

Abstract
PRDF1 and RIZ1 homology domain containing (PRDMs) are a subfamily of Krüppel-like zinc finger proteins controlling key processes in metazoan development and in cancer. PRDMs exhibit unique dualities: (a) PR domain/ZNF arrays—their structure combines a SET-like domain known as a PR domain, typically found in methyltransferases, with a variable array of C2H2 zinc fingers (ZNF) characteristic of DNA-binding transcription factors; (b) transcriptional activators/repressors—their physiological function is context- and cell-dependent; mechanistically, some PRDMs have a PKMT activity and directly catalyze histone lysine methylation, while others are rather pseudomethyltransferases and act by recruiting transcriptional cofactors; (c) oncogenes/tumor suppressors—their pathological function depends on the specific PRDM isoform expressed during tumorigenesis. This duality is well known as the ‘Yin and Yang’ of PRDMs and involves a complex regulation of alternative splicing or alternative promoter usage, to generate full-length or PR-deficient isoforms with opposing functions in cancer. In conclusion, once their dualities are fully appreciated, PRDMs represent a promising class of targets in oncology by virtue of their widespread upregulation across multiple tumor types and their somatic dispensability, conferring a broad therapeutic window and limited toxic side effects. The recent discovery of a first-in-class compound able to inhibit PRDM9 activity has paved the way for the identification of further small molecular inhibitors able to counteract PRDM oncogenic activity.
Keywords: epigenetic regulation, methyltransferase, PRDM, pseudomethyltransferase, transcription factor, yin-yang
Introduction—PRDM proteins: protein domains, origin, and evolution
PRDF1 and RIZ1 homology domain containing (PRDM) proteins represent an evolutionary recent branch of the methyltransferase superfamily. This subfamily contains 19 members (in humans) that control key biological processes, ranging from lineage specification to carcinogenesis [1–5]. All PRDM proteins share a characteristic structure that brings together a SET [Su(var)3–9, Enhancer-of-zeste and Trithorax]-like domain called a PR [PRDF1 (positive regulatory domain I-binding factor 1) domain, a RIZ1 (retinoblastoma protein-interacting zinc finger gene 1)] domain, and a variable array of Cys2-His2 (C2H2) zinc fingers (ZNF). Notable exceptions to this general structure include rare splice variants of PRDM7 and PRDM11 that are C2H2 zinc finger-deficient [3] and several PR-less isoforms, containing only the C-terminal zinc finger domains [2]. A Q-rich, unstructured domain is uniquely present at the C terminus of PRDM10. Consistent with the conserved role of this domain in transcriptional activation [6], its deletion leads to downregulation of PRDM10-regulated genes [7] (Fig. 1).
Fig. 1.
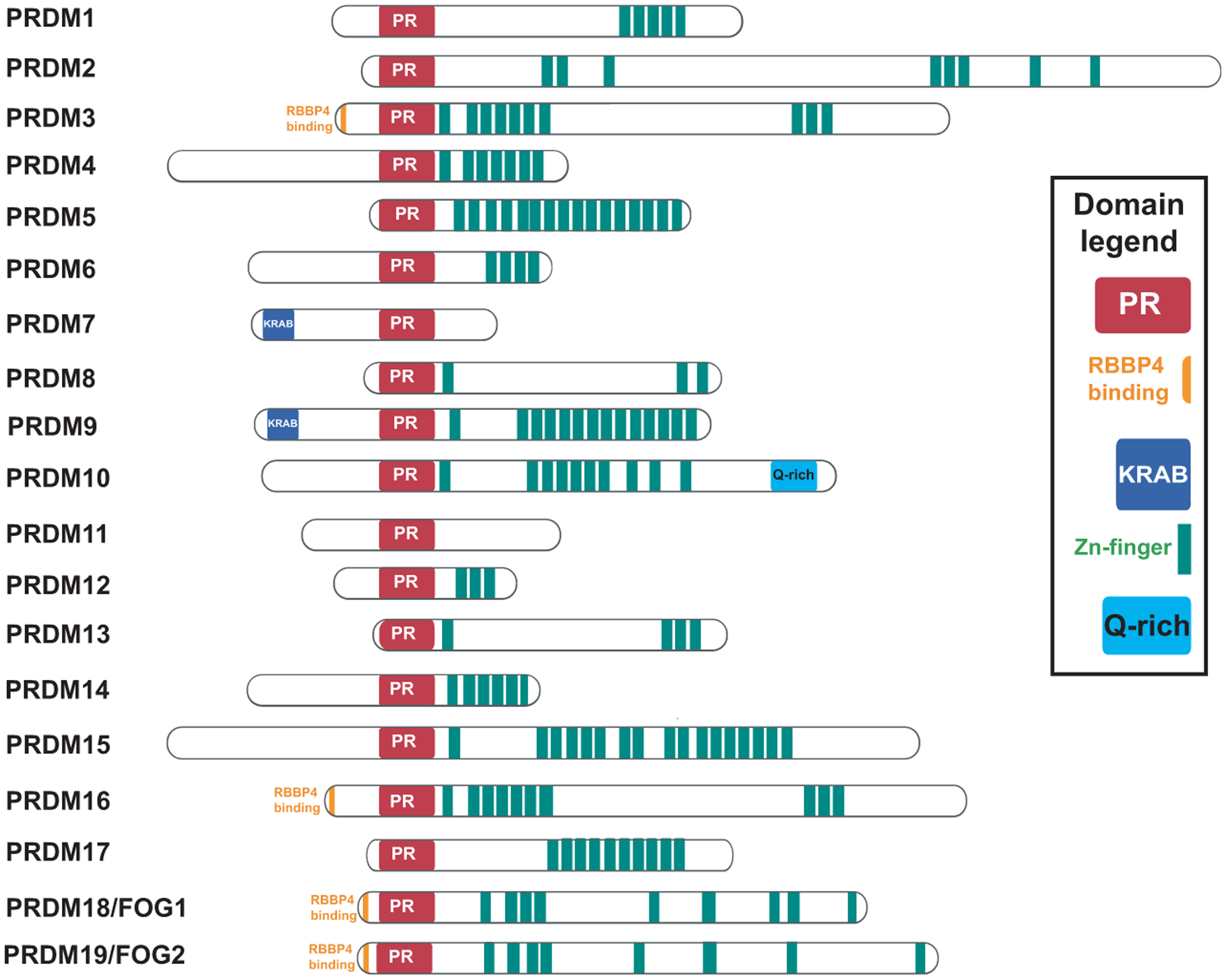
Domain structure of human PRDMs. PR domains are highlighted in red and C2H2 zinc fingers in green. Additional relevant domains are depicted including KRAB domains in blue, Q-rich regions in purple, and RBBP4 binding domain in orange. FOG-1 and FOG-2 proteins have been renamed respectively PRDM18 and PRDM19.
PRDMs first appeared in nematodes (two PRDMs) and insects (three PRDMs) and expanded across vertebrates. Given the high sequence homology between PR and SET domains, it is likely that PRDMs have evolved from the more ancient SET-containing protein lysine methyltransferases (PKMTs) (e.g., KMT2A/MLL1, SUV39H1) [3]. Intriguingly, despite the similarity between these two domains, only few PRDMs are endowed with an intrinsic methyltransferase activity. Flaws in experimental strategies/design, lack of knowledge of the actual cofactors or substrates, and evolutionary constraints are all plausible explanations for the enzymatically inactive PRDMs. The clear correlation between the distance to the root and methyltransferase activity, with fast-evolving PRDMs being more likely to lose activity, is nonetheless in favor of the latter explanation [8]. In the remainder of this review, we refer to these as pseudomethyltransferases in analogy with the more well-characterized pseudokinases [9].
It is also conceivable that the acquisition of stretches of zinc fingers, often involved in protein–protein and protein–nucleic acid interactions, during the evolutionary process allowed PRDMs to acquire novel functions essential for adaptation [3]. Indeed, despite the variability associated with enzymatic activity in PRDMs, their new DNA-binding capabilities, mediated by the ZNF domains, allowed them to bind to chromatin in a sequence-specific manner and to regulate transcription of target genes. Such regulation can be direct or indirect through tethering of other epigenetic factors. Indirect histone methylation, among other post-translational modifications, has been reported extensively for most PRDM proteins through recruitment of active enzymes to specific target sites [1–3].
In the following section, we examined the primary and tertiary structures of several PKMTs and PRDMs to highlight potentially critical amino acids for the conservation vs. loss of enzymatic activity during the SET-to-PR domain evolution. A special note should be made for Friend of GATA proteins, or FOGs, which are a group of proteins sharing structural homology with PRDMs (namely the PR domain and zinc finger array) [10]. FOGs are known to interact with the GATA transcription factor family and play important roles in development and disease [11]. In addition to their noncanonical C2HC ZNFs, similar to PRDMs, FOGs contain C2H2 ZNFs. In particular, ZNF1, ZNF5, ZNF6, and ZNF9 of FOG-1, and ZNF1, ZNF5, ZNF6, and ZNF8 of FOG-2 physically interact with the GATA-1N-finger, but not the GATA C-finger [12–14]. It is nonetheless unclear whether the remaining ZNFs can directly bind DNA or whether FOGs’ functions are entirely mediated by protein–protein interactions with different GATA family members [13,15]. We here propose to call the human FOGs, PRDM18 (FOG-1), and PRDM19 (FOG-2), respectively (Fig. 1).
PR domain: Structural aspects
PRDMs potentially function as methyltransferases through their PR domain, given its high homology with the SET domain [1]. However, experimental evidence of direct methyltransferase activity has been demonstrated for only a few members of the family that show stereoselectivity for the universal methyl donor SAM. Direct enzymatic activity has been demonstrated only for PRDM2 (H3K9me) [16], PRDM7 (H3K4me) [17], and PRDM9 (H3K4me3, H3K9me1/3, H3K18me1, H3K36me3, and H4K20me1/2) [18–22]. Controversial studies about PRDM3/PRDM16 methyltransferase activity have been reported, and it is not clear whether they act as H3K4me1 [23] or H3K9me1 methyltransferases [24]. Besides, an isolated study reported PRDM8 enzymatic activity (H3K9me), but further confirmations of the results are necessary [25].
