

Abstract
Changes in the regulatory landscape of chemical safety assessment call for the use of New Approach Methodologies (NAMs) including read-across to fill data gaps. One critical aspect of analogue evaluation is the extent to which target and source analogues are metabolically similar. In this study, a set of 37 structurally diverse chemicals were compiled from the EPA ToxCast inventory to compare and contrast a selection of metabolism in silico tools, in terms of their coverage and performance relative to metabolism information reported in the literature. The aim was to build understanding of the scope and capabilities of these tools and how they could be utilised in a read-across assessment. The tools were Systematic Generation of Metabolites (SyGMa), Meteor Nexus, BioTransformer, Tissue Metabolism Simulator (TIMES), OECD Toolbox, and Chemical Transformation Simulator (CTS). Performance was characterised by sensitivity and precision determined by comparing predictions against literature reported metabolites (from 44 publications). A coverage score was derived to provide a relative quantitative comparison between the tools. Meteor, TIMES, Toolbox, and CTS predictions were run in batch mode, using default settings. SyGMa and BioTransformer were run with user-defined settings, (two passes of phase I and one pass of phase II). Hierarchical clustering revealed high similarity between TIMES and Toolbox. SyGMa had the highest coverage, matching an average of 38.63% of predictions generated by the other tools though was prone to significant overprediction. It generated 5,125 metabolites, which represented 54.67% of all predictions. Precision and sensitivity values ranged from 1.1–29% and 14.7–28.3% respectively. The Toolbox had the highest performance overall. A case study was presented for 3,4-Toluenediamine (3,4-TDA), assessed for the derivation of screening-level Provisional Peer Reviewed Toxicity Values (PPRTVs), was used to demonstrate the practical role in silico metabolism information can play in analogue evaluation as part of a read-across approach.
Keywords: in silico tools, metabolism, read-across, TIMES, Meteor Nexus, OECD Toolbox, CTS, SyGMa, BioTransfomer, PPRTV
1.0. Introduction
Read-across is a well-established data gap filling technique within analogue and category approaches [1–3]. Much technical guidance already exists for developing read-across assessment though acceptance remains an ongoing issue [3]. Previous reviews describing read-across frameworks have discussed the steps involved in developing a read-across including identifying analogues and assessing their suitability [1–4]. One of the considerations that comes through from many of these frameworks is the importance of evaluating the metabolic similarity of the source analogues relative to the target [5]. Indeed, whilst there is a qualitative description of the sorts of information that informs metabolism as detailed in the OECD guidance on grouping [6], challenges remain in terms of the level of information required to justify metabolism similarity and how. A case in point was that from Ball et al., [7]; who found that metabolic similarity was a critical component in their unsuccessful read-across justification to waive the need for a screening level prenatal development study. Recent work over the last few years has revisited means of identifying analogues with the recognition that chemical structural similarity is insufficient to identify and evaluate analogue suitability [8,9] but a systematic means to really explore this is rather lacking. Indeed, metabolic dissimilarity can lead to conflicts in endpoint data for structurally similar substances e.g. a difference in metabolism was the reason tributylamine was not included in the OECD tertiary amines category [10]. Probably the most recent advances in this direction are those from the Mario Negri Institute and the Laboratory of Mathematical Chemistry (LMC). Gadaleta et al., (2020) from the Mario Negri Institute proposed an automated read-across workflow to facilitate evaluation of metabolic similarity in conjunction with physicochemical similarity and structural similarity [11]. Candidate source analogues for a target of interest are identified from a source database of substances with associated endpoint point data. In their publication, two databases were utilised for illustration, the DILrank dataset, a collection of 1,036 FDA-approved drugs divided into four classes according to their potential for causing drug-induced liver injury (DILI) [12,13] and the EPA’s Toxicity Reference Database (ToxRefDB) [14] which includes in vivo study data for 667 substances. Candidate analogues were then searched for on the basis of 3 similarity considerations; up to 10 analogues for structural similarity and up to 5 analogues each for metabolic and biological similarity. Structural similarity relied on using MACCS fingerprints [15] and computing a Tanimoto coefficient [16] whereas biological similarity relied on a weighted biological similarity metric proposed by Russo et al [17] underpinned by High Throughput Screening (HTS) assay data taken from PubChem (https://pubchemdocs.ncbi.nlm.nih.gov/pug-rest) and compiled into biological binary fingerprints. The metabolic similarity component used one specific metabolism tool, Systematic Generation of Metabolites (SyGMa), arguably chosen more for pragmatic and practical reasons rather than from any systematic evaluation of its performance. In this case, one cycle of Phase I metabolism was considered and a metabolic similarity (MS) score was calculated based on the number of common and exclusive metabolic pathways for a target-analogue pair. Analogues returned could additionally be filtered based on common scaffolds or functional groups. The 3 sets of analogues would then be combined together and ranked based on their presence in one of more list; for example an analogue found in all three lists would be assigned a maximum suitability. In the final step, the read-across prediction would be made by averaging the activities of analogues included in the combined similarity list. The Laboratory of Mathematical Chemistry (LMC), University As. Zlatarov, Bulgaria [18,19] proposed a framework within the OECD Toolbox to provide an end-user with a means to compare analogues on the basis of their simulated maps. This makes use of the Tissue Metabolism Simulator (TIMES) platform which already has this capability for comparing experimental metabolic maps with predicted ones for a specific chemical of interest. In addition, the newly developed Metabolic similarity functionality implemented in TIMES itself allows simulated metabolic maps to be compared with respect to different similarity criteria. The similarity criteria include commonality of simulated transformations, commonality in reactivity or structural pattern as well as generation of common metabolites. For more details of the specific criteria and how they can be applied, the reader is referred to Kuseva et al., 2021 [20]. However, of note was a specific case study where two structurally similar substances conflicted with respect to their Ames mutagenicity outcomes. Chemical 1 – 2-propenylurea, was Ames positive whereas Chemical 2 – 2-propene-1-amine, was Ames negative. These chemicals were additionally mechanistically similar with respect to their interactions with DNA. Chemical 2 was a metabolite of chemical 1 and both produced a common active metabolite (2-propenal). Closer examination of their metabolic pathways identified that there were differences in the quantity of their common reactive metabolite (chemical 1 produced far more of the active metabolite compared with chemical 2) which could plausibly explain the discordant Ames outcomes. The analysis showed how important it is to evaluate the metabolic dissimilarity between candidate analogues.
Our motivation is to generalise a similar approach and incorporate this as part of the overall Generalised Read-Across (GenRA) workflow that evaluates analogues on the basis of both structural and/or biological properties (bioactivity) to make toxicity predictions [21]. However, as a first step we needed to gain a better understanding of the coverage and performance of existing metabolism prediction tools on the basis of a set of substances of interest. The selected metabolism predictions tools considered in this evaluation were Systematic Generation of Metabolites (SyGMa), Meteor Nexus, BioTransformer, Tissue Metabolism Simulator (TIMES), OECD Toolbox, and Chemical Transformation Simulator (CTS). Two of these tools are commercial in nature (Meteor Nexus, TIMES) whereas the remainder are publicly available and/or open source. Our aim was to do a targeted performance assessment on our set of chemicals. Targeted in the sense that these substances were additionally being tested through an in vitro primary human hepatocyte screen to determine intrinsic clearance and the metabolites were then being identified using liquid-chromatography mass spectrometry as part of a non-targeted/suspect screening approach. A separate analysis is still ongoing to process this data stream with the intention of comparing the in vitro results with those from in silico tools (Boyce et al, in prep). Here, a dataset of 37 chemicals from the ExpoCast project [22] were compiled and profiled through the complement of different in silico metabolism tools to generate metabolite predictions. The ToxCast library was used to capture some diversity in the types of chemicals as well as their uses and occurrence in the environment [22]. The predicted metabolites along with their monoisotopic masses and molecular formulae were used as part of the non-targeted analysis to generate a suspect screening list. The substances were searched for in the literature to find articles reporting any experimental studies where metabolism had been measured. Metabolites identified by in vitro human metabolism studies were ideally prioritised over other studies but metabolites identified by in vitro or in vivo studies using humans, rats, mice or dogs were included. The reported metabolites in these studies were compared with the predicted metabolites. Based on the inferences from the performance and scope gained from the outputs from the different tools, we applied one or more of these tools to a case study chemical that was being evaluated as part of an existing Provisional Peer Review Toxicity Value (PPRTV) assessment [23]. In the report, 3,4-TolueneDiamine (3,4-TDA) was being subjected to a read-across approach and in silico metabolism tools were one facet of evaluating candidate analogues. Herein, we summarise aspects of that part of the assessment to show how metabolic similarity between substances can be compared in an objective manner using a fingerprint representation of transformations either those provided by the tools themselves or by constructing a bespoke transformation profile using the structural information from the parent and metabolites themselves. The latter whilst only being illustrated for this substance is intended as a template for how this approach might be generalised and applied at scale. It complements efforts by Gadaleta et al., [11] and offers a quantitative basis of comparison further to what is proposed by the LMC group as far as what has been published and implemented in the OECD Toolbox [18] and TIMES [19,20].
