

Abstract
Examples of anaerobic oxidation of aldehydes in hydrothermal solutions are reported. The reaction using iron(iii) nitrate as the oxidant occurs under mild hydrothermal conditions and generates carboxylic acids in good yields. This method differs from previous studies which use atmospheric oxygen as the oxidant.
Efficient anaerobic oxidation of aldehydes using iron(iii) nitrate under hydrothermal conditions is reported.
Oxidation of aldehydes is considered one of the most important organic transformations. It is a fundamental reaction that is not only critical for biological metabolism but is also involved in the synthesis of chemicals during industrial processes.1,2 For example, aldehydes can be biologically oxidized to carboxylic acids by aldehyde dehydrogenases, which inactivates and detoxifies the aldehydes to allow for easier excretion from the body.3,4 Industrially, aldehyde oxidation has been used in the manufacturing of cosmetic products, plasticizers, fibers, and biomass-derived chemicals.5,6 Because of its widespread processes and applications, efficient and green methods for oxidizing aldehydes to carboxylic acids are a pressing need.
Traditionally, aldehydes are oxidized stoichiometrically, often using hazardous oxidants such as the Cr(vi)-based Jones reagent, the Ag(i)-based Tollen's reagent, the Cu(ii)-based Fehling's reagent, or permanganate-based catalysts.7–10 These conventional methods also generate stoichiometric amounts of waste by-products, which are often toxic and expensive to recycle.5 To alleviate these problems, chemists have focused on developing “greener” processes for catalytic oxidation of aldehydes. For example, previous studies have used water as an environmentally friendly solvent for oxidation of aldehydes,1,2,11–13 which prevents the involvement of harmful organic solvents. However, this method could suffer from the necessity for large amounts of additives and expensive or toxic catalysts. Recently, studies have focused on finding alternative catalysts that are more naturally abundant and low-cost. For example, metal salts such as iron and copper have been applied to the oxidation of aldehydes as catalysts in aqueous solutions.8,11
Currently, most investigations on aldehyde oxidations focus on aerobic oxidation, i.e., using molecular oxygen (O2) to oxidize aldehydes into the corresponding carboxylic acids.2,8,11,14 Autoxidation of aldehydes with O2 also takes place at ambient conditions, which usually involves a free radical chain reaction to form peracid, followed by the Baeyer–Villiger oxidation.15,16 In comparison, fewer studies have explored the anaerobic oxidation pathway for aldehydes, especially in catalyst-free aqueous environments. Hydrothermal systems, however, may provide a unique environment for anaerobic organic redox reactions. Our recent research on alcohols, carboxylic acids, and amides have shown that organic oxidations can readily occur in the presence of metal salts such as Cu(ii) under O2-absent hydrothermal conditions.17–19 In those reactions, water serves as a green solvent, while Cu(ii) acts as an efficient oxidant. The metal-promoted hydrothermal reactions also mimic natural geochemical processes on Earth, which provides the new “geomimicry” concept of using Earth-abundant metals as the oxidant/reductant for green organic reactions.18,20,21 In this study, we investigated the oxidation of aldehydes to carboxylic acids in an anaerobic and mild hydrothermal environment, using simple Cu(ii) and non-toxic Fe(iii) salts as the oxidizing agent. The optimal reaction condition and the substrate scope were both studied.
