

Burst suppression is a stereotypical EEG pattern that can be induced by general anaesthesia. Liou et al. show that creating a seizure focus in rodent neocortex alters the spatial homogeneity of isoflurane-induced burst suppression in a highly reproducible manner, allowing the spatial limits of the epileptic focus to be mapped.
Keywords: burst suppression, seizure focus, calcium imaging, multielectrode array
Abstract
Burst suppression is an electroencephalogram pattern of globally symmetric alternating high amplitude activity and isoelectricity that can be induced by general anaesthetics. There is scattered evidence that burst suppression may become spatially non-uniform in the setting of underlying pathology. Here, we induced burst suppression with isoflurane in rodents and then created a neocortical acute seizure focus with injection of 4-aminopyridine (4-AP) in somatosensory cortex. Burst suppression events were recorded before and after creation of the focus using bihemispheric wide-field calcium imaging and multielectrode arrays. We find that the seizure focus elicits a rapid alteration in triggering, initiation, and propagation of burst suppression events. Compared with the non-seizing brain, bursts are triggered from the thalamus, initiate in regions uniquely outside the epileptic focus, elicit marked increases of multiunit activity and propagate towards the seizure focus. These findings support the rapid, widespread impact of focal epilepsy on the extended brain network.
Introduction
Seizures are abnormal electrophysiological events inextricably linked to the underlying circuitry of the brain (Goldberg and Coulter, 2013; Scharfman, 2016). Indeed, the presence of an epileptic focus has a profound influence on neuronal circuitry (Smith and Schevon, 2016). Not only are long-term pathological circuits formed, which cause epileptogenesis and seizure initiation (Sutula, 2002), but the presence of the epileptic focus also causes rapid modulation in normal local and long-range neuronal connections (Blumenfeld, 2014; Liou et al., 2018). Animal models of acute focal epilepsy, in particular, provide a method for analysing short-term alterations in brain circuitry since the ictal events can be triggered pharmacologically in one small area of the brain in the setting of healthy cortical architecture.
Although much work has focused on the imbalance of excitation and inhibition and the increase in synaptic transmission just before and during seizures (Casillas-Espinosa et al., 2012; Kaila et al., 2014), less is understood about the impact of seizures on widespread network interactions during the interictal period. Functional imaging with PET and functional MRI have shown far reaching depression in both cortical metabolism (Sarikaya, 2015) and blood flow in human patients (Chaudhary et al., 2013). However, little data exists on the balance of excitation and inhibition during the interictal period. To probe neuronal network interactions, we used burst suppression, which is known to elicit a stereotypical electrophysiological pattern throughout the brain, and investigated the alterations in the burst suppression pattern after creating a localized neocortical epileptic focus.
Burst suppression is an EEG pattern typically characterized by alternating periods of high voltage activity (burst) with inter-burst refractory periods (suppression) (Swank and Watson, 1949). Burst suppression can be observed under general anaesthesia, but can also occur in pathological conditions such as anoxia, coma or cortical deafferentation (Brown et al., 2010; Purdon et al., 2015). EEG and single unit studies have indicated that burst generators may be located within the neocortex and represent a state of hyperexcitability caused by a reduction in cortical inhibition and preserved glutamatergic excitation (Detsch et al., 2002; Lukatch et al., 2005; Kroeger and Amzica, 2007; Ferron et al., 2009). Triggers for bursts, however, can be caused by intrinsic rhythmicity in either cortex or thalamus, subthreshold sensory input or micromechanical stimulation (Steriade et al., 1994; Detsch et al., 2002; Hudetz and Imas, 2007; Kroeger and Amzica, 2007).
Burst suppression is thought to represent a spatially homogeneous brain state in both the burst and suppression phases (Clark and Rosner, 1973), mediated by callosal connections (Lazar et al., 1999). However, rare scattered reports in human patients have demonstrated that pathological abnormalities in the brain may induce spatial inhomogeneity in burst suppression events (Lazar et al., 1999; Lewis et al., 2013).
In this study, we investigated the relationship between burst suppression and an acute neocortical seizure focus in a rodent model. Burst suppression was induced with isoflurane anaesthesia and an epileptic focus was created with neocortical injection of 4-aminopyridine (4-AP). Using bulk calcium imaging and multielectrode array recordings from the bilateral cortex and the thalamus, we examined how the presence of an epileptic focus impacts the cortical-subcortical network underlying burst suppression events and its spatial and temporal homogeneity. We find that the acute seizure focus causes a dramatic alteration in the spatiotemporal dynamics of burst suppression events and that these inhomogeneities may actually be useful in identifying the location of the focus. The clinical relevance and impact of these studies could be dramatic since several treatment options exist for localized epilepsy that are only useful if the spatial topography of the focus is established.
Materials and methods
Animal preparation
All experimental procedures were approved by the Weill Cornell Medical College Animal Care and Use Committee following the National Institutes of Health guidelines. Adult male Sprague–Dawley rats (200–350 g) were anaesthetized with isoflurane in 70% N2:30% O2, 4% induction, and 1–2% maintenance, an anaesthesia depth that has been shown to induce burst suppression (Kroeger and Amzica, 2007). Isoflurane concentration was titrated until burst suppression rhythm was generated and then kept stable. Body temperature was maintained at 37°C with a regulated heating blanket (Harvard Apparatus). Heart rate, pO2, and the end tidal carbon dioxide (EtCO2) were monitored with a small animal capnography (Surgivet) and were sustained throughout the experiment (heart rate: 250–300 beats/min, pO2 > 90%, EtCO2 ∼ 25–28 mmHg). The head was fixed in a stereotaxic frame.
Focal epilepsy model and cortical local field potential recording
4-AP (Sigma-Aldrich, 15 mM, 0.5 μl), a potassium channel blocker, was injected 300–500 μm below the cortical surface through a glass electrode (50–100 µm tip opening) using a Nanoject II injector (Drummond Scientific). 4-AP was injected in the somatosensory cortex. The injection site’s local field potential (LFP) was simultaneously recorded through the 4-AP electrode. The LFP was amplified, band-pass filtered (1–500 Hz) using a Grass amplifier, digitized using CED Power 1401 and recorded via a PC running Spike2 software (Cambridge Electronic Design).
Calcium dye staining
The calcium indicator Oregon Green 488 BAPTA-AM (OGB-1, Life Technologies) was used for wide-field recording of neuronal activity. Convection enhanced delivery was used to bulk load the entire neocortex with OGB-1 (Ma et al., 2014). In brief, 50 µg of OGB-1 was diluted in 5 µl of DMSO-F127 then in 50 µl of artificial CSF. Eight microlitres of OGB-1 solution was injected in the neocortex via a glass electrode (50–100 µm opening) placed ∼1 mm below the brain surface at the speed of 100 nl/min, using a micro-pump (WPIa). A ∼5 × 8 mm craniotomy window was then opened over each stained hemisphere between bregma and lambda, and the exposed brain was covered with silicon oil (12 500 centistoke) to preserve cortical moisture.
