

Abstract
Background
Depressive disorders often begin during childhood or adolescence. There is a growing body of evidence supporting effective treatments during the acute phase of a depressive disorder. However, little is known about treatments for preventing relapse or recurrence of depression once an individual has achieved remission or recovery from their symptoms.
Objectives
To determine the efficacy of early interventions, including psychological and pharmacological interventions, to prevent relapse or recurrence of depressive disorders in children and adolescents.
Search methods
We searched the Cochrane Depression, Anxiety and Neurosis Review Group's Specialised Register (CCDANCTR) (to 1 June 2011). The CCDANCTR contains reports of relevant randomised controlled trials from The Cochrane Library (all years), EMBASE (1974 to date), MEDLINE (1950 to date) and PsycINFO (1967 to date). In addition we handsearched the references of all included studies and review articles.
Selection criteria
Randomised controlled trials using a psychological or pharmacological intervention, with the aim of preventing relapse or recurrence from an episode of major depressive disorder (MDD) or dysthymic disorder (DD) in children and adolescents were included. Participants were required to have been diagnosed with MDD or DD according to DSM or ICD criteria, using a standardised and validated assessment tool.
Data collection and analysis
Two review authors independently assessed all trials for inclusion in the review, extracted trial and outcome data, and assessed trial quality. Results for dichotomous outcomes are expressed as odds ratio and continuous measures as mean difference or standardised mean difference. We combined results using random‐effects meta‐analyses, with 95% confidence intervals. We contacted lead authors of included trials and requested additional data where possible.
Main results
Nine trials with 882 participants were included in the review. In five trials the outcome assessors were blind to the participants' intervention condition and in the remainder of trials it was unclear. In the majority of trials, participants were either not blind to their intervention condition, or it was unclear whether they were or not. Allocation concealment was also unclear in the majority of trials. Although all trials treated participants in an outpatient setting, the designs implemented in trials was diverse, which limits the generalisability of the results. Three trials indicated participants treated with antidepressant medication had lower relapse‐recurrence rates (40.9%) compared to those treated with placebo (66.6%) during a relapse prevention phase (odds ratio (OR) 0.34; 95% confidence interval (CI) 0.18 to 0.64, P = 0.02). One trial that compared a combination of psychological therapy and medication to medication alone favoured a combination approach over medication alone, however this result did not reach statistical significance (OR 0.26; 95% CI 0.06 to 1.15). The majority of trials that involved antidepressant medication reported adverse events including suicide‐related behaviours. However, there were not enough data to show which treatment approach results in the most favourable adverse event profile.
Authors' conclusions
Currently, there is little evidence to conclude which type of treatment approach is most effective in preventing relapse or recurrence of depressive episodes in children and adolescents. Limited trials found that antidepressant medication reduces the chance of relapse‐recurrence in the future, however, there is considerable diversity in the design of trials, making it difficult to compare outcomes across studies. Some of the research involving psychological therapies is encouraging, however at present more trials with larger sample sizes need to be conducted in order to explore this treatment approach further.
Plain language summary
Treatments for preventing the recurrence of depression in children and adolescents
Many children and adolescents diagnosed with a depressive disorder will experience a relapse or recurrence of their symptoms. Little is known about what treatment approach works best to prevent this from occurring, once a child or adolescent has initially remitted or recovered from a depressive episode. This review aimed to determine the efficacy of early interventions, including psychological, social and pharmacological interventions to prevent relapse or recurrence of depressive disorders in children and adolescents.The review included nine studies that assessed the efficacy of antidepressant medication and psychological therapies in reducing the risk of a future depressive episode in children and adolescents. Trials varied in their quality and methodological design, limiting conclusions that could be drawn from the result. Overall, the review found that antidepressant medication reduces the chance that children and adolescents will experience another episode of depression, compared with a pill placebo. Psychological therapies also look promising as a treatment to prevent future depressive episodes, however given the aforementioned issues concerning trial quality and design, along with the small number of trials included in the review, it is unclear how effective these therapies are at present.
Summary of findings
Summary of findings for the main comparison. Medication compared to placebo for preventing relapse and recurrence of a depressive disorder in children and adolescents.
| Medication compared to placebo for preventing relapse and recurrence of a depressive disorder in children and adolescents | ||||||
| Patient or population: patients with preventing relapse and recurrence of a depressive disorder in children and adolescents Settings: outpatient Intervention: medication Comparison: placebo | ||||||
| Outcomes | Illustrative comparative risks* (95% CI) | Relative effect (95% CI) | No of participants (studies) | Quality of the evidence (GRADE) | Comments | |
| Assumed risk | Corresponding risk | |||||
| Placebo | Medication | |||||
| Number relapsed‐recurred | 667 per 1000 | 405 per 1000 (265 to 561) | OR 0.34 (0.18 to 0.64) | 164 (3 studies) | ⊕⊕⊕⊝ moderate2 | |
| Suicide‐related behaviours | 12 per 1000 | 13 per 1000 (2 to 85) | OR 1.02 (0.14 to 7.39) | 164 (3 studies) | ⊕⊕⊝⊝ low2,3 | |
| Drop‐outs | 259 per 1000 | 263 per 1000 (117 to 494) | OR 1.02 (0.38 to 2.79) | 164 (3 studies) | ⊕⊕⊕⊝ moderate3,4 | |
| *The basis for the assumed risk (e.g. the median control group risk across studies) is provided in footnotes. The corresponding risk (and its 95% confidence interval) is based on the assumed risk in the comparison group and the relative effect of the intervention (and its 95% CI). CI: Confidence interval; OR: Odds ratio | ||||||
| GRADE Working Group grades of evidence High quality: Further research is very unlikely to change our confidence in the estimate of effect. Moderate quality: Further research is likely to have an important impact on our confidence in the estimate of effect and may change the estimate. Low quality: Further research is very likely to have an important impact on our confidence in the estimate of effect and is likely to change the estimate. Very low quality: We are very uncertain about the estimate. | ||||||
1 For allocation concealment, three trials contained unclear risk of bias. In more than one trial there was insufficient evidence to rate blinding of participants and/or interviewers. 2 In all three trials there was 'unclear' risk of bias pertaining to allocation concealment, and in two trials there was insufficient information to deduce if assessors and participants were adequately blinded to treatment condition. 3 Total number of events is less than 300. 4 All trials adequately reported on number of drop‐outs and reasons.
Background
Description of the condition
It is well established that depressive disorders are highly recurrent (Belsher 1988). Indeed, for approximately 50% of those who suffer from depression, their illness will follow a chronic, relapsing course associated with considerable disability and impairment (Crown 2002). Furthermore, research suggests that in many individuals, depressive episodes show a worsening pattern over the course of repeated episodes, characterised by increased severity, frequency, autonomy (i.e. episodes are less clearly precipitated by psychosocial stress) and lack of responsiveness to initially effective treatments (Kendler 2000; Post 1992). Despite advances in the treatment of depression, research shows that the long‐term outcome for those who experience multiple episodes has altered little over the last 20 years (Kennedy 2003). In sum, it appears that for many people the first or initial episode of depression acts as a gateway to a relapsing form of the illness that is associated with considerable disability over an individual's lifespan.
In a review of epidemiological studies, estimates of prevalence ranged between 0.4% and 2.5% for major depressive disorder (MDD) in children and 0.4% and 8.3% for MDD in adolescents, and between 0.6% and 1.7% for dysthymic disorder (DD) in children and 1.6% and 8.0% for DD in adolescents (Birmaher 1996). Whereas in MDD, depressed mood must be present for at least two weeks, DD is characterised by a persistent and long‐term depressed or irritable mood, with the mean episode lasting between three to four years (Nobile 2003). A more recent meta‐analysis put the prevalence of depressive disorders in children (aged under 13 years) at 2.8% and in adolescents (aged 13 to 18 years) at 5.7% (Costello 2006). Depressive disorders tend to have their onset in adolescence or early adulthood (up to 25 years) (Kessler 2005; Rutter 1995), suggesting that interventions that have the potential to reduce relapse are particularly critical in this age group, and may be able to influence a critical change to the lifetime course of the disorder. This is especially important given the high level of continuity between depressive disorders in childhood/adolescence and adulthood (Harrington 1990; Lewinsohn 1999), and the fact that early onset of depression is associated with significant reductions in 'human capital' (i.e. educational and vocational attainment) in affected individuals (Berndt 2000).
While the terminology 'first episode' is now frequently used in the area of psychosis (McGorry 2006), and is the basis of much research in early interventions, it is not yet widely used in the area of depression. However, it may have the potential to serve a similar purpose as it has in psychosis and drive the area of early interventions (Allen 2007; Hetrick 2008). Relapse rates after a first episode of depression in those with no depression history are 20% to 30% compared with 70% to 80% for those who have experienced two or more episodes (Keller 1984). In children and adolescents, relapse rates range between 34% and 75% within one to five years after a first depression episode (Kennard 2006). As such, it is critical to aid recovery and relapse following the first initial episode of depression in this population. In addition, it is important to note that the aetiology of early versus late‐onset of depression may differ (Jaffee 2002), making it especially important to consider the risk factors and developmental experiences most akin to the expression of depression in various age groups.
The criteria used to define both a relapse and a recurrence of a depressive episode also vary within the literature. For example, Frank 1991 describes 'relapse' as "a return of symptoms satisfying full syndrome criteria for an episode that occurs during a period of remission, but before recovery", and 'recurrence' as "the appearance of a new episode of MDE, occurring during recovery". However, often within trials the terms relapse and recurrence are used interchangeably, and rarely are both 'relapse' and 'recurrence' measured as simultaneous outcome measures. For this reason, we will follow the terminology used by Vittengl 2007 and refer to a future depressive episode as a relapse‐recurrence.
Description of the intervention
A range of interventions have been tested for preventing relapse and recurrence in adults with depression. Studies in this population suggest medication is effective in preventing relapse, but only during the period within which it is being taken (Geddes 2003; Keller 2005; Rapaport 2004; Simon 2004). Cognitive behavioural therapy (CBT) (Fava 1998; Hensley 2004) and, more recently, mindfulness‐based cognitive therapy (MBCT), have shown longer‐lasting effects (Ma 2004; Teasdale 2000), some of which are comparable to that achieved by medication alone (Segal 2010). Some trials have also found that there is a greater reduction in relapse rate after continuation therapy in individuals who have an earlier onset of depressive disorder (Jarrett 2001).
There are a variety of other psychological therapeutic approaches, such as psychodynamic approaches, family therapy, interpersonal therapy (IPT), acceptance and commitment therapy (ACT) and extended behavioural activation (both considered as third‐wave CBT approaches) (Churchill 2010; Martell 2010) that have been used in the treatment of depression in children and adolescents, and could therefore potentially be used for preventing relapse and recurrence. There are a range of psychodynamic therapy approaches, but they are based on the proposal that an individual's biological and temperamental vulnerabilities, early attachment relationships and childhood experiences lead to susceptibilities to depression with therapy aiming to develop insight into these processes. There are a range of different family therapy methods and each has a different emphasis on causative and maintaining factors, and different therapeutic targets and outcomes. However, all work on the premise that family relationships are an important factor in psychological health (Fisher 2010). Interpersonal approaches are based on the premise that depressive symptoms are due to the disruption of close personal relationships (Weissman 2007). Third‐wave CBT targets the individual’s relationship with cognitions and emotions, focusing primarily on the function of cognitions such as thought suppression or experiential avoidance (an attempt or desire to suppress unwanted internal experiences, such as emotions, thoughts and bodily sensations) (Hoffman 2008); and extended behavioural activation builds on behavioural activation targeting avoidant coping patterns but formulating and accomplishing behavioural goals.
According to a number of international guidelines, it is now standard practice for children and adolescents to receive CBT as a first‐line intervention for depression, with pharmacotherapy reserved for the treatment of more persisting, relapsing and chronic forms of depression (AACAP 2007; Cheung 2007; McDermott 2010; NICE 2005).
However, it is clear that what is considered effective for adults may not be effective in younger populations, as in the case of tricyclic antidepressants (TCAs) (Hazell 2002). Recent reviews highlight uncertainty about the risk‐benefit ratio of selective serotonin re‐uptake inhibitors (SSRIs) (Bridge 2007; Hetrick 2007) and suggest the effects of CBT and other psychotherapies are modest at best in this younger population (Weisz 2006). Further, there have been inconsistent findings regarding the combined use of SSRIs and CBT with adolescents (Clarke 2005; Goodyer 2007; March 2004; Melvin 2006).
It is also of interest that continuation or maintenance phase treatments that are undertaken following adequate response to treatment are not always the same as that in the acute phase. For example, some randomised controlled trials (RCTs) involve participants switching from pharmacotherapy in the acute phase, to CBT in the maintenance phase, while some have required participants to receive the same modality of therapy across treatment phases (Dobson 2008).
Some studies undertaken in children and adolescents have found that treatment with fluoxetine (after initial acute treatment with the same medication) significantly delayed the return of symptoms (Emslie 2004). Meta‐analyses suggest that psychological therapies are effective for treating depressive symptoms in the short term in children and adolescents, however they are no better than treatment as usual at six‐month follow‐up (Wantanabe 2007). Booster CBT sessions have not been shown to reduce the rate of recurrence compared to assessment sessions, but have accelerated recovery for adolescents who remained depressed at the end of the acute phase of treatment (Clarke 1999). However, in another small study, booster CBT sessions resulted in relapse rates of 6% compared to 50% in a comparison group who had no continued therapy (Kroll 1996). It should be noted that the majority of trials conducted in this population have utilised CBT, reflecting the bias towards this type of CBT in the literature. As such, it makes it difficult at present to determine the relative benefits of other forms of psychological therapy in reducing relapse rates in this population
How the intervention might work
Continuation or maintenance phase psychotherapy that is undertaken following adequate response to treatment has a different focus from that administered during an acute phase of depression. Psychotherapy undertaken during a relapse prevention phase tends to focus on addressing any residual symptoms of depression, which have been shown to increase the chances of a relapse, on affect regulation and on self management skills needed to promote recovery (Segal 2010). CBT and MBCT are more commonly described in the relapse prevention literature. While acute phase CBT aims to reduce depressive symptomology, continuation or maintenance phase CBT aims to prevent relapse following a reduction in symptoms, often in the presence of minimal or residual symptoms. Relapse to a depressive episode has been associated with a return to negative thinking styles, such as through ruminative thoughts or avoidance (Lau 2004). CBT targets the negative thoughts that might be maintaining residual depression symptoms with the aim of modifying these into more adaptive and helpful thoughts. MBCT is different from CBT in that there is little emphasis on changing the content of thoughts, rather the focus is on changing awareness of and relationship to those negative thoughts that might be maintaining the residual symptoms of depression (Teasdale 2000).
Common elements of CBT and MBCT relapse prevention programmes include: 1) de‐centring techniques in order to learn that negative thoughts and emotions are transient; 2) mood monitoring techniques that allow individuals to identify maladaptive thinking styles, indicators of relapse, or both; and 3) lifestyle modification to reduce stress and reinforce behaviours that promote well being, health‐enhancing behaviours and personal growth, such as meditation, yoga and exercise
Other aforementioned therapies broadly aim to help improve a persons self esteem, help the individual cope with past and ongoing conflicts, improve interpersonal relationships with others, and to accept and to understand themselves.
Until recently, a widely held belief was that dysfunction in serotonergic neurons and their targets may underlie depressive symptomatology (van Praag 1987). The dopaminergic system has also been implicated, given its association with reward and appetitive motivation, whereby depression is characterised by a diminished ability to experience pleasure. Serotonin does have modulatory effects on dopamine, either increasing or decreasing its activity depending on the concomitant action of other neurotransmitters and the receptor subtype it is acting on. Antidepressant medications work by affecting the release, or uptake, of various neurotransmitters. For example, TCAs affect the reuptake of serotonin, norepinephrine and, to a lesser extent, dopamine; SSRIs cause an initial inhibition of the reuptake of serotonin; newer generation antidepressants such as serotonin‐norepinephrine reuptake inhibitors (SNRIs) target the noradrenaline and dopamine systems to a greater degree than the SSRIs, though most also have an effect on the serotonergic system (Healy 1997).
Why it is important to do this review
The provision of effective interventions at the early stage of depression is important in order to reduce the likelihood of recurrent episodes, which have been demonstrated to occur more frequently as the illness progresses (Kessing 2004). There is compelling evidence that intervention in this early stage may prevent the development of cognitive factors associated with recurrent episodes (Kendler 2000; Lewinsohn 1999; Ma 2004). Any early intervention approach to depressive disorders must have a strong emphasis on relapse prevention as a primary outcome of interest. To have a truly significant impact, they must not only reduce the acute symptoms associated with depressive disorder, but should also aim to prevent or alter the development of underlying vulnerability factors. These factors can determine the likelihood of relapse and of developing a chronic depressive disorder following the first episode. Despite the compelling argument for early interventions to prevent relapse, there have been few specific studies of relapse prevention for the initial stages of depression. Given that for many individuals the first or initial episodes occur during childhood or adolescence, studies in this population are relevant to understand how best to change the trajectory of depressive disorders throughout the life span.
Given the uncertainties and inconsistencies regarding effective treatment of depressive disorders to prevent relapse during the early stage of the disorder, a systematic review of the literature is warranted. It is also timely given the current Cochrane Reviews published on the prevention and treatment of children and adolescents (Cox 2012; Hetrick 2012; Merry 2011) and will add to our knowledge of effective treatment at each stage of the illness.
Objectives
The objective of the review was to examine the impact of early interventions on the likelihood of relapse and recurrence of depression in children and adolescents. Early interventions include pharmacological and psychological interventions as described in the How the intervention might work section.
The protocol for this review stipulated that studies involving participants of any age, who had experienced a case level of a depressive disorder, would be included in the review. However, as outlined in the Background section, depression commonly emerges before the age of 25 years (Kessler 2005). As such, trials involving adults have not been included.
Methods
Criteria for considering studies for this review
Types of studies
Randomised controlled trials (RCTs) and cluster RCTs, irrespective of publication status, including unpublished abstracts and reports of any intervention to prevent relapse or recurrence from MDD or DD were included in the review.
Trial designs
It was anticipated that two potential trial designs were likely to be encountered: 1) participants who had responded or remitted from an episode of MDD or DD during an acute phase of treatment are then recruited into a trial and randomised into an intervention to prevent relapse/recurrence; 2) participants who undergo acute treatment of a depressive episode and go on (without re‐randomisation) to receive either controlled interventions with long‐term follow‐up or enter a phase when participants are free to seek any intervention of their choice (including no intervention) and are followed up in what is termed naturalistic long‐term follow‐up, with measures of relapse/recurrence collected for both. Considerable heterogeneity was likely within the second type of trial. For example, trials may have contained participants who had remitted or responded, and those who had not; some psychological studies may have used 'booster sessions' during the follow‐up phase; and pharmacological studies may have included participants who were continued on acute medication, or switched to a different medication, and subsequently followed up.
In the first type of trial design, 'continuation' and 'maintenance' phases of treatment vary depending on the individual trial design. We have described them as reported by trial authors (see Characteristics of included studies for each individual trial and the definition/length of each of these phases). Generally, when participants are in a maintenance phase, they have achieved the desired level of remission from their depressive symptoms, again as defined by each individual trial.
Both designs are included; however, given that in the second type of trial participants are not re‐randomised, it is more accurately described as an observational study with regard to the continuation/maintenance (relapse prevention) phase for depression. As such these studies are not included in meta‐analysis but are described narratively and included in the discussion to ensure that 1) as much data as possible is available in a field where very little research has been undertaken to guide practice; 2) to highlight the diversity of studies undertaken that attempt to answer questions about effective interventions for relapse prevention; and 3) to highlight the difficulties in undertaking high‐quality research in this area, particularly with regard to recruiting sufficient numbers of participants into trials such that re‐randomisation can take place.
There were no date or language restrictions.
Types of participants
Depression commonly begins in childhood and adolescence and, as a result, depression in this population is likely to be in the early phase of illness. Based on this rationale, trials involving children and adolescents up to the age of 25, who had responded or remitted from an episode of MDD or DD, were included.
All participants were required to be diagnosed with MDD or DD by a clinician using a diagnostic system (Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV‐TR), APA 2000 or International Classification of Diseases (ICD‐10), WHO 2007). Criteria for response/remission often vary between trials. Criteria for response or remission must have been based either on a clinical interview confirming absence of depressive symptoms for a specified time period, or on a score below a specified cut‐off point on a validated and standardised assessment tool.
Given the difficulties in defining recovery and relapse in DD, it was our intention to treat MDD separately within the review. MDD or DD must have been the primary diagnosis, but comorbidity was permitted, except for psychosis or bipolar disorder. However, there were no instances where we were required to treat MDD and DD separately. In future reviews, if this situation arises, we will follow these criteria.
Trials of participants with an intellectual quotient (IQ) of less than 70, organic brain injury or a serious medical condition were excluded.
Types of interventions
Experimental interventions
Any type of pharmacotherapy or psychological therapy was included.
Pharmacological interventions
Categories of pharmacotherapy included were TCAs, SSRIs and newer antidepressants (which include norepinephrine reuptake inhibitors (SNRIs), norepinephrine reuptake inhibitors (NRIs), norepinephrine dopamine disinhibitors (NDDIs) and tetracyclic antidepressants (TeCAs)), mood stabilisers, anxiolytic medications and other medications. Rather than being a homogeneous group based on mechanisms of action, they are classed together because they are modified versions of first and second generation antidepressants (Olver 2001). This categorisation is an update from the original protocol based on the rapid development of newer antidepressants since its publication, and to ensure consistency of this review with other reviews of antidepressant medication in children and adolescents (such as Hetrick 2007).
Psychological interventions
Categories of psychological therapy included were CBT‐based, psychodynamic, family, interpersonal and supportive/non‐directive, and other. Trials that included a combination of psychological therapy and pharmacotherapy were also included.
Comparator interventions
The experimental intervention groups were compared with: placebo control, other active interventions such as medication, psychotherapy or a combination of the two, and no treatment or treatment as usual (TAU). Although no trials were retrieved within which the active intervention was compared to a waiting list or attention placebo, in future updates of this review we will include these comparators if they arise.
Types of outcome measures
Primary outcomes
1. Prevention of a second or next episode was measured by:
the number of participants who met the criteria for relapse (as defined by trial authors on a scale of depression symptoms or by diagnosis using DSM or ICD criteria (APA 2000; WHO 2007); or
the number of participants who were readmitted or re‐presented to a service for treatment.
Relapse and recurrence were defined variously by trial authors, and as we could not obtain individual patient data in order to impose a consistent criteria, we extracted and documented data based on the criteria used by trial authors. As stated in the Background section, for clarity we will use the term relapse‐recurrence to describe these data, an approach adopted by Vittengl 2007. We extracted data for relapse from the last time point reported individually by trial authors. This varied based on the individual trial design.
2. In the protocol for this review, suicide‐related behaviour (both ideation and attempt) was specified as a secondary outcome. However, due to the concern that taking antidepressant medications may potentially result in suicidal behaviour, we made a decision to include such behaviours as a primary outcome (Hetrick 2007).
Secondary outcomes
3. Time to relapse 4. Functioning, including overall functioning, social, academic/occupational functioning and quality of life measured on a standardised and validated assessment scale (e.g. the Children's Global Assessment Scale (C‐GAS; Shaffer 1983) 5. Depressive symptoms measured on any standardised, validated and reliable rating scale (e.g. the Beck Depression inventory (BDI; Beck 1969); the Children's Depression Rating Scale‐Revised (CDRS‐R; Poznanski 1996); and the Hamilton Rating Scale for Depression (HAM‐D; Hamilton 1960)). 6. Drop‐outs 7. Emergence of secondary morbidity, including emergence of secondary co‐morbid conditions and a switch to bipolar disorder 8. Adverse outcomes (these include psychological and physiological adverse outcomes as reported by individual trial authors)
Search methods for identification of studies
CCDAN's Specialised Register (CCDANCTR)
The Cochrane Collaboration's Depression, Anxiety and Neurosis Group (CCDAN) maintains two clinical trials registers at their editorial base in Bristol, UK. A references register and a studies‐based register. The CCDANCTR‐References Register contains over 30,000 reports of trials in depression, anxiety and neurosis. Approximately 65% of these reports have been tagged and coded to individual trials. The coded trials are held in the CCDANCTR‐Studies Register and records are linked between the two registers through the use of unique Study ID tags. Coding of trials is based on the EU‐Psi coding manual. Please contact the CCDAN Trials Search Co‐ordinator for further details. References to trials for inclusion in the Group's Registers are collated from routine, generic searches of MEDLINE (1950 ‐ ), EMBASE (1974 ‐ ) and PsycINFO (1967 ‐ ), quarterly searches of the Cochrane Central Register of Controlled Trials (CENTRAL) and review‐specific searches of additional databases. Reports of trials are also sourced from international trials registers c/o the World Health Organization’s trials portal (ICTRP), drug companies, the handsearching of key journals, conference proceedings and other (non‐Cochrane) systematic reviews and meta‐analyses.