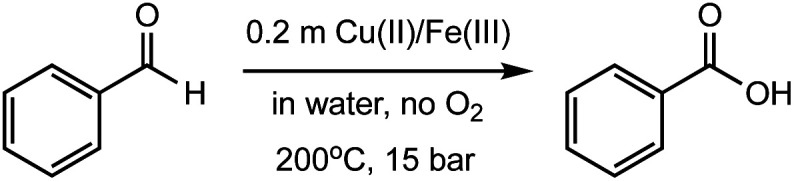
Hydrothermal experiments were conducted in sealed fused silica tubes under an aqueous condition of 200 °C and 15 bar (Psat, calculated using SUPCRT92)22 in the absence of O2, following a previously developed method.17,23,24 We started our investigation by examining the oxidation of benzaldehyde (compound 1) under hydrothermal conditions using various Cu(ii) and Fe(iii) salts, including CuCl2, CuSO4, Cu(OAc)2, Cu(NO3)2, FeCl3, Fe2(SO4)3, and Fe(NO3)3 (Table 1). In pure water without additives, only 4% of benzaldehyde reacted at 200 °C after 24 h, forming benzoic acid with a yield of ∼3% (entry 1). This slow reaction indicates that benzaldehyde is relatively inert under the anaerobic hydrothermal conditions, which is consistent with a previous study reporting <1% conversion for benzaldehyde at 250 °C after 36 h.25 In the presence of CuCl2, CuSO4, and Cu(OAc)2, both the benzaldehyde conversion (6–9%) and the acid yield (5–8%) at 200 °C were slightly increased after 24 h (entries 2–4). However, their effects were not as prominent as Cu(NO3)2, which increased the yield to 10% and 26% after 0.5 and 2 h, respectively (entries 5 and 7). NaNO3 was also tested and gave a yield of 10% after 2 h (entry 9), suggesting that the aldehyde oxidation could be driven by the nitrate ions, but not as strong as the copper–nitrate complex. In the presence of FeCl3 and Fe2(SO4)3, the acid yields were 10–13% at 200 °C after 24 h (entries 10 and 11), which were similar to those with CuCl2 and CuSO4. The most dramatic effect comes from Fe(NO3)3, which converted 64% of benzaldehyde with a boosted yield of 60% at 200 °C only after 0.5 h (entry 15). Increasing the reaction time to 2 h allowed Fe(NO3)3 to fully oxidize benzaldehyde, reaching the highest acid yield (98%, entry 17) among all the conditions studied. Halving the starting concentration of Fe(NO3)3 lowered the yield to 90% (entry 18), while decreasing the reaction temperature also reduced the yield significantly (entries 12–14). Interestingly, combining FeCl3 with NaNO3 exhibited a very high yield as that with Fe(NO3)3 (entries 19 and 20), which suggests the key of this aldehyde oxidation is having both Fe(iii) and nitrate ions present. Additionally, nitrates of redox-neutral metals such as Mg(NO3)2 and Ca(NO3)2 were also investigated (entries 21 and 22), which showed a significantly lower yield than that of Fe(NO3)3 or Cu(NO3)2. These results further indicate that both the redox metals and nitrate ions play an oxidizing role in this reaction.
Investigation of oxidation of benzaldehyde using copper(ii) and iron(iii) salts at different reaction conditions.
| ||||
|---|---|---|---|---|
| Entry | Condition | Additive | Conversion | Benzoic acid yielda |
| 1 | 200 °C, 24 h | None | 4% | 3% |
| 2 | 200 °C, 24 h | 0.2 m CuCl2 | 8% | 7% |
| 3 | 200 °C, 24 h | 0.2 m CuSO4 | 9% | 8% |
| 4 | 200 °C, 24 h | 0.2 m Cu(OAc)2 | 6% | 5% |
| 5 | 200 °C, 0.5 h | 0.2 m Cu(NO3)2 | 12% | 10% |
| 6 | 200 °C, 1 h | 0.2 m Cu(NO3)2 | 25% | 24% |
| 7 | 200 °C, 2 h | 0.2 m Cu(NO3)2 | 30% | 26% |
| 8 | 200 °C, 2 h | 0.1 m Cu(NO3)2 + 0.1 m NaNO3 | 15% | 13% |
| 9 | 200 °C, 2 h | 0.2 m NaNO3 | 11% | 10% |
| 10 | 200 °C, 24 h | 0.2 m FeCl3 | 14% | 13% |
| 11 | 200 °C, 24 h | 0.1 m Fe2(SO4)3 | 11% | 10% |
| 12 | 100 °C, 2 h | 0.2 m Fe(NO3)3 | 39% | 38% |
| 13 | 140 °C, 2 h | 0.2 m Fe(NO3)3 | 59% | 57% |
| 14 | 170 °C, 1 h | 0.2 m Fe(NO3)3 | 62% | 59% |
| 15 | 200 °C, 0.5 h | 0.2 m Fe(NO3)3 | 64% | 60% |
| 16 | 200 °C, 1 h | 0.2 m Fe(NO3)3 | >99% | 94% |
| 17 | 200 °C, 2 h | 0.2 m Fe(NO3)3 | >99% | 98% |
| 18 | 200 °C, 2 h | 0.1 m Fe(NO3)3 | >99% | 90% |
| 19 | 200 °C, 2 h | 0.2 m FeCl3 + 0.2 m NaNO3 | >99% | 97% |
| 20 | 200 °C, 2 h | 0.2 m FeCl3 + 0.6 m NaNO3 | >99% | 98% |
| 21 | 200 °C, 2 h | 0.2 m Mg(NO3)2 | 23% | 21% |
| 22 | 200 °C, 2 h | 0.2 m Ca(NO3)2 | 9% | 8% |
Yield determined by gas chromatography.
