

Abstract
Human endogenous retroviruses represent approximately 8% of our genome. Most of these sequences are defective except for a few genes such as the ancestral retroviral HEMO envelope gene (Human Endogenous MER34 ORF), recently characterized by our group. In this study, we characterized transcriptional activation of HEMO in primary tumors from The Cancer Genome Atlas (TCGA) and in metastatic tumors from a Gustave Roussy cohort. Pan‐cancer detection of the HEMO protein in a series of patient samples validated these results. Differential gene expression analysis in various TCGA datasets revealed a link between HEMO expression and activation of Wnt/β‐catenin signaling, in particular in endometrial cancer. Studies on cell models led us to propose that the Wnt/β‐catenin pathway could act as an upstream regulator of this retroviral endogenous sequence in tumor condition. Characterization of transcriptomic profiles of both HEMOLow and HEMOHigh tumors suggested that activation of HEMO is negatively associated with immune response signatures. Taken together, these results highlight that HEMO, as an endogenous retroviral envelope protein specifically expressed in tumors, represents a promising tumor biomarker and therapeutic target.
Keywords: cancer, ERVMER34‐1, HEMO, HERV, TCGA, Wnt/β‐catenin
Human endogenous retroviruses represent approximately 8% of our genome. Among these sequences, HEMO (Human Endogenous MER34 ORF) is the oldest retroviral envelope gene, endogenized 100 million years ago. Using in silico and experimental pan‐cancer approaches, we showed that the HEMO protein is a tumor biomarker whose expression can be regulated by the Wnt/β‐catenin pathway.
Abbreviations
- ADC
adenocarcinoma
- APC
adenomatosis polyposis coli
- C
non-tumor control adjacent tissues
- CTNNB1
catenin beta 1
- ERVMER34‐1
endogenous retrovirus medium‐reiteration‐family‐34 member 1
- FFPE
formalin‐fixed, paraffin‐embedded
- GO
gene ontology
- GSEA
gene set enrichment analysis
- GTEx
Genotype‐Tissue Expression
- HEMO
human endogenous MER34 ORF
- Her2
human epidermal growth factor receptor 2
- HERV
human endogenous retroviruses
- Kid-N
kidney normal tissues
- MOSCATO
molecular screening for cancer treatment optimization
- N
normal tissues
- SCC
squamous cell carcinoma
- T
tumor tissues
- TCGA
The Cancer Genome Atlas
1. Introduction
Human endogenous retroviruses (HERVs) originate from ancient retroviral germline invasions by infectious retroviruses, that recurrently occurred in vertebrates, since their origin more than 450 million years ago [1, 2, 3, 4, 5]. Infection and entry of the retrovirus into the cell are usually driven by an interaction between the retroviral envelope protein and a membrane protein used as a receptor on the target cell. The RNA retroviral genome is then reverse‐transcribed and integrated as a DNA proviral copy into the chromosome, with the retroviral gag‐pol‐env genes flanked by two LTR sequences (5′ and 3′ long terminal repeats). These relatively frequent events in a retroviral infectious cycle usually concern somatic cells and facilitate spreading of the retroviruses to other cells and individuals, the most representative of current infectious retroviruses in humans being HIV and HTLV‐I retroviruses. However, infections can also target germline cells in adults or embryonic cells at an early stage in utero and consequently fix the retroviral genome in cells of the descendants, as an endogenous retrovirus (ERV). Although rare at the individual level, these events have accumulated during the course of vertebrate evolution. Endogenized retroviral sequences were thereafter transmitted in a Mendelian way. Today, in humans, these stably inherited sequences occupy up to 8% of the genome [6] and are classified by sequence homology into HERV families, each corresponding to the initial infectious retrovirus [7]. As a result of genetic alterations, the majority of HERVs are highly defective, frequently recombined as solo LTRs or even scattered as fragments of retroviral sequences in the chromosomes. Nevertheless, due to selective advantage of some retroviral functions for the host physiology, a few retroviral envelope (env) genes retained their coding capacity, being ‘coopted’ by the host and distinctively preserved from genetic drift. This can be illustrated by the well‐known example of syncytins, essential in mammalian placental development because they drive cellular fusion in syncytiotrophoblast formation and possibly influence feto‐maternal immune tolerance [8, 9, 10, 11].
Besides their expression during embryogenesis, HERV sequences (as coding elements or fragments of noncoding sequences) are on the whole silenced by epigenetic mechanisms in adult tissues [12, 13, 14]. Transcriptional activation of these sequences has been reported in several diseases such as cancer [15, 16, 17], autoimmune [18, 19, 20], or neurological disorders [21, 22, 23], underlying the ambivalent role of HERVs. Diverse external and host factors have been proposed as causative agents of this dysregulation [24]. Host factors include epigenetic modifications of DNA or histone, as well as cellular transcription factors since LTR sequences contain numerous transcription factor binding sites [25].
Contribution of several signaling pathways in tumorigenesis has been reported, including Wnt signaling, known for its critical role in cell–cell communication during embryonic development and adult tissue homeostasis [26]. Hyperactivation of the canonical Wnt/β‐catenin pathway has been associated with human diseases such as cancer. Indeed, alterations of its components leading to aberrant signaling are found in many solid and hematological tumors [27, 28, 29, 30].
Our group previously reported the characterization of the HEMO gene (Human Endogenous MEdium‐Reiteration‐frequency‐family‐34 ORF, or Human ERVMER34 ORF) which is part of a MER34 provirus, endogenized about 100 million years ago. At that time, infection of an ancestral mammal by a ‘MER34 retrovirus’ led to the integration of several proviral copies as a multigene family. All the MER34 proviral gag, pol, and env sequences became defective, except for one envelope gene that was kept under selection pressure until the present time, further suggesting a beneficial role during mammal evolution [31]. This integration event took place long before the integration events of the two human syncytin‐1 and syncytin‐2 genes, which were endogenized in the primate lineage less than 50 million years ago. Therefore, the HEMO gene is the oldest captured and still full‐length endogenous retroviral envelope (env) gene identified in the human genome (GRCh38, Ensembl annotation: ERVMER34‐1). Due to genomic alterations, HEMO lost its initial 5′ LTR promoter and is transcribed from a cellular CpG island located near the env gene. Remarkably, HEMO is expressed at high levels in placenta and stem cells and is a membrane protein that can be secreted into the blood as a shed protein [31].
In this study, we analyzed the expression of the endogenous retroviral sequence HEMO in primary and metastatic tumors by combining in silico analyses of RNAseq data and pan‐cancer detection at the protein level in patient samples. We also investigated its regulation and proposed the Wnt/β‐catenin pathway as an upstream regulator of this retroviral endogenous sequence in tumor condition.
2. Materials and methods
2.1. RNAseq datasets collection
Uniformly processed RNAseq data from GTEx (Genotype‐Tissue Expression project) for normal tissues (N: 5825 samples) and from TCGA (The Cancer Genome Atlas) for human tumor (T: 10181 samples) and non‐tumor control adjacent tissues (C: 722 samples) from a total of 33 different anatomic sites were downloaded from Recount2 resource (https://jhubiostatistics.shinyapps.io/recount/) [32].
Gene expression was quantified and reported in TPM unit (Transcripts Per Million of reads). Profile of HEMO expression was obtained using ENSEMBL_Id ENSG00000226887 (ERVMER34‐1). TCGA clinical data including tumor stage, grade, histological type, and molecular subtype were downloaded using r tcgabiolinks [33]. Corresponding mutation data were retrieved from cBioPortal for Cancer Genomics (http://www.cbioportal.org). Downstream transcriptomic data analyses were performed on tumor samples for which tumor nuclei exceeds 60%. RNAseq data (644 samples) collected from the clinical trial MOSCATO (MOlecular Screening for CAncer Treatment Optimization) led by Gustave Roussy were also used in our study [34]. Tumor cellularity of these samples ranged from 10% to 95%. Read quality control was performed with Trim galore (version 0.4.4) (https://www.bioinformatics.babraham.ac.uk/projects/trim_galore/), with the settings ‘‐q 20 ‐‐stringency 3 ‐‐gzip ‐‐length 20 ‐‐paired’. Afterward, the reads were pseudo‐mapped to the human transcriptome (GENCODE v27) with kallisto (version 0.44.0) [35] quant mode with the settings ‘‐‐bias ‐‐rf‐stranded’. Finally, to get gene‐level expression results, the package tximport (version 1.16.0) [36] was used.
2.2. Differential gene expression analysis
deseq2 R package (v1.26) was used to perform differential gene expression analysis between HEMOHigh and HEMOLow tumors. Samples of TCGA datasets were considered as HEMOHigh or HEMOLow if their expression level were, respectively, superior to 90th percentile or inferior to 10th percentile of global HEMO expression in the dataset. Eight cohorts with more than 300 samples were selected for differential gene expression analysis (BLCA, BRCA, CESC, COAD, HNSC, LUAD, LUSC, and UCEC). We corrected for the platform as a source of batch effect in differential gene expression analysis. Low‐expressed genes were filtered out before analysis. Differentially expressed genes were selected using the following cutoffs: FDR‐adjusted (Benjamini and Hochberg procedure) P‐value < 0.01, log2 fold change (|log2FC|) > 1.5, and ‘baseMean’ (the per‐gene mean value of normalized counts) > 50.
2.3. Gene set enrichment analysis
Gene set enrichment analysis was performed using r (v3.6.3) package clusterprofiler (v3.14.3). Genes ranking was performed using the decreasing log2FC value. This ranked list and gene sets from the REACTOME and GO (Gene Ontology) Biological Process databases were, respectively, retrieved from the C2 and C5 MsigDB v7.2 collections using the msigdbr (v7.2.1) package, and provided for the analysis carried out with 106 permutations. A gene set was considered significantly enriched when adjusted P‐value was < 0.05.
2.4. Biological samples
All patient samples were obtained with written informed consent. The study methodologies conformed to the standards set by the Declaration of Helsinki. The study methodologies were approved by the local ethics committee. Formalin‐fixed, paraffin‐embedded (FFPE), and frozen samples of tumor and non‐tumor control adjacent tissues were obtained from the Biological Resource Centre (BB‐0033‐00074) and the Department of Pathology and Laboratory Medicine of GRCC (Gustave Roussy Cancer Campus/Research Agreements RT09916 and RT14017).
2.5. Immunohistochemistry assays
Sections (4 μm) of FFPE tumor, and non‐tumor adjacent tissues were deparaffinized in xylene and rehydrated. We used Bond Leica automated immunostainer (Leica Microsystèmes, Nanterre, France) for HEMO immunostaining and Ventana Benchmark Ultra (Roche Diagnostics, Meylan, France) automated immunostainer for cytokeratins 5/6, CDX2, or p63/HEMO immunostaining. Preparation and staining steps are summarized in Table S1. For all protocols, sections were counterstained with hematoxylin. A single representative whole tumor or normal tissue section from each patient was digitized using a slide scanner (VS120; Olympus Life Science, Waltham, MS, USA).
Detailed protocols of immunohistochemistry assays (Table S1) are provided in Supporting Information.
2.6. Cell lines, culture conditions, and FH535 treatment
Colorectal adenocarcinoma cell lines Caco‐2, HCT116, and SW480 were kindly gifted by F. Jaulin (Gustave Roussy). Lung cell lines NCIH520 (squamous carcinoma), HCC827 (adenocarcinoma), and ovary cell line OVMANA (clear cell adenocarcinoma) were kindly gifted by L. Friboulet (Gustave Roussy). Cells were grown at 37 °C with 5% CO2 in DMEM for Caco‐2 and SW480, Mc Coy's 5A medium for HCT116, and RPMI for NCIH520, HCC827, and OVMANA, supplemented with 10% heat‐inactivated fetal bovine serum, 100 μg·mL−1 streptomycin, and 100 U·mL−1 penicillin (all reagents were from Thermo Fisher Scientific, Illkirch, France). FH535 was purchased from Sigma‐Aldrich (St Quentin‐Fallavier, France) and dissolved in DMSO at 50 mm. Cells were seeded in 12‐well plates (1 × 105 and 2 × 105 cells/well for Caco‐2 and NCIH520) and treated 24 h after seeding with 40 μm of FH535 for 72 and 24 h for Caco‐2 and NCIH520, respectively.
2.7. siRNA transfection
Caco‐2 and NCIH520 cells were seeded as described above and transfected 24 h after seeding using Lipofectamine™ RNAiMAX reagent (Thermo Fisher Scientific) and CTNNB1 or Non‐targeting siRNA (SMART pool, ON‐TARGET Plus; Horizon Discovery, Cambridge, UK). Transfected cells were grown 2 days before analysis.
