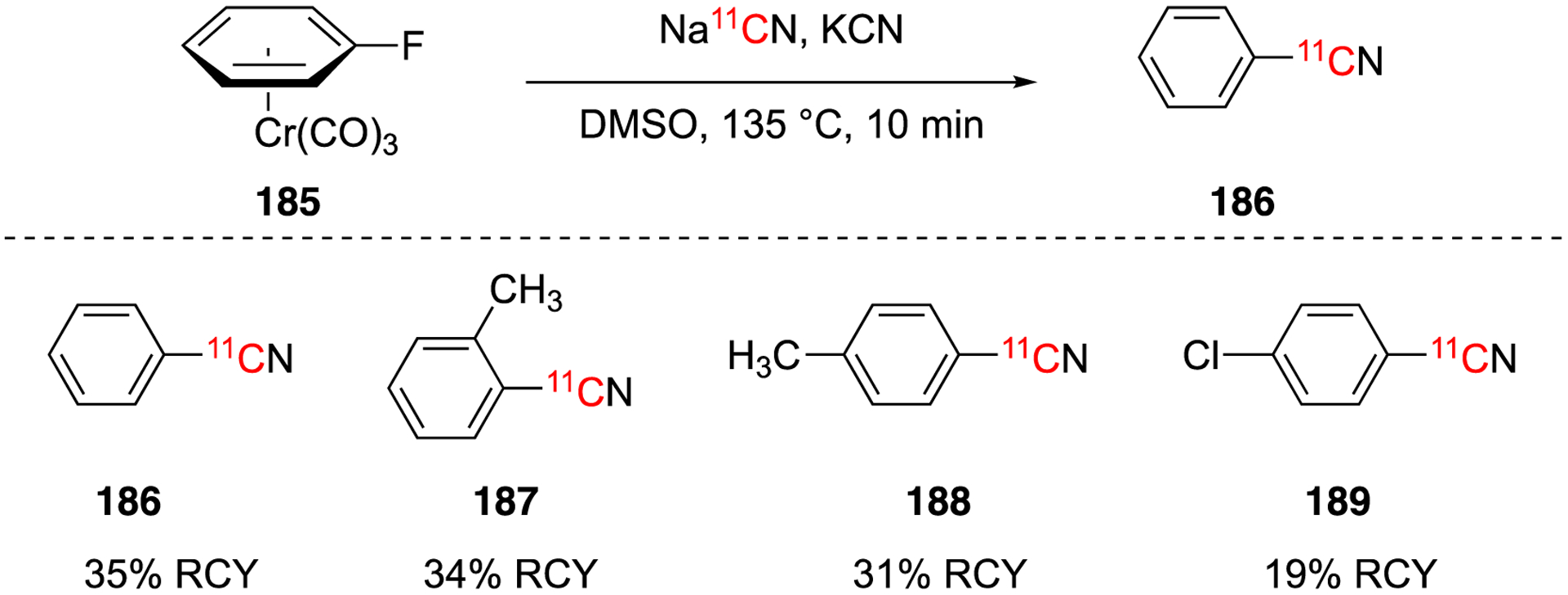
Expanded scope of aromatic [11C]cyanation using arenechromium tricarbonyl
5.2. Palladium-Mediated [11C]cyanation
In 1991, a symposium abstract published the first example of combining the previously developed radiolabeling arenetricarbonyl chromium complexes by nucleophilic displacement [11C]cyanide and the carbon-carbon bond formation reactions using palladium(0) to obtain aryl nitriles.114 The nucleophilic displacement of aryl halides was investigated with arene chromium complexes with and without palladium(0) catalyst (Fig. 28). It was found for the aryl fluorine substrates, the conditions without palladium(0) gave product as observed previously. Aryl chlorides could also undergo the cyanation reaction, whereas aryl bromides gave no product without palladium(0) present. Interestingly, the trend is reversed when palladium(0) is added. Aryl fluorides give no product while aryl chlorides and aryl bromides give high yields.
Fig. 28.
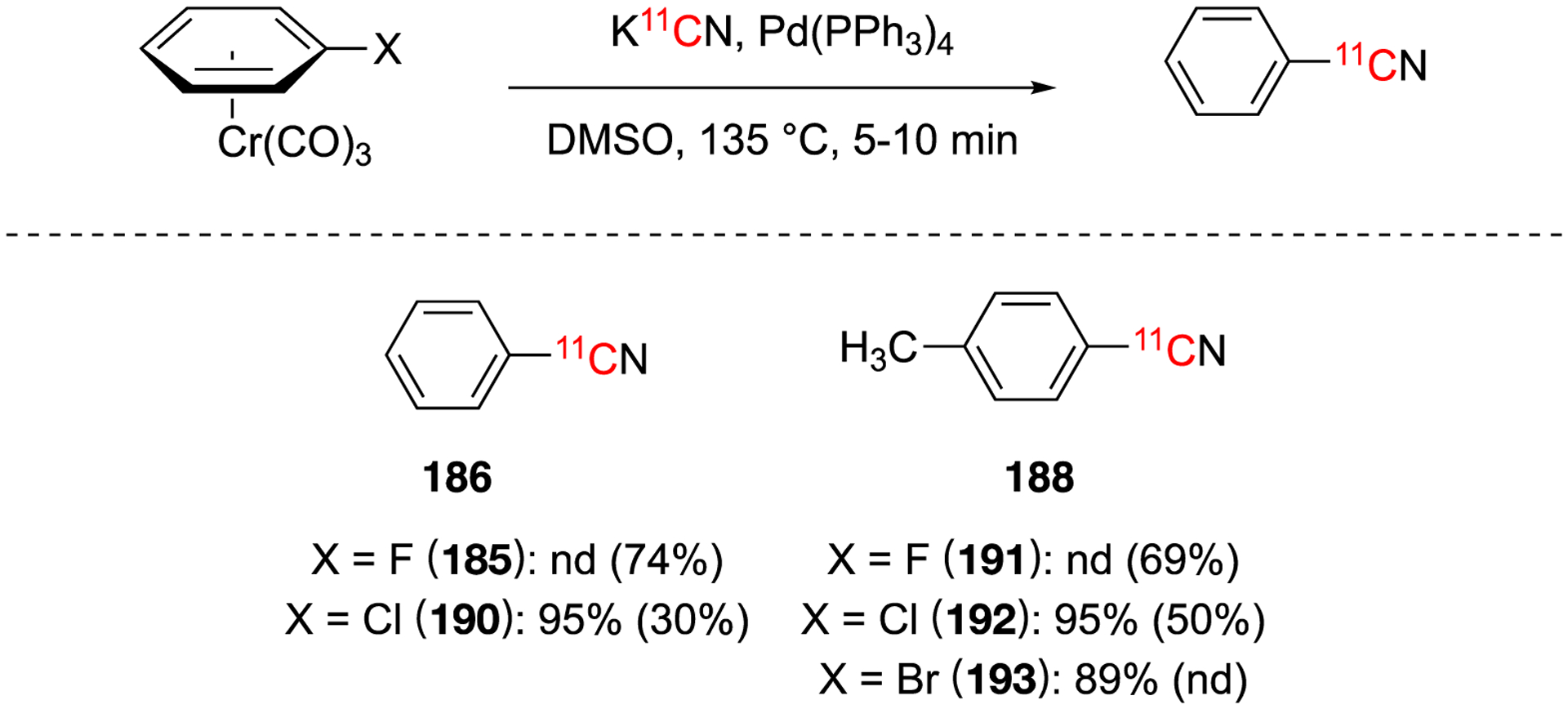
Substrate scope using arene chromium complex substrates and with (or without) palladium(0) catalyst.
Starting with the aryl halides and palladium(0) catalyst, a similar trend was observed when both arene chromium complexes and palladium(0) were used: aryl fluorides and aryl chlorides gave poor yields while aryl bromides and aryl iodides gave high yields (Fig. 29). Typical conditions used K11CN in DMSO at 135 °C for 5 min (with palladium(0) present) or 10 min (without palladium(0) present), followed by purification via Sep-Pak cartridge to obtain decay-corrected radiochemical yields of 49–80%, decay-corrected.114 The authors also found that decomplexation of the chromium complex were not necessary under these conditions.
Fig. 29.
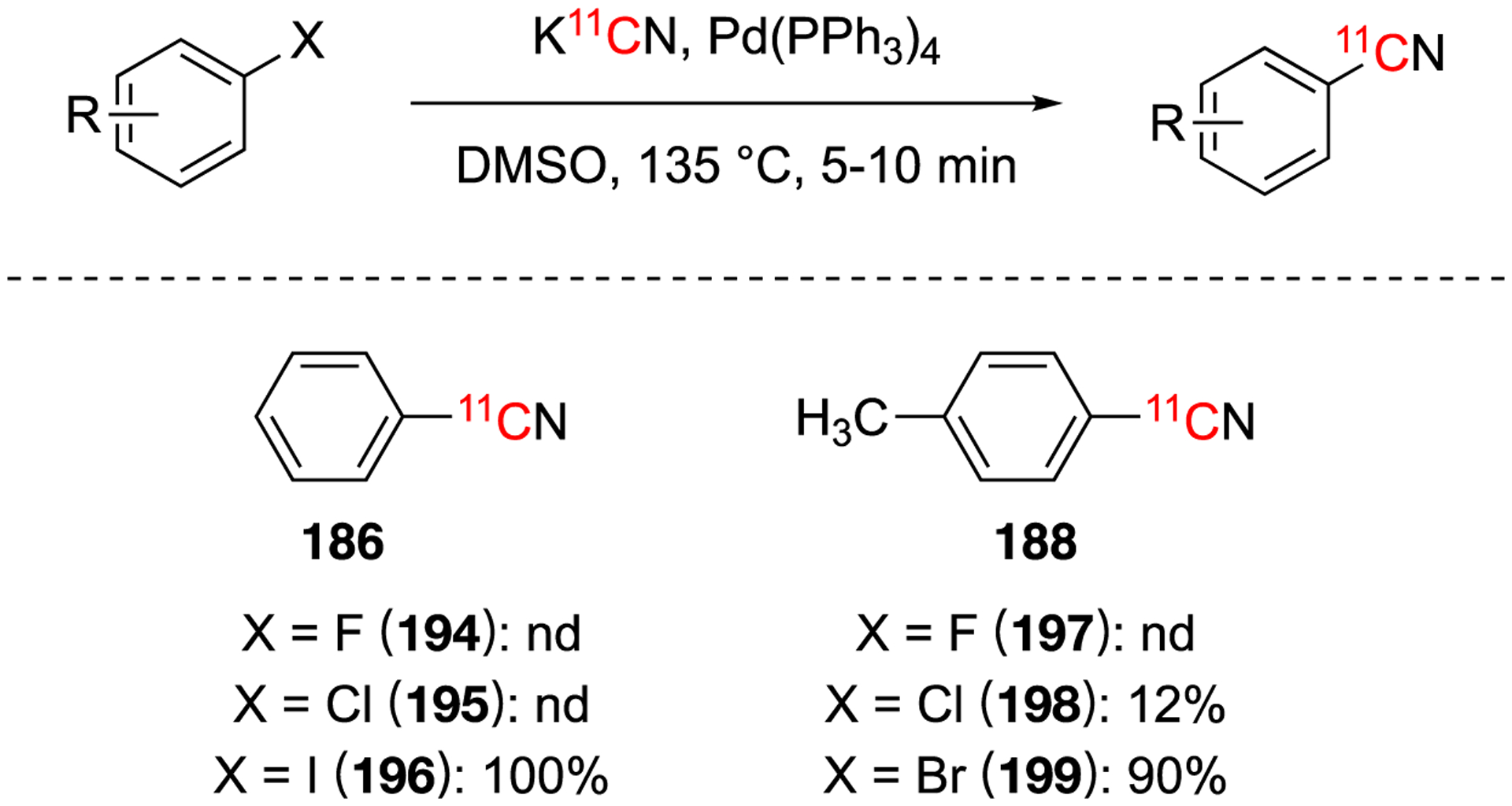
Substrate scope using aryl halide substrates and with palladium(0) catalyst.
In 1994, the relationship between reaction conditions (palladium(0) assisted, nucleophilic substitution of chromium complexes or a combined approach) and the leaving group were explored.115 In the Pd(0) assisted reactions, it was found that aryl iodides were the best substrates, followed by aryl bromides. Aryl chlorides gave a lower yield and aryl fluorides were not reactive (Fig. 30).115 The control reaction without Pd(0), gave no desired product. High yields were obtained when the Pd(0) complex was added as a solid in the last possible time in the synthetic procedure. Using a solution of the metal complex, prepared in advance, gave a decreased RCY, likely due to the rapid oxidation of the Pd(0) complex.115 This limitation could be circumvented if the reaction was performed under argon in an anaerobic conditions.
Fig. 30.
Palladium(0)-mediated [11C]cyanation of aromatic compounds.
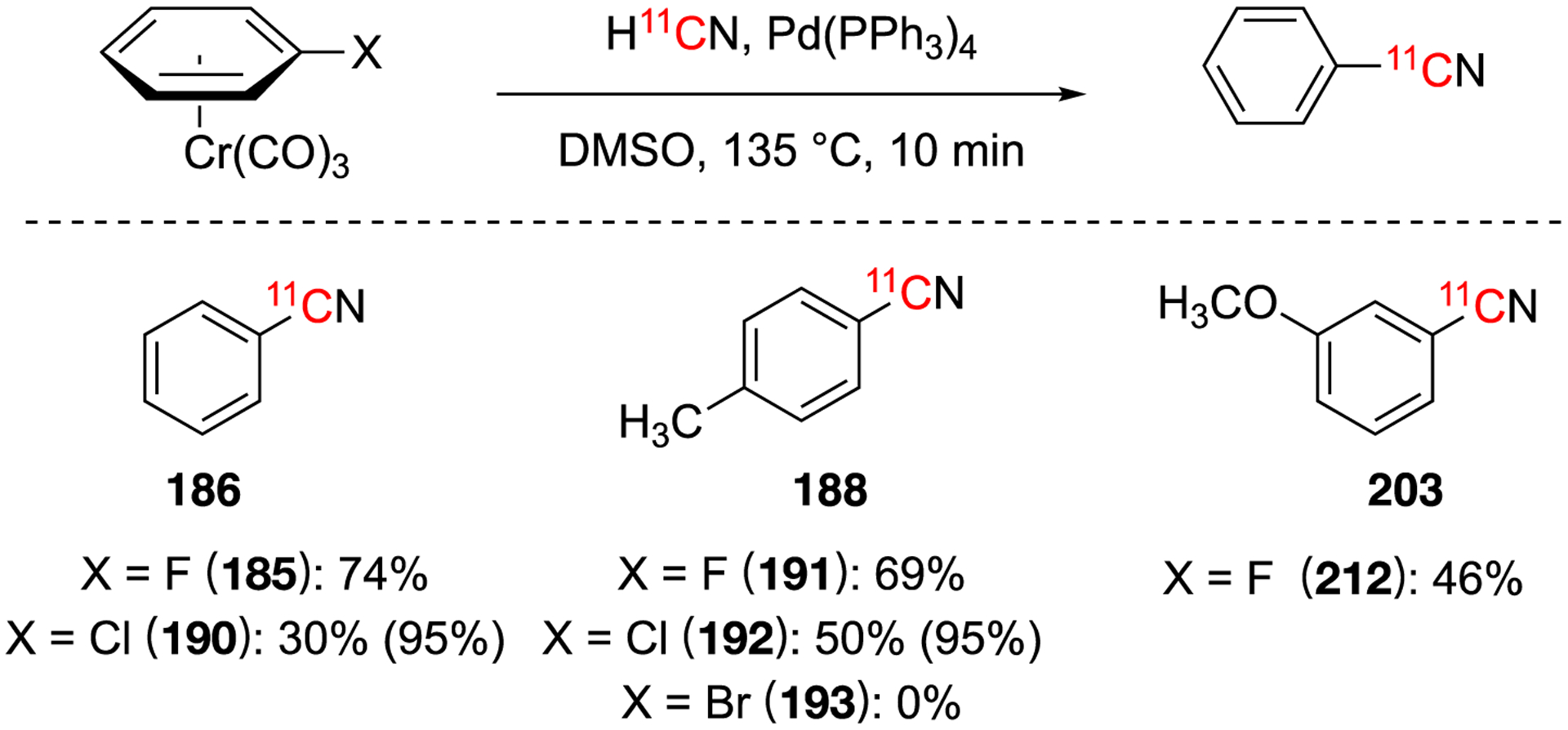
The halogen leaving group for the nucleophilic substitution of arene(tricarbonyl)chromium complexes was found to have the opposite trend of the Pd(0) assisted reactions. Aryl fluorides provided the best results, followed by aryl chlorides, and no product was determined with the aryl bromides (Fig. 31).115 Optimization led to an improvement of the original report of [11C]benzonritile (74% versus 35%).113 The decomplexation of the chromium complex was observed during the reaction conditions. The chromium complexes are known to be stable in crystalline form, but unstable in solution. It is probable that performing these reaction under anaerobic conditions would increase the yield, but then the decomplexation step would be required; therefore increasing the overall time of the reaction and likely not significantly improving the overall yield.115 Combining arene (tricarbonyl)chromium complexes with Pd(0) assisted reactions increased the yield of the reaction by facilitating the oxidative addition (Fig. 31 in parenthesis). This approach could lead to high yield products with the stable aryl chloride substrates.115
Fig. 31.