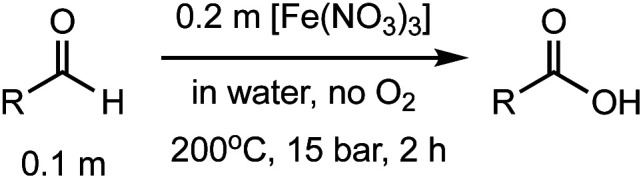
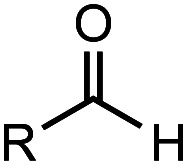
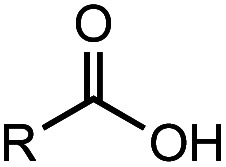
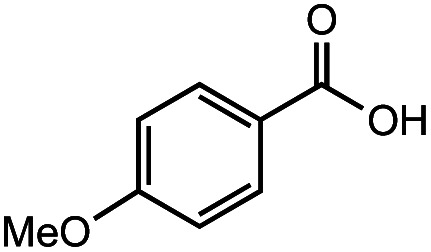
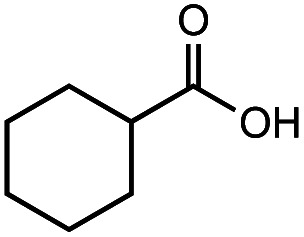
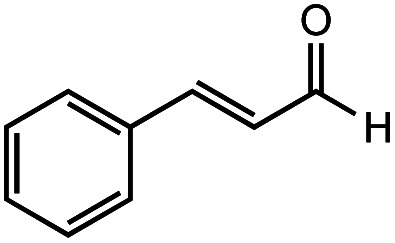
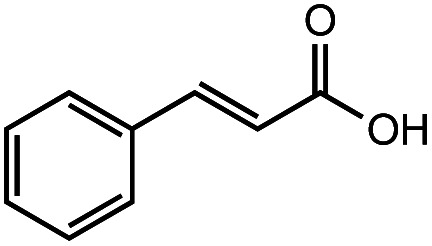
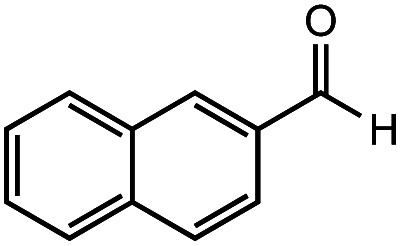
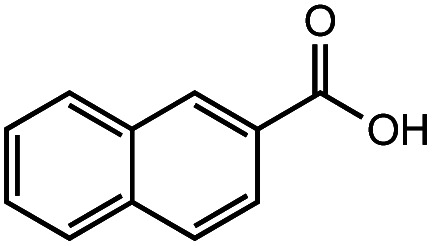
After identifying the optimal reaction conditions, we then tested with a group of 9 different functionalized aldehydes to examine the scope of this method (Table 2). All of these experiments were conducted using 2 equiv. Fe(NO3)3 at 200 °C and 15 bar for 2 h. Compared to the 98% yield from benzaldehyde, the aromatic aldehyde with an electron-withdrawing group (compound 2) gave a yield of 82%, whereas the ones with electron-donating groups such as p-tolualdehyde (compound 6) and 4-methoxybenzaldehyde (compound 7) gave a much lower yield of 54% and 46%, respectively. However, the relatively low yields from the electron-rich aldehydes are mainly due to the less-complete conversion of the starting material, which is expected to increase at longer reaction times. Halogen-substituted aldehydes such as 2-bromobenzaldehyde (compound 3) and 4-bromobenzaldehyde (compound 4) gave an acid yield of 47% and 66%, respectively, indicating the tolerance of this method for halogens and also a potential steric effect. Non-aromatic aldehydes such as hydrocinnamaldehyde (compound 5) and cyclohexanecarboxaldehyde (compound 8) also gave a moderate yield of 45% and 58%, respectively. Unsaturated aldehyde cinnamaldehyde (compound 9) also worked with this method, but the yield (30%) was lower than that of hydrocinnamaldehyde. The result suggests that the C C double bond potentially interferes with the oxidation, which seems consistent with the finding from another study on silver-catalyzed aldehyde oxidation.2 In addition, other aldehydes such as 2-naphthaldehyde (compound 10) resulted in a 41% yield under the same experimental conditions, demonstrating this method is also applicable to fused-ring aldehyde structures.