Optical recording
A CCD camera (Dalsa camera in Imager 3001, Optical Imaging; or J-MC023MGSY, Lighting Mind Inc.) using a tandem lens (50 × 50 mm) arrangement was focused 300–400 μm below the cortical surface. A 470 nm LED light (Thorlabs) was used as the illumination source for calcium-sensitive dye. The illumination was directed using fibre-optic light guides. A 510-nm long pass filter was placed before the camera to record the calcium fluorescence. Images were acquired with a framerate of 55 Hz.
Analysis of optical data
The calcium imaging data were calculated as dF/F, where F was the baseline illumination when no epileptiform activity was occurring and dF was the signal change during epileptiform activity as identified by the LFP. A 1-Hz high pass filter was applied to the calcium data in order to remove the glial contribution (Daniel et al., 2015).
The calcium data during burst suppression were then normalized to the peak amplitude of each individual pixel. The time point for when the trace reached 0.5 was defined as the onset time of that pixel. The pixel with the earliest onset time was defined as the initiation site.
To quantify the spatial extent of the neuronal component of the ictal events and bursts, we employed a seed trace initiated correlation method (White et al., 2011). For each ictal event or burst, a seed trace was selected from a small region of interest, corresponding to the 4-AP injection and LFP recording site. The correlation coefficients between the seed trace and the trace from every individual pixel in the surrounding image were calculated. A heat map was generated using the correlation coefficient at each pixel. A threshold at 50% of the maximal correlation coefficients was used to determine the spatial extent of the imaged signals.
Bilateral cortical multielectrode recording of LFP and multiunit activity
The multielectrode arrays used in this study were a customized pair of 5 × 10 grids with interelectrode distance 400 µm (Blackrock Microsystems Inc.). Animals were prepared analogously except for the addition of a bilateral durotomy to prevent damage to the multielectrode arrays. Each of the two grids were implanted into each hemisphere respectively by using a 0.5 mm pneumatic implanter (Blackrock Microsystems Inc, Salt Lake City, UT). The long side of both grids (10 electrodes) aligned with the animal’s anterior-posterior axis on both exposed hemispheres. The grids’ anterior edges met the animal’s somatosensory cortices. Raw electrical signal was 0.3 Hz to 7.5 kHz band-passed and 30 kHz digitally sampled with 16-bit precision (range ± 8 mV, resolution 0.25 μV). Raw data were subsequently separated into multiunit activity (MUA, 500 to 3000 Hz, 512-order zero-phase shift, window-based FIR filter) and LFP bands (500 Hz low pass, 90 order zero-phase shift, window-based FIR filter). The LFP band was further down-sampled to 1 kHz. After animal stabilization, a 2-min baseline recording was obtained before 4-AP injection. The baseline signals were used to calculate root mean square (RMS) of each signal band, estimated by robust statistics, ?>/0.6745 (Quiroga et al., 2004). Both bands were also visually inspected. Noisy channels with MUA band RMS > 8 μV or with excessive paroxysmal artefacts were discarded.
Multiunit spikes were detected from the MUA band (500–3000 Hz) by a threshold crossing method (Quiroga et al., 2004). Threshold was set to be −4.5 RMS (Schevon et al., 2012; Smith et al., 2016; Liou et al., 2017, 2018). The detection refractory period was set to 1 ms to avoid noise-induced false repeated detection. Detection results were further visually reviewed to remove artefacts. For each channel, its instantaneous multiunit firing rate was estimated by convolving the multiunit spike train with a 10-ms Gaussian kernel (Bokil et al., 2010; Liou et al., 2017), sampled every 1 ms.
Detection of ictal events
Ictal events were detected by visually reviewing the LFP signal recorded at the 4-AP injection site, either via the injection pipette in the case of calcium imaging studies or the electrode that is closest to the injection site in multielectrode array studies. Ictal events were defined as large amplitude, rhythmic epileptiform episodes which evolved continuously and lasted >5 s, commonly presented with a significant initial LFP discharge followed by low voltage fast activity (Ma et al., 2009; Zhao et al., 2009, 2011; Liou et al., 2018). Ictal events were required to have at least a 20-s interictal interval to be considered two separate episodes. The periods in between are therefore named ‘interictal periods’.
Analysing general anaesthetic-induced burst suppression rhythm
The continuous isoflurane-induced background burst suppression rhythm was temporally divided into burst and suppression phases (hereafter abbreviated as ‘burst’ and ‘suppression’, respectively). The LFPs recorded at 4-AP injection sites served as a reference for this division, supplemented by either calcium imaging or multielectrode recordings to avoid falsely identifying 4-AP ictal activity, which is focal and unilateral, from general anaesthetic-induced bursts, which are instead general and bilateral. Candidates of burst were first systematically searched by screening non-ictal periods by a threshold crossing method, looking for periods when LFP signals go beyond ± 3 RMS. Candidate periods which were temporally close to each other (<500-ms apart) were merged. All burst candidates were further visually reviewed, manually merged, removed, or separated. Bursts that occurred during interictal periods were labelled as interictal bursts, whereas those that occurred before 4-AP injection were labelled baseline bursts. Suppression was defined as electrically inactive periods (less than ± 3 RMS) that were both immediately preceded and followed by bursts. Burst suppression ratio, defined as the fraction of time spent in suppression (Vijn and Sneyd, 1998), was used as an indicator of overall cortical activity.
Detecting and analysing local neural activity during bursts
Bursts which were associated with local massive multiunit firings (≥2 neighbouring electrodes with instantaneous multiunit firing rate ≥50 spikes/s) and spiky LFP fluctuations (≥500 μV within 70 ms) (Noachtar et al., 1999) were considered bursts with local paroxysmal neural activities. They were further classified into ipsilateral and contralateral ones according to which hemisphere the paroxysmal neural activities were observed.
For each burst with local paroxysmal neural activity, multiunit spikes during the period from −35 to +35 ms with respect to the timing of peak instantaneous multiunit firing rate were further extracted to test if neurons were activated synchronously or sequentially in space, i.e. like traveling waves (Ma et al., 2013; Smith et al., 2016). Multivariate linear regression was applied to test the traveling wave hypothesis (Liou et al., 2017)
| (1) |
where ?> was multiunit spike index, ?> was the timing of multiunit spikes, ?> and ?> were their physical positions along the anterior-posterior and coronal axes respectively, ?>s were the determined regression coefficients. Least squares regression was applied to minimize ?>. An F-test with α = 0.05 was used to distinguish the traveling wave condition from the null hypothesis, i.e. ?>. The estimated velocity of a neuronal traveling wave was obtained by the equations
| (2) |
| (3) |
and speed was ?>.