2.8. RNA extraction and RT‐qPCR
For RNA extraction from tissues, OCT‐frozen tumor and adjacent non‐tumor samples were sectioned with a cryostat. Non‐OCT‐frozen samples were disrupted with a mortar. 10 sections of 50 μm or 20–30 mg of tissue fragment were used. After mechanical disruption with glass beads, RNA extraction was performed with ReliaPrep™ RNA Tissue Miniprep System (Promega, Charbonnières‐les‐Bains, France). For cell lines, RNeasy Isolation Kit (Qiagen, Courtaboeuf, France) was used according to the manufacturer's instructions and treated with DNase I (Ambion, Thermo Fisher Scientific). RNA quality and concentration were assessed using a NanoDrop ND‐1000 spectrophotometer (Thermo Fisher Scientific). Reverse transcription was performed with 1 μg RNA using M‐MLV reverse transcriptase and random hexamers (Thermo Fisher Scientific). RT‐qPCR experiments were run on an ABI Prism 7000 sequence detection system with SYBR green PCR master mix reagent (Qiagen) and specific primers (Table S2). All analyses were carried out in triplicate, and transcript levels were normalized to the level of housekeeping gene RPLP0 using the ΔΔCT method.
2.9. Protein extraction and western blot analysis
For protein extraction from tissues, 50 mg of frozen tumor or adjacent non‐tumor samples was quickly lysed by mechanical disruption with glass beads in 200 μL RIPA buffer (Thermo Fisher Scientific) supplemented with Halt™ Protease Inhibitor Cocktail (Thermo Fisher Scientific). For protein extraction of cell lines, cells were briefly washed with PBS and then lysed in IP lysis buffer (25 mm Tris/HCl pH 7.4, 150 nm NaCl, 1% NP‐40, 1 mm EDTA) supplemented with Halt™ Protease Inhibitor Cocktail (Thermo Fisher Scientific). After removing cell debris by a centrifugation step (10 min, 4 °C at 14 000 g ), protein lysates were titrated with Pierce™ BCA Protein Assay Kit (Thermo Fisher Scientific). Samples or cell lysates were analyzed by SDS/PAGE on gradient precast gels under reducing conditions (NuPAGE™ Novex 4–12% Bis‐Tris gels; Thermo Fisher Scientific) and transferred onto nitrocellulose membranes using a dry transfer system (iBlot2; Thermo Fisher Scientific). After blocking in PBS containing 0.1% Tween‐20, and 5% non‐fat milk, membranes were incubated overnight at 4 °C with primary antibodies (anti‐HEMO mouse mAb 2F7 [31]; anti‐β‐catenin, BD biosciences, Le Pont de Claix, France, #610153, 1 : 1000; and anti‐GAPDH HRP‐conjugated, Antibodies‐online #ABIN398425, 1 : 3000). For HEMO and β‐catenin staining, membranes were then incubated with anti‐mouse HRP‐conjugated secondary antibody (GE Healthcare, Buc, France, 1 : 5000) for 45 min at room temperature. Protein detection was performed by using enhanced chemiluminescence reagents (Pierce™ ECL plus; Thermo Fisher Scientific) and ImageQuant LAS400 camera system (GE Healthcare).
2.10. Statistical analyses
All statistical analyses were performed using prism Software, v6.2 (GraphPad, https://www.graphpad.com/scientific‐software/prism/).
3. Results
3.1. HEMO activation is detected by in silico analysis in solid tumors
In order to estimate HEMO expression in tumors, we analyzed the RNAseq‐based transcriptome of cancer samples retrieved from TCGA (The Cancer Genome Atlas) cohorts. HEMO expression levels observed in tumors were then compared to those of TCGA's non‐tumor adjacent tissues (‘Control’) as well as non‐diseased (‘Normal’) tissues from the GTEx (Genotype‐Tissue Expression) project (lists in Table S3). As shown in Fig. 1A, global activation of the HEMO gene was observed in several TCGA tumor cohorts (orange boxes), frequently associated with high heterogeneity between samples in a given cohort. Evidence of high‐expressing cases was highlighted for a series of solid tumors, as for instance the carcinomas of endometrium (UCEC). In contrast, low levels of HEMO expression were generally observed in GTEx normal tissues (white boxes), in agreement with our previous results based on microarray data [31]. Most of the TCGA's control adjacent tissues (green boxes) displayed HEMO levels similar to those of GTEx tissues, with some discrepancies in cohorts such as breast, uterus, and prostate, reflecting a possible precancerous state in corresponding adjacent TCGA tissues.
Fig. 1.
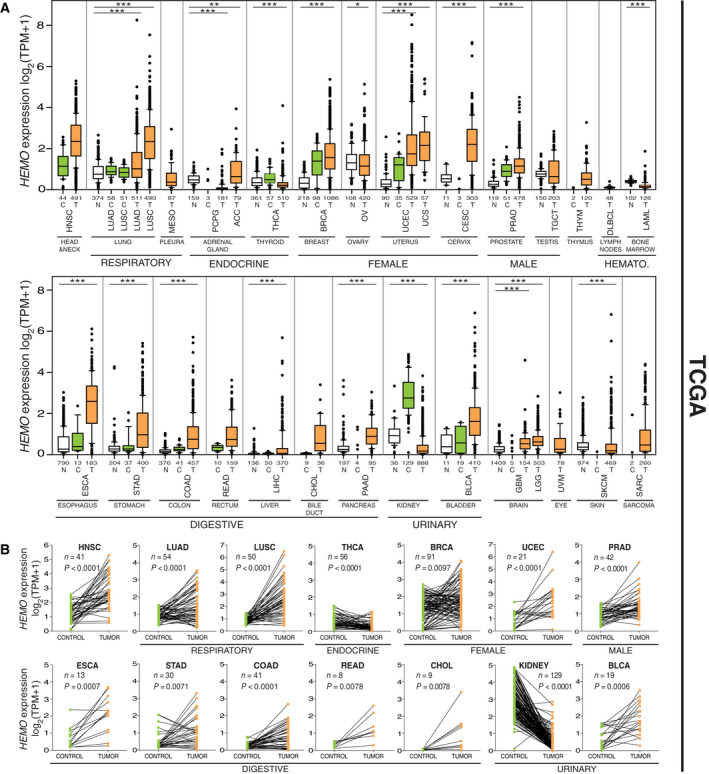
HEMO is overexpressed in primary solid tumors. (A) Boxplots of normalized (TPM) and log2‐transformed expression of HEMO in tumor (‘T’, orange boxplots) and control (‘C’, green boxplots) adjacent tissue samples retrieved from TCGA (The Cancer Genome Atlas)‐Recount2. The names of the cohorts are given in Table 1. White boxes correspond to basal expression in normal tissues (‘N’) from GTEx (Genotype‐Tissue Expression)‐Recount2. The number of samples analyzed in each group (N, C, T) is shown on the x axis (see also Table S3). The P‐values of pairwise comparisons between each group (T, C, N) are given in Table S3 and the (N, T) P‐values are shown as asterisks above the boxplots: *P < 0.05; **P < 0.01; ***P < 0.001, Mann–Whitney U‐test. Cohorts are grouped according to body systems. The breast cohort, which consisted primarily of female samples, include 12 male samples with similar heterogenous HEMO expression levels (Table S4). HEMATO., hematological tumors. Data are shown as mean with 25–75th percentile range (box) and 10–90th percentile range (whiskers). Mild outliers are depicted as black dots. (B) Matched HEMO gene expression analysis in pairs (Control + Tumor) from TCGA (LIHC cohort not depicted due to nonsignificant P‐value). n, number of pairs in each cohort; P, P‐values (Wilcoxon matched‐pairs test).
However, in kidney samples aggregated from the three cohorts KIRC, KIRP, and KICH, we could observe an unexpectedly high level of HEMO expression in TCGA's control compared to both GTEx and tumor samples, and this pattern was observed in the three individual kidney cohorts (Fig. S1E). A similar expression profile, although to a much lesser extent, was also noted for the thyroid (THCA) samples. The HEMO transcripts were not activated in ovarian serous adenocarcinoma (OV), which corresponds to the only histological subtype represented in the TCGA's ovarian cohort. This result is in line with our previous findings that demonstrated a specific upregulation of HEMO expression in clear cell and endometrioid ovarian adenocarcinoma but not in serous and mucinous histotypes [31]. Unlike a large proportion of solid tumors, hematological tumors (DLBCL and LAML) did not exhibit any HEMO activation.
Overall, to quantify HEMO activation in the different tumors, the fold change between the medians of normal‐GTEx samples and tumor‐TCGA samples was calculated for each individual cohort, based on TPM values (T/N median fold change in Table 1). However, because the kidney is a critical organ for survival and has a low but significant basal level of HEMO expression (white box), it is necessary to take this into account when evaluating the use of HEMO as a therapeutic target in non‐renal tumors. Therefore, we calculated for each cancer type, the fold change between the medians of kidney normal‐GTEx, and each tumor‐TCGA cohort (T/Kid‐N median fold change in Table 1).
Table 1.
Relative expression level of HEMO in tumor samples compared to normal tissue samples. Median fold change (based on TPM values), between the medians of normal‐GTEx and tumor‐TCGA samples for each cohort (T/N), and between the medians of kidney normal‐GTEx and other tumor‐TCGA samples (T/Kid‐N). KI (CH, RC, RP): Kidney cohort aggregated from KICH (Kidney Chromophobe), KIRC (Kidney Renal Clear Cell Carcinoma) and KIRP (Kidney Renal Papillary Cell Carcinoma). Fold changes > 2 are in bold.
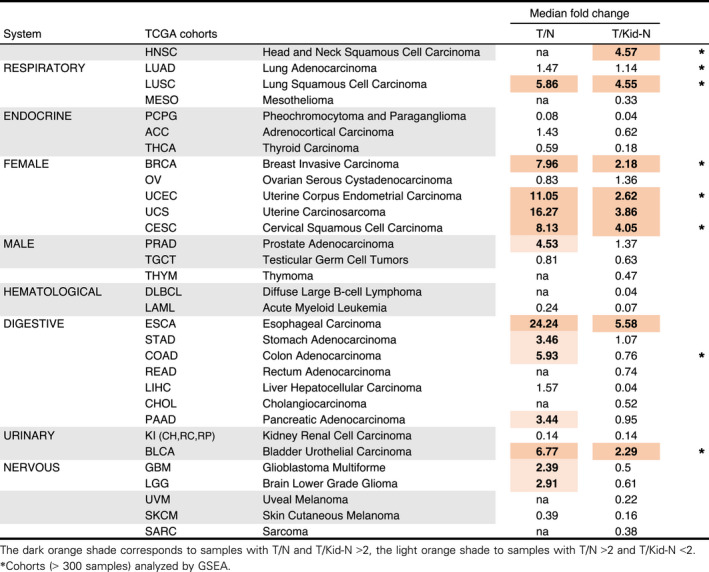
Accordingly, cohorts of the lung (LUSC), breast (BRCA), uterus (UCEC, UCS), cervix (CESC), esophagus (ESCA), and bladder (BLCA) showed significant tumor activation of HEMO, clearly above the level of HEMO expression in healthy kidney (median fold changes T/N and T/Kid‐N > 2). Although the T/N value was not available in the case of the head and neck tumors (HNSC), they most likely belong to the same group. Finally, tumors of prostate (PRAD), stomach (STAD), colon (COAD)—and likely rectum (READ)—pancreas (PAAD) and at a reduced level tumors of brain (GBM, LGG) can be grouped into a second category of tumors with lower but significant HEMO activation (T/N > 2, T/Kid‐N < 2).
To further verify specific activation of HEMO in tumors, pairs of samples from the TCGA database were individually compared in representative cohorts, as illustrated in Fig. 1B. HEMO mRNA level was significantly enhanced in tumors compared with matched control adjacent tissues, with less pronounced effect for BRCA (breast) samples as mentioned for the boxplot. As noticed above, unexpected high values in TCGA's control samples were observed for the kidney as well as a significant decrease of expression in almost all sample pairs, a pattern similarly observed at a reduced level in the THCA (thyroid) samples.
Given that most of these TCGA cancer samples correspond to primary tumors (apart from the TCGA's skin ‘SKCM’ cohort), we decided to go further with the transcriptome analysis of metastatic tumors (Fig. 2). In this regard, we measured HEMO expression in RNAseq data retrieved from the institutional clinical trial MOSCATO (MOlecular Screening for CAncer Treatment Optimization) [34] differently processed compared to Recount data (see Materials and methods). In this study encompassing metastatic or locally advanced tumor samples, HEMO transcripts were also detected in a significant proportion of tumors, with similar heterogeneity within a specific tumor site. Although absolute values could not be directly compared, and despite smaller number of samples, the rank order of the cohorts based on their HEMO expression was on the whole conserved between analyses of primary (TCGA) and metastatic (MOSCATO) tumors. Highest median values were observed in metastatic tumors of head and neck, cervix, and pancreas, while thyroid and kidney metastatic tumors remained at low levels.
Fig. 2.