It is worth noting that metabolism tools and approaches to predicting metabolism have been discussed in some detail in excellent reviews or indeed by the tool developers themselves in primary articles. Here we summarise the main conceptual basis of each of these different tools. That said, our study draws attention to the complement of different tools and which tools or combination of tools best meets our use contexts.
Examples of papers of particular note include reviews and primary work by Kirchmair and colleagues [24–26].
The main objectives of the study are to:
Compile a set of ToxCast chemicals of interest
Run predictions for the selected chemicals through a selection of (liver) metabolism prediction tools
Calculate Molecular Formula (MF), Monotonic Mass (MW) and compile metabolism predictions for the purposes of a related analysis of non-targeted screening using a suspect screening list
Review the literature to find articles reporting any metabolism studies to extract information on experimental metabolites generated (in vitro or in vivo in human, rat, mice, dog species) for the 37 substances.
Compare and contrast overall performance and coverage of the predictions relative to those reported in the literature on the basis of sensitivity/specificity on a ‘global’ basis as well as on a ‘local’ basis using predefined chemical clusters. Global being, overall performance for the entire dataset rather than subset (‘local’) within smaller chemical groups.
Demonstrate application of the metabolism predictions for a case study chemical that has undergone assessment by a read-across approach for which metabolic similarity was a key consideration.
Figure 1 shows a workflow for how the study fits into a larger study being undertaken. The overall aim of the broader study is to investigate the concordance between in vivo, in vitro and in silico metabolism information and how these information streams can be integrated to support the evaluation of analogues for read-across. Specifically, new in vitro data is being generated through in vitro human hepatocyte experiments to determine intrinsic clearance. Analytical liquid-chromatography spectroscopy is being applied for the detection of molecular species and non-targeted analysis for metabolite identification.
Figure 1.

Workflow of the larger study of which this study focuses on the components marked in red text.
1.1. Selected metabolism tools
1.11. BioTransformer
BioTransformer (https://bitbucket.org/wishartlab/biotransformer3.0jar/downloads/) was developed by the Wishart lab of the University of Alberta and includes a metabolite prediction tool (BMPT) and metabolite identification tool (BMIT) (Djombou-Feunang et al, 2019). As stated in Djoumbou Feunang et al. [27], the BMPT consists of five independent prediction modules: Enzyme Commission-based (EC-based) transformer, CYP450 (phase 1) transformer, Phase II transformer, human gut microbial transformer, and environmental microbial transformer. For the prediction of metabolites, BioTransformer implements two approaches, a rule-based or knowledge-based approach, and a machine learning approach. BioTransformer’s knowledge-based system consists of three major components: (1) a biotransformation database (called MetXBioDB) containing detailed annotations of experimentally confirmed metabolic reactions, (2) a reaction knowledgebase containing generic biotransformation rules, preference rules, and other constraints for metabolism prediction, and (3) a reasoning engine that implements both generic and transformer-specific algorithms for metabolite prediction and selection. The BMPT machine learning system uses a set of random forest and ensemble prediction models for the prediction of CYP450 substrate selectivity, and for the Phase II filtering of molecules. BioTransformer’s Metabolite Identification Tool builds on the BMPT to identify specific metabolites using mass spectrometry (MS) data, namely accurate mass or chemical formula information.
1.12. Meteor Nexus
Meteor Nexus is a commercially available metabolite prediction software that is part of a larger suite of tools developed by Lhasa Ltd. Meteor generates metabolites using a knowledge-based expert system, which identifies structural patterns within the parent compound and correlates these patterns to encoded biotransformations. Based on end-user needs, metabolite predictions can be generated using one of three algorithms: Absolute/Relative reasoning, Static Scoring, or Site of Metabolism. The Site of Metabolism (SoM) algorithm is the default setting. This builds upon the Static Scoring approach by modifying the static scores to account for structural differences between the parent compound and experimentally observed transformations. The Static Scoring algorithm assigns a likelihood statistic to each biotransformation, which is carried forward to predict the probability of a metabolite’s formation. A database of experimentally observed biotransformations is used to determine the ‘Occurrence Ratio’ of a biotransformation (i.e., the ratio between a specific biotransformation’s occurrences relative to the total number of biotransformations), which is multiplied by 1,000 to return the ‘Static Score’. As part of the algorithm, potential transformations are identified for the parent compound by using a database of experimentally observed biotransformations. These potential transformations are used to construct SoM fingerprints by iteratively constructing substructures ranging from 0 to 8 bonds from the SoM. A new subset of biotransformations are then selected from the database based on similarities between the SoM fingerprints and molecular mass (i.e., a ‘Molecular Mass Similarity Threshold’ set to a default of 70% of the parent’s molecular mass). Tanimoto coefficients are then calculated between parent fingerprint and the matched compounds, and the top N matches are preserved (N is set to 8 as default but can be adjusted between 6 – 12). The top matches are checked for the predicted biotransformation, and the Static Score is adjusted up or down depending on whether the biotransformation is observed for that compound.
Metabolites predicted by the SoM scoring are further filtered using ‘Relative’ of ‘Top N’ filters. The ‘Relative’ filter drops metabolites below a set percentage of the maximum score within that prediction grouping, and the ‘Top N’ filter drops metabolites that are outside of the top N statics scores (‘Top N’).
1.13. Chemical Transformation Simulator (CTS)
The Chemical Transformation Simulator (CTS) (https://qed.epacdx.net/cts/gentrans/input/) is a transformation prediction WebApp developed and maintained by the US EPA to predict transformation (abiotic and biological) and physiochemical properties of organic compounds. The simulator consists of three modules: Chemical Editor (CE), Physicochemical Properties (PCP) module, and Reaction Pathway Simulator (RPS). RPS provides a selection of reaction libraries that allows the user to generate predictions under specified conditions. These reaction libraries include abiotic hydrolysis, abiotic reduction, direct photolysis, and human phase 1 metabolism. The EPA maintains these libraries, with exception of the human phase 1 metabolism, which was developed by ChemAxon and represents 156 cytochrome p450 biotransformations. The reaction libraries provide a rule-based system for generating predictions, and further refine the results by identifying likely metabolites through predicted reaction rate. The generation of predictions and assignment reaction rates are handled through Metabolizer, which is a plugin for ChemAxon. As part of Metabolizer’s algorithm, a ranking between 1–7 is assigned to each reaction (1 representing a slow reaction and 7 representing the fastest) and the “ %production” of each transformation is determined be comparing the reaction rate to all other competing reactions. This calculation is propagated further to determine “%accumulation”, which represents the relative rate at which the compound is formed minus the rate at which it is converted to additional species. Compounds with a global “%accumulation” of at least 10% are considered “Likely” and all other transformations are considered “Unlikely”.
1.14. TIMES
Tissue Metabolism Simulator (TIMES) is a commercially available toxicity prediction software developed by the Laboratory of Mathematical Chemistry (LMC) at Bourgas ‘Prof. Assen Zlatarov’ University, Bulgaria [28]. Substances are profiled by a set of alerts underpinned by local QSAR models. Metabolism is simulated irrespective of whether alerting groups are identified in the target parent substance itself. The parent and all its transformation products are subsequently profiled for their toxicity potential by a set of alerts underpinned by local QSAR models. The metabolism modules are system and endpoint specific (e.g., mammalian lung, rat liver) and use a hybrid approach to predict metabolite maps. The approach includes a knowledge-based expert system to predict biotransformations, and optimisation procedures to adjust the transformation probabilities in a way to reproduce documented (observed) metabolism and metabolic activation of chemicals (endpoint specific data). In this respect, probabilities of transformations should be assumed to be correlated with formal rate constants of chemical reactions. When constructing metabolite maps, each biotransformation encoded in the module is applied to the starting molecule in order of their assigned probability. This process occurs iteratively for each predicted metabolite and generates branches with compounding probabilities for each biotransformation required to reach the predicted metabolite. Controlled propagation of a metabolic map terminates when a minimum probability of occurrence of a metabolite is reached. In turn, the probability of occurrence of a metabolite is estimated as a product of the probabilities of the transformations from the parent chemical to the considered metabolite. Quantitative assessment of feasibility of transformations confines the propagation of the generated metabolic maps and thus, results in the simulation of a manageable number of metabolites that are subsequently used for QSAR predictions within the same modelling platform. The knowledge-base of each metabolism module comprise biotransformations that include a parent fragment, transformation product, forbidden fragments, and probability. The parent fragment and transformation product define the structure change that the parent molecule will undergo, and the forbidden fragments act as inhibitory masks that prevent application of the transformation if these structures are present in the parent. Probabilities are assigned 1 for non-rate determining transformations (i.e., abiotic transformations of highly reactive groups or intermediates), whereas rate-determining transformations are assigned probabilities based on the above described optimisation procedure and adjusted by expert-knowledge.