Recording thalamic activity
To investigate thalamocortical interaction during burst suppression, we recorded bilateral thalamic and cortical activity simultaneously. Four glass electrodes were used to record the LFPs from the 4-AP injection site, the homotopic contralateral cortical area (mirror focus), and both ipsilateral and contralateral thalami. The thalamic electrodes were placed in the ventrobasal region: 2.6 mm posterior to Bregma, 2.8 mm lateral to the midline, and 5.8–6.0 mm ventral to the surface of the cortex on either side to target both the ventroposterolateral and ventral posterolateral thalamic nuclei (Paz et al., 2013). The RMS of LFP signal during the suppression section was calculated. The onset time of each burst at each recording site was defined as the moment when the LFP signal reached ± 3 RMS from the mean amplitude of the signal recorded during suppression. In a subgroup of experiments, tetrodotoxin (TTX) (0.3 mM, 1 µl) was injected into either the contralateral or ipsilateral thalamus ∼30 min after the onset of the first 4-AP ictal event. These injections targeted the ventrobasal nuclei of the thalamus. Given the volume of fluid injected, and the known diffusion of TTX in the thalamus we projected that a 1–1.5 mm3 volume of the thalamus was electrically silenced (Sacchetti et al., 1999). During these experiments, bipolar cortical stimulation was used to elicit ictal events, which served as a confirmation test that the TTX was not blocking cortical neurons’ ability to generate action potentials. The bipolar electrode (1 mm apart) was placed across the 4-AP injection electrode and square-wave electrical stimulation (1mA in amplitude and 1 ms in duration) was applied using a stimulation isolator and Master-9 (A.M.P.I. Jerusalem, Israel). The location of the TTX injection in the thalamus was confirmed in a pilot study with injection of 1 µl blue dye RH-1692 (Optical Imaging Ltd).
Data availability
The authors confirm that the data supporting the findings of this study are available within the article.
Results
General anaesthetic-induced burst suppression
With 1–2% isoflurane, LFP electrode recordings demonstrated a burst suppression pattern (Fig. 1A), with each burst having an amplitude of 0.62 ± 0.17 mV, duration of 1.76 ± 0.87 s, and a frequency of 13.97 ± 6.80 events/min [mean ± standard deviation (SD), n = 210 bursts, from seven animals]. The average burst suppression ratio was 64% ± 10%. Using calcium imaging, the bursts showed a symmetrical but variable bilateral propagation pattern. The bursts appear to initiate from the neocortex in bilateral mirror foci and propagated throughout both hemispheres in the whole field of view. Various initiation and propagation patterns were observed (Fig. 1A and B). However, no preferred onset and propagation pattern was identified (Fig. 1C), as no pixel was found to have consistently earlier or later onset time and distribution of cortical initiation site was spatially uniform (Fig. 1C). One-way ANOVA test indicates that no pixels show earlier or delayed onset time compared with any other (n = 51, left region of interest: F = 0.78, P = 0.87, right region of interest: F = 0.91; P = 0.65). In agreement with the calcium imaging results, bilateral microelectrode array recordings revealed a similar spatial homogeneity in burst patterns from all electrodes (Fig. 1D). Interestingly, in comparison to the dramatic LFP dynamics, the difference between bursts and suppressions was relatively minor in the MUA band. Mean multiunit firing rate remained very low during bursts (0.12 ± 0.06 spikes/s/electrode), only 33% higher than that during suppressions (0.09 spikes/s/electrode, t-test, n = 30 and 28, P = 0.02), suggesting that the bursts consist primarily of subthreshold membrane potential changes.
Figure 1.

The spatiotemporal dynamics of burst suppression in non-epileptic cortex. (A) Example of the onset of two bursts in wide field calcium imaging. Left: The field of view shows both hemispheres in one preparation. Two vertical red boxes indicate two regions of interest (ROI), from which the onset time is calculated. Right: The LFP recording of isoflurane induced burst suppression. Two blue lines indicate the onset (t = 0 ms) of two bursts (I and II). (B) The spatiotemporal evolution of calcium fluorescence intensity during the two bursts showed in A. Both bursts show bilaterally symmetric onset and propagation patterns. (C) The box-and-whisker plots of the onset time of calcium signal from each pixel in two vertical regions of interest (left and right) (n = 51 interictal bursts). For each box, the central mark indicates the median, and the bottom and top edges of the box indicate the 25th and 75th percentiles, respectively. The whiskers extend to the most extreme data points not considered outliers, and the outliers are plotted individually using the plus symbol. (D) Multi-electrode array recording of bursts in normal cortex. Grey circles show locations of each electrode, missing circles indicate non-functional electrodes. Colours demonstrate electrodes whose LFP traces shown on the right. The histograms below show each hemisphere’s multiunit spike counts every 100 ms. Bursts occur intermittently with globally symmetric spread of LFP but are with sparse MUA. Pink shaded vertical bar is expanded and displayed in E. (E) The array recording of two bursts with higher temporal resolution.
Burst pattern during the interictal period
After ∼10 min post injection, focal cortical 4-AP elicited stereotypical, repetitive ictal events (Fig. 2A). Ictal events lasted on average 39.18 ± 19.59 s with interictal intervals of 118.22 ± 97.40 s (n = 116 ictal events from seven animals). Wide field calcium imaging demonstrated that 4-AP induced a focal epileptiform event, as the calcium increases associated with ictal events were spatially constrained to a localized area around the 4-AP injection site with an average area of 8.35 ± 2.43 mm2 (n = 7 animals). Ictal events also elicit localized increases in LFP activity and MUA in a region 2 mm around the 4-AP injection site as demonstrated in a prior publication (Liou et al., 2018).
Figure 2.

Initiation of burst phase during interictal periods avoids the 4-AP injection zone. (A) Neuronal calcium signals are focal during ictal events. Left: Correlation coefficient heat map shows the spatial location of pixels with highest correlation with a seed at the 4-AP injection site. Right: The LFP recording and simultaneously recorded calcium traces from four different locations, showing the focal propagation of ictal event (grey box section) but global propagation of interictal bursts. (B) The LFPs recorded from the multielectrode array. The LFP traces from four different locations shows the global propagation of interictal bursts. (C) Calcium surges during interictal bursts. Top row shows example of ipsilaterally-initiating interictal bursts in a single animal. Left: The black top trace represents the LFP at the 4-AP injection site. Bottom traces represent wide-field calcium imaging data from four regions of interest depicted in the first panel on the right. Note the onset occurs at the green region of interest. Panels on the right correspond to calcium imaging of exposed bilateral cortex at three time points (each 20-ms apart) denoted as dashed vertical lines at left. Bottom row shows the same for contralaterally-initiating interictal bursts in a separate animal (blue region of interest). Note that while interictal bursts propagate globally, the initiation of interictal bursts does not occur in the 4-AP injection site. (D) Schematic showing four cortical zones. Zone 1: 4-AP injection, zone 2: ipsilateral surround, zone 3: contralateral homotopic, and zone 4: contralateral surround. Circles show multifocal initiation sites from one animal. (E) Bar graph showing spatial distribution of initiation sites of all interictal bursts detected with calcium imaging across five animals (36 interictal bursts). Note that the majority of events were detected outside Zone 1, i.e. not in the 4-AP injection zone.
