

Abstract
Skin is composed of diverse cell populations that cooperatively maintain homeostasis. Upregulation of the Nuclear factor-kappa B pathway may lead to the development of chronic inflammatory disorders of the skin, but its role during the early events remains unclear. Through analysis of single cell RNA sequencing data via iterative Random Forest Leave One Out Prediction, an explainable artificial intelligence method, we identified an immunoregulatory role for a unique Prx1+ fibroblast subpopulation. Disruption of Ikkb-NF-kB under homeostatic conditions in these fibroblasts paradoxically induced skin inflammation due to the overexpression of C-C Motif Chemokine Ligand 11 (CCL11, or eotaxin-1) characterized by eosinophil infiltration and a subsequent Th2 immune response. Because the inflammatory phenotype resembled that seen in human atopic dermatitis (AD), we examined validated human AD skin samples and found that human AD fibroblasts also over-expressed CCL11 and that perturbation of Ikkb-NFkB in primary human dermal fibroblasts upregulates CCL11. We also demonstrated that monoclonal antibody treatment against CCL11 was effective in reducing the eosinophilia and Th2 inflammation in a mouse model. Taken together, the murine model and human AD specimens point to dysregulated Prx1+ fibroblasts as a previously unrecognized etiologic factor that may contribute to the pathogenesis of AD and suggest targeting CCL11 as a way to treat AD-like skin lesions.
One sentence summary:
Disruption of the Ikkb-NF-kB axis in Prx1+ fibroblasts alters skin homeostasis, causing an inflammatory disorder that mimics human atopic dermatitis.
Introduction
Skin disorders affect 85 million people in the U.S. and globally amount to 41.6 million years lost due to disease and living with disability (1), representing a critical public health burden. The factors that initiate pathogenesis of psoriasis and atopic dermatitis (AD) include disruption of the epithelial barrier and altered immune responses (2, 3). However, despite recent advances, clinical therapies often fail to achieve adequate resolution of symptoms in moderate to severe cases (4). Because the skin must respond rapidly to physical and microbial challenges, resident cells in the skin must respond quickly to perturbations to maintain a fine balance between pro- and anti-inflammatory processes (5). Thus, an improved understanding of cutaneous homeostasis is essential to better understand the early events in dermatologic diseases and to develop therapeutics targeting these early events.
Fibroblasts are the principal cell type in the dermis and exhibit considerable heterogeneity, including scar-inducing subsets marked by En1 (6), Prrx1 (7), or Dpp4 (6, 8). Recent single cell RNA sequencing (scRNA-seq) studies on human AD skin have revealed fibroblast subsets with unexpected immuno-modulatory transcriptomes (9, 10), suggesting a potential role of these cells in the pathogenesis of inflammatory skin disorders. Synovial fibroblasts have been proposed to exhibit a pro-inflammatory phenotype to mediate joint destruction following induction of an autoimmune response in rheumatoid arthritis (11), whereas tumor-associated fibroblasts have been linked to immune-suppression and poor survival outcomes (12). In each of these cases, fibroblasts play a secondary role of responding to their microenvironment to modulate the immune response. Whether skin fibroblasts contribute to immune homeostasis in skin inflammatory disorders remains unknown.
Nuclear factor-kappa B (NF-kB) is a master inflammatory transcription factor that regulates immune-responsive genes (13). Canonical NF-kB proteins and their activator, the inhibitor of nuclear factor kappa B kinase complex, are ubiquitously expressed by cells including fibroblasts (14). In many dermatologic inflammatory conditions, upregulation of NF-kB activity is directly linked to pro-inflammatory events (15). Initiating events are not thought to be directly caused by NF-kB but rather by the response of cells to pro-inflammatory signals that activate NF-kB and upregulate the pathogenic process (16).
Here, we utilized a genetically modified mouse model in which NF-kB activation in fibroblastic cells was blocked by lineage-specific deletion of the NF-kB regulator, inhibitor of nuclear factor kappa-B kinase subunit beta (Ikkb). Inhibition of NF-kB in fibroblastic cells led to the formation of skin lesions, indicating that NF-kB played a key role in maintaining homeostasis and preventing excessive inflammation. With the use of scRNA-seq, we identified that paired related homeobox-1 (Prx1) positive (Prx1+) cells containing Ikkb deletion played a key role in immune disruption by overexpressing the eosinophilic chemokine CCL11, which led to an inflammatory response. These results suggest that Prx1+ cells are essential for suppressing myeloid inflammation initiated by Ikkb under basal conditions and that the lack of Ikkb leads to skin inflammatory disease characterized by early eosinophilia and a type 2 immune response frequently seen in atopic dermatitis-like lesions.
Results
To investigate the role of NF-kB activation in skin fibroblasts, we deleted Ikkb, a gene needed for activation of the canonical NF-kB pathway (17), in Prx1Cre+.Ikkbf/f mice. Prx1-lineage cells are present exclusively in the ventral mesenchyme (fig. S1), consistent with their embryological origin (18, 19). We first examined NF-kB p65 nuclear localization, indicative of its activation, by immunofluorescence in vimentin+ fibroblasts and perilipin+ adipocytes in adult Prx1Cre+.Ikkbf/f and control littermates. 17% of fibroblasts and 20% of adipocytes from the wildtype mice had NF-kB nuclear localization under standard physiologic conditions, whereas less than 4% had nuclear NF-kB in the Ikkb-deleted mice (Fig. 1, A and B). We observed cutaneous lesions in the ventral but not dorsal skin of the Ikkb-deleted mice (Fig. 1C). The affected skin demonstrated hair loss, thickening, scaling, erythema or focal crusting with scab formation (n=6) that was not present in wildtype mice (n=6). The histology of skin from 4 out of 6 Prx1Cre+.Ikkbf/f mice demonstrated hyperkeratotic scale comprised of ortho- or para-keratin and acanthosis associated with spongiosis, which was pronounced in three experimental mice and not present in wildtype mice (Fig. 1D). In the experimental mice, inflamed lesions had a prominent inflammatory infiltrate containing lymphocytes, eosinophils and occasional neutrophils in all skin compartments (Fig. 1D), similar to those seen in human AD (20). Consistent with the distribution of skin lesions, Prx1-lineage cells were found in ventral but not dorsal skin, and their cell percentage found in the ventral dermis of control and Ikkb-deleted mice was similar (fig. S1, A to C). Epidermal and dermal widths were significantly increased in ventral but not dorsal skin of Prx1Cre+.Ikkbf/f mice compared to control group (P<0.05; Fig. 1, E and F and fig. S1D). The skin with Ikkb deletion had decreased strength as measured by reduced stiffness and mechanical load to failure (Fig. 1G), despite the histological evidence of fibroplasia and increased width of the dermis. The results suggest that the skin anomaly caused by the gene deletion is more complex than fibroplasia, possibly reflecting thickened dermis due to edema caused by inflammatory infiltrates.
Fig. 1.
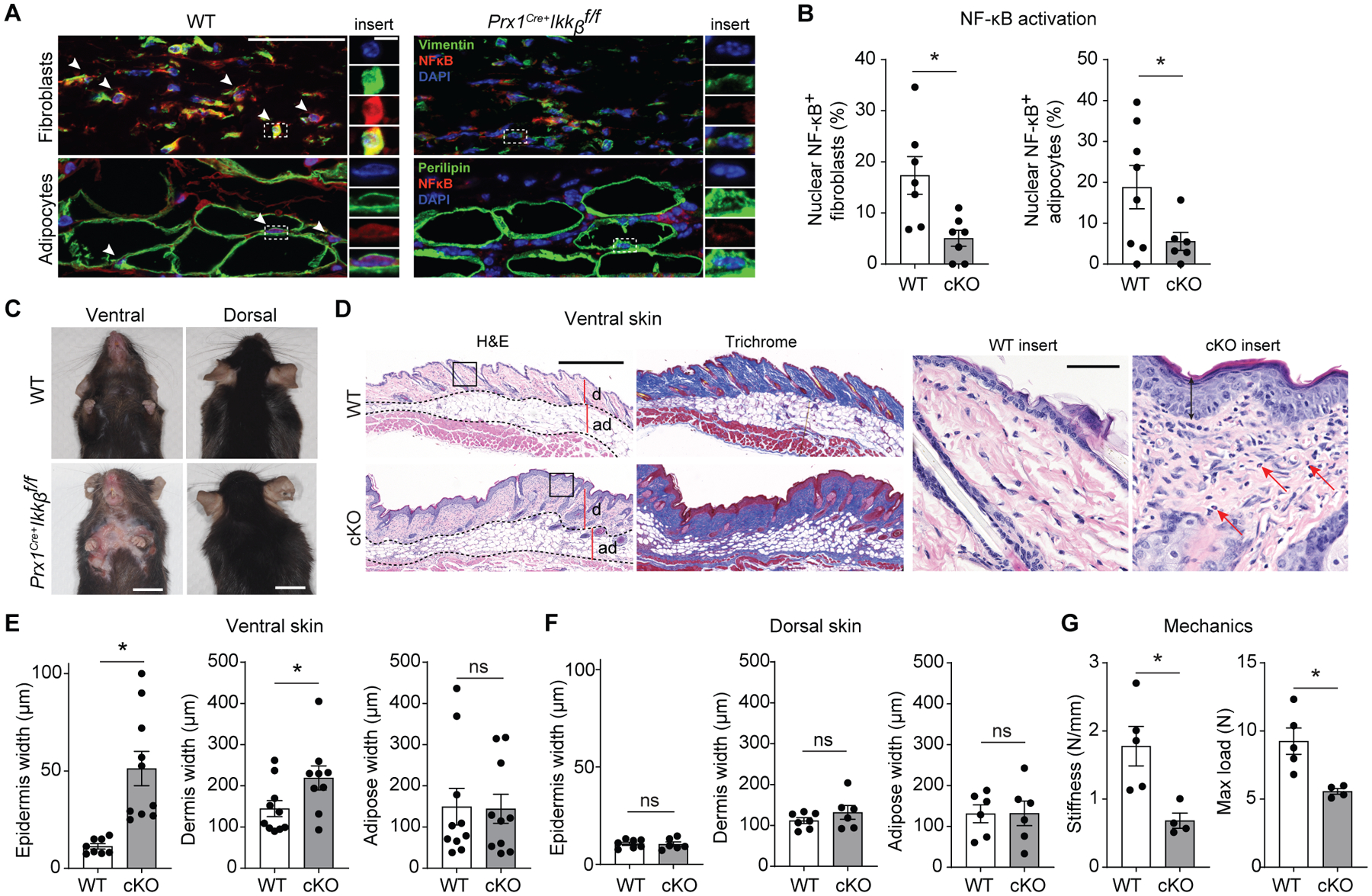
Preventing basal NF-kB activation in Prx1+ mesenchymal cells leads to ventral skin anomalies. A. Representative immunofluorescent images of ventral skin stained with NF-kB (p65) antibody and vimentin (fibroblast marker) or perilipin (adipocyte marker) in 16-week-old control (WT) or experimental (Prx1Cre+Ikkbf/f) mice. White arrows point to fibroblasts or adipocytes with nuclear NF-kB expression. Scale bar, 50um; insert scale bar, 5um). B. Quantification of double-positive vimentin+/nuclear NF-kB+ fibroblasts per total vimentin+ cells (left), and perilipin+/nuclear NF-kB+ adipocytes per total perilipin+ cells (right), comparing between WT and Prx1Cre+Ikkbf/f (conditional knockout, cKO) group. n=7–8 mice in each group. C. Representative photograph images of ventral and dorsal skin in 16–20-week-old WT and Prx1Cre+Ikkbf/f mice; scale bar, 1cm. D. Hematoxylin and eosin (H&E) and trichrome images of ventral skin from WT or cKO mice; d, dermis, ad, adipose layer. Scale bar, 0.5mm. Insert, red arrows point to eosinophils. Scale bar, 50um. E and F. Epidermis, dermis and adipose width in ventral (E) or dorsal (F) skin of WT and cKO mice. n=6–10 mice in each group. H. Mechanical testing of ventral skins from WT and cKO mice; stiffness and maximum load until failure (tear) were quantified. n=4–5 each. Data are represented as mean ± SEM of biological replicates. Animal experiments were repeated two to three times. *P<0.05; Student’s t-test comparing WT to cKO groups.