Details of CCDAN's generic search strategies can be found on the Group's website.
Electronic searches
The CCDANCTR was searched from inception up until 1 June 2011 by the Trials Search Co‐ordinator using the following terms:
CCDANCTR‐Studies
Diagnosis = (depress* or dysthymi* or "adjustment disorder*" or "mood disorder*" or "affective disorder*" or "affective symptoms") And Notes Field ="relapse prevention"
CCDANCTR‐References
Title/Abstract/Keyword = (depress* or dysthymi* or "adjustment disorder*" or "mood disorder*" or "affective disorder*" or "affective symptoms") And Free‐text = (maintenance* or maintain* or continu* or discontinu* or prevent* or relaps* or prophyla* or recur* or recrudesc* or ((first or prior or index) and (episod* or onset or inciden* or diagnos* or refer*))) And Title/Keyword/Abstract: (adolesc* or preadolesc* or pre‐adolesc* or boy* or girl* or child* or infant* or juvenil* or minors or school* or pediatri* or paediatri* or pubescen* or students or teen* or young or youth*)
We conducted an additional search of MEDLINE, EMBASE, PsycINFO and CENTRAL in June 2009, when the CCDANCTR was out of date due to a changeover of staff at the editorial base. Search strategies can be found in Appendix 1.
Searching other resources
Reference lists
We reviewed the reference lists of included trials and other reviews retrieved in the search.
Personal communication
In order to ensure that as many RCTs as possible were identified, we contacted the authors of the included trials and other experts in the field to ascertain if they knew of any published or unpublished RCTs in the area, which were not yet identified.
Data collection and analysis
Selection of studies
Two review authors independently selected trials for possible inclusion in the study. Firstly, we independently reviewed the titles and abstracts of trials identified from the search. Secondly, two review authors (MS, SH, GC or SD) independently examined the full text of all studies considered to be of possible relevance. Each review author compiled a list of studies, which they believed met the inclusion criteria. We compared the contents of each review author's list and discussed any discrepancies. Any disagreement was resolved by discussion and consensus between all of the review authors.
Data extraction and management
At least two review authors (GC, CF, OA or MP) independently extracted data using specially developed data extraction forms. We collected information on the following.
Participants (including summary information where applicable): age, gender, how the diagnosis was made, length of untreated illness, length of index episode, number of previous episodes, age of onset, baseline severity of depression, setting (inpatient versus outpatient), suicide‐related behaviours/level of suicidal ideation/risk of suicide, child medical illness, child co‐morbid conditions (physical and mental, Axis I and II) and country. We also aimed to collect information on index of socio‐economic status (SES) including any specifying household income, family employment, neighbourhood SES etc. and family factors including any specifying of number of parents residing at home, family employment/education/family history of physical and mental illness, however this information was not routinely reported in publications, and is included where possible.
Interventions and comparisons: description of medication including method of delivery, dose, length of treatment, intended and actual dose received, and/or description of psychological intervention including type, whether it was delivered to groups or individuals, was manualised, who delivered the intervention and for how long, and the actual number of sessions attended. Information on other adjunctive interventions was also to be collected.
Outcome measures: description of measures used.
Results: point estimates and measures of variability, and frequency counts for dichotomous variables.
One review author (GC) compiled all comparisons and entered outcome data into Review Manager software for meta‐analysis (RevMan 2011). Two research assistants, who are not named as authors, performed double‐data entry to ensure accuracy of results. We obtained missing data from trial authors wherever possible, and we have noted in the table of Characteristics of included studies where this was provided.
Main comparisons intended in the review
Antidepressant medication versus pill placebo
Antidepressant medication versus psychological therapy
Combination therapy (medication plus psychological therapy) versus psychological therapy alone
Combination therapy versus antidepressant medication alone
Psychological therapy versus no treatment or TAU
Assessment of risk of bias in included studies
Two review authors (GC, CF, OA or MP) independently assessed the risk of bias of the included trials using a descriptive approach as advocated in Chapter 8 of the Cochrane Handbook for Systematic Reviews of Interventions (Higgins 2008). For the following items, we noted a description of methods and made a judgement about the resulting risk of bias of 'low risk', 'unclear risk' or 'high risk' in accordance with the updated guidance and software from The Cochrane Collaboration:
1. Adequate sequence generation? 2. Allocation concealment? 3. Blinding? (participant ratings and clinician ratings) 4. Incomplete outcome data addressed? 5. Free of selective reporting? (Please note ‐ according to Chapter 8 of the Cochrane Handbook, most reviews will receive an 'unclear' judgement for this item, as study protocols are rarely available). To assess reporting bias, we recorded which of the review outcomes were available with usable data from each included trial, as well as noting which of the review outcomes were only reported in terms of whether there were significant differences between groups. Additionally, we compiled the other outcomes (not collected for the review) reported by the trialists in the paper publication(s). 6. Free of other bias?
Measures of treatment effect
For dichotomous outcomes, such as 'number relapsed‐recurred', results from each trial are expressed as an odds ratio (OR) with 95% confidence intervals (CI) and combined in meta‐analysis. Although the protocol for the review stipulated that we would express relapse rates as a risk ratio (RR), the OR has more favourable mathematical properties (Higgins 2008a section 12.5.4.4).
Continuous outcomes, such as symptom measures, are presented either as a mean difference (MD) when absolute values of post‐treatment means and standard deviations (SD) were given, using the same rating scale across studies, and standardised mean difference (SMD) when different scales were used to measure the same outcomes and then combined for meta‐analysis. Confidence intervals are presented at 95% across all meta‐analyses.
Unit of analysis issues
Studies with multiple treatment groups
Where a study had more than one active treatment arm, we extracted the appropriate arms for each of our main comparisons. Originally, if more than one comparison was relevant, we planned to include both in the comparison, with subtotals, rather than totals allowed in the meta‐analysis, so that double‐counting of data did not occur. There was one trial (Clarke 1999) in which three treatment arms were compared against each other within a single meta‐analysis; CBT versus assessment every four months versus assessment every 12 months. The Cochrane Handbook advises "To include a study with more than two intervention groups in a meta‐analysis, the recommended approach is usually to combine relevant groups to create a single pair‐wise comparison" (Chapter 16, section 16.5). As the two assessment conditions were felt to constitute a similar intervention, we combined these to represent an 'assessment only' condition and compared to CBT for dichotomous outcomes. However, it was not possible to obtain additional data concerning the mean and SD for assessment only conditions and therefore we performed subgroup analyses for the continuous outcomes of functioning and depressive symptoms.
Cluster‐randomised trials
No cluster RCTs or cross‐over trials were included, however if they are located in future updates, they will be included in the review. For cluster RCTs, we will apply an intraclass correlation (ICC) for the sample in order to take into account the effect of the clustering. In the first instance we will use the ICC reported in the publication or, if necessary, contact authors for this information. If we are unable to obtain this information, we will calculate an ICC estimate using the average of the ICCs obtained from the other studies included in the analysis.
Cross‐over trials
For cross‐over trials, if the appropriate data for a paired t‐test analysis are not available and cannot be obtained from trial authors, we will take all measurements from the intervention periods before and after cross‐over and all measurements from intervention periods before and after cross‐over and analyse these as if the trial were a parallel‐group trial. This approach gives rise to a unit of analysis error that results in confidence intervals that are likely to be too wide, and thus the trial will receive too little weight, with the possible consequence of disguising clinically important heterogeneity. However, given that this analysis is conservative, in that studies are under‐weighted rather than over‐weighted, it will be tolerated in this review.
Dealing with missing data
We obtained missing data from trial authors wherever possible. We intended to clearly document in the review where missing data were imputed where necessary (e.g. calculating SDs from standard errors and P values); however, we did not need to perform these calculations. Where available, we used intention‐to‐treat data and documented a note of the methods used for imputing missing data (such as last observation carried forward (LOCF) or other types of modelling).
Assessment of heterogeneity
Clinical homogeneity is satisfied when participants, interventions and outcome measures are considered to be similar. For trials that were clinically heterogeneous, or presented insufficient information for pooling, a descriptive analysis is presented. For trials that are clinically heterogeneous or present insufficient information for pooling, we performed a descriptive analysis. We assessed statistical homogeneity using the I2 statistic.
The Cochrane Handbook recommends using a range for I2 and states that “Thresholds for the interpretation of I2 can be misleading, since the importance of inconsistency depends on several factors". A rough guide to interpretation is as follows:
0% to 40%: might not be important;
30% to 60%: may represent moderate heterogeneity;
50% to 90%: may represent substantial heterogeneity;
75% to 100%: considerable heterogeneity.
Although we intended to perform sensitivity analyses on outcomes where heterogeneity was interpreted as being of clinical importance, this was not possible due to the paucity of data available for pooling.
Assessment of reporting biases
We assessed reporting bias through the 'Risk of bias' process and noted when: a) a trial failed measure a review outcome that we assessed would likely have been measured; b) a trial stated that it measured a review outcome but did not report the results or data; c) a trial stated that it measured a review outcome and reported a result but not the data for meta‐analysis. We sought to assess trial protocols where published/available in the first instance and also sought clarification from study authors in the case of suspicion of reporting bias.
We also intended to assess publication bias using a funnel plot for the primary outcomes relating to relapse and suicide‐related outcomes. However, funnel plot asymmetry may be due to reasons other than publication bias and is difficult to assess in the case of a small number of trials. As this review contained nine trials, and not all trials reported data on the primary outcomes, we did not conduct a funnel plot analysis. In future reviews, if the number of trials and data available permits, we will use a funnel plot to assess publication bias.
Data synthesis
For all meta‐analyses, we used a random‐effects model (DerSimonian 1986). The random‐effects method incorporates an assumption that various studies estimate a different, yet related, intervention effect.
Subgroup analysis and investigation of heterogeneity
Originally we intended to perform subgroup analyses on trials that included children and adolescents versus those that included participants of any age who had experienced a first episode of depression. However, as the search did not yield any trials of the latter type, we could not perform this analysis. We also intended to perform subgroup analyses on trials that contained children versus those that contained adolescents, but the nature of the trials included in the review did not contain enough data to allow for this subgroup analysis.
The protocol for this review also stipulated that we would analyse data from the two types of anticipated trial designs separately and this was done where applicable
During the review process it became apparent that within the two types of trial design that we had anticipated, there was considerable diversity. In trials where participants who had responded or remitted from an episode of MDD or DD during an acute phase of treatment were re‐randomised into a continuation or maintenance phase, re‐randomisation commonly occurred either early (after an acute phase) or late (after either a continuation and/or maintenance phase). Due to the variability in the length of treatment before re‐randomisation, we felt that it was important to perform subgroup analyses based on time of re‐randomisation (early or late).
Sensitivity analysis
Originally, we intended to undertake sensitivity analyses to assess the effect of risk of bias that may be introduced due to the decisions made in the process of undertaking the review. In psychiatry trials it is important to investigate the impact of assumptions made in various imputation methods used to account for missing data, such as analysis using LOCF and observed cases (OC). However, as there were limited data contained in trials, we were unable to perform these analyses.
Results
Description of studies
Results of the search
The original search in June 2009 yielded 2092 results. We ran two further updated searches: one in November 2010 which yielded 670 results, and another in June 2011 which yielded 102 results. We assessed the full‐text articles of 19 trials for inclusion into the review and, of these, nine trials were eligible for inclusion. Three trials (Cheung 2008; Emslie 2004; Emslie 2008) provided data suitable for at least one outcome in the meta‐analysis. Figure 1 shows the flow of records through the inclusion process.
1.
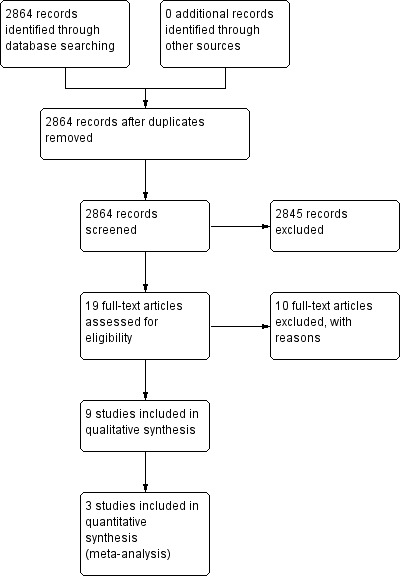
Study flow diagram.
Included studies
Design
All trials were RCTs and fell under the two main designs described in the methods section of this review. The first type of design was executed by four trials; participants who had responded or remitted from an episode of MDD or DD during an acute phase of treatment were entered into a continuation or maintenance phase. During this phase, all participants were re‐randomised to an intervention to prevent relapse‐recurrence (Cheung 2008; Emslie 2004; Emslie 2008; Kennard 2008). The second type of design was executed by five trials; participants underwent acute treatment of a depressive episode, then entered either controlled or naturalistic long‐term follow‐up, or a continuation or maintenance phase (or both), with measures of relapse‐recurrence collected at follow‐up. Entry into the follow‐up, continuation and/or maintenance phase was not based on response or remission status in these trials (Clarke 1999; Emslie 1998; Renaud 1998; TADS; TORDIA). There are a number of subtle differences between these designs and, for clarity, we will discuss trials within the two designs separately.
Prevention of relapse or recurrence after response or remission during acute treatment
Four trials contained continuation or maintenance phases (or both) specifically designed to prevent relapse‐recurrence after initial response, and involved only those participants who had responded or remitted after an acute phase of treatment (Cheung 2008; Emslie 2004; Emslie 2008; Kennard 2008). All trials re‐randomised participants on entrance to the continuation or maintenance phase. In two trials, acute phase treatment lasted 12 weeks (Emslie 2008; Kennard 2008), at which point participants entered a maintenance phase and were re‐randomised, potentially to a new treatment arm. In the other two trials, acute phase treatment lasted between 9 and 12 weeks, followed by a continuation phase of between 10 (Emslie 2004) and 24 weeks (Cheung 2008), after which participants entered a maintenance phase and again, were re‐randomised into a potentially new treatment arm. These maintenance phases lasted between 36 (Emslie 2004) and 52 weeks (Cheung 2008). For clarity, the maintenance period after which participants have been re‐randomised to a treatment arm will be called the 'relapse‐prevention' phase.
During the acute phase of treatment, one trial openly treated all participants with sertraline (Cheung 2008) and two others treated participants with fluoxetine (Emslie 2008; Kennard 2008). Only participants who responded to treatment during this acute phase entered the relapse‐prevention phase. One trial involved treating participants with either fluoxetine or placebo during the acute phase (Emslie 2004); participants who responded to treatment with fluoxetine were re‐randomised during the acute phase. Participants who responded to a placebo during the acute phase continued to be treated with a placebo during the relapse‐prevention phase and were not compared in statistical analysis with the re‐randomised participants.
During the relapse prevention phase, three trials compared medication with a placebo pill (Cheung 2008; Emslie 2004; Emslie 2008) and one trial compared a combination of psychotherapy and medication with medication alone (Kennard 2008). Medication trials all involved SSRIs; three trials administered fluoxetine (Emslie 2004; Emslie 2008; Kennard 2008) and one involved sertraline (Cheung 2008). In the three trials containing fluoxetine, medication doses varied between 10 and 60 mg/day depending on response to medication during the acute or continuation phase of treatment, and was administered by a child psychiatrist during clinic visits. In the trial by Cheung 2008, sertraline was administered at a dose of between 25 and 200 mg/day depending on response, by the treating clinician.
The psychotherapy intervention utilised by Kennard 2008 was individual, CBT‐based and developed specifically for relapse‐prevention. It focused on the symptoms that remain residual following adequate response to acute treatment, and aimed to promote current strengths to enhance well being. Participants attended between 8 and 11 sessions during the relapse‐prevention phase; a minimum of three family sessions were also written into the protocol. Fidelity of sessions was checked using the Cognitive Therapy Rating Scale and 100% were rated as acceptable. Therapists were doctoral or master's level psychologists (Kennard 2008).
Sample sizes in the acute phase of treatment ranged from 66 (Kennard 2008) to 219 participants (Emslie 2004), and in the relapse‐prevention phase ranged from 22 (Cheung 2008) to 102 participants (Emslie 2008). Three trials contained both children and adolescents between the ages of 7 and 18 years (Emslie 2004; Emslie 2008; Kennard 2008) and one contained adolescents between 13 and 19 years (Cheung 2008).
Acute treatment with long‐term follow‐up of relapse‐recurrence
Five trials involved acute treatment of depression with either a long‐term follow‐up of relapse‐recurrence, or a continuation or maintenance phase (or both).
Within this design, two trials conducted a naturalistic follow‐up of relapse in participants who had responded to acute treatment and did not re‐randomise participants to a separate relapse prevention treatment at any point (Emslie 1998; Renaud 1998). The Emslie 1998 trial involved an acute phase of eight weeks, where participants were treated with either fluoxetine or placebo. During the one‐year follow‐up period, participants were able to continue on their medication, receive no medication, or a different medication. Published data at follow‐up described the number of participants who experienced a relapse‐recurrence of a depressive episode, categorised by treatment during the follow‐up period. For the purposes of this review, we obtained data from the study authors regarding relapse of those who had responded to the acute treatment. The trial by Renaud 1998 randomised participants to between 12 and 16 weeks of acute phase CBT, systemic behavioural Family Therapy (SBFT) or non‐directive supportive therapy. One and two‐year follow‐up analysis of remission and relapse‐recurrence of depressive episode was split by whether participants were 'rapid responders', 'intermediate responders' or 'initial non‐responders', rather than by treatment group assignment. An associated publication also presented data on overall relapse‐recurrence rates but, again, did not split this analysis by participants' initial acute treatment group assignment.
Two trials treated participants during an acute phase, and then tailored treatment in a continuation or maintenance phase, depending on response status after acute treatment (TADS; TORDIA). In the TADS trial, participants were randomised to receive 12 weeks of acute treatment consisting of medication only (fluoxetine), psychotherapy only (CBT sessions), a combination of medication and psychotherapy (fluoxetine and CBT), or placebo. After acute treatment, participants who had been in the active treatment arms (participants in the placebo arm were offered open active treatment) entered a continuation phase which lasted an additional six weeks (weeks 12 to 18) and then a maintenance phase that lasted a further 18 weeks (weeks 18 to 36). During the continuation and maintenance phases, for participants who were receiving psychotherapy, the number of CBT sessions varied depending on response status; full responders at week 12 (defined as those with a Clinical Global Impression (CGI) score of 1 or 2) received three sessions over six weeks, compared with partial responders (defined as those with a CGI score of 3) who received six sessions over as many weeks. During the maintenance phase both partial and full responders received three booster sessions. In terms of those receiving medication, a flexible dosing schedule was adopted, with participants able to receive up to 60 mg/day depending on their CGI‐I severity score. At 12 weeks, non‐responders did not continue in the trial as per protocol, and were offered alternative treatment. In the TORDIA trial, participants were randomised to 12 weeks acute treatment involving either medication only (venlafaxine or another SSRI) or medication combined with psychotherapy (CBT). After 12 weeks, participants who responded to treatment were given the option of continuing in their treatment arm for an additional 12 weeks or receiving open treatment; non‐responders could either remain in blind continuation treatment, or open treatment up to 24 weeks along with responders. At no point did any re‐randomisation occur, and data were presented separately for responders and non‐responders in order to investigate relapse after response or remission at 12 weeks acute treatment.
In one trial (Clarke 1999) participants were randomly assigned to one of three acute 12‐week treatment conditions: adolescent group CBT, adolescent group CBT combined with a parent group or wait‐list control. After the acute phase, participants who had received one of the CBT conditions were re‐randomised to one of three maintenance/relapse prevention conditions: booster CBT sessions, assessments every four months or assessments every 12 months. This randomisation occurred in participants regardless of response status. At follow‐up, data were presented based on group assignment for those who had recovered, and subsequently had a relapse‐recurrence of a depressive episode during the relapse prevention phase.
Four trials utilised CBT (Clarke 1999; Renaud 1998; TADS; TORDIA). The CBT sessions generally involved core components of the approach such as mood monitoring, improving social skills, activity scheduling, reducing negative thinking and cognitive restructuring. Renaud 1998 administered 12 to 16 CBT sessions during the acute phase only, and the remaining three trials involved the use of CBT during the acute, continuation and/or maintenance phases. The Clarke 1999 trial involved 16 group CBT sessions over as many weeks in the acute phase, and six booster CBT sessions thereafter (one every four months). The TORDIA trial involved CBT sessions every week for 12 weeks during the acute phase and every other week for two months and then monthly thereafter in the proceeding 12 weeks. TADS administered 15 individual sessions over 12 weeks of acute treatment. The schedule of CBT sessions during the continuation/maintenance phase is as described above based on response status. The CBT sessions administered during the continuation/maintenance phases emphasised generalisation training in order to implement the skills learnt during earlier sessions, and on relapse prevention.
Whether a parent component was offered as part of the CBT package was specified by Renaud 1998. The Clarke 1999 trial involved two types of CBT; one with a parent component and one without. In the TORDIA trial, parents were offered psychoeducation by a nurse or psychiatrist around the symptoms, causes and effects of depression and in TADS, between one and three conjoint sessions could occur between parent and adolescents, with psychoeducation around depression discussed. CBT sessions were delivered by therapists with a median of 10 years' clinical experience (Renaud 1998), graduate psychology or social work students, master's or doctoral level clinicians (Clarke 1999), and therapists with a master's degree in a mental health field (TORDIA). TADS did not give details on who delivered the intervention. The trial by Renaud 1998 also contained a condition where SBFT was delivered. This approach involves problem solving and communication within the family unit.
Three trials involved medication; two used fluoxetine (Emslie 1998; TADS) and in the TORDIA trial, venlafaxine and an SSRI (either paroxetine or citalopram) was used. In the TORDIA trial, at 12 weeks on entry to the maintenance phase, the mean dose in the venlafaxine group was 205.4 mg (SD = 33.1) and in the SSRI group was 33.8 mg (SD = 9.3).
For trials that involved a continuation/maintenance phase, sample sizes ranged from 64 (Clarke 1999) to 144 participants (TORDIA). For trials that involved a long‐term follow‐up, sample sizes were 74 (Emslie 1998) and 100 (Renaud 1998).
Setting
Eight trials were carried out in the USA and one (Cheung 2008) took place in Canada.
Outcomes
The prevention of a second or next episode of depression was defined by the trials using both standardised assessment tools and clinical judgements. In the trial by Cheung 2008 relapse‐recurrence status was based on the clinical judgement of the physician who assessed both depressive symptoms and level of impairment. Although participants' HAM‐D scores were available to treating physicians, these were used as a guide and relapse was not formally defined by them. Emslie 2004 formally measured relapse‐recurrence as a one time CDRS‐R score of ≥ 40, with a two‐week history of clinical deterioration or relapse‐recurrence in the opinion of the physician. Emslie 2008 and Kennard 2008 used a similar criteria. They further specified that if a participant was deemed likely to relapse on the basis of clinical judgement if treatment was not altered, even if they scored less than 40 on the CDRS‐R, then they were recorded as having relapsed‐recurred. In the TADS trial, relapse‐recurrence was not formally measured rather they examined those who had 'failed to maintain response status'. Response to treatment was defined in two ways. Firstly, participants with a CGI‐I score of 1 or 2 were defined as 'full responders' and those with a score of 3 were 'partial responders'. Once full response status was achieved, 'sustained response' status was measured. This was defined as maintaining a 'full response' (CGI‐I score of 1 or 2) over two consecutive ratings. Once a participant had maintained a 'sustained response', they continued to be rated as such unless their CGI‐I score dropped to between 3 and 7 points and they were then classified as a 'failed to maintain' response status. For the purposes of analyses within this review, data concerning relapse‐recurrence in the group of the number of participants who experienced a sustained response by week 12 have been extracted. Of these participants, those who 'failed to maintain' a sustained response at weeks 18 and 36 are classified as relapsed‐recurred. The TORDIA measured relapse‐recurrence as at least two consecutive weeks with probable or definite depressive disorder (with a score of 3 or 4 on the Adolescent Longitudinal Interview Follow‐Up Evaluation).