[11C]Cyanation on tricarbonylchromium complexes of arenes (with Pd(PPh3)4.
Aryl nitriles can be converted into other useful functional groups including amides under 11C-labeling conditions in an one-pot procedure (Fig. 32).116 In anhydrous conditions the Pd(0) [11C]cyanation step could be performed rapidly (<2 min) with good yields in the presence of sodium percarbonate. The hydrolysis could be performed in the same pot by the addition of water to dissolve the sodium percarbonate which releases hydrogen peroxide. This method allows the authors to synthesize the three targeted compounds in useful yields of 45–66% decay-corrected yield in 25–35 min.116
Fig. 32.
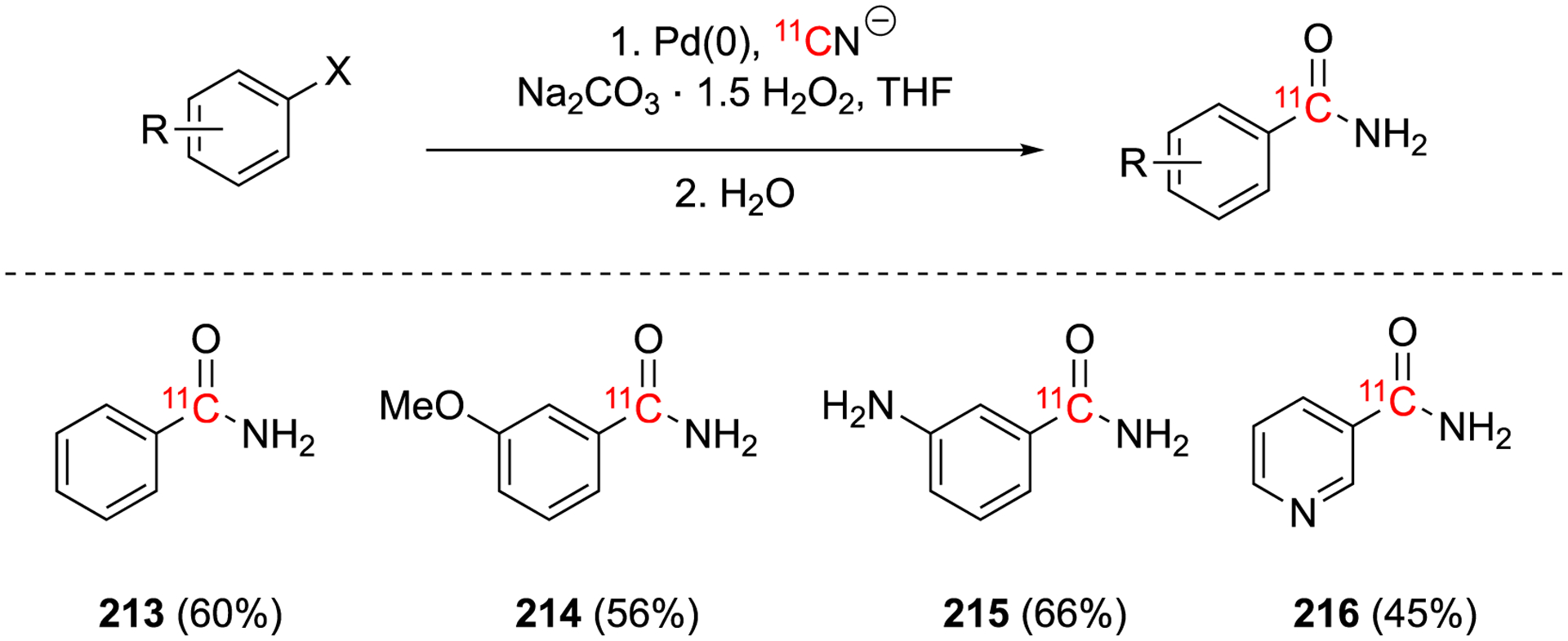
[11C]Cyanation of benzamide compounds as potential radiotracers.
Benzamide (213), methoxybenzamide (214), aminobenzamide (215), and nicotinamide (216) are known to be potent inhibitors of poly (ADPribose) synthetase (Fig. 32), a common target for anticancer drugs and could be used to evaluate the amount DNA damage that occurs during treatments with radiotherapy or chemotherapy.116 Compounds 214, 215, and 216 were studied in vivo in a rhesus monkey model. Purification optimization to remove Pd(0) from the reaction mixture was analyzed by ICP-AES. The preliminary studies investigated the initial distribution and kinetics of the potential tracers. It was found that the blood-clearance was too rapid, the brain uptake was too low, and there was rapid fixation with high uptake in the liver, kidney and lymph nodes with slow washout.
In 2015, a modified Pd(0) method was investigated where the active catalyst is generated in situ using biaryl phosphine ligands.117 These biaryl phosphine ligands offer several advantages from the traditional ligands, such as milder conditions, shorter reaction times, ideal solvents, and reduced addition of base.117 There are three general steps the reaction needs to undergo to form the desired product: oxidative addition (OA), transmetalation I, and reductive elimination (RE). Previous [11C]cyanation methodologies performed all three transformations after the 11CN− has been introduced to the reaction resulting in higher temperatures and longer reaction times (Fig. 33, pathway a).115–116 Biaryl phosphine ligands (L) allow the reaction to be performed at ambient temperature in under 5 min (Fig. 33, pathway b).117 While using a biaryl phosphine ligand offered some iprovements on the current procedures, the Pd(0) catalyst and ligand were added separately, required to form the active complex and perform OA after end of bombardment (EOB) while the 11CN− was in solution. These steps were independent of the 11CN− and it was proposed that they could occur prior to EOB to save on the overall reaction time (Fig. 33, pathway c).117 Complex A is a stable intermediate that can be isolated for some simple aryl groups, but this isolation can be tedious. This work generates complex A in situ prior to EOB, without the requirement to isolate complex A (Fig. 33, pathway d).117 Pathways c and d allow for the OA to occur before EOB resulting in an unnecessary loss of radioactivity. Then the TM and RE can occur post EOB resulting in a more efficient timeline.
Fig. 33.
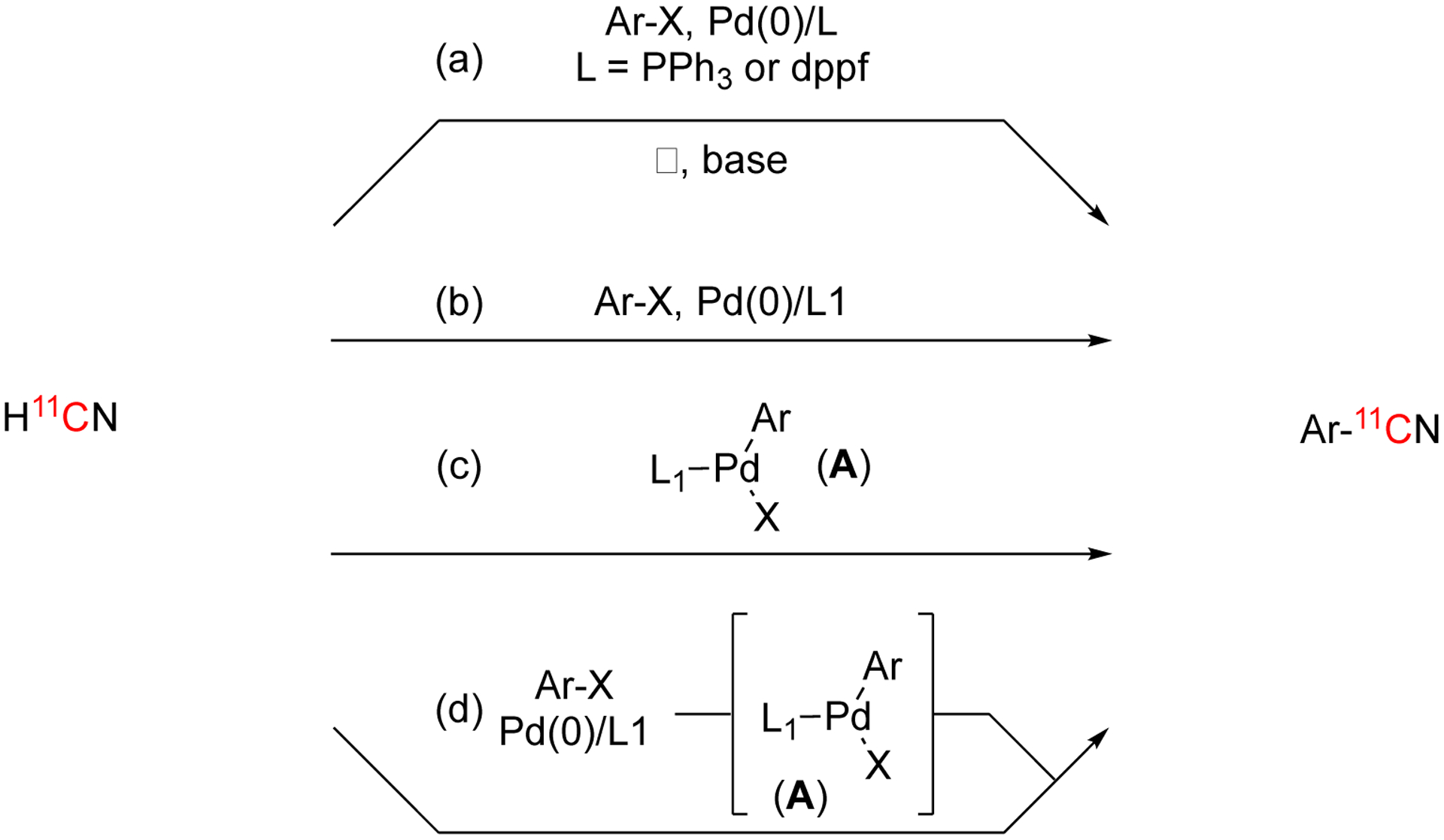
Synthetic strategies for Pd(0) mediated [11C]cyanation.
Optimization lead to the conditions of a preformed Pd(0)-L complex, aryl halide or pseudohalides in THF or toluene at room temperature for 30 min then H11CN in THF was added to the mixture and allowed to react for 1–5 min. The crude yields of reaction was found to be robust for common functional group, such as phenols, amides, and heterocycles, and small amounts of water. Commercial Pd(PPh3) could be used with K2CO3 in DMF at 100 °C for 5 min, but lower yields were observed in most cases.117 To show the versatility of this method, three known [11C]radiolabeled pharmaceuticals (217-219) were isolated in good non-decay corrected (ndc) RCY (10–20%) in 12–23 min with good molar activity (Fig. 34). This method is an improvement upon earlier Pd(0) methods in terms of yield, overall reaction time, and breadth of scope. A limitation is the Pd(0) catalyst used has to be synthesized prior and stored in an inert atmosphere at −20 °C due to their susceptibility to oxygen and moisture.
Fig. 34.
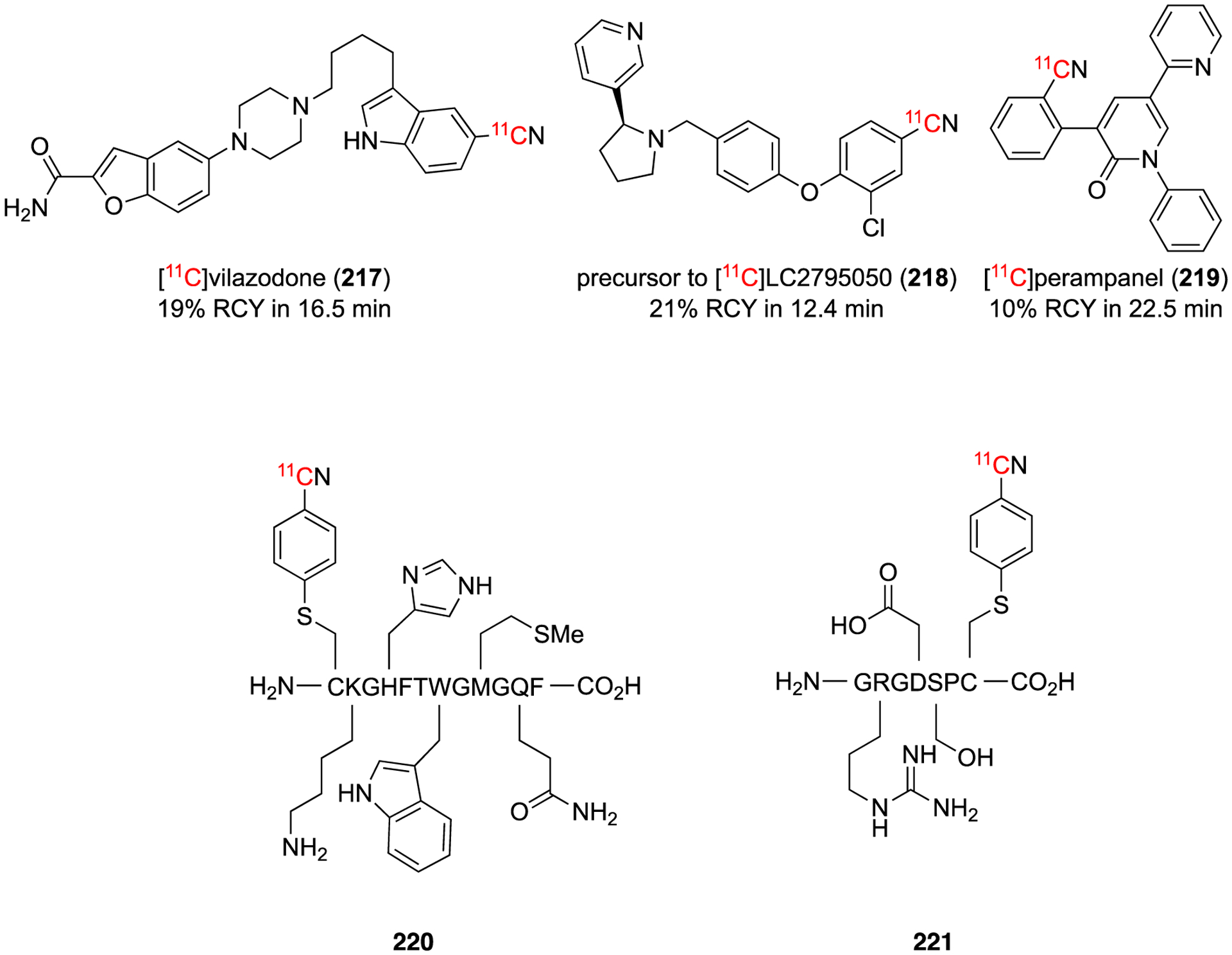
Synthesis of [11C]radiolabeled pharmaceuticals and peptides via Pd(0) method.