Investigation of substrate scope under an anaerobic hydrothermal condition of 200 °C, 15 bar after 2 h.
| ||||
|---|---|---|---|---|
| Comp# |
|
|
Conversion | Acid yielda |
| 1 |
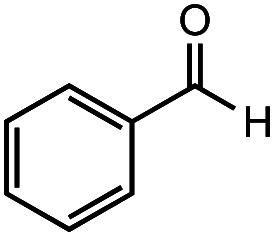
|
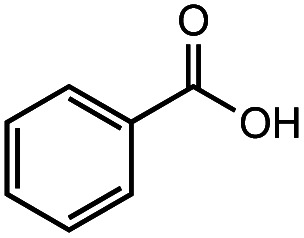
|
>99% | 98% |
| 2 |
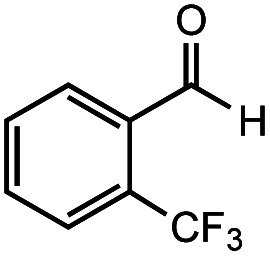
|
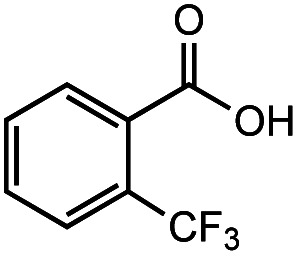
|
83% | 82% |
| 3 |
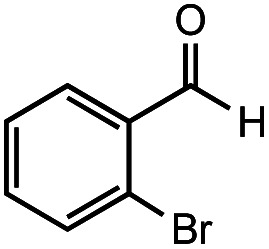
|
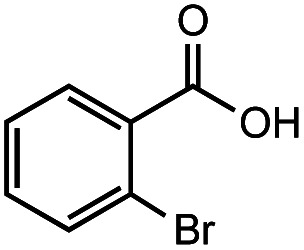
|
56% | 47% |
| 4 |
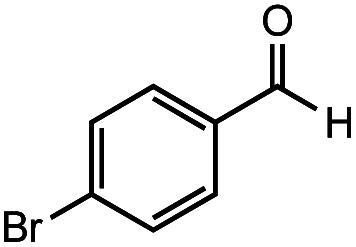
|
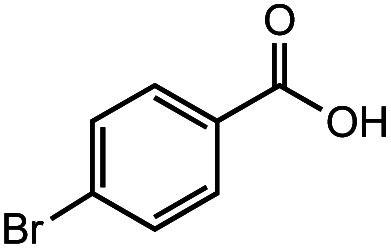
|
68% | 66% |
| 5 |
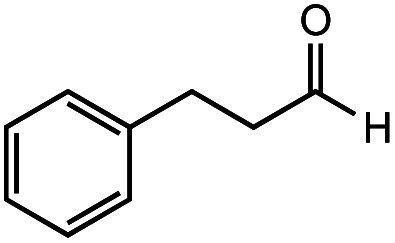
|
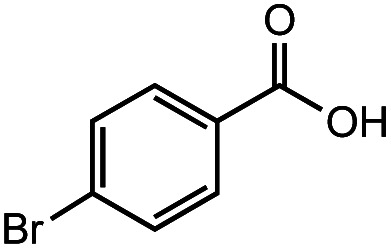
|
66% | 45% |
| 6 |
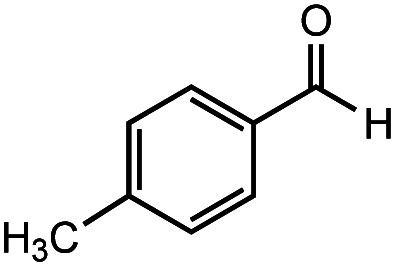
|
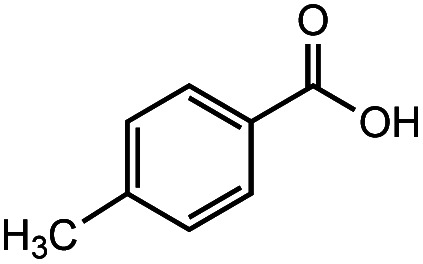
|
55% | 54% |
| 7 |
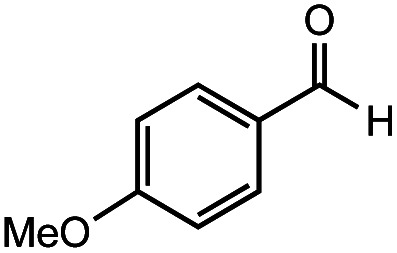
|
|
63% | 46% |
| 8 |
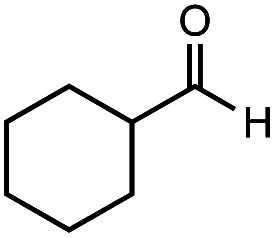
|
|
66% | 58% |
| 9 |
|
|
48% | 30% |
| 10 |
|
|
42% | 41% |
Yield determined by gas chromatography.