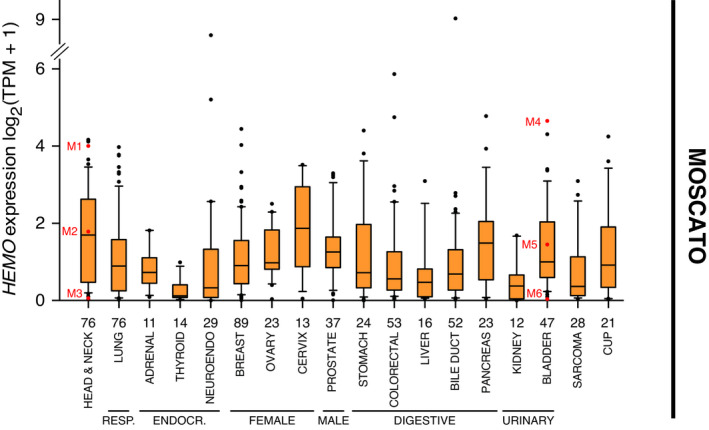
HEMO is overexpressed in metastatic tumors. Boxplots of normalized (TPM) and log2‐transformed HEMO expression in metastatic tumors from MOSCATO dataset (n = 644 samples), differently processed compared to Recount data (see Materials and methods). The organ names correspond to the localization of the primary tumor. The number of samples analyzed in each group is shown on the x axis. Samples marked in red (M1 to M6) are those selected for anti‐HEMO staining (Fig. 3). RESP., respiratory system; ENDOCR., endocrine system; NEUROENDO., neuro‐endocrine tumor; CUP, carcinoma of unknown primary. Data are shown as mean with 25–75th percentile range (box) and 10–90th percentile range (whiskers). Mild outliers are depicted as black dots.
To investigate whether HEMO is a marker of cancer progression, we stratified samples by tumor stage and grade, in the TCGA cohorts where the increase of HEMO expression in tumors was the highest compared to normal tissues (Fig. S2). Only a few significant associations were found between HEMO expression and each parameter, as in the HNSC (Head and Neck) and PAAD (pancreas) cohorts in which HEMO expression was positively correlated with high tumor grades (G2‐G3 and G3, respectively). Further analysis of histological types indicated that in the esophagus tumors, HEMO was preferentially expressed in the squamous cell carcinoma subtype (SCC) compared to the adenocarcinoma subtype (ADC). Of note, this difference was also observed for the lung tumors, between the lung squamous cell carcinoma (LUSC) and lung adenocarcinoma (LUAD) cohorts in Fig. 1A. Finally, stratification of breast tumor samples by their molecular signature revealed that HEMO was poorly activated in the Her2+ subgroup.
3.2. Tumor expression of other HERV‐env genes
Next, we wanted to investigate if other well‐known endogenous retroviral envelope genes were also overexpressed in tumor condition. Among the still coding envelope genes of the human genome [37, 38], four have specific characterized transcripts and a reliable Ensembl annotation (Table S5). We analyzed the syncytin‐1 (ERVW‐1) and syncytin‐2 (ERVFRD‐1) transcripts (which are known to encode identified placental proteins), ERVV‐2 (a placental specific transcript), and the more ubiquitous ERV3‐1 gene (encoding a putative soluble envelope protein).
As shown in Fig. S1, and by contrast with HEMO, the syncytin‐1 and ERVV‐2 envelope coding sequences displayed almost undetectable expression and were poorly activated in tumor conditions. High level of ERVW‐1 expression was detected in normal testis, but was not specifically enhanced in testis cancer (P‐value in Table S3). In the case of ERVV‐2, we could observe a few samples with high level of expression, notably in renal tumors (see also Fig. S1E), which need further investigations to confirm detection at the protein level.
Strikingly, the syncytin‐2 (ERVFRD‐1) sequence, which had a basal level of expression in most normal tissues, was slightly repressed in control adjacent tissues, and even more repressed in tumor conditions, except for the kidney, where we could observe faint ERVFRD‐1 activation in control adjacent tissues, as for HEMO. This was confirmed in the three individual kidney cohorts (Fig. S1E), but was not a general case for the other HERV‐env sequences.
Nevertheless, we noticed that syncytin‐1 (ERVW‐1) and syncytin‐2 (ERVFRD‐1) were activated in acute myeloid leukemia (LAML).
Finally, ERV3‐1 did not clearly appear as a tumor‐specific transcript since most of the cohorts displayed similar expression in normal tissues, control, and tumor samples, with sporadic activation or inactivation of the sequence (Fig. S1D). Still, it can be noted that its highest median value is observed in the LAML cohort.
Analysis of other full‐length envelope coding genes could not be performed, mainly due to the multigenic nature of some HERV family, such as in the HERV‐K(HML‐2) family [39], preventing the clear identification of the few coding copies.
3.3. HEMO is detected at the protein level
To confirm the in silico data, pan‐cancer immunohistochemistry analyses were performed on primary tumor FFPE samples from Gustave Roussy (n = 126, in Table 2) with a specific anti‐HEMO monoclonal antibody [31]. Evidence of weak to strong staining was found in different tumor tissues, while on the contrary, some other tumors were deprived of HEMO protein, as shown in a representative sampling of tumor tissues (Fig. 3). In our samples, the HEMO protein was not detectable in available adjacent non‐tumor tissues. When present, staining was generally scattered within the tumor area. At the cell level, HEMO localized to the cytoplasmic and membrane compartments, as expected for a membrane protein (see also staining in cancer cell lines, Fig. S3C) and in agreement with our previous results [31].
Table 2.
Distribution of HEMO expression in pan‐cancer cohort as measured by immunohistochemistry. HEMO expression was classified as strong, weak, or negative according to the strength and the prevalence of the staining within the sample. Samples include primary and metastatic (M: number in brackets) tumors. ‘Organ’ corresponds to the primary localization.
| System | Organ | Tumor type | Strong | Weak | Negative |
|---|---|---|---|---|---|
| Head and neck | Squamous carcinoma | 9 | 9 | 17 | |
| RESPIRATORY | Lung | Adenocarcinoma | 7 | 2 (M = 1) | |
| Squamous carcinoma | 1 | 2 | 1 | ||
| FEMALE | Breast | Carcinoma | 3 | 7 (M = 3) | 13 (M = 3) |
| Endometrium | Adenocarcinoma | 3 | 2 | 6 | |
| Cervix | Squamous carcinoma | 3 | |||
| MALE | Prostate | Adenocarcinoma | 3 | ||
| DIGESTIVE | Esophagus | Adenocarcinoma | 2 | 1 | 1 |
| Squamous carcinoma | 1 | 0 | 1 | ||
| Stomach | Adenocarcinoma | 1 | 1 | 2 | |
| Colon | Adenocarcinoma | 4 (M = 1) | 4 | 2 | |
| Rectum | Adenocarcinoma | 6 (M = 1) | 1 | ||
| Skin | Melanoma | 1 (M = 1) | 1 (M = 1) | 9 (M = 9) | |
| % of Total (126 cases) | 30% | 22% | 48% | ||
Fig. 3.
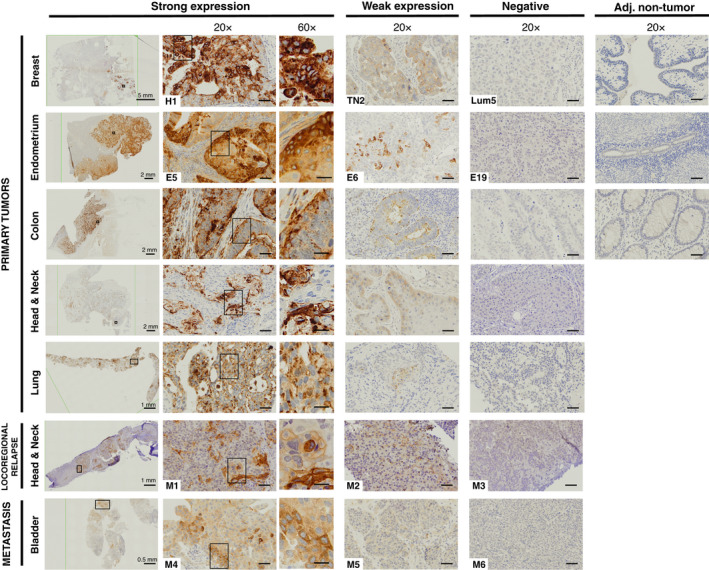
HEMO protein is detected in various tumor tissues. Examples are shown of strong, weak, and absent HEMO expression in cancer samples and of no detection in adjacent non‐tumor samples (staining with anti‐HEMO 2F7 mAb). Left column (0.4–2×): scale bar indicated in mm, black frame defined localization of 20× view. 20× columns: scale bars correspond to 50 μm, black frame defined localization of 60× view. 60× column: scale bars correspond to 20 μm. Numbers displayed on breast (H1, TN2, Lum5) and endometrium (E5, E6, E19) samples correspond to numbers used in Fig. 4. Numbers displayed on locoregional relapse samples and metastatic samples (MOSCATO M1 to M6) correspond to those marked in red in Fig. 2.
As reported in Table 2, strong HEMO staining was detected in 30% of this limited pan‐cancer cohort, with most of the cases found in head and neck squamous carcinoma (9/32), lung carcinoma (8/13), breast carcinoma (3/23), endometrium adenocarcinoma (3/11), esophagus (3/6), colon adenocarcinoma (4/10), and rectum adenocarcinoma (6/7), thereby confirming the tendency predicted by TCGA data.
In addition, in order to precisely compare RNAseq results and degree of protein detection in the same sample, tumors included in the MOSCATO cohort were also stained with anti‐HEMO antibody (Fig. 3, bottom). As shown for locoregional relapse of head and neck tumors and bladder metastases, we found a clear correlation between the amount of HEMO protein and the expression levels estimated from transcriptomic data of Fig. 2 (M1–M6 samples).
To get further into the unambiguous HEMO identification, we analyzed its expression in frozen tumor samples of breast, ovary, endometrium, and esophagus by RT‐qPCR and western blot analysis (Fig. 4). As for the TCGA results, control adjacent tissues displayed some slight variation in HEMO transcription levels. Heterogeneity with multiple high values of HEMO expression was observed in tumor samples. Consistently with these transcription results, we clearly detected by western blot a band with an apparent molecular mass similar to what is expected for the glycosylated full‐length HEMO protein in corresponding tumor lysates. Such band was not observed in HEMO negative tumors or surrounding non‐tumor tissues (Fig. 4B). Taken together, these results support the highly specific expression of the endogenous retroviral HEMO envelope in solid tumors.
Fig. 4.
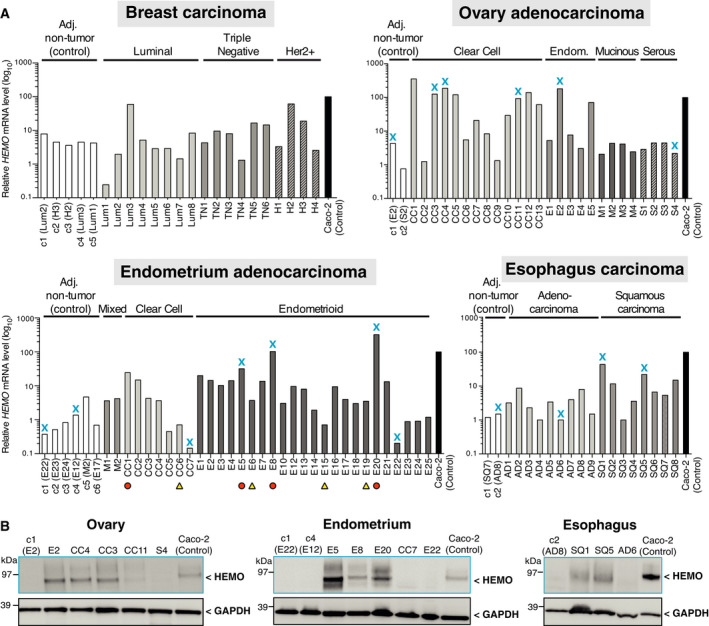
Detection of HEMO transcripts and corresponding protein in breast, ovary, endometrium and esophagus tumor samples. (A) Expression of HEMO measured by RT‐qPCR. Amount of mRNA was normalized to RPLP0 transcript level. Caco‐2 mRNA was used as positive control for HEMO expression. Bar plots represent mean expression of two independent technical replicates. Blue cross marks indicate samples analyzed by western blot in part B. Orange circles and yellow triangles, respectively, correspond to HEMOStrong and HEMOWeak/Neg samples selected for Fig. 6. Adj., adjacent; Endom., endometrioid. (B) Detection of glycosylated full‐length protein HEMO in corresponding lysates of ovary (left), endometrium (middle) and esophagus (right) tumors. Caco‐2 lysate was used as positive control for HEMO molecular weight. GAPDH was measured as a control of cell lysate protein loading. Results shown are representative of three independent technical replicates.