On the other hand, indirect activity has been reported for PRDM1 (G9a/H3K9me2, PRMT5/H4R3me2s) [26,27], PRDM3 (EZH2/H3K27me3) [28], PRDM5 (G9a/H3K9me2) [29], PRDM6 (H4K20me3, G9a/H3K9me2) [30,31], PRDM12 [32–35], and PRDM14 (EZH2/K27me3) [36,37], suggesting that PRDMs are able to recruit histone ‘writers’ through protein–protein interactions. Furthermore, PRDMs are able to recruit histone ‘erasers’ (i.e., demethylases and deacetylases). Examples of the latter include PRDM1 recruitment of the histone demethylase KDM4A to remove repressive H3K9me3 marks in neural development [38] or HDACs to deacetylate histone H3 and repress C-MYC transcription during B-cell differentiation [39]. The indirect and direct methyltransferase evidence is summarized in Fig. 2.
Fig. 2.
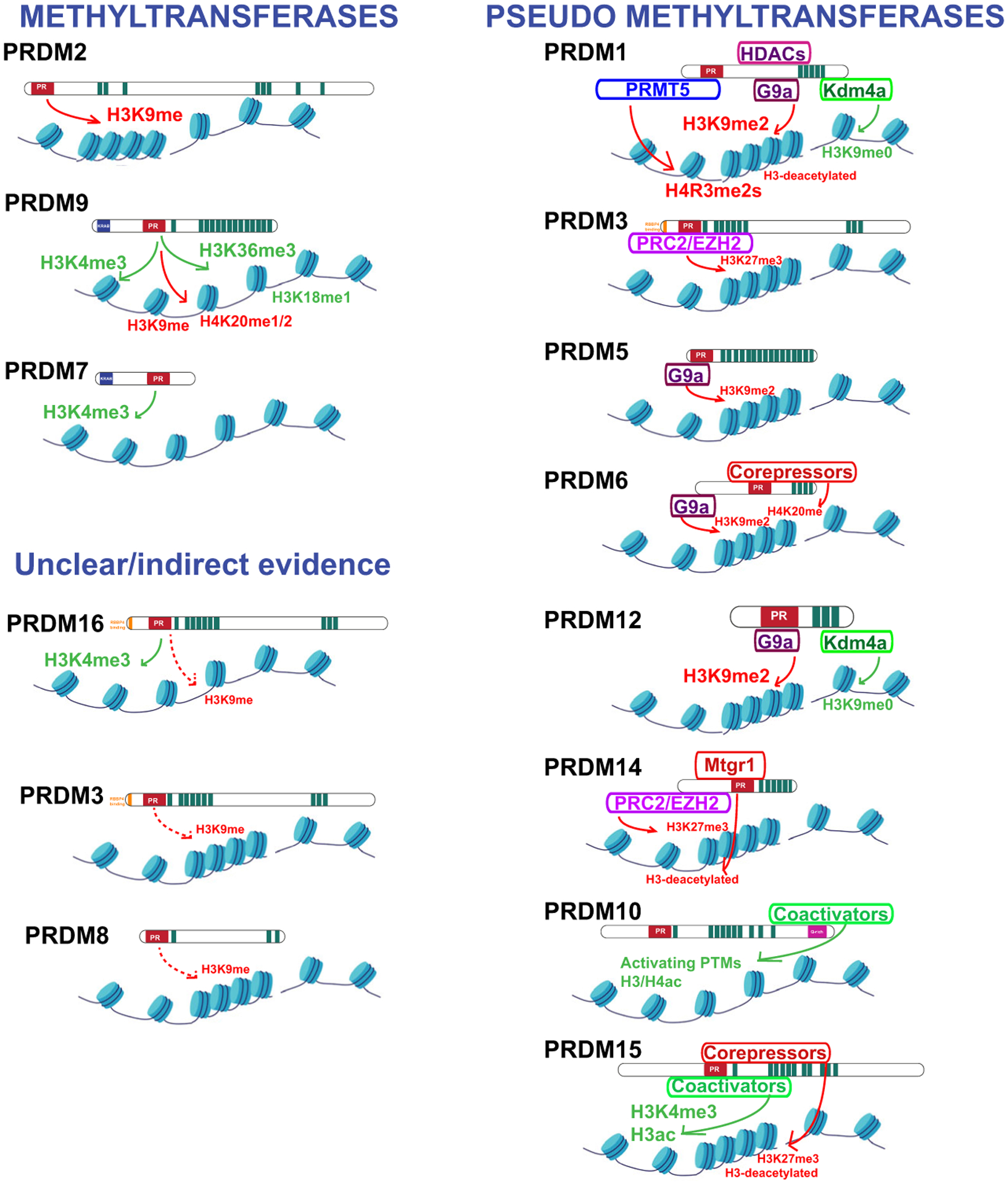
Summary of the experimental evidence of PRDM’s methyltransferase activity. PRDMs have been classified into three categories: (a) methyltransferases (PRDM2, PRDM7, PRDM9, and PRDM16); (b) methyltransferases with unclear/indirect evidence (PRDM3, PRDM8); and (c) pseudomethyltransferases (PRDM1, PRDM3, PRDM5, PRDM6, PRDM10, PRDM14, PRDM15).
Following the crystallization of SET domain proteins, extensive efforts have been made in the last decade to crystalize PRDM family members. Nonetheless, the presence of tandem ZNF arrays, technically difficult to purify and crystalize, has limited the success to crystallization of the PR domain only. More importantly, lack of knowledge regarding the substrates, if any, of different PR domains has further restricted all available structures to the inactive/unbound state, lacking both SAM and peptides/substrates, in their respective pockets (Fig. 3). It was not until 2013 that the first X-ray structure of PRDM9 in complex with a histone H3 peptide and SAH was reported by the Structural Genomics Consortium (PDB code 4C1Q) [18]. This was a seminal paper, highlighting how the PR/SET domain topology is similar to other SET domains, with the conserved PR domain fold surrounded by the pre- and post-SET regions. The construct used in this structure has a truncated post-SET domain fragment that was folded in the presence of SAH and a histone H3 peptide (H3K4me2).
Fig. 3.
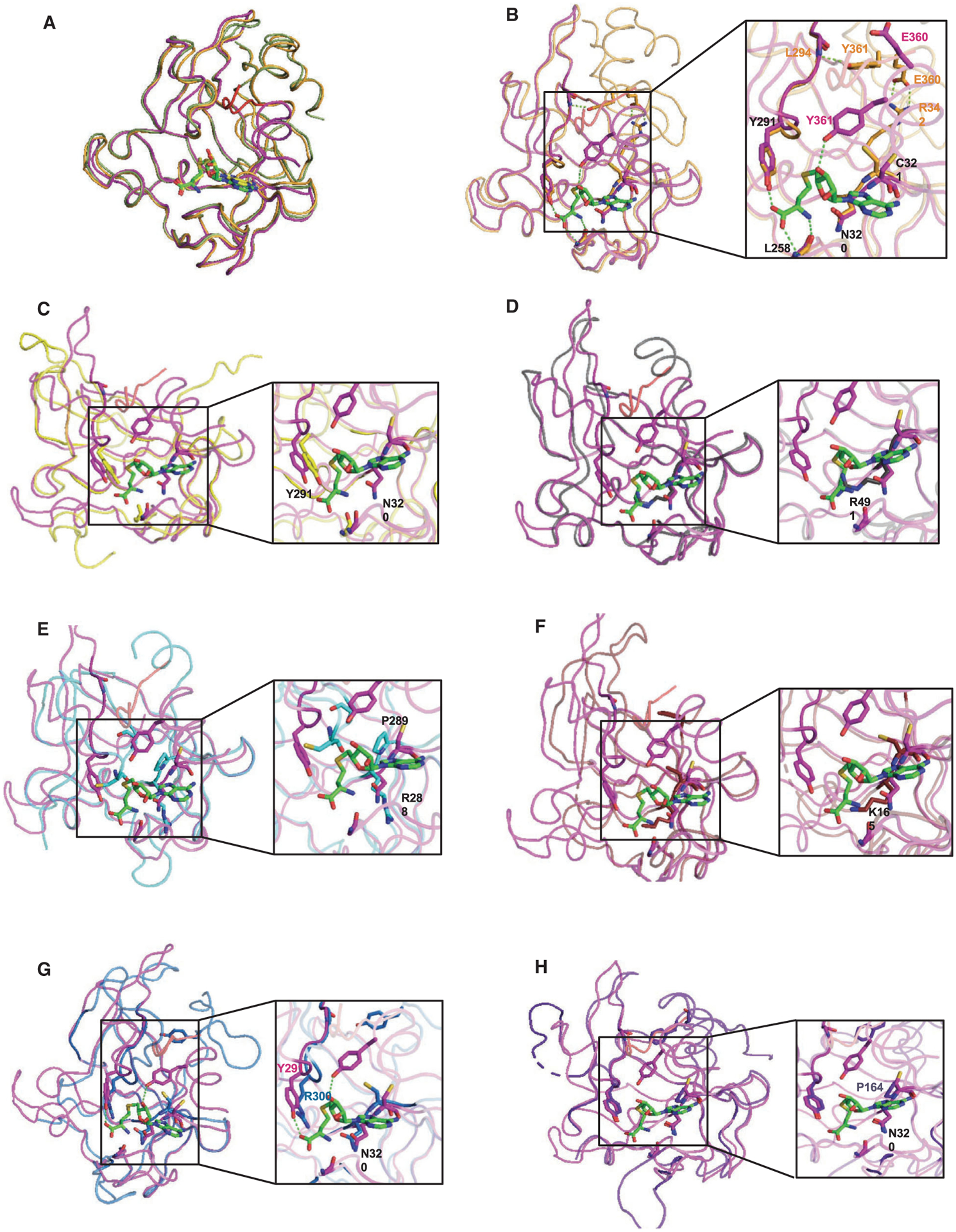
3D molecular models highlighting PR/SET domain interactions. (A) Comparison of the PR/SET domain of mPRDM9 (magenta) in complex with H3K4me2 peptide (red) and SAH (green) (PDB code 4C1Q), unbound hPRDM9 (orange) (PDB code 4IJD), and SAM (yellow) and bound hPRDM9 (green) (PDB code 6NM4). (B) Superposition of the PR/SET domain of mPRDM9-SAH (magenta) onto unbound hPRDM9 (orange). (C) PRDM2 (yellow) (PDB code 2QPW). (D) PRDM4 (gray) (PDB code 3DB5). (E) PRDM10 (cyan) (PDB code 3IHX). (F) PRDM12 (red) (PDB code 3EP0). (G) PRDM14 (blue) (PDB code 5ECJ). (H) PRDM1 (purple) (PDB code 3DAL). Residues interacting with SAH are highlighted. PYMOL (https://pymol.org/2/), an open-source molecular visualization system, has been used to generate the reported structures.