As mentioned above, data obtained from authors of the Emslie 1998 trial were based on the number of participants who relapsed‐recurred after responding to treatment at 12 weeks. Relapse‐recurrence was defined as a return to a depressive episode which occurred whilst in a period of remission, before recovery. Renaud 1998 did not measure relapse‐recurrence, and reported on clinical remission and clinical recovery. However the associated publication by Birmaher 1998 measured both relapse and recurrence in participants based on outcomes from the Kiddie‐Schedule for Affective Disorders and Schizophrenia in School Age Children Present and Lifetime Version (K‐SADS‐PL). Relapse was defined as an episode of MDD during a period of remission, and recurrence as a an episode of MDD during recovery. However, these data were not split by treatment group and therefore were not suitable for meta‐analysis. Clarke 1999 reported on rates of recurrence to a unipolar episode of depression. Assessments used the Longitudinal Interval Follow‐Up Evaluation (LIFE) and defined recovery as a period of eight weeks symptom‐free. The authors do not explicitly state whether recurrence is based on this outcome.
Three trials reported on the mean time in which participants relapsed‐recurred (Cheung 2008; Emslie 1998; TORDIA) and one reported the median (Emslie 2008).
Functional outcomes were reported in four trials, two of which administered the C‐GAS (Emslie 1998; Kennard 2008) and two the Global Assessment of Functioning (GAF) (Clarke 1999; Emslie 2004). Due to the nature of data reported, the C‐GAS scores in the Emslie 1998 publication could not be utilised in meta‐analysis.
The majority of studies included clinician‐rated or self rated depressive symptoms (or both) using standardised and validated assessment tools. The CDRS‐R was utilised by Emslie 1998, Emslie 2004, Emslie 2008 and Kennard 2008, and the HAM‐D by Cheung 2008 and Clarke 1999. The BDI was also administered by Clarke 1999.
Measures of suicidal ideation and or attempt tended to be reported as part of 'adverse events'. Although the TADS trial utilised the Suicidal ideation Questionnaire‐Junior High School Version (SIQ‐JR) as an outcome measure, data were not available for the subset of participants who had responded to treatment at 12 weeks, and thus formed the 'relapse prevention' subset that this review focused on. All trials apart from Clarke 1999, Emslie 1998 and Renaud 1998 reported incidences of suicidal behaviours in their adverse events data.
Adverse events were reported in trials that involved medication (Cheung 2008; Emslie 2004; Emslie 2008; Kennard 2008; TADS; TORDIA).
The protocol for the review aimed to collect data on emergence of secondary morbidity or switch to bipolar disorder. This outcome was rarely reported in trials, although one reported instances of psychosis or mania as part of their adverse events (Cheung 2008).
The number of participants who dropped out were routinely reported in trials.
We contacted all lead authors of included studies where additional data were needed, three of whom were unable to provide additional data. Additional data were obtained for Emslie 1998, Emslie 2008 and Kennard 2008; the content of these data is explained in the notes section for each individual study. For a full description of each of the included studies please see the Characteristics of included studies section.
Excluded studies
We excluded 10 studies from the review; eight investigated treatment in the acute phase of a depressive episode only (Birmaher 1998; Birmaher 2000; Eli 1986; Eli 1995; Emslie 2009; GlaxoSmithKline 1997; GlaxoSmithKline 2001; TADS(acute phase)), one was not a RCT (Franchini 2006) and one trial did not measure relapse in participants (ADAPT). For exclusion reasons for individual trials see Characteristics of excluded studies.
Risk of bias in included studies
Allocation
The majority of studies specified the method of random sequence generation used to allocate participants to a treatment group; in general, this tended to be either through a computer‐generated random number sequence (Cheung 2008; Emslie 2004; Emslie 2008; TADS) or Efron's biased coin toss (Renaud 1998; TORDIA). In many of the trials, it was unclear whether allocation concealment was accomplished, and publications contained insufficient information in order to make a judgement.
Blinding
Just over half of the studies stated that outcome assessors were blind to participant treatment group (Cheung 2008; Clarke 1999; Kennard 2008; TADS). In the TORDIA trial, although independent evaluators were intended to be blind to treatment condition, the authors acknowledge that in 64 cases (out of a possible 334) the blind was not achieved. In two trials (Emslie 2004; Emslie 2008), although it is likely that outcome assessors were blind to condition, there was insufficient information to make a judgement.
In terms of participants remaining blind to the condition to which they were allocated, there was a high risk that participants were aware of the intervention they were receiving. Only one trial clearly stated that participants were blind to treatment condition (Cheung 2008). As the majority of studies involved a form of psychotherapy, it is likely that participants would have been aware that they had been assigned to receive psychotherapy due to the nature of the intervention itself.
Incomplete outcome data
The majority of trials stated that they used an intention‐to‐treat analysis in order to deal with missing data (Cheung 2008; Clarke 1999; Emslie 2008; Renaud 1998; TADS; TORDIA). The Emslie 1998 study executed a naturalistic follow‐up, and thus presented data only from those who were available to participate one year after receiving treatment, and the Emslie 2008 trial did not provide enough information in order to make a valid judgement.
Selective reporting
There was some evidence of reporting bias; however, the majority of trials reported on outcomes specified in their methods. Although Emslie 2008 report time to relapse in graph form, no mean time to relapse was reported, and depression severity as measured by CDRS‐R endpoint scores was also not presented by treatment group. In addition, the TORDIA trial reported CDRS‐R endpoint scores in graph form only, which did not permit us to extract meaningful data on this outcome. Clarke 1999 did not report the reasons for participants dropping out across groups, meaning that the effect of a specific treatment approach on drop‐out rate could not be considered.
Other potential sources of bias
Other potential sources of bias varied across trials. There were some baseline imbalances in depression severity and in the rate of co‐morbidity between treatment groups in some trials. In the Clarke 1999 trial there was a baseline imbalance regarding BDI score between the annual assessment group and the booster session groups. Emslie 2004 reported baseline imbalances relating to age and height, with participants who received fluoxetine being older and taller than those in the placebo group. The trial was also funded by the drug company Elli Lilly. Emslie 2008 noted that participants in the fluoxetine group had higher levels of co‐morbid anxiety compared with those in the placebo group, and in the TADS trial participants in the combination therapy group showed higher levels of suicidal ideation compared with those in the CBT or medication only groups. See Figure 2 and Figure 3 for the 'Risk of bias' graphs and refer to each individual study's 'Risk of bias' assessment in the Characteristics of included studies.
2.
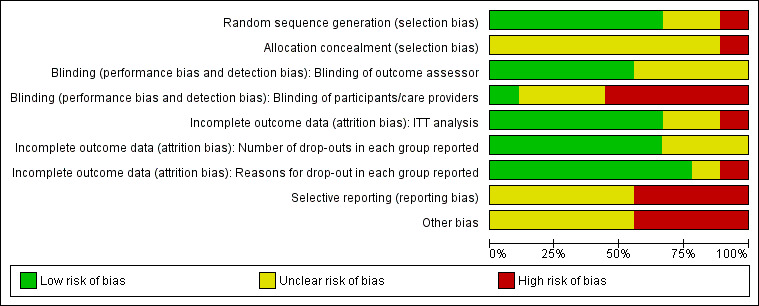
'Risk of bias' graph: review authors' judgements about each risk of bias item presented as percentages across all included studies.
3.
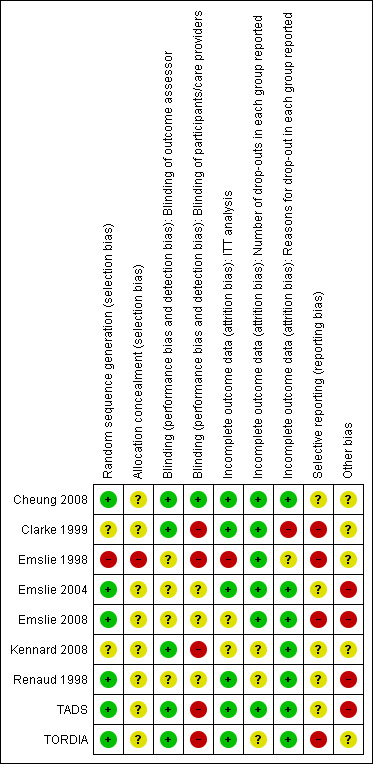
'Risk of bias' summary: review authors' judgements about each risk of bias item for each included study.
Effects of interventions
See: Table 1
Given the paucity of studies recruiting and randomising participants who had achieved some level of remission during acute phase treatment for depression, only two comparison could be made: 1) Antidepressant medication alone versus placebo; 2) Combination therapy versus antidepressant medication alone.
1. Medication versus placebo
1.1 Prevention of a second or next depressive episode (number relapsed‐recurred)
All trials in this comparison involved only participants who had responded or remitted after an acute phase of treatment (Analysis 1.1). Three studies (Cheung 2008; Emslie 2004; Emslie 2008) containing a total of 164 participants compared medication with a placebo pill during a relapse prevention phase. Of these, one re‐randomised participants early (Emslie 2008) after 12 weeks of acute treatment with a total of 102 participants, and the remaining two trials re‐randomised participants late after an acute and continuation phase, and contained a total of 62 participants. In the Emslie 2008 study in which re‐randomisation occurred early, there was evidence of an effect favouring the use of medication over placebo in order to prevent a next episode of depression (odds ratio (OR) 0.32; 95% confidence interval (CI) 0.14 to 0.73). In the two studies in which re‐randomisation occurred late (Cheung 2008; Emslie 2004) there was an effect favouring the use of medication to prevent the next episode of depression (OR 0.37; 95% CI 0.13 to 1.05). When considering these three studies together, there is evidence of an effect favouring medication over placebo in preventing the next episode of depression as measured by relapse‐recurrence rates (OR 0.34; 95% CI 0.18 to 0.64). The Cheung 2008 trial reported relapse rates 52 weeks after response or remission had been achieved, the Emslie 2004 trial after 32 weeks and the Emslie 2008 trial after 24 weeks.
1.1. Analysis.
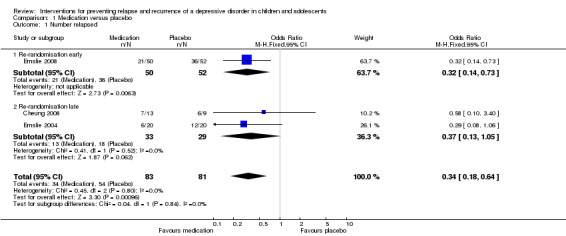
Comparison 1 Medication versus placebo, Outcome 1 Number relapsed.
1.2 Suicide‐related behaviours
In the trial by Emslie 2008 which involved early randomisation of participants, there was no statistically significant difference in the number of suicide‐related events in participants receiving placebo compared with medication (OR 3.18; 95% CI 0.13 to 79.96). The sole event was a suicide attempt of one participant in the medication arm. In the two trials where randomisation occurred at a late stage, there was also no statistically significant difference in the number of suicide‐related events reported by participants administered medication, compared with placebo (OR 0.32; 95% CI 0.01 to 8.26). In Emslie 2004, a single participant in one of the two placebo arms experienced suicidal ideation, while a participant in the other placebo arm reported self injurious behaviour. The study by Cheung 2008 relied on spontaneous report of suicide‐related events and none were reported. Overall, there was no statistically significant difference in suicide‐related behaviours reported for those receiving medication compared with placebo (OR 1.02; 95% CI 0.14 to 7.39). See Analysis 1.2.
1.2. Analysis.
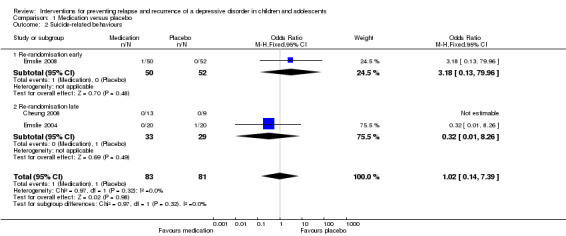
Comparison 1 Medication versus placebo, Outcome 2 Suicide‐related behaviours.
1.3 Time to relapse‐recurrence
Cheung 2008 report time to relapse‐recurrence as 29.3 weeks for participants in the medication group and 16.4 weeks for participants treated with placebo. Emslie 2004 report time to relapse‐recurrence as 180.7 days for medication and 71.2 days for placebo‐treated participants. Emslie 2008 reports median time to relapse‐recurrence, this being 14 weeks for the placebo group, and greater than 24 weeks (i.e. beyond time frame of the study) for the fluoxetine group. In the trial by Emslie 1998, mean time (standard deviation (SD)) to relapse‐recurrence was 195.9 (75.3) days for participants treated with medication and 187.9 (94.6) days for participants treated with placebo.
1.4 Functioning
One trial contained data suitable for this comparison and reported mean change in Global Assessment of Functioning (GAF) scores from the start to the end of the maintenance phase. There was no statistically significant difference in the level of functioning between participants treated with medication and those who received a placebo (standardised mean difference (SMD) 0.04; 95% CI ‐0.59 to 0.68).
1.5 Clinician‐rated depressive symptoms
Three trials (Cheung 2008; Emslie 2004; Emslie 2008) contained data suitable for this comparison. Data from the Children's Depression Rating Scale‐Revised (CDRS‐R) (Emslie 2004; Emslie 2008) and Hamilton Rating Scale for Depression (HAM‐D) (Cheung 2008) were included. We performed subgroup analyses depending on early (Emslie 2008) or late (Cheung 2008; Emslie 2004) re‐randomisation of participants. In the trial where participants were re‐randomised early after the acute phase of treatment, there was evidence of an effect favouring medication in producing lower levels of clinician‐rated depressive symptoms compared with placebo (SMD ‐0.47; 95% CI ‐0.86 to ‐0.07). In the two trials that re‐randomised participants at a late stage, after a period of continuation treatment, there was no statistically significant difference between medication and placebo in levels of depressive symptoms (OR 0.37; 95% CI 0.13 to 1.05). Overall there was no statistically significant difference in levels of depressive symptoms in those participants who had been treated with medication compared with placebo (SMD ‐0.07; 95% CI ‐0.68 to 0.55). See Analysis 1.4.
1.4. Analysis.
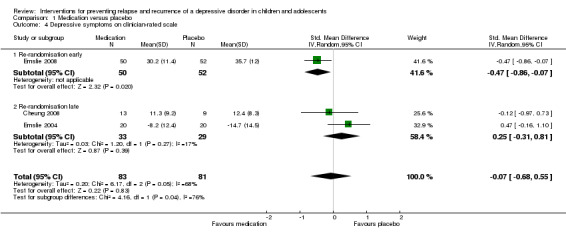
Comparison 1 Medication versus placebo, Outcome 4 Depressive symptoms on clinician‐rated scale.
1.6 Self rated depressive symptoms
No data were reported for this outcome.
1.7 Drop‐outs
In the Emslie 2008 trial, there was no statistically significant difference in the number of participants who dropped out of the medication arm of the trial compared with the placebo arm (OR 2.03; 95% CI 0.73 to 5.67). In the two late‐randomised trials (Cheung 2008; Emslie 2004) there was also no statistically significant difference in drop‐out rates between the two groups (OR 0.57; 95% CI 0.18 to 1.76). Overall, there was no statistically significant difference in drop‐out rates between those receiving medication and those who had switched to a placebo (OR 1.02; 95% CI 0.38 to 2.79). See Analysis 1.5.
1.5. Analysis.
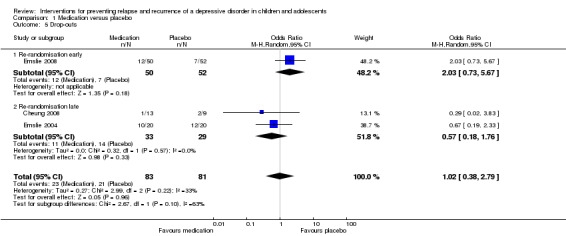
Comparison 1 Medication versus placebo, Outcome 5 Drop‐outs.
1.8 Emergence of co‐morbidity or switch to bipolar disorder
In the trial by Cheung 2008, three participants were recorded as experiencing psychosis and mania under adverse events and these occurred during the acute phase only. Emergence of co‐morbidity or switch to bipolar was not systematically measured. Emslie 2004 report that 70% of participants in the medication group and 60% of participants in the placebo group experienced 'any adverse event' during the trial, and Emslie 2008 report that "adverse events were similar between the two groups, and there were no discontinuations due to physical adverse events during continuation treatment" pg. 464.
1.9 Adverse events
Cheung 2008 report that six participants discontinued the trial due to adverse events, however these were all during the acute phase of treatment. No serious adverse events were reported.
2. Combination treatment (medication plus psychological therapy) versus medication alone
2.1 Prevention of a second or next depressive episode (number relapsed‐recurred)
The trial by Kennard 2008 involved only participants (N = 46) who had responded after an acute phase of treatment. There was a greater rate of relapse in those who received medication alone compared to combination therapy, but the difference did not reach statistical significance (OR 0.26; 95% CI 0.06 to 1.15). Relapse rates were reported after 24 weeks of treatment following initial response or remission. It should be noted that this effect differs from that reported in the paper, and the authors provided us with additional unpublished data in order to conduct this analysis.
2.2 Suicide‐related behaviours
There were comparable suicide‐related behaviours reported in those receiving medication alone compared with combination treatment (OR 0.52; 95% CI 0.04 to 6.22). Overall, one out of 22 participants in the combination group were recorded as experiencing a suicide‐related event compared with two out of 22 participants in the medication group.
2.3 Time to relapse‐recurrence
There were no data suitable for this outcome.
2.4 Functioning
The trial by Kennard 2008 containing 46 participants had data suitable for this outcome. There was no evidence of an effect of combination treatment in improving functioning more than medication alone (mean difference (MD) 1.30; 95% CI ‐4.42 to 7.02).
2.5 Clinician‐rated depressive symptoms
The trial by Kennard 2008 also contained data suitable for this outcome and is based on the CDRS‐R assessment tool. There was no evidence of an effect to suggest that combination therapy was superior to medication alone in reducing depressive symptoms (MD ‐6.20; 95% CI ‐12.96 to 0.56).
2.6 Self rated depressive symptoms
No data were reported for this outcome.
2.7 Drop‐outs
The trial by Kennard 2008 found no statistically significant difference between drop‐out rates in combination compared with medication alone (OR 1.11; 95% CI 0.20 to 6.15).
2.8 Emergence of co‐morbidity or switch to bipolar disorder
No data were reported for this outcome.
2.9 Adverse events
Kennard 2008 report four serious adverse events that occurred during the trial, three of which were suicide‐related behaviours, and one was hospitalisation for diabetic ketoacidosis.
Narrative results
The following results are based on those trials included in the review where participants underwent acute treatment of a depressive episode, and then entered (without re‐randomisation) a continuation/maintenance relapse prevention stage with measures of relapse/recurrence collected at longer‐term follow‐up. Two of these studies (Emslie 1998; Renaud 1998) followed up participants after a naturalistic period of time during which participants were able to receive any treatment (or no treatment) as they chose. In Clarke 1999 participants were re‐randomised into relapse prevention conditions, however, this was not based on response, i.e. all participants who entered the acute phase of the trial were re‐randomised regardless of response status. In TADS and TORDIA all participants were re‐randomised to a continuation/maintenance phase regardless of response status with outcomes analysed by response status.
In TADS 147 participants who had achieved full response at 12 weeks received either medication or psychological therapy. One out of 32 failed to maintain this full response through weeks 18 to 26 in the cognitive behavioural therapy (CBT) group; 14 out of 54 failed to maintain this full response in the medication group; and seven out of 61 in the combination group (CBT plus medication) failed to maintain this full response.
In TORDIA 153 participants were classed a responders at the end of the acute phase. Of these 20 out of 86 failed to maintain this response at 12‐week follow‐up in the combination group and 10 out of 67 failed to maintain response in the medication group.
Only the trial by Clarke 1999 compared a psychological therapy with no treatment. This was a three‐armed trial comparing psychological therapy (booster CBT sessions) to frequent assessments (every four months) and annual assessments (every 12 months). The assessment only conditions were combined and compared to the CBT booster condition. At 12 months four out of 15 in the booster CBT group compared with two out of 25 in the assessment only group had relapsed. At 24 months five out of 14 in the booster CBT group compared with three out of 23 in the assessment only group had relapsed. In terms of clinician and self rated depressive symptoms and functioning, there was no difference between the groups at any time point.
Discussion
Summary of main results
In this review we have presented data from nine published trials on the efficacy of pharmacological and psychological interventions to prevent relapse‐recurrence after a first episode of depressive disorder in children and adolescents. There were few trials that targeted relapse prevention in children and adolescents and within these there were a limited amount of data that could be pooled for meta‐analysis. At present, it is difficult to draw conclusions as to the most effective treatment approach to adopt when aiming to prevent a second or next episode of depression in this population.
The results from this review suggest that medication can be effective in preventing relapse‐recurrence of depression in children and adolescents when compared with placebo. This result is based on three trials (Cheung 2008; Emslie 2004; Emslie 2008), in which between six and 12 months of treatment was undertaken during a controlled relapse prevention phase. In contrast, levels of clinician‐reported depressive symptoms and levels of functioning were not differentiated by treatment group. It is difficult to know the significance of these conflicting results given the low participant numbers and the difficulties in interpreting cross‐sectional endpoint data. An ongoing caveat to the longer‐term use of medication in this age group is uncertainty about the long‐term effects of antidepressant medication on the developing brain.
This review identified limited data based on just a single study (TADS), which suggest that psychological therapy may be superior to medication in producing lower relapse‐recurrence rates of depression. However, given the paucity of data available for this outcome, and the fact that the relapse prevention phase of the trial was not randomised, these results should be interpreted with caution.
We were only able to meta‐analyse the results of one trial in order to assess the effect of combining medication with psychological therapy when compared with medication alone to prevent relapse‐recurrence. The trial results reported by Kennard 2008 in their publication differ from those obtained through the analysis in this review and this is likely due to the variance in statistical techniques used. The populations sampled within the additional trials which looked at the effect of combination therapy versus medication alone reported in the narrative results were diverse, again making it difficult to draw firm conclusions regarding the differing effects of these treatment approaches. The TORDIA trial was specifically targeting children and adolescents defined as 'treatment resistant'. Thus the treatment approach that may be most effective for these participants may be different from that needed for those with mild to moderate depression, who are undertaking a first‐line treatment. It should be noted that the Kennard 2008 trial implemented a much more rigorous experimental design, and administered CBT that was specifically focused on relapse prevention. Given the sound experimental platform on which the trial was executed, the results from the trial as reported in the original publication, which favour combination therapy over medication alone, should be seriously considered.
Overall, this review has highlighted the need for more trials to be conducted in the area of relapse prevention for depression in children and adolescents. There is the potential for methodologically rigorous trials to be undertaken, that look at a variety of treatment approaches and are reflective of best practice guidelines. Whilst psychological therapies are recommended as a first‐line treatment option for children and adolescents with mild to moderate depression (McDermott 2010; NICE 2005), the majority of trials identified through our search used medication to treat depression in the first instance, and only one trial systematically compared psychotherapy to medication in a relapse prevention phase. There is scope for more innovative designs, such as those that compare combination treatment during an acute phase and subsequently re‐randomise participants to each monotherapy alone, to be investigated. Furthermore, in other mental disorders, such as psychosis, it is apparent that the content of the psychotherapy during a relapse prevention phase needs to specifically target signs for relapse and so forth (Alvarez‐Jimenez 2011), rather than being a generic form of acute phase psychotherapy.
Overall completeness and applicability of evidence
In general, there were few trials suitable for inclusion in the review. We were unable to assess whether different relapse prevention treatment approaches were more effective for children compared with adolescents, as no trials could be located where data were compared separately across these age groups. The trials included in the review were diverse in terms of design, and due to these differences subgroup analyses were required which reduced the total number of participants in comparisons; this thus limits the conclusions we could draw from them. Furthermore, the time points at which relapse‐recurrence data were collected varied; this review has combined them all into meta‐analysis at one time point only, which again could affect the overall results.
It is also important to note that the severity of depression in participants varied across trials. For example, the TORDIA trial included 'treatment resistant' participants, whereas TADS did not. As a result, it may be the case that different treatments are more effective for some participants compared with others, depending on stage of illness, severity of previous symptomology and age (Jaffee 2002; Kessler 2005). We were unable to conduct analyses of this type within this review. Where data are available, future reviews would benefit from assessing treatment approach based on depression severity where possible, in order to elucidate what treatment is most effective at different ends of the spectrum.