1.15. OECD QSAR Toolbox: Metabolism Profilers
The OECD QSAR Toolbox is a publicly available hazard assessment software developed by LMC and distributed by OECD (Organisation for Economic Co-operation and Development) and ECHA (European Chemical Agency) [29]. The software was designed to aid in chemical risk assessment by filling data gaps through in silico predictions, which are generated via read-across, trend analysis, and/or QSAR models. For purposes of predicting metabolites for the target substance, only the ‘Input’ and ‘Profiling’ features of the full Toolbox workflow are used. As part of the profiling step, the Toolbox includes nine metabolism simulation modules developed by LMC and five databases of observed metabolic maps. These metabolism modules are donated from the commercially available TIMES and CATALOGIC software but do exhibit differences in the biotransformation reactions used to generate metabolites (e.g., the in vivo Rat metabolism simulator within TIMES (v.2.30.1.11) and QSAR Toolbox (v4.5) use 622 and 671 generalised biotransformation reactions, respectively). The simulation of possible metabolites, both in the commercial (TIMES/CATALOGIC) and freely available (Toolbox) software is tissue-specific. Additionally, the QSAR Toolbox does not generate phase II metabolites, as the main purpose of the metabolism modules within the Toolbox are to generate potentially active metabolites (i.e. Phase I metabolites). Then, endpoint specific knowledge (profiles) are applied for searching alerting functionalities in the up- and downstream comparisons against source analogues.
1.16. Systematic Generation of potential Metabolites (SyGMa)
Systematic Generation of potential Metabolites (SyGMa) is a knowledge-based metabolite prediction software developed in 2008 by Lars Ridder and Markus Wagener [30]. The knowledge-base comprises 144 reaction rules, which were generated through a combination of expert knowledge and statistical analysis of phase I and phase II human metabolic reactions. Each prediction is accompanied by a probability score (i.e., SyGMa score), which represents the accuracy of the associate rule (e.g., the rule associated with primary alcohol oxidation correctly predicted 58 of a total of 85 and was assigned a SyGMa score of ~68%). SyGMa is available as a Python package and provides user control over which rulesets are applied (phase I or phase II) to parent compounds.
2.0. Materials and Methods
2.1. Chemical structure data
A selection of 37 structurally diverse chemicals from the ExpoCast project were used in this study. Chemical information including DSSTox Substance ID (DTXSID), CAS registry number, Simplified Molecular Input Line Entry System (SMILES), QSAR-ready SMILES [31] and International Chemical Identifier Key (InChIKey) were queried against the EPA CompTox Chemicals Dashboard [32,33] using the batch search capability and downloaded as a CSV file. The InChIKey is a 27 character hashed version of InChI which is suited for Internet and database querying and is based on a SHA-256 hash of the InChI character string. The RDKit package within Python (version 2020.03; Landrum; rdkit.org) was used to convert the information in the CSV files into required input formats for processing by the different metabolism prediction tools.
2.2. Profiling substances through the metabolism prediction software
The RDKit package in Python was used to convert QSAR-ready SMILES into chemical table files — .mol files for each starting substance and a SDF containing data on all substances. These files were used to read chemical information into four of the metabolite prediction software tools: BioTransformer, Chemical Transformation Simulator (CTS), and Nexus Meteor (Meteor). SyGMa, is available as a Python package (https://pypi.org/project/SyGMa/) and predictions for this software were generated by directly reading the QSAR-ready SMILES. Table 1 summarises the six in silico metabolism tools used in the study.
Table 1.
Summary of the six in silico metabolism tools used in this study
| Tool Name | SyGMa | TIMES | OECD Toolbox | Nexus (Meteor) | BioTransformer | CTS |
|---|---|---|---|---|---|---|
| Developer | Lars Ridder lars.ridder@esciencecenter.nl | Laboratory of Mathematical Chemistry (LMC) | LMC, OECD, ECHA | Lhasa Ltd | Wishart Group | EPA |
| Availability | Freely available under GNU GPL | Commercial | Freely available | Commercial | Freely available under GNU LGPL | Freely available |
| Install via pip install SyGMa or https://pypi.org/project/SyGMa/ | http://oasis-lmc.org/products/software/times.aspx | qsartoolbox.org | https://www.lhasalimited.org/products/meteor-nexus.htm | https://bitbucket.org/wishartlab/biotransformer3.0jar/src/master/ | https://qed.epa.gov/cts/ | |
| Interface | API | GUI | GUI/WebAPI locally deployed | GUI (Single substance)/CLI (Batch) | API/CLI | Web GUI/WebAPI : https://qed.epa.gov/cts/rest/#/ |
| Implementation Language | Python | Delphi | Delphi | Java | Java | |
| Operating System | OS Independent | Windows | Windows | Windows | OS Independent | OS Independent |
| Reference | https://doi.org/10.1002/cmdc.200700312 | https://doi.org/10.1002/cmdc.200700312 | ||||
| Customisable metabolism options | Yes | Yes | No | Yes | Yes | Limited |
| Available options | Human, Rat (Liver) | Rat liver (in vivo), Rat liver (S9, in vitro), Lung (mammal), Gut (mammal), Abiotic, Skin (mammal) | Rat liver (in vivo), Rat liver (S9, in vitro), Lung (mammal), Gut (mammal), Abiotic, Skin (mammal) | Mammals (Dog, Mouse, Rat, Human) | Human (Liver, Gut), Microbial | Human |
Due to differences in software features, additional ad hoc steps were needed to be taken for selected software. For the CTS web application, only ten substances could be processed at a time, such that four SD files were needed. For the TIMES software, the rat (S9, in vitro) and rat (in vivo) modules provided separate readouts, so these results were processed separately. The two metabolism modules provided in the OECD Toolbox could be combined into a single report, but for consistency with the TIMES software, the results from the rat (S9, in vitro) and rat (in vivo) were processed separately. For Meteor Nexus, a command line batch processing was possible but only for individual MDL MOL files. Indeed, Meteor is only able to process up to 100 MDL MOL files at a time. Table 2 provides a description of relevant information and settings used for the prediction software.
Table 2:
Settings used to process the 37 substances through the selected metabolism prediction tools
| Model (version) | Availability | Module/Species | Prediction Settings | Input | Output |
|---|---|---|---|---|---|
| BioTransformer (3.0) |
Free (http://biotransformer.ca ) |
Human | Cyp450: 2 steps Phase II: 1 step |
SDF | .csv |
| CTS (1.0) |
Free (https://qed.epacdx.net/cts/ ) |
Human Phase 1 Metabolism | Maximum number of generations: 3 | SDF | .csv (All) |
| Meteor (3.1.0, Nexus 2.2.2) |
Commercial (https://www.lhasalimited.org/ ) |
Mammal | Default | .mol | .xls |
| Toolbox (4.5) |
Free (https://qsartoolbox.org/ ) |
Rat (S9, in vitro), Rat (in vivo) |
Default | .txt | .csv |
| TIMES (2.30.1.11) |
Commercial (http://oasis-lmc.org ) |
Rat (S9, in vitro), Rat (in vivo) | Default | .txt | .txt |
| SyGMa (1.1.1) |
Free (https://sygma.readthedocs.io ) |
Human | Phase I: 2 steps Phase II: 1 step |
QSAR-ready SMILES | Python Data Structure |
2.3. Literature Review
Experimentally confirmed metabolites were identified by querying structured databases and the literature for relevant studies for each of the 37 parent substances. The search first targeted selected databases with curated lists of reported metabolites. The Human Metabolome Database (https://hmdb.ca/), EPA CompTox Chemicals Dashboard (https://comptox.epa.gov/dashboard) [33], and Drug Bank (https://go.drugbank.com/) were used for this initial search since these databases are manually curated and provide literature citations for reported biotransformations. If no hits were retrieved from these database queries, a search in Pubmed for relevant literature using the AbstractSifter (v4) [34] was undertaken. Queries using AbstractSifter were performed by combining chemical identifiers (chemical name and DTXSID) with relevant subject queries keywords (metabolite, metabolism, pharmacokinetics, clearance, and excretion). Articles from the resulting query were rank ordered based on the total frequency count of three sifter terms present within each abstract. The sifter terms were a combination of the chemical identifiers and subject query keywords used to generate the search, and the final selection was optimised for each parent compound to prioritise relevant articles (i.e., articles that generated and/or reported novel metabolites). To confirm relevancy, each article identified by AbstractSifter was manually reviewed. If no suitable primary articles were sourced from the databases or the AbstractSifter query, then both Google Scholar and Web of Science were manually searched using the same terms. At all steps of the literature review process, metabolites were sourced from primary articles which included in vitro and in vivo data generated from different species (dog, bovine, mouse, rat, and human). In cases where multiple primary articles were identified with overlapping pathways, the most recent articles with rat and human metabolism were recorded. The full list of articles per substances are captured in the Supplementary information (see Table 1 Supplementary Information).
2.4. Processing reported Metabolism data
Experimentally reported metabolites from the literature were stored in the DSSTox database [32] by drawing the structures into the DSSTox ChemReg application (EPA’s internal chemical registration system), which generates chemical information from the drawn structures and assigns chemical identifiers (DTXSID and DTXCID). Transformation relationships were captured using ‘Predecessor’ and ‘Successor’ annotations, and the associated publications were included in the comments section. In cases where Markush structures (these describe a compound class by generic notation) were reported, the Markush structure was stored in the DSSTox Database and child structures were enumerated using MarvinJS (https://chemaxon.com/products/marvin-js). InChIKeys were generated from the Markush children and associated with parent Markush’s DTXSID.