This pioneer work later led to the identification of the first-in-class PRDM inhibitor, MRK-740 [40]. This small molecule is selective for PRDM9 over other SET/PR domain proteins and binds to PRDM9 through a large SAM interaction interface. This mode of binding is based on disrupting the active conformation of the post-SET region, exposing SAM to interact with the inhibitor. Furthermore, MRK-740 is cell-active and was shown to inhibit PRDM9 activity on chromatin, as measured by the ability of the compound to reduce the levels of H3K4me3 at PRDM9 binding regions [40].
Solving the crystal structure of additional PR domains is key to understanding the main differences between active and pseudomethyltransferases. Notably, inclusion of the post-SET region might be critical in such crystallography experiments. Indeed, the transition between the PRDM9 active and inactive states is followed by clear conformational changes in the structure and more precisely in the post-SET domain. The unbound PRDM9 structure shows a closed conformation in which the post-SET region binds across the PR/SET domain, blocking both the peptide and SAH binding cleft [18]. This autoinhibitory conformation was further confirmed with the recently published PRDM9-MRK-740-SAM structure [40]. Superposition of both unbound and inhibitor-bound PRDM9 structures shows no difference in the global conformation; however, the SAH-bound structure adopts a different conformation with a fully folded post-SET domain and a clear translocation of the helix α2 allowing the transition to the active state (Fig. 3A). A closer examination of the PR domain in PRDM9 shows that tyrosine 357 (Y357), glutamate 360 (E360), and tyrosine 361 (Y361) residues are directly involved in the substrate recognition site. E360 stabilizes the closed conformation by forming a salt bridge with arginine 342 (R342), whereas Y361 blocks the binding cleft by forming a hydrogen bond with leucine 294 (L294). Both residues rotate in the SAM/substrate-bound conformation, with Y361 making direct contacts to stabilize SAM. On the other hand, asparagine 320 (N320) and cysteine 321 (C321) residues at position N320 and C321 are crucial for SAM binding (Figs 3B and 4). Interestingly, Y357 is mutated to a serine in PRMD7. This mutation negatively impacts the catalytic efficiency, as demonstrated by the fact that a simple mutation back to tyrosine restores catalytic levels observed for PRDM9 [17] (Fig. 4).
Fig. 4.
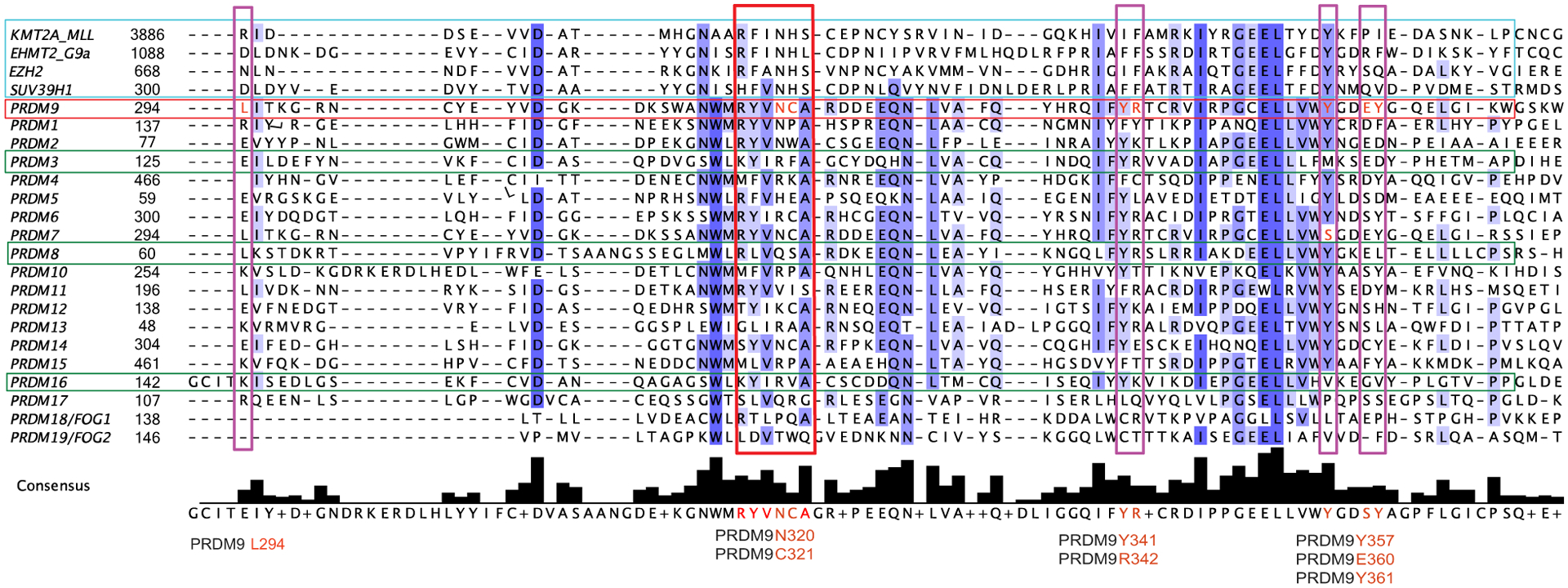
Sequence alignment of PRDMs’ PR domains and representative human canonical SET domain containing methyltransferases (KMT2/MLL, EHMT2/G9a, Su(var)3–9H1, and EZH2 highlighted in the turquoise box). The green boxes refer to the PRDMs with demonstrated methyltransferase activity (PRDM9, PRDM3, and PRDM16). The red box highlights the exclusive PR domain motif that is associated with putative methyltransferase activity. The purple boxes denote key amino acidic residues critical for methyltransferase activity (identified in Fig. 3). The alignment has been generated by using the T-Coffee multiple sequence alignment algorithm (https://www.ebi.ac.uk/Tools/msa/tcoffee/) accessible through Jalview open-source software (http://www.jalview.org/). The amino acid sequences were downloaded from the publicly accessible database UniProt (https://www.uniprot.org/); their accession numbers are as follows: Q03164 (KMT2A_MLL), Q96KQ7 (EHMT2_G9A), Q15910 (EZH2), O43463 (SUV39H1), O75626 (PRDM1), Q13029 (PRDM2), Q03112 (PRDM3), Q9UKN5 (PRDM4), Q9NQX1 (PRDM5), Q9NQX0 (PRDM6), Q9NQW5 (PRDM7), Q9NQV8 (PRDM8), Q9NQV7 (PRDM9), Q9NQV6 (PRDM10), Q9NQV5 (PRDM11), Q9H4Q4 (PRDM12), Q9H4Q3 (PRDM13), Q9GZV8 (PRDM14), P57071 (PRDM15), Q9HAZ2 (PRDM16), Q9H9D4 (PRDM17), Q8IX07 (PRDM18/FOG1), and Q8WW38 (PRDM19/FOG2).
Based on strict sequence alignment, and on the confirmed catalytic activity of PRDM9 [18–22], one would predict that only PRDM2, PRDM7, and potentially PRDM14 are active KMTs (Fig. 4). In Fig. 3, we superimposed publicly available structures of other PRDM proteins to that of PRDM9’s PR domain to infer potential catalytic activity. The caveat is that none of these structures contain either SAM/SAH or peptides/substrates, making it difficult to predict subsequent conformational changes. Nonetheless, we do note that N320, which is conserved in the SET domain of active PKMTs (SUV39H1, EZH2, G9a, and KMT2A) and in the active PRDM2, is substituted by either arginine or lysine in PRDM4, PRDM10, and PRDM12. (Figs 3D–F and 4). We predict that such changes would prevent efficient SAM binding. In PRDM2, for which a methyltransferase activity was reported, the corresponding asparagine is conserved, as well as the tyrosine at position 291, forming a hydrogen bond with SAM (Fig. 3C and 4). Comparing the PRDM9 structure with that of PRDM14 is interesting, as most residues interacting with SAM are conserved, but no catalytic activity has been reported to date (Figs 3G and 4). Finally, C321 is substituted by a proline (P164) in PRDM1 and PRDM10 resulting in loss of the hydrogen bonds observed in the PRDM9 structure and may consequently disrupt SAM binding (Figs 3E,H and 4).
In summary, specific members of the PRDM family are active KMTs, while others are devoid of direct methyltransferase activity (i.e., pseudomethyltransferases) and act as docking proteins for enzymatically active epigenetic modifiers. As such, defining the PRDM interactome is paramount to comprehensively understand their role as epigenetic modifiers. As a caveat to our analysis, most PRDM structures published so far lack the post-SET domain, which has a great impact on the enzymatic conformational changes. Thus, it will be important for the field to obtain extended structures that include this region, in order to gain deeper insights on the mechanistic aspects of PRDMs’ activation. Last, understanding how certain PR domains bind SAM, while others do not will be important in order to develop PRDM-binding small molecules inhibitors or degraders for therapeutic applications.