In 2017, the same group applied this new method development to label unprotected peptides with [11C]cyanide.118 The two approaches prior to this method was either to attach a prosthetic group that could be radiolabeled later or radiolabel the prosthetic group and then conjugate to the peptide. The method reduces the previous two step approaches to a one step, avoiding the need for a prosthetic group. Using conditions similar to the initial publication,117 a [11C]benzonitrile group can be installed on a cysteine reside in the presence of other nucleophilic functional groups (220 and 221).118
In 2018, the substrate scope was extended to include arylboron compounds using Pd(II) complexes.119 The optimal conditions consisted of NH411CN with excess NH3 and PdCl2(PPh3)2 in DMF at 100 °C for 5 min. These conditions were used to label a range of arylboronic acids and arylboronic esters with good RCC. The reaction was automated to produce [11C]Cetrozole (224) and [11C]YM511 (225) in 45% and 33% decay corrected RCY, respectively, in 28 min from the generation of 11CH4 (Fig. 35). All reagents are bench-stable and readily available. Using NH411CN as the cyanide source eliminates the need to remove the excess NH3, saving time in the synthesis. This method uses a relatively large amount of starting precursor (20 μmol compared to <10 μmol of related methods) which can be difficult to achieve for some complex structures.119
Fig. 35.
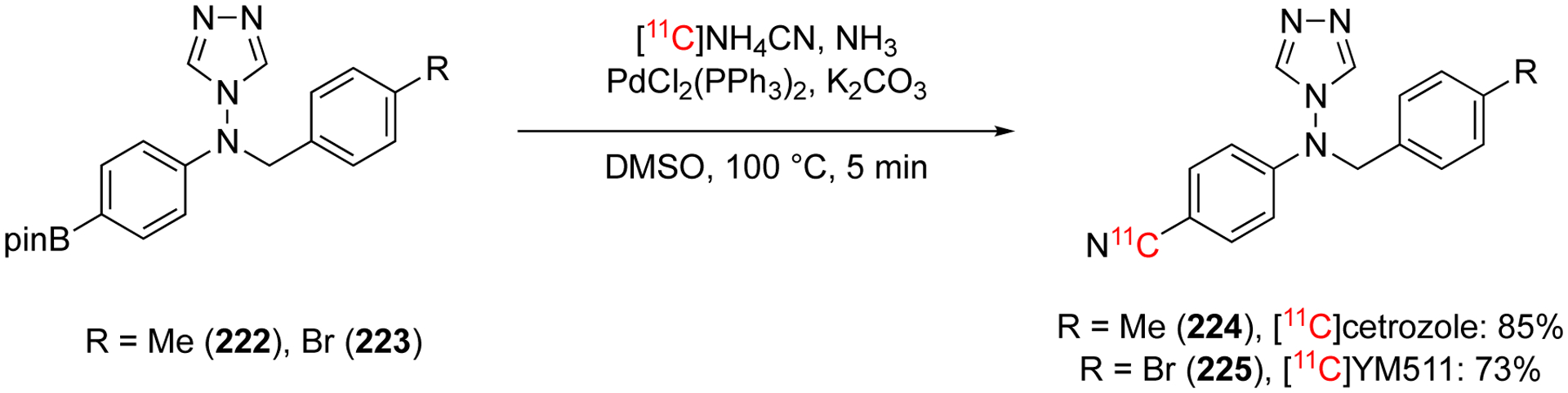
Automated synthesis [11C]cetrozole and [11C]YM511.
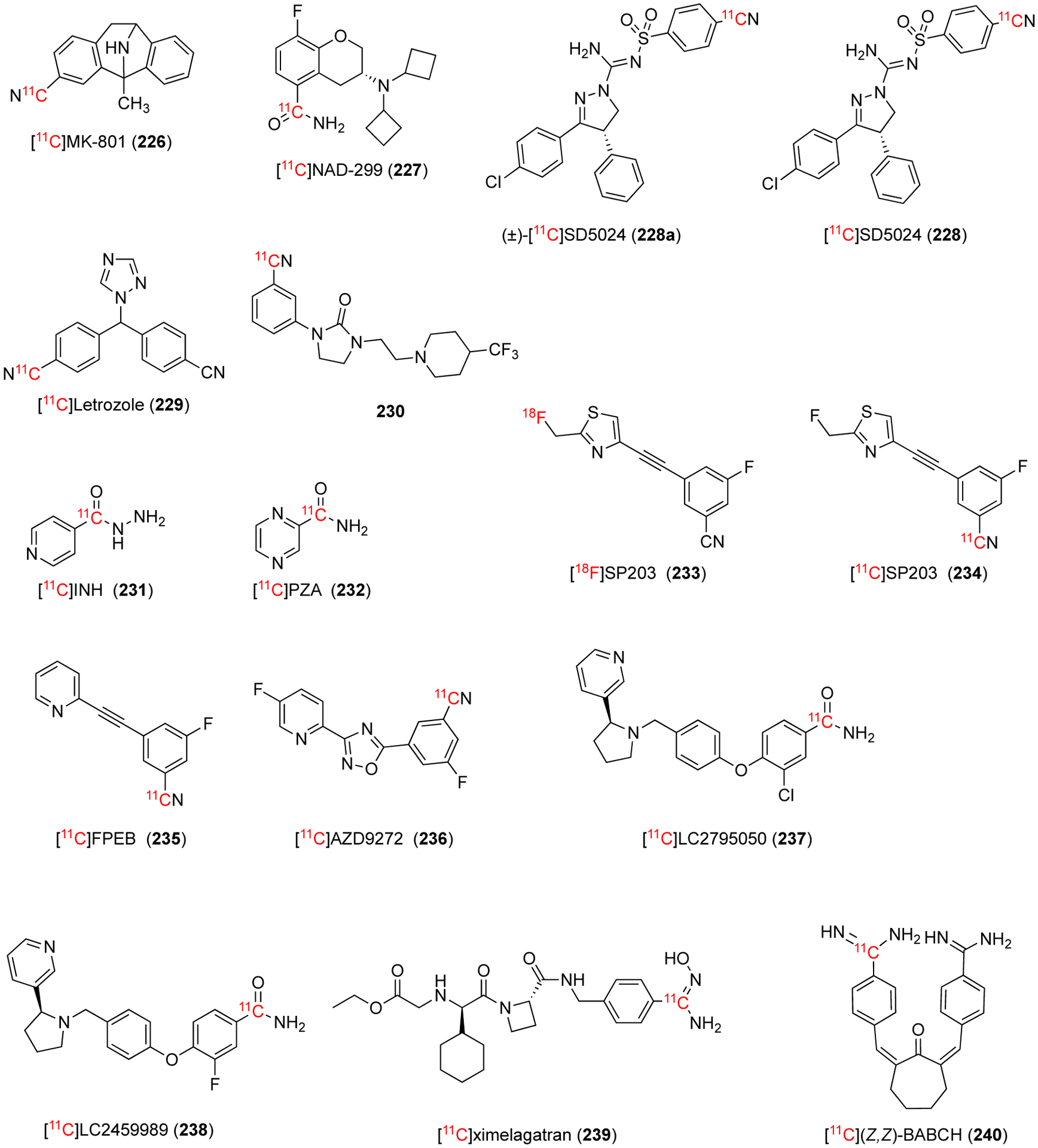
The general Pd-mediated [11C]cyanation reaction conditions that were developed in 1994 by Andersson and co-workers has been used in many imaging studies (Fig. 36).115 These general conditions consisted of an aryl halide (X = I or Br), Pd(PPh3)4 in DMF or DMSO at relatively high temperatures (>100 °C) for ~5 min. In 1998, [11C]MK-801 (226) was synthesized from an aryl iodide precursor in an overall 37% DCY in 32 min from end of bombardment (Fig. 36).120 [11C]MK-801 (226) was used in an in vitro investigation of rat brain to analyze the specific and high-affinity binding to the N-methyl-D-aspartate (NMDA) receptor. An in vivo experiment in a Rhesus monkey did not show an improvement over the known [18F]methyl-MK-801 and the preliminary data was not sufficient to validate [11C]MK-801 (226) as a useful tracer for studying the NMDA receptor.120
Fig. 36.
Radiotracers developed based on Pd-mediated cyanation.
In 1999, [11C]NAD-299 (227) was synthesized over two steps in 20–40% DCY in 40–45 min (Fig. 36). The benzonitrile intermediate was synthesized from the aryl triflate with Pd2(dba)3CHCl3 complex with extra dppf ligand in NMP at 80 °C for 3 min.121 When the conventional Pd(PPh3)4 catalyst was used, the benzonitrile intermediate had a 20% incorporation of [11C]cyanide as opposed to a nearly quantitive yield when Pd2(dba)CHCl3 with dppf was used.121 In 1999, [11C]NAD-299 (227) was found in vitro to demonstrate high binding to the 5-HT1A receptors using human brain postmortem analyzed by autoradiography techniques.121 In 2002, the same group analyzed [11C]NAD-299 (227) in a cynomolgus monkey for metabolite evaluation. After 45 min post injection, 49% of radioactivity was from unchanged radioligand, a much slower metabolism than the currently used [carbonyl-11C]WAY-100635 which has 25% remaining at the same time point.122
In 2008, a new radioligand investigated the imaging of cannabinoid subtype-1 (CB1) receptors in vivo with PET.123 This radioligand, later commonly referred to as (±)-[11C]SD5024 (228a, Fig. 36), was synthesized as a racemic mixture in a 36% DCY in an overall time of 30 min using Pd(PPh3)4 in DMSO at 110 °C from the aryl bromide precursor. When attempting to isolate the higher affinity enantiomer from the enantiomerically pure precursor, complete racemization was observed likely due to KOH used in K11CN formation.123 They found exchanging the base to KH2PO4 allowed for both enantiomers to be isolated in high e.e. All three isomers were analyzed in a cynomolgus monkey and it was found that the binding assays matched the studies conducted with the naturally abundant isotope. Namely, the racemic compound had a mixture of specific and non-specific binding, the high affinity isomer ([11C]SD5024 (228)) was responsible for the specific and the enantiomer (+) was the result of the non-specific binding.123 In 2014, the compound was compared in vitro and in vivo to other known CB1 receptor ligands previously used in humans.124 The dissociation constant and lipophilicity was measured in vitro in the human brain. Specific binding was measured in vivo in monkeys. Studied the kinetics in healthy subjects. [11C]SD5024 (228) had favorable or improved results the other known CB1 receptors in all sections analyzed.124 In 2016, [11C]SD5024 (228) was used to analyze the efficacy of three drug candidates in non-human primate brain in vivo.125
In 2009, a high-affinity aromatase inhibitor, letrozole, was labelled with carbon-11 (Fig. 36).126 [11C]Letrozole (229) was synthesized from the aryl bromide precursor with Pd(PPh3)4 in DMSO at 110 °C for 5 min with an overall synthesis of 60 min. [11C]Letrozole (229) was not a promising PET radiotracer for brain aromatase due to the lack of specific binding and saturability in regions known to have a high concentration of aromatase.
In 2009, compound 230 (Fig. 36) was synthesized as a potent and selective antagonist of the dopamine D3 receptor.127 Compound 230 was synthesized from the aryl iodide precursor with Pd2dba3, dppf ligand, and KHCO3 as the base in either NMP at 80 °C for 3 min or in DMF at 110 °C for 5 min. Analysis in a non-human primate model showed no specific signal for the dopamine D3 receptor.127
In 2010, multiple tuberculosis chemotherapeutics were 11C-labeled using Pd-mediated method (Fig. 36) and analyzed in baboons in vivo.128 [11C]isoniazid (INH) was synthesized in three steps starting from the iodopyridine with Pd(PPh3)4 in DMSO for 5 min to give [11C]cyanopyridine in 90% crude RCC. The final product, [11C]INH (231) was synthesized in a 45–50% DCY in 50 min over three steps. [11C]pyrazinamide (PZA, 232) was synthesized with the same conditions as [11C]INH (231) to provide an overall 50–55% DCY in 45 min. The preliminary kinetics and metabolites for the potential radiotracers were analyzed in baboons.128
In 2012, a known effective high-affinity and selective radioligand for imaging metabotropic 5 receptors (mGluR5), [18F]SP203, was labelled with carbon-11 (Fig. 36).129 Both aryl bromides and aryl iodides were used to synthesize the desired product with Pd(PPh3)4 in THF at 80 °C for 4 min. Aryl bromide precursor gave poor reproducibility (20–52% DCY) while the aryl iodide provides a nearly quantitive decay-corrected yield.129 The imaging study showed similar results for the new [11C]SP203 (233) compared to the known [18F]SP203 in terms of uptake and stability. For metabolites, a known metabolic pathway is the defluorination where the fluorine-18 is labelled, indicated by bone uptake, a process avoids radioactivity accumulation in bone.129 In 2017, another mGluR5 ligand FPEB was labelled with carbon-11 and compared with [11C]SP203 (233) (Fig. 36). The same trend was observed with [11C]FPEB (235) having more favorable binding and kinetic results than [11C]SP203 (233).130
In 2013, another selective mGluR5 antagonist, [11C]AZD9272 (236, Fig. 36), was prepared from an aryl bromide precursor with Pd(PPh3)4 in DMSO at 135 °C for 5 min to give an overall DCY of 50% in 45 min.131 In vivo analysis in non-human primates provided favorable results and additional studies shown an improvement upon the known mGluR5 antagonists.131–134
In 2013, [11C]LY2795050 (237) was synthesized and evaluated as selective κ-opioid receptor (KOR) antagonist (Fig. 36).135 [11C]LY2795050 (237) was synthesized over two steps from the aryl iodide precursor with Pd2dba3 in DMF at 80 °C for 5 min, followed by hydrolysis to give an overall 12% RCY. [11C]LY2795050 (237) was found to have favorable imaging properties in non-human primates.135 In 2014, the derivative [11C]LY2459989 (238) was synthesized in similar conditions to [11C]LY2795050 (237) and found to have a higher binding affinity for KOR in vitro and in vivo (Fig. 36).136
Aside from methodology development publications, there were several examples of Pd-mediated [11C]cyanation that were not used in an imaging study or investigated further after the initial study. Two examples, shown in Fig. 36, were made with the standard Pd-mediated method developed in 1994 using Pd(PPh3)4. In 2008, [11C]Ximelagatran (239) intermediate was synthesized in DMSO at 135 °C for 4 min to yield the Ar-11CN compound. Further elaboration let to the final compound in an overall decay-corrected yield of 27% in 45 min.137 In 2001, the two step [11C](Z,Z)-BABCH (240) was synthesized in a 55% RCY in 55 min via Pd-mediated [11C]cyanation.138
5.3. Copper-Mediated [11C]cyanation
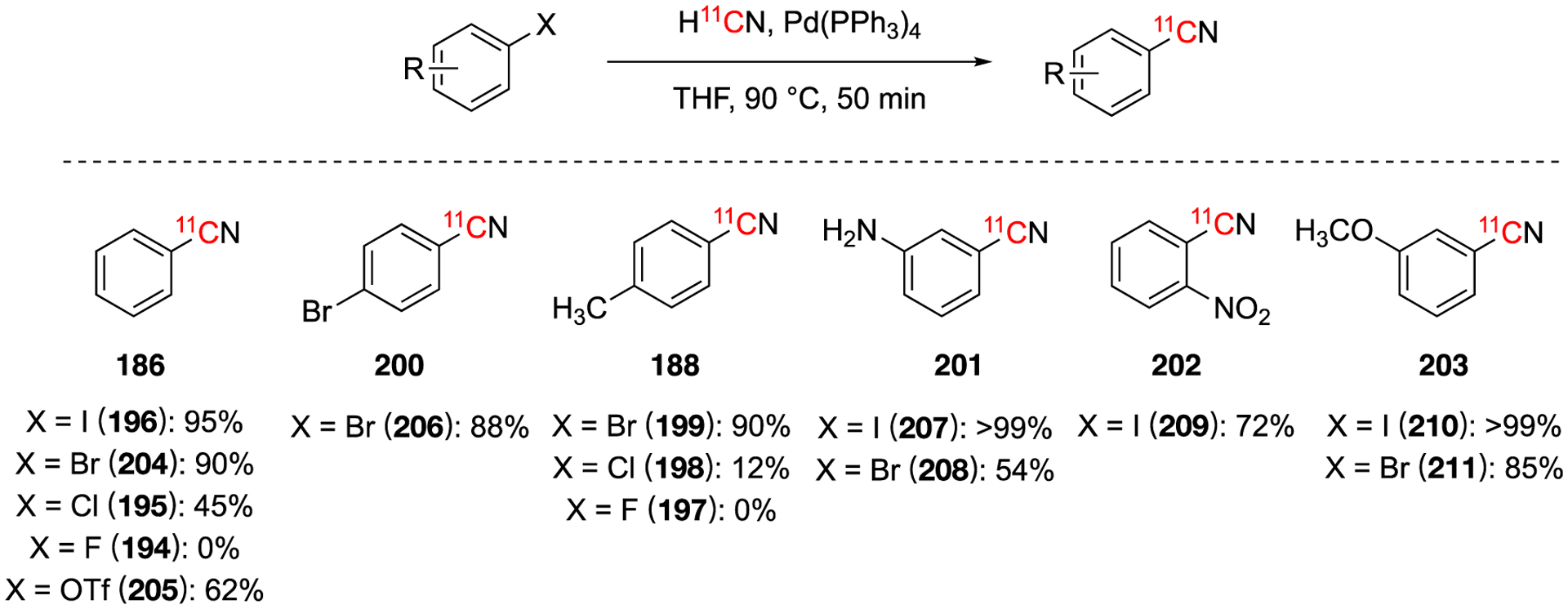
A reoccurring limitation for the previous sections is the requirement for inert and anhydrous conditions. Additionally, palladium has a lower allowable amount (10 μg/day) when compared to other first row metals such as chromium and copper (parenteral 1070 μg/day and 340 μg/day, respectively).139 While copper(II) salts are known to be sensitive to atmospheric oxygen, the reaction conditions are generally more forgiving than the related palladium-mediated methods.