3.4. HEMO upregulation is linked to Wnt/β‐catenin signaling in endometrial tumors
To further characterize endometrial tumors expressing HEMO and identify potential co‐expression signatures in these tumors, we performed differential gene expression analysis between HEMOHigh and HEMOLow tumors from TCGA's UCEC cohort (Table S6). Patients were separated into ‘High’ or ‘Low’ HEMO expression groups, using 90th and 10th percentiles as cutoffs (size: 53 patients in each group). Next, Gene Set Enrichment Analysis (GSEA) was performed using REACTOME and Gene Ontology (GO) Biological Process (BP) databases (Table S7).
It revealed that among enriched gene sets, several pathways were related to Wnt/β‐catenin signaling, (see also results in paragraph 3.5), with four of them (marked with an asterisk) belonging to the top 20 most significant GO gene sets, listed in Fig. 5A. Enrichment plots of ‘GO canonical Wnt signaling pathway’ (number 2) and ‘GO negative regulation of Wnt signaling pathway’ (number 14) are illustrated in Fig. 5B, with positive Normalized Enrichment Score (NES) and robust adjusted P‐value (< 0.001).
Fig. 5.
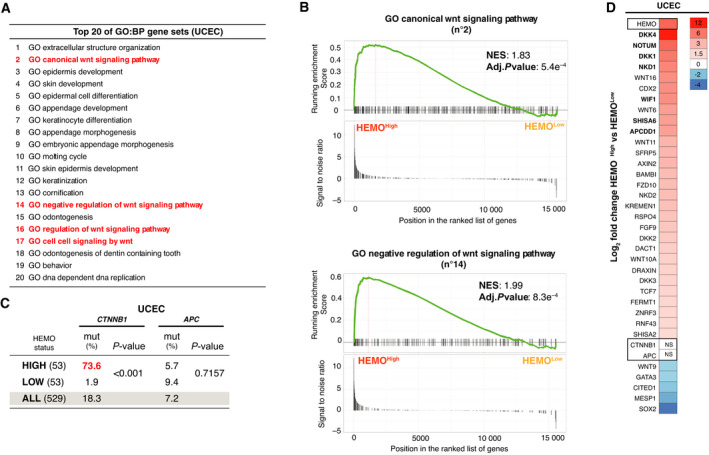
HEMO upregulation is associated with an active Wnt/β‐catenin pathway in UCEC cohort. (A) Top 20 GO (Gene Ontology) BP (Biological Process) gene sets with the highest significance in UCEC cohort, extracted from Table S7. In red: Wnt signaling pathways. (B) GSEA enrichment plots showing ‘GO canonical Wnt signaling pathway’ and ‘GO negative regulation of Wnt signaling pathway’ enriched signatures between UCEC HEMOHigh vs HEMOLow tumors. NES: Normalized Enrichment Score. (C) Mutational status of CTNNB1 and APC in UCEC HEMOHigh and HEMOLow tumors, 53 cases of each (P‐value, Fisher's exact test). For comparison, mutational status of the total UCEC cohort is indicated. (D) Heatmap for the significant Wnt‐related genes differentially expressed between HEMOHigh and HEMOLow tumors. Color gradation is representative of Log2 fold change. The differential level of HEMO expression is also indicated at the top of the heatmap (LogFC = 7). For all depicted genes, adjusted P‐value is < 0.01 (except for APC and CTNNB1, NS: nonsignificant). In bold, common upregulated genes found in COAD cohort (Fig. S4B).
Such associations with active Wnt/β‐catenin signaling led us to analyze in each subgroup of UCEC tumors (HEMOHigh and HEMOLow) the mutational status (Fig. 5C) and the expression level (Fig. 5D) of two key genes of this pathway, namely CTNNB1 (β‐catenin coding gene) and APC (adenomatous polyposis coli gene). Alterations in these genes are indeed frequently found in endometrial tumors and lead to a constitutive activation of the Wnt/β‐catenin pathway [30]. In the UCEC TCGA cohort, 18.3% of the samples are mutated in CTNNB1, and 7.2% in APC gene (Fig. 5C). Importantly and convincingly, we found that 73.6% (39 of 53) of HEMOHigh versus 1.9% (1 of 53) of HEMOLow tumors harbored stabilizing mutations in CTNNB1 (P < 0.0001), known to drive a drastic activation of the Wnt/β‐catenin pathway, which was consistent with the GSEA results. Conversely, no association with APC mutations was observed (Fig. 5C). None of these two genes were differentially expressed (Fig. 5D). A heatmap of the relative changes in Wnt/β‐catenin signaling‐related gene expression (Fig. 5D) showed that most genes involved in this pathway were upregulated in HEMOHigh tumors with a noticeable upregulation of DKK4 (LogFC = 12), a member of the Wnt inhibitor DKK family [40].
Following these findings, the expression of a series of genes upregulated in the UCEC HEMOHigh versus HEMOLow tumors was examined by RT‐qPCR in our endometrium tumor samples, selected among those of Fig. 4 with corresponding HEMO ‘Strong’ and ‘Weak/Neg’ expression level (indicated by orange circles and yellow triangles). As shown in Fig. 6A, AXIN2, DKK4, and CDX2 were found to be significantly upregulated in HEMOStrong compared to HEMOWeak/Neg tumors. Given that these genes are considered as negative regulators of the Wnt/β‐catenin pathway, their overexpression could indicate a negative response developed to counteract abnormal activation of the pathway, as already described [41]. Immunohistochemistry analyses by HEMO and β‐catenin co‐staining were performed on the endometrial tumor samples (Fig. 6C). In HEMOStrong tumors, HEMO colocalized in specific tumor cells in which nuclear β‐catenin was observed (white arrow in sample E5), whereas in HEMO negative cells, either in the same sample or in HEMOWeak/Neg tumors (sample E15), the β‐catenin was mainly localized to the cell membrane.
Fig. 6.
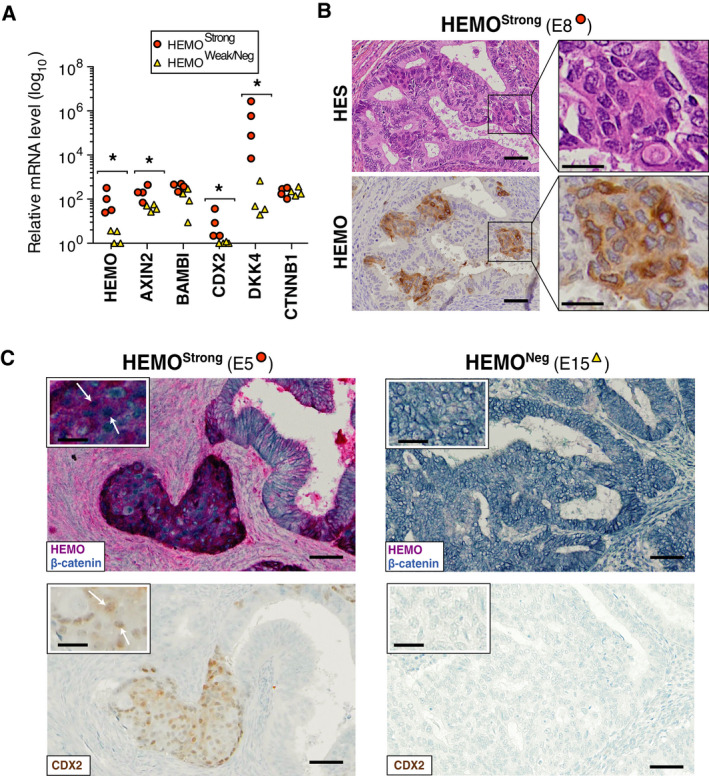
HEMO is detected in morular metaplasia of endometrium adenocarcinoma displaying activation of the Wnt/β‐catenin pathway. (A) Transcript levels of representative Wnt‐related genes overexpressed in HEMOHigh UCEC tumors measured by RT‐qPCR on endometrium adenocarcinoma samples, classified as HEMOStrong (n = 4 samples) or HEMOWeak/Neg (n = 4 samples) according to their HEMO expression level, previously estimated by RT‐qPCR in Fig. 4. Amount of mRNA was normalized to RPLP0 expression level (*P < 0.05, Mann–Whitney test). (B) HES and immunohistochemical detection of HEMO (2F7 mAb) in morular metaplasia of endometrium adenocarcinoma. Left, magnification: 20×, scale bar: 50 μm; right, magnification: 60×, scale bar: 20 μm. (C) Co‐staining of HEMO (red) and β‐catenin (blue) in examples of HEMOStrong (left) and HEMONeg (right) endometrial adenocarcinoma (top) and CDX2 nuclear staining (bottom). Morular foci displayed nuclear β‐catenin and CDX2 staining (as shown by white arrows). Of note, nuclear β‐catenin staining was detected in 3 out of the 4 HEMOStrong samples (1 undetermined) and was not detected in the 4 HEMOWeak/Neg samples. Magnification: 20×, scale bar: 50 μm; insert, magnification: 40×, scale bar: 20 μm. Samples are identified as in Fig. 4.
Nuclear localization of β‐catenin reflects its stabilization induced by mutations of either CTNNB1 or APC and results in activation of the signaling pathway, therefore without any up‐ or downregulation of CTNNB1 expression, as observed in Fig. 6A. CTNNB1 mutations and subsequent nuclear accumulation of β‐catenin have been already reported in endometrial adenocarcinoma and in particular in foci of morular metaplasia [42, 43]. Strikingly, we noticed that HEMO prominent areas in endometrium adenocarcinoma matched with morules clearly identified in HES (Fig. 6B) and positively stained with CDX2 (Fig. 6C), known as being expressed in these typical elements [44].
Taken together, these results clearly demonstrate that HEMO is preferentially found in tumors with an active Wnt/β‐catenin pathway. Therefore, in addition to cell morphological modifications, this pathway could participate in activation of HEMO in tumors.
3.5. HEMO upregulation and Wnt/β‐catenin signaling in other tumors
Next, we sought to determine if HEMOHigh tumors from TCGA cohorts other than UCEC (marked with an asterisk in Table 1) displayed a similar relationship with the Wnt/β‐catenin pathway. The Gene Set Enrichment Analyses (Tables [Link], [Link]) performed on HNSC, LUAD, LUSC, BRCA, CESC, COAD, and BLCA cohorts showed that signatures related to the Wnt/β‐catenin signaling pathway were also enriched in BRCA, CESC, and COAD and to a lesser extent in LUSC and BLCA cohorts (Table 3). Of note, no GO nor REACTOME gene sets were found to be enriched in the HNSC cohort under our conditions of GSEA (adj. P‐value < 0.05).
Table 3.
Wnt‐related pathways are enriched in HEMOHigh tumors in TCGA cohorts. For each TCGA cohort, normalized enrichment score (NES, bold) and adjusted P‐value (in brackets) of GSEA (performed on Gene Ontology Biological Process and REACTOME databases) are given when the Wnt‐related pathway is significantly enriched. RESP., respiratory; Green: terms illustrated on enrichment plots of Fig. 5B and Fig. S4A.
| RESP. | FEMALE | DIGESTIVE | URINARY | ||||
|---|---|---|---|---|---|---|---|
| LUSC | BRCA | UCEC | CESC | COAD | BLCA | ||
| GO canonical Wnt signaling pathway* | 1.42 (0.017) | 1.45 (0.042) | 1.83 (5.4e‐4) | 1.56 (0.011) | |||
| GO negative regulation of Wnt signaling pathway* | 1.99 (8.3e‐4) | 1.63 (0.019) | |||||
| GO regulation of Wnt signaling pathway* | 1.41 (0.012) | 1.62 (2.3e‐3) | 1.77 (1.0e‐3) | 1.59 (3.3e‐3) | |||
| GO cell signaling by Wnt* | 1.52 (4.7e‐3) | 1.70 (1.0e‐3) | 1.48 (6.0e‐3) | ||||
| GO negative regulation of canonical Wnt signaling pathway | 1.90 (2.2e‐3) | ||||||
| GO beta catenin TCF complex assembly | 1.87 (0.019) | ||||||
| REACTOME negative regulation of TCF dependent signaling by Wnt ligand antagonists | 1.87 (0.015) | ||||||
| REACTOME signaling by Wnt in cancer | 2.13 (6.3e‐4) | ||||||
| REACTOME Wnt ligand biogenesis and trafficking | 1.79 (0.047) | ||||||
| REACTOME formation of the beta catenin TCF transactivating complex | 2.00 (2.0e‐3) | ||||||
| REACTOME TCF dependent signaling in response to Wnt | 1.64 (0.013) | ||||||
| REACTOME signaling by Wnt | 1.48 (0.036) | ||||||
Terms belonging to the top 20 list of Fig. 5A.