One of our primary outcomes of interest was the time in which children and adolescents relapsed. This was not always reported and varied based on days, weeks or months to relapse. Routine reporting of these types of data is of paramount importance, as identifying times in which a child or adolescent is at a 'higher risk' of relapse may also be important in developing effective prevention strategies targeting this time course. Data of this type are important to obtain and include in the review given the potential for withdrawal symptoms from discontinuation of medication in placebo groups to be mistaken as relapse, thus overestimating the effect of medication in preventing relapse
In addition, it was not stated how many times a participant had a actually relapsed, therefore we are currently unaware whether the relapse data reported refers to just one relapsing episode or many. This type of information would be important in determining the chronicity of a depressive episode at a within‐subjects level.
A limited number of treatment approaches have been tested to prevent relapse of depression in children and adolescents and, predominantly, pharmacotherapy has been utilised. This is disappointing given the high prevalence of depressive disorders in this population, coupled with the observation that future episodes become more debilitating and impact negatively on functioning and well being. Furthermore, despite mindfulness‐based cognitive therapy (MBCT) being identified as an effective relapse prevention treatment for depression in adults, no trials that we are aware of have been carried out in children and adolescents, therefore we are unable to draw any conclusions about the potential benefits of MBCT for this population.
Quality of the evidence
The quality of evidence contained in the review varied depending on the type of design. Trials in which participants were re‐randomised after they responded to an initial course of treatment (Cheung 2008; Emslie 2004; Emslie 2008; Kennard 2008) are the most robust and methodologically rigorous trials to draw conclusions from. They allow participants to be drawn once again from a random sample, thus reducing sampling bias and allowing treatments administered only during the relapse prevention phase to be assessed. Trials in which a long‐term follow‐up of relapse‐recurrence is recorded and treatment group remains the same across time are not necessarily comparable to the aforementioned trials. We acknowledged the diversity in these designs and, as such, chose to perform subgroup analyses within the meta‐analysis. However, one disadvantage of doing so was that the sample sizes from which we were able to assess treatment efficacy was reduced. The number of children and adolescents who respond to initial treatment, and thus are eligible for inclusion into a relapse‐prevention randomised controlled trial (RCT), is much smaller than the original sample size. In some instances, after accounting for attrition and response rates, approximately 60% of children and adolescents from an original sample will continue through to be randomised into a relapse prevention phase (Emslie 2008; Kennard 2008). In some studies this figure is even lower, at 34.2% of the original randomised sample (Emslie 2004). It is a balancing act for researchers to assess the relative merits in implementing this type of design into a relapse prevention trial. However, the sound methodological platform that it allows evidence to be drawn from may outweigh these shortcomings.
Two of the trials contained in this review (TADS; TORDIA) are both large‐scale and well‐designed RCTs. However, it was difficult to extract data from these trials based on our outcomes of interest, and neither re‐randomised participants based on response status after the acute stage of treatment. Both studies stated that they aimed to prevent relapse, however limited data were contained in the publications in order for sound judgements to be drawn regarding treatment effectiveness. TADS described response in various ways, however published few data concerning participants who maintained a full response (and thus did not relapse) in comparison with those who did not. The addition of self reported outcomes and suicide‐related outcomes for this subset of participants would have increased our knowledge of what interventions are most effective and potentially protective in the long term.
Potential biases in the review process
There was the potential for bias to arise when trials that executed very different designs were combined in meta‐analysis. We attempted to keep this bias to a minimum by performing subgroup analyses on different trial designs. As relapse and recurrence can only be assessed after a child or adolescent has achieved a period free from depressive symptoms, the sample size of many trials was small, limiting the effect that they exerted within a meta‐analysis. Furthermore, the variety of ways in which relapse and recurrence were defined made it difficult to interpret results regarding the efficacy of any single treatment approach.
Every effort was made to obtain additional information from authors of included studies, however not all authors were able to provide the data that we asked for. On some occasions, the data used in this review were from a subset of participants contained within a trial, the results of which had not been reported in the original publication.
Agreements and disagreements with other studies or reviews
This is the first systematic review and meta‐analysis conducted on interventions to prevent relapse and recurrence of a depressive disorder in children and adolescents. The results are consistent with trials that suggest medication is more effective than placebo in preventing a next episode of depression. However, beyond this specific comparison, the best treatment approach remains unclear. Some trials suggest that CBT is no more beneficial than assessment only in preventing recurrence of depression (Clarke 1999). Others have concluded that CBT and, to a lesser degree, a combination of CBT and medication, is more effective than medication alone in preventing relapse after initial response to acute treatment (TADS). At this stage, there is not enough evidence to conclude what type of treatment is most effective. Individual trials undertaken within this area, as highlighted in this review, also have mixed results regarding effective relapse prevention treatments.
Authors' conclusions
Implications for practice.
There is limited evidence that continued medication is more effective than placebo in preventing the next episode of depression once a child or adolescent has initially responded to medication during acute phase treatment. However, at present no study has investigated the most effective treatment course after a child or adolescent has responded to acute phase psychotherapy, or a combination of medication and psychotherapy. It is unclear whether continued treatment with medication is needed in this instance, or whether psychotherapy in isolation would be sufficient. Given that psychotherapy is recommended as a first‐line treatment in children and adolescents with depression, this sequenced approach to treatment is an avenue to be explored.
Acute phase and relapse‐prevention phase cognitive behavioural therapy (CBT) are markedly different, with the former focusing on reducing depressive symptoms, while the latter places emphasis on maintaining response. As such, the structure of each will be different and dependent on whether the individual has received any psychotherapy in the acute phase or not. If they have not, time may need to be spent consolidating the basic skills of CBT and then integrating these into a relapse prevention context. However, if initial CBT has been undertaken, then more emphasis can be placed on pure relapse prevention elements such as mood monitoring and self management skills. Indeed, there is a suggestion from one small pilot study (Kennard 2008) that specifically formulated relapse‐prevention CBT may be beneficial. Research trials investigating the effects of CBT in the relapse prevention stage should consider designing the core elements of the psychotherapy based on patients' previous exposure to the approach.
Implications for research.
This review has highlighted two very different designs used to study relapse prevention interventions for depression in children and adolescents. As noted above, there are difficulties in retaining sample sizes large enough to power an effect though a continuation or maintenance phase of treatment, in order to examine relapse rates over longer periods of time. However, in order to develop evidence‐based interventions for this phase of depressive illness, it is essential that the most methodologically rigorous designs are implemented within the context of a randomised controlled trial (RCT). Re‐randomising participants into treatment group at the point of response and the beginning of the relapse prevention phase is one way to achieve this outcome. Alternatively, there may need to be a second phase of recruitment that occurs before the relapse prevention phase, in order to boost sample size numbers with participants who have achieved the same level of response as those in the initial recruitment, and combine them at that point.
This review has also highlighted the paucity of psychological therapies that have been tested to reduce the rate of relapse in depression in children and adolescents. No trials of mindfulness‐based CBT were retrieved; this is surprising given the effectiveness of this approach in the adult population. Further research utilising this treatment approach would be advantageous and further our knowledge as to the effectiveness of this intervention across the age range.
There were no trials suitable for inclusion in which participants were between 18 and 25 years of age. Given the expanding definition of youth, and the indication that the majority of depressive disorders emerge before 25 years, more research is needed to assess the effect of relapse‐prevention interventions in this older age group. Furthermore, no comparisons could be made between treatment approaches for children versus adolescents, or either group compared with adults. Future research should endeavour to investigate the most effective treatment approach across the lifespan.
What this review has not considered in depth is relapse rates in continuation and maintenance treatment based on the initial type of acute treatment that children and adolescents receive. Given arguments that highlight the merit of medication in allowing this population to engage in therapy, future reviews may focus more broadly on the treatment approaches offered at different stages of depression, and the most effective combination of treatments over time.
The difficulties encountered in extracting comparable relapse or recurrence data (or both) across trials should also be considered by researchers embarking on this field. For many children and adolescents, relapse into a depressive episode will not be a single event, and the more chronic and persistent depression becomes for an individual, the higher likelihood that multiple relapses will occur. The current survival models of relapse utilised in many trials investigating relapse of depression assume that each individual has only one relapse event. Future research should endeavour to present data based on raw relapse rate, and state where applicable if an individual has experienced more than one relapse event over the course of treatment. By doing so, it may be possible to investigate changes in the number of relapse events per se, and some interventions may be more effective based on this outcome measure.
Depressive disorder in children and adolescents is rarely a straightforward, uni‐dimensional illness. It is also important to consider the fact that comorbid mental illnesses, especially anxiety and substance use disorders, are common among this population with depressive disorders. Therefore any meaningful intervention package should seek to address comorbidity and include assessment of recovery from these other illnesses among its outcomes. Furthermore, focusing on the early stage of depression, as this review has done, has highlighted the lack of data available concerning the emergence of other psychiatric disorders, such as psychosis or mania. When relapse occurs, it may signal the beginning of a co‐morbid disorder. For instance, individuals may respond to treatment for anxiety or depression but subsequently develop psychosis. There is an emerging research field suggesting that for a small group of adolescents and young people, a depressive disorder may be an early sign of an emerging bipolar or psychotic disorder (Thompson 2003; Yung 2003). It is therefore necessary to assess for such disorders as potential outcomes in intervention studies and in follow‐up data collection.
Feedback
Comments on discontinuation effects from J Jureidini, 20 December 2012
Summary
I believe that withdrawal/ discontinuation effects contributed to outcomes in a way that exaggerates the apparent effect of antidepressants. I have previously critiqued claims that venlafaxine prevented relapse (Journal of Clinical Psychiatry 2008, 69:865–866), based on the fact that the data more supported a withdrawal effect in those that discontinued than a protective effect on those that continued.
I note that authors did not report (presumably because they did not have access to) survival curves or individual data. However the mean times to relapse (see ‘1.3 Time to relapse‐recurrence’; placebo relapses occur earlier on average) are consistent with what was found in the venlafaxine data; that is an overrepresentation of very early ‘relapse’ in the placebo group that we argued was more likely to represent discontinuation.
Discontinuation confounds and potentially invalidates the studies I propose that the review should include some discussion of the possibility that conflating discontinuation with relapse might exaggerate the apparent benefit of antidepressants.
Reply
Thank you for the comment. You are correct that we did not have access to individual patient data and could not undertake to analyse data using survival curves. We were able to extract data on time to relapse‐recurrence as reported in the results, section 1.3. We have reported mean time to relapse‐recurrence for Cheung 2008, and have not reported an estimate of variance as the trial authors do not state this. It should be noted that they do report a median time to relapse‐recurrence of 10 weeks in the placebo group. These estimates are both lower than the mean time, and suggest that there may have been some skew in the data. However, given we do not have access to individual patient data we cannot know the time of greatest risk of relapse‐recurrence. Both estimates presented by the trial authors (median of 10 weeks; mean of 16.4 weeks) are inconsistent with the end of the taper, which was four weeks for those randomised to placebo.
For the trials that tested the efficacy of fluoxetine against placebo in an RCT design, none used a taper period for those randomised to placebo. Trial authors state this is due to discontinuation effects being unlikely with fluoxetine given the relatively long half‐life.
Emslie 2008 reports a median time to relapse‐recurrence; and we note an error in our write up [now corrected] in the Description of studies under the 'Outcomes' subheading where we state four trials reported mean time in which participants relapsed‐recurred. The median time to relapse‐recurrence in Emslie 2008 was 14 weeks in the placebo group; Emslie 2004 reports a mean of 71.2 days (10 weeks) and a standard error of 9.5, which equates to a standard deviation of 42 days (6 weeks). While there is relatively large standard deviations in Emslie 2004, and the suggestion of skewed data in Emslie 2008 (median time to relapse is reported); again without access to individual patient data we cannot know the time of greatest risk of recurrence. Time to recurrence was well beyond when you would expect discontinuation effects.
Without more data, including discussion on the issue of discontinuation would be going beyond the data that is available to us from the trials, and presented in the review.
Contributors
Sarah Hetrick, Georgina Cox and Mark Phelan
Further comments on discontinuation effects from J Jureidini, 23 August 2013
Summary
In their reply to my comment of 20 December 2012, Hetrick et al acknowledge that discontinuation effects could have played a part in exaggerating the apparent benefit of continuing on antidepressants. They could hardly conclude otherwise, given the existing studies that show that withdrawal effects from stopping antidepressants can be mistaken for relapse.They note that they were unable to obtain data to resolve this uncertainty. Faced with a plausible hypothesis, with no data to confirm or refute it, they inexplicably conclude that their review should remain silent on the issue. Surely they should modify their review to note the inability to exclude a withdrawal effect as the explanation for their findings.
I agree with the conflict of interest statement: I certify that I have no affiliations with or involvement in any organization or entity with a financial interest in the subject matter of my feedback.
Reply
Thank you for the ongoing discussion. Given we have not extracted and included data relevant to disentangling the effects of medication withdrawal and relapse, a lengthy discussion of this issue would go beyond the data that is available. However, given it is clinically important to ensure that clinicians are clear with clients who are discontinuing on medication that it can be difficult to differentiate between withdrawal symptoms and relapse of depression, we will include a comment in the section of the discussion “Overall completeness and applicability of the evidence" highlighting that this data is not available and that the possibility that a withdrawal effect may explain the apparent benefit of antidepressant medication for preventing relapse.
Contributors
Sarah Hetrick, Georgina Cox.
What's new
| Date | Event | Description |
|---|---|---|
| 24 February 2014 | Feedback has been incorporated | Further comments added on previous feedback |
History
Protocol first published: Issue 4, 2008 Review first published: Issue 11, 2012
| Date | Event | Description |
|---|---|---|
| 6 March 2013 | Feedback has been incorporated | One item of feedback incorporated along with an author response |
| 1 November 2008 | Amended | Headings activated. |
Acknowledgements
Many thanks to the CCDAN editorial team for ongoing support and advice.
Thanks to headspace Research Assistants Lauren Colautti and Alan Bailey for double‐checking all of the numerical data for the review and the characteristics of studies table.
Orygen Research Centre is thankful for the support of the Colonial Foundation.
Appendices
Appendix 1. Search strategies (MEDLINE, EMBASE, PsycINFO and CENTRAL) to June 2009
Concept 1: Depression
| MEDLINE (1950 ‐ ) | EMBASE (1980 ‐ ) | PsycINFO (1809 ‐ ) | CENTRAL |
| 1. depression/ | 1. depression/ or agitated depression/ or atypical depression/ or depressive psychosis/ or dysthymia/ or endogenous depression/ or involutional depression/ or major depression/ or masked depression/ or melancholia/ or "mixed anxiety and depression"/ or "mixed depression and dementia"/ or mourning syndrome/ or organic depression/ or postoperative depression/ or premenstrual dysphoric disorder/ or pseudodementia/ or puerperal depression/ or reactive depression/ or recurrent brief depression/ or seasonal affective disorder/ | 1. exp major depression/ or atypical depression/ |
#1. depression/ |
| 2. mood disorders/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or dysthymic disorder/ or seasonal affective disorder/ | 2. mood disorder/ | 2. affective disorders/ or seasonal affective disorder/ or affective psychosis/ | #2. mood disorders/ or depressive disorder/ or depression, postpartum/ or depressive disorder, major/ or dysthymic disorder/ or seasonal affective disorder/ |
| 3. adjustment disorders/ | 3. adjustment disorder/ | 3. adjustment disorders/ | #3. depressi*.ti,ab. |
| 4. or/1‐3 | 4. or/1‐3 | 4. ((affective or mood or adjustment) adj dis$).ti,ab. | #4. (affective NEAR/2 dis*) |
| 5. (depress$ adj3 (patient$ or symptom$ or disorder$)).ti,ab. | #5. (mood NEAR/2 dis*) | ||
| 6. or/1‐5 | #6. (depress$ NEAR/3 (patient* or symptom* or disorder*)) | ||
| #7. (#1 or #2 or #3 or #4 or #5 or #6) |
Concept 2: Children or First Onset
| MEDLINE | EMBASE | PsycINFO | CENTRAL |
| 5. adolescent/ or child/ or child, Preschool/ or infant/ | 5. exp adolescent/ or exp child/ or exp adolescence/ or exp childhood/ | 7. (child$ or infant$ or juvenil$ or minors or school$ or p?ediatri$ or adolesc$ or teen$ or young or youth$).mp. | #8. (child* or infant* or juvenil* or minors or school* or pediatri* or paediatric* or adolesc* or teen* or young or youth*) |
| 6. (child$ or infant$ or juvenil$ or minors or school$ or p?ediatri$ or adolesc$ or teen$ or young or youth$).ti,ab. | 6. (child$ or infant$ or juvenil$ or minors or school$ or p?ediatri$ or adolesc$ or teen$ or young or youth$).ti,ab. | 8. ((first or prior or index) adj (episod$ or onset or inciden$ or diagnos$ or refer$)).ti,ab. | #9. (first or prior or index) NEAR/3 (episod* or onset or inciden* or diagnos* or refer*) |
| 7. ((first or prior or index) adj (episod$ or onset or inciden$ or diagnos$ or refer$)).ti,ab. | 7. ((first or prior or index) adj (episod$ or onset or inciden$ or diagnos$ or refer$)).ti,ab. | 9. or/7‐8 | #10. (#8 or #9) |
| 8. or/5‐7 | 8. or/5‐7 |
Concept 3: Recurrence/Relapse prevention
| MEDLINE | EMBASE | PsycINFO | CENTRAL |
| 9. recurrence/ | 9. recurrent disease/ or relapse/ | 9. (recur$ or relaps$ or recrudesc$).mp. | #11. (recur* or relaps* or recrudesc* or maintenance or prophyla* or continuation) |
| 10. (recur$ or relaps$ or recrudesc$).ti,ab. | 10. (recur$ or relaps$ or recrudesc$).ti,ab. | 10. (maintenance$ or prophyla$ or contin$ or discontinue$).ti,ab. | #12. (prevent* NEAR/7 (recur* or relaps* or remis* or episode*)) |
| 11. (maintenance$ or prophyla$ or prevent$ or continu$ or discontinu$).ti,ab. | 11. (maintenance$ or prophyla$ or continuation).ti,ab. | 11. (prevent$ adj7 (recur$ or relaps$ or remis$ or episode$)).ti,ab. | #13. (#11 or #12) |
| 12. or/9‐11 | 12. (prevent$ adj7 (recur$ or relaps$ or remis$ or episode$)).ti,ab. | 12. or/9‐11 | |
| 13. or/9‐12 |
Concept 4: Human RCTs
| MEDLINE | EMBASE | PsycINFO |
| 13. randomized controlled trials as topic/ | 14. randomized controlled trial/ | 13. treatment effectiveness evaluation/ |
| 14. randomized controlled trial.pt. | 15. phase 3 clinical trial/ or phase 4 clinical trial/ | 14. clinical trials/ |
| 15. controlled clinical trial.pt. | 16. double blind procedure/ | 15. placebo/ |
| 16. randomi#ed.ab. | 17. single blind procedure/ | 16. mental health program evaluation/ |
| 17. placebo$.ab. | 18. triple blind procedure/ | 17. mental health program evaluation/ |
| 18. randomly.ab. | 19. randomization/ | 18. placebo$.tw. |
| 19. trial.ti. | 20. controlled study/ | 19. random$.tw. |
| 20. or/13‐19 | 21. placebo/ | 20. randomi#ed controlled trial$.tw. |
| 21. (animals not (humans and animals)).sh. | 22. placebo$.tw. | 21. (clinical adj3 trial$).tw. |
| 22. 20 not 21 | 23. random$.tw. | 22. (research adj3 design).tw. |
| 24. randomi#ed controlled trial$.tw. | 23. (evaluat$ adj3 stud$).tw. | |
| 25. (clinical adj3 trial$).tw. | 24. (prospectiv$ adj3 stud$).tw. | |
| 26. ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$)).tw. | 25. ((singl$ or doubl$ or trebl$ or tripl$) adj3 (blind$ or mask$ or dummy)).tw. | |
| 27. or/13‐25 | 26. or/13‐25 | |
| 28. ((animal or nonhuman) not (human and (animal or nonhuman))).de. | 27. (animal not ((human or inpatient or outpatient) and animal)).po. | |
| 29. 27 not 28 | 28. 26 not 27 |
Combining concepts
| MEDLINE | EMBASE | PsycINFO | CENTRAL |
| 23. (4 and 8 and 12 and 22) | 30. (4 and 8 and 13 and 29) | 29. (6 and 9 and 12 and 28) | #7 and #10 and #13 |
Data and analyses
Comparison 1. Medication versus placebo.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number relapsed | 3 | 164 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.34 [0.18, 0.64] |
| 1.1 Re‐randomisation early | 1 | 102 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.14, 0.73] |
| 1.2 Re‐randomisation late | 2 | 62 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.37 [0.13, 1.05] |
| 2 Suicide‐related behaviours | 3 | 164 | Odds Ratio (M‐H, Fixed, 95% CI) | 1.02 [0.14, 7.39] |
| 2.1 Re‐randomisation early | 1 | 102 | Odds Ratio (M‐H, Fixed, 95% CI) | 3.18 [0.13, 79.96] |
| 2.2 Re‐randomisation late | 2 | 62 | Odds Ratio (M‐H, Fixed, 95% CI) | 0.32 [0.01, 8.26] |
| 3 Functioning (C‐GAS/GAF) | 1 | Std. Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 3.1 Continuation/maintenance treatment for responders only | 1 | Std. Mean Difference (IV, Random, 95% CI) | 0.0 [0.0, 0.0] | |
| 4 Depressive symptoms on clinician‐rated scale | 3 | 164 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.07 [‐0.68, 0.55] |
| 4.1 Re‐randomisation early | 1 | 102 | Std. Mean Difference (IV, Random, 95% CI) | ‐0.47 [‐0.86, ‐0.07] |
| 4.2 Re‐randomisation late | 2 | 62 | Std. Mean Difference (IV, Random, 95% CI) | 0.25 [‐0.31, 0.81] |
| 5 Drop‐outs | 3 | 164 | Odds Ratio (M‐H, Random, 95% CI) | 1.02 [0.38, 2.79] |
| 5.1 Re‐randomisation early | 1 | 102 | Odds Ratio (M‐H, Random, 95% CI) | 2.03 [0.73, 5.67] |
| 5.2 Re‐randomisation late | 2 | 62 | Odds Ratio (M‐H, Random, 95% CI) | 0.57 [0.18, 1.76] |
1.3. Analysis.
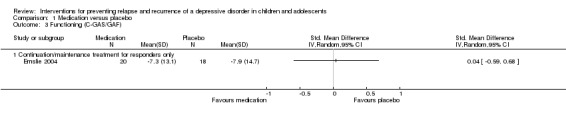
Comparison 1 Medication versus placebo, Outcome 3 Functioning (C‐GAS/GAF).
Comparison 2. COMB (med + psych) versus med.
| Outcome or subgroup title | No. of studies | No. of participants | Statistical method | Effect size |
|---|---|---|---|---|
| 1 Number relapsed | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected | |
| 2 Suicide‐related behaviours | 1 | Odds Ratio (M‐H, Fixed, 95% CI) | Totals not selected | |
| 3 Functioning (C‐GAS) | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 4 Depressive symptoms on clinician‐rated scale | 1 | Mean Difference (IV, Random, 95% CI) | Totals not selected | |
| 5 Drop‐outs | 1 | Odds Ratio (M‐H, Random, 95% CI) | Totals not selected |
2.1. Analysis.
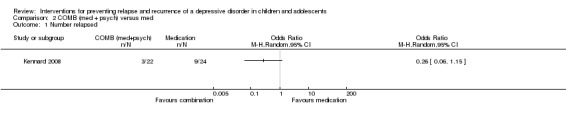
Comparison 2 COMB (med + psych) versus med, Outcome 1 Number relapsed.
2.2. Analysis.
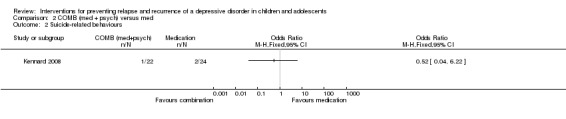
Comparison 2 COMB (med + psych) versus med, Outcome 2 Suicide‐related behaviours.
2.3. Analysis.
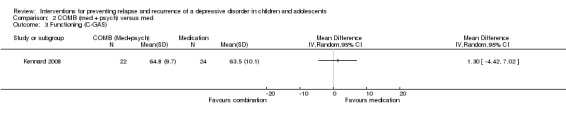
Comparison 2 COMB (med + psych) versus med, Outcome 3 Functioning (C‐GAS).