We also proposed a tentative mechanism for this Fe(iii)-involved aldehyde oxidation. As shown in Scheme 1, aldehydes are expected to undergo either the hydration followed by oxidation pathway or the oxidation followed by hydration pathway to form a radical cation intermediate. After losing a proton, the radical cation could be subsequently oxidized by Fe(NO3)3 to form a carbocation before the corresponding acid is produced. This reaction mechanism is also based on proposed mechanisms in previous studies, where Cu(ii) has been found as an oxidant for the oxidation of benzylalcohol and benzaldehyde under similar hydrothermal conditions.17,20 In the present study, however, the electron-donating –CH3 and –CH3O substituted benzaldehyde resulted in a lower conversion than that of the –CF3 substituted benzaldehyde (Table 2), which does not seem to support the formation of a positively charged intermediate. It is thus more likely that the hydrate formation is the rate-determining step, since the substituent effect on hydrate formation could be opposite to that on oxidation.20,26 More ring-substituted structures and detailed kinetics studies are needed to further pinpoint the reaction mechanisms.
Scheme 1. Proposed mechanism for anaerobic oxidation of aldehydes to carboxylic acids with Fe(NO3)3 as the oxidant.

In addition, we performed thermodynamic calculations for the anaerobic oxidation of aldehyde under a range of hydrothermal conditions. Using acetaldehyde as a model aldehyde, results show that the logarithm of equilibrium constant (log Keq) of oxidation of acetaldehyde to acetic acid in pure water was between 0 and 1 under the hydrothermal condition and slowly increased with temperature (Fig. S1, ESI†). In contrast, the log Keq for Fe(NO3)3-promoted acetaldehyde oxidation was orders of magnitude higher, which suggests the reaction is highly favorable when Fe(NO3)3 is present (Fig. S1, ESI†). Although the actual log Keq values for the aromatic aldehydes could be quite different from that of acetaldehyde, the increasing trend of log Keq by the addition of Fe(NO3)3 may hold true for other aldehyde structures. The results of geochemical modeling are consistent with our experimental observations.
In conclusion, we reported the first examples of oxidation of aldehydes with Fe(NO3)3 in water under anaerobic hydrothermal conditions. The oxidation is green and relatively clean, with relatively high yields of carboxylic acids achieved within hours. Different aldehyde structures were tested to show a good versatility of the reaction. This method also represents one of the “geomimicry” approaches that use Earth-abundant geological materials for organic synthesis and green chemistry applications. Future investigation on the reaction mechanism and the potential effects of other metal salts is anticipated.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We acknowledge the support from the National Science Foundation (OCE-2042213) and the Michigan Space Grant Consortium/NASA (80NSSC20M0124) for this work.
Electronic supplementary information (ESI) available: Detailed experimental materials and methods, thermodynamic calculations, and spectroscopy. See DOI: 10.1039/d1ra08444e
Notes and references
- Zhang Y. Cheng Y. Cai H. He S. Shan Q. Zhao H. Chen Y. Wang B. Green Chem. 2017;19:5708–5713. doi: 10.1039/C7GC02983G. [DOI] [Google Scholar]