APC and CTNNB1 mutations were then screened in HEMOHigh and HEMOLow groups of these TCGA cohorts (Table S8). Particularly in the COAD cohort, the prevalence of APC mutations was significantly higher (P = 0.0016) in HEMOHigh compared with HEMOLow group (67.4% vs 32.6%). Thus, this increase could be linked to enrichment in the Wnt‐related pathways reported in Table 3. As shown in the heatmap of Fig. S4B, and similarly to UCEC, COAD HEMOHigh tumors displayed absence of differential expression of CTNNB1 and APC, as well as an upregulation of members of the DKK gene family and NOTUM. However, in the COAD cohort, GSEA revealed enrichment in REACTOME pathways, such as ‘formation of the beta catenin TCF transactivating complex’ or ‘TCF dependent signaling in response to Wnt’ (Fig. S4A). These were not shared by the UCEC cohort, mainly due to some differences found in their profiles of upregulated Wnt/β‐catenin signaling‐related genes observed in the two heatmaps.
Overall, these results suggest that HEMO activation in tumor condition and, in particular, in endometrial tumors may involve Wnt/β‐catenin signaling.
3.6. HEMO expression is modulated by the Wnt pathway in cell models
To specifically address whether regulation of HEMO expression is modulated by the Wnt/β‐catenin signaling pathway in tumors, inhibition of this pathway was performed on cell models. Among six cell lines which endogenously express HEMO (NCIH520, HCC827, OVMANA, Caco‐2, SW480, and HCT116), the lung squamous cell carcinoma NCIH520 and colorectal adenocarcinoma Caco‐2 cell lines were selected since they showed the highest HEMO expression levels (Fig. S3). Besides, Caco‐2 cells are known to harbor somatic mutations in both CTNNB1 and APC [45], thus constituting an example of cells under an active Wnt/β‐catenin pathway. Conversely, the NCIH520 model does not exhibit such mutations.
First, cells were treated with FH535, a chemical inhibitor of transcription mediated by the TCF/β‐catenin complex [46]. Expression of HEMO, CTNNB1, and the Wnt/β‐catenin target gene AXIN2, was measured by RT‐qPCR (Fig. 7). We found that treatment by FH535 significantly decreased HEMO transcript level in Caco‐2 and NCIH520 cells while expression of ASCT2 was not reduced (Fig. 7A,D). This result was further confirmed by western blot analysis with a strong reduction of the corresponding HEMO protein in both cell lines (Fig. 7C,F, left). In contrast, transcription levels of CTNNB1 and its target gene AXIN2 decreased with varying degrees of downregulation in the two cell lines. Whether it is associated with diverse toxicity effects of the FH535 drug on carcinoma cell lines [46] or with differential genetic background of these two cell lines needs to be further investigated.
Fig. 7.
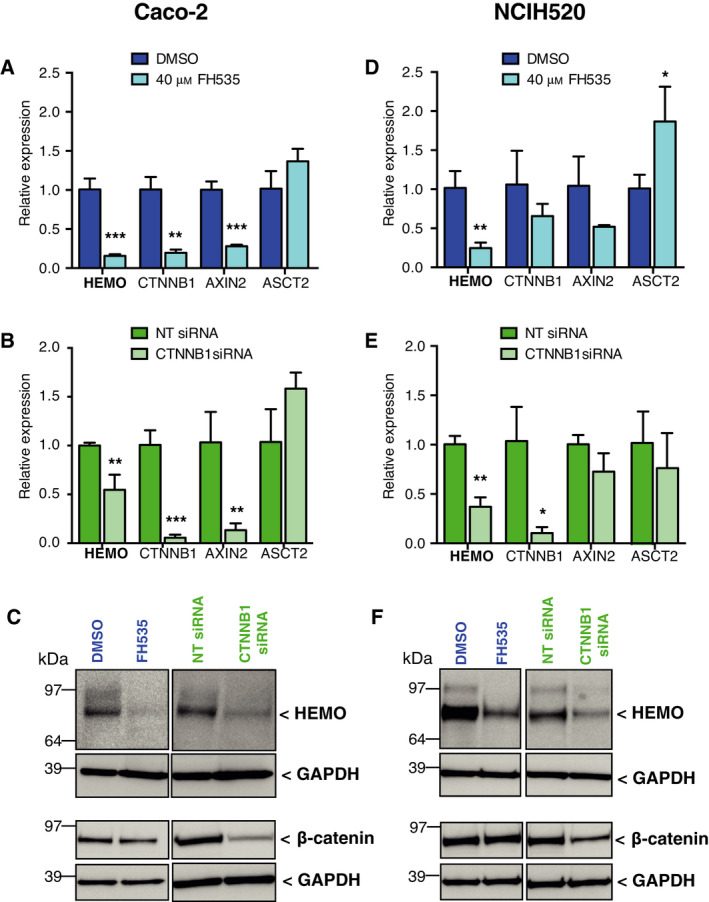
HEMO expression decreases after Wnt/β‐catenin inhibition in Caco‐2 (A–C) and NCIH520 (D–F) cells. Inhibition was performed with 40 μm of FH535 (A: Caco‐2, 72 h; D: NCIH520, 24 h) or by CTNNB1 silencing with siRNAs (B, E). Amount of mRNAs was normalized to housekeeping gene RPLP0 transcript level. Bar plots represent mean relative expression of three independent RT‐qPCR experiments ± SD. (*P < 0.05; **P < 0.01; ***P < 0.001, unpaired Student's t‐test). Corresponding HEMO and β‐catenin protein amount was assessed by western blot (C, F). GAPDH was measured as a control of cell lysate protein loading. NT: Non‐targeting; ASCT2: neutral amino acid transporter.
Using a more specific approach, the same cell lines were then transfected with a pool of small interfering RNAs directed against CTNNB1. The efficacy of this siRNA was first validated, as shown by the significant reduction of CTNNB1 mRNA levels (Fig. 7B,E) and of the corresponding β‐catenin protein (Fig. 7C,F, right). In that condition, a significant downregulation of HEMO transcription was observed in the Caco‐2 and NCIH520 cell lines, which resulted into an extensive decrease of the HEMO protein. Concerning AXIN2, responses to inhibition by siRNA were consistent with those observed with the drug. Overall, these experiments, carried out in two independent cell lines, provided evidence that the Wnt/β‐catenin signaling pathway participates in the regulation of HEMO expression.
3.7. HEMO activation is associated with keratinization and immune signatures
Besides identification of Wnt/β‐catenin signaling as a putative regulator of HEMO expression, GSEA performed on the TCGA cohorts (enriched pathways listed in Table S7) led us to identify other gene expression patterns associated with HEMO activation.
Remarkably, we could identify that epidermis development or keratinization gene sets were strongly enriched in HEMOHigh tumors compared to HEMOLow tumors in the BRCA, UCEC, CESC, LUSC, and BLCA cohorts (as already depicted in the top 20 list for the UCEC cohort in Fig. 5A). Of note, absence of these gene sets was observed in the COAD cohort. As shown in Fig. 8A illustrating the distribution of all the GSEA gene sets for the seven cohorts, these epidermis‐related pathways (orange dots), belonged to the most significantly enriched pathways (Adj. P‐value < 0.01, NES > 2). The pathways common to at least six cohorts are listed in Fig. 8B. To confirm these bioinformatics data, we performed immunohistochemistry assays (Fig. 8C). The HEMO protein was detected in squamous and keratinizing nests of endometrium adenocarcinoma, highlighted by CK5/6 and p63‐positive staining (Fig. 8C, top). Comparison of HEMO‐positive regions with corresponding HES unveiled that this protein was frequently expressed in areas of squamous differentiation, sometimes associated with abnormal keratinization, as shown for tumors of breast, endometrium, head and neck, or esophagus tumors (Fig. 8C, middle). In contrast, HEMO was undetectable in normal squamous tissue such as exocervix, or in squamous and keratinized tissue such as skin (Fig. 8C, bottom). This observation ruled out possible non‐specific staining on these tissues, with high content of keratins in the case of skin.
Fig. 8.
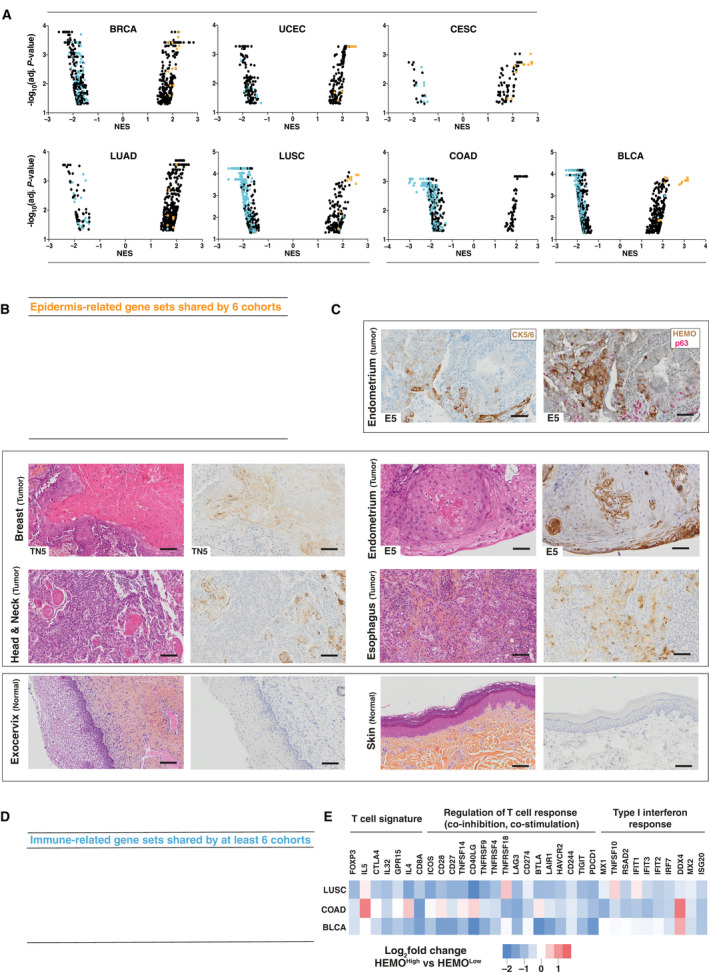
HEMO activation in tumors is linked to epidermis and immune‐related signatures. (A) GSEA (Gene Ontology Biological Process and REACTOME) pathway distribution for HEMOHigh vs HEMOLow tumors in TCGA cohorts. Of note, for each cohort, these two groups displayed similar tumor cellularity. Immune response‐ and epidermis‐related gene sets are, respectively, demarcated as blue and orange dots. NES: Normalized Enrichment Score. (B) List of epidermis‐related gene sets shared by six cohorts (absence of such gene sets in the COAD cohort). (C) Top: Endometrium adenocarcinoma sample stained with CK5/6 (left) or HEMO (brown) and p63 (pink) (right), magnification: 15×, scale bar: 100 μm. Middle: pairs of HES and anti‐HEMO staining in differentiated squamous tumors of breast, endometrium, Head and Neck with peculiar localization of HEMO in squamous pearls and esophagus. Bottom: pairs of HES and anti‐HEMO staining in normal tissue of exocervix and skin. Magnification: 10×, scale bar: 100 μm. When specified, numbers correspond to samples analyzed in Fig. 4. (D) List of immune‐related gene sets shared by 6 or 7 cohorts. (E) Heatmap displaying differential expression between HEMOHigh and HEMOLow tumors, of immune‐related genes belonging to ‘T‐cell (Treg and CD8)’, ‘Regulation of immune response’ and ‘Type I interferon response’ signatures established by [47]. PDCD1 was also added on this figure. Color gradation is representative of Log2 fold change.
Lastly, GSEA results revealed a substantial depletion of immune‐related gene sets in HEMOHigh tumors of several TCGA cohorts (Fig. 8A, blue dots) with numerous pathways reaching very high significance (Adj. P‐value < 0.001, NES < −2). Interestingly, pathways belonging to both innate and adaptive immune responses were commonly under‐represented in HEMOHigh groups of several TCGA cohorts (Fig. 8D).
Negative association with these immune signatures was particularly noticeable in LUSC, COAD, and BLCA cohorts as shown in their top 10 most significant GO:BP and REACTOME gene sets (Table S9). Indeed, a heatmap of representative genes of immune features such as ‘T cell’, ‘Regulation of T‐cell response’, and ‘Type I interferon response’ established by [47] mainly showed their underexpression in HEMOHigh tumors (Fig. 8E).
4. Discussion
Altogether, the presented results indicate that the endogenous retrovirus‐derived HEMO envelope gene is specifically upregulated in solid primary tumors. Indeed, demonstration of the concordance between HEMO transcriptional activation and detection of the corresponding protein in a series of tumor tissues (by immunohistochemistry and clear identification of the protein by western blot) allowed us to validate the relevance of measuring HEMO expression via large scale high throughput RNAseq data.
These analyses revealed that HEMO is highly activated in head and neck, lung, breast, endometrium, cervix, esophagus, and bladder tumors, whereas it is faintly or not detected in normal tissues. Activation was also observed, although to a lower level, in cohorts such as prostate, stomach, colorectal, pancreatic, or brain tumors.