To better understand the progression of the skin anomaly caused by Ikkb deletion, we tracked the lesion development throughout postnatal growth from young (4 weeks old) to adult (20 weeks old) phase. In the Ikkb-deleted mice, alopecia developed as early as 10 weeks, always preceded ulceration (Fig. 2, A and B) and progressed without resolution (Fig. 2C). Because the initial clinical phenotype was hair loss, we tested if the hair follicle niche was affected by the Ikkb deletion. The number of hair follicles and CD34+ hair bulges were not changed by Ikkb deletion (Fig. 2D), ruling out hair follicle niche disruption as an etiologic factor for the hair loss and alopecia.
Fig. 2.
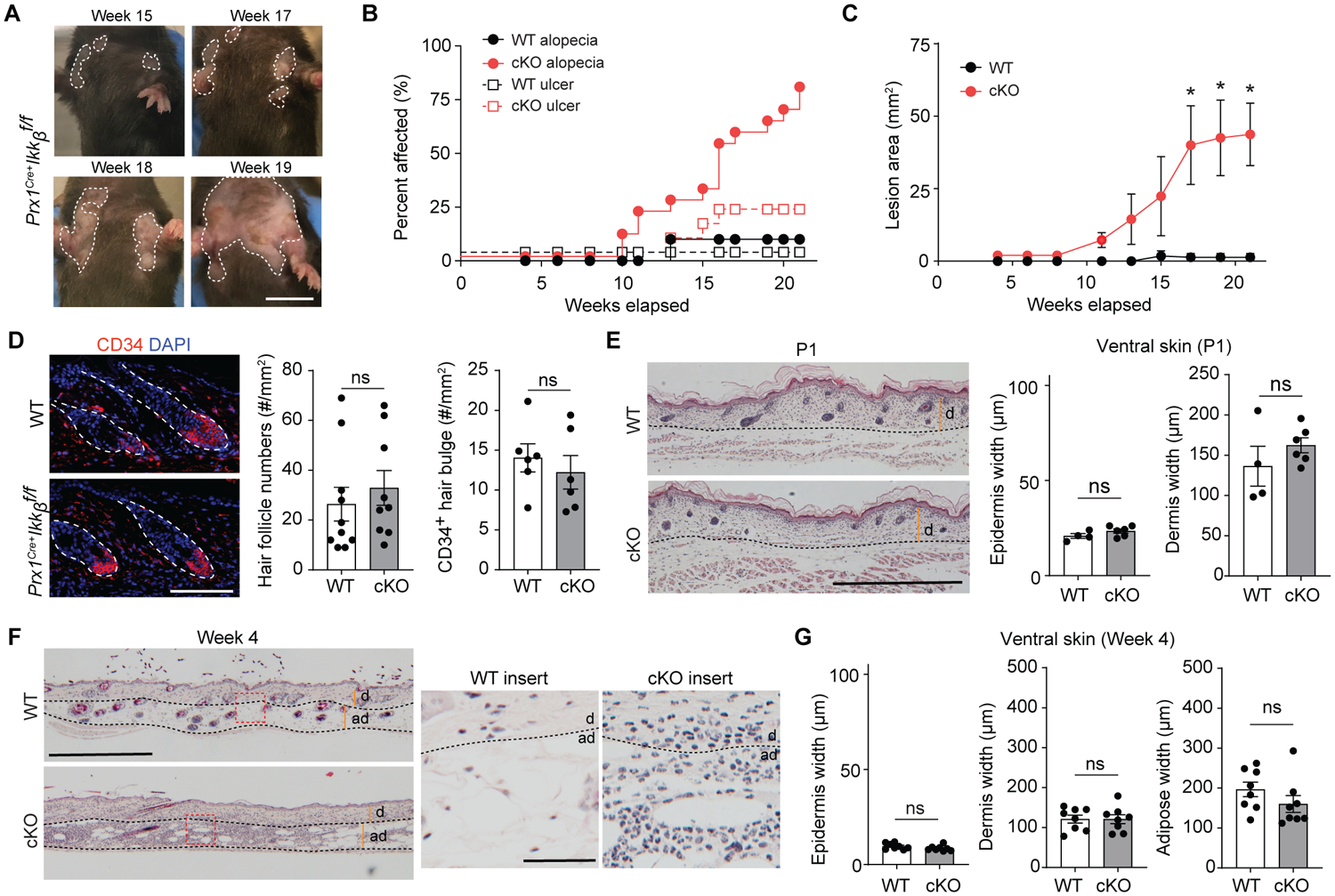
Progressive dermatitis by Ikkb deletion worsens with aging. A. Representative images of clinical lesion from Prx1Cre+Ikkbf/f (cKO) mice over 15- to 19-week period in age. Scale bar, 1cm. B. Quantification of percent control (WT) or cKO animals affected by alopecia (solid line) and/or ulceration (dashed line) in function of age over 4- to 21-week time points. C. Lesion area quantified in mm2 over the observational period. D. Representative immunofluorescent images of CD34+ hair follicle bulge from WT or cKO groups (left, dotted line) and quantification of hair follicle numbers from H&E images or by CD34+ staining (right) in 20-week-old mice. Scale bar, 50um. E. H&E images of ventral skin in 1-day old (P1) neonatal WT and cKO mice (left) and quantification of epidermis and dermis thickness in each group (right). Scale bar, 0.5mm. F. H&E images of ventral skin in 4-week-old young WT or cKO mice (left; scale bar, 0.5mm), with insert images showing overt inflammatory infiltrate in the skin of cKO mice (right; scale bar, 50um). G. Quantification of epidermis, dermis and adipose layer thickness between WT versus cKO mice at 4 weeks of age. Dashed lines demarcate dermal (d) and adipose (ad) borders in E and F. Data are represented as mean ± SEM of biological replicates. Animal experiments were repeated three times. n=6–10 each. *P<0.05, ns = not significant; Student’s t-test comparing WT to cKO groups.
To investigate early cellular events that may be responsible for the skin anomaly in adult mice, we examined neonatal (1 day after birth, P1) and young (4 weeks after birth) mice, time points at which no gross skin lesions were present. Histologically, the skin of P1 mice in control and Ikkb deletion groups showed no signs of acanthosis or fibrosis (Fig. 2E). Interestingly, 4-week-old mice with the Ikkb gene deletion exhibited an abundant subcutaneous and dermal leukocyte infiltration with multi-lobed or kidney shaped nuclei, consistent with myeloid cells (Fig. 2F), and an absence of dermal and epidermal thickening (Fig. 2G). We therefore sought to better characterize the type of inflammation and found significantly elevated numbers of myeloperoxidase (MPO)+ neutrophils and F4/80+ myeloid cells in both young and adult mice with Ikkb deletion, whereas the total number of CD4+ T cells was unchanged (P<0.05; Fig. 3, A and B and fig. S2A). Ikkb deletion did not alter the number of inflammatory cells in dorsal skin (fig. S2B), and there were no significant differences in MPO+ cells in the ventral skin of control and Ikkb-deleted P1 mice (P>0.05; Fig. 3C). Similar results were obtained by flow cytometry analysis; Gr-1high cells (equivalent to Ly6g+ neutrophils) and F4/80+ myeloid cells were significantly higher in the Ikkb-deleted mice whereas CD3+ T cell numbers did not change (P<0.05; Fig. 3, D and E). To rule out the possibility that the inflammation was caused by external irritation from the solid corn bedding, young mice were housed with soft paper bedding, which did not reduce skin inflammation (Fig. 3F). Taken together, Ikkb deletion in Prx1+ fibroblastic cells causes myeloid inflammation in the skin that precedes the clinical anomalies found in adulthood.
Fig 3.
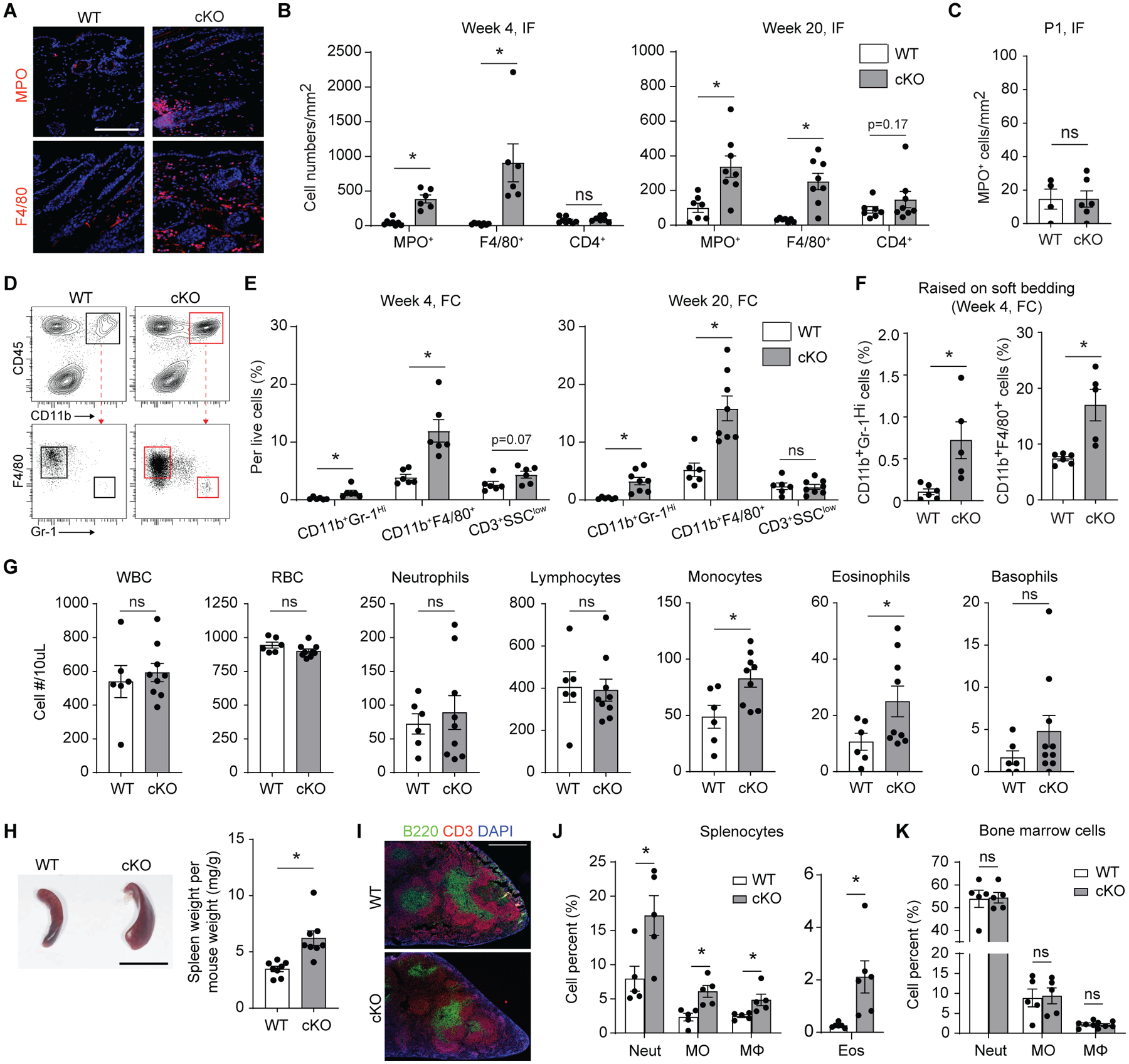
Skin inflammation in Ikkb-deleted mice is characterized by excessive myeloid cell infiltration. A. Representative immunofluorescent images with myeloperoxidase antibody (MPO, a neutrophil marker) and F4/80 antibody (a pan-macrophage marker) in 4-week-old wildtype (WT) or Prx1Cre+Ikkbf/f (cKO) mice, scale bar, 50um. B. Quantification of immunopositive cells for MPO, F4/80, or CD4 expression in 4-week (left) or 20-week-old (right) mice between WT and cKO groups; IF = immunofluorescence experiment. n=6–8 each. C. Quantification of MPO+ cells in the ventral skin of 1-day-old (P1) mice between WT and cKO groups. n=4–6 each. D. Representative flow cytometry plot on myeloid cell gating (live, CD45+CD11b+ cells) to analyze Gr-1Hi neutrophils and F4/80+ myeloid cells in 4-week-old mice. E. Quantification of neutrophils (CD45+CD11b+F4/80−Gr-1Hi), F4/80+ myeloid cells (CD45+CD11b+F4/80+Gr-1−) and CD3 T cells (SSClowCD45+CD3) per total live cells in 4-week (left) or 20-week-old mice (right) by flow cytometry analysis (FC). F. Quantification of neurophils and F4/80+ myeloid cells from 4-week- old mice that were weaned in soft paper cage bedding. G. Quantification of circulating hematopoetic cells from retro-orbital blood samples of the control (WT) or Prx1Cre+Ikkβf/f (cKO) mice aged at 20 weeks old by complete blood count/differential analysis. WBC: white blood cell, RBC: red blood cell, n=6–9 each. H. Representative micrograph of spleen from 20-week-old WT and cKO mice (left; scale bar, 1cm). Right, quantification of spleen weight per weight. I. Spleen cryosections stained for T cells (CD3 antibody) and B cells (B220 antibody) demonstrating intact germinal centers in both WT and cKO mice. Scale bar, 0.5mm. J and K. Quantification of neutrophils (Neut; CD45+CD11b+Gr-1Hi), monocytes (MO; CD45+CD11b+CD115+), macrophages (MΦ; CD45+CD11b+F4/80+) and eosinophils (Eos; CD45+CD11b+Siglec-F+) per total live cells from splenocyte preparations (J) and from bone marrow preparations (K) of WT and cKO mice; n=5–6 each. Data are represented as mean ± SEM of biological replicates. Animal experiments were repeated three times. *P<0.05, ns = not significant; Student’s t-test comparing WT to cKO groups.