2.4. Analysis.
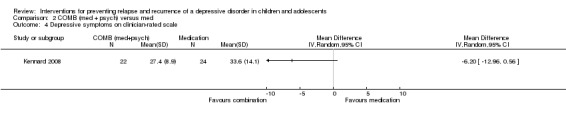
Comparison 2 COMB (med + psych) versus med, Outcome 4 Depressive symptoms on clinician‐rated scale.
2.5. Analysis.
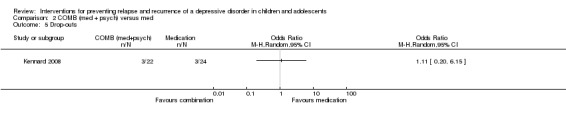
Comparison 2 COMB (med + psych) versus med, Outcome 5 Drop‐outs.
Characteristics of studies
Characteristics of included studies [ordered by study ID]
Cheung 2008.
| Methods | Design: treatment of individuals after remission/recovery from an acute episode of depression to prevent relapse/recurrence Phases: acute phase: participants treated with sertraline for 12 weeks. Continuation phase: participants that responded to acute phase treated with sertraline for 24 weeks. Maintenance phase: participants who maintained response during continuation phase, randomised to receive either treatment with sertraline or placebo for 52 weeks. Comparison groups: sertraline versus placebo Duration of relapse prevention (termed maintenance in trial) phase: 52 weeks Follow‐up assessment point of relapse prevention: from the maintenance phase, participants assessed weekly for first 4 weeks, then assessed every 2 weeks up to week 52 Funded by: the Canadian Institute of Health Research (CIHR) |
|
| Participants |
Acute phase N = 93 Maintenance phase N = 22 Child and adolescent or adolescent: adolescent (13 to 19 years) Depression diagnoses (DSM or ICD) included: MD as determined by a clinical interview using the K‐SAD‐PL and scoring > 16 on the first 17 items of the HAM‐D Criteria for remission/response: 2 consecutive HAM‐D scores of < 9 and greater than a 50% reduction in HAM‐D score within 12 weeks Criteria for relapse: defined by clinical judgement of treating physician or if an intervention beyond that permitted by the study protocol was required Are those at risk of suicide excluded from the trial? Not stated Suicide risk: not stated Baseline severity of depression (acute phase): mean (SD) HAM‐D score: total = 20.7 (3.9); sertraline = 21.3 (4.1); placebo = 19.9 (3.8) Length of index episode: not stated Number of previous episodes (% of participants): total = 14%; sertraline = 23%; placebo = 0% Age of onset: not stated Comorbidity of the participants (by group): comorbid anxiety disorder: total = 23%; sertraline = 23%; placebo = 22% Mean (SD) age: at maintenance phase: sertraline = 15.2; placebo = 16.3 Sex (M:F): sertraline = 3:10; placebo = 2:7 Family SES: not stated Setting: outpatient mood disorders clinics in 3 tertiary care centres Psychiatric diagnoses excluded: past or current hypomanic or manic episodes, current psychotic symptoms, substance dependence in the last 3 months Country: Canada |
|
| Interventions |
Medication N = 13 Name (class and type): sertraline (SSRI) Dose (mg/day)/length: 25 to 200 mg/day, depending on response. During maintenance phase no treatment changes were permitted. Delivered how: by treating clinician every 2 weeks Placebo N = 9 Delivered how: by treating clinician every 2 weeks Content: participants previously treated with sertraline who were randomised to receive placebo pill intervention had their sertraline tapered by 25% of the initial dose every week for the first 4 weeks of the maintenance phase. During maintenance phase no treatment changes were permitted. |
|
| Outcomes | Prevention of second or next episode defined by: clinical judgement based on depressive symptoms and functional impairment Suicide‐related behaviours: reported as part of adverse events Time to relapse: recorded as weeks in table 1 Functioning: not a trial outcome Depressive symptoms on a clinician rated scale: HAM‐D |
|
| Notes | *All demographics describe the 22 participants included at the baseline of the maintenance phase, unless otherwise specified. 6 participants discontinued the study during the acute and continuation phases due to adverse effects. Reasons for drop‐out were: agitation and hostility (n = 2), psychosis (n = 1) and mania (n = 3). |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was conducted by the study pharmacist using a computer‐generated randomisation schedule". pg. 390 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Low risk | "...clinicians and research staff remained blinded to treatment during the randomisation phase". pg 390 |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | Low risk | "Participants...remained blinded to treatment during the randomisation phase". pg 390 |
| Incomplete outcome data (attrition bias) ITT analysis | Low risk | "Subjects who were randomised and received at least one dose of treatment/placebo were included in the analyses". pg 391 Imputation: all drop‐outs considered as recurred in analysis (Figure 1. pg. 390) |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Low risk | Number randomised in maintenance phase: sertraline = 13; placebo = 9; total = 22 Number of drop‐outs during maintenance phase: sertraline = 1; placebo = 2; total = 3 Number analysed post‐maintenance phase: sertraline = 13; placebo = 9; total = 22 |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | Sertraline: 1 participant lost to follow‐up Placebo: 1 participant lost to follow‐up and 1 withdrew consent |
| Selective reporting (reporting bias) | Unclear risk | Authors reported on specified outcomes. No access to protocol. |
| Other bias | Unclear risk | Small sample size limits generalisation. Only included adolescents with MDD and did not capture the full spectrum of depressive disorders present in this age group. |
Clarke 1999.
| Methods | Design: acute treatment of a depressive episode with long‐term follow‐up of relapse/recurrence Phases: acute phase: participants randomly assigned to either group CBT, group CBT with a separate parent group or wait‐list control for 8 weeks. Maintenance phase: participants randomly assigned to either assessments every 4 months with booster session, assessments every 4 months only, or assessments every 12 months only for 24 months Comparison groups: acute phase: group CBT versus group CBT + parent CBT versus wait‐list control. Maintenance phase: assessment and booster at 4 months versus assessment only at 4 months versus assessment only at 12 months. Duration of relapse prevention phase: 24 months (2 years) Follow‐up assessment point of relapse prevention: 12 and 24 months Funded by: National Institute of Mental Health (NIMH) |
|
| Participants |
Acute phase N = 123 Maintenance phase N = 64 Child and adolescent or adolescent: adolescent (14 to 18 years) Depression diagnoses (DSM or ICD) included: current DSM‐III‐R diagnosis of MDD or dysthymia as determined by clinical interview using the K‐SADS‐E Criteria for remission/response: for acute phase: no longer meeting DSM‐III‐R criteria for either major depression or dysthymia for 2 weeks preceding post‐treatment assessment. For maintenance phase: 8 weeks or more of minimal or absent depression symptoms. Criteria for relapse: not stated Are those at risk of suicide excluded from the trial? Not stated in exclusion criteria, however participants were excluded on the basis of needing immediate, acute treatment Suicide risk: not stated Baseline severity of depression: mean (SD) HAM‐D score: adolescent CBT = 13.0 (5.3); adolescent CBT + parent = 15.1 (6.0); wait‐list = 14.5 (5.9) Length of index episode: not stated Number of previous episodes: not stated Age of onset: not stated Comorbidity of the participants (by group): at post‐treatment assessment, 23.6% had a current comorbid anxiety disorder, and 23.6% had a non‐affective disorder in the past Mean (SD) age: at maintenance phase: 16.2 (1.3) Sex (M:F): total at post‐treatment assessment: 28:68 Family SES: not stated Setting: 2 sites (Eugene and Portland, Oregon) Psychiatric diagnoses excluded: mania/hypomania, panic disorder, generalised anxiety disorder, conduct disorder, psychoactive substance abuse/dependence, schizophrenia Country: USA |
|
| Interventions |
Psychotherapy (acute phase) N = 45 Name: Adolescent Coping With Depression Course (CWD‐A; Clarke 1990). Skills taught include mood monitoring, improving social skills increasing pleasant activities, decreasing anxiety, reducing negative thinking, improving communication and conflict resolution. # sessions/length: 16 2‐hour sessions over 8 weeks Manualised (Y/N): yes Individual or group: group (up to 10 people) Parent involvement: no Fidelity check: yes. Videotaped sessions of adolescent and parent sessions independently rated for protocol compliance by group leader. Mean therapist compliance 90.5% across 72 rated sessions. Delivered by: advanced graduate psychology or social work students, or master's or doctoral‐level clinicians Psychotherapy (acute phase) N = 42 Name: Adolescent Coping With Depression Course (CWD‐A; Clarke 1990) plus parent component. Details as above. # sessions/length: 16 2‐hour sessions over 8 weeks Manualised (Y/N): yes Individual or group: group (up to 10 people) Parent involvement: yes. Parents received separate but parallel sessions reviewing the content in the adolescent course, and 2 joint sessions with the adolescent. Fidelity check: details as above Delivered by: details as above Control (acute phase) N = 36 Name: wait‐list Content: at the conclusion of the acute phase, adolescents in the wait‐list condition were offered their choice of treatment and no longer included in the study Psychotherapy (maintenance phase) N = 24 Name: booster CBT. Based on relapse prevention in addictive disorder (e.g. Marlatt & Gordon 1985). Adolescents could choose from 6 booster protocols: pleasant events, social skills and communication, relaxation, cognitions, negotiation and problem solving, maintaining gains and setting goals) # sessions/length: 6 sessions, 1 every 4 months. Attendance for booster sessions not formally collected but estimated at less than 50%. Manualised (Y/N): yes Individual or group: group Parent involvement: yes. Therapist worked with adolescent and parent to determine which booster sessions would be most appropriate. Fidelity check: no information Delivered by: advanced graduate psychology or social work students, or master's or doctoral‐level clinicians Assessment only (maintenance phase) N = 16 Name: frequent assessments (FA); assessments once every 4 months Delivered by: interviewers with a bachelor's or master's degree in psychology or social work Assessment only (maintenance phase) N = 24 Name: annual assessments (AA); assessments once every 12 months Delivered by: interviews with a bachelor's or master's degree in psychology or social work |
|
| Outcomes | Prevention of second or next episode defined as: not clearly stated in the publication Suicide‐related behaviours: not measured Time to relapse: not measured Functioning: GAF Depressive symptoms on a clinician‐rated scale: HAM‐D Depressive symptoms on a self rated scale: BDI |
|
| Notes | Requested additional data from authors on 15 March 2011. Reply received on 16 March 2011; unable to provide requested data due to staffing resources. Annual and frequent assessment data have been combined for dichotomous outcomes |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "Eligible participants were randomly assigned to 1 of 3 conditions". pg. 273 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Low risk | "Interviewers were blind to the participants' conditions". pg. 274 |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | High risk | Unlikely as psychotherapy was being administered |
| Incomplete outcome data (attrition bias) ITT analysis | Low risk | "It was used to examine outcome effects on all 123 participants randomly assigned to conditions (an "intent‐to‐treat" sample)". pg. 274 |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Low risk | Number randomised at acute phase: adolescent only CBT = 45; adolescent + parent CBT = 42; wait‐list = 36; total = 123 Number of drop‐outs during acute phase: adolescent only CBT = 8; adolescent + parent CBT = 10; wait‐list = 9; total = 96 Number randomised at maintenance phase: booster CBT = 24; FA = 16; AA = 24; total = 64 Number of drop‐outs during maintenance phase: booster CBT = 7; FA = 6; AA = 5; total = 18 Number analysed at maintenance phase: booster CBT = 17; FA = 10; AA = 19; total = 46 *Means (SD) for HAM‐D, BDI and GAF presented in Table 2 represent observed cases only (reported above). RER planned comparisons based on ITT analysis (n = 63). |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | High risk | Authors report reasons for some participants not completing booster sessions including: being recovered and not interested in additional treatment, seeing a non‐study therapist, being unable to schedule or cancelling the appointment, or moving out of area |
| Selective reporting (reporting bias) | High risk | Drop‐out reasons for each group not reported |
| Other bias | Unclear risk | Acute‐phase attrition varied across 2 study sites (31% versus 15%). Baseline BDI score imbalance for AA compared with FA and booster CBT group. |
Emslie 1998.
| Methods | Design: acute treatment of a depressive episode with 12‐month follow‐up of relapse/recurrence Phases: acute phase: 8 weeks. Maintenance phase: participants given the option of continuing on blind study medication or treated openly for a further 12 months. Comparison groups: fluoxetine versus no medication versus other medication Duration of relapse prevention phase: 12 months (published trial data included both patients who had and those that had not responded to acute phase treatment) Follow‐up assessment point of relapse prevention: 6 and 12 months from post‐acute phase Funded by: National institute of Mental Health |
|
| Participants |
Acute phase N = 96 Maintenance phase N = 87 Child and adolescent or adolescent only or first episode population: child and adolescent (7 to 18 years) Depression diagnoses (DSM or ICD) included: MDD diagnosed by clinical interview using DSM‐III‐R K‐SADS depressive items and a Children’s Depression Rating Scale‐Revised (CDSR‐R) score of > 40 Criteria for remission/response: remission defined as a relatively asymptomatic period (MDD K‐LIFE rating of 1 or 2) for at least 14 days. Recovery was defined as an asymptomatic period of at least 60 days. Are those at risk of suicide excluded from the trial? Suicide risk not stated in exclusion criteria Suicide risk: not stated Baseline severity of depression (at start of relapse‐prevention phase): CDRS‐R mean (SD) score: fluoxetine = 38.4 (14.8); placebo = 47.1 (17.0) Mean (SD) length of index episode (weeks) at start of acute phase: fluoxetine = 14.6 (9.7); placebo = 13.7 (7.5) Mean (SD) number of previous episodes at start of acute phase: fluoxetine = 1.7 (0.7); placebo = 1.8 (0.8) Mean (SD) age of onset: fluoxetine = 10.6 (2.7); placebo = 11.0 (2.6) Comorbidity included: fluoxetine = none 7; dysthymia 20; anxiety disorders 32; ADHD 16; ODD/CD 13. Placebo = none 11; dysthymia 14; anxiety disorders 22; ADHD 13; ODD/CD 16 Age: fluoxetine = 12.2 (2.7); placebo = 12.5 (2.6) Family SES: fluoxetine: 1 to 2 = 29.2%; 3 = 33.3%; 4 to 5 = 37.5%. Placebo: 1 to 2 = 33.3%; 3 = 37.5%; 4 to 5 = 29.2%. Setting: outpatient What psychiatric diagnoses were excluded: bipolar I and II; psychotic depression; independent sleep‐wake disorder; alcohol and other substance abuse; anorexia nervosa; bulimia nervosa; previous adequate treatment with fluoxetine; at least 1 first‐degree relative with bipolar I disorder Country: USA |
|
| Interventions |
Fluoxetine Acute N = 48 Maintenance N = 34 Name (class and type): fluoxetine (SSRI) Dose (mg/day)/length: 20 mg/day during acute phase depending on response with a 1‐week placebo run‐in prior to the acute phase. During maintenance phase no treatment changes were permitted Delivered how: by treating clinician every 2 weeks Placebo Acute phase N = 48 Maintenance phase N = 40 |
|
| Outcomes | Prevention of second or next episode defined as: an MDD K‐LIFE rating of 5 or greater for 14 days. Relapse is defined as an episode occurring after remission but before recovery and recurrence is defined as an episode of depression after recovery. Recovery is defined as minimal symptoms for a period of 60 days as defined on the K‐LIFE as an MDD score of ≤ 2. Suicide‐related behaviours: not measured Time to relapse: obtained from authors Functioning: C‐GAS Depressive symptoms on a clinician‐rated scale: CDRS‐R |
|
| Notes | On behalf of Dr Emslie, Taryn Mayes provided additional data for 1997/1998 trial. Data are split by responders at 12 weeks (n = 74), based on original treatment arm assignment (fluoxetine versus placebo) and do not take into account the course of treatment that occurred in the relapse prevention phase. Due to the uncontrolled nature of the relapse prevention phase, a decision was made to use data based on responders at 12 weeks and initial acute phase treatment. The data reported in the 1998 publication analyses participants based on whether participants received medication, no medication or other medication during the 1‐year follow‐up period, and only recurrence rather than relapse data are presented. These data are not part of the meta‐analysis in this review. |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | High risk | Participants were not re‐randomised following acute phase |
| Allocation concealment (selection bias) | High risk | Not possible as participants not re‐randomised |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Unclear risk | "Patients were followed for 12 months following the end of acute treatment. Treatment was not controlled and information collected was primarily a naturalistic follow‐up of patients completing the acute trial" pg. 34 (Emslie 1998) |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | High risk | "On exiting the acute treatment trial, patients were given the option of continuing blind on study medication or being treated openly. Most non‐responders were treated openly with fluoxetine" pg. 34 (Emslie 1998) |
| Incomplete outcome data (attrition bias) ITT analysis | High risk | "Ninety‐six subjects were randomised in the acute phase of the study...eighty‐seven subjects completed the 1‐year naturalistic follow‐up" pg. 35 (Emslie 1998) |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Low risk | Number randomised at acute phase: fluoxetine = 48; placebo = 48; total = 96 Number of drop‐outs during acute phase: fluoxetine = 14; placebo = 22; total = 36 Number continuing onto maintenance phase: recovered participants: fluoxetine = 47; no medication = 22; other medication = 5; non‐recovered participants: adequate trial = 11; inadequate trial = 2 Number of drop‐outs during maintenance phase: *naturalistic follow‐up. Of original 96 participants, 87 completed maintenance phase. Number analysed at maintenance phase: recovered participants = 74; non‐recovered participants = 13 |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Unclear risk | 96 participants randomised in acute phase of which 36 dropped out. At 1‐year follow‐up results presented for 86 participants. |
| Selective reporting (reporting bias) | High risk | Adverse outcomes during maintenance phase not reported |
| Other bias | Unclear risk | Not enough information to make a clear judgement |
Emslie 2004.
| Methods | Design: treatment of individuals after remission/recovery from an acute episode of depression to prevent relapse/recurrence Phases: acute phase: participants treated with either fluoxetine or pill placebo for 9 weeks. Titration phase: dose of fluoxetine adjusted depending on response based on CDRS‐R score for next 10 weeks. Maintenance phase: fluoxetine participants defined as remitted randomised to continue with fluoxetine (F/F) or switch to pill placebo (F/P) for 32 weeks. Participants who responded in the pill placebo group continued to receive pill placebo (P/P). Comparison groups: fluoxetine (F/F) versus placebo (F/P) versus pure pill placebo (P/P) Duration of relapse prevention phase: 32 weeks (7.5 months) Follow‐up assessment point of relapse prevention: week 19 defined as baseline for maintenance phase. Assessments taken at weeks 19, 23, 27, 31, 35, 43 and 51. Funded by: Eli‐Lilly and Company, Indianapolis, USA |
|
| Participants |
Acute phase N = 219 Maintenance phase N = 75 Child and adolescent or adolescent: child and adolescent (8 to 13 years) Depression diagnoses (DSM or ICD) included: a primary DSM‐IV diagnosis of MDD for at least 4 weeks, and a score of > 40 on the CDRS‐R and ≥ 4 on the CGI Criteria for remission/response: CDRS‐R total score of ≤ 28 Criteria for relapse: 2 criteria for the purpose of analysis: i) a one time CDRS‐R score of > 40, with a 2‐week history of clinical deterioration or relapse in the opinion of the physician; ii) a one time CDRS‐R score of > 40, with a history of 2 weeks of clinical deterioration Are those at risk of suicide excluded from the trial? Those with 'serious suicidal risk' were excluded from the study Suicide risk: not stated Baseline severity of suicide: not stated Baseline severity of depression (acute phase): CDRS‐R mean score (SD): fluoxetine = 57.1 (9.9); placebo = 55.1 (11.8) Length of index episode: duration of current episode at baseline of acute phase (mean, weeks): fluoxetine = 60.44; placebo = 61.29 Number of previous episodes: % first episode of depression at acute phase: fluoxetine = 79.8%; placebo = 78.2% Age of onset (years): fluoxetine mean (SD) = 10.41 (2.92); placebo mean (SD) = 10.26 (3.11) Comorbidity of the participants (by group at acute phase): ADHD: fluoxetine = 14.7%, placebo = 13.6%; ODD: fluoxetine = 15.6%, placebo = 15.5%; conduct disorder: fluoxetine = 2.8%, placebo = 0.9% Sex (M:F): F/F = 13.45 (2.38); F/P = 11.65 (2.48) Family SES: not reported Setting: outpatient Psychiatric diagnoses excluded: DSM‐IV defined disorders: bipolar I or II disorder, sleep‐wake disorder, psychotic depression (lifetime), anorexia (lifetime), bulimia (lifetime) borderline personality disorder, or substance abuse disorder (within the past 6 months) Country: USA |
|
| Interventions |
Medication N = 20 Name (class and type): fluoxetine (SSRI) Dose (mg/day)/length: 20 to 60 mg/day depending on response Delivered how: by child psychiatrist during regular visits to the clinic Placebo N = 20 Content: pill placebo. Due to fluoxetine's long half life, tapering is generally not necessary. Participants switched directly from fluoxetine to placebo. Delivered how: by a child psychiatrist during regular visits to the clinic *NB: 35 participants who responded to pill placebo during the continuation phase remained on placebo for the maintenance phase, however were not included in re‐randomisation |
|
| Outcomes | Prevention of second or next episode defined as: 2 criteria for the purpose of analysis: i) a one time Children's Depression Rating Scale‐Revised (CDRS‐R) score of ≥ 40, with a 2‐week history of clinical deterioration or relapse in the opinion of the physician; ii) a one time Children's Depression Rating Scale‐Revised (CDRS‐R) score of ≥ 40, with a history of 2 weeks of clinical deterioration Suicide‐related behaviours: reported as part of adverse events Time to relapse: reported as days Functioning: GAF Depressive symptoms on a clinician rated scale: CDRS‐R |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "After 9 weeks of acute treatment, patients... according to a computer‐generated randomisation scheme. After 19 weeks of treatment, patients...were randomly reassigned to maintenance treatment with their current dose of fluoxetine or with placebo". Figure 1, pg. 1398 |
| Allocation concealment (selection bias) | Unclear risk | Not stated |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Unclear risk | "A double‐blind, placebo controlled study". pg. 1397 |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | Unclear risk | "A double‐blind, placebo controlled study". pg. 1397 |
| Incomplete outcome data (attrition bias) ITT analysis | Low risk | "Analyses were conducted on an intent‐to‐treat basis unless otherwise specified". pg. 1399 NB: mean reported outcome measures calculated on observed rather than ITT participant totals |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Low risk | Number randomised at acute phase: fluoxetine = 109; placebo = 110; total = 219 Number of drop‐outs during acute phase: fluoxetine = 19; placebo = 42; total = 61 Number randomised at maintenance phase: F/F = 20; F/P = 20; P/P = 35; total = 75 Number of drop‐outs during maintenance phase: F/F = 10; F/P = 12; P/P = 18; total = 40 Number analysed at maintenance phase: varies based on analysis (see ITT analysis note above) |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | F/F: 1 participant experienced an adverse event (agitation), 6 relapsed and 3 made a decision to discontinue F/P: 12 participants relapsed P/P: 2 participants experienced an adverse event (hyperkinesia and infection), 6 relapsed, 2 decided to discontinue, 3 discontinued due to a protocol requirement, 3 lost to follow‐up and 2 reported a satisfactory response |
| Selective reporting (reporting bias) | Unclear risk | Authors reported on specified outcomes. No access to protocol. |
| Other bias | High risk | Funding provided by the drug company (Elli‐Lilly). Baseline imbalances in age and height. |
Emslie 2008.