2.5. Compiling in silico predictions and literature data
The output files containing the results from each software tool were processed in a series of steps: i) data were read into a DataFrame using the Python Pandas package; ii) SMILES for each metabolite were standardised using RDKit to remove stereochemistry; iii) standardised SMILES were converted into InChIKeys using RDKit; and iv) a DataFrame was constructed where columns captured metabolite InChIKeys, SMILES, parent DTXSIDs, and the software that generated the prediction.
Results from the literature review were stored in a DataFrame with columns that represented the DTXSID of the parent substance, the DTXSID of the Markush parent, the metabolite InChI key derived from the Markush child or discrete structure of non-Markush metabolites, a binary category to indicate if the metabolite is a Markush, and a binary category to indicate that the metabolite is represented in literature.
DataFrames constructed from the prediction software results and summarised literature were sequentially merged using an outer join on the parent DTXSIDs and InChI keys. A truncated example of the resulting DataFrame is provided in Table 3. Columns with either a 0 or 1 serve as a bit vector to indicate whether the metabolite is present within the corresponding dataset.
Table 3.
Sample (truncated) summary table to illustrate how prediction information was recorded for subsequent analysis.
| Parent_DTXSID | Metabolite_INCHIKEY | TB_iv | TB_ivt | Meteor | BioTransformer | TIMES_InVivo | TIMES_InVitro | SyGMa | CTS | Reported | Metabolite DTXSID | Markush |
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| DTXSID0020151 | UKPSIRZGSFJNRP-UHFFFAOYSA-N | 0 | 0 | 0 | 1 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | GNPWZLYNRQMTAP-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | YJEAVKWPGDUAFC-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | JIFDDBBEZMXNFM-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | HNGQRLUIWXZSSX-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | WYORKAVSTBSIMG-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | OEGPRYNGFWGMMV-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | UADYGGYNNBDDQR-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | CVRGLSNUBFHEQZ-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | KQBFNLRVOIBBDR-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | AVTYLLXTBWFTSS-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | IIGNZLVHOZEOPV-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | ZGCWLHMESZKYFN-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | GEYHQZXETQUQQO-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
| DTXSID0020151 | IHFPLGRHJDIZLM-UHFFFAOYSA-N | 0 | 0 | 0 | 0 | 0 | 0 | 1 | 0 | 0 | NA | False |
2.6. Assessing the similarity and performance of the software predictions
Model performance was compared using four metrics: similarity, coverage, precision, and sensitivity. These metrics provide inter-model comparisons (coverage) and empirical evaluations relative to reported metabolites (precision and sensitivity). The data used for these calculations are described in the section 2.5. Intersecting metabolites for model-model and model-reported comparisons were determined by subsetting the compiled DataFrame, where the rows between the respective columns both contain the value of ‘1’.
Model similarity was determined by identifying the intersection of predicted metabolites between two models. For this calculation, the columns corresponding to the prediction software (i.e., TIMES (ivt), CTS, Meteor, BioTransformer, etc) were used as bit vectors to calculate the Jaccard distance between each model pairing. Hierarchical clustering using the Ward criterion was used to calculate a linkage matrix and quantify similarity.
Coverage represents the percent of overlapping predictions that one model has for another, and is calculated using:
This formula provides a non-associative similarity value, in which the percent overlap can be determined with respect to model A or model B (see results Table 4 from which these percentages are computed). Here, the table of all metabolites generated by all tools and reported metabolites was first used to generate a total metabolite count for each tool. For example, across all 9539 metabolites, there were 236 metabolites generated by the Toolbox (ivt). If we look across each tool, a ‘match count’ can be computed e.g. for metabolite X for parent Y, if 3 of the tools predicted metabolite X, the corresponding match count would be 3. The sum of the match counts for a specific tool could then be computed to generate an unique sum across all metabolites. Matching predictions between 2 software tools could then be determined to compute a coverage score. For example, there were a total of 236 metabolites generated by the Toolbox (iv) of which 90 were matching between the Toolbox (iv) and Meteor; thus 90/236*100 would give rise to a coverage score of 38.14% in terms of the proportion that the Toolbox (iv) ‘covered’ Meteor predictions. In contrast, there were 714 predicted metabolites for Meteor of which 90 were matching with the Toolbox (ivt), thus 90/714*100 would give rise to a coverage score of 12.6% to reflect the proportion of Meteor predictions that the Toolbox (ivt) covered. The distinction over other similarity calculations (e.g., Tanimoto/Jaccard index) allowed the coverage calculation to identify instances where high coverage was non-reciprocal due to one model having a significantly greater number of predictions.
Table 4.
Coverage Matrix
| Model | Total | Unique | TB_ivt | TB_iv | Meteor | BioTransformer | TIMES_InVivo | TIMES_InVitro | SyGMa | CTS | Reported |
|---|---|---|---|---|---|---|---|---|---|---|---|
| TB_ivt | 194 | 0 | 194 | 113 | 80 | 62 | 113 | 194 | 107 | 109 | 56 |
| TB_iv | 236 | 4 | 113 | 236 | 90 | 46 | 232 | 117 | 108 | 99 | 57 |
| Meteor | 714 | 414 | 80 | 90 | 714 | 102 | 153 | 98 | 240 | 94 | 67 |
| BioTransformer | 3464 | 2553 | 62 | 46 | 102 | 3464 | 114 | 84 | 840 | 82 | 39 |
| TIMES_InVivo | 570 | 120 | 113 | 232 | 153 | 114 | 570 | 158 | 295 | 99 | 70 |
| TIMES_InVitro | 283 | 20 | 194 | 117 | 98 | 84 | 158 | 283 | 162 | 109 | 65 |
| SyGMa | 5215 | 4018 | 107 | 108 | 240 | 840 | 295 | 162 | 5215 | 153 | 73 |
| CTS | 472 | 254 | 109 | 99 | 94 | 82 | 99 | 109 | 153 | 472 | 46 |
| Reported | 822 | 708 | 56 | 57 | 67 | 39 | 70 | 65 | 73 | 46 | 822 |
Sensitivity indicates how well a model predicts all reported metabolites and is calculated by determining the percent of true predictions (i.e., predicted metabolites that are reported in literature) relative to all metabolites reported in literature. This metric is calculated using:
In cases where the predicted metabolites represent enumerations of a Markush structure reported in literature, the predicted metabolites are counted as a single match.
Precision indicates the percent of true predictions relative to all predictions generated by a model, and is calculated using:
2.7. Investigation of the chemical space: coverage of the models
The relative coverage of each prediction tool was evaluated using the chemical classification information generated from the freely available web application ClassyFire [35]. This rule-based approach to classify chemicals, is based on structural features derived from ChemOnt, a manually curated list of over 4000 chemical classifications. The text-based classification descriptors generated by ClassyFire are human-readable which permits overarching taxonomic categories for each chemical grouping to be readily identified.
ClassyFire descriptors were generated using the web application (http://classyfire.wishartlab.com/) for each parent compound on the basis of their SMILES. The resulting text file containing chemical classifications for each parent compound were converted into bit vectors, where each bit corresponded to the presence or absence of a classification assignment. A pairwise distance matrix using Jaccard distance was then calculated. Hierarchical clustering using Ward’s criterion was then performed and the tree was cut to derive a practical set of clusters by visual inspection of the dendrogram.
2.8. Case study for 3,4-TDA
Predictions were generated for 3,4-TDA and its candidate analogues [23] in the same way as described already though for practical reasons only Meteor and TIMES had been considered in the PPRTV assessment. Predictions from Meteor Nexus were carried forward to highlight how similarity in biotransformation pathway could be objectively quantified. Here the biotransformations for each of the candidate analogues were converted into a pivoted dataframe with unique biotransformation pathways as columns, presence or absence of a metabolite as a value and rows as the target and candidate analogue and their respective metabolites. This was represented as a fingerprint which was then converted into a squareform distance matrix using the pairwise Jaccard distance. A heatmap was then created to summarise the pairwise similarity between target and candidate source analogues with respect to biotransformation pathways. A representation of the presence of exact metabolites was also derived to show the overlap in the experimental and simulated metabolites. The similarity in biotransformation was not feasible to construct for all the software tools since some like the OECD Toolbox do not provide information on the predecessor and successor relationships as part of their exportable tabular summary information to enable the metabolic trees to be reproduced.
2.9. Data analysis software and code
Data processing was conducted using the Anaconda distribution of Python 3.8 and associated libraries. Jupyter Notebooks, scripts and datasets are available on github at https://github.com/g-patlewicz/in_silico_metabolism and at the EPA FTP website at https://gaftp.epa.gov/Comptox/CCTE_Publication_Data/CCED_Publication_Data/PatlewiczGrace/CompTox-metabolism
3.0. Results and Discussion
3.1. Assessing overall metabolic predictions and performance
A total of 9139 metabolites were generated for the 37 substances. Of the metabolism tools, SyGMa simulated the most number of metabolites relative to the other tools. The maximum number of metabolites predicted was 739 for SyGMa whereas the median number of metabolites was 91. BioTransformer had a maximum number of 538 metabolites with a median of 52. The other tools had a lower median number of metabolites ranging from 5–11 metabolites. Figure 2 shows the distribution of the number of metabolites simulated by each of the tools relative to the reported Literature information.