In 1997, the first example of a copper(I)-mediated [11C]cyanation was reported.140 This method adapted the known Rosenmund-von Braun reaction to carbon-11 by synthesizing a Cu11CN. Cu(I) salts are known to not require an inert atmosphere and/or anhydrous conditions. Additionally, these salts are known to react with other aromatic nucleophilic substitutions and have been used with other isotopes, including PET and SPECT radiopharmaceutics. This long history and versatility made it a new approach when extending from palladium-mediated methods. Cu11CN was generated in situ by adding an aqueous solution of CuSO4 to H11CN and headed at 60 °C for 5 min. Next, the precursor was added in DMF and heated at 180 °C for 5 min (Fig. 37). The benzonitrile products were converted into tetrazoyls, benzamides, or benzoic acids in conditions that were amenable to carbon-11. The authors found that trapping H11CN without a base present, gave more optimal yields than trapping with KOH to make K11CN. It was hypothesized that the excess KOH needed, disrupted the Cu reaction that needed to occur. This reaction worked well for a variety of substrates and the benzonitriles were able to be converted into other function groups with modified conditions (Fig. 37).140
Fig. 37.
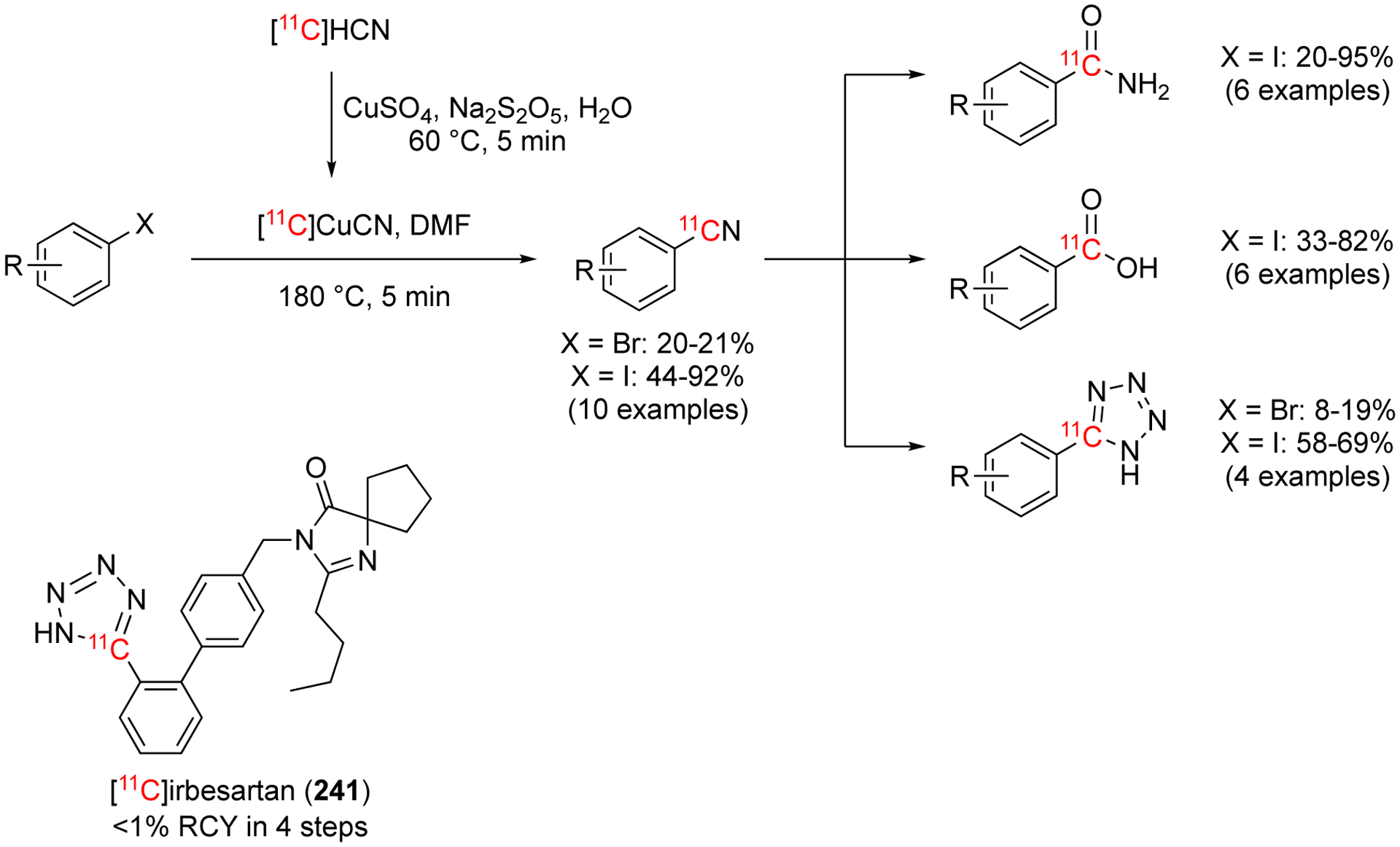
[11C]Cyanation using Cu11CN with representative decay-corrected yields.
This method was used in a one-pot, four steps method from H11CN via Cu11CN to radiolabel [tetrazoyl-11C]irbesartan (241), a non-peptidic angiotensin II antagonist (Fig. 38).141 Due to the lengthy synthesis, a relatively low RCY was obtained (<1% non-decay corrected) in 39 min. A preliminary study was performed in dogs and it was found this radiotracer was not suitable for studying myocardial angiotensin II receptors with PET.141
Fig. 38.
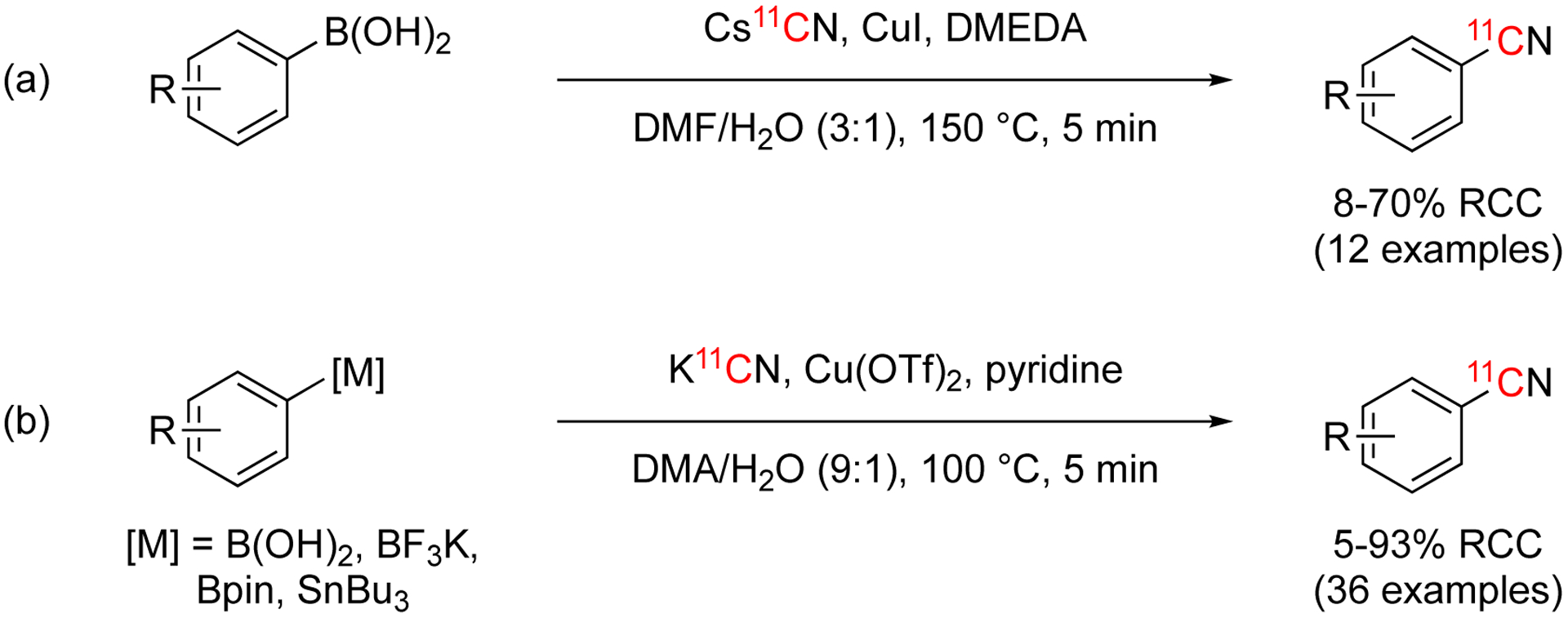
Copper-mediated [11C]cyanation of arenes using organometallic reagents.
In 2017, a copper-mediated [11C]cyanation of arylboronic acids was developed (Fig. 38, reaction a).142 The following year, this method was extended further to include other arylboron derivates and arylstannanes (Fig. 38, reaction b).143 These methodologies improved the limitation with the previous Cu(I) method of generating Cu11CN in situ prior to introducing the precursor, saving time in the overall reaction.140 Additionally, using more active organometallic reagents as precursors help the conditions to be more mild and higher yielding than using aryl halides as the starting materials. The optimal conditions for both methodologies were similar the general format of copper source, base, DMF-type solvent, water, and >100 °C for 5 min (Fig. 38). The substrate scope was similar for both conditions with high yields observed. Both methods were able to be automated and the desired product isolated.142–143
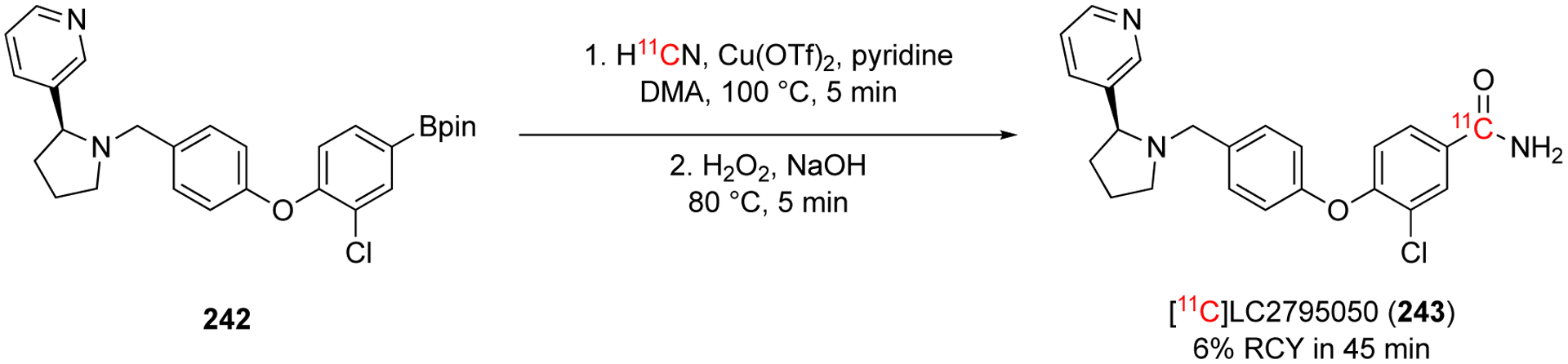
The 2018 [11C]cyanation method, developed by the Sanford/Scott group, was used to radiolabel [11C]LY2795050 (243) for clinical PET imaging (Fig. 39).143 Previous methods to synthesize [11C]LY2795050 (243) used Pd(0) catalysts. This approach is a complementary method that avoids using expensive catalyst that require inert and anhydrous conditions to obtain high yields. The authors were able to the final product in 6% RCY, non-decay corrected and were able to perform preclinical studies in rodents and in nonhuman primates. This method was able to be initiated into clinical imaging studies.143–144
Fig. 39.
Synthesis of [11C]LY2795050 (243).
There have been several potential tracers synthesized with Cu11CN. In 2001, [11C](Z,Z)-BABCH (244), a serine protease inhibitor was synthesized with Cu11CN as a first attempt.138 A RCY of 46% was obtained and could not be improved with further optimization (Fig. 40, 244). Higher yields were obtained with Pd(PPh3)4 (see above). In 2006, a potential glutamate 5 receptor (mGluR5) antagonist, [11C]LY2232645 (245), was synthesized using Cu11CN in a RCY of 2.5% in 26 min (Fig. 40, 245).145 In 2013, a series of [11C]methyl- and [11C]cyano-substrates were evaluated as potential orexin receptors ligands in vitro in rat models.146 Fig. 40, 246 was synthesized via Cu11CN in 2.8% decay-corrected radiochemical yield in 34 min since end of bombardment. In 2021, PET tracer [11C]MTP38 (247) for phosphodiesterase 7 was synthesized using similar method with ≥99.4% radiochemical purity and molar activity of 38.6 ± 12.6 GBq/μmol.147 [11C]MTP38 was used for evaluating PDE7 expression in the brain of rodents and monkeys.
Fig. 40.
[11C]CuCN used to synthesize potential radiotracers.
5.4. Other aromatic [11C]cyanation methodology
In 1990, a method based on Reissert-Kaufmann Reaction was developed to extend [11C]cyanation reactions to functional groups that were base-sensitive by trapping the H11CN on a silica gel column.93 This trapping method, avoiding using NaOH or KOH which could hinder later reactions. The method was used to synthesize methyl 2-[11C]-cyano-isonicotinate (249) in a 32% RCY via a Reissert-Kaufmann Reaction (Fig. 41). The following year, the same research group used the solid-state support method to synthesize 2-[11C]cyano-isonicotinic acid hydrazide (250) in 10% RCY in 35 min from end of bombardment.148 This compound is a potential radiotracer to diagnose tuberculoma.
Fig. 41.
Synthesis of 2-[11C]cyano-isonicotinic acid hydrazide via a solid-state support.