During the interictal periods, the burst suppression pattern continued, as before the 4-AP injection, but with a different onset and propagation pattern. Interictal bursts had an average amplitude 0.71 ± 0.21 mV, duration of 1.83 ± 0.82 s, and a frequency of 14.70 ± 7.26 events/min (mean ± SD, n = 194 bursts, from seven animals). Although interictal bursts still propagated globally (Fig. 2B), they now appeared to initiate focally and unilaterally in the cortex and then spread globally, but not symmetrically (Fig. 2C). The initiation site of the interictal bursts occurred mostly in the cortical area surrounding the seizure focus, and rarely in the focus. We quantified this observation further by dividing the cortex into quadrants: zone 1, ipsilateral epileptic focus zone; zone 2, ipsilateral surround; zone 3, contralateral homotopic; and zone 4, contralateral surround (Fig. 2C). We confirmed that interictal bursts appeared to initiate predominantly in zones 2–4, avoiding zone 1, the site of the seizure focus (Fig. 2C and D). Of the 36 interictal bursts analysed in five animals, only one arose first in the quadrant containing the seizure focus (Fig. 2D), which was a statistically significant non-random distribution [χ2 (3, n = 36) = 20.44, P < 0.001].
Comparison of burst-associated cortical neural activity before and after seizure focus establishment
Compatible with our calcium imaging study, multielectrode array recordings showed that anaesthetic-induced burst suppression continued globally after creation of the 4-AP ictal focus, which was established at the anterior edge of the left multielectrode grid (Fig. 3A). Bursts during the interictal periods (interictal bursts), on average, occurred every 3.55 s (quartiles: 1.38–4.51 s, n = 766). They were more frequent than bursts during the baseline periods (baseline bursts), which occurred, on average, every 5.6 s (quartiles: 2.6–5.96 s, n = 30) (unpaired t-test, P = 0.003). Interictal bursts also lasted longer than baseline bursts (1.08 versus 0.77 s, unpaired t-test, P = 0.01). Longer bursts with more frequent occurrence translated to a decreased burst suppression ratio (from 0.82 to 0.71, U-test, P < 0.001), suggesting an increase in cortical excitability following the establishment of the seizure focus. In addition, of the 766 interictal bursts, we observed 115 (15%) interictal bursts associated with local paroxysmal neural activity (massive multiunit firing with spiky LFP fluctuations, see ‘Materials and methods’ section for definition details). In contrast, no baseline bursts was associated with such MUA (0/30, chi-square test, P < 0.001).
Figure 3.

Ipsilateral focal neural activity during interictal bursts. (A) Cartoon illustrates locations of working electrodes. Coloured circles indicate the physical locations of the electrodes from which the LFPs (right) are recorded. The bottom graph shows ipsilateral hemisphere’s multiunit spike counts every 100 ms. Red boxes indicate samples of interictal bursts with focal neural activity at the ipsilateral cortex. (B) The averaged multiunit (MU) firing rate over the ipsilateral cortex during interictal bursts-associated ipsilateral paroxysmal neural events. (C) Spatial distribution of LFP power during the ictal events (n = 56) versus ipsilateral paroxysmal neural events (n = 58). Notice their spatial dissociation. (D) Quantification of data in C: distribution of LFP power along the anterior-posterior axis in ipsilateral cortices. The thin, light-coloured lines correspond to each ictal (red) or ipsilateral paroxysmal neural event (green), and the thick, dark-coloured lines are the averages.
Fifty-eight paroxysmal events arose from the cortex ipsilateral to the 4-AP focus, but none of them co-localized with the focus (samples shown in Fig. 3A, red boxes). Although 4-AP was injected at the multielectrode grid’s anterior edge (Fig. 3A, left), the neural activity was instead found at the array centre (Fig. 3A–C), away from the ictal focus. Anterior-posteriorly, the point of maximal neural activity was 2 mm away from the 4-AP injection site (Fig. 3B and C). The spatial separation from the seizure focus and temporal dissociation from the ictal activity confirmed that these paroxysmal neural events were not part of the 4-AP ictal events (Fig. 3D). Instead, it suggested that cortical excitability within the ipsilateral surround had been enhanced during the interictal periods, compatible with our calcium imaging study results.
The local paroxysmal neural activity at the ipsilateral surround arose sequentially in space (Fig. 4). Specifically, in a time scale of milliseconds, neural activity appeared to emerge first in a region distant from the seizure focus and then subsequently propagated to the proximal regions (Figs. 4A and B). Linear regression analysis (α = 0.05) confirmed 28 traveling waves propagating from the ipsilateral surround toward the ictal focus. These waves travelled at an average speed of 26.2 cm/s (Fig. 4C, F-test, P < 0.001). Directions of individual traveling wave were highly similar (Fig. 4D, circular SD = 31 degrees, Rayleigh test, n = 28, Z = 23.0, P < 0.001), confirming that the ipsilateral surrounds were persistently more active than the seizure focus during the interictal periods.
Figure 4.

Ipsilateral focal neural activity formed traveling waves propagating towards the epileptic focus. (A) Left: Cartoon illustrates locations of working electrodes and coloured electrodes indicate sources for LFP traces on the right. Thin lines indicate each event. Thick lines indicate averages. The bottom graph shows ipsilateral hemisphere’s averaged multiunit spike counts every 1 ms. (B) The averaged spatiotemporal evolution of LFPs and MUA. The timing of each frame is indicated by the grey vertical bars in A. Notice the sequential appearance of multiunit (MU) spikes and LFP discharges (left bottom to top right). (C) Linear regression (data from the black-marked electrodes, right inlet, gathered from all ipsilateral neural events) shows that multiunit spikes (blue dots) were detected sequentially in space, propagating toward the epileptic focus (orange arrow, right inlet). Vertical axis of the raster plot (left inlet) indicates each electrode’s position along the propagation path (orange arrow). Horizontal axis indicates timing of multiunit spikes. (D) Estimated velocity of each individual traveling wave. Notice the consistent propagation direction.
We observed 57 interictal bursts in which events of local paroxysmal neural activity occurred in the contralateral hemisphere (Fig. 5A). Their anterior-posterior spatial distribution, however, was different from those at the ipsilateral cortices (Fig. 5B and C). The point of maximal activity was found at the anterior edge of the multielectrode grid, topologically homotopic to the ictal focus (Fig. 5B and C). The occurrence of contralateral neural activity during interictal periods and its spatial dissociation with the seizure focus, again, confirming these paroxysmal events were not ictal in nature (Fig. 5D). Unlike ipsilateral bursts, there was no consistent traveling wave behaviour in the contralateral events.
Figure 5.

Contralateral focal neural activity during interictal bursts. (A) Left: Cartoon illustrates locations of working electrodes and coloured electrodes indicate sources for LFP traces on the right. Thin lines indicate each event. Thick lines indicate averages. The bottom graph shows ipsilateral hemisphere’s averaged multiunit spike counts every 1 ms. (B) The averaged multiunit (MU) firing rate over the contralateral cortex during interictal bursts-associated contralateral paroxysmal neural events. (C) Spatial distribution of LFP power during the ictal events (n = 56) versus contralateral paroxysmal neural events (n = 57). Note that both were concentrated at the anterior edges of multielectrode grids. (D) Quantification of data in C: distribution of LFP power along the coronal axis in the anterior portion of the multielectrode grids. Data gathered from the electrodes in black. The thin, light-coloured lines correspond to each ictal (red) or contralateral paroxysmal neural event (green), and the thick, dark-coloured lines are the averages.