Since Prx1+ mesenchymal progenitors that line the bone marrow are critical for the maintenance of hematopoiesis (21), we tested if Ikkb deletion in Prx1-lineage cells had an impact on systemic inflammation. We found a significant increase in circulating monocytes and eosinophils from the peripheral blood whereas other cell metrics, including total red blood cell (RBC) count, were largely unchanged (P<0.05; Fig. 3G). Splenomegaly was always present in the Ikkb deletion group (Fig. 3H) with normal architecture of splenic germinal centers (Fig. 3I). There was a significant increase in myeloid splenocyte counts (neutrophils, monocytes, macrophages and eosinophils) (P<0.05; Fig. 3J), indicating systemic inflammation without abnormal erythropoiesis. A similar increase in myeloid cell numbers was found in axillary lymph nodes of Ikkb-deleted adult mice (fig. S3). However, the number of myeloid cells in the bone marrow was unchanged (Fig. 3K), suggesting that myelopoiesis was not affected by Ikkb deletion. Combined with the finding that dorsal skin was unaffected by Ikkb deletion, the lack of change in myelopoiesis suggests that changes in circulating myeloid cells were likely the result of severe skin inflammation and not vice versa.
Prx1-lineage cells in the adult skin include dermal fibroblasts and subcutaneous adipocytes (19, 22). We therefore generated mice that had Ikkb-specific deletion in adipocytes (Adipoq-Cre+.Ikkbf/f) and pan-fibroblasts (Col1a2CreER+.Ikkbf/f) and examined whether skin lesions developed. Adipocyte-specific deletion of Ikkb failed to elicit myeloid inflammation in the skin, spleen, or bone marrow of young mice (Fig. 4, A to C), indicating that skin adipocytes were not the source of immune dysregulation caused by the NF-kB inhibition. In contrast, induced Ikkb deletion in Col1a2+ fibroblasts resulted in histological changes characterized by a mild increase in myeloid and eosinophilic cells, as well as enhanced F4/80+ cell numbers without inducing myeloid cell changes in the spleen or bone marrow (Fig. 4, D to F). These results rule out skin adipocytes as the source of immune dysregulation caused by the NF-kB inhibition. We further tested if postnatal Prx1+ (pnPrx1+) cells are responsible for maintaining immune homeostasis. In neonatal mice, pnPrx1+ cells represent early mesenchymal progenitors, and lineage tracing demonstrated that they became skin fibroblasts and subcutaneous adipocytes at 4 weeks (Fig. 4G). Induced deletion of Ikkb in P1 mice elicited mild skin inflammation at 4 weeks (Fig. 4H and I) whereas induced Ikkb deletion in 8-week-old mice failed to produce inflammation in mice aged 12 or 24 weeks despite the similar pnPrx1+ cell frequency for each induction time points (fig. S4). These results indicate that there is a critical perinatal growth period during which immune homeostasis is established in the skin and that Prx1+ skin fibroblasts, not adipocytes, are essential for this to occur through basal Ikkb activity.
Fig. 4.
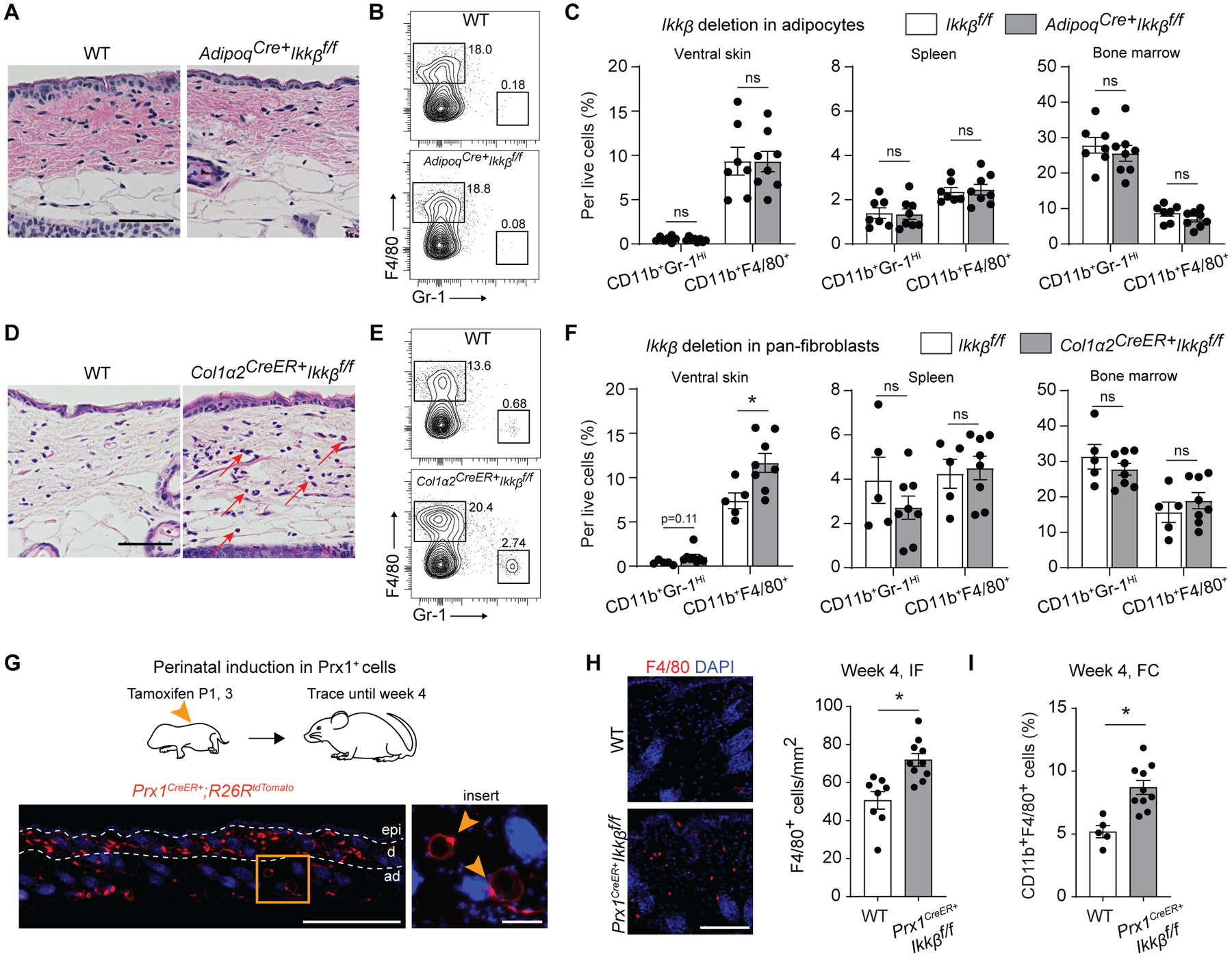
Ikkb deletion in skin fibroblasts, not adipocytes, is responsible for myeloid inflammation during perinatal growth. A. H&E images of ventral skin in 4-week-old control group (WT) or adipocyte-specific Ikkb deletion (Adipoq-Cre+Ikkbf/f) mice. Scale bar, 50um. B. Flow cytometry analysis of neutrophils (CD45+CD11b+Gr-1Hi) and F4/80+ myeloid cells (CD45+CD11b+F4/80+) from enzymatically digested ventral skin of WT or Adipoq-Cre+Ikkbf/f mice. C. Quantification of neutrophils and F4/80+ myeloid cells per total live cell count in ventral skin (left), spleen (middle), and bone marrow (right) comparing WT versus adipocyte-specific deletion of Ikkb group. n=7–8 each. D. H&E images of ventral skin in 4-week-old WT or experimental mice that had induced deletion of Ikkb in Col1a2-expressing fibroblasts (Col1a2CreERT+Ikkbf/f). Mice received tamoxifen twice at P1 and P3 (50ug/dose) and were euthanized at 4 weeks of age. Red arrows point at monocytic and/or eosinophilic cells. Scale bar, 50um. E. Flow cytometry plot for neutrophils and F4/80+ myeloid cells in WT or mice with Ikkb-deleted in Col1a2+ fibroblasts. Tamoxifen was administered at P1 and P3 (intragastric, 50ug/dose). F. Quantification of neutrophils and F4/80+ myeloid cells per total live cell count in ventral (left), spleen (middle) and bone marrow (right) comparing WT versus Ikkb deletion in Col1a2-expressing pan-fibroblasts. n=5–8 each. G. Perinatal induction of dTomato reporter gene in postnatal Prx1+ cells (Prx1CreERT+R26RdTomato), labeling dermal mesenchymal cells and some subcutaneous adipocytes (insert, arrows; scale bar, 20um) after 4-week tracing period. Scale bar, 0.5mm. H. Immunofluorescent images of F4/80+ cells in the ventral skin of 4-week-old WT mice and Prx1CreERT+Ikkbf/f mice that had perinatal deletion of Ikkb (left), and quantification of F4/80+ cells per mm2 (right). IF=immunofluorescence analysis. n=8–10 each. Both WT and Prx1CreERT+Ikkbf/f mice received tamoxifen. Scale bar, 50um. I. Flow cytometry analysis (FC) for F4/80+ myeloid cells from ventral skin of WT and Prx1CreERT+Ikkbf/f mice. n=5–10 each. Data are represented as mean ± SEM of biological replicates. Animal experiments were repeated three times. *P<0.05, ns=not significant; Student’s t-test comparing WT to each respective experimental group.
To investigate the transcriptomic shift caused by the Ikkb deletion in Prx1-lineage cells, we performed scRNA-seq on skin cells from 4-week-old Prx1Cre+.Ikkbf/f mice (4,485 cells) and controls (6,599 cells). We applied iterative Random Forest Leave One Out Prediction (iRF-LOOP) (23), an explainable-AI method, to the combined samples by using each cell’s gene expression as a feature vector to create a predictive cell expression network. Next, Markov Clustering (MCL) (24) was applied at 1.30 inflation to the top 0.4% of edges in this network to create 316 separate clusters of cells (fig. S5), in which the gene expression profile of each cell within a cluster was highly predictive of the other cells in that cluster (data file S1). Cell types were probabilistically assigned to each cell via their marker genes by applying the CellAssign R package (25). This cell typing was projected onto the iRF-LOOP+MCL clusters and visualized using Cytoscape (26) (Fig. 5A). This high-granularity separation of the cells enabled us to identify group-based distributions between the control and Ikkb-deleted conditional knockout (cKO) groups (Fig. 5B).
Fig. 5.