6. Summary and outlook
In summary, numerous organic molecules, including biologically important molecules such as [11C]α-amino acids (e.g. [11C]alanine, [11C]tyrosine [11C]methionine, [11C]glycine), [11C]sugars (e.g. 2-deoxy-D-[1-11C]glucose, D-[1-11C]glucose, D-[1-11C]mannose), neurotransmitters (e.g. [11C]dopamine, [11C]norepinephrine, [11C]GABA) have been synthesized from H11CN in useful radiochemical yields. 11C-cyanation reactions can be classified in three broad categories. In the first category (Fig. 42), 11C-cyanide is incorporated into organic molecules to produce [11C]nitriles, which can be further modified to generate [11C]amines, [11C]aldehydes, [11C]indoles, [11C]carboxylic acids, [11C]amides, [11C]hydrazides, [11C]amidoximes, [11C]imidamides and [11C]tetrazoles. Some of the conditions used for the secondary transformations have poor chemoselectivity and require judicious selection of reagents, protecting groups and reaction conditions.
Fig. 42.
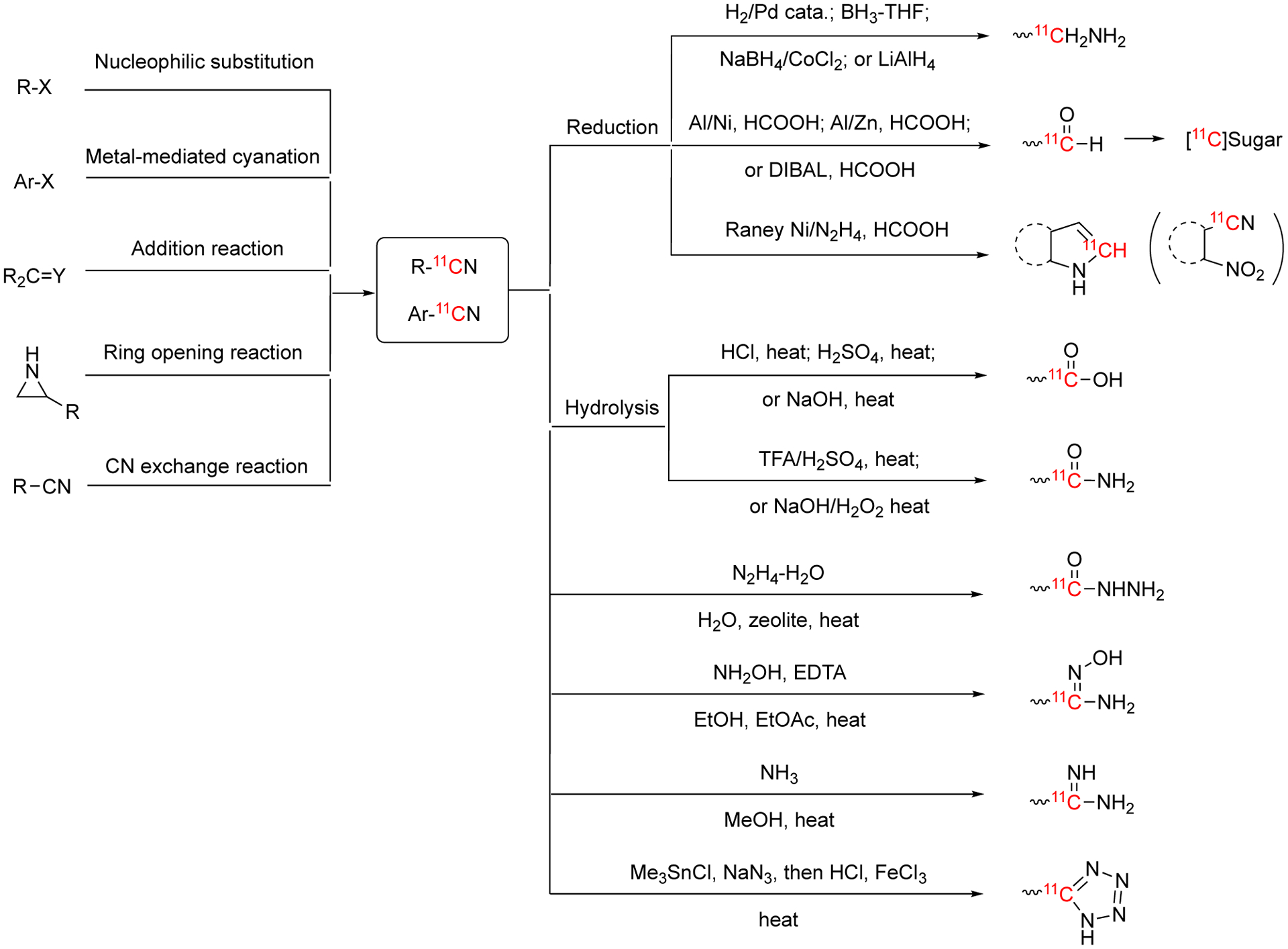
H11CN reactions via [11C]nitriles
In the second category, 11C cyanide is incorporated into organic molecules to generate other types of 11C-labeled intermediates different from nitriles (Fig. 43). For example, the reaction of H11CN with ketones in the presence of ammonium carbonate generates [11C]hydantoins which can be hydrolyzed to produce amino acids. Or the reaction of H11CN with dialkylthexylborane generates the cyanide adduct which upon further chemical transformation produces the corresponding [11C]dialkylketone.
Fig. 43.
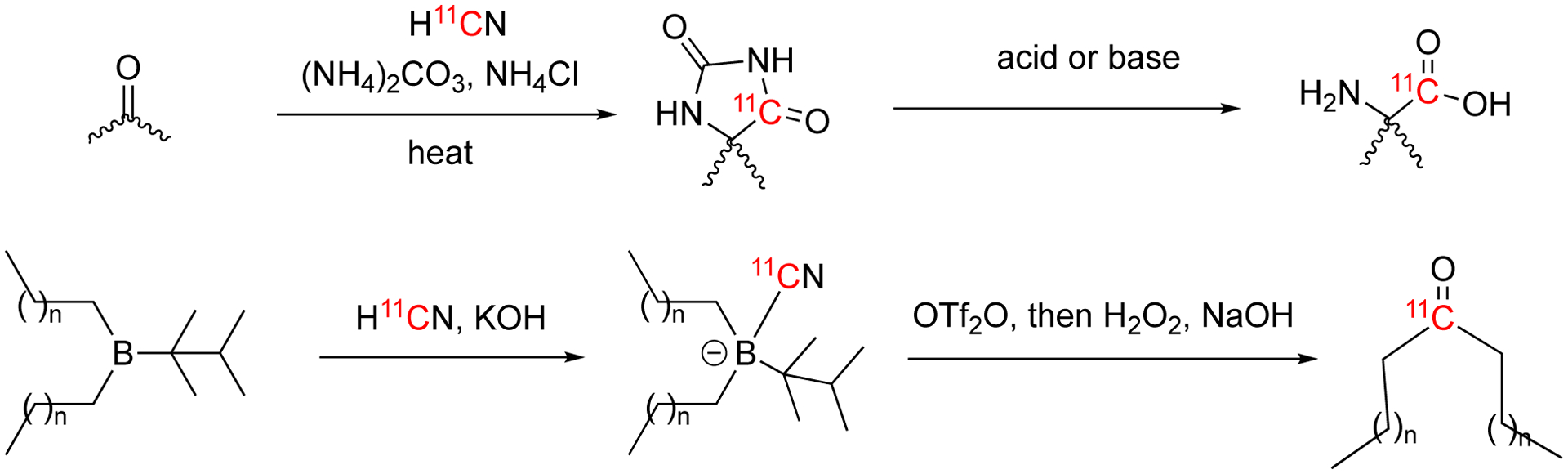
Reactions via [11C]hydantoins and [11C]cyanide
In the third category, [11C]cyanide is first transferred to a secondary 11C synthon (e.g. [11C]cyanate, [11C]thiocyanate, and [11C]cyanogen bromide) (Fig. 44). Starting from these secondary synthons, the 11C labeled N-substituted (thio)urea, (thio)cyanate esters, and guanidines can be obtained by the reactions of [11C](thio)cyanate and [11C]cyanogen bromide with corresponding precursors.
Fig. 44.
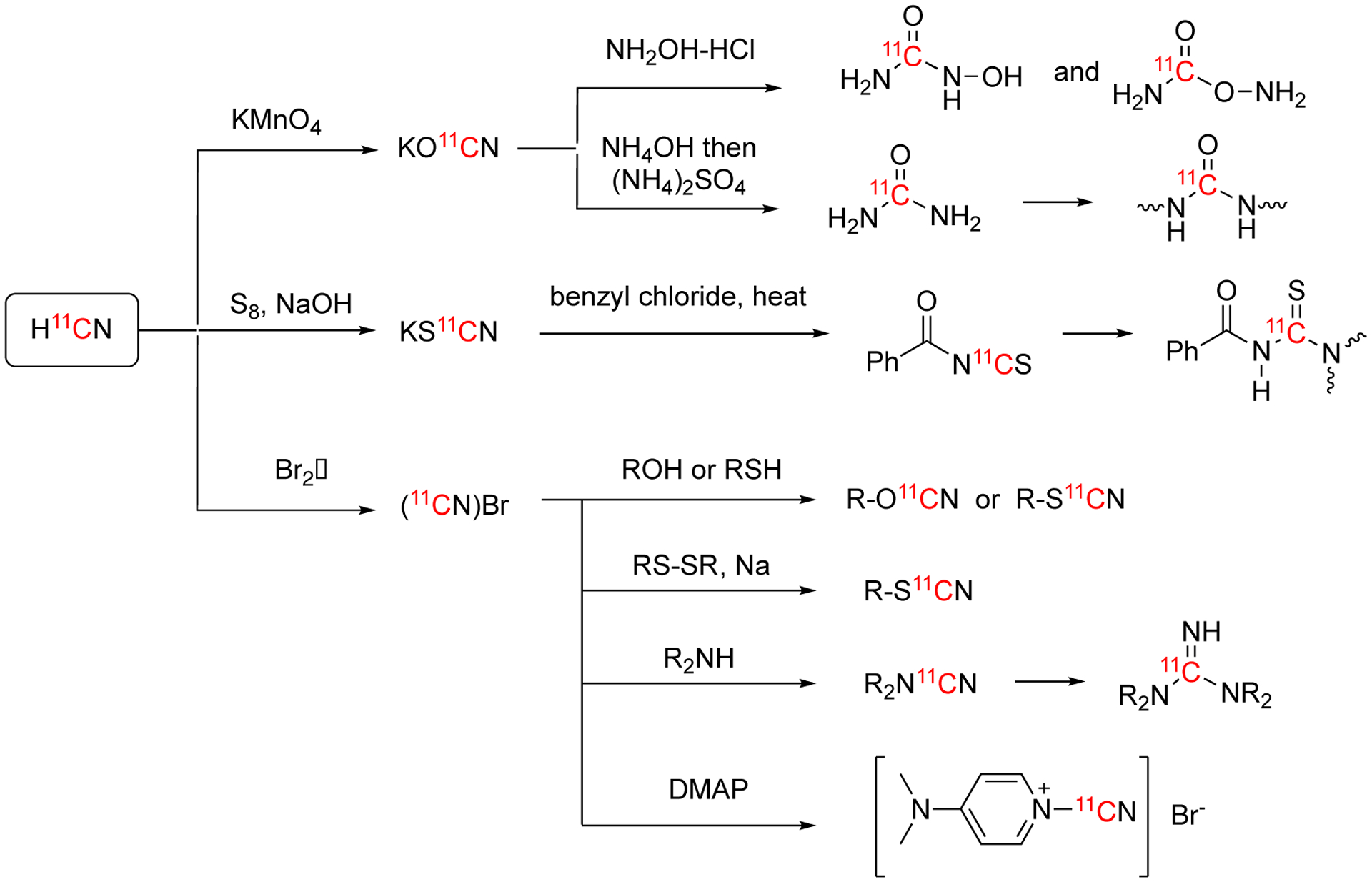
Summary of [11C]cyanations and related reactions (continue).
These reactions and their products illustrate the versatility of H11CN to generate diverse radiolabeled molecules. Nevertheless, many opportunities and challenges remain in order to harness the full potential of H11CN labeling in PET radiochemistry, such as for example:
In our experience, the production of H11CN is not as robust and reproducible as the production of 11CO2 11CH4 and 11CH3I. Variability in the dryness and flow of NH3 gas can result in poor conversion from 11CH4 to H11CN and the transfer of the H11CN through gas lines can also be problematic. Advances in instrumentation to reliably produce H11CN with high molar activity would greatly facilitate the future use of H11CN in PET radiochemistry.
Many 11C-cyanations developed in 20th century to produce labeled amino acids and sugars employed carrier-added 11CN−. These methods may need to be further optimized to produce no carrier added tracers.
Many 11C-cyanation publications prior to 1960s reported only the radiochemistry and did not include the evaluation of the radiolabeled compounds in vitro or in vivo. Further, evaluation of some of these compounds may result in useful tracers.
Even though this review described several examples of multistep reactions in which the nitrile group is further converted into other groups such as amine, amide or carboxylic acid, etc, it remains challenging to do this selectively in complex molecules containing other reactive functional groups. As such, advances in organic chemistry that allow efficient, selective and fast conversion of nitriles into other functional groups would further the utility of H11CN in PET tracer radiochemistry.
Finally, we believe that the rapid growth of PET radiopharmaceuticals is generating a demand for rapid development of new radiolabeled compounds. This field could greatly benefit from the increased use of H11CN labeling and therefore we are optimistic to witness increased use and development of 11C-cyanation in coming years.
Fig. 11.
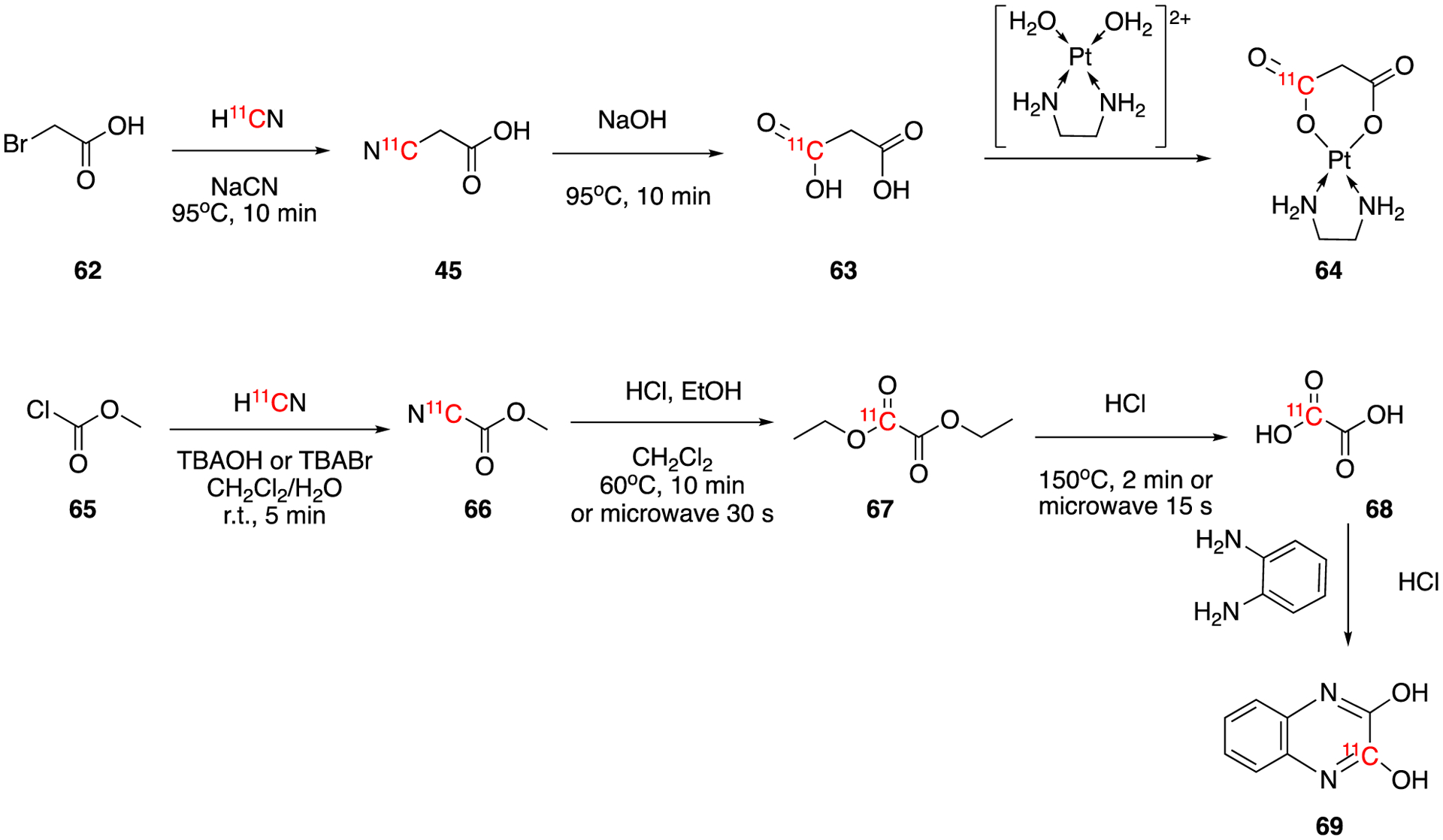
Synthesis of (ethylenediamine)(1-[11C]malonate)platinum(ll) (64), [11C]oxalic acid (68), and [11C]-2,3-dihydroxyquinoxalin (69).