| Methods | Design: treatment of individuals after remission/recovery from an acute episode of depression to prevent relapse/recurrence Phases: acute phase: participants treated with fluoxetine for 12 weeks. Maintenance phase: participants who had an adequate response to fluoxetine randomised to either continue with fluoxetine or receive placebo for 6 months. Comparison groups: fluoxetine versus placebo Duration of relapse prevention phase: 24 weeks (6 months) Follow‐up assessment point of relapse prevention: week 12 defined as baseline for maintenance phase. Every other week from week 12 to 16 and monthly from week 16 to 36, with 2 additional visits if needed. Funded by: National Institute of Mental Health (NIMH) |
|
| Participants |
Acute phase N = 168 Maintenance phase N = 102 Child and adolescent or adolescent: child and adolescent (7 to 18 years) Depression diagnoses (DSM or ICD) included: a primary DSM‐IV diagnosis of MDD for at least 4 weeks, and a score of > 40 on the CDRS‐R and ≥ 4 on the CGI Criteria for remission/response: CGI improvement score of 1 (very much improved) or 2 (much improved) and a decrease of at least 50% on the CDRS‐R score or a CGI improvement score of 1 or 2 and a CDRS‐R score of ≤ 28 Criteria for relapse: a one time CDRS‐R score of ≥ 40, with worsening of depressive symptoms for at least 2 weeks, or a clinical determination that there was significant clinical deterioration suggesting that full relapse would be likely without altering treatment, even if the CDRS‐R score was < 40 Are those at risk of suicide excluded from the trial? Those with 'severe suicidal ideation requiring inpatient treatment' were excluded from the study Suicide risk: not stated Baseline severity of suicide: measured according to FDA criteria. For participants entering the maintenance phase: death wishes mean = 38; suicidal ideation mean = 34; suicide plans mean = 10. Baseline severity of depression at acute phase: CDRS‐R mean score (SD): for participants who entered the maintenance phase: fluoxetine = 23.3 (3.9); placebo = 22.4 (4.4) Length of index episode: not stated Number of previous episodes: reported number of episodes at baseline of maintenance phase: mean = 1.3; SD = 0.5 Age of onset (years): for participants who entered the maintenance phase mean = 10.5; SD = 2.8 Comorbidity of the participants: for participants who entered the maintenance phase: fluoxetine = 36%; placebo = 15.4% Mean (SD) age at baseline of maintenance phase: 11.5 (2.8) Sex (M:F) of participants who entered the continuation phase: 65:37 Family SES: not stated Setting: general child and adolescent psychiatry outpatient clinic Psychiatric diagnoses excluded: lifetime history of any psychotic disorder (including psychotic depression), bipolar disorder, anorexia nervosa or bulimia, alcohol or substance abuse within the past 6 months and serve suicidal ideation requiring inpatient treatment Country: USA |
|
| Interventions |
Medication N = 50 Name (class and type): fluoxetine (SSRI) Dose (mg/day)/length: during the acute phase, participants received 10 to 40 mg/day depending on response. Participants who were re‐randomised to the fluoxetine group continued to receive the same dose as in the acute phase. Delivered how: by child psychiatrist during regular visits to the clinic Placebo N = 52 Content: pill placebo. Fluoxetine was not tapered given its half life. Participants switched directly from fluoxetine to placebo. Delivered how: by a child psychiatrist during regular visits to the clinic |
|
| Outcomes | Prevention of second or next episode defined as: relapse rate based on Childrens Depression Rating Scale‐Revised (CDRS‐R) Time to relapse: median time to relapse could only be calculated for placebo group. By 24 weeks, less than 50% of those in F/F group had relapse and thus median time cannot be calculated. Functioning: not measured Depression symptoms on clinician or self rated scale: CDRS‐R |
|
| Notes | Additional data (CDRS‐R endpoint scores based on LOCF) provided by authors on 12 May 2011 Serious adverse events (SAEs): fluoxetine: 1 suicide attempt; placebo: 2 SAEs (both medical procedures) 1 person in the fluoxetine group experience 'rebound activation' |
|
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "Randomization was accomplished by a computer implementation of the minimization method in order to accommodate stratification by response category (remission versus adequate clinical response), gender, and age (participants age 12 or under and those age 13 and over )". pg. 460 |
| Allocation concealment (selection bias) | Unclear risk | "This was a single site, double blind, randomised discontinuation trial". pg. 460 |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Unclear risk | As above |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | Unclear risk | As above |
| Incomplete outcome data (attrition bias) ITT analysis | Unclear risk | No information. Appears that an ITT analysis was used but not stated. |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Low risk | Number randomised at acute phase: 168 Number of drop‐outs during acute phase: 49 Number randomised at maintenance phase: fluoxetine = 50; placebo = 52; total = 102 Number of drop‐outs during maintenance phase: fluoxetine = 12; placebo = 7 Number analysed at maintenance phase: fluoxetine = 50; placebo = 52; total = 102 |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | In the fluoxetine group, 12 participants discontinued: 1 had an adverse events, 8 withdrew consent (of which 1 had other time commitments, 2 refused medication, 1 was feeling better, 1 risk of placebo, 2 had additional treatment, 1 had family issues), 1 was lost to follow‐up and 2 were non‐adherent In the placebo group 7 participants discontinued: 6 withdrew consent (2 refused medication, 1 was feeling better, 1 sought additional treatment and 1 had family issues) and 1 participant was non‐adherent |
| Selective reporting (reporting bias) | High risk | Do not report CDRS‐R endpoint scores and mean time to relapse |
| Other bias | High risk | "...the rate of anxiety disorders was found to be significantly different between the fluoxetine and placebo groups....". pg. 462 |
Kennard 2008.
| Methods | Design: treatment of individuals after remission/recovery from an acute episode of depression to prevent relapse/recurrence Phases: acute phase: participants treated with fluoxetine for 12 weeks. Maintenance phase: participants who had an adequate treatment response to fluoxetine randomised to either continue with fluoxetine (antidepressant medication management (MM)) or antidepressant MM + CBT (MM + CBT) for 6 months. Comparison groups: MM + CBT versus MM Duration of relapse prevention phase: 24 weeks (6 months) Follow‐up assessment point of relapse prevention: week 12 defined as baseline for maintenance phase. Every other week from week 12 to 16 and monthly from week 16 to 36, with additional visits allowed when needed. Funded by: National Institute of Mental Health (NIMH) |
|
| Participants |
Acute phase N = 66 Maintenance phase N = 46 Child and adolescent or adolescent: child and adolescent (11 to 18 years) Depression diagnoses (DSM or ICD) included: a primary DSM‐IV diagnosis of MDD for at least 4 weeks, based on the K‐SADS‐PL, and a score of ≥ 40 on the CDRS‐R Criteria for remission/response: CGI improvement score of 1 (very much improved) or 2 (much improved) and a decrease of at least 50% on the CDRS‐R score Criteria for relapse: a one time CDRS‐R score of ≥ 40, with a 2‐week symptom worsening based on patient and parent report or clinical history or clinical deterioration in which the CDRS‐R score was < 40, but the clinician noted significant deterioration that would suggest full relapse if the patients treatment was not altered Are those at risk of suicide excluded from the trial? Those with 'severe suicidal ideation (active ideation with plan and intent) requiring inpatient treatment' were excluded from the study Suicide risk: N/A as excluded from the study Baseline severity of depression: at baseline of maintenance phase: CDRS‐R mean score (SD): MM = 26.7 (5.1); MM + CBT = 26.5 (5.4) Length of index episode: mean (SD) number of weeks: MM = 35.0 (27.4); MM + CBT = 28.0 (25.1) Number of previous episodes (mean (SD)): MM = 1.2 (0.41); MM + CBT = 1.3 (0.56) Age of onset in years (mean (SD)): MM = 13.3 (2.2); MM + CBT = 13.5 (1.9) Number of comorbidity of the participants (mean (SD)): MM = 1.04 (0.90); MM + CBT = 0.86 (0.83) Age (mean (SD)): MM = 14.4 years (2.2); MM + CBT = 14.3 years (1.7) Sex (M:F): MM = 12:12; MM + CBT = 12:10 Family SES raw score (mean (SD)): MM = 44.9 (14.2); MM + CBT = 48.3 (13.2) Setting: general child and adolescent psychiatry outpatient clinic Psychiatric diagnoses excluded: lifetime history of any psychotic disorder (including psychotic depression), bipolar disorder, anorexia nervosa or bulimia, alcohol or substance abuse within the past 6 months and suicidal ideation requiring inpatient treatment Country: USA |
|
| Interventions |
Medication (acute phase) N = 66 Name (class and type): fluoxetine (SSRI) Dose (mg/day)/length: 10 to 40 mg/day depending in response for 12 weeks Delivered how: pharmacotherapy visits with a child and adolescent psychiatrist weekly for weeks 1 to 4 and every other week for weeks 5 to 12 Medication management + psychotherapy (MM + CBT) N = 22 Medication name (class and type): fluoxetine (SSRI) Dose (mg/day)/length: 10 to 40 mg/day depending on response Delivered how: by child psychiatrist during regular visits to the clinic Psychotherapy name: relapse‐prevention (RP) CBT. Aims to target residual symptoms that remain after adequate treatment response, and identify and enhance current strengths to promote well being. # sessions/length: 8 to 11 sessions over 6 months (weekly for 4 weeks, biweekly for 2 months and optional booster sessions for 3 months) Manualised (Y/N): yes Individual or group: individual Parent involvement: a minimum of 3 family sessions were included in the protocol Fidelity check: all sessions were audio‐taped. 20.8% of tapes were rated by master's or doctorate‐level therapists on the Cognitive Therapy Rating Scale (Rush 1998). 100% were rated as acceptable. Delivered by: 1 doctoral‐level psychologist, 1 master's level psychologist and 1 post‐doctoral research fellow Medication management N = 24 Content: medication administered as above |
|
| Outcomes | Prevention of second or next episode defined as: a one time Children's Depression Rating Scale‐Revised (CDRS‐R; Poznanski 1996) score of ≥ 40, with a 2‐week symptom worsening based on patient and parent report or clinical history or clinical deterioration in which the CDRS‐R score was ≤ 40, but the clinician noted significant deterioration that would suggest full relapse if the patient's treatment was not altered Suicide‐related behaviours: reported as part of adverse events Time to relapse: not reported Functioning: C‐GAS Depressive symptoms on a clinician rated scale: CDRS‐R |
|
| Notes | On behalf of Betsy Kennard, Taryn Mayes provided additional data concerning relapse rates (both ITT and observed cases data). ITT data used in meta‐analysis. | |
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Unclear risk | "randomisation was stratified by two levels of response: adequate responders, as previously defined...and remitters... Randomization was also stratified by age." pg. 1397‐8 |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Low risk | "Primary outcome measures were completed at weeks 12 (randomisation baseline), 24, and 36 by IEs who were blind to treatment assignment". pg. 1398 |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | High risk | Unlikely as psychotherapy was being administered |
| Incomplete outcome data (attrition bias) ITT analysis | Unclear risk | "A Cox proportional hazards regression, with adjustment for CDRS‐R total score at randomisation and for the hazard of relapsing by age across the trial (e.g. absorbing age in the model), was used to compare time to relapse between participants in the MM group and those in the CBT group". pg. 1398 |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Unclear risk | Number randomised at acute phase: total = 66 Number of drop‐outs during acute phase: 26 Number randomised at maintenance phase: MM = 24; MM + CBT = 22; total = 46 Number of drop‐outs during maintenance phase: MM = 3; MM + CBT = 3; total = 6 Number analysed at maintenance phase: unclear |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | In MM group, 1 dropped out due to a suicide attempt and 2 withdrew consent for additional treatment. In the MM + CBT group, 3 withdrew consent; 1 was feeling better and no longer wanted intervention and 2 sought additional treatment. |
| Selective reporting (reporting bias) | Unclear risk | Some inaccuracies reporting drop‐out data |
| Other bias | Unclear risk | Not enough information to make a clear judgement |
Renaud 1998.
| Methods | Design: acute treatment of a depressive episode with long‐term follow‐up of relapse/recurrence Phases: acute phase: participants received CBT for 12 to 16 weeks. Continuation phase: participants received 2 to 4 booster sessions over 2 to 4 months. Maintenance/follow‐up phase: 2 years. Comparison groups: acute phase: CBT versus Systematic Behavioural Family Therapy (SBFT) versus non‐directive supportive treatment (NDST). Continuation phase: follow‐up analysis split into rapid responders (RR), intermediate responders (IR) and initial non‐responders (INR) across all groups. Duration of relapse prevention phase: 24 months Follow‐up assessment point of relapse prevention: 12 and 24 months Funded by: National Institute of Mental health (NIMH) |
|
| Participants |
Acute phase N = 107 randomised, 100 still in study in session 2; data are based on the 100 participants only Maintenance phase N = 100 Child and adolescent or adolescent: adolescent Depression diagnoses (DSM or ICD) included: DSM‐III‐R defined MD using the K‐SADS‐P and E Criteria for remission/response: absence of MDD combined with a BDI score of less than 9 for 3 consecutive treatment sessions and sustained throughout any remaining treatment sessions Criteria for relapse: the onset of a new depressive episode over the follow‐up period Are those at risk of suicide excluded from the trial? Suicide risk not specified in exclusion criteria. However, participants that made a suicide attempt were removed from the study. Suicide risk: not measured Baseline severity of suicide: not measured Baseline severity of depression at acute phase: no data reported at baseline. At 6 weeks: BDI mean score (SD): RR = 21.8 (5.9); IR = 24.2 (7.7); INR = 28.8 (10.3) Length of index episode: not reported Number of previous episodes: not reported Age of onset (years): not reported Comorbidity of the participants (by group at acute phase): dysthymic disorder RR = 19.4%, IR = 21.6%, INR = 27.8%; anxiety disorder RR = 29.0%, IR = 35.3%, INR = 33.3%; disruptive disorder RR = 25.8%, IR = 13.7%, INR = 27.8% Age in years (mean (SD)): RR = 15.2 (1.4); IR = 15.9 (1.3); INR = 15.3 (1.4) Sex: RR: M = 29%; IR: M = 27.5%; INR: M = 11.1% Family SES: RR: 37.7 (15.7); IR: 43.6 (11.7); INR = 34.1 (10.1) Setting: outpatient; recruited from the Child and Adolescent Mood and Anxiety Disorder Clinic at Western Psychiatric Institute and Clinic, Pittsburgh Psychiatric diagnoses excluded: no clear exclusion criteria, however authors state all participants were: nonpsychotic, non bipolar, without obsessive‐compulsive disorder, eating disorder, substance abuse or ongoing physical or sexual abuse Country: USA |
|
| Interventions |
Psychotherapy (CBT) N = 37 Name: CBT # sessions/length: acute phase: 12 to 16 sessions delivered over 12 to 16 weeks Manualised (Y/N): yes Individual or group: individual Parent involvement: not specified Fidelity check: yes. Sessions were videotaped and rated for adherence using the Cognitive Therapy Rating Scale (Vallis 1986). More than 90% of treatment sessions were rated as acceptable. Delivered by: therapists with a median of 10 years clinical experience Psychotherapy (SBFT) N = 35 Name: Systemic Behaviour Family Therapy (SBFT) # sessions/length: acute phase: 12 to 16 sessions delivered over 12 to 16 weeks Manualised (Y/N): yes Individual or group: individual Parent involvement: yes Fidelity check: yes. Sessions were videotaped and rated for adherence using the Cognitive Therapy Rating Scale (Vallis 1986). More than 90% of treatment session were rated as acceptable. Delivered by: therapists with a median of 10 years clinical experience |
|
| Outcomes | Prevention of second or next episode defined as: relapse or recurrence based on the presence of a period of MDD as defined by the K‐SADS All other outcomes not reported by treatment group assignment |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | "To ensure comparability among the groups, the Begg and Iglewicz modification of the Efron biased coin toss was used". pg. 878 (Brent 1997 publication) |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Unclear risk | No information |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | Unclear risk | No information. As participants were receiving 1 of 3 psychologically based treatments it is possible that participants may not have been aware to which group they had been allocated. |
| Incomplete outcome data (attrition bias) ITT analysis | Low risk | "Data from all 100 subjects were analysed, consistent with our previous report and the overall approach of "intent to treat", defined as "the inclusion of eligible subjects regardless of compliance with the protocol". pg. 1186 (Renaud 1998 publication) |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Unclear risk | Number randomised at acute phase: CBT = 37; SBFT = 35; NST = 35; total = 107 Number of drop‐outs during acute phase: CBT = 7; SBFT = 11; NST = 11; total = 29 Number analysed at maintenance phase: *at the end of the acute phase, participants were subdivided into rapid responders (RR), intermediate responders (IR) and initial non‐responders (INR): RR = 31; IR = 51; INR = 18; total = 100 |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | 4 did not return for treatment, 8 dropped out, 7 were removed for clinical reasons and 10 were discovered to have pre‐existing conditions that violated the inclusion criteria and were thus removed from the study |
| Selective reporting (reporting bias) | Unclear risk | Authors reported on specified outcomes. No access to protocol. |
| Other bias | High risk | Authors state that an ITT analysis was carried out on all 107 participants in the Brent 1997 publication, however in the Renaud 1998 publication it states that analysis was carried out on 100 randomised participants (see ITT analysis details above) |
TADS.
| Methods | Design: acute treatment of a depressive episode with long‐term follow‐up of relapse/recurrence Phases: acute phase: participants treated with either fluoxetine (FLX), CBT or fluoxetine + CBT (COMB) for 12 weeks (stage I) Continuation phase: participants in CBT and COMB group received 3 to 6 CBT booster sessions depending on remission status. Participants in FLX group received 2 to 4 office visits depending on remission status for 6 weeks (weeks 12 to 18; stage II) Maintenance phase: participants in CBT/COMB groups received 3 sessions and FLX group followed up in a medication visit 3 times over 18 weeks (weeks 18 to 36; stage III) Comparison groups: FLX versus COMB versus CBT Duration of relapse prevention phase: 24 weeks (continuation phase: 6 weeks; maintenance phase: 18 weeks) Follow‐up assessment point of relapse prevention: weeks 12, 18 and 36 Funded by: National Institute of Mental Health (NIMH) |
|
| Participants |
Acute phase N = 327 (FLX, COMB, CBT) Continuation phase (12 weeks) N = 242 Maintenance phase (18 weeks) N = 210 Child and adolescent or adolescent: adolescent (12 to 17 years) Depression diagnoses (DSM or ICD) included: current DSM‐IV defined MDD, with a score of ≥ 45 on the CDRS‐R Criteria for remission/response: remission for Kennard (2009): a CDRS‐R score of ≤ 28 (split into acute remitters at week 12 and continuation remitters at week 18). Response for Rohde (2008): full responders; a CGI score of 1 or 2. Partial responders; a CGI score of 3. Criteria for relapse: Rohde (2008): once a participant had experienced 'sustained response' they were classed at subsequent assessments as "Failed to maintain" if they were given a CGI score of 3 to 7 Are those at risk of suicide excluded from the trial? Yes. Participants excluded if deemed ‘high risk’ because of a suicide attempt requiring medical attention within 6 months. Also excluded on the basis of having a clear intent or active plan to commit suicide, or suicidal ideation accompanied by a disorganised family unable to guarantee adequate safety monitoring. Suicide risk: N/A as excluded Baseline severity of suicide: measured using the Suicidal ideation Questionnaire‐Junior High School Version (SIQ‐JR; Reynolds 1987). Adjusted mean (SD): FLX = 21.81 (15.68) CBT = 21.91 (16.28) COMB = 27.33 (18.51) Baseline severity of depression at acute phase: CDRS‐R mean (SD) score: FLX = 58.96 (10.16); CBT = 59.58 (9.21); COMB = 60.75 (11.58) Length of index episode (at start of acute phase; weeks (range)): FLX = 38; CBT = 52; COMB = 48 Number of previous episodes: not reported Age of onset (mean years (SD)): 13.3 (2.16) Comorbidity of the participants (by group at acute phase). Any psychiatric disorder (%): FLX = 43.12; CBT = 58.18; COMB = 55.66 Age in years (mean (SD)): FLX = 14.50 (1.57); CBT = 14.62 (1.50); COMB = 14.6 (1.48) Sex (M:F): FLX = 50:59; CBT = 50:61; COMB = 47:60 Family SES: not reported by treatment group Setting: set over 13 outpatient sites Psychiatric diagnoses excluded: current or past diagnosis of bipolar disorder, severe conduct disorder, current substance abuse or dependence, pervasive developmental disorder(s), thought disorder or psychiatric disorders requiring out of protocol treatments Country: USA |
|
| Interventions |
Psychotherapy Acute phase N = 111 Continuation phase N = 76 Maintenance phase N = 66 Name: CBT # sessions/length: acute phase: 15 sessions over 12 weeks. Continuation phase: partial responders received 6 and full responders received 3 sessions over 6 weeks. Maintenance phase: 3 booster sessions every 6 weeks for 18 weeks. Manualised (Y/N): yes Individual or group: individual Parent involvement: yes. CBT programme involved 1 to 3 conjoint sessions and parent psychoeducation sessions Fidelity check: not reported Delivered by: not reported Medication Acute phase N = 109 Continuation phase N = 80 Maintenance phase N = 66 Name (class and type): fluoxetine (SSRI) Dose (mg/day)/length: 10 to 60 mg/day depending on response Delivered how: monitoring of status and medication effects occurred during 20 to 30‐minute visits to a study psychiatrist. Clinician also offered general encouragement about the effectiveness of pharmacotherapy for MDD. Combination medication + psychotherapy Acute phase N = 107 Continuation phase N = 86 Maintenance phase N = 78 Content: medication and psychotherapy delivered as described above |
|
| Outcomes | Prevention of second or next episode defined as: those who 'failed to maintain' response, defined as 'relapsed'. FLX: 14/54 (plus 2 unknown response status). CBT: 1/32 (plus 1 unknown response status). In table 2 of publication. All other outcomes not reported for the subset of participants who responded to treatment at 12 weeks |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Eligible participants were randomly assigned...using a computerized stratified randomisation, a 1:1:1:1 treatment allocation ratio, permuted blocking (first block size = 4, with subsequent random block sizes of 4 and 8) within each striatum, and site and sex stratification variables”. pg. 808 (under heading "Randomization and Blinding"), 2004. |
| Allocation concealment (selection bias) | Unclear risk | “Participants were randomly assigned...at the coordinating centre”. pg.448‐449 (under heading "Stage 1 Participants and Procedures"), 2008 |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Low risk | “TADS used 2 primary measures of depression status assessed...by an independent evaluator blind to condition”. pg. 448 (under heading "Stage 1 Participants and Procedures"), 2008 |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | High risk | “Participants and all study staff remained masked in the ‘pills only’ condition (fluoxetine therapy and placebo) until the end of stage 1 (week 12). Patients and treatment providers in the combination and CBT conditions were aware of treatment assignment”. Pg. 1133 (under heading "Methods"), 2007. |
| Incomplete outcome data (attrition bias) ITT analysis | Low risk | “The primary analyses of remission rates...were conducted using an “intention to treat” (ITT) approach in which the analysis included all participants randomised to treatment regardless of protocol adherence and/or treatment completion” (under heading "Data Analysis"), 2009 |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Low risk | Number randomised: CBT: 111; fluoxetine: 109; fluoxetine + CBT: 107; total: 327 Number of drop‐outs during intervention: CBT: 41; fluoxetine: 38; fluoxetine + CBT: 23; total: 102 Number drop‐outs in follow‐up (18 weeks; continuation phase): CBT: 21; fluoxetine: 37; fluoxetine + CBT: 15; total: 73 Number drop‐outs in follow‐up (36 weeks; maintenance phase): CBT: 25; fluoxetine: 21; fluoxetine + CBT: 23; total: 69 Number analysed post‐intervention: CBT: 111; fluoxetine: 109; fluoxetine + CBT: 107; total: 327 Number analysed follow‐up 1 (18 weeks; continuation phase): CBT: 111; fluoxetine: 109; fluoxetine + CBT: 107; total: 327 Number analysed follow‐up 2 (36 weeks; maintenance phase): CBT: 111; fluoxetine: 109; fluoxetine + CBT: 107; total: 327 |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | 84/327 exited the study because of loss of follow‐up or withdrawal of consent (N = 21 for COMB, N = 32 for FLX, N = 31 for CBT) 96/327 discontinued treatment before week 36 due to premature termination or non‐response at the end of stage 1 (N = 25 for COMB, N = 39 for FLX, N = 32 for CBT), and this discontinuation was decided by the study physician |
| Selective reporting (reporting bias) | Unclear risk | Not enough information to make a judgement |
| Other bias | High risk | Suicidal ideation: COMB participants had an excess of suicidal ideation at baseline relative to FLX or CBT groups |
TORDIA.