Our data indicate that an acute neocortical seizure focus can have a rapid, widespread impact on network excitability during the interictal period, which in turn impacts the homogeneity of the burst suppression pattern.
Thalamic activity precedes interictal bursts
After demonstrating the impact of the epileptic focus on distant neocortical areas, we further investigate if the seizure focus could impact subcortical structures within the extended network, namely the thalamus. To test this, we simultaneously recorded interictal bursts from bilateral thalami, targeting both the ventrobasal nuclei, as well as bilateral neocortices surrounding the 4-AP injection and contralateral homotopic cortex (Fig. 6A). Before 4-AP injection, the bursts showed synchronized activity between cortex and thalamus. The bursts initiated from either the thalamus or the cortex. (Fig. 6A and C). After 4-AP injection, all bursts during the interictal period appeared to initiate in the thalamus before spreading to the cortex with a delay of 10.33 ± 3.13 ms (n = 295 bursts in six animals, unpaired t-test, P < 0.001) (Fig. 6B and C). Ictal events, on the other hand, remained predominantly cortical with minimal involvement in the thalamus after the initial spike (Fig. 6A). Our data indicate that the establishment of a seizure focus induces the thalamus to trigger interictal bursts and that the neocortical initiation site of these occurred in the surround, avoiding the focus.
Figure 6.

Simultaneous bilateral cortical and thalamic LFP recordings suggest thalamic origin of bursts. (A) Left: Cartoon illustrates experimental set-up with bilateral thalamic and cortical LFP electrodes. Middle: Example of LFP traces from one animal showing burst suppression induced with isoflurane before 4-AP injection. Colours correspond to arrows in the cartoon. Note that the simultaneous activity in cortex and thalamus. The initiation of two bursts (single and double asterisks) are expanded below. Note, normal bursts initiate from either the cortex (single asterisk) or the thalamus (double asterisk). Right: Example of LFP traces showing 4-AP-induced ictal event and interictal bursts (IBs). Note that interictal bursts appear to involve both the thalamus and the cortex equally while ictal activity initiates and evolves predominantly in the neocortex. The initiation of two interictal bursts (single and double asterisks) are expanded below. Note, interictal bursts initiate from thalamus and spread in a delayed fashion to the 4-AP injection site. (B) Averaged LFP traces of interictal bursts during interictal period. Bold traces represent the averaged LFPs from the same locations (n = 295 bursts) and interictal bursts are shown in lighter shade. Black vertical lines denote event initiation. Note thalamic onset of interictal bursts (CT = contralateral thalamus; IT = ipsilateral thalamus) precedes cortical onset bilaterally (CC = contralateral cortex, 4-AP injection site) with delayed activation of the epileptic focus. (C) Averaged thalamus-cortex delay during anaesthesia-induced burst suppression events (before 4-AP injection) and interictal bursts (post 4-AP). Note, before the seizure focus is induced, interictal bursts arise from both the thalamus and the cortex. After seizure focus is created, most interictal bursts arise from the thalamus.
To confirm a causal role of the thalamus in triggering interictal bursts, we injected TTX, a potent sodium channel blocker, into the ventrobasal nucleus of the thalamus to suppress its electrical activity. After TTX injection, burst frequency significantly decreased from 1.8 ± 0.85 events/min to 0.44 ± 0.35 events/min (n = 6 animals, one-tailed unpaired t-test, P = 0.0084) (Fig. 7A and B). Thalamic TTX injection also abolished ictal events (Fig. 7C), leaving little spontaneous cortical activity that could be observed. However, cortical circuits remained excitable, as electrical stimulation at the 4-AP ictal focus could still trigger a full-blown focal ictal event (Fig. 7C). Our selective thalamic inactivation experiment therefore confirmed that focal ictal events produced persistent changes in thalamocortical loop dynamics, favouring thalamic over cortical burst initiations.
Figure 7.
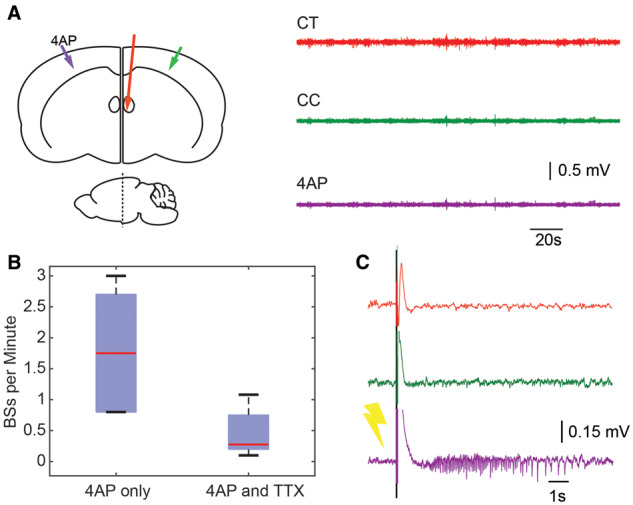
Thalamic inactivation downregulates both ictal events and interictal bursts. (A) Left: Cartoon shows experimental set up-for TTX injection in contralateral thalamus (red line). Right: LFP traces correspond to coloured arrows in one animal after TTX injection in the contralateral thalamus. Note the lack of activity on all channels. (B) A box-and-whisker plot showing bursts in spikes/min before and after TTX injection. 4-AP only condition elicited a mean of 1.8 ± 0.85 spikes/min while after TTX injection, this was reduced to 0.44 ± 0.35 spikes/min (n = 6 animals). (C) Stimulation of the cortex at the 4-AP injection site. A 2-ms, 10-mA single stimulus delivered at the 4-AP/seizure initiation site successfully induces an ictal event in spite of thalamic inactivation with TTX.
Discussion
In this study, we show that the establishment of an acute neocortical seizure focus causes a rapid alteration of general anaesthesia-induced burst suppression. Changes of burst initiation and propagation patterns suggested that a focal ictal event’s impacts are widespread and long-lasting, affecting both cortical and subcortical structures and extending well into non-ictal periods. As hypothesized, we find that burst suppression can be used as a method to help uncovering pathological alterations. These findings not only indicate a potential new role for burst suppression in clinical neurophysiology but also shed light on the rapid widespread impact of an epileptic focus on the extended associated network.