ZNF arrays and DNA-binding selectivity
C2H2-type ZNF motifs are among the most prominent structural domains present in chromatin-bound proteins, enabling protein–protein and protein–nucleic acid interactions. Their structure is defined by spacing constraints between cysteines and histidines (X2-C-X2,4-C-X12-H-X3-,5-H), which coordinate a centrally located zinc ion and adopt a two-stranded antiparallel β-sheet and an α-helix [41]. The α-helix of each ZNF, often dubbed the recognition helix, is able to engage in sequence-specific contacts with DNA, with each ZNF recognizing approximately three bases. Several groups have attempted to map the cistrome for PRDMs in different cell lines; however, the low expression levels of most PRDMs, their high cell-specific expression, and the lack of specific ChIP-grade antibodies have often impaired these efforts.
As a general rule, most PRDMs are direct sequence-specific chromatin regulators, which utilize between three and four ZNFs to make direct contact with DNA motifs spanning 7 to 15 bases, with the exception of PRDM9 isoforms, which recognize very long motifs using the entirety of their ZNF arrays [42]. We have summarized the experimental evidence of PRDMs’ DNA-binding motifs in Fig. 5. In addition, we have used an online prediction tool (http://zf.princeton.edu/ [43,44] to calculate the DNA bases potentially recognized by each ZNF array present at the C terminus of PRDM family members.
Fig. 5.

Summary of predicted and experimentally demonstrated PRDM zinc finger binding sites. A comparative analysis has been performed between predicted binding sites by using Princeton online tool (http://zf.princeton.edu/) and either experimentally demonstrated binding sites or binding sites deposited in JASPAR database.
PRDM1, also known as BLIMP-1, was originally discovered for reducing beta-interferon expression in the context of viral infection [45] and for its role in B-cell maturation [46]. Later, it was identified as a tumor suppressor in B-cell lymphomas [47–51], where its silencing alone is insufficient to induce tumorigenesis, since it requires additional mutations for lymphoma to develop [52]. The PRDM1 gene has two alternative promoters, transcribing two isoforms (i.e., PRDM1α and PRDM1β) with opposite functions, but both still able to bind to DNA (a phenomenon described as ‘Yin and Yang’) [53]. The PRDM1 cistrome has been well characterized in multiple cell lines by ChIP-sequencing [54–57], and a C/T-rich consensus characterized by two consecutive [ACTTTC] repeats is consistent with the predicted consensus of ZNF1–4.
PRDM2, also known as RIZ, was discovered in the mid-90s as a result of its ability to bind retinoblastoma (Rb) and was hypothesized to play a role in neuronal development [58]. It was first reported to have tumor suppressive properties in breast cancer [59], lymphoma [60], and leukemia [61,62]. A PR-less oncogenic isoform has also been identified. Similar to other PRDMs (i.e., PRDM1α/β, PRDM3/EVI1, and PRDM16/MEL1), these two isoforms (named RIZ1 and RIZ2) have opposing functions in cancer and are expressed in different contexts [63–68]. While the predicted ZNF recognition motif is [TCxTCxGAGGxACT], whether PRDM2 binds directly to DNA remains to be explored.
The short isoform of PRDM3, known as EVI1, is a potent oncogene in several leukemia subtypes [69]. PRDM3/EVI1 has two sets of ZNFs, one at the N- and one at the C terminus of the protein, comprised of seven and three ZNF tandem repeats, respectively (Fig. 1). Previous studies have demonstrated that the PRDM3/EVI1 N-terminal ZNF domain recognizes a GATA-like motif [GAC/TA] X0–6 [GAT/CA], while the C terminus binds to an ETS-like motif [GAA/TGAT/G], respectively [70–75]. The predicted consensus is consistent with the experimental data.
PRDM4 is a nonessential transcriptional regulator during development, as Prdm4 null mice are born healthy and in a Mendelian ratio [76]. Using a combination of SELEX (systematic evolution of ligands by exponential enrichment) and ChIP-seq, the binding site for PRDM4 has been determined to be [CTGTTTC] [76], consistent with the predicted consensus of ZNF2–4.
PRDM5 is a well-established tumor suppressor gene in a multitude of cancer types, whose expression is epigenetically silenced through CpG methylation [77,78]. The proposed mechanism for PRDM5’s tumor suppressor function involves its ability to negatively regulate WNT signaling through transcriptional regulation of WNT reporters and WNT-responsive genes. This was initially reported in Zebrafish development [79] and has since been replicated in the context of tumorigenesis [77,80,81]. Besides being often deregulated in cancer, PRDM5 is also highly expressed in mouse ES cells (mESCs). While PRDM5 is dispensable for the self-renewal potential of mESCs, it transcriptionally regulates genes important for cell differentiation and development. PRDM5 binds to a [GGxGCxCCxG] consensus [82], consistent with the predicted consensus at the extreme C terminus of the protein (ZNF14–16).
PRDM6 is a poorly characterized member of the family; a study on the role of PRDM6 in vascular-associated smooth muscle cells reported physical interactions with the histone methyltransferase EHMT2/G9a and with class I histone deacetylases, mediated through the PR domain [31]. There is no evidence of direct DNA binding by PRDM6, but it is predicted to recognize a G/T-rich motif with its four ZNFs [TGTxGTTxC].
PRDM8 is known to play a role in the development of the central nervous system, testes, and hematopoietic cells [25,83–85]. Its role in cancer appears to be context-dependent, with some studies describing PRDM8 as a tumor suppressor [86], while others reporting an oncogenic function [87]. Direct DNA binding has not been tested, but is predicted to recognize a short [AxTxGxACC] motif with its three ZNFs.
PRDM9 has been extensively studied for its role in meiotic recombination. PRDM9 binds first to chromatin and labels specific target sites, through deposition of the H3K4me3 and H3K36me3 histone marks [18,19,21,22,88]. Subsequently, proteins responsible for the formation of double-stranded breaks and for meiotic recombination are recruited to these sites [89–91]. Overall, the PRDM9-binding motif identified in vivo matches very well with those predicted in vitro. However, PRDM9 is unique among other PRDMs as several alleles are present within the same species. These alleles bind to different consensus sites in the genome and drive meiotic recombination at different hot spots [42].
The function of PRDM10 has recently been characterized in early embryonic development [7]. Specifically, PRDM10 mediates its function through direct binding to a [A/GA/GTGGT/AxxG/CxxCC] motif, weakly consistent with the predicted consensus of ZNF4–8. Importantly, its ZNFs are essential to PRDM10’s transcriptional activity, which is mediated by the recruitment of coactivators by both the PR- and Q-rich domains [7] (Fig. 1).
Mutations in PRDM12 have been linked to congenital insensitivity to pain. These mutations occur in a homozygous fashion and affect the development of sensory neurons destined to differentiate into nociceptors [92,93]. Patient mutations span the entire protein, including the PR domain and the ZNF array [92]. No direct evidence has demonstrated PRDM12-DNA binding; however, the three ZNFs are predicted to bind a G/T-rich motif [GTGTGTT].
PRDM13 is expressed in the dorsal neural tube [94] and retina [95] where it plays a role in lineage specification. There is no evidence of direct DNA binding, but rather, PRDM13 has been shown to interact with basic helix–loop–helix transcriptional activators to repress transcription of ventral neural tube specification genes (e.g., Olig1 and Olig2) [96] and Tlx1/3. Since TLX1/3 expression would specify the excitatory neuronal lineage, PRDM13-mediated repression promotes differentiation toward the inhibitory lineage. The four ZNFs are predicted to bind a [CxAGTCCGC] motif, but this has not been validated experimentally.
PRDM14 plays a key role in germ cell specification and in the maintenance of embryonic stem cells’ naïve pluripotency [97–101]. PRDM14’s function is mediated by direct transcriptional regulation of downstream effectors. These have been determined by ChIP-sequencing in relevant cell types (i.e., mouse and human ES and germ cells). The data collected by multiple groups have identified a conserved motif [GGTTAGA/GGACCC]. Unfortunately, in silico prediction tools are not able to consistently corroborate the experimental data, suggesting that PRDM14 might use noncanonical modalities to bind DNA or use the ZNFs in a nonsequential order [102–104].
The first functional studies characterized PRDM15 as gatekeeper of naïve pluripotency in mESCs [105]. In mESCs, PRDM15 operates upstream of the WNT and MAPK signaling pathways via transcriptional regulation of critical genes in both pathways, namely Rspo1 and Spry1 among others. This role seems to be bypassed in vivo, at least partially, as PRDM15KO mice implant normally and die at mid-gestation due to growth delay and forebrain malformations reminiscent of holoprosencephaly (HPE) and microcephaly [106]. Indeed, loss-of-function mutations affecting the ZNF domains of PRDM15 have been recently reported in patients with a syndromic from of HPE [106,107]. One particular mutation (C844Y) in ZNF15 appears to abolish PRDM15 binding to its target genes, many of which are important for anterior/posterior patterning and forebrain development, and its ability to regulate their expression [106]. The predicted consensus is recognized by ZNF10–15 and matches the experimentally validated binding motif [105,106].
In a ‘Yin and Yang’ fashion, PRDM16/MEL1 expresses two isoforms, one longer (PRDM16) comprising the PR domain and one shorter (MEL1), which lacks the PR domain and acts as a dominant-negative isoform in cancer [108]. MEL1 binding to chromatin has been mapped by multiple experimental methods. Earlier reports identified two motifs, similar to those of PRDM3/EVI1, by DNA-binding activity by the cyclic amplification and selection of target (CASTing) technique [108]. Later attempts, however, did not confirm these findings by ChIP-seq, and rather identified binding sites consistent with those of EBF and C/EBP at regulatory regions of brown adipose tissue genes (e.g., Ucp1, Ppara) in brown adipocytes [109] and of LHX2, SOX2/3, NEUROD1, TBR2, and MEIS1 at regulatory regions of cell migration and progenitor amplification genes (e.g., Nrg1/Itga6 and Mycn/Jag1, respectively) in mouse E15.5 cortex [110]. Similar to PRDM14, PRDM16 might also bind DNA in a noncanonical function, utilizing a combination of the two ZNF arrays (one more centrally located and the second at the C terminus) in a nonconsecutive fashion, or rely on other DNA-binding factors to engage with chromatin. Interestingly, the in silico prediction of the binding preference for the three ZNF at the C terminus of PRDM16/MEL1 is consistent with a [CATCc/tTC] motif identified by the earlier study [108].