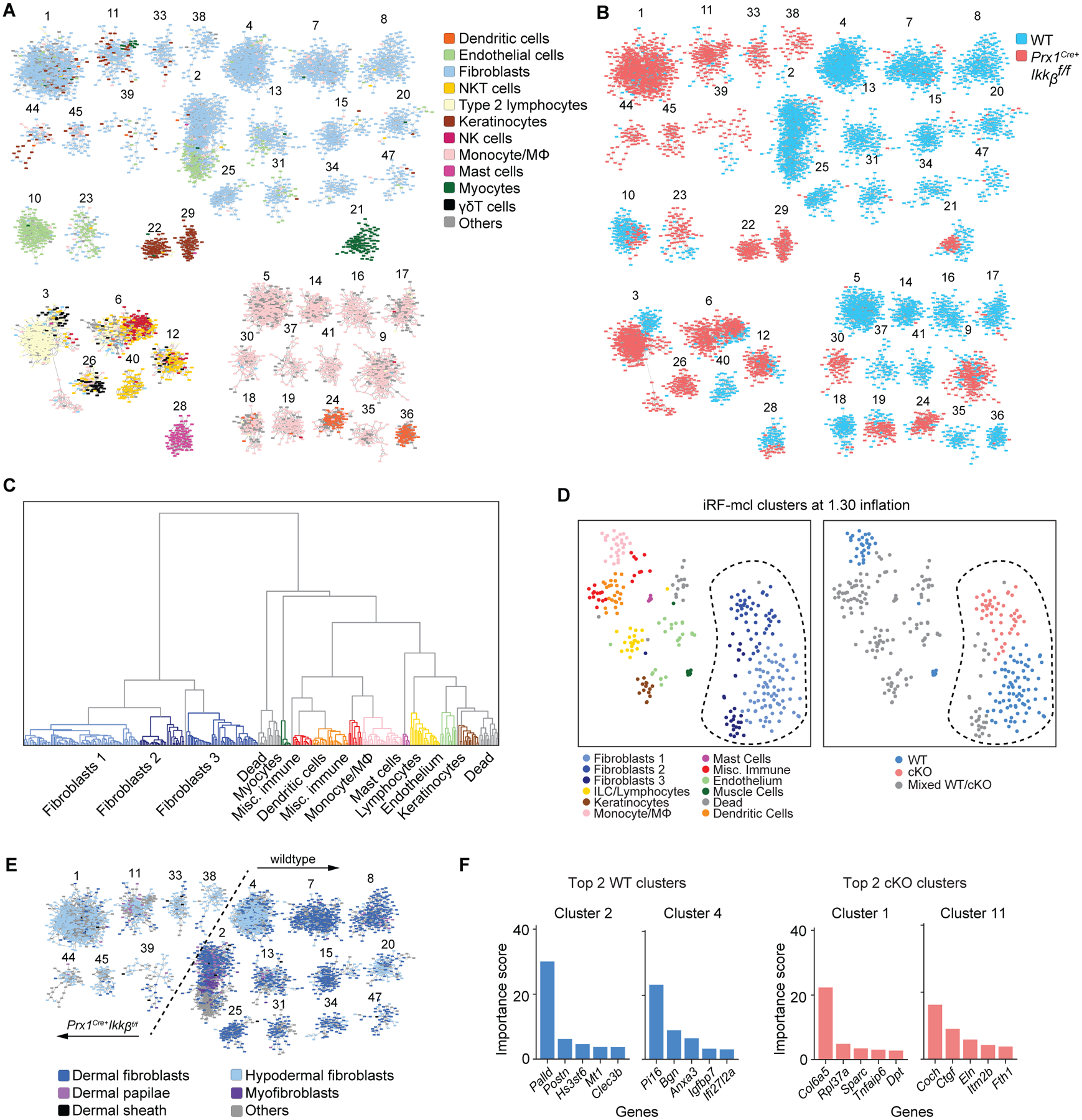
Single cell RNA sequencing reveals marked changes in fibroblast transcriptome by the Ikkb gene deletion. A. iRF-MCL (iterative random forest-Markov clustering) of 11084 cells from enzymatically digested ventral skin of WT and Prx1Cre+Ikkbf/f mice with cell type designation based on putative gene expression. Clusters with >50 cells are shown. B. WT (blue) and cKO (Prx1Cre+Ikkbf/f, red) group designation demonstrating even distribution of sample (3566 cells from WT, 2510 cells from cKO are shown). C. Hierarchical partitioning of Markov clusters and cell type designation, using Ward’s linkage method between Pearson correlations of mean expression vectors of the clusters. The groups generated by this partitioning have been t-SNE projected in Figure 5D and share the same color legend. D. t-SNE plot with cell type designation (left) and WT/cKO designation (right) demonstrating clear separation of fibroblast clusters (encircled in dashed line) derived from Ikkb-deleted group. Mixed WT/cKO cluster threshold was determined at <0.90 cutoff. E. Fibroblast subtyping based on (36) demonstrating a transcriptomic shift towards that of hypodermal fibroblasts when Ikkb is deleted (left of the dashed line). F. Importance score for genes that determined the formation of top 2 clusters from WT and cKO groups as characterized in A and B.
To determine inter-cluster relatedness, we performed hierarchical clustering analysis with Ward’s linkage method (27) on the mean expression vectors of each iRF-LOOP+MCL cluster and projected this separation onto a t-distributed stochastic neighbor embedding (t-SNE) plot of these vectors. Among the three major fibroblast groups identified by the hierarchical clustering (Fig. 5C), fibroblast group 2 was largely found in the Ikkb deletion group (Fig. 5D), indicating a substantial change in the transcriptome in fibroblasts caused by the Ikkb deletion. Fibroblasts from the control group had subtypes similar to those based on Joost et al. (28), while the majority of fibroblasts from the Ikkb-deleted group exhibited a transcriptome profile similar to hypodermal fibroblasts (Fig. 5E). Furthermore, we used each iRF-LOOP+MCL cluster identity as a class label and utilized iRF to obtain an ordered list of genes predictive of each cluster. The largest fibroblast clusters were predicted by the expression of Palld, a microfilament gene ubiquitously expressed in mesenchymal cells (29) and by Pi16, a gene that regulates neuropathic pain (30) (Fig. 5F). In contrast, the top two fibroblast clusters from the Ikkb-deleted mice were predicted by Col6a5 and Coch, which are highly upregulated in human AD and in the inner ear, respectively (31, 32) (Fig. 5F). These results suggest that the fibroblast phenotypes in the Ikkb deletion and control groups are functionally distinct.
We next hypothesized that Ikkb deletion in Prx1-lineage fibroblasts caused a severe immune cell infiltration by aberrant chemokine expression. Pairwise differential expression comparisons between all fibroblast clusters in the control and Ikkb deletion groups were examined using the DESeq2 R package (33), resulting in 88 such comparisons (data file S2). Several chemokines were upregulated in fibroblasts with Ikkb deletion, including Ccl7, Cxcl12, Ccl8, Ccl19 and Ccl11 (Fig. 6A). Among these, three chemokines (Cxcl12, Ccl11, and Ccl7) were upregulated in Ikkb-deleted fibroblasts with high Prrx1 unique molecular identifier (UMI) count (constituting 42% of fibroblasts) compared to those in control group (48% of fibroblasts) (Fig. 6A and fig. S6A; full data in data file S3). Gene ontology analysis revealed an upregulation of biological processes related to cell migration, cytokine response, and inflammatory response in the Prx1hi fibroblasts of the Ikkb-deleted group (Fig. 6B). Projecting gene expression heatmaps of Cxcl12, Ccl11, and Ccl7 onto the t-SNE visualization of the iRF-LOOP+MCL clusters revealed that Cxcl12 and Ccl7 are highly expressed by endothelial cells and macrophages, respectively. In contrast, Ccl11 expression was exclusive to fibroblasts and enhanced in the Ikkb deletion clusters (Fig. 6C and fig. S6B). Upregulation of Ccl11 in fibroblasts from the Ikkb-deleted mice was verified by the Seurat pipeline (34) (Fig. 6D and fig. S6, C to H). Immunofluorescence experiments demonstrated that CCL11+ cells with fibroblastic morphology were enriched in the ventral skin of the Ikkb-deleted group and were localized to the border between the dermis and subcutaneous adipose layer (Fig. 6E). Because CCL11 is a potent chemoattractant for eosinophils, we quantified the number of eosinophils in ventral skin and found that Ikkb deletion caused a marked increase in F4/80+Siglec-F+ eosinophil numbers (Fig. 6F). Because the F4/80+ myeloid cells we detected in Fig. 3 could be either macrophages or eosinophils, we examined the co-expression of markers associated with classically or alternatively activated macrophages (CD80 and CD206, respectively) and Siglec-F for eosinophilic identity, and found that over 80% of F4/80+ cells were identified as eosinophils in the ventral skin of Ikkb deletion mice (Fig. 6G). Thus, Ikkb deletion in skin fibroblasts causes aberrant expression of CCL11, which is associated with overt eosinophilia in early skin inflammation.
Fig. 6.