Figure 2.

Summary boxplot to depict the distribution of the total number of metabolites predicted per substance across the different in silico tools and those metabolites reported in the literature. The ‘box’ in the plot depicts the 25th percentile (Q1 or first quartile), 50th percentile (or median) and the 75% percentile (Q3 or third quartile). The whiskers on the box depict the minimum and the maximum values in the dataset excluding outliers. These are computed for minimum as Q1 – 1.5*IQR whereas the maximum is computed as Q3 + 1.5*IQR. The IQR is known as the interquartile range – the width between Q1 and Q3.
Meteor experienced an apparent combinatorial explosion in the number of metabolites predicted for decabromodiphenyl ether (DTXSID9020376; CASRN 1163–19-5). A total of 382 metabolites were predicted for this structure in contrast to the other tools; in fact, no metabolites were simulated by BioTransformer, CTS or SyGMa. In the literature, 543 metabolites were reported for this substance if their Markush structures were enumerated fully. BioTransformer did not simulate metabolites for very low molecular weight (=< 60 mol/g) substances in the dataset or very high molecular weight (MW) (> 900) substances, specifically, acetone (DTXSID8021482; CASRN 67–64-1), isopropanol (DTXSID7020762; CASRN 67–63-0), and 1,1’-oxybis[2,3,4,5,6-pentabromobenzene (DTXSID9020376, CASRN 1163–19-5). In contrast, SyGMa generated 739 substances for this very high MW substance. There were a handful of other substances for which BioTransformer did not produce any metabolites for specifically, Curcumin (DTXSID8031077, CASRN 458–37-7), beta-Hexachlorocyclohexane (DTXSID7020685, CASRN 319–85-7), Dieldrin (DTXSID9020453, CASRN 60–57-1), Sulindac (DTXSID4023624, CASRN 38194–50-2) and CP-122721 (DTXSID9047251, CASRN 145742–28-5). These last three substances are pesticide actives which made it all the more surprising that BioTransformer did not produce any metabolites since its underlying reactant database is very much enriched by drugs and pesticides. The Toolbox in vitro module was not able to generate predictions for benzoic acid (DTXSID6020143: CASRN 65–85-0) whereas the Toolbox in vivo module was the only tool not able to generate predictions for Bisphenol A (DTXSID7020182, CASRN 80–05-7) and Zileuton (DTXSID9023752, CASRN 111406–87-2).This is not surprising given that Phase II metabolites are not generated by the Toolbox in keeping with its intended purpose. CTS did not generate predictions for decabromodiphenyl ether (DTXSID9020376: CASRN 1163–19-5) and beta-Hexachlorocyclohexane (DTXSID7020685; CASRN 319–85-7). The number of predicted metabolites CTS produced were generally low in number reflecting in part that it only produced CYP450 Phase I transformations. Meteor and TIMES produced metabolites for all substances in the dataset. SyGMa had significantly greater number of total predictions (5215) relative to the other software, BioTransfomer was the next highest with a total count of 3464 metabolites. There were 3 substances where both SyGMa and BioTransformer exceeded 200 predicted metabolites, namely: 1-amino-5-azotoluene (DTXSID1020069, CASRN 97–56-3), Benzyl butyl phthalate (DTXSID3020205, CASRN 85–68-7) and Haloperidol (DTXSID4034150, CASRN 52–86-8). These large numbers indicate that both SyGMa and BioTransformer are susceptible to overprediction, though this could be useful for specific decision contexts where a high sensitivity is desirable. This tendency of generating larger numbers of metabolites is due to the manner in which these tools were derived as well as their intended purpose. For instance SyGma is considered a suitable tool for producing as extensive a set of metabolites as possible to facilitate the analytical spectroscopy undertaken as part of non-targeted screening studies [30]. Its underlying training dataset (MDL Metabolite) is very much biased towards drugs and drug like substances. BioTransformer’s training sets are also enriched with pharmaceuticals and pesticides. On the other hand, TIMES and/or Toolbox models generated substantially fewer metabolites for the same substances – within TIMES, the simulation of metabolites has been closely coupled with the prediction of (eco)toxicological properties as part of the same modelling platform – such that the metabolic pathways produced have been trained to help inform the chemical safety assessment for specific toxicity endpoints such as Ames or in vivo micronucleus. The rat S9 and liver metabolism simulators are effectively those used to help inform the prediction of these endpoints. The underlying training sets are also more enriched by cosmetic, food ingredient and industrial chemicals and to a much lesser extent by drug candidates.
Table 4 shows the coverage matrix for the metabolites generated from each of the software tools relative to the metabolites reported.
As seen in Table 4, the Toolbox (ivt) had no unique predictions, in that all its predictions overlapped with the other software. SyGMa had the highest numbers of predictions (5215) with 4018 being unique to SyGMa. Over half of those predictions were captured by the Toolbox (ivt) and both of the TIMES modules. BioTransfomer had the second highest number of predictions (3464) with over half of those predictions (2553) being unique to BioTransformer.
Model performance was determined by calculating sensitivity and precision for each model (shown in the Radar charts in Figure 3), and the overall performance of each model was calculated by taking the average between these values. The Toolbox (ivt) model scored highest with a precision of 28.9% and sensitivity of 21.9%, followed by the in vitro module of TIMES (23% precision and 25.5% sensitivity) and the in vivo module of TIMES (12.3% precision and 27.9% sensitivity). Meteor and SyGMa both had relatively higher sensitivities (23.1% and 28.3%, respectively) but exhibited relatively low precision (<10%). BioTransformer and CTS showed the lowest sensitivity (14.7% and 17.9%, respectively). The rankings are: Toolbox (ivt) (25.4), TIMES (ivt) (24.3), Toolbox (iv) (23.45), TIMES (iv) (20.1), Meteor (16.3), SyGMa (14.9), CTS (13.8), and BioTransformer (7.9). The low numbers are not altogether surprising – for 2 of the substances, no metabolites were reported at all. In addition, the low numbers could also be due to limitations of the actual experimental studies in terms of what metabolites might have been the object of investigation, what was being detected e.g. Phase II metabolites not being identified and how (in terms of the analytical techniques used). Species-specific considerations will also have had an impact on the model performance. Most of the available experimental data ultimately identified were in vivo rat studies whereas some of the software tools (a case in point being SyGMa, BioTransformer and CTS) focus on human metabolism. Note not all pathways are expected to be conserved between species. This might also explain why TIMES and the Toolbox performed so well as their simulators were rat S9 or liver based and optimised for toxicity prediction. For some of the substances, reported studies had been conducted using rat liver microsomes or human microsomes. Indeed, the manner in which metabolites are generated in vitro is significantly impacted by test system, study design as well as analytical methods. Overall, the performance characteristics at minimum help provide additional context for the predictions generated.
Figure 3.

Sensitivity and Precision radar charts
Furthermore, the median number of reported metabolites is 6 while the median number of predictions generated by each model range from 5 to 91. These values indicate that some of the models generally overpredict (such as BioTransformer and SyGMa) and hence yield much lower precision values relative to their sensitivity scores.
3.2. Evaluating overall model coverage relative to each other
A bit-vector of metabolite predictions was constructed for each model and used to perform a hierarchical clustering analysis using Ward’s criterion and a Jaccard distance metric (Figure 4).
Figure 4.

Dendrogram of the software tools on the basis of their metabolite predictions.
Figure 4 shows the dendrogram produced from the clustering exercise.
As noted from the dendrogram in Figure 4, TIMES (iv and ivt) and Toolbox (iv and ivt) have the highest degree of similarity. These results are to be expected, as the Toolbox derives its modules from TIMES, with the greatest difference being that Toolbox does not generate Phase II predictions. The remaining models showed high degrees of dissimilarity, with Jaccard similarities < 0.1. This was also observed in the associated heatmap of the pairwise similarity matrix, Figure 5.
Figure 5.

Pairwise similarity matrix of the different software tools
The largest coverage value was identified between Toolbox (ivt) and TIMES (ivt), in which Toolbox (ivt) predicted 58.25% of the metabolites generated by TIMES (ivt). SyGMa had the largest average coverage (38.63%) amongst models, followed by BioTransformer (34.03%) which are attributed to the higher number of total predictions.
3.3. Evaluating overall local metabolite performance on the basis of predefined clusters
A hierarchical clustering using Ward’s criterion was performed for the 37 parent substances using ClassyFire [35] taxonomic representations expressed as bit vectors. Based on the dendrogram (Figure 1 - Supplemental), a cut was made to create five clusters.