A last note of interest for FOG-1: Although FOG proteins are known to mediate their transcriptional effects by protein–protein interaction with GATA proteins, it is intriguing to note that our in silico analysis (Fig. 5, bottom) predicts the first three ZNFs of FOG-1 to bind to palindromic [GATA] motifs. Whether FOG-1 is able to directly contact DNA, or whether its ZNFs are able in any way to interfere with GATA binding to chromatin, remains to be investigated.
PRDMs in Cancer
Historically, PRDM proteins have been associated with tumor suppressor functions. Most PRDMs are either silenced or deleted in cancer through an array of genetic (i.e., polymorphisms, frameshift/inactivating mutations, and chromosomal deletion) and epigenetic (i.e., DNA methylation and transcriptional silencing) mechanisms. For instance, genomic regions containing PRDM1 (6q21-q22.1) [49], PRDM2 (1p36) [111], and PRDM4 (12q23-q24.1) [112] are frequently deleted in cancer. Frameshift and inactivating mutations identified in PRDM1, PRDM2, PRDM3, PRDM8, and PRDM11 have been linked to transformation [1]. In addition, an increasing number of studies have reported downregulation of several PRDMs at the transcriptional level via a variety of epigenetic mechanisms involving DNA methylation, histone post-translational modifications, and micro-RNAs, leading to deregulation of the local chromatin structure at PRDM-associated regulatory regions. The underlying molecular mechanisms have been extensively characterized elsewhere [1,4,113].
From a therapeutic perspective, PRDMs have been hard to drug: (a) First, the majority are associated with a tumor suppressive activity (i.e., deleted, mutated, or not expressed in cancer); and (b) their ‘Yin and Yang’ regulation entails that the oncogenic isoforms (i.e., PRDM1β, EVI1, MEL1) are deprived of the druggable PR domain. Nonetheless, more recent studies have uncovered another category of oncogenic PRDMs in cancer, which are expressed as full length, hence retaining the druggable PR domain. Among the PRDMs showing an oncogenic function, extensive molecular characterization has been provided only for PRDM9, for PRDM14, and recently for PRDM15, while PRDM6 and PRDM10 still lack proper functional validation and demonstration of their pivotal role in carcinogenesis.
We dedicate the next section to discussing these maverick PRDMs and the associated molecular mechanisms linked to cancer.
PRDM9: from meiosis to tumorigenesis
PRDM9 contains an N-terminal Krüppel-associated box (KRAB) domain involved in protein–protein interactions, an SSXRD nuclear localization signal, a PR domain with intrinsic methyltransferase activity, and a set of zinc fingers at the C terminus providing direct site-specific DNA binding [114].
PRDM9 was first characterized as a methyltransferase that catalyzes H3K4me3, followed by additional evidence of its expanded substrate specificity toward other histone lysines [18–22] and potentially nonhistone lysines (i.e., automethylation activity) [115].
As discussed in a previous paragraph, over the last decade, PRDM9 has been established as a major epigenetic regulator of meiotic progression. Typically, PRDM9 is expressed in germ cells. However, a number of recent studies have reported its re-expression in cancer. This is not uncommon, as hundreds of cancer/testis (C/T) genes have been documented to be normally expressed in germ cells and aberrantly reactivated in a variety of tumor types [116]. In a pan-cancer analysis of The Cancer Genome Atlas (TCGA), comprised of data from 32 different cancer types, the upregulation of PRDM9, as opposed to neighboring healthy tissues, was reported in about 20% of cases [117,118]. Another meta-analysis of clinical datasets from different tumor types revealed the increased expression of a number of meiotic cancer genes, including PRDM9, in lymphoma and leukemia cell lines [119]. In addition, a recent analysis of a TCGA exome sequencing datasets identified PRDM9 as one of the most mutated genes in the PRDM family. A high mutation rate not localized in any specific hot spot exceeds 10% in uterine corpus endometrial carcinoma, 14% in lung adenocarcinoma, and 15% in skin cutaneous melanoma [2].
Although these studies do not characterize the mutations nor provide any functional evidence, other reports have associated PRDM9 genetic variants with cancer. For instance, exome sequencing data from families with children affected by B-cell precursor acute lymphoblastic leukemia (B-ALL) revealed a significant excess of rare allelic forms of PRDM9 [120]. This observation was confirmed in an independent cohort of children with B-ALL, where the excess was found in aneuploid and infant B-ALL patients. The latter findings suggest the involvement of PRDM9 in genomic instability and aneuploidy in cancer. Indeed, PRDM9 expression levels correlate with the abundance of somatic structural variants (SVs) in several tumor types, with PRDM9-expressing cancers exhibiting a higher number of such breakpoints. Importantly, these SVs are enriched within known PRDM9 binding sites, reinforcing the potential link between PRDM9 expression and genomic instability in cancer [118].
Altogether, while mounting evidence places PRDM9 as a new oncogenic member of the family, in-depth functional studies are needed to characterize its involvement in tumorigenesis. Notably, the recent development of the first-in-class inhibitor for PRDM9 [40] makes the validation of an oncogenic function for this enzyme a high-priority task. Indeed, as a C/T gene, PRDM9 would be an ideal therapeutic target: Not only do C/T genes’ germ line-restricted expression profiles make them outstandingly safe targets, but also they are additionally good biomarkers and targets for immunotherapy given their documented immunogenicity in cancer.
PRDM14: an ideal target in oncology with links to stem cell biology
Mechanistic studies have elucidated PRDM14’s association with EZH2/H3K27me3, a key repressive histone mark associated with gene silencing. While this association is essential to maintain ESC pluripotency in a naïve state, this association remains to be demonstrated in cancer [37]. The full-length (PR-plus) isoform of PRDM14 is oncogenic, and its genetic locus is often amplified in several tumor types. In particular, PRDM14 expression is upregulated in 25% of lymphoid neoplasms and increased copy-number variants have been identified in breast cancer [121–123]. Ectopic expression of PRDM14 in breast cancer cell lines increases their proliferation and resistance to standard chemotherapeutic combinations, while siRNA knockdown has the opposite effect [123]. Nandi et al., based on the statistic that women with diabetes have a 20% higher risk of developing breast cancer, identified MiR-424 as a key regulator of cancer cell stemness as its overexpression under hyperglycemic conditions resulted in reduced stem cell activity. Particularly, miR-424 regulates cdc42, which, in turn, activates PRDM14 via PAK1 and STAT4, thus highlighting a role for the miR-424–cdc42–prdm14 axis in breast cancer progression in diabetic patients [124]. This study hints at a crucial role of PRDM14 in integrating extracellular signals with epigenetic modifiers.
PRDM14 function has been particularly well characterized in lymphoid leukemias and T-ALL mouse models, demonstrating full penetrance and accelerated disease onset due to expansion of hematopoietic stem cells, lymphoid progenitors, and blockade of pro-differentiating cellular programs [125]. Specifically, in a PRDM14-induced T-ALL model, NOTCH1 is activated through a RAG-recombinase complex, enabled by a PRDM14-induced permissive chromatin state [125,126]. In addition, upregulation of critical genes in pluripotency, tumor initiation, epithelial-to-mesenchymal transition, and other onco-pathways such as WNT and Ras/MAPK was noted in lymphoblastic leukemia. These latter findings have been validated in other cancers including non-small-cell lung carcinoma [127].
Currently, no studies have identified a PR-less isoform or an isoform with tumor suppressor function.
PRDM15: a context-dependent regulator of multiple oncogenic signaling pathway
PRDM15 is one of the least characterized members of the PRDM family. It was first identified as a candidate gene for a particular phenotype of Down syndrome known as bipolar affective disorder, which maps to human chromosome 21q22.3 [128]. Similar to PRDM9, although to a lesser extent, PRDM15 expression in adult somatic cells is rather low, albeit ubiquitous. This is consistent with its essential role during embryogenesis [106], while being dispensable in adult mice [129]. Intriguingly, PRDM15 expression is upregulated in several subtypes of B-cell lymphomas, as opposed to healthy tissues [129]. Such a selective expression profile, characteristic of most PRDM proteins, is well suited for designing safe therapeutic strategies against oncogenic members of this family. Our studies provide extensive functional and mechanistic characterization of the oncogenic role of PRDM15 in B-cell lymphomas. We provide evidence that perturbation of PRDM15 expression, either genetically using knockout models or pharmacologically using antisense oligonucleotides (AONs), delays tumorigenesis and selectively kills established B-cell lymphomas, both in vitro and in vivo [129]. Mechanistically, depletion of PRDM15 blocks metabolic processes, mainly glycolysis and mTOR signaling in tumor cells, inducing a ‘metabolic crisis’ that is detrimental to their survival, while sparing normal tissues that rely less on these pathways. In addition to its expression profile, the role of PRDM15 as a transcription factor able to orchestrate a broad transcriptional program to sustain tumor survival makes it a unique and promising therapeutic target. Indeed, our data show that, as opposed to the widely reported resistance to mTOR or AKT inhibitors, due to rapid signaling rewiring, PRDM15 depletion has long-lasting effects on tumor growth inhibition. Eμ-myc mice lacking the Prdm15 gene live twice as long as their Prdm15 WT counterparts. Intriguingly, the KO mice do not develop B-cell lymphomas, but some of them tend to eventually develop either T-cell or less-characterized non-B/non-T-cell malignancies. This is consistent with the fact that B-cell lymphoma cells in vitro fail to rewire their signaling or survive PRDM15 depletion.