The epileptic focus as a disruptor of burst suppression homogeneity
Burst suppression is believed to arise from generators located within the neocortex and represents a state of hyperexcitability caused by a reduction in cortical inhibition in the setting of preserved glutamatergic excitation (Detsch et al., 2002; Lukatch et al., 2005; Kroeger and Amzica, 2007; Ferron et al., 2009). Both the bursting and suppression phases have traditionally been viewed as spatially homogeneous events, as recorded by scalp EEG (Clark and Rosner, 1973). Indeed, intracellular recordings reveal that most cortical neurons, if not all, depolarize during these events (Steriade and Contreras, 1995; Amzica, 2009). The periodicity of these events is thought to be caused either by fluctuations in intracellular calcium or energy depletion and restoration (Kroeger and Amzica, 2007; Ching et al., 2012). Ictal events, likewise, are similarly periodic events and result in periods of profound hyperactivity and hypermetabolism followed by an interictal refractory period (Duncan, 1992). However, focal neocortical epilepsy is a spatially inhomogeneous event and as a result, metabolic and neurotransmitter depletion are similarly spatially non-uniform. Ictal termination is followed by a prolonged depolarization block characterized by channel inactivation, as well as metabolic, neurotransmitter and pH alterations that must return to a critical threshold before seizure initiation can reoccur (Ayala et al., 1970; Duffy and MacVicar, 1999; Xiong et al., 2000; Pinto et al., 2005). The inhomogeneity of the post-ictal cortical energy state provides an opportunity to survey the network activity of the brain and burst suppression appears to be a suitable mechanism to perform such a probe. Our results show that bursts appear to arise preferentially in areas of the brain outside the epileptic focus with few events occurring within the focus. These results are consistent with a recent study in patients with medically intractable epilepsy underwent implantation of subdural electrodes (Lewis et al., 2013). Burst suppression was induced with propofol and marked spatial inhomogeneity in both the spatial extent and timing of events around the epileptic focus was demonstrated. The authors hypothesized that local variations in cerebral metabolism may explain this phenomenon but do not directly implicate the neighbouring epileptic focus in this variation. In our study we clearly show a direct link between the epileptic focus and the spatial non-uniformity of burst suppression events. Theoretically, one could use burst suppression in patients with refractory chronic epilepsy as a technique for localizing their epileptic focus, as part of the preoperative work-up in preparation for either surgical resection or implantation of a closed loop abortive device such as the responsive neurostimulator. Although burst suppression would require the administration of general anaesthesia, it would be much less invasive and far less risky, than the implantation of subdural grid and strip electrodes or stereo-EEG, the current state of the art in ictal onset zone localization. Whether a post-ictal state or merely an interictal state is required to reproduce our findings is unclear from the 4-AP model since the ictal events occur so frequently. However, the preliminary results from the human study referenced above indicates that the interictal state may be sufficient.
The epileptic focus alters cortico-cortical and thalamocortical interactions
In addition to creating spatial non-uniformity during bursts, the epileptic focus appeared to induce other, more subtle changes. Not only did we record massive MUA at cortical initiation sites, but the trigger of the events, which was both cortical and thalamic at baseline, became predominantly thalamic after the epileptic focus was created. Intracellular recordings of cortical neurons demonstrate that neurons are hyperpolarized during suppression and bursts consist of synaptic depolarizations, occasionally crowned by action potentials (Steriade et al., 1994). Burst suppression places the cortex in a state of hyperexcitability from reduced cortical inhibition, while the duration of each event appears to be caused by depletion of extracellular calcium stores (Kroeger and Amzica, 2007; Amzica, 2009; Ferron et al., 2009). Ictal events, on the other hand, have been shown to create a penumbra of ‘veto’ inhibition in the surrounding brain caused by local recruitment of interneurons (Trevelyan et al., 2006, 2007; Schevon et al., 2012; Liou et al., 2018). Such disseminated upregulation of inhibition may play a role in limiting spatial spread of seizures. It is possible that metabolic exhaustion and temporary dysfunction of inhibitory cells results in increased MUA in the surrounding cortex during the interictal period. Another alternative is the occurrence of post-inhibitory rebound excitation, an intrinsic property of cortical neurons (Steriade and Contreras, 1995; Castro-Alamancos and Connors, 1996; Steriade and Timofeev, 1997), might be responsible for the interictal hyperexcitability. Moreover, rebound excitation has been shown to rely heavily on thalamic input and rebound spike-bursts invariably occur first in thalamic cells prior to cortical cells (Grenier et al., 1998). Given that sensory input, presumably via thalamocortical afferents, has been shown to trigger bursts (Kroeger and Amzica, 2007), it is possible that the primary role of the thalamus in triggering interictal bursts in our model arises from the intrinsic mechanisms of rebound excitation. This mechanism would also explain the acuity of the alterations we find in the thalamocortical network as opposed to the chronic increases in thalamocortical excitability that have been shown to occur following a rodent neocortical stroke model of epilepsy (Paz et al., 2013).
In addition to triggering the interictal bursts, thalamocortical afferents may also trigger the onset of seizures, since silencing the ventral posterolateral thalamic and ventroposterolateral nuclei markedly decreased the rate of seizures. Compatible with our findings, Paz et al. (2013) also showed that optogenetically suppressing thalamic activity in a post-stroke seizure model reduces seizure occurrence. Ictal onset requires synchronized activity of a large population of neurons and the interictal bursts appear to act as a trigger for ictal onsets. Our previous study showed that most spontaneous ictal events in the anaesthetized 4-AP model initiate with a first spike (Ma et al., 2013) and the first spike has a similar propagation pattern as interictal bursts. These initial spikes may represent a thalamocortical oscillatory event, sufficient to trigger a seizure. With the thalamus blocked, cortical stimulation triggered ictal events and appeared to be a substitute for the thalamic trigger. Thus, the thalamus may be critical for seizure generation under anaesthetic conditions.
Interictal bursts and interictal spikes
It is important to emphasize that the isoflurane-induced bursts during interictal periods result from a complex interplay between general anaesthetics and the seizure focus, which is different from the scenario during classic interictal spikes. First, although interictal spikes may arise from a different population of cells than ictal events, they are often regionally associated and can be used clinically to identify the epileptic focus during human epilepsy surgery recorded with intraoperative electrocorticography (Hufnagel et al., 2000). In contrast, in our study, the interictal-burst related LFP spikes and massive increase in MUA during interictal bursts are spatially distinct from the 4-AP seizure focus. Second, although some focal interictal spikes can quickly spread throughout the entire cortical mantle via mechanisms that probably involve subcortical structures, a process called ‘secondary bisynchrony’ (Tukel and Jasper, 1952; Blume and Pillay, 1985), interictal spikes associated with focal epilepsy tend to arise from the neocortex (de Curtis and Avanzini, 2001). In contrast, the interictal bursts we recorded under anaesthesia represented a complex neural dynamic coordinated via the thalamocortical loop: triggered from the thalamus, emerging in a localized area of the cortex and then rapidly spreading throughout both hemispheres. Finally, although interictal spikes occur in a wide range of brain status, the interictal bursts dynamics we described here are specific to appropriate anaesthesia depth. Indeed, in a similar model of 4-AP injection in unanaesthetized animals, interictal bursts such as the ones we report here are not recorded (unpublished observations).