Furthermore, in cohorts such as breast, uterus, or prostate, we noticed, between normal and tumor conditions, an intermediate level of activation in the control adjacent tissues, suggesting a possible precancerous state or local inflammatory response [48].
However, a special case was the renal tumors, where both a strong activation in control adjacent tissues and a very clear decrease in expression in tumor tissues were observed in the kidney cohorts. This unexpected level of HEMO expression in tumor adjacent tissues (highest median value of all tumor samples in Fig. 1) could not simply represent a precancerous intermediate state. The kidney is a vital organ, where tissue damage reactivates specific pathways to repair renal function, in particular the Wnt/β‐catenin signaling pathway usually involved in nephron morphogenesis during embryogenesis [49, 50]. In tumor conditions, control adjacent tissues of the kidney could be engaged in active regenerative processes to compensate for the destruction of renal parenchyma, with consecutive HEMO activation dependent on this pathway. On the other side, the HEMO downregulation observed in the renal tumor cells could result from specific epigenetic modifications or from tumor amplification of renal cells not expressing HEMO. Indeed, single‐cell RNAseq analyses of normal tissue of the Human Protein Atlas project (https://proteinatlas.org) showed preferential HEMO (ERVMER34‐1) expression in collecting duct cells. Corresponding tumors represent only 1% of the renal cell carcinomas [51] and are not present in the TCGA consortium.
In addition, we showed that HEMO expression is maintained in metastatic tumors and is associated with high tumor grade in head and neck tumors and possibly in the smaller pancreatic cohort. Interestingly, it was recently reported that HEMO (ERVMER34‐1) belongs to a gene signature predicting recurrence of colon adenocarcinoma based on the comparison of mRNAs, lincRNA, and miRNA transcriptomic profiles of recurrent and non‐recurrent tumors [52]. These results unveil its putative interest as a prognostic factor.
Furthermore, in the cohorts analyzed in this study, HEMO's activation in solid tumors made this retroviral envelope gene stand out from others such as syncytin‐1 (ERVW‐1) and ERVV‐2 for which we mainly found low expression, from syncytin‐2 (ERVFRD‐1) usually inactivated in tumor samples, and from ERV3‐1 which mainly shows non‐tumor‐specific activation. However, since syncytins and ERVV‐2 sequences are mostly undetectable in normal tissues, their occasional activation in some tumors, as well as the ERVW‐1 expression in testicular cells, could be of interest for specific targeted therapy. In contrast to HEMO, syncytin‐1 [53] and syncytin‐2 were specifically increased in hematological LAML tumors.
Characterization of co‐expression signature associated with HEMO activation in TCGA datasets led us to hypothesize that its expression could be linked to the Wnt/β‐catenin pathway, in particular in endometrium tumors, where we found a clear association between the presence of CTNNB1 mutations and a high level of HEMO expression (73.6%). Altered Wnt/β‐catenin signaling, as a result of gene alterations such as those found in APC or in CTNNB1 (β‐catenin), has been reported to drive tumorigenesis in numerous tumors including endometrial cancer [30]. For patients with low‐grade and early‐stage endometrioid endometrial tumors, these mutations have been associated with a higher risk of recurrence [54, 55]. From a histological standpoint, constitutive activation of the Wnt/β‐catenin pathway with abnormal nuclear accumulation of β‐catenin is preferentially observed in morules of endometrial carcinomas [42]. In our endometrium tumor samples, colocalization of HEMO and nuclear β‐catenin in specific morular metaplasia therefore confirmed the initial hypothesis indicating a close link between the Wnt/β‐catenin pathway and HEMO activation. Moreover, the in vitro inhibition of this pathway clearly provided evidence that Wnt/β‐catenin signaling is involved in HEMO regulation. Data from the ENCODE project supported the hypothesis that the HEMO promoter could be bound by TCF/β‐catenin partners or by transcription factors of the Wnt signaling pathway [56]. Chromatin‐immunoprecipitation assays in human cell lines having high levels of HEMO expression or targeted CRISPR experiments to modify the HEMO promoter or knock‐out specific members of the Wnt signaling pathway will help to establish whether HEMO can be considered as a bona fide downstream direct or indirect target of this pathway.
Interestingly, it has been previously shown that HEMO is expressed in placenta and embryonic stem cells (ESC) [31] in which Wnt signaling is known to be activated and plays a key role in stem cell maintenance [57] or in placental development and differentiation [58]. Thus, epigenetic modifications leading to hypomethylation of the HEMO CpG‐rich promoter and activation of Wnt signaling could be considered as two concomitant events resulting in HEMO expression in tumors, stem cell, and placenta (Fig. 9).
Fig. 9.
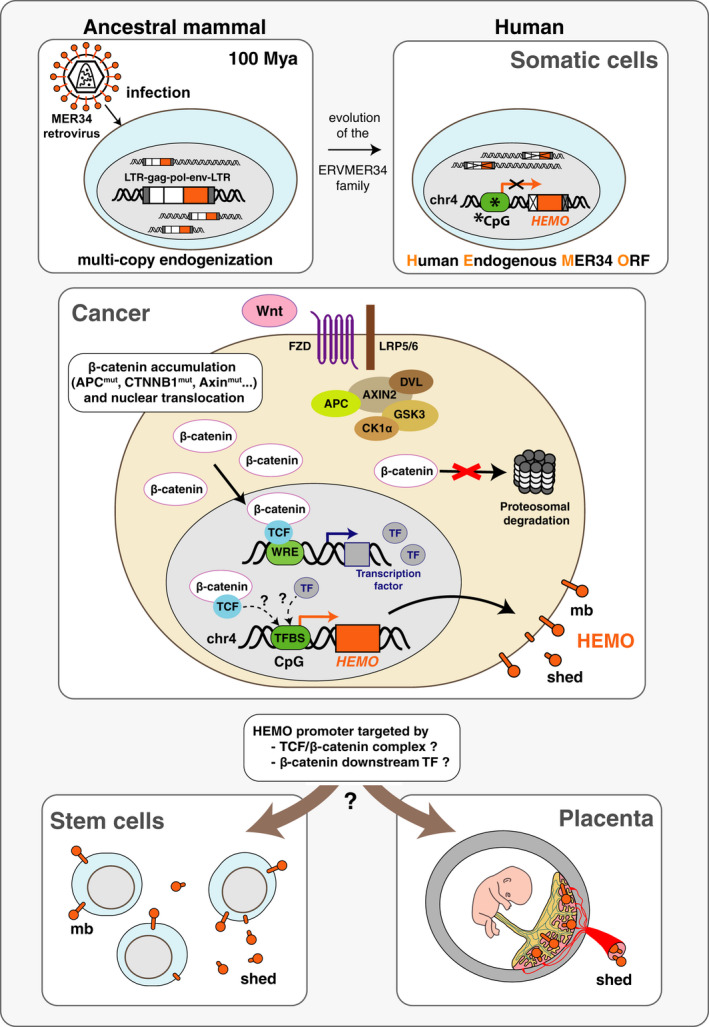
Model for the regulation of HEMO expression in cancer. A ‘MER34 retrovirus’ was endogenized in an ancestral mammal genome about 100 mya, as a multi‐copy ERV family. Following genetic evolution, only one env ORF remains present in human genome on chromosome 4, having lost its 5′ LTR and being transcribed from a cellular CpG‐rich promoter. This sequence was previously described as HEMO (Human Endogenous MER34 ORF) [31]. Methylation of its promoter could participate in the HEMO silencing in somatic cells (*CpG methylation). In cancer, besides epigenetic modifications, such as hypomethylation of the HEMO promoter, we propose that the Wnt/β‐catenin pathway is an upstream regulator of HEMO expression. Activation of this signaling pathway, due to genetic or epigenetic alterations, is frequently observed in tumors and results in the accumulation of β‐catenin and its subsequent translocation to the nucleus of the tumor cell. Once in the nuclear compartment, β‐catenin associates with TCF to form a complex that binds WREs (‘Wnt Responsive Elements’) and thus activates transcription of numerous target genes. Among them, transcription factors could in turn activate HEMO transcription. Alternatively, HEMO could directly be activated by the β‐catenin/TCF complex. These mechanisms could also drive HEMO activation in stem cells and placenta where this gene is expressed as membrane and shed HEMO proteins [31]. mb, membrane; LTR, long terminal repeat; TF, transcription factor, TFBS, transcription factor binding site.
In addition, we identified a specific pattern of HEMO expression in squamous tumors. Even though the Wnt pathway has been reported to play a role in squamous transdifferentiation [59], the specific underlying mechanisms leading to activation of HEMO in squamous (+/−keratinizing) tumor areas remained unclear and need to be further explored.
Lastly, in silico analysis provided evidence of a link between HEMO and immune signatures. Tumors with high HEMO transcript levels were found to be negatively associated with immune features such as regulation of T‐cell response or interferon response, particularly in lung, colon, and bladder tumors. This finding was unexpected as there is an accumulation of evidence showing endogenous retroviral products as triggers of antiviral immune response [60, 61, 62]. Nevertheless, HEMO belonging to an ERV with a degenerated 3′ LTR sequence, its contribution to dsRNA formation [63] that elicits in turn viral mimicry should be minimal. Furthermore, given that active Wnt/β‐catenin signaling has been documented as being a determinant factor of immune exclusion in tumors [64, 65, 66], frequent activation of this pathway in HEMOHigh tumors may actually account for the observed downregulation of tumor immune response.
On the other hand, as a retroviral envelope protein, HEMO could still carry a functional immunosuppressive domain (ISD) that modulates the immune response [67]. Given that the shedding cleavage of the protein occurs inside the ISD sequence, regulation of the shedding process may have important consequences on the immunosuppressive function, both in physiological and tumor conditions.
5. Conclusion
Overall, our results strengthen the potential of HEMO as a tumor biomarker and therapeutic target. Because this retroviral envelope protein has the unique property of being shed and secreted into the blood, it is a promising serum marker for the detection and follow‐up of HEMO‐positive tumors, even in renal tumors where its expression in control adjacent tissues could be activated in the early stages of tumor development. In healthy individuals, the HEMO shed protein is filtered by the kidney, as we could not detect the protein in the urine, even in the 3rd trimester of pregnancy when its level in the serum is high ([31] and our unpublished results). Therefore, it could also be used as a urinary marker in case of renal injury or bladder carcinoma. In tumor sites other than kidney, depending on the relative levels of HEMO expression, tumors could be targeted by systemic anti‐HEMO therapy or solely by intra‐tumoral treatment to protect renal function. Given that this sequence codes for a membrane protein, HEMO is an interesting target for anti‐tumor strategies such as antibody‐drug‐conjugates (ADC) or oncolytic viruses in particular in tumors in which Wnt signaling is altered and where efficacy of immune checkpoint blockade is limited [68, 69].
Conflict of interest
The authors declare no conflict of interest.
Author contributions
AK was involved in all experimental strategy and design, generated and analyzed data and was involved in the writing/editing of the manuscript. AB designed and performed western blotting and qPCR, was involved in all data analyses and in writing/editing of the manuscript. OB participated in the design of the immunohistochemistry experiments and performed them. KDA initiated bioinformatics analysis on RNAseq data. BJ performed bioinformatics Gene Set Enrichment Analysis on RNAseq data. CM provided access to the MOSCATO cohort and participated in discussions. J‐YS participated in the design and analysis of immunohistochemistry experiments. TH supervised the study. OH conceptualized and directed the project, was involved in all strategy, design, analysis of data and writing/editing of the manuscript. All authors revised and approved the final version of the paper.
Supporting information
Fig. S1. Expression level of HERV‐env genes in tumors of the TCGA cohorts. Boxplots of normalized (TPM) and log2‐transformed expression of HERV‐env genes in tumor (‘T’, orange) and control (‘C’, green) samples retrieved from TCGA‐Recount2. White boxes correspond to basal expression in normal (‘N’) tissues from GTEx‐Recount2. Data are shown as mean with 25–75th percentile range (box) and 10–90th percentile (whiskers). Mild outliers are depicted as black dots. P‐values are shown for the pairwise (N,T) comparison: *, p < 0.05; **, p < 0.01; ***, p < 0.001, Mann‐Whitney U‐test (see Table S3). (A‐D) Expression level of the 4 HERV‐env genes: ERVW‐1 (ENSG00000242950), ERVFRD‐1 (ENSG00000244476), ERVV‐2 (ENSG00000268964) env and ERV3‐1 (ENSG00000213462) genes. P‐values for pairwise comparisons between each group (T, C, N) are given in Table S3. HEMATO: Hematological tumors. (E) Expression level of HEMO and the 4 HERV‐env genes in the three kidney TCGA cohorts KICH, KIRC and KIRP.