While the crystal structure of PRDM15 has yet to be solved and potential PRDM15-specific inhibitors may take a while to develop, the use of AONs or PRDM15 degraders may represent interesting alternatives. As an alternative consideration, our studies analyzing the therapeutic potential of drugging the PRDM15 transcriptional network suggest that combination therapy targeting glycolysis and the PI3K/AKT/mTOR signaling axis, both of which have FDA-approved drugs used in the clinic, is an exciting therapeutic strategy for B-cell lymphomas.
PRDM6 and PRDM10: unexplored oncogenes
Unlike the previously reported oncogenic PRDMs, more work needs to be done to better appreciate the significance of those that are less well characterized, including PRDM6 and PRDM10. PRDM6 has been recently identified as a novel driver gene in class 4 medulloblastoma, a class that still lacks a comprehensive analysis of its driver genes. A novel oncogenic function of PRDM6 has been identified thanks to cis expression structural alteration mapping, a method able to infer enhancer hijacking events. In particular, PRDM6 maps 600 kb downstream of the recurrently mutated gene SNCAIP and its expression was 20-fold higher, much more than any other gene within the proximal topologically associated domains (TADs) (SNCAIP included). By integrating NGS and topological data, the structural variants that disrupt the SNCAIP locus led to novel super-enhancer interactions with neighboring TADs, most notably with the PRDM6 promoter [130]. These data suggest that there is more to be discovered about PRDM6 in cancer, considering that it appeared to be fundamental in very specific tumor types, thus strengthening the idea that PRDMs function in an inherently context-dependent manner. Broad bioinformatic analyses of publicly available datasets might corroborate this hypothesis of context specificity.
PRDM10 is a sequence-specific transcription factor essential for preimplantation embryos and mouse embryonic stem cell homeostasis as loss of Prdm10 results in dramatic cell growth inhibition. Indeed, Prdm10 regulates global translation in early embryonic development and mESC homeostasis, acting as the transcription factor of the core component of the eukaryotic translation initiation factor 3 [7]. Aside from basic studies in mESCs, the oncogenic role of PRDM10 has been identified in undifferentiated pleomorphic sarcoma as a consistent proportion of patients are characterized by novel gene fusions involving PRDM10 as the 3′ partner and either MED12 or CITED2 at the 5′ [131]. CADM3 was found to be consistently upregulated both in PRDM10-rearranged soft tumors and in fibroblasts overexpressing a CITED2-PRDM10 fusion protein. ATAC-seq data show enrichment at PRDM10 binding sites of the CITED2-PRDM10 fusion protein, suggesting that the N terminus partner might increase chromatin accessibility to PRDM10 [132]. Hitherto, little is known about the mechanisms by which PRDM10 contributes to cancer evolution, the affected signaling pathways, and whether it has direct or indirect methyltransferase activity.
PRDMs interactome: a missing link
A systematic identification of the interactome for each PRDM in different cell types and organs is still missing and would certainly shed light on the function of specific family members, in particular those lacking a catalytic activity. A key example of the utility of such an approach comes from PRDM9, where the KRAB domain is able to establish protein–protein interactions with multiple proteins important to further modify chromatin and advance toward the phases of meiotic recombination. In particular, CXXC1, EWSR1, EHMT2, and CDYL are all direct PRDM9-KRAB interactors. In addition, PRDM9 is able to interact with the meiotic cohesin REC8 protein and the SYCP1/ SYCP3 synaptonemal complex subunits. Such interactions are partially mediated by DNA looping and, in addition to PRDM9 catalytic activity, are important to ensure meiosis [133].
A second example of how mass spectrometry approaches have been instrumental in characterizing the repressive function of a PRDM protein is the characterization of the PRDM14 interactome. Two groups reported the interaction between PRDM14 and components of the polycomb complex, PRC2 (e.g., EZH2). This interaction is important for PRDM14 to repress genes critical to the activity of the fibroblast growth factor receptor signaling. It also maintains hypomethylated chromatin by preventing the expression of DNA methyltransferases (i.e., Dnmt3a, Dnmt3b, and Dnmt3l) that would lead to a primed epiblast-like state if expressed [97] and, finally, represses essential developmental targets during iPSC reprogramming [37]. A later report characterized the interaction between PRDM14 and the Mtgr1 transcription corepressor [134]. Mtgr1 is a member of the ETO family of transcriptional corepressors [135] that binds tightly to the pre-SET region and SET domain of PRDM14, occupying overlapping genomic loci.
An additional effort toward identifying the PRDM interactome has recently characterized the relevant interactions that distinguishes between the long and short isoforms of PRDM3 and PRDM16 [136]. The authors focused on the duality of PRDMs and asked what features enable the tumor suppressive functions of the long isoforms. To address this question, they performed co-immunoprecipitation followed by mass spectrometry of GFP-tagged long or short (PR-less) isoforms of both PRDM3 and PRDM16. They found that the long isoforms had significantly enriched interactions with proteins associated with the nucleosome remodeling and deacetylase (NuRD) complex (including Rbbp4, Mta1, Mta2, and Chd4) compared with the truncated proteins. Immunoprecipitation of endogenous human RBBP4 pulled down the full-length, but not the short, isoforms of both proteins, highlighting that they are indeed binding partners. Specifically, the first 12 amino acids of the N termini of the full-length isoforms bind to RBBP4 in the same pocket where histone H3 can also bind. In fact, all three proteins contain conserved lysine and arginine residues that are likely key to their interactions with RBBP4. More intriguingly, related proteins, including PRDM18/FOG-1 and PRDM19/FOG-2, maintain these residues and have been reported to bind to RBBP4 too [137,138]. Overall, Ivanochko et al. provide a mechanism by which the long isoforms of PRDM3 and PRDM16 are specifically able to recruit transcriptional corepressor complexes, a function that is lost in their short isoforms. The group proposes that these PRDMs provide a ‘tether’ for the complex—with their ZNF arrays bound to DNA, the PRDMs can bring the NuRD complex to chromatin by interacting with RBBP4 and other protein members. When the truncated isoforms are expressed, this interaction is lost or significantly weakened and transcriptional repression is released, allowing expression of potential oncogenes. It would be worthwhile exploring the transcriptomes of cells with these alternative isoforms to discern the downstream targets of the NuRD/PRDM interaction. Similarly, ChIP-sequencing of both PRDM isoforms in different cellular contexts would also provide insight into their altered capacity to bind to target genes and potentially regulate their transcription [136]. The methodology used by the Arrowsmith laboratory could be used as a framework for future studies of other PRDM interactomes. While the path to targeting PRDMs may take a while, it would be particularly advantageous to identify physical interactors of the oncogenic PRDMs that could be easier to target or which selective inhibitors are already available.
Beyond the structure of PRDMs: therapeutic perspectives and final remarks
We here propose that PRDMs play a fundamental role integrating the upstream signals from oncodrivers or signaling pathways, by either establishing oncogenic transcriptional programs or supporting chromatin remodeling toward a pro-oncogenic phenotype. For instance, the long latency time associated with PRDM1/Blimp1 LOF-induced tumorigenesis [52] supports the hypothesis that PRDM1/Blimp-1 acts as a gatekeeper for oncogenesis. Developmental studies have uncovered the highly tissue-specific expression of PRDM proteins and their involvement in lineage specification and tumorigenesis. The key to understanding their involvement would require a better characterization of the upstream regulatory mechanisms, currently representing one of the biggest gaps in the literature. In particular, promoter hypermethylation, noncoding RNAs, and other epigenetic mechanisms have been postulated to be pivotal for upstream PRDM regulation. This is, indeed, concordant with the current context-dependent hypothesis of PRDMs’ function.
From a functional perspective, PRDMs can be inhibited through different strategies, by tackling either the putative catalytic PR domain or the zinc fingers. ZNF inhibitors have not been translated to the clinic given the lack of protein specificity as this domain is present in multiple proteins mediating DNA-binding interactions [139]. Moreover, there is a lack of understanding on whether it would be more efficient to target ZNFs responsible for protein–nucleic acid interactions or those mediating protein–protein binding.
Recently, MRK-740, a potent, selective, and cell-active probe targeting PRDM9, has been identified as the first-in-class PRDM inhibitor [40]. Yet, there is a pressing need to expand pharmacological research on small molecules targeting other PRDMs, considering their noncatalytic domains involved in noncanonical signaling. By drawing similarities from pseudokinases, several members of the PRDM family of proteins may be classified as pseudomethyltransferases as they are not able to exert primary methyltransferase activity. Paradoxically, the PR domain lacks the most conserved motif H/RxxNHxC that has been repeatedly demonstrated to be essential for methyltransferase activity in SET domain containing proteins [140]. A recent and promising idea that is developing traction in the field is that of designing proteolysis targeting chimera, hetero-bifunctional molecules linking a compound to an E3 ubiquitin ligase to promote proteasomal degradation via formation of E3 tertiary structures. Identifying small molecule binders for PRDMs would open up new possibilities to develop tool compounds and future clinical molecules [141].
Alternatively, AONs can be a valuable strategy to tackle oncogenic PRDMs by directly downregulating the PRDM mRNA levels or by causing the skipping of essential domains. AONs, also known as antisense oligonucleotides (ASOs) or splice-switching antisense oligonucleotides, are short, single-stranded oligonucleotides able to modify RNA expression by several mechanisms such as splice-switching and modulation of protein expression. Thanks to improvements in backbone composition and pharmacokinetic properties, several AONs have been translated to the clinic to treat neurological diseases such as Duchenne muscular dystrophy and spinal muscular atrophy [142]. However, their role in cancer therapy has been hampered by several issues, including selective delivery to tumor cells, plasticity, and clonal heterogeneity of cancer. A new generation of delivery tools including nanoparticles, lipid micelles, and direct chemical conjugation will hopefully solve some of these issues in the near future.