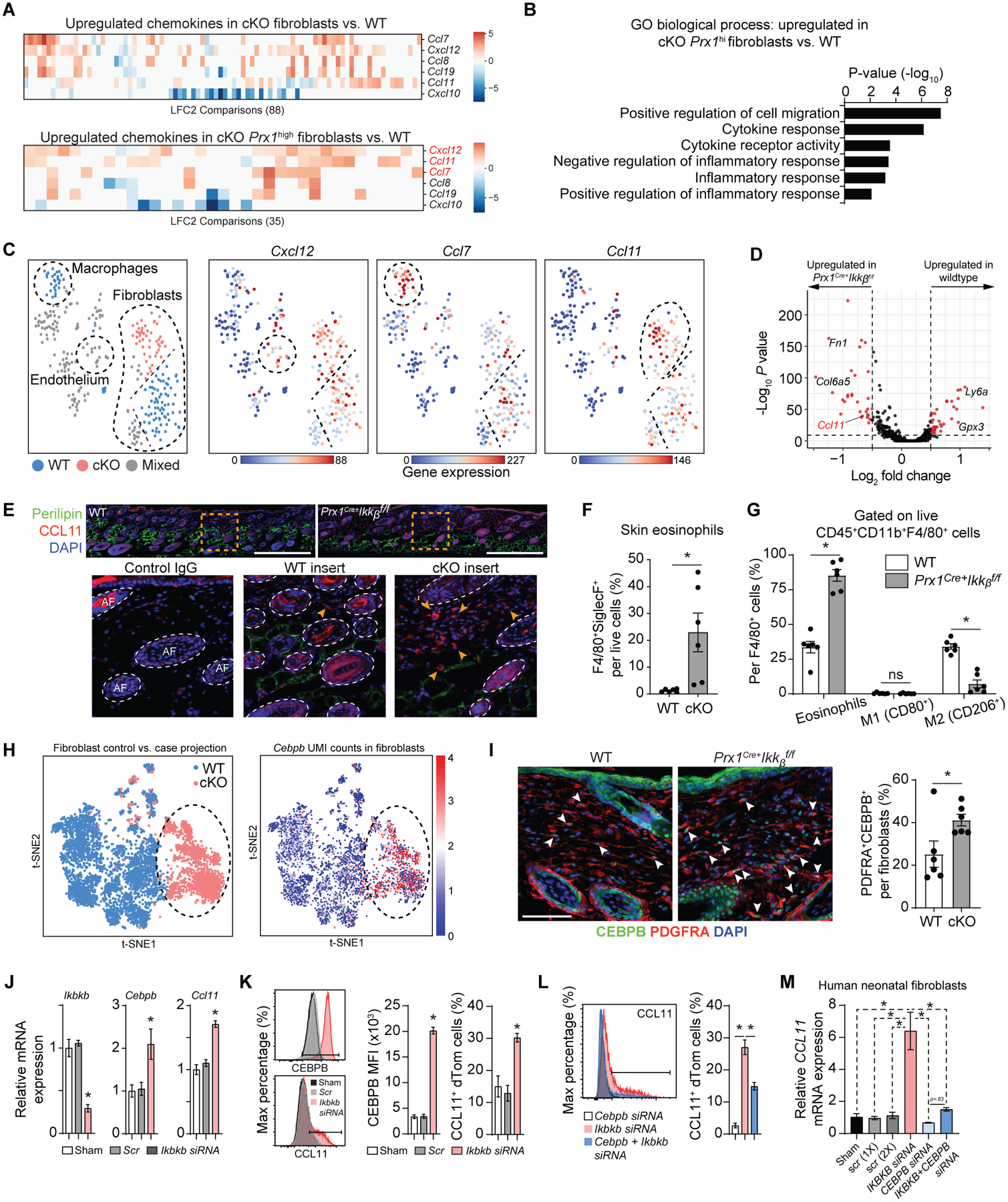
Ikkb deletion causes excessive Ccl11 expression by the fibroblasts and leads to subsequent eosinophilia. A. Pairwise comparisons of chemokine genes from each fibroblast clusters (top) and from Prx1high fibroblasts, comparing WT to Prx1Cre+Ikkbf/f (cKO) group. Notable upregulated chemokine genes were Cxcl12, Ccl11, and Ccl7. LFC2: log2 fold-change. B. Gene ontology (GO) analysis for biological process related to chemotaxis and inflammation based on genes upregulated in Prx1high fibroblasts from cKO group compared to that of WT. C. tSNE plots of iRF-MCL clusters with Cxcl12, Ccl7 and Ccl11 gene expression with notable upregulation of Ccl11 in fibroblast clusters derived from cKO group. D. Volcano plot of genes upregulated in WT and cKO groups using each fibroblast as a datapoint through single cell analysis pipeline established by Seurat and DESeq2 packages. E. In vivo validation of CCL11 protein upregulation by immunofluorescence with CCL11 antibody in ventral skin of 4-week-old WT and cKO mice. Scale bar, 200um. Insert images (orange box) show CCL11+ cells at the border of dermis and subcutaneous layers, as indicated by the arrows. Scale bar: 50um. Representative images are from n=7 each. Isotype IgG control shows autofluorescence in hair follicles. F. Flow cytometry analysis of eosinophil count (CD45+CD11b+F4/80+Siglec-F+) per total live cells, comparing WT versus cKO groups. G. Percentage of Siglec-F+ eosinophils, classically activated macrophages (M1, CD80+), and alternatively activated macrophages (M2, CD206+) per total CD45+CD11b+F4/80+ cells. H. t-SNE plot for 5,136 fibroblasts (3,609 WT and 1,527 cKO) in the scRNA-seq dataset, comparing both WT versus cKO (left) and Cebpb UMI expression (normalized and log10-transformed) (right). I. Immunofluorescence with antibodies specific to CEBPB and PDGFRA in 4-week-old WT and experimental mice (left) and quantification of CEBPB+PDGFRA+ fibroblasts. Scale bar, 50um. J. Quantitative real-time PCR (RT-qPCR) for Ikkb, Cebpb and Ccl11 mRNA in skin fibroblasts with no treatment (sham), scrambled siRNA control (Scr), or Ikkb siRNA (20nM). Cells were isolated from ventral skin of 2-week-old Prx1Cre+R26RdTomato mice and stimulated with low-dose IL4 (5ng/ml) for 12–16h before analysis. K. Flow cytometry analysis of Prx1+ fibroblasts for mean fluorescent intensity of CEBPB (left) or CCL11+ fibroblast numbers (right). Cells were gated for live/dTomato+ expression. L. Quantification of CCL11+ fibroblasts after knockdown with Cebpb siRNA alone, Ikbkb siRNA alone, and Cebpb+Ikbkb siRNA. M. Quantification of CCL11 mRNA by RT-qPCR with IKBKB knockdown alone or IKBKB+CEBPB knockdown in human neonatal fibroblasts without IL4 treatment. F to G. n=6 each. J to M. n=3 each. Data are represented as mean ± SEM of biological replicates. In vitro experiments were repeated twice, and animal experiments were repeated three times. *P<0.05, ns: not significant; Student’s t-test comparing WT to cKO groups (F and G) or one-way ANOVA followed by post-hoc test (J to M).
We next explored possible mechanisms by which Ikkb-deletion prevents NF-kB activation to paradoxically induce inflammation. ScRNA-seq data pointed to Cebpb (CCAAT/enhancer-binding protein beta), a basic leucine zipper domain transcription factor implicated in inflammation (35), as highly upregulated in fibroblasts with Ikkb-deletion (Fig. 6H). Increased CEBPB protein expression by the fibroblasts with Ikkb-deletion was validated by immunofluorescence studies (Fig. 6I). To test direct modulation of Cebpb and Ccl11 expression, we examined Prx1+ fibroblasts from 2-week-old Prx1Cre+.RosadTomato mice and found that Ikkb knockdown with small interfering RNA (siRNA) enhanced CEBPB and CCL11 mRNA and protein expression in vitro (Fig. 6, J and K). We then used a publicly available ChIP-seq database [Signaling Pathways Project, (36)] and noted high binding scores of CEBPB to CCL11 promoter/enhancer region in mesenchymal stromal cells (table S1), suggesting direct transcriptional regulation. Concomitant knockdown of Ikkb and Cebpb by siRNA significantly reduced CCL11 upregulation as noted by the diminished numbers of CCL11+Prx1+ fibroblasts compared to Ikkb knockdown alone (P<0.05; Fig. 6L), indicating that CEBPB is an important mechanistic link between Ikkb deletion and CCL11 upregulation in these cells. To show relevance with human fibroblasts, we examined CCL11 expression after IKBKB and CEBPB knockdown in two independent neonatal human fibroblast cell lines and found analogous results (Fig. 6M and fig. S6I).
We next examined the alteration in immune cell profiles by Ikkb deletion in fibroblasts. Within scRNA-seq data, type 2 lymphocytes (Th2) constituted the largest proportion of lymphocytes in mice with Ikkb deletion, and their absolute numbers were also elevated (Fig. 7A). The largest leukocyte cluster (Cluster 3) was mostly derived from the Ikkb deleted group, , and the formation of this cluster by iRF-LOOP+MCL was predicted by transcripts representing type-2 immunity (Il5, Areg and Il13) (Fig. 7B and fig. S7A). Seurat analysis verified this result and identified type-2 cytokines (Il5, Areg, Il13), Il1rl1 (encoding for ST2 receptor) and transcription factor Gata3 as upregulated transcripts in Ikkb-deleted group (Fig. 7, C and D and fig. S7, B to F). Other notable findings included myeloid cells from the cKO mice exhibiting increased pro-inflammatory Il1b gene and reduced pro-resolving Tgfb1 gene signatures (Fig. 7E), and mast cells from the cKO group with increased expression of Hdc and Fcer1a (fig. S8, A to C), which are genes encoding for histamine decarboxylase and the Fc-IgE receptor, respectively, that are implicated in pruritus (37). Additionally, serum IgE concentration was significantly higher in mice with Ikkb-deletion (P<0.05; fig. S8D), a characteristic finding in patients with AD (20). Based on the scRNA-seq results, we next hypothesized that early skin lesions are characterized by Th2 in addition to eosinophils, which worsens as mice age from 4 weeks (young) to 20 weeks (adult). We found that high eosinophil counts from young Ikkb-deleted mice were reduced with time (Fig. 7F). In contrast, CD3+CD4+IL4+ Th2 cells increased significantly as the mice aged (P<0.05; Fig. 7F) although there was no overall increase in CD4+ lymphocytes (Fig. 3B). Because type-2 immunity and eosinophilia are classic signs of human atopic dermatitis (20), we investigated if CCL11 is overexpressed by the fibroblasts in human AD lesions. Using RNAScope, we examined validated human AD skin with sex- and age-matched control and found that AD skin had a significant increase in the absolute and relative numbers of CCL11-expressing fibroblasts (CCL11+PDGFRA+) per total PDGFRA+ fibroblasts (P<0.05; Fig. 7, G and H and fig. S9A). Moreover, human AD skin had significantly higher numbers of CCL11+CEBPB+ cells (P<0.05; Fig. 7J and fig. S9B). This was consistent with our murine model of dysregulated fibroblasts as a source of CCL11 for early disease progression in AD-like lesions. We further examined published scRNA-seq data from human AD skin (10) for potential chemokine dysregulation by PRRX1+ mesenchymal cells and found that IKBKB was downregulated whereas CEBPB and CCL11 were upregulated (P<0.05; table S3 and data file S4), in agreement with our observations.
Fig. 7.
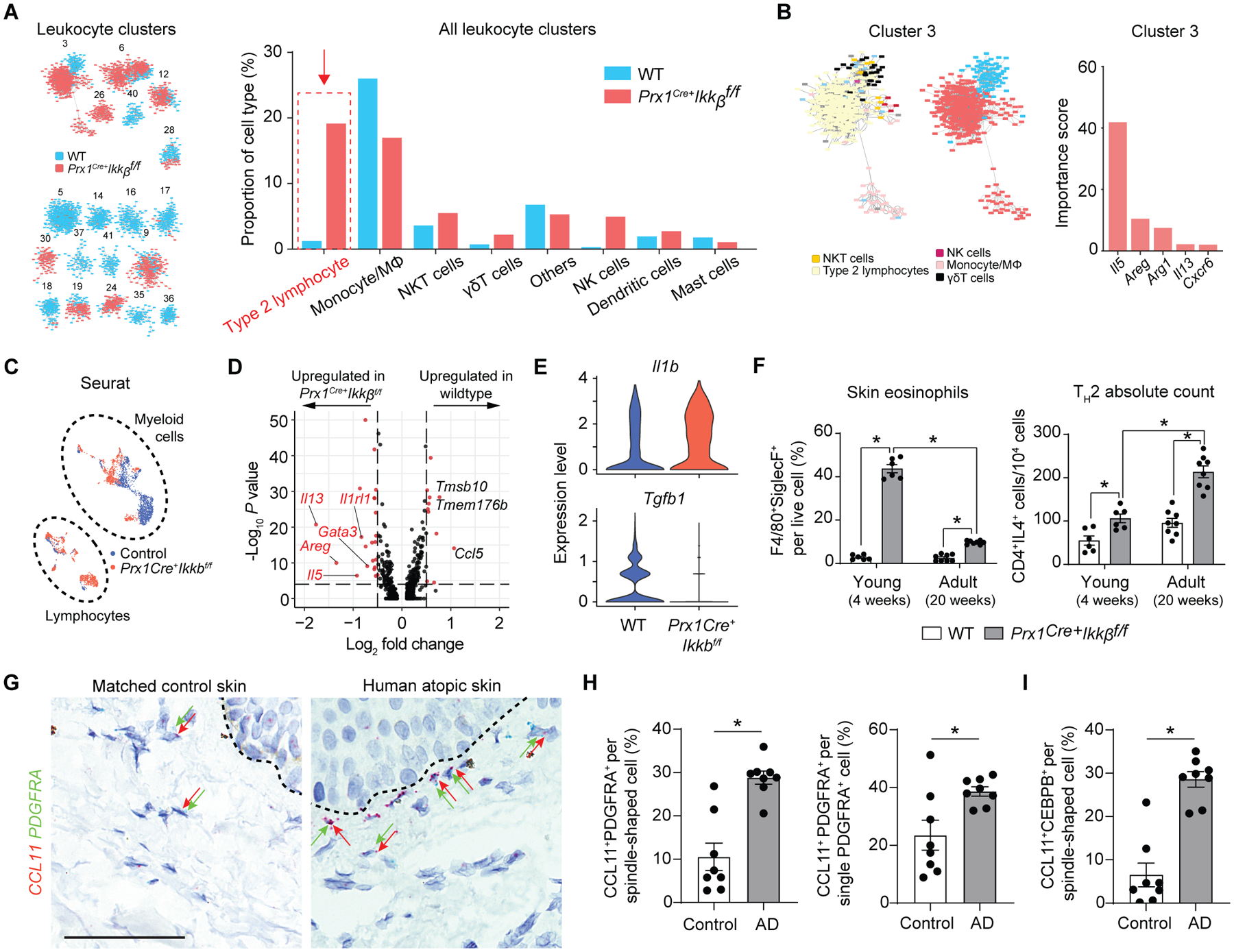
Ikkb deletion in fibroblasts causes a shift in inflammatory phenotype towards type-2 immune response, mimicking atopic skin in humans. A. Leukocyte frequency distribution between control (WT) and Prx1Cre+Ikkbf/f (cKO) groups demonstrating marked increase in Th2 proportion in cKO group. B. Importance scores of genes that determined the largest immune cell cluster (Cluster 3) that is predominated by cells of cKO group. Il5, Areg and Il13 are classic type 2 immune cytokines. C. Seurat clustering of immune cells from WT (blue) and cKO (red) mice; myeloid and lymphoid cell designation was determined by their putative gene expression as shown in fig. S7. D. Volcano plot of genes upregulated in lymphocyte population of WT and cKO groups using Seurat and DESeq2 packages. E. Violin plot of Il1b and Tgfb1 in myeloid cell population between WT and cKO mice. F. Flow cytometry analysis of skin eosinophils and CD3+CD4+IL4+ Th2 cells from young (4-week-old) and adult (20-week-old) WT and cKO mice. n=6–8 each. G. Representative RNAScope images of human control and atopic skin specimens detecting CCL11 (red) and PDGFRA (green dots) signals in dermis. Dashed line demarcates epidermal-dermal junction. Black arrows point to some PDGFRA+ fibroblasts and red arrows to CCL11+ cells. Scale bar, 50um. H. Quantification of double CCL11+PDGFRA+ cells per total spindle-shaped cells (left) and percentage of CCL11+ expressing fibroblasts by normalizing to total PDGFRA+ cells. I. Quantification of CCL11+CEBPB+ spindle-shaped cells in matched control versus human AD skin. n=8 each. Data are represented as mean ± SEM of biological replicates. Animal experiments were repeated three times, and RNAScope was repeated twice. *P<0.05; F, two-way ANOVA followed by Tukey’s post-hoc test; H and I, Student’s t-test.