- Liu M. Wang H. Zeng H. Li C.-J. Sci. Adv. 2015;1:e1500020. doi: 10.1126/sciadv.1500020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu X. Chai S. Wang P. Zhang C. Yang Y. Yang Y. Wang K. Cancer Lett. 2015;369:50–57. doi: 10.1016/j.canlet.2015.08.018. [DOI] [PubMed] [Google Scholar]
- Singh S. Brocker C. Koppaka V. Chen Y. Jackson B. C. Matsumoto A. Thompson D. C. Vasiliou V. Free Radical Biol. Med. 2013;56:89–101. doi: 10.1016/j.freeradbiomed.2012.11.010. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yu H. Ru S. Zhai Y. Dai G. Han S. Wei Y. ChemCatChem. 2018;10:1253–1257. doi: 10.1002/cctc.201701599. [DOI] [Google Scholar]
- Gorbanev Y. Y. Klitgaard S. K. Woodley J. M. Christensen C. H. Riisager A. ChemSusChem. 2009;2:672–675. doi: 10.1002/cssc.200900059. [DOI] [PubMed] [Google Scholar]
- Chakraborty D. Gowda R. R. Malik P. Tetrahedron Lett. 2009;50:6553–6556. doi: 10.1016/j.tetlet.2009.09.044. [DOI] [Google Scholar]
- Liu M. Li C. J. Angew. Chem. 2016;128:10964–10968. doi: 10.1002/ange.201604847. [DOI] [Google Scholar]
- Bal B. S. Childers W. E. Pinnick H. W. Tetrahedron. 1981;37:2091–2096. doi: 10.1016/S0040-4020(01)97963-3. [DOI] [Google Scholar]
- Menger F. M. Lee C. J. Org. Chem. 1979;44:3446–3448. doi: 10.1021/jo01333a051. [DOI] [Google Scholar]
- Yu H. Ru S. Dai G. Zhai Y. Lin H. Han S. Wei Y. Angew. Chem. 2017;129:3925–3929. doi: 10.1002/ange.201612225. [DOI] [PubMed] [Google Scholar]
- Kitanosono T. Masuda K. Xu P. Kobayashi S. Chem. Rev. 2018;118:679–746. doi: 10.1021/acs.chemrev.7b00417. [DOI] [PubMed] [Google Scholar]
- Dai P.-F. Qu J.-P. Kang Y.-B. Org. Lett. 2019;21:1393–1396. doi: 10.1021/acs.orglett.9b00101. [DOI] [PubMed] [Google Scholar]
- Liu K.-J. Fu Y.-L. Xie L.-Y. Wu C. He W.-B. Peng S. Wang Z. Bao W.-H. Cao Z. Xu X. He W.-M. ACS Sustainable Chem. Eng. 2018;6:4916–4921. doi: 10.1021/acssuschemeng.7b04400. [DOI] [Google Scholar]
- Clinton N. A. Kenley R. A. Traylor T. G. J. Am. Chem. Soc. 1975;97:3746–3751. doi: 10.1021/ja00846a031. [DOI] [Google Scholar]
- Vanoye L. Aloui A. Pablos M. Philippe R. Percheron A. Favre-Réguillon A. de Bellefon C. Org. Lett. 2013;15:5978–5981. doi: 10.1021/ol401273k. [DOI] [PubMed] [Google Scholar]
- Fu X. Jamison M. Jubb A. M. Liao Y. Aspin A. Hayes K. Glein C. R. Yang Z. Chem. Commun. 2020;56:2791–2794. doi: 10.1039/C9CC09825A. [DOI] [PubMed] [Google Scholar]
- Fu X. Liao Y. Aspin A. Yang Z. ACS Earth Space Chem. 2020;4:1596–1603. doi: 10.1021/acsearthspacechem.0c00150. [DOI] [Google Scholar]
- Liao Y. Aspin A. Fu X. Yang Z. ACS Earth Space Chem. 2021;5:2021–2031. doi: 10.1021/acsearthspacechem.1c00113. [DOI] [Google Scholar]
- Yang Z. Hartnett H. E. Shock E. L. Gould I. R. J. Org. Chem. 2015;80:12159–12165. doi: 10.1021/acs.joc.5b02109. [DOI] [PubMed] [Google Scholar]
- Bockisch C. Lorance E. D. Shaver G. Williams L. B. Hartnett H. E. Shock E. L. Gould I. R. Green Chem. 2019;21:4159–4168. doi: 10.1039/C9GC00636B. [DOI] [Google Scholar]
- Johnson J. W. Oelkers E. H. Helgeson H. C. Comput. Geosci. 1992;18:899–947. doi: 10.1016/0098-3004(92)90029-Q. [DOI] [Google Scholar]
- Yang Z. Fu X. JoVE. 2018:e58230. doi: 10.3791/58230. [DOI] [Google Scholar]
- Fu X. Liao Y. Glein C. R. Jamison M. Hayes K. Zaporski J. Yang Z. ACS Earth Space Chem. 2020;4:722–729. doi: 10.1021/acsearthspacechem.0c00009. [DOI] [Google Scholar]
- Fecteau K. M. Gould I. R. Glein C. R. Williams L. B. Hartnett H. E. Shock E. L. ACS Earth Space Chem. 2019;3:170–191. doi: 10.1021/acsearthspacechem.8b00130. [DOI] [Google Scholar]
- Wiberg K. B. Richardson W. H. J. Am. Chem. Soc. 1962;84:2800–2807. doi: 10.1021/ja00873a031. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.