Fig. 22.
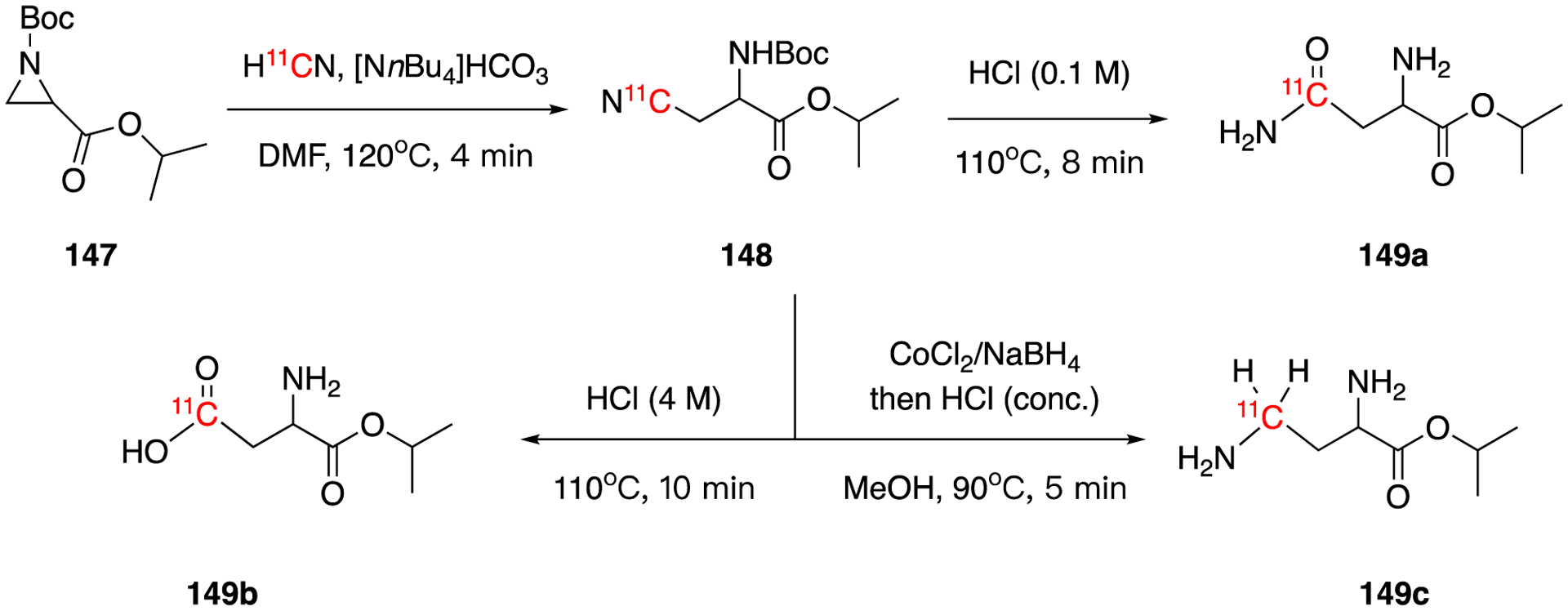
Synthesis of DL-[4-11C]asparagine (174), DL-[4-11C]aspartic acid (175), and DL-2,4-diamino[4-11C]butyric acid (176).
Acknowledgement
We acknowledge the financial support of NIH R21NS120139 (PB), R01NS114066 (PB), and MGH Fund for Medical Discovery (YPZ). We thank the reviewers for their valuable comments and corrections.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- 1.Fowler JS; Wolf AP, Working against Time: Rapid Radiotracer Synthesis and Imaging the Human Brain. Acc. Chem. Res 1997, 30 (4), 181–188. [Google Scholar]
- 2.Phelps ME, Positron emission tomography provides molecular imaging of biological processes. Proceedings of the National Academy of Sciences 2000, 97 (16), 9226–9233. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Willmann JK; van Bruggen N; Dinkelborg LM; Gambhir SS, Molecular imaging in drug development. Nature Reviews Drug Discovery 2008, 7 (7), 591–607. [DOI] [PubMed] [Google Scholar]
- 4.Piel M; Vernaleken I; Rösch F, Positron Emission Tomography in CNS Drug Discovery and Drug Monitoring. J. Med. Chem 2014, 57 (22), 9232–9258. [DOI] [PubMed] [Google Scholar]
- 5.Victor WP, Considerations in the Development of Reversibly Binding PET Radioligands for Brain Imaging. Current Medicinal Chemistry 2016, 23 (18), 1818–1869. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Chansaenpak K; Vabre B; Gabbaï FP, [18F]-Group 13 fluoride derivatives as radiotracers for positron emission tomography. Chem. Soc. Rev 2016, 45 (4), 954–971. [DOI] [PubMed] [Google Scholar]
- 7.Preshlock S; Tredwell M; Gouverneur V, 18F-Labeling of Arenes and Heteroarenes for Applications in Positron Emission Tomography. Chem. Rev 2016, 116 (2), 719–766. [DOI] [PubMed] [Google Scholar]
- 8.van der Born D; Pees A; Poot AJ; Orru RVA; Windhorst AD; Vugts DJ, Fluorine-18 labelled building blocks for PET tracer synthesis. Chem. Soc. Rev 2017, 46 (15), 4709–4773. [DOI] [PubMed] [Google Scholar]
- 9.Miller PW; Long NJ; Vilar R; Gee AD, Synthesis of 11C, 18F, 15O, and 13N Radiolabels for Positron Emission Tomography. Angew. Chem. Int. Ed 2008, 47 (47), 8998–9033. [DOI] [PubMed] [Google Scholar]
- 10.Liang SH; Vasdev N, Total Radiosynthesis: Thinking Outside ‘the Box’. Aust. J. Chem 2015, 68 (9), 1319–1328. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Deng X; Rong J; Wang L; Vasdev N; Zhang L; Josephson L; Liang SH, Chemistry for Positron Emission Tomography: Recent Advances in 11C-, 18F-, 13N-, and 15O-Labeling Reactions. Angew. Chem. Int. Ed 2019, 58 (9), 2580–2605. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Surti S, Update on Time-of-Flight PET Imaging. Journal of Nuclear Medicine 2015, 56 (1), 98–105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Cherry SR; Jones T; Karp JS; Qi J; Moses WW; Badawi RD, Total-Body PET: Maximizing Sensitivity to Create New Opportunities for Clinical Research and Patient Care. Journal of Nuclear Medicine 2018, 59 (1), 3–12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Brugarolas P; Comstock J; Dick DW; Ellmer T; Engle JW; Lapi SE; Liang SH; Parent EE; Kishore Pillarsetty NV; Selivanova S; Sun X; Vavere A; Scott PJH, Fifty Years of Radiopharmaceuticals. Journal of Nuclear Medicine Technology 2020, 48 (Supplement 1), 34S–39S. [PubMed] [Google Scholar]
- 15.Antoni G, Development of carbon-11 labelled PET tracers—radiochemical and technological challenges in a historic perspective. J. Labelled Compd. Radiopharm 2015, 58 (3), 65–72. [DOI] [PubMed] [Google Scholar]
- 16.Kilian K; Pękal A; Juszczyk J, Synthesis of C-methionine through gas phase iodination using Synthra MeI synthesis module. Nukleonika 2016, 61 (1), 29–33. [Google Scholar]
- 17.Dahl K; Halldin C; Schou M, New methodologies for the preparation of carbon-11 labeled radiopharmaceuticals. Clinical and Translational Imaging 2017, 5 (3), 275–289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Rotstein BH; Liang SH; Holland JP; Collier TL; Hooker JM; Wilson AA; Vasdev N, 11CO2 fixation: a renaissance in PET radiochemistry. Chem. Commun 2013, 49 (50), 5621–5629. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Kealey S; Gee A; Miller PW, Transition metal mediated [11C]carbonylation reactions: recent advances and applications. J. Labelled Compd. Radiopharm 2014, 57 (4), 195–201. [DOI] [PubMed] [Google Scholar]
- 20.Rahman O, [11C]Carbon monoxide in labeling chemistry and positron emission tomography tracer development: scope and limitations. J. Labelled Compd. Radiopharm 2015, 58 (3), 86–98. [DOI] [PubMed] [Google Scholar]
- 21.Rotstein BH; Liang SH; Placzek MS; Hooker JM; Gee AD; Dollé F; Wilson AA; Vasdev N, 11C=O bonds made easily for positron emission tomography radiopharmaceuticals. Chem. Soc. Rev 2016, 45 (17), 4708–4726. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Taddei C; Pike VW, [11C]Carbon monoxide: advances in production and application to PET radiotracer development over the past 15 years. EJNMMI Radiopharmacy and Chemistry 2019, 4 (1), 25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Bragg RA; Sardana M; Artelsmair M; Elmore CS, New trends and applications in carboxylation for isotope chemistry. J. Labelled Compd. Radiopharm 2018, 61 (13), 934–948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Nielsen DU; Neumann KT; Lindhardt AT; Skrydstrup T, Recent developments in carbonylation chemistry using [13C]CO, [11C]CO, and [14C]CO. J. Labelled Compd. Radiopharm 2018, 61 (13), 949–987. [DOI] [PubMed] [Google Scholar]
- 25.Taddei C; Gee AD, Recent progress in [11C]carbon dioxide ([11C]CO2) and [11C]carbon monoxide ([11C]CO) chemistry. J. Labelled Compd. Radiopharm 2018, 61 (3), 237–251. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Lu S; Siméon FG; Telu S; Cai L; Pike VW, Chapter Four - The chemistry of labeling heterocycles with carbon-11 or fluorine-18 for biomedical imaging. In Adv. Heterocycl. Chem, Scriven EFV; Ramsden CA, Eds. Academic Press: 2020; Vol. 132, pp 241–384. [Google Scholar]
- 27.Fukumura T; Mori W; Ogawa M; Fujinaga M; Zhang M-R, [11C]phosgene: Synthesis and application for development of PET radiotracers. Nuclear Medicine and Biology 2020. [DOI] [PubMed] [Google Scholar]
- 28.Eriksson J; Antoni G; Långström B; Itsenko O, The development of 11C-carbonylation chemistry: A systematic view. Nuclear Medicine and Biology 2020. [DOI] [PubMed] [Google Scholar]
- 29.Wilson TC; Cailly T; Gouverneur V, Boron reagents for divergent radiochemistry. Chem. Soc. Rev 2018, 47 (18), 6990–7005. [DOI] [PubMed] [Google Scholar]
- 30.Derdau V, New trends and applications in cyanation isotope chemistry. J. Labelled Compd. Radiopharm 2018, 61 (14), 1012–1023. [DOI] [PubMed] [Google Scholar]
- 31.Dubrin J; MacKay C; Pandow ML; Wolfgang R, Reactions of atomic carbon with π-bonded inorganic molecules. J. Inorg. Nucl. Chem 1964, 26 (12), 2113–2122. [Google Scholar]
- 32.Ache HJ; Wolf AP, Reactions of Energetic Carbon Atoms with Nitrogen Molecules [1]. In Radiochimica Acta, 1966; Vol. 6, p 32. [Google Scholar]
- 33.Finn RD; Christman DR; Ache HJ; Wolf AP, The preparation of Cyanide-11C for use in the synthesis of organic radiopharmaceuticals II. The International Journal of Applied Radiation and Isotopes 1971, 22 (12), 735–744. [DOI] [PubMed] [Google Scholar]
- 34.Christman DR; Finn RD; Karlstrom KI; Wolf AP, Production of Carrier-Free H11CN for Medical Use and Radiopharmaceutical Syntheses. IX. Journal of Nuclear Medicine 1973, 14 (11), 864–866. [PubMed] [Google Scholar]
- 35.Christman DR; Finn RD; Karlstrom KI; Wolf AP, The production of ultra high activity 11C-labeled hydrogen cyanide, carbon dioxide, carbon monoxide and methane via the 14N(p,α)11C reaction (XV). The International Journal of Applied Radiation and Isotopes 1975, 26 (8), 435–442. [Google Scholar]
- 36.Niisawa K; Ogawa K; Saito J; Taki K; Karasawa T; Nozaki T, Production of no-carrier-added 11C-carbon disulfide and 11C-hydrogen cyanide by microwave discharge. The International Journal of Applied Radiation and Isotopes 1984, 35 (1), 29–33. [Google Scholar]
- 37.Christman DR; Hoyte RM; Wolf AP, Organic Radiopharmaceuticals Labeled with Isotopes of Short Half-Life I: 11C-1-Dopamine Hydrochloride. Journal of Nuclear Medicine 1970, 11 (8), 474–478. [PubMed] [Google Scholar]
- 38.Fowler JS; Ansari AN; Atkins HL; Bradley-Moore PR; MacGregor RR; Wolf AP, Synthesis and Preliminary Evaluation in Animals of Carrier-Free 11C-1-Dopamine Hydrochloride: X. Journal of Nuclear Medicine 1973, 14 (11), 867–869. [PubMed] [Google Scholar]
- 39.Fowler JS; MacGregor RR; Ansari AN; Atkins HL; Wolf AP, Radiopharmaceuticals. 12. New rapid synthesis of carbon-11-labeled norepinephrine hydrochloride. J. Med. Chem 1974, 17 (2), 246–248. [DOI] [PubMed] [Google Scholar]
- 40.Fowler JS; MacGregor RR; Wolf AP; Ansari AN; Atkins HL, Radiopharmaceuticals. 16. Halogenated dopamine analogs. Synthesis and radiolabeling of 6-iododopamine and tissue distribution studies in animals. J. Med. Chem 1976, 19 (3), 356–360. [DOI] [PubMed] [Google Scholar]
- 41.Winstead MB; Podlesny EJ; Winchell HS, Preparation and scintigraphic evaluation of carbon-11-labeled 2-N-phenethylaminoalkanenitriles. Nuklearmedizin 1978, 17 (6), 277–282. [PubMed] [Google Scholar]
- 42.Iwata R; Ido T; Takahashi T; Nakanishi H; Iida S, Optimization of [11C]HCN production and no-carrier-added [1–11C]amino acid synthesis. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1987, 38 (2), 97–102. [DOI] [PubMed] [Google Scholar]
- 43.Takahashi T; Någren K; Aho K, An alternative synthesis of DL-[1-11C]Alanine from [11C]HCN. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1990, 41 (12), 1187–1191. [Google Scholar]
- 44.Iwata R; Ido T; Tada M, On-column preparation of 1-aminocyclopentane-1-[11C]carboxylic acid. Appl. Radiat. Isot 1995, 46 (9), 899–905. [Google Scholar]
- 45.Huang C; McConathy J, Radiolabeled Amino Acids for Oncologic Imaging. Journal of Nuclear Medicine 2013, 54 (7), 1007–1010. [DOI] [PubMed] [Google Scholar]
- 46.Bjurling P; Antoni G; Watanabe Y; Langstrom B, Enzymatic-synthesis of carboxy-C-11-labeled L-tyrosine, L-DOPA, L-tryptophan and 5-hydroxy-L-tryptophan. Acta Chem. Scand 1990, 44 (2), 178–182. [Google Scholar]
- 47.Bjurling P; Långström B, Synthesis of 1- and 3-11C-labelled L-lactic acid using multi-enzyme catalysis. J. Labelled Compd. Radiopharm 1990, 28 (4), 427–432. [Google Scholar]
- 48.Drandarov K; Schubiger PA; Westera G, Automated no-carrier-added synthesis of [1-11C]-labeled d- and l-enantiomers of lactic acid. Appl. Radiat. Isot 2006, 64 (12), 1613–1622. [DOI] [PubMed] [Google Scholar]
- 49.Maeda M; Koga Y; Fukumura T; Kojima M, [11C]octopamine synthesis using [11C]cyanide: Chemical and enzymatic approaches for the [11C]cyanohydrin synthesis. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1990, 41 (5), 463–469. [DOI] [PubMed] [Google Scholar]
- 50.Studenov AR; Szalda DE; Ding Y-S, Synthesis of no-carrier-added C-11 labeled D- and L-enantiomers of phenylalanine and tyrosine for comparative PET Studies. Nuclear Medicine and Biology 2003, 30 (1), 39–44. [DOI] [PubMed] [Google Scholar]
- 51.Cai H; Mangner TJ; Muzik O; Wang M-W; Chugani DC; Chugani HT, Radiosynthesis of 11C-Levetiracetam: A Potential Marker for PET Imaging of SV2A Expression. ACS Medicinal Chemistry Letters 2014, 5 (10), 1152–1155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Proceedings of the 25th Annual Meeting. Journal of Nuclear Medicine 1978, 19 (6), 662–759.351153 [Google Scholar]
- 53.Abstracts. Second international symposium on radiopharmaceuticals chemistry. J. Labelled Compd. Radiopharm 1979, 16 (1), 1–118. [Google Scholar]
- 54.Shiue C-Y; MacGregor RR; Lade RE; Wan C-N; Wolf AP, A new synthesis of 2-deoxy-d-arabino-hexose. Carbohydr. Res 1979, 74 (1), 323–326. [Google Scholar]
- 55.MacGregor RR; Fowler JS; Wolf AP; Shiue C-Y; Lade RE; Wan C-N, A Synthesis of 2-Deoxy-D-[1-11C]Glucose for Regional Metabolic Studies: Concise Communication. Journal of Nuclear Medicine 1981, 22 (9), 800–803. [PubMed] [Google Scholar]