To test if the aberrant upregulation of CCL11 by Ikkb-deleted fibroblasts is responsible for downstream events of inflammation, we performed an in vivo neutralization experiment by injecting a monoclonal CCL11 antibody into 2-week-old mice (Fig. 8A) and examined the effect on preventing eosinophilia and Th2 responses. Antibody treatment effectively reduced the number of eosinophil infiltrations by >50% in the skin of the Ikkb-deleted mice (Fig. 8B–D). Subsequently, the number of Th2 cells was significantly abrogated when CCL11 was inhibited in the skin of Ikkb-deleted mice (P<0.05; Fig. 8, E to F), demonstrating a therapeutic effect of CCL11 neutralization on the degree of eosinophilia and Th2 response in early atopic dermatitis-like skin lesions.
Fig. 8.
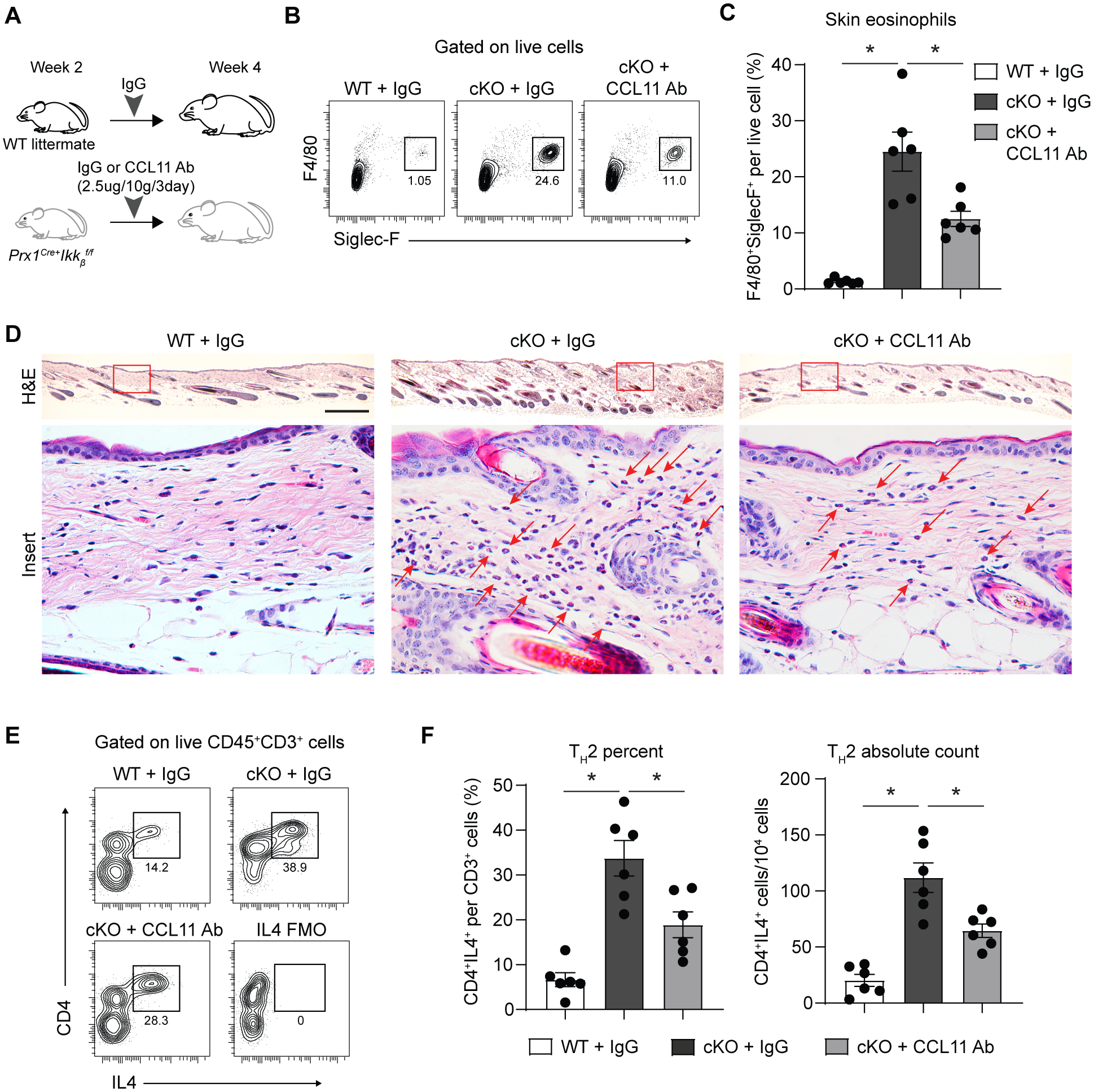
CCL11 neutralizing antibody treatment in young experimental mice with Ikkb deletion reduces eosinophil infiltration and Th2 cell numbers. A. Experimental design for CCL11 neutralization in young (2–4-weeks-old) mice. B. Flow cytometry plot of eosinophils (F4/80+Siglec-F+) from enzymatically digested ventral skin of control (WT) that received isotype IgG, Prx1Cre+Ikkbf/f (cKO) mice that received either isotype IgG or CCL11 neutralizing antibody. C. Quantification of eosinophils per total live cells comparing WT+IgG, cKO+IgG and cKO+CCL11 neutralizing antibody group. D. H&E images of ventral skin from WT mice that received control IgG, cKO mice that received IgG or CCL11 neutralizing antibody. Scale bar, 0.5mm. Red arrows point to eosinophils. Insert scale bar, 50um. E. Flow cytometry plot of Th2 cells (CD4+IL4+) on live CD45+CD3+ gated cells. F. Quantification of Th2 percent per total CD3+ T cells (left) and that of Th2 absolute count per 104 cells (right). Data are represented as mean ± SEM of biological replicates. Animal experiments were repeated three times. *P<0.05; one-way ANOVA followed by Tukey’s post-hoc test. Ab: antibody, FMO: fluorescence-minus-one control.
Discussion
Epithelial barrier disruption and immune dysregulation are thought to be the primary etiologic factors in chronic inflammatory skin conditions such as atopic dermatitis (2, 20), based on the classical experimental models utilizing tape-stripping, ectopic allergen, and/or gene deletion for immune function (38). Here, we show that skin fibroblasts, in addition to their traditional role in skin repair, are also critical regulators of immune homeostasis during early perinatal growth, which is regulated by Ikkb under basal physiologic conditions. Fibroblasts in which activation of the canonical NF-kB pathway is blocked by Ikkb gene deletion resulted in a skin phenotype that mirrors human atopic dermatitis-like skin lesions that exhibit eosinophilia and large numbers of type 2 immune cells (2, 20, 39). The lesion was specific to Prx1+ fibroblast-enriched ventral skin, an area where mice have limited ability to directly scratch. Because regular corn bedding can be abrasive to murine skin (40), we housed mice in a soft bedding material to reduce exogenous irritation but found it had no rescue effect. Thus, our mouse model likely reflects an “inside-out” mechanism of AD (20), where dysregulation of endogenous fibroblasts initiates a subsequent immune cascade, as opposed to external stimulation (“outside-in”, for example, byscratching). The AD-like lesions we observed in Prx1-driven Ikkb-deleted mice is corroborated by a recent study with Nestin-driven Ikkb in facial mesenchymal cells, both of which target phenotypically similar fibroblastic cells (41). Although the latter study did not identify any mechanisms that led to the dermal lesions, Nestin+ and Prx1+ fibroblastic cells are thought to have several similarities (42). Moreover, we found that Ikkb deletion in adipocytes had minimal effect on subcutaneous inflammation in young mice as it did in another study that examined 16-weeks-old adult mice (43). We showed that Ikkb-deleted fibroblasts aberrantly expressed CCL11 to initiate an eosinophilic and Th2 inflammation, and that blocking CCL11 effectively reduced eosinophilia and type 2 immune response. Collectively, these data demonstrate that Prx1+ fibroblasts may have a unique role in maintaining homeostasis during development and that disruption of Ikkb in these cells may lead to the formation of dermal lesions that share many similarities to human AD. Moreover, the results suggest that blocking CCL11 is a potentially important therapeutic approach to treating early inflammatory skin conditions.
Pediatric patients have a propensity for atopic dermatitis and other inflammatory skin conditions (44), suggesting a disease mechanism linked to early development. We were able to detect histologic changes prior to the gross formation of skin lesions in mice, providing new insight into the etiologic factors. Skin inflammation caused by Ikkb deletion occurred during perinatal development. The changes were not due to age-dependent reduction in pnPrx1+ cells, as their frequency in the skin dermis was similar in both neonatal and adult mice. Rather, these findings suggest that early perinatal growth is a critical time period during which skin fibroblasts with basal NF-kB activity are essential for their immuno-regulatory function, consistent with the AD susceptibility for children (20, 44, 45). Our findings highlight the previously unknown immune-regulatory function of young fibroblasts without which skin immunity fails to stabilize and causes severe inflammatory skin condition that closely mimics human atopy.
NF-kB activation is critical in modulating inflammatory immune responses (13), and its inhibition in chronic inflammatory conditions such as diabetes and autoimmunity alleviates the disease outcome (46, 47). However, we demonstrate that preventing NF-kB activation in skin fibroblasts paradoxically induces severe skin inflammation, contrary to the expected anti-inflammatory outcome. Despite the well-established pro-inflammatory role of NF-kB, there are recent examples where conditional deletion of Ikkb interferes with homeostasis. This has been shown in hepatocytes for glucose homeostasis (48) and for steady-state function of migratory dendritic cells (49). Thus, our results further strengthen the concept that in addition to mediating inflammatory responses, NF-kB plays an essential role in maintaining homeostasis under physiologic conditions. In the dermis, NF-kB specifically is needed in Prx1+ fibroblasts to prevent the overexpression of factors that generate a dermal inflammatory condition. For example, CEBPB was highly upregulated when Ikkb was deleted or knocked down in Prx1+ fibroblasts in vivo and in vitro. CEBPB enhances transcription of inflammatory cytokines (50) and is predicted to directly bind to Ccl11 promoter region in stromal cells (table S1). Moreover, CEBPB may interact with STAT proteins to indirectly upregulate CCL11 in fibroblasts (51). Importantly, double knockdown of Ikkb and Cebpb rescued Ccl11 upregulation induced by Ikkb knockdown alone, suggesting a modulatory role of Ikkb-NF-kB axis on CEBPB and subsequent upregulation of CCL11 expression by neonatal fibroblasts.