These were merged with ClassyFire taxonomic descriptors to construct 5 groups that represented broad chemical categories namely: Group 0: Nitrobenzene compounds; Group 1: Benzyl methyl ethers; Group 2: Carboxylic acid/ketone; Group 3: Variable functional groups (combination of small molecules and aromatic systems, weakest grouping of the five groups) and Group 4: Poly chlorinated ring structures. The precision and sensitivity were determined for each chemical grouping for each model. Toolbox (ivt) displayed a general, well-rounded precision and sensitivity across the four of the five groups with a precision ranging from 26.8% up to 46.2% and sensitivity from 20.6% to 35.3%. BioTransformer and Meteor showed low sensitivity for Group 4 (polychlorinated ring structures) (1.7% and 3.4% respectively), and BioTransformer had a low precision (0.3%) for the same group. Toolbox (ivt) appeared to have similar performance to TIMES (ivt) with the highest precision and sensitivity for Group 2, 46.2% and 35.3% respectively. Figure 6 summarises the precision and sensitivity across the 5 groups.
Figure 6.

Radar plots summarising precision and sensitivity for the 5 clusters
3.4. Case study for 3,4-TDA
Provisional Peer-Reviewed Toxicity Values (PPRTVs) are derived by the U.S. Environmental Protection Agency (U.S. EPA) to assist the Superfund Program. PPRTVs are derived after a review of the relevant scientific literature using established EPA guidance on human health toxicity value derivations. A PPRTV is intended to provide support for the hazard and dose-response assessment pertaining to chronic and subchronic exposures of substances of concern, to present the major conclusions reached in the hazard identification and derivation of the PPRTVs, and to characterise the overall confidence in these conclusions and toxicity values. 3,4-Toluenediamine (3,4-TDA), also known as 3,4-diaminotoluene (DTXSID9024930; CASRN 496–72-0), belongs to the class of compounds known as anilines and is an ortho (o)-substituted compound. There are no potentially relevant subchronic or chronic studies or developmental or reproductive toxicity studies for 3,4-TDA either in humans or animals for non-cancer and cancer endpoints following oral or inhalation exposures. In the absence of relevant data, a read-across approach following the Wang et al. [5] approach was performed in order to identify candidate source analogues to 3,4-TDA for noncancer repeated-dose toxicity. Data for candidate analogues are then retrieved and an evaluation of their similarity with respect to structural, metabolic and toxicity-like contexts is performed to select the most suitable analogue both from a toxicological and chemical perspective. In the assessment for 3,4-TDA, the following candidate analogues were identified: 2,3-TDA (DTXSID4027494; CASRN 2687–25-4), 2,4-TDA (DTXSID4020402; CASRN 95–80-7), 2,5-TDA (DTXSID6029123; CASRN 95–70-5) and 2,6-TDA (DTXSID4027319; CASRN 823–40-5) which are all positional isomers. All analogues share a common basic structure, which comprises a benzene ring, 2 amino groups and a methyl group differing only in the position of the amino functional groups. The candidate analogues were considered suitable analogues for 3,4-TDA based on their similarities in structural and physicochemical properties as well as common structural alerts as identified by the profilers within the OECD Toolbox (as described in the main report, [23]). No toxicokinetic data was identified for 3,4-TDA. However, information was available for candidate analogues 2,4-TDA, 2,5-TDA, and 2,6-TDA suggesting that these compounds are rapidly and extensively absorbed following oral exposure, and are rapidly eliminated in the urine, which is a predominant route of excretion. A full summary of all the ADME information collected is available in tabular form in the main report [23]. This permitted a qualitative comparison to be made of the similarities between candidate analogues. Major metabolic steps for the TDA analogues are acetylation of amino groups and ring hydroxylation, with some evidence for oxidation of the methyl groups in rats and mice exposed via intraperitoneal (i.p.) administration (references include [36–43]).
In the absence of in vivo toxicokinetic data on 3,4-TDA, a selection of available software tools, specifically the in vivo and in vitro rat metabolic simulators available within the TIMES and Meteor Nexus were used to predict metabolites for the target compound and analogues. Predicted metabolites for the TDA isomers did reveal some overlap in terms of metabolites for the individual TDA compounds across the different tools. Commonality between the metabolite predictions with available experimental data from in vivo rodent studies, provided confidence in the in silico results. Meteor and TIMES (ivt) showed most similarity to each other in terms of the actual predicted metabolites, although TIMES (iv) was more similar to the reported metabolites. Given the low number of reported metabolites for the analogues with data, the pairwise similarities between the metabolites simulated and those experimentally found were otherwise very low (see pairwise similarity matrix in Supplementary Figure 2 and the dendrogram in Supplementary Figure 3). Table 5 shows the counts of metabolites for all the TDAs across the tools and what were reported. The full set of metabolites predicted and reported for each of the TDAs are tabulated in the main report [23].
Table 5:
Counts of metabolites across the in silico tools and what had been reported
| Source | DTXSID | Expt_Metab | Meteor | TIMES_InVivo | TIMES_InVitro |
|---|---|---|---|---|---|
| 2,3-TDA | DTXSID4027494 | 0 | 14 | 8 | 19 |
| 2,4-TDA | DTXSID4020402 | 9 | 19 | 5 | 15 |
| 2,5-TDA | DTXSID6029123 | 1 | 18 | 2 | 15 |
| 2,6-TDA | DTXSID4027319 | 4 | 8 | 4 | 11 |
| 3,4-TDA | DTXSID9024930 | 0 | 15 | 8 | 18 |
Experimental metabolism information was only available for 3 of the TDAs. As an illustration, a comparison of the low overlap between predicted metabolites from TIMES (ivt), Meteor and the experimentally reported metabolites is shown in the venn diagram in Figure 7 for 2,4-TDA.
Figure 7.

Overlap of the metabolites simulated (using Meteor and TIMES (ivt)) and experimental for 2,4-TDA
The metabolites predicted that overlapped with those reported are shown in Table 6. Although limited in number, the predicted metabolites do at least reflect the main expected transformation pathways as noted earlier.
Table 6.
Experimental metabolites that were predicted by TIMES, Meteor for 2,4-TDA
| Parent: 2,4-TDA |
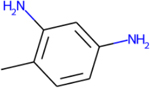
|
| Metabolite |
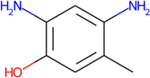
|
| Metabolite |
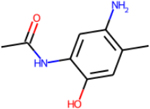
|
| Metabolite |
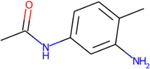
|
Instead of looking at the overlap in actual metabolites between tools vs reported data, the corresponding metabolic pathway transformations were extracted from Meteor in an effort to evaluate metabolic similarity between the TDAs. Figure 8 does reveal a consistent pattern of pathway transformations and a high degree of similarity between 3,4-TDA and its candidate analogues with regards to the Meteor pathway predictions.
Figure 8.

Similarity of transformation pathways as generated by Meteor for the TDA analogues.
There is also concordance between the in silico results and the major pathways expected for this group of compounds (N-acetylation, ring hydroxylation, and oxidation of methyl groups). Importantly, no outstanding differences in the predicted metabolic profiles between the target and analogues were noted. The metabolic tree for 2,4-TDA is displayed in Figure 9 to illustrate the relationship of the predicted metabolites for this candidate analogue that correspond to the pathway transformations shared among the TDAs.
Figure 9.

Metabolite tree for 2,4-TDA
In vivo data demonstrated toxicokinetic commonalities among the analogues, particularly with respect to metabolism pathways, and according to in silico predictions, a similar metabolism pattern is expected for 3,4-TDA. Therefore, the candidate analogues were considered suitable analogues for 3,4-TDA based on their toxicokinetic properties. Further, the in silico analysis provides a means for making similarity comparisons between the target and analogues, adding to the weight of evidence in support of the selection of a source analogue for read-across.
The limitations of this approach are two fold – comparing the similarity of the actual metabolites generated could be expected to be low since the actual metabolites from the different TDAs will lead to structurally similar metabolites but not necessarily the same substances; using transformation pathways offers a more realistic means of comparison but this level of information is not provided from all software tools. However, even similarity in transformation pathways presents only a single criterion in characterising metabolic similarity (or indeed its dissimilarity) fully. A question remains of how granular or specific that comparison ought to be – more general transformations are likely to result in higher similarities than characterising transformations in more granular form within a hierarchy of transformations e.g. hydrolysis vs ester hydrolysis vs carboxylic ester hydrolysis. Other considerations that could be evaluated include common reactivity (structural) patterns of the metabolites as well as their own structural similarity e.g. do the predicted metabolites across analogue share a certain degree of structural similarity or reactivity similarity. Another avenue for investigation could be encoding similarity in the sequence of transformation e.g. is the shape of the metabolic tree or network comparable across two or more analogues. Extracting metrics from metabolic trees which in of themselves are effectively graph networks could be an additional complement in the rationalisation of metabolic similarity and the extent to which this informs similarity of the endpoint of interest, in this case repeat dose toxicity. Further work will focus on approaches to codify the similarity of transformations using a custom specific fingerprint representation, an illustrative example is shown for how such a fingerprint could be constructed using all the in silico predictions for 3,4-TDA as part of the supplementary information. On the basis of structural similarity of the parent substances using Morgan fingerprints [44] and calculating a Jaccard distance; 3,4-TDA was found to be most similar to 2,5-TDA (Jaccard index of 0.41) (range of similarities were 0.28–0.41) whereas on the basis of metabolic similarity using the custom fingerprint, all candidate analogues were equally similar (range 0.54–0.57).