Altogether, PRDMs represent a family of proteins with invaluable therapeutic potential, as several members show oncogenic functions either by gain-of-function mutation, upregulation, or isoform switching. Data on targeting PRDM15 in B-cell lymphoma [129] provide hope that targeting oncogenic PRDMs, and thus entire transcriptional programs, is a promising strategy to overcome resistance and selectively target cancerous cells. Furthermore, genetic studies on PRDMs have shown that they are crucial for embryo development, but become partially or virtually dispensable for somatic homeostasis during adulthood. Their expression is, however, reactivated during oncogenesis, which makes them safe therapeutic targets in adults. Oncogenesis is a selfish development: It reinstates developmental programs that were meant to be permanently turned off in adulthood, giving cells a growth advantage that paradoxically kills its host. Such processes lead to dedifferentiation and acquisition of aberrant stem cell-like properties. A notable example of how this can be exploited is the treatment of acute promyelocytic leukemia, where pro-differentiating agents that promote terminally differentiation of cancer cells have revolutionized treatment options [143]. Similarly, inducing differentiation through manipulation of master transcriptional regulators, such as PRDMs, is an exciting strategy for cancer therapy.
Acknowledgements
Research reported in this publication was supported in part by the National Cancer Institute of the NIH (R01CA249204, R01CA248984, and R01HD102614) and ISMMS seed fund to EG. The authors gratefully acknowledge the use of the services and facilities of the Tisch Cancer Institute supported by the NCI Cancer Center Support Grant (P30 CA196521). MS was supported by an NCI training grant (T32CA078207).
Abbreviations
- AON
antisense oligonucleotides
- B-ALL
B-cell acute lymphoblastic leukemia
- C2H2
Cys2-His2
- FOG
Friends Of GATA
- KO
knockout
- KRAB
Krüppel-associated box
- mESC
mouse embryonic stem cell
- miR
MicroRNA
- ncRNA
noncoding RNA
- NuRD
nucleosome remodeling and deacetylase
- Pak1
p-21-activated kinase
- PKMT
protein lysine methyltransferases
- PR
PRDF1-RIZ
- PRDM
PRDF1 and RIZ1 homology domain containing
- SAH
S-adenosyl homocysteine
- SAM
S-adenosyl methionine
- SELEX
systematic evolution of ligands by exponential enrichment
- SET
Su(var)3–9, Enhancer-of-zeste and Trithorax
- shRNA
short hairpin RNA
- siRNA
short interference RNA
- SV
somatic structural variants
- TAD
topologically associated domain
- T-ALL
T-cell acute lymphoblastic leukemia
- ZNF
zinc finger
Footnotes
Conflict of interest
EG is cofounders and scientific advisors of IMMU-NOA Pte. Ltd. EG has served on advisory board for Lion TCR Pte. Ltd. The rest of the authors declare no competing financial interests.
References
- 1.Mzoughi S, Tan YX, Low D & Guccione E (2016) The role of PRDMs in cancer: one family, two sides. Curr Opin Genet Dev 36, 83–91. [DOI] [PubMed] [Google Scholar]
- 2.Sorrentino A, Federico A, Rienzo M, Gazzerro P, Bifulco M, Ciccodicola A, Casamassimi A & Abbondanza C (2018) PR/SET domain family and cancer: novel insights from the cancer genome atlas. Int J Mol Sci 19, 3250. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Vervoort M, Meulemeester D, Behague J & Kerner P (2016) Evolution of Prdm genes in animals: insights from comparative genomics. Mol Biol Evol 33, 679–696. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Fog CK, Galli GG & Lund AH (2012) PRDM proteins: important players in differentiation and disease. BioEssays 34, 50–60. [DOI] [PubMed] [Google Scholar]
- 5.Leszczynski P, Śmiech M, Parvanov E, Watanabe C, Mizutani K-I & Taniguchi H (2020) Emerging roles of PRDM factors in stem cells and neuronal system: cofactor dependent regulation of PRDM3/16 and FOG1/2 (Novel PRDM Factors). Cells 9, 2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Arnold CD, Nemčko F, Woodfin AR, Wienerroither S, Vlasova A, Schleiffer A, Pagani M, Rath M & Stark A (2018) A high-throughput method to identify trans-activation domains within transcription factor sequences. EMBO J 37, e98896. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Han BY, Seah MKY, Brooks IR, Quek DHP, Huxley DR, Foo C-S, Lee LT, Wollmann H, Guo H, Messerschmidt DM et al. (2020) Global translation during early development depends on the essential transcription factor PRDM10. Nat Commun 11, 3603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Fumasoni I, Meani N, Rambaldi D, Scafetta G, Alcalay M & Ciccarelli FD (2007) Family expansion and gene rearrangements contributed to the functional specialization of PRDM genes in vertebrates. BMC Evol Biol 7, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Kung JE & Jura N (2019) Prospects for pharmacological targeting of pseudokinases. Nat Rev Drug Discov 18, 501–526. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Clifton MK, Westman BJ, Thong SY, O’Connell MR, Webster MW, Shepherd NE, Quinlan KG, Crossley M, Blobel GA & Mackay JP (2014) The identification and structure of an N-terminal PR domain show that FOG1 is a member of the PRDM family of proteins. PLoS One 9, e106011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Chang AN, Cantor AB, Fujiwara Y, Lodish MB, Droho S, Crispino JD & Orkin SH (2002) GATA-factor dependence of the multitype zinc-finger protein FOG-1 for its essential role in megakaryopoiesis. Proc Natl Acad Sci USA 99, 9237–9242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Fox AH, Liew C, Holmes M, Kowalski K, Mackay J & Crossley M (1999) Transcriptional cofactors of the FOG family interact with GATA proteins by means of multiple zinc fingers. EMBO J 18, 2812–2822. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Tevosian SG, Deconinck AE, Cantor AB, Rieff HI, Fujiwara Y, Corfas G & Orkin SH (1999) FOG-2: A novel GATA-family cofactor related to multitype zinc-finger proteins Friend of GATA-1 and U-shaped. Proc Natl Acad Sci USA 96, 950–955. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Liew CK, Simpson RJY, Kwan AHY, Crofts LA, Loughlin FE, Matthews JM, Crossley M & Mackay JP (2005) Zinc fingers as protein recognition motifs: structural basis for the GATA-1/friend of GATA interaction. Proc Natl Acad Sci USA 102, 583–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Crispino JD, Lodish MB, Thurberg BL, Litovsky SH, Collins T, Molkentin JD & Orkin SH (2001) Proper coronary vascular development and heart morphogenesis depend on interaction of GATA-4 with FOG cofactors. Genes Dev 15, 839–844. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Kim KC, Geng L & Huang S (2003) Inactivation of a histone methyltransferase by mutations in human cancers. Cancer Res 63, 7619–7623. [PubMed] [Google Scholar]
- 17.Blazer LL, Lima-Fernandes E, Gibson E, Eram MS, Loppnau P, Arrowsmith CH, Schapira M & Vedadi M (2016) PR domain-containing protein 7 (PRDM7) Is a histone 3 lysine 4 trimethyltransferase. J Biol Chem 291, 13509–13519. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Wu H, Mathioudakis N, Diagouraga B, Dong A, Dombrovski L, Baudat F, Cusack S, de Massy B & Kadlec J (2013) Molecular basis for the regulation of the H3K4 methyltransferase activity of PRDM9. Cell Rep 5, 13–20. [DOI] [PubMed] [Google Scholar]
- 19.Hayashi K, Yoshida K & Matsui Y (2005) A histone H3 methyltransferase controls epigenetic events required for meiotic prophase. Nature 438, 374–378. [DOI] [PubMed] [Google Scholar]
- 20.Koh-Stenta X, Joy J, Poulsen A, Li R, Tan Y, Shim Y, Min J-H, Wu L, Ngo A, Peng J et al. (2014) Characterization of the histone methyltransferase PRDM9 using biochemical, biophysical and chemical biology techniques. Biochem J 461, 323–334. [DOI] [PubMed] [Google Scholar]
- 21.Eram MS, Bustos SP, Lima-Fernandes E, Siarheyeva A, Senisterra G, Hajian T, Chau I, Duan S, Wu H, Dombrovski L et al. (2014) Trimethylation of histone H3 lysine 36 by human methyltransferase PRDM9 protein. J Biol Chem 289, 12177–12188. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Powers NR, Parvanov ED, Baker CL, Walker M, Petkov PM & Paigen K (2016) The meiotic recombination activator PRDM9 trimethylates both H3K36 and H3K4 at recombination hotspots in vivo. PLoS Genet 12, e1006146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Zhou B, Wang J, Lee SY, Xiong J, Bhanu N, Guo Q, Ma P, Sun Y, Rao RC, Garcia BA et al. (2016) PRDM16 suppresses MLL1r leukemia via intrinsic histone methyltransferase activity. Mol Cell 62, 222–236. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Pinheiro I, Margueron R, Shukeir N, Eisold M, Fritzsch C, Richter FM, Mittler G, Genoud C, Goyama S, Kurokawa M et al. (2012) Prdm3 and Prdm16 are H3K9me1 methyltransferases required for mammalian heterochromatin integrity. Cell 150, 948–960. [DOI] [PubMed] [Google Scholar]
- 25.Eom GH, Kim K, Kim S-M, Kee HJ, Kim J-Y, Jin HM, Kim J-R, Kim JH, Choe N, Kim K-B et al. (2009) Histone methyltransferase PRDM8 regulates mouse testis steroidogenesis. Biochem Biophys Res Commun 388, 131–136. [DOI] [PubMed] [Google Scholar]
- 26.Gyory I, Wu J, Fejer G, Seto E & Wright KL (2004) PRDI-BF1 recruits the histone H3 methyltransferase G9a in transcriptional silencing. Nat Immunol 5, 299–308. [DOI] [PubMed] [Google Scholar]