Fig. S2. Association between HEMO expression and tumor stage, grade, histological type or molecular subtype. Boxplots of HEMO expression in TCGA tumors stratified by clinical/pathologic stages, neoplasm histologic grade, histological type and molecular subtype. Statistical significance was evaluated by the Mann‐Whitney U‐test for comparison of two groups, and by the Kruskal‐Wallis test for comparison of more than 2 groups (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Significant p‐values are in red. Data are shown as mean with 25–75th percentile range (box) and 10–90th percentile (whiskers). Mild outliers are depicted as black dots. Cohorts are grouped according to body systems as in Fig. 1. ADC: Adenocarcinoma, ADSQ: Adenosquamous, BAC: Bronchoalveolar Carcinoma, NOS: Not Otherwise Specified, SC: Squamous Carcinoma, SCC: Squamous Cell Carcinoma, SPP: Solid Pattern Predominant.
Fig. S3. Comparison of HEMO expression within NCIH20, HCC827, OVMANA, Caco‐2, SW480 and HCT116 cell lines. Cell lines were selected among HEMO high expressing cases reported in [70]. Comparison was based on RT‐qPCR and western blot analysis (A), mutational status of CTNNB1 and APC (B) and immunohistochemistry analysis (anti‐HEMO staining with 2F7 mAb) (C). Std: protein standard. Magnification: 40X, scale bar: 20 μm.
Fig. S4. HEMO upregulation is associated with active Wnt/β‐catenin pathway in COAD cohort. (A) GSEA enrichment plots showing ‘REACTOME formation of the beta catenin TCF transactivating complex’ (n°25 in Table S7) and ‘REACTOME TCF dependent signaling in response to Wnt’ (n°71 in Table S7) enriched signatures between COAD HEMOHigh and HEMOLow tumors. NES: Normalized Enrichment Score. (B) Heatmap for the significant Wnt‐related genes differentially expressed between HEMOHigh and HEMOLow tumors. Color gradation is representative of Log2 fold change. The differential level of HEMO expression is also indicated at the top of the heatmap (LogFC = 8). For all depicted genes (except for APC and CTNNB1) adjusted p‐value < 0.01. In bold, common upregulated genes found in UCEC cohort (Fig. 5D).
Table S1. Summary of immunohistochemistry protocols.
Table S2. List of the primers.
Table S3. List and size of the cohorts investigated in this study from GTEx and TCGA databases.
Table S4. Expression level of HERV‐env genes in male breast tumor samples.
Table S5. List of retroviral sequences annotated in the human genome (Ensembl).
Table S6. List of differentially‐expressed genes associated with HEMO in 8 TCGA cohorts (BLCA, BRCA, CESC, COAD, HNSC, LUAD, LUSC, UCEC).
Table S7. Results of Gene Set Enrichment Analysis (GSEA) performed on REACTOME and GO Biological process databases.
Table S8. Mutational status of CTNNB1 and APC in HEMOHigh and HEMOLow tumors of representative TCGA cohorts.
Table S9. Top 10 GO:BP and Top 10 REACTOME gene sets for LUSC, COAD and BLCA cohorts.
Acknowledgements
We would like to thank the Breast (S. Delaloge and M. C. Mathieu), Digestive (M. Ducreux and P. Dartigues), Head & Neck (I. Breuskin and O. Casiraghi) and Gynecological (A. Leary and C. Genestie) clinical committees for providing us access to tumor tissues through the Gustave Roussy Biological Resource Centre (Gustave Roussy, Villejuif, France) and its staff (C. Machavoine), and M. Ngo‐Camus for clinical data management of the MOSCATO cohort (DITEP, Gustave Roussy). We thank E. Solary for its support, both scientific and administrative as former Director of Research at Gustave Roussy. We thank M. Deloger and D. Gautheret (Gustave Roussy Bioinformatics core facility, AMMICa, UMS CNRS3655) for bioinformatics assistance and helpful discussions and F. Jaulin (Gustave Roussy, UMR1279) and L. Friboulet (Gustave Roussy, U981) for the gift of cell lines. We thank H. Rocheteau (PETRA platform, AMMICa, UMS CNRS3655), E. Etui, HM. Lin and S. Piterboth (Department of Pathology and Laboratory Medicine, Gustave Roussy) for their contribution to the histological analyses, and M. Koster for technical support. We thank C. Lavialle for critical reading of the manuscript. A. Kasperek was supported by a fellowship from ‘Course of Excellence in Oncology—Fondation Philanthropia’.
Data accessibility
The original RNAseq data that support the findings in this study are openly available in Recount2 resource (https://jhubiostatistics.shinyapps.io/recount/), and the processed analyses are available in Tables [Link], [Link] in the supplementary material of this article.
References
- 1. Mager DL & Stoye JP (2015) Mammalian endogenous retroviruses. Microbiol Spectr 3, MDNA3‐0009‐2014. [DOI] [PubMed] [Google Scholar]
- 2. Feschotte C & Gilbert C (2012) Endogenous viruses: insights into viral evolution and impact on host biology. Nat Rev Genet 13, 283–296. [DOI] [PubMed] [Google Scholar]
- 3. Dewannieux M & Heidmann T (2013) Endogenous retroviruses: acquisition, amplification and taming of genome invaders. Curr Opin Virol 3, 646–656. [DOI] [PubMed] [Google Scholar]
- 4. Johnson WE (2019) Origins and evolutionary consequences of ancient endogenous retroviruses. Nat Rev Microbiol 17, 355–370. [DOI] [PubMed] [Google Scholar]
- 5. Aiewsakun P & Katzourakis A (2017) Marine origin of retroviruses in the early Palaeozoic Era. Nat Commun 8, 13954. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6. Lander ES, Linton LM, Birren B, Nusbaum C, Zody MC, Baldwin J, Devon K, Dewar K, Doyle M, FitzHugh W et al. (2001) Initial sequencing and analysis of the human genome. Nature 409, 860–921. [DOI] [PubMed] [Google Scholar]
- 7. Vargiu L, Rodriguez‐Tomé P, Sperber GO, Cadeddu M, Grandi N, Blikstad V, Tramontano E & Blomberg J (2016) Classification and characterization of human endogenous retroviruses; mosaic forms are common. Retrovirology 13, 7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8. Mi S, Lee X, Li X‐P, Veldman GM, Finnerty H, Racie L, LaVallie E, Tang X‐Y, Edouard P, Howes S et al. (2000) Syncytin is a captive retroviral envelope protein involved in human placental morphogenesis. Nature 403, 785–789. [DOI] [PubMed] [Google Scholar]
- 9. Blond JL, Besème F, Duret L, Bouton O, Bedin F, Perron H, Mandrand B & Mallet F (1999) Molecular characterization and placental expression of HERV‐W, a new human endogenous retrovirus family. J Virol 73, 1175–1185. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10. Blaise S, de Parseval N, Bénit L & Heidmann T (2003) Genomewide screening for fusogenic human endogenous retrovirus envelopes identifies syncytin 2, a gene conserved on primate evolution. Proc Natl Acad Sci USA 100, 13013–13018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11. Lavialle C, Cornelis G, Dupressoir A, Esnault C, Heidmann O, Vernochet C & Heidmann T (2013) Paleovirology of ‘syncytins’, retroviral env genes exapted for a role in placentation. Philos Trans R Soc Lond B Biol Sci 368, 20120507. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12. Grundy EE, Diab N & Chiappinelli KB (2021) Transposable element regulation and expression in cancer. FEBS J, doi: 10.1111/febs.15722 [DOI] [PubMed] [Google Scholar]
- 13. Hurst TP & Magiorkinis G (2017) Epigenetic control of human endogenous retrovirus expression: focus on regulation of long‐terminal repeats (LTRs). Viruses 9, 130. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14. Geis FK & Goff SP (2020) Silencing and transcriptional regulation of endogenous retroviruses: an overview. Viruses 12, 884. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15. Bannert N, Hofmann H, Block A & Hohn O (2018) HERVs new role in cancer: from accused perpetrators to cheerful protectors. Front Microbiol 9, 178. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16. Kassiotis G & Stoye JP (2017) Making a virtue of necessity: the pleiotropic role of human endogenous retroviruses in cancer. Philos Trans R Soc Lond B Biol Sci 372, 20160277. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17. Gonzalez‐Cao M, Iduma P, Karachaliou N, Santarpia M, Blanco J & Rosell R (2016) Human endogenous retroviruses and cancer. Cancer Biol Med 13, 483–488. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18. Brodziak A, Ziółko E, Muc‐Wierzgoń M, Nowakowska‐Zajdel E, Kokot T & Klakla K (2012) The role of human endogenous retroviruses in the pathogenesis of autoimmune diseases. Med Sci Monit 18, RA80–RA88. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19. Levet S, Charvet B, Bertin A, Deschaumes A, Perron H & Hober D (2019) Human endogenous retroviruses and type 1 diabetes. Curr Diab Rep 19, 141. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20. Volkman HE & Stetson DB (2014) The enemy within: endogenous retroelements and autoimmune disease. Nat Immunol 15, 415–422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 21. Küry P, Nath A, Créange A, Dolei A, Marche P, Gold J, Giovannoni G, Hartung H‐P & Perron H (2018) Human endogenous retroviruses in neurological diseases. Trends Mol Med 24, 379–394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22. Balestrieri E, Matteucci C, Cipriani C, Grelli S, Ricceri L, Calamandrei G & Sinibaldi Vallebona P (2019) Endogenous retroviruses activity as a molecular signature of neurodevelopmental disorders. Int J Mol Sci 20, 6050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23. Dolei A, Ibba G, Piu C & Serra C (2019) Expression of HERV genes as possible biomarker and target in neurodegenerative diseases. Int J Mol Sci 20, 3706. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24. Zhang M, Liang JQ & Zheng S (2019) Expressional activation and functional roles of human endogenous retroviruses in cancers. Rev Med Virol 29, e2025. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25. Ito J, Sugimoto R, Nakaoka H, Yamada S, Kimura T, Hayano T & Inoue I (2017) Systematic identification and characterization of regulatory elements derived from human endogenous retroviruses. PLOS Genet 13, e1006883. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26. MacDonald BT, Tamai K & He X (2009) Wnt/β‐catenin signaling: components, mechanisms, and diseases. Dev Cell 17, 9–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27. Zhan T, Rindtorff N & Boutros M (2017) Wnt signaling in cancer. Oncogene 36, 1461–1473. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28. Polakis P (2012) Wnt signaling in cancer. Cold Spring Harb Perspect Biol 4, a008052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29. Prosperi JR, Luu HH & Goss KH (2011) Dysregulation of the Wnt pathway in solid tumors. In Targeting the Wnt Pathway in Cancer (Goss K & Kahn M, eds), pp. 81–128. Springer, New York, NY. [Google Scholar]
- 30. Bugter JM, Fenderico N & Maurice MM (2021) Mutations and mechanisms of WNT pathway tumour suppressors in cancer. Nat Rev Cancer 21, 5–21. doi: 10.1038/s41568-020-00307-z [DOI] [PubMed] [Google Scholar]
- 31. Heidmann O, Béguin A, Paternina J, Berthier R, Deloger M, Bawa O & Heidmann T (2017) HEMO, an ancestral endogenous retroviral envelope protein shed in the blood of pregnant women and expressed in pluripotent stem cells and tumors. Proc Natl Acad Sci USA 114, E6642–E6651. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32. Collado‐Torres L, Nellore A, Kammers K, Ellis SE, Taub MA, Hansen KD, Jaffe AE, Langmead B & Leek JT (2017) Reproducible RNA‐seq analysis using recount2. Nat Biotechnol 35, 319–321. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33. Colaprico A, Silva TC, Olsen C, Garofano L, Cava C, Garolini D, Sabedot TS, Malta TM, Pagnotta SM, Castiglioni I et al. (2016) TCGAbiolinks: an R/Bioconductor package for integrative analysis of TCGA data. Nucleic Acids Res 44, e71. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34. Massard C, Michiels S, Ferté C, Le Deley M‐C, Lacroix L, Hollebecque A, Verlingue L, Ileana E, Rosellini S, Ammari S et al. (2017) High‐throughput genomics and clinical outcome in hard‐to‐treat advanced cancers: results of the MOSCATO 01 trial. Cancer Discov 7, 586–595. [DOI] [PubMed] [Google Scholar]
- 35. Bray NL, Pimentel H, Melsted P & Pachter L (2016) Near‐optimal probabilistic RNA‐seq quantification. Nat Biotechnol 34, 525–527. [DOI] [PubMed] [Google Scholar]