4. Conclusions
An important consideration in evaluating analogue suitability in read-across is metabolic similarity. In this study, a set of 37 structurally diverse substances were selected from the ExpoCast project and processed through a selection of metabolism prediction tools. This was one component of a broader study to evaluate the concordance between in silico predictions and in vitro data relative to reported metabolism in the literature. The predicted metabolites were compared across 6 different tools to better understand their basis as well as gauge their performance and coverage. SyGMa had the highest average coverage (38.63%) and the largest number of metabolites predicted (5215). This was followed by TIMES (iv) which had an average coverage of 34.03% but generated far fewer metabolites (570). The precision and sensitivity values ranged from 1.4–29% and 14.7–28.3% respectively highlighting the challenges of comparing predictions to reported data. Afterall, the basis for the tools were different (Phase I human only (CTS) or human Phase I & Phase II together (BioTransformer, SyGMa), in vitro or in vivo rat models applied separately (TIMES, Toolbox) and the reported data was very heterogenous in study type. Of all the tools evaluated, the Toolbox (ivt) appeared to have the overall best performance across all the substances evaluated. At the local chemistry domain level whereupon the substances evaluated could be grouped into 5 main clusters of similar substances and across those clusters, the Toolbox (ivt) was the overall best performing for all the clusters with exception to group 4. Although the set of substances evaluated was quite limited, the study was informative in terms of highlighting the coverage of the tools for these substances of interest. For the purposes of informing our non-targeted approach to help in the identification of metabolites, then SyGMa appears to be the most useful given its high coverage and sensitivity. However, given our primary motivation was to better understand the scope and coverage for the purposes of a read-across assessment, then TIMES (ivt) appears to be most promising since its average coverage (28.44%), precision (23%) and sensitivity (25.55%) were all similar, moreover functionality implemented in TIMES allows simulated metabolic maps to be compared for similarity based on a number of different criteria (as discussed in Kuseva et al., [20]). This is certainly valuable when evaluating a single substance in detail for a risk assessment application. Exporting a summary tabular report with the same level of detail is not currently feasible within TIMES which would be needed for evaluating a large number of substances such as what is ultimately desired for use in the GenRA approach [2,21]. Two tools were applied to 3,4-TDA that had undergone a PPRTV assessment [23]. The insights gained from the evaluating the tools apriori were informative when structuring an objective means of summarising the prediction information available to justify metabolic similarity across the candidate analogues which in turn informed the selection of the best candidate analogue for read-across. Further work will focus on scaling this approach to a larger number of substances and evaluating other means of structuring information from metabolism tools to inform metabolic similarity.
Supplementary Material
Highlights.
Metabolic similarity is a key consideration in analogue evaluation for read-across
A set of ToxCast chemicals were profiled through a selection of 6 in silico metabolism prediction tools
Their performance and coverage were evaluated relative to reported literature information
The similarity between tools was evaluated using clustering approaches
The insights derived were helpful for how predicted metabolism can be applied in the read-across of 3,4-Toluenediamine
Acknowledgments
This work was presented at the 19th International Workshop on QSARs in Environmental and Health Sciences (QSAR2021) meeting held virtually from the 7–9 June 2021.
Footnotes
Disclaimer
The views expressed in this article are those of the authors and do not necessarily reflect the views or policies of the U.S. Environmental Protection Agency. Mention of trade names or commercial products does not constitute endorsement or recommendation for use.
ToxRefDB v2 referenced in Watford et al (2019) contains data for more than 667 substances.
References
- [1].Escher SE; Kamp H; Bennekou SH; Bitsch A; Fisher C; Graepel R; Hengstler JG; Herzler M; Knight D; Leist M; Norinder U; Ouédraogo G; Pastor M; Stuard S; White A; Zdrazil B; van de Water B; Kroese D. Towards Grouping Concepts Based on New Approach Methodologies in Chemical Hazard Assessment: The Read-across Approach of the EU-ToxRisk Project. Arch Toxicol 2019, 93 (12), 3643–3667. 10.1007/s00204-019-02591-7. [DOI] [PubMed] [Google Scholar]
- [2].Patlewicz G; Cronin MTD; Helman G; Lambert JC; Lizarraga LE; Shah I. Navigating through the Minefield of Read-across Frameworks: A Commentary Perspective. Computational Toxicology 2018, 6, 39–54. 10.1016/j.comtox.2018.04.002. [DOI] [Google Scholar]
- [3].Rovida C; Barton-Maclaren T; Benfenati E; Caloni F; Chandrasekera PC; Chesné C; Cronin MTD; Knecht JD; Dietrich DR; Escher SE; Fitzpatrick S; Flannery B; Herzler M; Bennekou SH; Hubesch B; Kamp H; Kisitu J; Kleinstreuer N; Kovarich S; Leist M; Maertens A; Nugent K; Pallocca G; Pastor M; Patlewicz G; Pavan M; Presgrave O; Smirnova L; Schwarz M; Yamada T; Hartung T. Internationalization of Read-across as a Validated New Approach Method (NAM) for Regulatory Toxicology. ALTEX 2020, 37 (4), 579–606. 10.14573/altex.1912181. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [4].Wu S; Blackburn K; Amburgey J; Jaworska J; Federle T. A Framework for Using Structural, Reactivity, Metabolic and Physicochemical Similarity to Evaluate the Suitability of Analogs for SAR-Based Toxicological Assessments. Regul Toxicol Pharmacol 2010, 56 (1), 67–81. 10.1016/j.yrtph.2009.09.006. [DOI] [PubMed] [Google Scholar]
- [5].Wang NCY; Jay Zhao Q; Wesselkamper SC; Lambert JC; Petersen D; Hess-Wilson JK Application of Computational Toxicological Approaches in Human Health Risk Assessment. I. A Tiered Surrogate Approach. Regulatory Toxicology and Pharmacology 2012, 63 (1), 10–19. 10.1016/j.yrtph.2012.02.006. [DOI] [PubMed] [Google Scholar]
- [6].OECD. Guidance on Grouping of Chemicals, Second Edition | en | OECD; https://www.oecd.org/publications/guidance-on-grouping-of-chemicals-second-edition-9789264274679-en.htm (accessed 2021 −08 −10). [Google Scholar]
- [7].Ball N; Bartels M; Budinsky R; Klapacz J; Hays S; Kirman C; Patlewicz G. The Challenge of Using Read-across within the EU REACH Regulatory Framework; How Much Uncertainty Is Too Much? Dipropylene Glycol Methyl Ether Acetate, an Exemplary Case Study. Regul Toxicol Pharmacol 2014, 68 (2), 212–221. 10.1016/j.yrtph.2013.12.007. [DOI] [PubMed] [Google Scholar]
- [8].Ellison CM; Enoch SJ; Cronin MTD A Review of the Use of in Silico Methods to Predict the Chemistry of Molecular Initiating Events Related to Drug Toxicity. Expert Opin Drug Metab Toxicol 2011, 7 (12), 1481–1495. 10.1517/17425255.2011.629186. [DOI] [PubMed] [Google Scholar]
- [9].Lester C; Reis A; Laufersweiler M; Wu S; Blackburn K. Structure Activity Relationship (SAR) Toxicological Assessments: The Role of Expert Judgment. Regulatory Toxicology and Pharmacology 2018, 92, 390–406. 10.1016/j.yrtph.2017.12.026. [DOI] [PubMed] [Google Scholar]
- [10].OECD. SIDS Initial Assessment Profile. Tertiary Amines; 2012. [Google Scholar]
- [11].Gadaleta D; Golbamaki Bakhtyari A; Lavado GJ; Roncaglioni A; Benfenati E. Automated Integration of Structural, Biological and Metabolic Similarities to Improve Read-Across. ALTEX 2020, 37 (3), 469–481. 10.14573/altex.2002281. [DOI] [PubMed] [Google Scholar]