Here we have focused on aberrant CCL11 expression as a potent etiologic factor for eosinophil recruitment. We did not capture eosinophils in the scRNA-seq experiments due to our initial exclusion of high side-scatter granulocytes through cell sorting. Nevertheless, using other approaches we observed that the eosinophil population made up to 20–40% of cells obtained from the skin of young Ikkb-deleted mice, and that eosinophil dominance was substantially reduced in 20-week-old mice. In contrast, Th2 population increased with age in adult Ikkb-deleted mice, consistent with the overt type-2 immune response seen in human AD (20). Upregulation of CCL11 had been implicated in human atopic skin (52), though the cellular source is not well understood. In results similar to our findings, dermal fibroblasts from human atopic skin have been shown to exhibit higher CCL11 mRNA expressioncompared to control groups in response to IL4 stimulation in vitro (53). The etiology of human AD has been elusive, possibly because of the difficulty in detecting the subclinical phase of AD. The current therapeutics for moderate to severe AD utilize topical immunosuppressive drugs or monoclonal antibody against IL4Rα (dupilumab) to suppress Th2 response (54). Our data suggest that alternative approaches to block eosinophilia through chemokine antagonists will reduce Th2 response. In this regard, clinical trials are ongoing using bertilimumab, a humanized antibody against CCL11, to treat bullous pemphigoid [NCT02226146], an autoimmune skin disorder characterized by eosinophilia (55). Neutralization of CCL11 in early AD may be a key to preventing emergence of clinical AD symptoms and possibly atopic march.
ScRNA-seq studies on human AD samples have recently been reported (9, 10). When PRRX1+ mesenchymal cells were specifically examined, results consistent with our studies were observed, including a decrease in IKBKB and increases in CEBPB and CCL11. In contrast, another human study had CCL11 values that were too low to analyze (9). We also found in human specimens that CEBPB was highly upregulated in AD skin and in mice with Ikkb deletion. These studies support an immuno-modulatory role of Prx1+ cells for eosinophil trafficking to the skin. Interestingly, fibroblasts in developing human skin exhibit more pronounced inflammatory gene signatures than adult skin (10), suggesting an age-dependent mechanism. Our data indicate that the Ikkb-NF-kB signaling axis plays a role in CEBPB-dependent CCL11 production in dermal fibroblasts. However, additional immune mechanisms may also contribute to lesion formation.
Our study has limitations. We found that reduced Ikkb-NF-kB signaling through gene deletion triggered AD-like lesion formation in mice and that human fibroblasts from AD-lesions had reduced IKBKB expression. However, the mechanisms that may be responsible for reduced Ikkb-NF-kB signaling in human dermal fibroblastic cells are unknown. One possibility is AD susceptibility due to genetic mutation in IKBKB, similar to that of filaggrin loss-of-function mutation (20). We were able to trigger pre-clinical etiologic events in mice and temporally follow the steps that occur prior to the formation of established dermal lesions. Whether the same temporal sequence of events occurs in human AD lesions is not known and will be difficult to establish because of their pre-clinical nature. However, the studies provide insight into the cellular dysregulation that can lead to a type-2 lesion in the skin.
In summary, there is similarity between our mouse model of AD-like lesions and human AD, demonstrating the participation of the immunoregulatory transcriptome of Prx1+ skin fibroblasts in lesion formation. Because AD-like lesions arise from Ikkb-NF-kB perturbation in murine fibroblasts, it will be important to investigate similar mechanism in the early stages of human AD.
Materials and Methods
Study design.
The objective of this study was to investigate pathogenic mechanisms by which perturbation of Ikkb-NF-kB signaling in skin fibroblasts causes an inflammatory skin disorder that closely mirrors human AD. Mice that lacked Ikkb gene in Prx1+ mesenchyme were examined from neonatal to adult stages to identify initial event that preceded skin lesion development. Fibrotic etiology was assessed by biomechanical testing. The extent and type of skin inflammation was examined by flow cytometry and immunofluorescence, and systemic inflammation was examined by complete blood count with differential analysis and by multicolor flow cytometry of splenic and bone marrow cells. To test if the inflammatory effects were due to Prx1-derived skin fibroblasts or adipocytes, mice with lineage-specific deletion of Ikkb in pan-fibroblasts or adipocytes were examined. Prx1+ fibroblasts were further examined by scRNA-seq and iRF-LOOP analyses, which revealed a dramatic shift in chemotactic transcripts by the skin fibroblasts that lacked Ikkb gene. Further bioinformatics and flow cytometry analyses validated CCL11 as a primary component responsible for early eosinophilia that later transitioned to type 2 immunity, both of which are implicated in human AD. To establish clinical linkage, we used vetted human skin specimens to investigate if human AD lesions overexpressed CCL11 in fibroblasts by using in situ hybridization. We further tested for translational value of targeting CCL11 by preclinical treatment with neutralizing antibody in young mice and examined its impact on early eosinophilia and type-2 immunity. The study was carried out following ARRIVE guidelines. Study size was calculated by power analysis using each animal as an experimental unit. Animals were randomly allocated to either control or experimental group. Results were analyzed in a blinded fashion, such that the researcher was unaware of group designations. Validation of Ikkb deletion in animal models and flow cytometry gating strategies are provided in fig. S10 and fig. S11, respectively.
Animal studies.
All animal experiments were carried out conforming to a protocol approved by the University of Pennsylvania Institutional Animal Care and Use Committee (IACUC #804855). The following mice were purchased from Jackson Laboratory: B6.Cg-Tg(Prrx1-cre)1Cjt/J (Prx1Cre), B6.Cg-Tg(Prrx1-cre/ERT2,-EGFP)1Smkm/J (Prx1CreER), B6.Cg-Gt(ROSA)26Sortm9(CAG-tdTomato)Hze/J (R26TdTomato), Tg(Adipoq-cre)1Evdr (Adipoq-Cre), Tg(Col1a2-cre/ERT,-ALPP)7Cpd/J (Col1a2-CreER). Floxed Ikkβ mice (Ikkβf/f) were obtained from M. Karin (University of California, San Diego). Ikkb-deletion by Cre-mediated recombination was validated as in fig. S10. Mice were housed in regular corn bedding and were euthanized at 1-day, 4-week, and 16–20-week-old time points. In another set of experiments, animals were housed in Cellu-nest™ (Shepherd Specialty Papers) soft bedding from birth to the 4th week euthanasia time point.
Statistical analysis.
Statistical analysis was performed using Prism software (GraphPad-ver.9). All data are expressed as the mean ± SEM. Power analysis with alpha=0.05 and 40% effect was used to determine the sample size. Unpaired Student’s t-test was performed in comparing two groups (wildtype versus Ikkb-deleted mice) under the assumption of normal Gaussian distribution. For experiments comparing among three or more groups, one-way analysis of variance (ANOVA) or two-way ANOVA followed by Tukey’s posthoc analysis was used as indicated in the figure legends. P<0.05 was considered statistically significant. The individual animal was used as a unit of measurement (n), replicated twice or thrice as indicated in the figure legends, and sex was not considered as a factor in the analyses.
Further details are provided in the Supplementary Materials.
Supplementary Material
Acknowledgement.
The authors thank M. Karin (UCSD) for providing transgenic mice. This research used resources of the Oak Ridge Leadership Computing Facility, which is a Department of Energy Office of Science User Facility supported under Contract DE-AC05-00OR22725.
Funding.
This study was supported by NIH grants R01-DE019108 (to D.T.G.), K08-DE027129 (to K.I.K.), R01- DE030415 (to K.I.K.), R01-DA051908 (to D.J., M.R.G., J.J.M.), R01-ES028114 (to J.T.S) and P30-AR069589 (to J.T.S.). M.L.H.and Y.H. are supported by T32-AR007465 (NIAMS), and M.L.H. is supported by the Waine C. Johnson Endowed Research Fellowship.
Footnotes
List of Supplementary Materials.
Data files S1 to S5.
Competing interest. None declared.
Data and materials availability.
All data associated with this study are found in the main text and the supplementary materials. ScRNA-seq data is available through Gene Expression Omnibus (GEO: GSE172226). Computer codes for scRNA-seq analyses are available at https://doi.org/10.5281/zenodo.5826191. Animals and human neonatal primary fibroblasts from this study can be made available via materials transfer agreement upon request to the corresponding authors.
Reference and notes
- 1.Karimkhani C, Dellavalle RP, Coffeng LE, Flohr C, Hay RJ, Langan SM, Nsoesie EO, Ferrari AJ, Erskine HE, Silverberg JI, Vos T, Naghavi M, Global Skin Disease Morbidity and Mortality: An Update From the Global Burden of Disease Study 2013. JAMA Dermatol 153, 406–412 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Weidinger S, Beck LA, Bieber T, Kabashima K, Irvine AD, Atopic dermatitis. Nat Rev Dis Primers 4, 1 (2018). [DOI] [PubMed] [Google Scholar]
- 3.Greb JE, Goldminz AM, Elder JT, Lebwohl MG, Gladman DD, Wu JJ, Mehta NN, Finlay AY, Gottlieb AB, Psoriasis. Nat Rev Dis Primers 2, 16082 (2016). [DOI] [PubMed] [Google Scholar]
- 4.Arkwright PD, Motala C, Subramanian H, Spergel J, Schneider LC, Wollenberg A, Atopic A Dermatitis Working Group of the Allergic Skin Diseases Committee of the, Management of difficult-to-treat atopic dermatitis. J Allergy Clin Immunol Pract 1, 142–151 (2013). [DOI] [PubMed] [Google Scholar]
- 5.Pasparakis M, Haase I, Nestle FO, Mechanisms regulating skin immunity and inflammation. Nat Rev Immunol 14, 289–301 (2014). [DOI] [PubMed] [Google Scholar]
- 6.Rinkevich Y, Walmsley GG, Hu MS, Maan ZN, Newman AM, Drukker M, Januszyk M, Krampitz GW, Gurtner GC, Lorenz HP, Weissman IL, Longaker MT, Skin fibrosis. Identification and isolation of a dermal lineage with intrinsic fibrogenic potential. Science 348, aaa2151 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Leavitt T, Hu MS, Borrelli MR, Januszyk M, Garcia JT, Ransom RC, Mascharak S, desJardins-Park HE, Litzenburger UM, Walmsley GG, Marshall CD, Moore AL, Duoto B, Adem S, Foster DS, Salhotra A, Shen AH, Griffin M, Shen EZ, Barnes LA, Zielins ER, Maan ZN, Wei Y, Chan CKF, Wan DC, Lorenz HP, Chang HY, Gurtner GC, Longaker MT, Prrx1 Fibroblasts Represent a Pro-fibrotic Lineage in the Mouse Ventral Dermis. Cell Rep 33, 108356 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Tabib T, Morse C, Wang T, Chen W, Lafyatis R, SFRP2/DPP4 and FMO1/LSP1 Define Major Fibroblast Populations in Human Skin. J Invest Dermatol 138, 802–810 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.He H, Suryawanshi H, Morozov P, Gay-Mimbrera J, Del Duca E, Kim HJ, Kameyama N, Estrada Y, Der E, Krueger JG, Ruano J, Tuschl T, Guttman-Yassky E, Single-cell transcriptome analysis of human skin identifies novel fibroblast subpopulation and enrichment of immune subsets in atopic dermatitis. J Allergy Clin Immunol 145, 1615–1628 (2020). [DOI] [PubMed] [Google Scholar]