| Methods | Design: treatment of individuals after remission/recovery from an acute episode of depression to prevent relapse/recurrence Phases: acute phase: 12 weeks treatment with either venlafaxine, another SSRI, venlafaxine + CBT or another SSRI + CBT maintenance phase (weeks 13 to 24): those who had responded to treatment continued in same blinded treatment arm. *NB: non‐responders also included in trial and were treated with open‐label treatment after 12 weeks, which could consist of a switch to another SSRI, augmentation or addition of CBT/another psychotherapy Comparison groups: SSRI versus venlafaxine versus SSRI + CBT versus venlafaxine + CBT Relapse prevention phase: 12 weeks Follow‐up assessment point of relapse prevention: 24 weeks post‐baseline Funded by: National Institute of Mental Health |
|
| Participants |
Acute phase N = 334 Maintenance phase Responders: N = 144 Non‐responders: N = 131 Child and adolescent or adolescent only or first episode population: adolescents only (12 to 18 years) What depression diagnoses (DSM or ICD) were included: DSM‐IV defined MDD, with a CDRS‐R total score of ≥ 40 and a CGI score of ≥ 4 Criteria for remission: at least 3 consecutive weeks without clinically significant depressive symptoms, corresponding to a score of 1 on the Adolescent Longitudinal Interval Follow‐Up Evaluation. Criteria for response: a CGI rating of ≤ 2 (much or very much improved) and a ≥ 50% decrease from baseline in CDRS‐R score. Criteria for relapse: at least 2 consecutive weeks with probable or definite depressive disorder (score of 3 or 4 on the Adolescent Longitudinal Interview Follow‐Up Evaluation) Are those at risk of suicide excluded from the trial? Not stated as an exclusion reason Suicide risk: mean (SD) SIQ‐Jr score at acute stage (all randomised participants): venlafaxine = 40.4 (22.6); SSRI = 42.8 (22.0); no CBT = 41.9 (21.1); CBT = 41.3 (23.5) Baseline severity of depression: mean (SD) CDRS‐R score at acute stage (all randomised participants): venlafaxine = 57.8 (10.1); SSRI = 59.9 (10.6); no CBT = 58.4 (9.7); CBT = 59.2 (11.0) Mean (SD) length of index episode in months at start of acute stage: (all randomised participants) venlafaxine = 21.4 (19.0); SSRI = 23.5 (21.6); no CBT = 22.6 (21.4); CBT = 22.3 (19.4) % first episode at start of acute stage (all randomised participants): venlafaxine = 73.0; SSRI = 74.8; no CBT = 73.5; CBT = 74.4 Age of onset in years (SD): at start of acute stage (all randomised participants) venlafaxine = 12.8 (2.4); SSRI = 12.6 (2.6); no CBT = 12.5 (2.6); CBT = 12.9 (2.4) Comorbidity of the participants included: anxiety (including PTSD), ADHD, oppositional/conduct, dysthymia Mean (SD) age: 15.9 (1.6) Sex (M:F): 101:233 Family SES: no information Setting: outpatient What psychiatric diagnoses were excluded: bipolar spectrum disorder, psychosis, pervasive developmental disorder, autism, eating disorders and substance abuse or dependence Country: USA |
|
| Interventions |
Medication (SNRI) Acute phase N = 83 Responders (blind) in maintenance phase N = 31 Name (class and type): venlafaxine (SNRI) Dose (mg/day)/length: weeks 1, 2, 3 and 4 to 6: 37.5 mg, 75 mg, 112.5 mg and 150 mg. Non‐responders at week 6 could receive 225 mg. Delivered how: by psychiatrists or master's degree prepared nurses working with the supervision of a psychiatrist. Medication sessions were 30 to 60 minutes and included monitoring of vital signs, adverse effects, safety and symptomatic response, and were weekly for the first 4 weeks, every other week during acute treatment and monthly during the continuation phase. *NB: family psychoeducation also provided by a nurse or psychiatrist. Discussion around symptoms of depression, causes, treatments and potential adverse effects. Medication (SSRI) Acute phase N = 85 Responders (blind) in maintenance phase N = 26 Name (class and type): SSRI Dose (mg/day)/length: 10 mg at week 1 and 20 mg for weeks 2 to 6. Non‐responders at week 6 could receive 40 mg. Delivered how: as above *NB: family psychoeducation also provided by a nurse or psychiatrist. Discussion around symptoms of depression, causes, treatments and potential adverse effects. Combination: psychotherapy + venlafaxine Acute phase N = 83 Responders (blind) in maintenance phase N = 36 Name (description): CBT. Focuses on cognitive restructuring, behavioural activation, emotional regulation, social skills and problem solving sessions. # sessions/length: acute phase: up to 12 sessions. Every other week for 2 months and monthly thereafter. Manualised (Y/N): yes Individual or group: individual Parent involvement: family psychoeducation provided by a nurse or psychiatrist. Discussion around symptoms of depression, causes, treatments and potential adverse effects. Fidelity check: Cognitive Therapy Rating Scale used by 3 raters: 94.9%, 94% and 93.9% of taped sessions were rated as acceptable Delivered by: therapists with at least a master's degree in a mental health field Medication: venlafaxine as above Combination: psychotherapy + SSRI Acute phase N = 83 Responders (blind) in maintenance phase N = 35 Psychotherapy: as above Medication: SSRI as above |
|
| Outcomes | Prevention of second or next episode defined as: at least 2 consecutive weeks with probable or definite depressive disorder (score of 3 or 4 on the Adolescent Longitudinal Interview Follow‐Up Evaluation) All other outcomes not reported for the subset of participants who responded to acute treatment |
|
| Notes | ||
| Risk of bias | ||
| Bias | Authors' judgement | Support for judgement |
| Random sequence generation (selection bias) | Low risk | “Randomization was balanced both within and across sites using a variation of Efron’s biased coin toss”. pg. 904 (Brent 2008) |
| Allocation concealment (selection bias) | Unclear risk | No information |
| Blinding (performance bias and detection bias) Blinding of outcome assessor | Low risk | “independent evaluators were blind to medication type. Independent evaluators were also blind to CBT treatment” NB: “In 64 cases, the blinding of the independent evaluator was compromised, most commonly because of participant disclosure of receiving CBT”. pg. 785 (Emslie 2010) |
| Blinding (performance bias and detection bias) Blinding of participants/care providers | High risk | “Participants were blind to medication type”. NB: unlikely that participants were blind if receiving adjunctive CBT treatment. pg. 785 (Emslie 2010) |
| Incomplete outcome data (attrition bias) ITT analysis | Low risk | Indicated ITT analysis in Figure 1 consort diagram |
| Incomplete outcome data (attrition bias) Number of drop‐outs in each group reported | Unclear risk | Number randomised to acute phase: venlafaxine: 83; venlafaxine + CBT: 83; SSRI alone: 85; SSRI with CBT: 83; total: 334 Number of drop‐outs during acute phase: venlafaxine: 22; venlafaxine + CBT: 30; SSRI alone: 25; SSRI with CBT: 25; total: 102 Number of responders in maintenance phase: venlafaxine: 34 (31 blind); venlafaxine + CBT: 40 (36 blind); SSRI alone: 31 (26 blind); SSRI with CBT: 39 (35 blind); total: 144 Number analysed post‐continuation (24 weeks): venlafaxine: 83; venlafaxine + CBT: 83; SSRI alone: 85; SSRI with CBT: 83; total: 334 |
| Incomplete outcome data (attrition bias) Reasons for drop‐out in each group reported | Low risk | Venlafaxine group: 19 had an inadequate response to medication, 1 had an adverse event Venlafaxine + CBT group: 1 had an adverse event, 1 had a family conflict SSRI alone: 2 withdrew due to lack of efficacy, 2 received paroxetine, 1 had ancillary treatment co‐morbidity, 2 were non‐compliant and 1 was lost to follow‐up SSRI + CBT: 3 had adverse events, 2 received paroxetine, 1 had ancillary treatment co‐morbidity, 2 were non‐compliant and 2 withdrew consent |
| Selective reporting (reporting bias) | High risk | CDRS‐R scores only reported in graph form Drop‐outs across blind treatment and open treatment not clearly reported |
| Other bias | Unclear risk | Not enough information to make a judgement |
AA: annual assessments; ADHD: attention deficit hyperactivity disorder; BDI: Beck Depression inventory; CBT: cognitive behavioural therapy; CD: Conduct Disorder; CDRS‐R: Children's Depression Rating Scale‐Revised; C‐GAS: Children’s Global Assessment Scale; CGI: Clinical Global Impression; DSM: Diagnostic and Statistical Manual of Mental Disorders; F/F: fluoxetine/fluoxetine; F/P: fluoxetine/placebo; FA: frequent assessments; FDA: Food and Drug Administration (US); FLX: fluoxetine; GAF: Global Assessment of Functioning; HAM‐D: Hamilton Rating Scale for Depression; ICD: International Classification of Diseases; INR: initial non‐responders; IR: intermediate responders; ITT: intention‐to‐treat; K‐SADS‐PL: Kiddie Schedule for Affective Disorder and Schizophrenia Present and Lifetime Version LIFE: Longitudinal Interval Follow‐Up Evaluation; MD: Mean Difference; MDD: major depressive disorder; MM: medication management (antidepressant); N/A: not applicable; NST: Nondirective Supportive Therapy; ODD: Oppositional Definant Disorder; P/P: placebo/placebo; PTSD: post‐traumatic stress disorder; RER: Random Effects Regression; RP: relapse‐prevention; RR: rapid responders ; SAE: serious adverse event; SBFT: Systemic Behaviour Family Therapy; SD: standard deviation; SES: socioeconomic status; SSRI: selective serotonin re‐uptake inhibitors
Characteristics of excluded studies [ordered by study ID]
| Study | Reason for exclusion |
|---|---|
| ADAPT | Does not measure relapse and is not specified as a relapse prevention study |
| Birmaher 1998 | 10‐week acute phase treatment only involving a treatment resistant population |
| Birmaher 2000 | Acute phase treatment only |
| Eli 1986 | Acute phase treatment only |
| Eli 1995 | Acute phase treatment only |
| Emslie 2009 | Acute phase treatment only |
| Franchini 2006 | Not a RCT |
| GlaxoSmithKline 1997 | Acute phase treatment only in adolescent population |
| GlaxoSmithKline 2001 | Acute phase treatment only in adolescent population |
| TADS(acute phase) | Acute phase treatment only in adolescent population |
RCT: randomised controlled trial
Differences between protocol and review
In the protocol for this review, suicide‐related behaviour (both ideation and attempt) was specified as secondary outcome. However, due to the concern that taking antidepressant medications may potentially result in suicidal behaviour, we made a decision to include such behaviours as a primary outcome.
For dichotomous outcomes, such as 'number relapsed', results from each trial are expressed as an odds ratio (OR) with 95% confidence intervals and combined in meta‐analysis. Although the protocol for the review stipulated that we would express relapse rates as a risk ratio (RR), ORs have more favourable mathematical properties.
Originally we intended to perform subgroup analyses on trials that included children and adolescents versus those that included participants of any age who had experienced a first episode of depression. However, as the search did not yield any trials of the latter type, we could not perform this analysis. We also intended to perform subgroup analyses on trials that contained children versus those that contained adolescents, but the nature of the trials included in the review did not contain enough data to allow for this subgroup analysis.
During the review process it became apparent that within the two types of trial design that we had anticipated, there was considerable diversity. In trials where participants who had responded or remitted from an episode of MDD or DD during an acute phase of treatment were re‐randomised into a continuation or maintenance phase, re‐randomisation commonly occurred either early (after an acute phase) or late (after either a continuation and/or maintenance phase). Due to the variability in the length of treatment before re‐randomisation, we felt that it was important to perform subgroup analyses based on time of re‐randomisation (early or late).
Originally, we intended to undertake sensitivity analyses to assess the effect of risk of bias that may be introduced due to the decisions made in the process of undertaking the review. In psychiatry trials it is important to investigate the impact of assumptions made in various imputation methods used to account for missing data, such as analysis using LOCF and OC. However, as there were limited data contained in trials, we were unable to perform these analyses.
Contributions of authors
All authors contributed to the protocol development.
Georgina Cox screened trials for inclusion in the second search phase, extracted data for included trials, entered data for meta‐analysis and was involved in writing all sections of the review.
Sarah Hetrick co‐ordinated the development of the protocol, screened articles for inclusion and was involved in writing all sections of the review.
Magenta Simmons screened trials for inclusion.
Caroline Fisher, Stefanie De Silva, Olaoluwa Akinwale and Mark Phelan screened trials for inclusion and extracted data for the review.
Sources of support
Internal sources
Orygen Youth Health ‐ Research Centre. Melbourne, Australia.
External sources
No sources of support supplied
Declarations of interest
There are no declarations of interest for authors.
Edited (no change to conclusions), comment added to review
References
References to studies included in this review
Cheung 2008 {published data only}
- Cheung A, Kusumakar V, Kutcher S, Dubo E, Garland J, Weiss M, et al. Maintenance study for adolescent depression. Journal of Child and Adolescent Psychopharmacology 2008;4:389‐94. [DOI] [PubMed] [Google Scholar]
Clarke 1999 {published data only}
Emslie 1998 {published data only}
- Emslie GJ. Depression in children and adolescents [conference abstract]. 9th Congress of the Association of European Psychiatrists. Copenhagen, Denmark. Sept 20‐24, 1998.
- Emslie GJ, Heiligenstein JH, Hoog SL, Ernest DE, VanHooy B, Brown E, et al. Fluoxetine for maintenance of recovery from depression in children and adolescents: a placebo‐controlled, randomized clinical trial [Abstract No. NR736]. 154th Annual Meeting of the American Psychiatric Association; 2001 May 5‐10; New Orleans, LA. Arlington, VA: American Psychiatric Association, 2001.
- Emslie GJ, Rush AJ, Weinberg WA, Kowatch RA, Carmody T, Mayes TL. Fluoxetine in child and adolescent depression: acute and maintenance treatment. Depression and Anxiety 1998;7:32‐9. [DOI] [PubMed] [Google Scholar]
- Emslie GJ, Rush AJ, Weinberg WA, Kowatch RA, Hughes CW, Carmody T. A double‐blind placebo controlled trial of fluoxetine in depressed children and adolescents [conference abstract]. Sixth World Congress of Biological Psychiatry, Nice, France. June 22‐27, 1997.
- Emslie GJ, Rush R, Weinberg WA, Kowatch WA, Hughes CW, Carmody T, et al. A double blind, randomized, placebo‐controlled trial of fluoxetine in children and adolescents with depression. Archives of General Psychiatry 1997;54(11):1031‐7. [DOI] [PubMed] [Google Scholar]
- Hughes CW, Emslie G, Kowatch R, Weinberg W, Rintelmann J, Rush AJ. Clinician, parent, and child prediction of medication or placebo in double‐blind depression study. Neuropsychopharmacology 2000;23(5):591‐4. [DOI] [PubMed] [Google Scholar]
- Kowatch RA, Carmody TJ, Emslie GJ, Rintelmann JW, Hughes CW, Rush AJ. Prediction of response to fluoxetine and placebo in children and adolescents with major depression: a hypothesis generating study. Journal of Affective Disorders 1999;54(3):269‐76. [DOI] [PubMed] [Google Scholar]
Emslie 2004 {published data only}
- Emslie GJ, Heiligenstein JH, Hoog SL, Wagner KD, Findling RL, McCracken JT, et al. Fluoxetine treatment for prevention of relapse of depression in children and adolescents: a double blind, placebo‐controlled study. Journal of the American Academy of Child & Adolescent Psychiatry 2004; Vol. 43, issue 11:1397‐405. [DOI] [PubMed]
- Emslie GJ, Heiligenstein JH, Wagner KD, Hoog SL, Ernest DE, Brown E, et al. Fluoxetine for acute treatment of depression in children and adolescents: a placebo‐controlled, randomized clinical trial. Journal of the American Academy of Child & Adolescent Psychiatry 2002;41(10):1205‐15. [DOI] [PubMed] [Google Scholar]
Emslie 2008 {published data only}
- Emslie GJ, Kennard BD, Mayes TL, Nightingale‐Teresi J, Carmody T, Hughes CW, et al. Fluoxetine versus placebo in preventing relapse of major depression in children and adolescents. American Journal of Psychiatry 2008;165:459‐67. [DOI] [PMC free article] [PubMed] [Google Scholar]
Kennard 2008 {published data only}
- Kennard BD, Emslie GJ, Mayes TL, Nightingale‐Teresi J, Nakonezny PA, Hughes JL, et al. Cognitive‐behavioral therapy to prevent relapse in paediatric responders to pharmacotherapy for major depressive disorder. Journal of the American Academy of Child & Adolescent Psychiatry 2008;47(12):1395‐404. [DOI] [PMC free article] [PubMed] [Google Scholar]
Renaud 1998 {published data only}
- Birmaher B, Brent DA, Kolko D, Baugher M, Bridge J, Holder D, et al. Clinical outcomes after short‐term psychotherapy for adolescents with major depressive disorder. Archives of General Psychiatry 2000;57:29‐36. [DOI] [PubMed] [Google Scholar]
- Brent DA, Birmaher B, Kolko D, Baugher M, Bridge J. Subsyndromal depression in adolescents after a brief psychotherapy trial: course and outcome. Journal of Affective Disorders 2001;63(1‐3):51‐8. [DOI] [PubMed] [Google Scholar]
- Brent DA, Holder D, Kolko D, Birmaher B, Baugher M, Roth C, et al. A clinical psychotherapy trial for adolescent depression comparing cognitive, family, and supportive therapy. Archives of General Psychiatry 1997;54(9):877‐85. [DOI] [PubMed] [Google Scholar]
- Brent DA, Kolko DJ, Birmaher B, Baugher M, Bridge J. A clinical trial for adolescent depression: predictors of additional treatment in the acute and follow‐up phases of the trial. Journal of the American Academy of Child & Adolescent Psychiatry 1999;38(3):253‐70. [DOI] [PubMed] [Google Scholar]
- Kolko DJ, Brent DA, Baugher M, Bridge J, Birmaher B. Cognitive and family therapies for adolescent depression: treatment specificity, mediation, and moderation. Journal of Consulting and Clinical Psychology 2000;68(4):603‐14. [PubMed] [Google Scholar]
- Renaud J, Brent DA, Baugher M, Birmaher B, Kolko D J, Bridge J. Rapid response to psychosocial treatment for adolescent depression: a two‐year follow‐up. Journal of the American Academy of Child & Adolescent Psychiatry 1998;37(11):1184‐90. [DOI] [PubMed] [Google Scholar]
- Weersing VR, Brent DA. Cognitive‐behavioral therapy for adolescent depression: comparative efficacy, mediation, moderation and effectiveness. Evidence‐based psychotherapies for children and adolescents. New York, NY: Guilford Press, 2003:135‐47. [Google Scholar]
TADS {published data only}
- Domino ME, Foster EM, Vitiello B, Kratochvil CJ, Burns BJ, Silva SG, et al. Relative cost‐effectiveness of treatments for adolescent depression: 36‐week results from the TADS randomized trial. American Academy of Child and Adolescent Psychiatry 2009;48(7):711‐20. [DOI] [PubMed] [Google Scholar]
- Kennard BD, Silva SG, Mayes TL, Hughes JL, Kratochvil CJ, Emslie GJ, et al. Assessment of safety and long‐term outcomes of initial treatment with placebo in TADS. American Journal of Psychiatry 2009;166(3):337‐44. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard BD, Silva SG, Tonev S, Rohde P, Hughes JL, Vitiello B, et al. Remission and recovery in the Treatment for Adolescents with Depression Study (TADS): acute and long‐term outcomes. Journal of the American Academy of Child & Adolescent Psychiatry 2009;48(2):186‐95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- March J, Silva S, Petrycki S, Curry J, Wells K, Fairbank J, et al. The Treatment for Adolescents with Depression Study (TADS): demographic and clinical characteristics. Journal of the American Academy of Child & Adolescent Psychiatry 2005;44(1):28‐40. [DOI] [PubMed] [Google Scholar]
- March J, Silva S, Vitiello B. The Treatment for Adolescents with Depression Study (TADS): methods and message at 12 weeks. Journal of the American Academy of Child & Adolescent Psychiatry 2006;45(12):1393‐403. [DOI] [PubMed] [Google Scholar]
- March JS, Silva S, Petrycki S, Curry J, Wells K, Fairbank J, et al. The Treatment for Adolescents With Depression Study (TADS): long‐term effectiveness and safety outcomes. Archives of General Psychiatry 2007;64(10):1132‐43. [DOI] [PubMed] [Google Scholar]
- Rohde P, Silva SG, Tonev ST, Kennard BD, Vitiello B, Kratochvil CJ, et al. Achievement and maintenance of sustained response during the Treatment for Adolescents with Depression Study continuation and maintenance therapy. Archives of General Psychiatry 2008;65(4):447‐55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Simons AD, Rohde P, Kennard BD, Robins M. Relapse and recurrence prevention in the Treatment for Adolescents with Depression Study. Cognitive and Behavioral Practice 2005;12(2):240‐51. [Google Scholar]
- Treatment for Adolescents with Depression Study Team. The Treatment for Adolescents with Depression Study (TADS): outcomes over 1 year of naturalistic follow‐up. American Journal of Psychiatry 2009;166(10):1141‐9. [DOI] [PubMed] [Google Scholar]
TORDIA {published data only}
- Emslie GJ, Mayes T, Porta G, Vitiello B, Clarke G, Wagner KD, et al. Treatment of Resistant Depression in Adolescents (TORDIA): week 24 outcomes. American Journal of Psychiatry 2010;167(7):782‐91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Vitiello B, Emslie G, Clarke G, Wagner KD, Asarnow JR, Keller MB, et al. Long‐term outcome of adolescent depression initially resistant to selective serotonin reuptake inhibitor treatment: a follow‐up study of the TORDIA sample. Journal of Clinical Psychiatry 2011;72(3):388‐96. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies excluded from this review
ADAPT {published data only}
- Goodyer I, Dubicka B, Wilkinson P, Kelvin R, Roberts C, Byford S, et al. Selective serotonin reuptake inhibitors (SSRIs) and routine specialist care with and without cognitive behaviour therapy in adolescents with major depression: randomised controlled trial. BMJ 2007;335(7611):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goodyer IM, Dubicka B, Wilkinson P, Kelvin R, Roberts C, Byford S, et al. A randomised controlled trial of cognitive behaviour therapy in adolescents with major depression treated by selective serotonin reuptake inhibitors. The ADAPT trial. Health Technology Assessment 2008;12(14):iii‐iv, ix‐60. [DOI] [PubMed] [Google Scholar]
Birmaher 1998 {published data only}
- Birmaher B, Waterman GS, Ryan ND, Perel J, McNabb J, Balach L, et al. Randomized, controlled trial of amitriptyline versus placebo for adolescents with "treatment‐resistant" major depression. Journal of the American Academy of Child & Adolescent Psychiatry 1998;37(5):527‐35. [DOI] [PubMed] [Google Scholar]
Birmaher 2000 {published data only}
- Birmaher B, Brent DA, Kolko D, Baugher M, Bridge J, Holder D, et al. Clinical outcome after short‐term psychotherapy for adolescents with major depressive disorder. Archives of General Psychiatry 2000;57(1):29‐36. [DOI] [PubMed] [Google Scholar]
Eli 1986 {published data only}
- Eli Lilly. Fluoxetine: fluoxetine versus placebo in adolescent depressed patients [Trial 236]. Eli Lilly ‐ Clinical Study Register (http://www.lillytrials.com) 1986.
Eli 1995 {published data only}
- Eli Lilly. Fluoxetine versus placebo in the acute treatment of major depressive disorder in children and adolescents [Trial 375]. Eli Lilly ‐ Clinical Study Register (http://www.lillytrials.com) 1995.
Emslie 2009 {published data only}
- Emslie GJ, Ventura D, Korotzer A, Tourkodimitris A. Escitalopram in the treatment of adolescent depression: a randomized placebo‐controlled multisite trial. Journal of the American Academy of Child & Adolescent Psychiatry 2009;48(7):721‐9. [DOI] [PubMed] [Google Scholar]
- Forest Laboratories. A double‐blind, fixed‐dose extension study of escitalopram in pediatric patients with major depressive disorder [SCT‐MD‐32/32A]. Forest Laboratories ‐ Clinical Study Register (http://www.forestclinicaltrials.com) 2007.
Franchini 2006 {published data only}
- Franchini L, Bongiorno F, Spagnolo C, Florita M, Santoro A, Dotoli D, et al. Psychoeducational group intervention in addition to antidepressant therapy as relapse preventive strategy in unipolar patients. Clinical Neuropsychiatry 2006; Vol. 3, issue 4:282‐5.
GlaxoSmithKline 1997 {published data only}
- GlaxoSmithKline. A multi‐center, double‐blind, placebo controlled study of paroxetine and imipramine in adolescents with unipolar major depression ‐ acute phase (29060/329). GSK ‐ Clinical Study Register (http://www.gsk‐clinicalstudyregister.com) 1997.