- [12].Chen M; Suzuki A; Thakkar S; Yu K; Hu C; Tong W. DILIrank: The Largest Reference Drug List Ranked by the Risk for Developing Drug-Induced Liver Injury in Humans. Drug Discovery Today 2016, 21 (4), 648–653. 10.1016/j.drudis.2016.02.015. [DOI] [PubMed] [Google Scholar]
- [13].Chen M; Vijay V; Shi Q; Liu Z; Fang H; Tong W. FDA-Approved Drug Labeling for the Study of Drug-Induced Liver Injury. Drug Discovery Today 2011, 16 (15), 697–703. 10.1016/j.drudis.2011.05.007. [DOI] [PubMed] [Google Scholar]
- [14].Watford S; Ly Pham L; Wignall J; Shin R; Martin MT; Friedman KP ToxRefDB Version 2.0: Improved Utility for Predictive and Retrospective Toxicology Analyses. Reprod Toxicol 2019, 89, 145–158. 10.1016/j.reprotox.2019.07.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [15].Durant JL; Leland BA; Henry DR; Nourse JG Reoptimization of MDL Keys for Use in Drug Discovery. J. Chem. Inf. Comput. Sci. 2002, 42 (6), 1273–1280. 10.1021/ci010132r. [DOI] [PubMed] [Google Scholar]
- [16].Willett P. Similarity Searching Using 2D Structural Fingerprints. Methods Mol Biol 2011, 672, 133–158. 10.1007/978-1-60761-839-3_5. [DOI] [PubMed] [Google Scholar]
- [17].Russo DP; Kim MT; Wang W; Pinolini D; Shende S; Strickland J; Hartung T; Zhu H. CIIPro: A New Read-across Portal to Fill Data Gaps Using Public Large-Scale Chemical and Biological Data. Bioinformatics 2017, 33 (3), 464–466. 10.1093/bioinformatics/btw640. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [18].Yordanova D; Kuseva C; Tankova K; Pavlov T; Chankov G; Chapkanov A; Gissi A; Sobanski T; Schultz TW; Mekenyan OG Using Metabolic Information for Categorization and Read-across in the OECD QSAR Toolbox. Computational Toxicology 2019, 12, 100102. 10.1016/j.comtox.2019.100102. [DOI] [Google Scholar]
- [19].Yordanova DG; Schultz TW; Kuseva CD; Mekenyan OG Assessing Metabolic Similarity for Read-across Predictions. Computational Toxicology 2021, 18, 100160. 10.1016/j.comtox.2021.100160. [DOI] [Google Scholar]
- [20].Kuseva CD; Yordanova D; Ivanova H. Criteria for Quantitative Assessment of Metabolic Similarity between Chemicals. II. Application to Human Health Endpoints. 2021, 19. 10.1016/j.comtox.2021.100173. [DOI] [Google Scholar]
- [21].Helman G; Shah I; Williams AJ; Edwards J; Dunne J; Patlewicz G. Generalized Read-Across (GenRA): A Workflow Implemented into the EPA CompTox Chemicals Dashboard. ALTEX 2019, 36 (3), 462–465. 10.14573/altex.1811292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [22].Wambaugh JF; Wang A; Dionisio KL; Frame A; Egeghy P; Judson R; Setzer RW High Throughput Heuristics for Prioritizing Human Exposure to Environmental Chemicals. Environ Sci Technol 2014, 48 (21), 12760–12767. 10.1021/es503583j. [DOI] [PubMed] [Google Scholar]
- [23].US EPA. Provisional Peer-Reviewed Toxicity Values for 3,5-Dinitroaniline.; 2021. [PubMed] [Google Scholar]
- [24].Kirchmair J; Göller AH; Lang D; Kunze J; Testa B; Wilson ID; Glen RC; Schneider G. Predicting Drug Metabolism: Experiment and/or Computation? Nat Rev Drug Discov 2015, 14 (6), 387–404. 10.1038/nrd4581. [DOI] [PubMed] [Google Scholar]
- [25].Kirchmair J; Williamson MJ; Tyzack JD; Tan L; Bond PJ; Bender A; Glen RC Computational Prediction of Metabolism: Sites, Products, SAR, P450 Enzyme Dynamics, and Mechanisms. J Chem Inf Model 2012, 52 (3), 617–648. 10.1021/ci200542m. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [26].Tyzack JD; Kirchmair J. Computational Methods and Tools to Predict Cytochrome P450 Metabolism for Drug Discovery. Chem Biol Drug Des 2019, 93 (4), 377–386. 10.1111/cbdd.13445. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [27].Djoumbou-Feunang Y; Fiamoncini J; Gil-de-la-Fuente A; Greiner R; Manach C; Wishart DS BioTransformer: A Comprehensive Computational Tool for Small Molecule Metabolism Prediction and Metabolite Identification. J Cheminform 2019, 11 (1), 2. 10.1186/s13321-018-0324-5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [28].Mekenyan OG; Dimitrov SD; Pavlov TS; Veith GD A Systematic Approach to Simulating Metabolism in Computational Toxicology. I. The TIMES Heuristic Modelling Framework. Curr Pharm Des 2004, 10 (11), 1273–1293. 10.2174/1381612043452596. [DOI] [PubMed] [Google Scholar]
- [29].Dimitrov SD; Diderich R; Sobanski T; Pavlov TS; Chankov GV; Chapkanov AS; Karakolev YH; Temelkov SG; Vasilev RA; Gerova KD; Kuseva CD; Todorova ND; Mehmed AM; Rasenberg M; Mekenyan OG QSAR Toolbox - Workflow and Major Functionalities. SAR QSAR Environ Res 2016, 27 (3), 203–219. 10.1080/1062936X.2015.1136680. [DOI] [PubMed] [Google Scholar]
- [30].Ridder L; Wagener M. SyGMa: Combining Expert Knowledge and Empirical Scoring in the Prediction of Metabolites. ChemMedChem 2008, 3 (5), 821–832. 10.1002/cmdc.200700312. [DOI] [PubMed] [Google Scholar]
- [31].Mansouri K; Grulke CM; Richard AM; Judson RS; Williams AJ An Automated Curation Procedure for Addressing Chemical Errors and Inconsistencies in Public Datasets Used in QSAR Modelling. SAR QSAR Environ Res 2016, 27 (11), 939–965. 10.1080/1062936X.2016.1253611. [DOI] [PubMed] [Google Scholar]
- [32].Grulke CM; Williams AJ; Thillanadarajah I; Richard AM EPA’s DSSTox Database: History of Development of a Curated Chemistry Resource Supporting Computational Toxicology Research. Comput Toxicol 2019, 12. 10.1016/j.comtox.2019.100096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [33].Williams AJ; Grulke CM; Edwards J; McEachran AD; Mansouri K; Baker NC; Patlewicz G; Shah I; Wambaugh JF; Judson RS; Richard AM The CompTox Chemistry Dashboard: A Community Data Resource for Environmental Chemistry. J Cheminform 2017, 9 (1), 61. 10.1186/s13321-017-0247-6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [34].Baker N; Knudsen T; Williams A. Abstract Sifter: A Comprehensive Front-End System to PubMed. F1000Res 2017, 6, Chem Inf Sci-2164. 10.12688/f1000research.12865.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [35].Djoumbou Feunang Y; Eisner R; Knox C; Chepelev L; Hastings J; Owen G; Fahy E; Steinbeck C; Subramanian S; Bolton E; Greiner R; Wishart DS ClassyFire: Automated Chemical Classification with a Comprehensive, Computable Taxonomy. J Cheminform 2016, 8, 61. 10.1186/s13321-016-0174-y. [DOI] [PMC free article] [PubMed] [Google Scholar]
- [36].Timchalk C; Smith FA; Bartels MJ Route-Dependent Comparative Metabolism of [14C]Toluene 2,4-Diisocyanate and [14C]Toluene 2,4-Diamine in Fischer 344 Rats. Toxicology and Applied Pharmacology 1994, 124 (2), 181–190. 10.1006/taap.1994.1022. [DOI] [PubMed] [Google Scholar]
- [37].Grantham PH; Mohan L; Benjamin T; Roller PP; Miller JR; Weisburger EK Comparison of the Metabolism of 2,4-Toluenediamine in Rats and Mice. J Environ Pathol Toxicol 1979, 3 (1–2), 149–166. [PubMed] [Google Scholar]
- [38].Waring RH; Pheasant AE Some Phenolic Metabolites of 2, 4-Diaminotoluene in the Rabbit, Rat and Guinea-Pig. Xenobiotica 1976, 6 (4), 257–262. 10.3109/00498257609151635. [DOI] [PubMed] [Google Scholar]
- [39].Wenker MAM. Absorption, Distribution, Metabolism and Excretion of 14C-Toluene-2,5-Diamine Sulphate in the Female Wistar Kyoto Rat after a Single Intravenous Dose. 2005. [Google Scholar]
- [40].Wenker MAM. Pharmacokinetics of 14C-Toluene-2,5-Diamine Sulphate in the Female Sprague-Dawley Rat after a Single Oral or Dermal Dose. 2005. [Google Scholar]
- [41].River Charles. Toxicokinetics of [14]C Toluene-2,5-Diamine Sulfate Following a Single Oral Administration to Sprague-Dawley Rats. (Study Number 420782). 2010. [Google Scholar]
- [42].SCCS (Scientific Committee on Consumer Safety). (2012). Opinion on Toluene-2,5-Diamine and Its Sulfate. (SCCS/1479/12). European Commission: Health and Consumers Scientific Committees. 2012. [Google Scholar]
- [43].Cunningham ML; Burka LT; Matthews HB Metabolism, Disposition, and Mutagenicity of 2,6-Diaminotoluene, a Mutagenic Noncarcinogen. Drug Metab Dispos 1989, 17 (6), 612–617. [PubMed] [Google Scholar]
- [44].Rogers D; Hahn M. Extended-Connectivity Fingerprints. J Chem Inf Model 2010, 50 (5), 742–754. 10.1021/ci100050t. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.