- 36. Soneson C, Love MI & Robinson MD (2015) Differential analyses for RNA‐seq: transcript‐level estimates improve gene‐level inferences. F1000Res 4, 1521. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37. de Parseval N, Lazar V, Casella J‐F, Benit L & Heidmann T (2003) Survey of human genes of retroviral origin: identification and transcriptome of the genes with coding capacity for complete envelope proteins. J Virol 77, 10414–10422. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38. Blaise S, de Parseval N & Heidmann T (2005) Functional characterization of two newly identified human endogenous retrovirus coding envelope genes. Retrovirology 2, 19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39. Xue B, Sechi LA & Kelvin DJ (2020) Human endogenous retrovirus K (HML‐2) in health and disease. Front Microbiol 11, 1690. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40. Cruciat C‐M & Niehrs C (2013) Secreted and transmembrane Wnt inhibitors and activators. Cold Spring Harb Perspect Biol 5, a015081. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41. Liu Y, Patel L, Mills GB, Lu KH, Sood AK, Ding LI, Kucherlapati R, Mardis ER, Levine DA, Shmulevich I et al. (2014) Clinical significance of CTNNB1 mutation and Wnt pathway activation in endometrioid endometrial carcinoma. J Natl Cancer Inst 106, dju245. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42. Saegusa M & Okayasu I (2001) Frequent nuclear β‐catenin accumulation and associated mutations in endometrioid‐type endometrial and ovarian carcinomas with squamous differentiation. J Pathol 194, 59–67. [DOI] [PubMed] [Google Scholar]
- 43. Chinen K, Kamiyama K, Kinjo T, Arasaki A, Ihama Y, Hamada T & Iwamasa T (2004) Morules in endometrial carcinoma and benign endometrial lesions differ from squamous differentiation tissue and are not infected with human papillomavirus. J Clin Pathol 57, 918–926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44. Houghton O, Connolly LE & McCluggage WG (2008) Morules in endometrioid proliferations of the uterus and ovary consistently express the intestinal transcription factor CDX2. Histopathology 53, 156–165. [DOI] [PubMed] [Google Scholar]
- 45. Ilyas M, Tomlinson IPM, Rowan A, Pignatelli M & Bodmer WF (1997) β‐Catenin mutations in cell lines established from human colorectal cancers. Proc Natl Acad Sci USA 94, 10330–10334. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46. Handeli S & Simon JA (2008) A small‐molecule inhibitor of Tcf/beta‐catenin signaling down‐regulates PPARgamma and PPARdelta activities. Mol Cancer Ther 7, 521–529. [DOI] [PubMed] [Google Scholar]
- 47. Rooney MS, Shukla SA, Wu CJ, Getz G & Hacohen N (2015) Molecular and genetic properties of tumors associated with local immune cytolytic activity. Cell 160, 48–61. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48. Aran D, Camarda R, Odegaard J, Paik H, Oskotsky B, Krings G, Goga A, Sirota M & Butte AJ (2017) Comprehensive analysis of normal adjacent to tumor transcriptomes. Nat Commun 8, 1077. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49. Guo Q, Kim A, Li B, Ransick A, Bugacov H, Chen XI, Lindström N, Brown A, Oxburgh L, Ren B et al. (2021) A β‐catenin‐driven switch in TCF/LEF transcription factor binding to DNA target sites promotes commitment of mammalian nephron progenitor cells. eLife 10, e64444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50. Kawakami T, Ren S & Duffield JS (2013) Wnt signalling in kidney diseases: dual roles in renal injury and repair: Wnt signalling in renal injury and repair. J Pathol 229, 221–231. [DOI] [PubMed] [Google Scholar]
- 51. Sui W, Matulay JT, Robins DJ, James MB, Onyeji IC, RoyChoudhury A, Wenske S & DeCastro GJ (2017) Collecting duct carcinoma of the kidney: disease characteristics and treatment outcomes from the National Cancer Database. Urol Oncol 35, 540.e13–540.e18. [DOI] [PubMed] [Google Scholar]
- 52. Jin LP, Liu T, Meng FQ & Tai JD (2020) Prognosis prediction model based on competing endogenous RNAs for recurrence of colon adenocarcinoma. BMC Cancer 20, 968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53. Sun YI, Zhu H, Song J, Jiang Y, Ouyang H, Huang R, Zhang G, Fan X, Tao R, Jiang J et al. (2016) Upregulation of leukocytic syncytin‐1 in acute myeloid leukemia patients. Med Sci Monit 22, 2392–2403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54. Kurnit KC, Kim GN, Fellman BM, Urbauer DL, Mills GB, Zhang W & Broaddus RR (2017) CTNNB1 (beta‐catenin) mutation identifies low grade, early stage endometrial cancer patients at increased risk of recurrence. Mod Pathol 30, 1032–1041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55. Myers A, Barry WT, Hirsch MS, Matulonis U & Lee L (2014) β‐Catenin mutations in recurrent FIGO IA grade I endometrioid endometrial cancers. Gynecol Oncol 134, 426–427. [DOI] [PubMed] [Google Scholar]
- 56. Dunham I, Kundaje A, Aldred SF, Collins PJ, Davis CA, Doyle F, Epstein CB, Frietze S, Harrow J, Kaul R et al. (2012) An integrated encyclopedia of DNA elements in the human genome. Nature 489, 57–74. doi: 10.1038/nature11247 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57. Van Camp JK, Beckers S, Zegers D & Van Hul W (2014) Wnt signaling and the control of human stem cell fate. Stem Cell Rev Rep 10, 207–229. [DOI] [PubMed] [Google Scholar]
- 58. Knöfler M & Pollheimer J (2013) Human placental trophoblast invasion and differentiation: a particular focus on Wnt signaling. Front Genet 4, 190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59. Saegusa M, Hashimura M & Kuwata T (2012) Sox4 functions as a positive regulator of β‐catenin signaling through upregulation of TCF4 during morular differentiation of endometrial carcinomas. Lab Invest 92, 511–521. [DOI] [PubMed] [Google Scholar]
- 60. Chiappinelli K, Strissel P, Desrichard A, Li H, Henke C, Akman B, Hein A, Rote N, Cope L, Snyder A et al. (2015) Inhibiting DNA methylation causes an interferon response in cancer via dsRNA including endogenous retroviruses. Cell 162, 974–986. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61. Roulois D, Loo Yau H, Singhania R, Wang Y, Danesh A, Shen S, Han H, Liang G, Jones P, Pugh T et al. (2015) DNA‐demethylating agents target colorectal cancer cells by inducing viral mimicry by endogenous transcripts. Cell 162, 961–973. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62. de Cubas AA, Dunker W, Zaninovich A, Hongo RA, Bhatia A, Panda A, Beckermann KE, Bhanot G, Ganesan S, Karijolich J et al. (2020) DNA hypomethylation promotes transposable element expression and activation of immune signaling in renal cell cancer. JCI Insight 5, e137569. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63. Dunn CA, Romanish MT, Gutierrez LE, van de Lagemaat LN & Mager DL (2006) Transcription of two human genes from a bidirectional endogenous retrovirus promoter. Gene 366, 335–342. [DOI] [PubMed] [Google Scholar]
- 64. Xue J, Yu X, Xue L, Ge X, Zhao W & Peng W (2019) Intrinsic β‐catenin signaling suppresses CD8+ T‐cell infiltration in colorectal cancer. Biomed Pharmacother 115, 108921. doi: 10.1016/j.biopha.2019.108921 [DOI] [PubMed] [Google Scholar]
- 65. Luke JJ, Bao R, Sweis RF, Spranger S & Gajewski TF (2019) WNT/β‐catenin pathway activation correlates with immune exclusion across human cancers. Clin Cancer Res 25, 3074–3083. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66. Li X, Xiang Y, Li F, Yin C, Li B & Ke X (2019) WNT/β‐catenin signaling pathway regulating T cell‐inflammation in the tumor microenvironment. Front Immunol 10, 2293. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67. Mangeney M, Renard M, Schlecht‐Louf G, Bouallaga I, Heidmann O, Letzelter C, Richaud A, Ducos B & Heidmann T (2007) Placental syncytins: genetic disjunction between the fusogenic and immunosuppressive activity of retroviral envelope proteins. Proc Natl Acad Sci USA 104, 20534–20539. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68. Trujillo JA, Luke JJ, Zha Y, Segal JP, Ritterhouse LL, Spranger S, Matijevich K & Gajewski TF (2019) Secondary resistance to immunotherapy associated with β‐catenin pathway activation or PTEN loss in metastatic melanoma. J Immunother Cancer 7, 295. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69. Kalbasi A & Ribas A (2020) Tumour‐intrinsic resistance to immune checkpoint blockade. Nat Rev Immunol 20, 25–39. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70. Klijn C, Durinck S, Stawiski EW, Haverty PM, Jiang Z, Liu H, Degenhardt J, Mayba O, Gnad F, Liu J et al. (2015) A comprehensive transcriptional portrait of human cancer cell lines. Nat Biotechnol 33, 306–312. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Fig. S1. Expression level of HERV‐env genes in tumors of the TCGA cohorts. Boxplots of normalized (TPM) and log2‐transformed expression of HERV‐env genes in tumor (‘T’, orange) and control (‘C’, green) samples retrieved from TCGA‐Recount2. White boxes correspond to basal expression in normal (‘N’) tissues from GTEx‐Recount2. Data are shown as mean with 25–75th percentile range (box) and 10–90th percentile (whiskers). Mild outliers are depicted as black dots. P‐values are shown for the pairwise (N,T) comparison: *, p < 0.05; **, p < 0.01; ***, p < 0.001, Mann‐Whitney U‐test (see Table S3). (A‐D) Expression level of the 4 HERV‐env genes: ERVW‐1 (ENSG00000242950), ERVFRD‐1 (ENSG00000244476), ERVV‐2 (ENSG00000268964) env and ERV3‐1 (ENSG00000213462) genes. P‐values for pairwise comparisons between each group (T, C, N) are given in Table S3. HEMATO: Hematological tumors. (E) Expression level of HEMO and the 4 HERV‐env genes in the three kidney TCGA cohorts KICH, KIRC and KIRP.
Fig. S2. Association between HEMO expression and tumor stage, grade, histological type or molecular subtype. Boxplots of HEMO expression in TCGA tumors stratified by clinical/pathologic stages, neoplasm histologic grade, histological type and molecular subtype. Statistical significance was evaluated by the Mann‐Whitney U‐test for comparison of two groups, and by the Kruskal‐Wallis test for comparison of more than 2 groups (*, p < 0.05; **, p < 0.01; ***, p < 0.001). Significant p‐values are in red. Data are shown as mean with 25–75th percentile range (box) and 10–90th percentile (whiskers). Mild outliers are depicted as black dots. Cohorts are grouped according to body systems as in Fig. 1. ADC: Adenocarcinoma, ADSQ: Adenosquamous, BAC: Bronchoalveolar Carcinoma, NOS: Not Otherwise Specified, SC: Squamous Carcinoma, SCC: Squamous Cell Carcinoma, SPP: Solid Pattern Predominant.
Fig. S3. Comparison of HEMO expression within NCIH20, HCC827, OVMANA, Caco‐2, SW480 and HCT116 cell lines. Cell lines were selected among HEMO high expressing cases reported in [70]. Comparison was based on RT‐qPCR and western blot analysis (A), mutational status of CTNNB1 and APC (B) and immunohistochemistry analysis (anti‐HEMO staining with 2F7 mAb) (C). Std: protein standard. Magnification: 40X, scale bar: 20 μm.
Fig. S4. HEMO upregulation is associated with active Wnt/β‐catenin pathway in COAD cohort. (A) GSEA enrichment plots showing ‘REACTOME formation of the beta catenin TCF transactivating complex’ (n°25 in Table S7) and ‘REACTOME TCF dependent signaling in response to Wnt’ (n°71 in Table S7) enriched signatures between COAD HEMOHigh and HEMOLow tumors. NES: Normalized Enrichment Score. (B) Heatmap for the significant Wnt‐related genes differentially expressed between HEMOHigh and HEMOLow tumors. Color gradation is representative of Log2 fold change. The differential level of HEMO expression is also indicated at the top of the heatmap (LogFC = 8). For all depicted genes (except for APC and CTNNB1) adjusted p‐value < 0.01. In bold, common upregulated genes found in UCEC cohort (Fig. 5D).
Table S1. Summary of immunohistochemistry protocols.
Table S2. List of the primers.
Table S3. List and size of the cohorts investigated in this study from GTEx and TCGA databases.
Table S4. Expression level of HERV‐env genes in male breast tumor samples.
Table S5. List of retroviral sequences annotated in the human genome (Ensembl).
Table S6. List of differentially‐expressed genes associated with HEMO in 8 TCGA cohorts (BLCA, BRCA, CESC, COAD, HNSC, LUAD, LUSC, UCEC).
Table S7. Results of Gene Set Enrichment Analysis (GSEA) performed on REACTOME and GO Biological process databases.
Table S8. Mutational status of CTNNB1 and APC in HEMOHigh and HEMOLow tumors of representative TCGA cohorts.
Table S9. Top 10 GO:BP and Top 10 REACTOME gene sets for LUSC, COAD and BLCA cohorts.
Data Availability Statement
The original RNAseq data that support the findings in this study are openly available in Recount2 resource (https://jhubiostatistics.shinyapps.io/recount/), and the processed analyses are available in Tables [Link], [Link] in the supplementary material of this article.