- 10.Reynolds G, Vegh P, Fletcher J, Poyner EFM, Stephenson E, Goh I, Botting RA, Huang N, Olabi B, Dubois A, Dixon D, Green K, Maunder D, Engelbert J, Efremova M, Polanski K, Jardine L, Jones C, Ness T, Horsfall D, McGrath J, Carey C, Popescu DM, Webb S, Wang XN, Sayer B, Park JE, Negri VA, Belokhvostova D, Lynch MD, McDonald D, Filby A, Hagai T, Meyer KB, Husain A, Coxhead J, Vento-Tormo R, Behjati S, Lisgo S, Villani AC, Bacardit J, Jones PH, O’Toole EA, Ogg GS, Rajan N, Reynolds NJ, Teichmann SA, Watt FM, Haniffa M, Developmental cell programs are co-opted in inflammatory skin disease. Science 371, (2021). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Croft AP, Campos J, Jansen K, Turner JD, Marshall J, Attar M, Savary L, Wehmeyer C, Naylor AJ, Kemble S, Begum J, Durholz K, Perlman H, Barone F, McGettrick HM, Fearon DT, Wei K, Raychaudhuri S, Korsunsky I, Brenner MB, Coles M, Sansom SN, Filer A, Buckley CD, Distinct fibroblast subsets drive inflammation and damage in arthritis. Nature 570, 246–251 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Liu T, Han C, Wang S, Fang P, Ma Z, Xu L, Yin R, Cancer-associated fibroblasts: an emerging target of anti-cancer immunotherapy. J Hematol Oncol 12, 86 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Hayden MS, Ghosh S, NF-kappaB in immunobiology. Cell research 21, 223–244 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Zhang Q, Lenardo MJ, Baltimore D, 30 Years of NF-kappaB: A Blossoming of Relevance to Human Pathobiology. Cell 168, 37–57 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Leung TH, Zhang LF, Wang J, Ning S, Knox SJ, Kim SK, Topical hypochlorite ameliorates NF-kappaB-mediated skin diseases in mice. J Clin Invest 123, 5361–5370 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Liu T, Zhang L, Joo D, Sun SC, NF-kappaB signaling in inflammation. Signal Transduct Target Ther 2, (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Oeckinghaus A, Hayden MS, Ghosh S, Crosstalk in NF-kappaB signaling pathways. Nature immunology 12, 695–708 (2011). [DOI] [PubMed] [Google Scholar]
- 18.Logan M, Martin JF, Nagy A, Lobe C, Olson EN, Tabin CJ, Expression of Cre Recombinase in the developing mouse limb bud driven by a Prxl enhancer. Genesis 33, 77–80 (2002). [DOI] [PubMed] [Google Scholar]
- 19.Durland JL, Sferlazzo M, Logan M, Burke AC, Visualizing the lateral somitic frontier in the Prx1Cre transgenic mouse. J Anat 212, 590–602 (2008). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Langan SM, Irvine AD, Weidinger S, Atopic dermatitis. Lancet 396, 345–360 (2020). [DOI] [PubMed] [Google Scholar]
- 21.Greenbaum A, Hsu YM, Day RB, Schuettpelz LG, Christopher MJ, Borgerding JN, Nagasawa T, Link DC, CXCL12 in early mesenchymal progenitors is required for haematopoietic stem-cell maintenance. Nature 495, 227–230 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Sanchez-Gurmaches J, Hsiao WY, Guertin DA, Highly selective in vivo labeling of subcutaneous white adipocyte precursors with Prx1-Cre. Stem Cell Reports 4, 541–550 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Cliff A, Romero J, Kainer D, Walker A, Furches A, Jacobson D, A High-Performance Computing Implementation of Iterative Random Forest for the Creation of Predictive Expression Networks. Genes (Basel) 10, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.van Dongen S, A cluster algorithm for graphs. Information Systems [INS], (2000). [Google Scholar]
- 25.Zhang AW, O’Flanagan C, Chavez EA, Lim JLP, Ceglia N, McPherson A, Wiens M, Walters P, Chan T, Hewitson B, Lai D, Mottok A, Sarkozy C, Chong L, Aoki T, Wang X, Weng AP, McAlpine JN, Aparicio S, Steidl C, Campbell KR, Shah SP, Probabilistic cell-type assignment of single-cell RNA-seq for tumor microenvironment profiling. Nat Methods 16, 1007–1015 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Shannon P, Markiel A, Ozier O, Baliga NS, Wang JT, Ramage D, Amin N, Schwikowski B, Ideker T, Cytoscape: a software environment for integrated models of biomolecular interaction networks. Genome Res 13, 2498–2504 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Ward J, Hierarchical Grouping to Optimize an Objective Function. Journal of the American Statistical Association 58, 236–244 (1963). [Google Scholar]
- 28.Joost S, Annusver K, Jacob T, Sun X, Dalessandri T, Sivan U, Sequeira I, Sandberg R, Kasper M, The Molecular Anatomy of Mouse Skin during Hair Growth and Rest. Cell stem cell 26, 441–457 e447 (2020). [DOI] [PubMed] [Google Scholar]
- 29.Parast MM, Otey CA, Characterization of palladin, a novel protein localized to stress fibers and cell adhesions. The Journal of cell biology 150, 643–656 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Singhmar P, Trinh RTP, Ma J, Huo X, Peng B, Heijnen CJ, Kavelaars A, The fibroblast-derived protein PI16 controls neuropathic pain. Proceedings of the National Academy of Sciences of the United States of America 117, 5463–5471 (2020). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Soderhall C, Marenholz I, Kerscher T, Ruschendorf F, Esparza-Gordillo J, Worm M, Gruber C, Mayr G, Albrecht M, Rohde K, Schulz H, Wahn U, Hubner N, Lee YA, Variants in a novel epidermal collagen gene (COL29A1) are associated with atopic dermatitis. PLoS Biol 5, e242 (2007). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Robertson NG, Cremers CW, Huygen PL, Ikezono T, Krastins B, Kremer H, Kuo SF, Liberman MC, Merchant SN, Miller CE, Nadol JB Jr., Sarracino DA, Verhagen WI, Morton CC, Cochlin immunostaining of inner ear pathologic deposits and proteomic analysis in DFNA9 deafness and vestibular dysfunction. Hum Mol Genet 15, 1071–1085 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Love MI, Huber W, Anders S, Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM 3rd, Hao Y, Stoeckius M, Smibert P, Satija R, Comprehensive Integration of Single-Cell Data. Cell 177, 1888–1902 e1821 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.McPeak MB, Youssef D, Williams DA, Pritchett CL, Yao ZQ, McCall CE, El Gazzar M, Frontline Science: Myeloid cell-specific deletion of Cebpb decreases sepsis-induced immunosuppression in mice. Journal of leukocyte biology 102, 191–200 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Ochsner SA, Abraham D, Martin K, Ding W, McOwiti A, Kankanamge W, Wang Z, Andreano K, Hamilton RA, Chen Y, Hamilton A, Gantner ML, Dehart M, Qu S, Hilsenbeck SG, Becnel LB, Bridges D, Ma’ayan A, Huss JM, Stossi F, Foulds CE, Kralli A, McDonnell DP, McKenna NJ, The Signaling Pathways Project, an integrated ‘omics knowledgebase for mammalian cellular signaling pathways. Sci Data 6, 252 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Hirasawa N, Expression of Histidine Decarboxylase and Its Roles in Inflammation. Int J Mol Sci 20, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Jin H, He R, Oyoshi M, Geha RS, Animal models of atopic dermatitis. J Invest Dermatol 129, 31–40 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Liu FT, Goodarzi H, Chen HY, IgE, mast cells, and eosinophils in atopic dermatitis. Clin Rev Allergy Immunol 41, 298–310 (2011). [DOI] [PubMed] [Google Scholar]
- 40.R. C. L. Carter NS, Management of Animal Care and Use Programs in Research, Education, and Testing. T. G. Weichbrod RH, Norton JN, Ed., Chapter 27. Feed and Bedding (CRC Press/Taylor & Francis, Boca Raton, Florida, ed. Second, 2018). [PubMed] [Google Scholar]
- 41.Nunomura S, Ejiri N, Kitajima M, Nanri Y, Arima K, Mitamura Y, Yoshihara T, Fujii K, Takao K, Imura J, Fehling HJ, Izuhara K, Kitajima I, Establishment of a Mouse Model of Atopic Dermatitis by Deleting Ikk2 in Dermal Fibroblasts. J Invest Dermatol 139, 1274–1283 (2019). [DOI] [PubMed] [Google Scholar]
- 42.Kfoury Y, Scadden DT, Mesenchymal cell contributions to the stem cell niche. Cell stem cell 16, 239–253 (2015). [DOI] [PubMed] [Google Scholar]
- 43.Park SH, Liu Z, Sui Y, Helsley RN, Zhu B, Powell DK, Kern PA, Zhou C, IKKbeta Is Essential for Adipocyte Survival and Adaptive Adipose Remodeling in Obesity. Diabetes 65, 1616–1629 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Chovatiya R, Silverberg JI, Pathophysiology of Atopic Dermatitis and Psoriasis: Implications for Management in Children. Children (Basel) 6, (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Kim J, Kim BE, Leung DYM, Pathophysiology of atopic dermatitis: Clinical implications. Allergy Asthma Proc 40, 84–92 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Ko KI, Syverson AL, Kralik RM, Choi J, DerGarabedian BP, Chen C, Graves DT, Diabetes-Induced NF-kappaB Dysregulation in Skeletal Stem Cells Prevents Resolution of Inflammation. Diabetes 68, 2095–2106 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Herrington FD, Carmody RJ, Goodyear CS, Modulation of NF-kappaB Signaling as a Therapeutic Target in Autoimmunity. J Biomol Screen 21, 223–242 (2016). [DOI] [PubMed] [Google Scholar]
- 48.Liu J, Ibi D, Taniguchi K, Lee J, Herrema H, Akosman B, Mucka P, Salazar Hernandez MA, Uyar MF, Park SW, Karin M, Ozcan U, Inflammation Improves Glucose Homeostasis through IKKbeta-XBP1s Interaction. Cell 167, 1052–1066 e1018 (2016). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Baratin M, Foray C, Demaria O, Habbeddine M, Pollet E, Maurizio J, Verthuy C, Davanture S, Azukizawa H, Flores-Langarica A, Dalod M, Lawrence T, Homeostatic NF-kappaB Signaling in Steady-State Migratory Dendritic Cells Regulates Immune Homeostasis and Tolerance. Immunity 42, 627–639 (2015). [DOI] [PubMed] [Google Scholar]
- 50.Rahman SM, Janssen RC, Choudhury M, Baquero KC, Aikens RM, de la Houssaye BA, Friedman JE, CCAAT/enhancer-binding protein beta (C/EBPbeta) expression regulates dietary-induced inflammation in macrophages and adipose tissue in mice. J Biol Chem 287, 34349–34360 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Zhou X, Hu H, Balzar S, Trudeau JB, Wenzel SE, MAPK regulation of IL-4/IL-13 receptors contributes to the synergistic increase in CCL11/eotaxin-1 in response to TGF-beta1 and IL-13 in human airway fibroblasts. Journal of immunology 188, 6046–6054 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Owczarek W, Paplinska M, Targowski T, Jahnz-Rozyk K, Paluchowska E, Kucharczyk A, Kasztalewicz B, Analysis of eotaxin 1/CCL11, eotaxin 2/CCL24 and eotaxin 3/CCL26 expression in lesional and non-lesional skin of patients with atopic dermatitis. Cytokine 50, 181–185 (2010). [DOI] [PubMed] [Google Scholar]
- 53.Gahr N, Folster-Holst R, Weichenthal M, Christophers E, Schroder JM, Bartels J, Dermal fibroblasts from acute inflamed atopic dermatitis lesions display increased eotaxin/CCL11 responsiveness to interleukin-4 stimulation. Br J Dermatol 164, 586–592 (2011). [DOI] [PubMed] [Google Scholar]
- 54.Shirley M, Dupilumab: First Global Approval. Drugs 77, 1115–1121 (2017). [DOI] [PubMed] [Google Scholar]
- 55.Amber KT, Valdebran M, Kridin K, Grando SA, The Role of Eosinophils in Bullous Pemphigoid: A Developing Model of Eosinophil Pathogenicity in Mucocutaneous Disease. Front Med (Lausanne) 5, 201 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Waltman L, van Eck NJ, A smart local moving algorithm for large-scale modularity-based community detection. Eur. Phys. J. B 86, (2013). [Google Scholar]
- 57.McInnes L, Healy J, Melville J, UMAP: Uniform Manifold Approximation and Projection for Dimension Reduction. arXiv:1802.03426 [stat.ML], (2018). [Google Scholar]
- 58.Vieth B, Parekh S, Ziegenhain C, Enard W, Hellmann I, A systematic evaluation of single cell RNA-seq analysis pipelines. Nature communications 10, 4667 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Wang T, Li B, Nelson CE, Nabavi S, Comparative analysis of differential gene expression analysis tools for single-cell RNA sequencing data. BMC Bioinformatics 20, 40 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
All data associated with this study are found in the main text and the supplementary materials. ScRNA-seq data is available through Gene Expression Omnibus (GEO: GSE172226). Computer codes for scRNA-seq analyses are available at https://doi.org/10.5281/zenodo.5826191. Animals and human neonatal primary fibroblasts from this study can be made available via materials transfer agreement upon request to the corresponding authors.