- Keller MB, Ryan ND, Strober M, Klein RG, Kutcher SP, Birmaher B, et al. Efficacy of paroxetine in the treatment of adolescent major depression: a randomized, controlled trial. Journal of the American Academy of Child & Adolescent Psychiatry 2001;40(7):762‐72. [DOI] [PubMed] [Google Scholar]
GlaxoSmithKline 2001 {published data only}
- GlaxoSmithKline. A randomized, multi‐center, 8‐week, double‐blind, placebo‐controlled, flexible‐dose study to evaluate the efficacy and safety of paroxetine in children and adolescents with major depressive disorder (MDD). GSK ‐ Clinical Study Register (http://www.gsk‐clinicalstudyregister.com) 2001.
TADS(acute phase) {published data only}
- Jacobs RH, Silva SG, Reinecke MA, Curry JF, Ginsburg GS, Kratochvil CJ, et al. Dysfunctional attitudes scale perfectionism: a predictor and partial mediator of acute treatment outcome among clinically depressed adolescents. Journal of Clinical Child & Adolescent Psychology 2009;38(6):803‐13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kennard B, Silva S, Vitiello B, Curry J, Kratochvil C, Simons A, et al. Remission and residual symptoms after short‐term treatment in the Treatment of Adolescents with Depression Study (TADS). Journal of the American Academy of Child & Adolescent Psychiatry 2006;45(12):1404‐11. [DOI] [PubMed] [Google Scholar]
- Kratochvil C, Emslie G, Silva S, McNulty S, Walkup J, Curry J, et al. Acute time to response in the Treatment for Adolescents with Depression Study (TADS). Journal of the American Academy of Child & Adolescent Psychiatry 2006;45(12):1412‐8. [DOI] [PubMed] [Google Scholar]
- Lewis CC, Simons AD, Silva SG, Rohde P, Small DM, Murakami JL, et al. The role of readiness to change in response to treatment of adolescent depression. Journal of Consulting and Clinical Psychology 2009;77(3):422‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
References to studies awaiting assessment
Oleichik 1998 {published data only}
- Oleichik IV, Artiouch VV. The treatment of cognitive symptoms of adolescent endogenous depressive disorders. 11th European College of Neuropsychopharmacology Congress, Paris, France. 31st October ‐ 4th November 1998.
References to ongoing studies
Goodyer 2011 {published data only}
- Goodyer IM, Tsancheva S, Byford S, Dubicka B, Hill J, Kelvin R, et al. Improving mood with psychoanalytic and cognitive therapies (IMPACT): a pragmatic effectiveness superiority trial to investigate whether specialised psychological treatment reduces the risk for relapse in adolescents with moderate to severe unipolar depression: study protocol for a randomised controlled trial. Trials 2011;12:175. [DOI] [PMC free article] [PubMed] [Google Scholar]
NCT00612313 {unpublished data only}
- Emslie GJ. Pediatric MDD: sequential treatment with fluoxetine and relapse prevention. ClinicalTrials.gov (http://www.clinicaltrials.gov) 2008; Vol. http://clinicaltrials.gov/show/NCT00612313 (accessed 23 November 2011).
Additional references
AACAP 2007
- American Academy of Child, Adolescent Psychiatry (AACAP). Practice parameters for the assessment and treatment of children and adolescents with depressive disorders. Journal of the American Academy of Child & Adolescent Psychiatry 2007;46(11):1503‐26. [DOI] [PubMed] [Google Scholar]
Allen 2007
- Allen NB, Hetrick SE, Simmons JG, Hickie IB. Early intervention in depressive disorders in young people: the opportunity and the (lack of) evidence. Medical Journal of Australia 2007;187(7):s15‐7. [DOI] [PubMed] [Google Scholar]
Alvarez‐Jimenez 2011
- Alvarez‐Jimenez M, Parker AG, Hetrick SE, McGorry PD, Gleeson JF. Preventing the second episode: a systematic review and meta‐analysis of psychosocial and pharmacological trials in first‐episode psychosis. Schizophrenia Bulletin 2011;37(3):619‐30. [DOI] [PMC free article] [PubMed] [Google Scholar]
APA 2000
- American Psychological Association. Diagnostic and Statistical Manual of Mental Disorders (DSM‐IV‐TR). 4th Edition. Washington, DC: American Psychiatric Association, 2000. [Google Scholar]
Beck 1969
- Beck AT, Ward CH, Mendelssohn MJ, Erbaugh J. An inventory for measuring depression. Archives of General Psychiatry 1961;4:561‐71. [DOI] [PubMed] [Google Scholar]
Belsher 1988
- Belsher G, Costello CG. Relapse after recovery from unipolar depression: a critical review. Psychological Bulletin 1988;104(1):84‐96. [DOI] [PubMed] [Google Scholar]
Berndt 2000
- Berndt ER, Koran LM, Finkelstein SN, Gelenberg AJ, Kornstein SG, Miller IM, et al. Lost human capital from early‐onset chronic depression. American Journal Psychiatry 2000;157(6):940‐7. [DOI] [PubMed] [Google Scholar]
Birmaher 1996
- Birmaher B, Ryan ND, Williamson DE, Brent DA, Kaufman J, Dahl RE. Childhood and adolescent depression: a review of the past 10 years: part 1. Journal of the American Academy of Child & Adolescent Psychiatry 1996;35(11):1427‐39. [DOI] [PubMed] [Google Scholar]
Bridge 2007
- Bridge JA, Iyengar S, Salary CB, Barbe RP, Birmaher B, Pincus HA, et al. Clinical response and risk for reported suicidal ideation and suicide attempts in paediatric antidepressant treatment a meta‐analysis of randomized controlled trials. JAMA 2007;297(15):1683‐96. [DOI] [PubMed] [Google Scholar]
Cheung 2007
- Zuckerbrot RA, Cheung AH, Zuckerbrot RA, Jensen PS, Ghalib K, Laraque D, et al. Guidelines for adolescent depression in primary care (GLAD‐PC): II treatment and ongoing management. Pediatrics 2007;120:e1313‐26. [DOI] [PubMed] [Google Scholar]
Churchill 2010
- Churchill R, Moore THM, Davies P, Caldwell D, Jones H, Lewis G, et al. Mindfulness‐based ’third wave’ cognitive and behavioural therapies versus treatment as usual for depression. Cochrane Database of Systematic Reviews 2010, Issue 9. [DOI: 10.1002/14651858.CD008705] [DOI] [PMC free article] [PubMed] [Google Scholar]
Clarke 1990
- Clarke GN, Lewinsohn PM, Hops H, Seeley JR. Instructors Manual for the Adolescent Coping With Depression Course. Eugene, OR: Castalia Press, 1990. [Google Scholar]
Clarke 2005
- Clarke G, Debar L, Lynch F, Powell J, Gale J, O'Connor E, et al. A randomized effectiveness trial of brief cognitive‐behavioral therapy for depressed adolescents receiving antidepressant medication. Journal of the American Academy of Child & Adolescent Psychiatry 2005;44:888‐98. [PubMed] [Google Scholar]
Costello 2006
- Costello JE, Erkanli A, Angold A. Is there an epidemic of child or adolescent depression?. Journal of Child Psychology and Psychiatry 2006;47(12):1263–71. [DOI] [PubMed] [Google Scholar]
Cox 2012
- Cox GR, Callahan P, Churchill R, Hunot V, Merry SN, Parker AG, et al. Psychological therapies versus antidepressant medication, alone and in combination for depression in children and adolescents. Cochrane Database of Systematic Reviews 2012. [DOI] [PubMed] [Google Scholar]
Crown 2002
- Crown WH, Finkelstein S, Berndt ER, Ling D, Poret AW, Rush AJ, et al. The impact of treatment‐resistant depression on health care utilization and costs. Journal of Clinical Psychiatry 2002;63(11):963‐71. [DOI] [PubMed] [Google Scholar]
DerSimonian 1986
- DerSimonian R, Laird N. Meta‐analysis in clinical trials. Controlled Clinical Trials 1986;7:177‐88. [DOI] [PubMed] [Google Scholar]
Dobson 2008
- Dobson KS, Dimidjian S, Kohlenberg RJ, Rizvi SL, Dunner DL, Jacobson NS, et al. Randomized trial of behavioral activation, cognitive therapy, and antidepressant medication in the prevention of relapse and recurrence in major depression. Journal of Consulting and Clinical Psychology 2008;76(3):468‐77. [DOI] [PMC free article] [PubMed] [Google Scholar]
Fava 1998
- Fava GA, Rafanelli C, Grandi S, Canestrari R, Morphy MA. Six‐year outcome for cognitive behavioral treatment of residual symptoms for major depression. American Journal of Psychiatry 1998;155:1443‐5. [DOI] [PubMed] [Google Scholar]
Fisher 2010
- Fisher CA, Hetrick SE, Rushford N. Family therapy for anorexia nervosa. Cochrane Database of Systematic Reviews 2010, Issue 4. [DOI: 10.1002/14651858.CD004780.pub2] [DOI] [PubMed] [Google Scholar]
Frank 1991
- Frank E, Prien RF, Jarrett RB, Keller MB, Kupfer DJ, Lavori PW, et al. Conceptualization and rationale for consensus definitions of terms in major depressive disorder. Remission, recovery, relapse, and recurrence. Archives of General Psychiatry 1991;48:851‐5. [DOI] [PubMed] [Google Scholar]
Geddes 2003
- Geddes JR, Carney SM, Davies C, Furukawa TA, Kupfer DJ, Frank E, et al. Relapse prevention with antidepressant drug treatment in depressive disorders: a systematic review. Lancet 2003;361(9358):653‐61. [DOI] [PubMed] [Google Scholar]
Goodyer 2007
- Goodyer I, Dubicka B, Wilkinson P, Kelvin R, Roberts C, Byford S, et al. Selective serotonin reuptake inhibitors (SSRIs) and routine specialist care with and without cognitive behaviour therapy in adolescents with major depression: randomised controlled trial. BMJ 2007;335(7611):142. [DOI] [PMC free article] [PubMed] [Google Scholar]
Hamilton 1960
- Hamilton M. A rating scale for depression. Journal of Neurology, Neurosurgery & Psychiatry 1960;23:56‐62. [DOI] [PMC free article] [PubMed] [Google Scholar]
Harrington 1990
- Harrington R, Fudge H, Rutter M, Pickles A, Hill J. Adult outcomes of childhood and adolescent depression. 1. Psychiatric status. Archives of General Psychiatry 1990;47(5):465‐73. [DOI] [PubMed] [Google Scholar]
Hazell 2002
- Hazell P, O'Connell D, Heathcote D, Henry D. Tricyclic drugs for depression in children and adolescents. Cochrane Database of Systematic Reviews 2002, Issue 2. [DOI: 10.1002/14651858.CD002317] [DOI] [PubMed] [Google Scholar]
Healy 1997
- Healy D. The Anti‐Depressant Era. Massachusetts: Harvard University Press, 1997. [Google Scholar]
Hensley 2004
- Hensley PL, Nadiga D, Uhlenhuth EH. Long‐term effectiveness of cognitive therapy in major depressive disorder. Depression and Anxiety 2004;20(1):1‐7. [DOI] [PubMed] [Google Scholar]
Hetrick 2007
- Hetrick S, Merry S, McKenzie J, Sindahl P, Proctor M. Selective serotonin reuptake inhibitors (SSRIs) for depressive disorders in children and adolescents. Cochrane Database of Systematic Reviews 2007, Issue 3. [DOI: 10.1002/14651858.CD004851.pub2] [DOI] [PubMed] [Google Scholar]
Hetrick 2008
- Hetrick SE, Parker AG, Purcell R, Hickie I, Yung AR, McGorry PD. Early identification and intervention in depressive disorders: toward a clinical staging model. Journal of Psychotherapy and Psychosomatics 2008;77(5):263‐70. [DOI] [PubMed] [Google Scholar]
Hetrick 2012
- Hetrick SE, McKenzie JE, Cox GR, Simmons M, Merry SN. Newer generation antidepressants for depressive disorders in children and adolescents. Cochrane Database of Systematic Reviews 2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
Higgins 2008
- Higgins JPT, Altman DG, on behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 8: Assessing risk of bias in included studies. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Higgins 2008a
- Higgins JPT, Altman DG, On behalf of the Cochrane Statistical Methods Group and the Cochrane Bias Methods Group. Chapter 12: Interpreting results and drawing conclusions. In: Higgins JPT, Green S (editors). Cochrane Handbook for Systematic Reviews of Interventions Version 5.0.2 [updated September 2009]. The Cochrane Collaboration, 2008. Available from www.cochrane‐handbook.org.
Hoffman 2008
- Hofmann SG, Asmundson GJG. Acceptance and mindfulness‐based therapy: new wave or old hat?. Clinical Psychology Review 2008;28(1):1‐16. [DOI] [PubMed] [Google Scholar]
Jaffee 2002
- Jaffee SR, Moffitt TE, Caspi A, Fombonne E, Poulton R, Martin J. Differences in early childhood risk factors for juvenile‐onset and adult‐onset depression. Archives of General Psychiatry 2002;59(3):215‐22. [DOI] [PubMed] [Google Scholar]
Jarrett 2001
- Jarrett RB, Kraft D, Doyle J, Foster BM, Eaves GG, Silver PC. Preventing recurrent depression using cognitive therapy with and without a continuation phase: a randomized clinical trial. Archives of General Psychiatry 2001;58:381‐8. [DOI] [PMC free article] [PubMed] [Google Scholar]
Keller 1984
- Keller MB, Klerman GL, Lavori PW, Coryell W, Endicott J, Taylor J. Long‐term outcomes of episodes of major depression: clinical and public health significance. JAMA 1984;252:788‐92. [PubMed] [Google Scholar]
Keller 2005
- Keller MB, Ruwe FKL, Janssens CJJG, Sitsen JMA, Jokinen R, Janczewski J. Relapse prevention with gepirone ER in outpatients with major depression. Journal of Clinical Pharmacology 2005;25(1):79‐84. [DOI] [PubMed] [Google Scholar]
Kendler 2000
- Kendler KS, Thornton LM, Gardner CO. Stressful life events and previous episodes in the etiology of major depression in women: an evaluation of the "kindling" hypothesis. American Journal of Psychiatry 2000;157(8):1243‐51. [DOI] [PubMed] [Google Scholar]
Kennard 2006
- Kennard BD, Emslie GJ, Mayes TL, Hughes JL. Relapse and recurrence in paediatric depression. Child and Adolescent Psychiatric Clinics of North America 2006;15:1057‐79. [DOI] [PubMed] [Google Scholar]
Kennedy 2003
- Kennedy N, Abbott R, Paykel ES. Remission and recurrence of depression in the maintenance era: long‐term outcome in a Cambridge cohort. Psychological Medicine 2003;33:827‐38. [DOI] [PubMed] [Google Scholar]
Kessing 2004
- Kessing LV, Hansen MG, Andersen PK. Course of illness in depressive and bipolar disorders. Naturalistic study, 1994‐1999. British Journal of Psychiatry 2004;185:372‐7. [DOI] [PubMed] [Google Scholar]
Kessler 2005
- Kessler RC, Berglund P, Demler O, Jin R, Merikangas KR, Walters EE. Lifetime prevalence and age‐of‐onset distributions of DSM‐IV disorders in the National Comorbidity Survey replication. Archives of General Psychiatry 2005;62:593‐602. [DOI] [PubMed] [Google Scholar]
Kroll 1996
- Kroll L, Harrington R, Jayson D. Pilot study of continuation cognitive‐behavioral therapy for major depression in adolescent psychiatric patients. Journal of the American Academy of Child & Adolescent Psychiatry 1996;35(9):1156‐61. [DOI] [PubMed] [Google Scholar]
Lau 2004
- Lau MA, Segal ZV, Williams JM. Teasdale's differential activation hypothesis: implications for mechanisms of depressive relapse and suicidal behaviour. Behavior Research and Therapy 2004;42(9):1001‐17. [DOI] [PubMed] [Google Scholar]
Lewinsohn 1999
- Lewinsohn PM, Allen NB, Seeley JR, Gotlib IH. First onset versus recurrence of depression: differential processes of psychosocial risk. Journal of Abnormal Psychology 1999;108:483‐9. [DOI] [PubMed] [Google Scholar]
Ma 2004
- Ma SH, Teasdale JD. Mindfulness‐based cognitive therapy for depression: replication and exploration of differential relapse prevention effects. Journal of Consulting and Clinical Psychology 2004;72(1):31‐40. [DOI] [PubMed] [Google Scholar]
March 2004
- March J, Silva S, Petrycki S, Curry J, Wells K, Fairbank J et al (Treatment for Adolescents with Depression Team). Fluoxetine, cognitive‐behavioral therapy, and their combination for adolescents with depression: Treatment for Adolescents with Depression Study (TADS) randomized controlled trial. JAMA 2004;292(7):807‐20. [DOI] [PubMed] [Google Scholar]
Marlatt & Gordon 1985
- Marlatt GA, Gordon JR. Relapse Prevention: Maintenance Strategies in the Treatment of Addictive Behaviours. New York: Guildford, 1985. [Google Scholar]
Martell 2010
- Martell CR, Dimidjian S, Herman‐Dunn R. Behavioral Activation for Depression: A Clinician’s Guide. New York: Guilford, 2010. [Google Scholar]
McDermott 2010
- McDermott B, Baigent M, Chanen A, Fraser L, Graetz B, Hayman N et al. beyondblue Expert Working Committee. Clinical practice guidelines: depression in adolescents and young adults. beyondblue: the National Depression Initiative: Melbourne 2010.
McGorry 2006
- McGorry PD, Hickie IB, Yung AR, Pantelis C, Jackson HJ. Clinical staging of psychiatric disorders: a heuristic framework for choosing earlier, safer and more effective interventions. Australian and New Zealand Journal of Psychiatry 2006;40:616‐22. [DOI] [PubMed] [Google Scholar]
Melvin 2006
- Melvin GA, Tonge BJ, King NJ, Heyne D, Gordon MS, Klimkeit EA. Comparison of cognitive‐behavioral therapy, sertraline, and their combination for adolescent depression. Journal of the American Academy of Child & Adolescent Psychiatry 2006;45:1151‐61. [DOI] [PubMed] [Google Scholar]
Merry 2011
- Merry SN, Hetrick SE, Cox GR, Brudevold‐Iversen T, Bir JJ, McDowell H. Psychological and educational interventions for preventing depression in children and adolescents. Cochrane Database of Systematic Reviews 2011, Issue 12. [DOI: 10.1002/14651858.CD003380.pub3] [DOI] [PubMed] [Google Scholar]
NICE 2005
- National Institute for Health and Clinical Excellence (NICE). Depression in children and young people: identification and management in primary, community and secondary care. The British Psychological Society: Leicester, UK 2005. [PubMed]
Nobile 2003
- Nobile M, Cataldo GM, Marino C, Molteni M. Diagnosis and treatment of dysthymia in children and adolescents. CNS Drugs 2003;17(13):927‐46. [DOI] [PubMed] [Google Scholar]
Olver 2001
- Olver JS, Burrows GD, Norman TR. Third‐generation antidepressants: do they offer advantages over the SSRIs?. CNS Drugs 2001;15(12):941‐54. [DOI] [PubMed] [Google Scholar]
Post 1992
- Post RM. Transduction of psychosocial stress into the neurobiology of recurrent affective disorder. American Journal of Psychiatry 1992;149(8):999‐1010. [DOI] [PubMed] [Google Scholar]
Poznanski 1996
- Poznanski EO, Mokros HB. Children's Depressions Rating Scale, Revised (CDRS‐R) Manual. Los Angeles: Western Psychological Services, 1996. [Google Scholar]
Rapaport 2004
- Rapaport MH, Bose A, Zheng H. Escitalopram continuation treatment prevents relapse of depressive episodes. Journal of Clinical Psychopharmacology 2004;65(1):44‐9. [DOI] [PubMed] [Google Scholar]
RevMan 2011 [Computer program]
- The Nordic Cochrane Centre, The Cochrane Collaboration. Review Manager (RevMan). Version 5.1. Copenhagen: The Nordic Cochrane Centre, The Cochrane Collaboration, 2011.
Reynolds 1987
- Reynolds WM. Suicidal Ideation Questionnaire‐Junior.. Odessa, FL: Psychological Assessment Resources, 1987. [Google Scholar]
Rush 1998
- Rush AJ, Koran LM, Keller MB, et al. The treatment of chronicdepression, part 1: study design and rationale for evaluating thecomparative efficacy of sertraline and imipramine as acute, crossover,continuation, and maintenance phase therapies. Journal of Clinical Psychiatry 1998;59:589‐97. [PubMed] [Google Scholar]
Rutter 1995
- Rutter M, Smith DJ. Psychosocial Disorders in Young People: Time Trends and Their Causes. Chichester, England: John Wiley & Sons, 1995. [Google Scholar]
Segal 2010
- Segal ZV, Bieling P, Young T, MacQueen G, Cooke R, Martin L, et al. Antidepressant monotherapy vs sequential pharmacotherapy and mindfulness‐based cognitive therapy, or placebo, for relapse prophylaxis in recurrent depression. Archives of General Psychiatry 2010;67(12):1256‐64. [DOI] [PMC free article] [PubMed] [Google Scholar]
Shaffer 1983
- Shaffer D, Gould MS, Brasic J, Ambrosini P, Fisher P, Bird H, et al. A children's global assessment scale (CGAS). Archive of General Psychiatry 1983;40(11):1228‐31. [DOI] [PubMed] [Google Scholar]
Simon 2004
- Simon JS, Aguiar LM, Kunz NR, Lei D. Extended‐release venlafaxine in relapse prevention for patients with major depressive disorder. Journal of Psychiatric Research 2004;38(3):249‐57. [DOI] [PubMed] [Google Scholar]
Teasdale 2000
- Teasdale JD, Segal ZV, Williams JMG, Ridgeway VA, Soulsby JM, Lau MA. Prevention of relapse/recurrence in major depression by mindfulness‐based cognitive therapy. Journal of Consulting and Clinical Psychology 2000;68(4):615‐23. [DOI] [PubMed] [Google Scholar]
Thompson 2003
- Thompson KN, Conus PO, Ward JL, Phillips LJ, Koutsogiannis J, Leicester S, et al. The initial prodrome to bipolar affective disorder: prospective case studies. Journal of Affective Disorders 2003;77(1):79‐85. [DOI] [PubMed] [Google Scholar]
Vallis 1986
- Vallis MT, Shaw, BF, Dobson KS. The Cognitive Therapy Scale: Psychometric properties.. Journal of Consulting and Clinical Psychology 1986;54(3):381‐85. [DOI] [PubMed] [Google Scholar]
van Praag 1987
- Praag HM, Kahn RS, Asnis GM, Wetzler S, Brown SL, Bleich A, et al. Denosologization of biological psychiatry or the specificity of 5‐HT disturbances in psychiatric disorders. Journal of Affective Disorders 1987;13:1‐8. [DOI] [PubMed] [Google Scholar]
Vittengl 2007
- Vittengl JR, Clark LA, Dunn TW, Jarrett RB. Reducing relapse and recurrence in unipolar depression: a comparative meta‐analysis of cognitive‐behavioral therapy's effects. Journal of Consulting and Clinical Psychology 2007;75(3):475‐88. [DOI] [PMC free article] [PubMed] [Google Scholar]
Wantanabe 2007
- Watanabe N, Hunot V, Omori IM, Churchill R, Furukawa TA. Psychotherapy for depression among children and adolescents: a systematic review. Acta Psychiatrica Scandinavica 2007;116:84‐95. [DOI] [PubMed] [Google Scholar]
Weissman 2007
- Weissman MM, Markowitz JC, Klerman GL. Clinician’s Quick Guide to Interpersonal Psychotherapy. New York: Oxford University Press, 2007. [Google Scholar]
Weisz 2006
- Weisz JR, McCarty CA, Valeri SM. Effects of psychotherapy for depression in children and adolescents: a meta‐analysis. Psychological Bulletin 2006;132:132‐49. [DOI] [PMC free article] [PubMed] [Google Scholar]
WHO 2007
- World Health Organization. International Classification of Diseases and Related Health Problems 10th Revision (ICD‐10) (accessible online: http://apps.who.int/classifications/apps/icd/icd10online/). Geneva: WHO, 2007. [Google Scholar]
Yung 2003
- Yung AR, Phillips LJ, Yuen HP, Francey SM, McFarlane CA, Hallgren M, et al. Psychosis prediction: 12‐month follow up of a high‐risk ("prodromal") group. Schizophrenia Research 2003;60(1):21‐32. [DOI] [PubMed] [Google Scholar]