Burst suppression as a treatment for status epilepticus
In clinical practice, general anaesthesia can be used as a last resort in the treatment of refractory status epilepticus and the goal is to induce burst suppression (Mirsattari et al., 2004; Phabphal et al., 2018). As the aetiology of burst suppression is a combination of deafferentation with the generation of a hyperexcitable, disinhibited cortex (Amzica, 2015), it may function to suppress seizures using a similar mechanism to the TTX we placed in the thalamus, which also caused deafferentation. However, burst suppression was not effective at eliminating 4-AP seizures, likely because this pharmacological model induces a powerful trigger for seizures, much more intense than exists in chronic human epilepsy. Nevertheless, our data raises the intriguing hypothesis that burst suppression may be effective as a treatment for status epilepticus not because it suppresses cortical activity, but because it causes deafferentation and alters oscillation in the thalamocortical loop.
Conclusion
We have demonstrated that the creation of an epileptic focus in the neocortex alters the spatial homogeneity of burst suppression induced by isoflurane anaesthesia in very specific ways. The trigger for bursts shifts uniquely to the thalamus, possibly from rebound excitation, and the events themselves initiate in cortical regions outside the epileptic focus. This rapid alteration in the extended brain network associated with an epileptic focus supports the widespread impact of focal epilepsy and may be useful to map the spatial limits of the epileptic focus as a potentially diagnostic tool in the treatment of human epilepsy.
Funding
This project was supported by the National Science Foundation (NSF-1264948) (H.M.), the National Key Research and Development Program of China (2017YFC0107200) (H.M.), NIH RO1 NS084142 (C.A.S.), and NIH RO1 NS095368 (C.A.S.).
Competing interests
The authors report no competing interests.
Glossary
Abbreviations
- 4-AP
4-aminopyridine
- LFP
local field potential
- MUA
multiunit activity
References
- Amzica F. Basic physiology of burst-suppression. Epilepsia 2009; 50 (Suppl 12): 38–9. [DOI] [PubMed] [Google Scholar]
- Amzica F. What does burst suppression really mean? Epilepsy Behav 2015; 49: 234–7. [DOI] [PubMed] [Google Scholar]
- Ayala GF, Matsumoto H, Gumnit RJ. Excitability changes and inhibitory mechanisms in neocortical neurons during seizures. J Neurophysiol 1970; 33: 73–85. [DOI] [PubMed] [Google Scholar]
- Blume WT, Pillay N. Electrographic and clinical correlates of secondary bilateral synchrony. Epilepsia 1985; 26: 636–41. [DOI] [PubMed] [Google Scholar]
- Blumenfeld H. What is a seizure network? Long-range network consequences of focal seizures. Adv Exp Med Biol 2014; 813: 63–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bokil H, Andrews P, Kulkarni JE, Mehta S, Mitra PP. Chronux: a platform for analyzing neural signals. J Neurosci Methods 2010; 192: 146–51. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown EN, Lydic R, Schiff ND. General anesthesia, sleep, and coma. New Engl J Med 2010; 363: 2638–50. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Casillas-Espinosa PM, Powell KL, O’Brien TJ. Regulators of synaptic transmission: roles in the pathogenesis and treatment of epilepsy. Epilepsia 2012; 53 (Suppl 9): 41–58. [DOI] [PubMed] [Google Scholar]
- Castro-Alamancos MA, Connors BW. Cellular mechanisms of the augmenting response: short-term plasticity in a thalamocortical pathway. J Neurosci 1996; 16: 7742–56. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chaudhary UJ, Duncan JS, Lemieux L. Mapping hemodynamic correlates of seizures using fMRI: A review. Hum Brain Mapp 2013; 34: 447–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ching S, Purdon PL, Vijayan S, Kopell NJ, Brown EN. A neurophysiological-metabolic model for burst suppression. Proc Natl Acad Sci USA 2012; 109: 3095–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clark DL, Rosner BS. Neurophysiologic effects of general anesthetics. I. The electroencephalogram and sensory evoked responses in man. Anesthesiology 1973; 38: 564–82. [PubMed] [Google Scholar]
- Daniel AG, Laffont P, Zhao M, Ma H, Schwartz TH. Optical electrocorticogram (OECoG) using wide-field calcium imaging reveals the divergence of neuronal and glial activity during acute rodent seizures. Epilepsy Behav 2015; 49: 61–5. [DOI] [PubMed] [Google Scholar]
- de Curtis M, Avanzini G. Interictal spikes in focal epileptogenesis. Progress in neurobiology 2001; 63: 541–67. [DOI] [PubMed] [Google Scholar]
- Detsch O, Kochs E, Siemers M, Bromm B, Vahle-Hinz C. Increased responsiveness of cortical neurons in contrast to thalamic neurons during isoflurane-induced EEG bursts in rats. Neurosci Lett 2002; 317: 9–12. [DOI] [PubMed] [Google Scholar]
- Duffy S, MacVicar BA. Modulation of neuronal excitability by astrocytes. Adv Neurol 1999; 79: 573–81. [PubMed] [Google Scholar]
- Duncan R. Epilepsy, cerebral blood flow, and cerebral metabolic rate. Cerebrovasc Brain Metabol Rev 1992; 4: 105–21. [PubMed] [Google Scholar]
- Ferron JF, Kroeger D, Chever O, Amzica F. Cortical inhibition during burst suppression induced with isoflurane anesthesia. J Neurosci 2009; 29: 9850–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Goldberg EM, Coulter DA. Mechanisms of epileptogenesis: a convergence on neural circuit dysfunction. Nat Rev Neurosci 2013; 14: 337–49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Grenier F, Timofeev I, Steriade M. Leading role of thalamic over cortical neurons during postinhibitory rebound excitation. Proc Natl Acad Sci USA 1998; 95: 13929–34. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hudetz AG, Imas OA. Burst activation of the cerebral cortex by flash stimuli during isoflurane anesthesia in rats. Anesthesiology 2007; 107: 983–91. [DOI] [PubMed] [Google Scholar]
- Hufnagel A, Dumpelmann M, Zentner J, Schijns O, Elger CE. Clinical relevance of quantified intracranial interictal spike activity in presurgical evaluation of epilepsy. Epilepsia 2000; 41: 467–78. [DOI] [PubMed] [Google Scholar]
- Kaila K, Ruusuvuori E, Seja P, Voipio J, Puskarjov M. GABA actions and ionic plasticity in epilepsy. Curr Opin Neurobiol 2014; 26: 34–41. [DOI] [PubMed] [Google Scholar]
- Kroeger D, Amzica F. Hypersensitivity of the anesthesia-induced comatose brain. J Neurosci 2007; 27: 10597–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lazar LM, Milrod LM, Solomon GE, Labar DR. Asynchronous pentobarbital-induced burst suppression with corpus callosum hemorrhage. Clin Neurophysiol 1999; 110: 1036–40. [DOI] [PubMed] [Google Scholar]