- 56.Vora MM; Boothe TE; Finn RD; Smith PM; Gilson AJ, Quality control procedures in the preparation of 2-deoxy-D-[1-11C] glucose radiopharmaceutical. J. Labelled Compd. Radiopharm 1983, 20 (3), 417–427. [Google Scholar]
- 57.Padgett HC; Barrio JR; MacDonald NS; Phelps ME, The Unit Operations Approach to the Applied to the Synthesis of [1-11C]2-Deoxy-d-Glucose for Routine Clinical Applications. Journal of Nuclear Medicine 1982, 23 (8), 739–744. [PubMed] [Google Scholar]
- 58.Elias H; Lotterhos HF; Weimer K, Notiz über die schnelle Synthese von [3-11C]β-Alanin. Chem. Ber 1972, 105 (11), 3754–3756. [Google Scholar]
- 59.Mestelan G; Crouzel C; Comar D, Synthesis and distribution kinetics in animals of [α-11C] 3,4-dimethoxyphenethylamine. International Journal of Nuclear Medicine and Biology 1977, 4 (3), 185–193. [DOI] [PubMed] [Google Scholar]
- 60.Stone-Elander S; Nilsson JLG; Blomqvist G; Ehrin E; Eriksson L; Garmelius B; Greitz T; Johnström P; Sjögren I; Widén L, 11C-2-Deoxy-d-glucose: synthesis and preliminary comparison with 11C-d-glucose as a tracer for cerebral energy metabolism in PET studies. European Journal of Nuclear Medicine 1985, 10 (11), 481–486. [DOI] [PubMed] [Google Scholar]
- 61.van Haver D; Rabi NA; Vandewalle M; Goethals P; Vandecasteele C, Routine production of 2-deoxy-D-[1-11C]glucose : An alternative. J. Labelled Compd. Radiopharm 1985, 22 (7), 657–666. [Google Scholar]
- 62.Winstead MB; Dischino DD; Munder NA; Walsh C; Winchell HS, Relationship of molecular structure to in vivo distribution of carbon-11-labeled compounds. European Journal of Nuclear Medicine 1980, 5 (2), 165–169. [DOI] [PubMed] [Google Scholar]
- 63.McPherson DW; Fowler JS; Wolf AP; Arnett CD; Brodie JD; Volkow N, Synthesis and Biodistribution of No-Carrier-Added [1-11C]Putrescine. Journal of Nuclear Medicine 1985, 26 (10), 1186–1189. [PubMed] [Google Scholar]
- 64.Ding Y-S; Antoni G; Fowler JS; Wolf AP; Langstrom B, Synthesis of L-[5-11C]ornithine. J. Labelled Compd. Radiopharm 1989, 27 (9), 1079–1090. [Google Scholar]
- 65.De Spiegeleer B; Goethals P; Slegers G; Gillis E; Van den Bossche W; De Moerloose P, Synthesis and Radiopharmaceutical Preparation of (Ethylenediamine) (1-Carbon-11-Malonate) Platinum(II) for PET Studies. Journal of Nuclear Medicine 1988, 29 (6), 1107–1113. [PubMed] [Google Scholar]
- 66.Thorell J-O; Stone-Elander S; Elander N, Preparation of [11C]diethyl oxalate and [11C]oxalic acid and demonstration of their use in the synthesis of [11C]-2,3-dihydroxyquinoxaline. J. Labelled Compd. Radiopharm 1993, 33 (11), 995–1005. [Google Scholar]
- 67.Takahashi T; Ido T; Hatano K; Iwata R; Nakanishi H, Synthesis of 1-11C-labeled fatty acid from [11C]HCN. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1990, 41 (7), 649–654. [Google Scholar]
- 68.Hassan M; Thorell J-O; Warne N; Stone-Elander S, 11C-labeling of busulphan. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1991, 42 (11), 1055–1059. [DOI] [PubMed] [Google Scholar]
- 69.Thorell J-O; Stone-Elander S; Elander N, Difunctional two-carbon molecules derived from [11C]cyanide. J. Labelled Compd. Radiopharm 1994, 34 (4), 383–390. [Google Scholar]
- 70.Zhang Z; Ding Y-S; Studenov AR; Gerasimov MR; Ferrieri RA, Novel synthesis of [1–11C]γ-vinyl-γ-aminobutyric acid ([1–11C]GVG) for pharmacokinetic studies of addiction treatment. J. Labelled Compd. Radiopharm 2002, 45 (3), 199–211. [Google Scholar]
- 71.Lee SJ; Fowler JS; Alexoff D; Schueller M; Kim D; Nauth A; Weber C; Kim SW; Hooker JM; Ma L; Qu W, An efficient and practical synthesis of [2-11C]indole via superfast nucleophilic [11C]cyanation and RANEY® Nickel catalyzed reductive cyclization. Organic & Biomolecular Chemistry 2015, 13 (46), 11235–11243. [DOI] [PubMed] [Google Scholar]
- 72.Gleede T; Riehl B; Shea C; Kersting L; Cankaya AS; Alexoff D; Schueller M; Fowler JS; Qu W, Investigation of SN2 [11C]cyanation for base-sensitive substrates: an improved radiosynthesis of L-[5-11C]-glutamine. Amino Acids 2015, 47 (3), 525–533. [DOI] [PubMed] [Google Scholar]
- 73.Antoni G; Omura H; Bergström M; Furuya Y; Moulder R; Roberto A; Sundin A; Watanabe Y; Långström B, Synthesis of L-2,4-Diamino[4-11C]butyric acid and its use in some In vitro and In vivo tumour models. Nuclear Medicine and Biology 1997, 24 (6), 595–601. [DOI] [PubMed] [Google Scholar]
- 74.Antoni G; Omura H; Ikemoto M; Moulder R; Watanabe Y; Långström B, Enzyme catalysed synthesis of L-[4–11C]aspartate and L-[5-11C]glutamate. J. Labelled Compd. Radiopharm 2001, 44 (4), 287–294. [Google Scholar]
- 75.Reid AE; Kim SW; Seiner B; Fowler FW; Hooker J; Ferrieri R; Babst B; Fowler JS, Radiosynthesis of C-11 labeled auxin (3-indolyl[1-11C]acetic acid) and its derivatives from gramine. J. Labelled Compd. Radiopharm 2011, 54 (8), 433–437. [Google Scholar]
- 76.Xu Y; Alexoff, D. L; Kunert, A. T; Qu W; Kim D; Paven M; Babst, B. A; Ferrieri, R. A; Schueller, M. J; Fowler, J. S, Radiosynthesis of 3-indolyl[1-11C]acetic acid for phyto-PET-imaging: An improved production procedure and formulation method. Appl. Radiat. Isot 2014, 91, 155–160. [DOI] [PubMed] [Google Scholar]
- 77.Lee S; Alexoff DL; Shea C; Kim D; Schueller M; Fowler JS; Qu W, Tetraethylene glycol promoted two-step, one-pot rapid synthesis of indole-3-[1-11C]acetic acid. Tetrahedron Lett 2015, 56 (3), 517–520. [Google Scholar]
- 78.Hayes RL; Washburn LC; Wieland BW; Sun TT; Turtle RR; Butler TA, Carboxyl-Labeled 11C-1-Aminocyclopentanecarboxylic Acid, a Potential Agent for Cancer Detection. Journal of Nuclear Medicine 1976, 17 (8), 748–751. [PubMed] [Google Scholar]
- 79.Hayes RL; Washburn LC; Wieland BW; Sun TT; Anon JB; Butler TA; Callahan AP, Synthesis and purification of 11C-carboxyl-labeled amino acids. The International Journal of Applied Radiation and Isotopes 1978, 29 (3), 186–187. [Google Scholar]
- 80.Sambre J; Vandecasteele C; Goethals P; Rabi NA; Van Haver D; Slegers G, Routine production of H11CN and [11C]-1-Aminocyclopentanecarboxylic acid. The International Journal of Applied Radiation and Isotopes 1985, 36 (4), 275–278. [Google Scholar]
- 81.Casey DL; Digenis GA; Wesner DA; Washburn LC; Chaney JE; Hayes RL; Callahan AP, Preparation and preliminary tissue studies of optically active 11C-D- and L-phenylalanine. The International Journal of Applied Radiation and Isotopes 1981, 32 (5), 325–330. [DOI] [PubMed] [Google Scholar]
- 82.Barrio JR; Keen RE; Ropchan JR; MacDonald NS; Baumgartner FJ; Padgett HC; Phelps ME, L-[1-11C]Leucine: Routine Synthesis by Enzymatic Resolution. Journal of Nuclear Medicine 1983, 24 (6), 515–521. [PubMed] [Google Scholar]
- 83.Schmall B; Conti PS; Bigler RE; Zanzonico PB; Robert Dahl J; Sundoro-Wu BM; Jacobsen JK; Lee R, Synthesis and quality assurance of [11C]alpha-aminoisobutyric acid (AIB), a potential radiotracer for imaging and amino acid transport studies in normal and malignant tissues. International Journal of Nuclear Medicine and Biology 1984, 11 (3), 209–214. [DOI] [PubMed] [Google Scholar]
- 84.Johnström P; Stone-Elander S; Ericson K; Mosskin M; Bergström M, 11C-labelled glycine: Synthesis and preliminary report on its use in the investigation of intracranial tumours using positron emission tomography. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1987, 38 (9), 729–734. [DOI] [PubMed] [Google Scholar]
- 85.Ropchan JR; Barrio JR, Enzymatic Synthesis of [1–11C]Pyruvic Acid, l-[1-11C]Lactic Acid and l-[1–11C]Alanine via dl-[1–11C]Alanine. Journal of Nuclear Medicine 1984, 25 (8), 887–892. [PubMed] [Google Scholar]
- 86.Fissekis JD; Nielsen CM; Tirelli S; Knott AF; Dahl JR, A remote control process for the routine synthesis of branched chain [1-11C]α amino acids. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1991, 42 (12), 1169–1176. [Google Scholar]
- 87.Shiue C-Y; Wolf AP, The syntheses of 1-[11C]-D-glucose and related compounds for the measurement of brain glucose metabolism. J. Labelled Compd. Radiopharm 1985, 22 (2), 171–182. [Google Scholar]
- 88.Maeda M; Nishimura S; Fukumura T; Kojima M, Enantioselective synthesis of 11C-labeled phenylethanolamine. J. Labelled Compd. Radiopharm 1988, 25 (3), 233–245. [Google Scholar]
- 89.Tada M; Oikawa A; Matsuzawa T; Itoh M; Fukuda H; Kubota K; Kawai H; Abe Y; Sugiyama H; Ido T; Ishiwata K; Iwata R; Imahori Y; Sato T, A convenient synthesis of D-[1-11C]glucopyranose and D-[1-11C]galactopyranose using diborane. J. Labelled Compd. Radiopharm 1989, 27 (1), 1–7. [Google Scholar]
- 90.Nishimura S; Yajima K; Harada N; Ogawa Y; Hayashi N, Automated synthesis of radiopharmaceuticals for PET - an apparatus for 1-C-11 labelled aldoses. J. Autom. Chem 1994, 16 (6), 195–204. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Xing JH; Brooks AF; Fink D; Zhang HB; Piert MR; Scott PJH; Shao X, High-Yielding Automated Convergent Synthesis of No-Carrier-Added C-11-Carbonyl -Labeled Amino Acids Using the Strecker Reaction. Synlett 2017, 28 (3), 371–375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Carroll VN; Truillet C; Shen B; Flavell RR; Shao X; Evans MJ; VanBrocklin HF; Scott PJH; Chin FT; Wilson DM, [11C]Ascorbic and [11C]dehydroascorbic acid, an endogenous redox pair for sensing reactive oxygen species using positron emission tomography. Chem. Commun 2016, 52 (27), 4888–4890. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Somawardhana CW; Sajjad M; Lambrecht RM, Novel solid-state support for rapid synthesis of radiopharmaceuticals labelled with [11C]-cyanide. J. Chem. Soc., Chem. Commun 1990, (5), 370–371. [Google Scholar]
- 94.Somawardhana CW; Sajjad M; Lambrecht RM, Solid state support for the synthesis of [1−11C]-putrescine. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1991, 42 (6), 555–558. [DOI] [PubMed] [Google Scholar]
- 95.Antoni G; Långström B, Synthesis of γ-amino[4-11C]butyric acid (GABA). J. Labelled Compd. Radiopharm 1989, 27 (5), 571–576. [Google Scholar]
- 96.Gillings NM; Gee AD, Synthesis of [4-11C]amino acids via ring-opening of aziridine-2-carboxylates. J. Labelled Compd. Radiopharm 2001, 44 (13), 909–920. [Google Scholar]
- 97.Winstead MB; Chern C.-i.; Lin T-H; Khentigan A; Lamb JF; Winchell HS, Synthesis and preliminary scintigraphic evaluation of in vivo distribution of 11C-hydroxyurea/isohydroxyurea and 11C-Cyanate. The International Journal of Applied Radiation and Isotopes 1978, 29 (7), 443–447. [DOI] [PubMed] [Google Scholar]
- 98.Emran AM; Boothe TE; Finn RD; Vora MM; Kothari PJ, Preparation of 11C-Urea from No-carrier-added 11C-Cyanide. The International Journal of Applied Radiation and Isotopes 1983, 34 (7), 1013–1014. [Google Scholar]
- 99.Boothe TE; Emran AM; Finn RD; Vora MM; Kothari PJ, Use of 11C as a tracer for studying the synthesis of [11C]urea from [11C]cyanide. The International Journal of Applied Radiation and Isotopes 1985, 36 (2), 141–144. [DOI] [PubMed] [Google Scholar]
- 100.Emran AM; Boothe TE; Finn RD; Vora MM; Kothari PJ, Use of liquid chromatography for separation and determination of carrier species associated with the synthesis of no carrier-added 11C-labelled compounds determination of the specific activity of [11C]urea. J. Radioanal. Nucl. Chem 1985, 91 (2), 277–284. [Google Scholar]
- 101.Emran AM; Boothe TE; Finn RD; Vora MM; Kothari PJ; Thomas Wooten J, Optimized production of high specific activity [11C]urea. The International Journal of Applied Radiation and Isotopes 1985, 36 (9), 739–740. [DOI] [PubMed] [Google Scholar]
- 102.Emran AM; Boothe TE; Finn RD; Vora MM; Kothari PJ, Use of 11C as a tracer for studying the synthesis of radiolabelled compounds—II: 2-[11C]-5,5-diphenylhydantoin from [11C]cyanide. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1986, 37 (10), 1033–1038. [Google Scholar]
- 103.Westerberg G; Langstrom B, Synthesis of C-11 cyanogen-bromide and (C-13)-cyanogen bromide, useful electrophilic labeling precursors. Acta Chem. Scand 1993, 47 (10), 974–978. [Google Scholar]