- Lewis LD, Ching S, Weiner VS, Peterfreund RA, Eskandar EN, Cash SSet al. Local cortical dynamics of burst suppression in the anaesthetized brain. Brain 2013; 136 (Pt 9): 2727–37. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou JY, Ma H, Wenzel M, Zhao M, Baird-Daniel E, Smith EHet al. Role of inhibitory control in modulating focal seizure spread. Brain 2018; 141: 2083–97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liou JY, Smith E, Bateman L, McKhann G, Goodman R, Greger Bet al. Multivariate regression methods for estimating velocity of ictal discharges from human microelectrode recordings. J Neural Eng 2017; 14: 044001. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lukatch HS, Kiddoo CE, Maciver MB. Anesthetic-induced burst suppression EEG activity requires glutamate-mediated excitatory synaptic transmission. Cereb Cortex 2005; 15: 1322–31. [DOI] [PubMed] [Google Scholar]
- Ma H, Zhao M, Harris S, Schwartz TH. Simultaneous multi-wavelength optical imaging of neuronal and hemodynamic activity. In: Neurovascular Coupling Methods; 2014. p. 237–49. [Google Scholar]
- Ma H, Zhao M, Schwartz TH. Dynamic neurovascular coupling and uncoupling during ictal onset, propagation, and termination revealed by simultaneous in vivo optical imaging of neural activity and local blood volume. Cereb Cortex 2013; 23: 885–99. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma H, Zhao M, Suh M, Schwartz TH. Hemodynamic surrogates for excitatory membrane potential change during interictal epileptiform events in rat neocortex. J Neurophysiol 2009; 101: 2550–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mirsattari SM, Sharpe MD, Young GB. Treatment of refractory status epilepticus with inhalational anesthetic agents isoflurane and desflurane. Arch Neurol 2004; 61: 1254–9. [DOI] [PubMed] [Google Scholar]
- Noachtar S, Binnie C, Ebersole J, Mauguiere F, Sakamoto A, Westmoreland B. A glossary of terms most commonly used by clinical electroencephalographers and proposal for the report form for the EEG findings. The International Federation of Clinical Neurophysiology. Electroencephalogr Clin Neurophysiol Suppl 1999; 52: 21–41. [PubMed] [Google Scholar]
- Paz JT, Davidson TJ, Frechette ES, Delord B, Parada I, Peng Ket al. Closed-loop optogenetic control of thalamus as a tool for interrupting seizures after cortical injury. Nat Neurosci 2013; 16: 64–70. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Phabphal K, Chisurajinda S, Somboon T, Unwongse K, Geater A. Does burst-suppression achieve seizure control in refractory status epilepticus? BMC Neurol 2018; 18: 46. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pinto DJ, Patrick SL, Huang WC, Connors BW. Initiation, propagation, and termination of epileptiform activity in rodent neocortex in vitro involve distinct mechanisms. J Neurosci 2005; 25: 8131–40. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Purdon PL, Sampson A, Pavone KJ, Brown EN. Clinical electroencephalography for anesthesiologists: part i: background and basic signatures. Anesthesiology 2015; 123: 937–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Quiroga RQ, Nadasdy Z, Ben-Shaul Y. Unsupervised spike detection and sorting with wavelets and superparamagnetic clustering. Neural Comput 2004; 16: 1661–87. [DOI] [PubMed] [Google Scholar]
- Sacchetti B, Lorenzini CA, Baldi E, Tassoni G, Bucherelli C. Auditory thalamus, dorsal hippocampus, basolateral amygdala, and perirhinal cortex role in the consolidation of conditioned freezing to context and to acoustic conditioned stimulus in the rat. J Neurosci 1999; 19: 9570–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sarikaya I. PET studies in epilepsy. Am J Nuclear Med Mol Imaging 2015; 5: 416–30. [PMC free article] [PubMed] [Google Scholar]
- Scharfman HE. The enigmatic mossy cell of the dentate gyrus. Nat Rev Neurosci 2016; 17: 562–75. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schevon CA, Weiss SA, McKhann G Jr, Goodman RR, Yuste R, Emerson RGet al. Evidence of an inhibitory restraint of seizure activity in humans. Nat Commun 2012; 3: 1060. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EH, Liou JY, Davis TS, Merricks EM, Kellis SS, Weiss SAet al. The ictal wavefront is the spatiotemporal source of discharges during spontaneous human seizures. Nat Commun 2016; 7: 11098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Smith EH, Schevon CA. Toward a mechanistic understanding of epileptic networks. Curr Neurol Neurosci Rep 2016; 16: 97. [DOI] [PubMed] [Google Scholar]
- Steriade M, Amzica F, Contreras D. Cortical and thalamic cellular correlates of electroencephalographic burst-suppression. Electroencephalogr Clin Neurophysiol 1994; 90: 1–16. [DOI] [PubMed] [Google Scholar]
- Steriade M, Contreras D. Relations between cortical and thalamic cellular events during transition from sleep patterns to paroxysmal activity. J Neurosci 1995; 15: 623–42. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Steriade M, Timofeev I. Short-term plasticity during intrathalamic augmenting responses in decorticated cats. J Neurosci 1997; 17: 3778–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutula T. Seizure-induced axonal sprouting: assessing connections between injury, local circuits, and epileptogenesis. Epilepsy currents 2002; 2: 86–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Swank RL, Watson CW. Effects of barbiturates and ether on spontaneous electrical activity of dog brain. J Neurophysiol 1949; 12: 137–60. [DOI] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Watson BO, Yuste R. Modular propagation of epileptiform activity: evidence for an inhibitory veto in neocortex. J Neurosci 2006; 26: 12447–55. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Trevelyan AJ, Sussillo D, Yuste R. Feedforward inhibition contributes to the control of epileptiform propagation speed. J Neurosci 2007; 27: 3383–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tukel K, Jasper H. The electroencephalogram in parasagittal lesions. Electroencephalogr Clin Neurophysiol 1952; 4: 481–94. [DOI] [PubMed] [Google Scholar]
- Vijn PC, Sneyd JR. I.v. anaesthesia and EEG burst suppression in rats: bolus injections and closed-loop infusions. Br J Anaesthesia 1998; 81: 415–21. [DOI] [PubMed] [Google Scholar]
- White BR, Bauer A Q, Snyder AZ, Schlaggar BL, Lee JM, Culver JP. Imaging of functional connectivity in the mouse brain. PLoS One 2011; 6: e16322. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xiong ZQ, Saggau P, Stringer JL. Activity-dependent intracellular acidification correlates with the duration of seizure activity. J Neurosci 2000; 20: 1290–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Ma H, Suh M, Schwartz TH. Spatiotemporal dynamics of perfusion and oximetry during ictal discharges in the rat neocortex. J Neurosci 2009; 29: 2814–23. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao M, Nguyen J, Ma H, Nishimura N, Schaffer CB, Schwartz TH. Preictal and ictal neurovascular and metabolic coupling surrounding a seizure focus. J Neurosci 2011; 31: 13292–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Data Availability Statement
The authors confirm that the data supporting the findings of this study are available within the article.