- 104.Westerberg G; Långström B, On-line production of [11C]cyanogen bromide. Appl. Radiat. Isot 1997, 48 (4), 459–461. [Google Scholar]
- 105.Westerberg G; Kärger W; Onoe H; Långström B, [11C]cyanogen bromide in the synthesis of 1,3-di(2-tolyl)-[11C]guanidine. J. Labelled Compd. Radiopharm 1994, 34 (8), 691–696. [Google Scholar]
- 106.Zhao AY; Brooks AF; Raffel DM; Stauff J; Arteaga J; Scott PJH; Shao X, Fully Automated Radiosynthesis of [11C]Guanidines for Cardiac PET Imaging. ACS Medicinal Chemistry Letters 2020, 11 (11), 2325–2330. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Westerberg G; Bergström M; Gustafson S; Lindqvist U; Sundin A; Långström B, Labelling of polysaccharides using [11C]cyanogen bromide. In Vivo and in vitro evaluation of 11C-hyaluronan uptake kinetics. Nuclear Medicine and Biology 1995, 22 (2), 251–256. [DOI] [PubMed] [Google Scholar]
- 108.Amano R; Crouzel C, Synthesis of no-carrier-added 11C-labelled methylamine. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1986, 37 (6), 541–543. [Google Scholar]
- 109.Jay M; Layton WJ; Digenis GA, Exchange between labelled cyanide and the nitrile function of acetonitrile. Tetrahedron Lett 1980, 21 (27), 2621–2624. [Google Scholar]
- 110.Kothari PJ; Finn RD; Kabalka GW; Vora MM; Boothe TE; Emran AM; Mohammadi M, Carbon-11 labeled dialkylketones: Synthesis of 9-[11C]heptadecan-9-one. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1986, 37 (6), 471–473. [DOI] [PubMed] [Google Scholar]
- 111.Johnström P; Bergman L; Varnäs K; Malmquist J; Halldin C; Farde L, Development of rapid multistep carbon-11 radiosynthesis of the myeloperoxidase inhibitor AZD3241 to assess brain exposure by PET microdosing. Nuclear Medicine and Biology 2015, 42 (6), 555–560. [DOI] [PubMed] [Google Scholar]
- 112.Balatoni JA; Adam MJ; Brule M; Hall LD, Labelled Compd J. Radiopharm 1986, 23, 1054. [Google Scholar]
- 113.Balatoni JA; Adam MJ; Hall LD, Synthesis of 11C-labeled aromatics using aryl chromium tricarbonyl intermediates. J. Labelled Compd. Radiopharm 1989, 27 (12), 1429–1435. [Google Scholar]
- 114.Andersson J; Malmborg P; Langstrom B, Labelled Compd J. Radiopharm 1991, 30, 144–145. [Google Scholar]
- 115.Andersson Y; Langstrom B, Transition metal-mediated reactions using C-11 cyanide in synthesis of C-11 labeled aromatic-compounds. J. Chem. Soc.-Perkin Trans 1 1994, (11), 1395–1400. [Google Scholar]
- 116.Andersson Y; Bergström M; Långström B, Synthesis of 11C-labelled benzamide compounds as potential tracers for poly(ADP-ribose) synthetase. Appl. Radiat. Isot 1994, 45 (6), 707–714. [DOI] [PubMed] [Google Scholar]
- 117.Lee HG; Milner PJ; Placzek MS; Buchwald SL; Hooker JM, Virtually Instantaneous, Room-Temperature [11C]-Cyanation Using Biaryl Phosphine Pd(0) Complexes. Journal of the American Chemical Society 2015, 137 (2), 648–651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Zhao W; Lee HG; Buchwald SL; Hooker JM, Direct 11CN-Labeling of Unprotected Peptides via Palladium-Mediated Sequential Cross-Coupling Reactions. Journal of the American Chemical Society 2017, 139 (21), 7152–7155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Zhang Z; Niwa T; Watanabe Y; Hosoya T, Palladium(II)-mediated rapid 11C-cyanation of (hetero)arylborons. Organic & Biomolecular Chemistry 2018, 16 (41), 7711–7716. [DOI] [PubMed] [Google Scholar]
- 120.Andersson Y; Tyrefors N; Sihver S; Onoe H; Watanabe Y; Tsukada H; Långström B, Synthesis of a 11C-labelled derivative of the N-methyl-D-aspartate receptor antagonist MK-801. J. Labelled Compd. Radiopharm 1998, 41 (6), 567–576. [Google Scholar]
- 121.Sandell J; Halldin C; Hall H; Thorberg S-O; Werner T; Sohn D; Sedvall G; Farde L, Radiosynthesis and autoradiographic evaluation of [11C]NAD-299, a radioligand for visualization of the 5-HT1A receptor. Nuclear Medicine and Biology 1999, 26 (2), 159–164. [DOI] [PubMed] [Google Scholar]
- 122.Sandell J; Halldin C; Chou Y-H; Swahn C-G; Thorberg S-O; Farde L, PET-examination and metabolite evaluation in monkey of [11C]NAD-299, a radioligand for visualisation of the 5-HT1A receptor. Nuclear Medicine and Biology 2002, 29 (1), 39–45. [DOI] [PubMed] [Google Scholar]
- 123.Donohue SR; Pike VW; Finnema SJ; Truong P; Andersson J; Gulyás B; Halldin C, Discovery and Labeling of High-Affinity 3,4-Diarylpyrazolines as Candidate Radioligands for In Vivo Imaging of Cannabinoid Subtype-1 (CB1) Receptors. J. Med. Chem 2008, 51 (18), 5608–5616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 124.Tsujikawa T; Zoghbi SS; Hong J; Donohue SR; Jenko KJ; Gladding RL; Halldin C; Pike VW; Innis RB; Fujita M, In vitro and in vivo evaluation of 11C-SD5024, a novel PET radioligand for human brain imaging of cannabinoid CB1 receptors. NeuroImage 2014, 84, 733–741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Hjorth S; Karlsson C; Jucaite A; Varnäs K; Wählby Hamrén U; Johnström P; Gulyás B; Donohue SR; Pike VW; Halldin C; Farde L, A PET study comparing receptor occupancy by five selective cannabinoid 1 receptor antagonists in non-human primates. Neuropharmacology 2016, 101, 519–530. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Kil K-E; Biegon A; Ding Y-S; Fischer A; Ferrieri RA; Kim SW; Pareto D; Schueller MJ; Fowler JS, Synthesis and PET studies of [11C-cyano]letrozole (Femara), an aromatase inhibitor drug. Nuclear Medicine and Biology 2009, 36 (2), 215–223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bennacef I; Salinas CA; Bonasera TA; Gunn RN; Audrain H; Jakobsen S; Nabulsi N; Weinzimmer D; Carson RE; Huang Y; Holmes I; Micheli F; Heidbreder C; Gentile G; Rossi T; Laruelle M, Dopamine D3 receptor antagonists: The quest for a potentially selective PET ligand. Part 3: Radiosynthesis and in vivo studies. Bioorganic & Medicinal Chemistry Letters 2009, 19 (17), 5056–5059. [DOI] [PubMed] [Google Scholar]
- 128.Liu L; Xu Y; Shea C; Fowler JS; Hooker JM; Tonge PJ, Radiosynthesis and Bioimaging of the Tuberculosis Chemotherapeutics Isoniazid, Rifampicin and Pyrazinamide in Baboons. J. Med. Chem 2010, 53 (7), 2882–2891. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 129.Siméon FG; Liow J-S; Zhang Y; Hong J; Gladding RL; Zoghbi SS; Innis RB; Pike VW, Synthesis and characterization in monkey of [11C]SP203 as a radioligand for imaging brain metabotropic glutamate 5 receptors. European Journal of Nuclear Medicine and Molecular Imaging 2012, 39 (12), 1949–1958. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Lohith TG; Tsujikawa T; Siméon FG; Veronese M; Zoghbi SS; Lyoo CH; Kimura Y; Morse CL; Pike VW; Fujita M; Innis RB, Comparison of two PET radioligands, [11C]FPEB and [11C]SP203, for quantification of metabotropic glutamate receptor 5 in human brain. Journal of Cerebral Blood Flow & Metabolism 2016, 37 (7), 2458–2470. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Andersson JD; Seneca N; Truong P; Wensbo D; Raboisson P; Farde L; Halldin C, Palladium mediated 11C-cyanation and characterization in the non-human primate brain of the novel mGluR5 radioligand [11C]AZD9272. Nuclear Medicine and Biology 2013, 40 (4), 547–553. [DOI] [PubMed] [Google Scholar]
- 132.Varnäs K; Juréus A; Finnema SJ; Johnström P; Raboisson P; Amini N; Takano A; Stepanov V; Halldin C; Farde L, The metabotropic glutamate receptor 5 radioligand [11C]AZD9272 identifies unique binding sites in primate brain. Neuropharmacology 2018, 135, 455–463. [DOI] [PubMed] [Google Scholar]
- 133.Nag S; Varnäs K; Arakawa R; Jahan M; Schou M; Farde L; Halldin C, Synthesis, Biodistribution, and Radiation Dosimetry of a Novel mGluR5 Radioligand: 18F-AZD9272. ACS Chemical Neuroscience 2020, 11 (7), 1048–1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Varnäs K; Cselényi Z; Arakawa R; Nag S; Stepanov V; Moein MM; Johnström P; Kingston L; Elmore CS; Halldin C; Farde L, The pro-psychotic metabotropic glutamate receptor compounds fenobam and AZD9272 share binding sites with monoamine oxidase-B inhibitors in humans. Neuropharmacology 2020, 162, 107809. [DOI] [PubMed] [Google Scholar]
- 135.Zheng M-Q; Nabulsi N; Kim SJ; Tomasi G; Lin S.-f.; Mitch C; Quimby S; Barth V; Rash K; Masters J; Navarro A; Seest E; Morris ED; Carson RE; Huang Y, Synthesis and Evaluation of 11C-LY2795050 as a κ-Opioid Receptor Antagonist Radiotracer for PET Imaging. Journal of Nuclear Medicine 2013, 54 (3), 455–463. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 136.Zheng M-Q; Kim SJ; Holden D; Lin S.-f.; Need A; Rash K; Barth V; Mitch; Navarro A; Kapinos M; Maloney K; Ropchan J; Carson RE; Huang Y, An Improved Antagonist Radiotracer for the κ-Opioid Receptor: Synthesis and Characterization of 11C-LY2459989. Journal of Nuclear Medicine 2014, 55 (7), 1185–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Airaksinen AJ; Andersson J; Truong P; Karlsson O; Halldin C, Radiosynthesis of [11C]ximelagatran via palladium catalyzed [11C]cyanation. J. Labelled Compd. Radiopharm 2008, 51 (1), 1–5. [Google Scholar]
- 138.Siméon F; Sobrio F; Gourand F; Barré L, Total synthesis and radiolabelling of an efficient rt-PA inhibitor: [11C] (Z,Z )-BABCH. A first route to [11C] labelled amidines. J. Chem. Soc., Perkin Trans 1 2001, (7), 690–694. [Google Scholar]
- 139.Q3D(R1) Guidelines for Elemental Impurities, International Council for Harmonisation of Technical Requirements for Pharmaceuticals for Human Use (ICH) https://database.ich.org/sites/default/files/Q3D-R1EWG_Document_Step4_Guideline_2019_0322.pdf (accessed 8//16/2021).
- 140.Ponchant M; Hinnen F; Demphel S; Crouzel C, [11C]copper(I) cyanide: a new radioactive precursor for 11C-cyanation and functionalization of haloarenes. Appl. Radiat. Isot 1997, 48 (6), 755–762. [Google Scholar]
- 141.Ponchant M; Demphel S; Hinnen F; Crouzel C, Radiosynthesis of [tetrazoyl-11C]irbesartan, a non-peptidic angiotensin II antagonist. European Journal of Medicinal Chemistry 1997, 32 (9), 747–752. [Google Scholar]
- 142.Ma L; Placzek MS; Hooker JM; Vasdev N; Liang SH, [11C]Cyanation of arylboronic acids in aqueous solutions. Chem. Commun 2017, 53 (49), 6597–6600. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 143.Makaravage KJ; Shao X; Brooks AF; Yang L; Sanford MS; Scott PJH, Copper(II)-Mediated [11C]Cyanation of Arylboronic Acids and Arylstannanes. Org. Lett 2018, 20 (6), 1530–1533. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 144.Yang L; Brooks AF; Makaravage KJ; Zhang H; Sanford MS; Scott PJH; Shao X, Radiosynthesis of [11C]LY2795050 for Preclinical and Clinical PET Imaging Using Cu(II)-Mediated Cyanation. ACS Medicinal Chemistry Letters 2018, 9 (12), 1274–1279. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 145.Mathews WB; Monn JA; Ravert HT; Holt DP; Schoepp DD; Dannals RF, Synthesis of a mGluR5 antagonist using [11C]copper(I) cyanide. J. Labelled Compd. Radiopharm 2006, 49 (9), 829–834. [Google Scholar]
- 146.Oi N; Suzuki M; Terauchi T; Tokunaga M; Nakatani Y; Yamamoto N; Fukumura T; Zhang M-R; Suhara T; Higuchi M, Synthesis and Evaluation of Novel Radioligands for Positron Emission Tomography Imaging of the Orexin-2 Receptor. J. Med. Chem 2013, 56 (16), 6371–6385. [DOI] [PubMed] [Google Scholar]
- 147.Obokata N; Seki C; Hirata T; Maeda J; Ishii H; Nagai Y; Matsumura T; Takakuwa M; Fukuda H; Minamimoto T; Kawamura K; Zhang M-R; Nakajima T; Saijo T; Higuchi M, Synthesis and preclinical evaluation of [11C]MTP38 as a novel PET ligand for phosphodiesterase 7 in the brain. European Journal of Nuclear Medicine and Molecular Imaging 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 148.Somawardhana CW; Sajjad M; Lambrecht RM, Radiopharmaceutical for differential diagnosis of tuberculoma: Synthesis of 2−[11C]cyano-isonicotinic acid hydrazide. International Journal of Radiation Applications and Instrumentation. Part A. Applied Radiation and Isotopes 1991, 42 (6), 559–562. [DOI] [PubMed] [Google Scholar]