- 27.Ancelin K, Lange UC, Hajkova P, Schneider R, Bannister AJ, Kouzarides T & Surani MA (2006) Blimp1 associates with Prmt5 and directs histone arginine methylation in mouse germ cells. Nat Cell Biol 8, 623–630. [DOI] [PubMed] [Google Scholar]
- 28.Yoshimi A, Goyama S, Watanabe-Okochi N, Yoshiki Y, Nannya Y, Nitta E, Arai S, Sato T, Shimabe M, Nakagawa M et al. (2011) Evi1 represses PTEN expression and activates PI3K/AKT/mTOR via interactions with polycomb proteins. Blood 117, 3617–3628. [DOI] [PubMed] [Google Scholar]
- 29.Duan Z, Person RE, Lee H-H, Huang S, Donadieu J, Badolato R, Grimes HL, Papayannopoulou T & Horwitz MS (2007) Epigenetic regulation of protein-coding and microRNA genes by the Gfi1-interacting tumor suppressor PRDM5. Mol Cell Biol 27, 6889–6902. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Wu Y, Ferguson JE, Wang H, Kelley R, Ren R, McDonough H, Meeker J, Charles PC, Wang H & Patterson C (2008) PRDM6 is enriched in vascular precursors during development and inhibits endothelial cell proliferation, survival, and differentiation. J Mol Cell Cardiol 44, 47–58. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Davis CA, Haberland M, Arnold MA, Sutherland LB, McDonald OG, Richardson JA, Childs G, Harris S, Owens GK & Olson EN (2006) PRISM/PRDM6, a transcriptional repressor that promotes the proliferative gene program in smooth muscle cells. Mol Cell Biol 26, 2626–2636. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Yildiz O, Downes GB & Sagerstrom CG (2019) Zebrafish prdm12b acts independently of nkx6.1 repression to promote eng1b expression in the neural tube p1 domain. Neural Dev 14, 5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Thelie A, Desiderio S, Hanotel J, Quigley I, Van Driessche B, Rodari A, Borromeo MD, Kricha S, Lahaye F, Croce J et al. (2015) Prdm12 specifies V1 interneurons through cross-repressive interactions with Dbx1 and Nkx6 genes in Xenopus. Development 142, 3416–3428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Yang CM & Shinkai Y (2013) Prdm12 is induced by retinoic acid and exhibits anti-proliferative properties through the cell cycle modulation of P19 embryonic carcinoma cells. Cell Struct Funct 38, 197–206. [DOI] [PubMed] [Google Scholar]
- 35.Matsukawa S, Miwata K, Asashima M & Michiue T (2015) The requirement of histone modification by PRDM12 and Kdm4a for the development of preplacodal ectoderm and neural crest in Xenopus. Dev Biol 399, 164–176. [DOI] [PubMed] [Google Scholar]
- 36.Taniguchi H, Hoshino D, Moriya C, Zembutsu H, Nishiyama N, Yamamoto H, Kataoka K & Imai K (2017) Silencing PRDM14 expression by an innovative RNAi therapy inhibits stemness, tumorigenicity, and metastasis of breast cancer. Oncotarget 8, 46856–46874. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Chan YS, Göke J, Lu X, Venkatesan N, Feng B, Su I-H & Ng H-H (2013) A PRC2-dependent repressive role of PRDM14 in human embryonic stem cells and induced pluripotent stem cell reprogramming. Stem Cells 31, 682–692. [DOI] [PubMed] [Google Scholar]
- 38.Prajapati RS, Hintze M & Streit A (2019) PRDM1 controls the sequential activation of neural, neural crest and sensory progenitor determinants. Development 146. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Yu J, Angelin-Duclos C, Greenwood J, Liao J & Calame K (2000) Transcriptional repression by blimp-1 (PRDI-BF1) involves recruitment of histone deacetylase. Mol Cell Biol 20, 2592–2603. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Allali-Hassani A, Szewczyk MM, Ivanochko D, Organ SL, Bok J, Ho JSY, Gay FPH, Li F, Blazer L, Eram MS et al. (2019) Discovery of a chemical probe for PRDM9. Nat Commun 10, 5759. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Klug A & Schwabe JW (1995) Protein motifs 5. Zinc fingers. FASEB J 9, 597–604. [PubMed] [Google Scholar]
- 42.Billings T, Parvanov ED, Baker CL, Walker M, Paigen K & Petkov PM (2013) DNA binding specificities of the long zinc-finger recombination protein PRDM9. Genome Biol 14, R35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Persikov AV, Osada R & Singh M (2009) Predicting DNA recognition by Cys2His2 zinc finger proteins. Bioinformatics 25, 22–29. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Persikov AV & Singh MD (2014) novo prediction of DNA-binding specificities for Cys2His2 zinc finger proteins. Nucleic Acids Res 42, 97–108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Keller AD & Maniatis T (1991) Identification and characterization of a novel repressor of beta-interferon gene expression. Genes Dev 5, 868–879. [DOI] [PubMed] [Google Scholar]
- 46.Turner CA Jr, Mack DH & Davis MM (1994) Blimp-1, a novel zinc finger-containing protein that can drive the maturation of B lymphocytes into immunoglobulin-secreting cells. Cell 77, 297–306. [DOI] [PubMed] [Google Scholar]
- 47.Lin Y, Wong K & Calame K (1997) Repression of c-myc transcription by Blimp-1, an inducer of terminal B cell differentiation. Science 276, 596–599. [DOI] [PubMed] [Google Scholar]
- 48.Mandelbaum J, Bhagat G, Tang H, Mo T, Brahmachary M, Shen Q, Chadburn A, Rajewsky K, Tarakhovsky A, Pasqualucci L et al. (2010) BLIMP1 is a tumor suppressor gene frequently disrupted in activated B cell-like diffuse large B cell lymphoma. Cancer Cell 18, 568–579. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Pasqualucci L, Compagno M, Houldsworth J, Monti S, Grunn A, Nandula SV, Aster JC, Murty VV, Shipp MA & Dalla-Favera R (2006) Inactivation of the PRDM1/BLIMP1 gene in diffuse large B cell lymphoma. J Exp Med 203, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Nie K, Zhang T, Allawi H, Gomez M, Liu Y, Chadburn A, Wang YL, Knowles DM & Tam W (2010) Epigenetic down-regulation of the tumor suppressor gene PRDM1/Blimp-1 in diffuse large B cell lymphomas: a potential role of the microRNA let-7. Am J Pathol 177, 1470–1479. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Tam W, Gomez M, Chadburn A, Lee JW, Chan WC & Knowles DM (2006) Mutational analysis of PRDM1 indicates a tumor-suppressor role in diffuse large B-cell lymphomas. Blood 107, 4090–4100. [DOI] [PubMed] [Google Scholar]
- 52.Calado DP, Zhang B, Srinivasan L, Sasaki Y, Seagal J, Unitt C, Rodig S, Kutok J, Tarakhovsky A, Schmidt-Supprian M et al. (2010) Constitutive canonical NF-kappaB activation cooperates with disruption of BLIMP1 in the pathogenesis of activated B cell-like diffuse large cell lymphoma. Cancer Cell 18, 580–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Gyory I, Fejer G, Ghosh N, Seto E & Wright KL (2003) Identification of a functionally impaired positive regulatory domain I binding factor 1 transcription repressor in myeloma cell lines. J Immunol 170, 3125–3133. [DOI] [PubMed] [Google Scholar]
- 54.Doody GM, Care MA, Burgoyne NJ, Bradford JR, Bota M, Bonifer C, Westhead DR & Tooze RM (2010) An extended set of PRDM1/BLIMP1 target genes links binding motif type to dynamic repression. Nucleic Acids Res 38, 5336–5350. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Mould AW, Morgan MA, Nelson AC, Bikoff EK & Robertson EJ (2015) Blimp1/Prdm1 functions in opposition to Irf1 to maintain neonatal tolerance during postnatal intestinal maturation. PLoS Genet 11, e1005375. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Kuo TC & Calame KL (2004) B lymphocyte-induced maturation protein (Blimp)-1, IFN regulatory factor (IRF)-1, and IRF-2 can bind to the same regulatory sites. J Immunol 173, 5556–5563. [DOI] [PubMed] [Google Scholar]
- 57.Doody GM, Stephenson S, McManamy C & Tooze RM (2007) PRDM1/BLIMP-1 modulates IFN-gamma-dependent control of the MHC class I antigen-processing and peptide-loading pathway. J Immunol 179, 7614–7623. [DOI] [PubMed] [Google Scholar]
- 58.Buyse IM, Shao G & Huang S (1995) The retinoblastoma protein binds to RIZ, a zinc-finger protein that shares an epitope with the adenovirus E1A protein. Proc Natl Acad Sci USA 92, 4467–4471. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.He L, Yu JX, Liu L, Buyse IM, Wang MS, Yang QC, Nakagawara A, Brodeur GM, Shi YE & Huang S (1998) RIZ1, but not the alternative RIZ2 product of the same gene, is underexpressed in breast cancer, and forced RIZ1 expression causes G2-M cell cycle arrest and/or apoptosis. Cancer Res 58, 4238–4244. [PubMed] [Google Scholar]
- 60.Steele-Perkins G, Fang W, Yang XH, Van Gele M, Carling T, Gu J, Buyse IM, Fletcher JA, Liu J, Bronson R et al. (2001) Tumor formation and inactivation of RIZ1, an Rb-binding member of a nuclear protein-methyltransferase superfamily. Genes Dev 15, 2250–2262. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Shadat NM, Koide N, Khuda II-E, Dagvadorj J, Tumurkhuu G, Naiki YO, Komatsu T, Yoshida T & Yokochi T (2010) Retinoblastoma protein-interacting zinc finger 1 (RIZ1) regulates the proliferation of monocytic leukemia cells via activation of p53. Cancer Invest 28, 806–812. [DOI] [PubMed] [Google Scholar]
- 62.Pastural E, Takahashi N, Dong W-F, Bainbridge M, Hull A, Pearson D, Huang S, Lowsky R, DeCoteau JF & Geyer CR (2007) RIZ1 repression is associated with insulin-like growth factor-1 signaling activation in chronic myeloid leukemia cell lines. Oncogene 26, 1586–1594. [DOI] [PubMed] [Google Scholar]
- 63.Abbondanza C, De Rosa C, D’Arcangelo A, Pacifico M, Spizuoco C, Piluso G, Di Zazzo E, Gazzerro P, Medici N, Moncharmont B et al. (2012) Identification of a functional estrogen-responsive enhancer element in the promoter 2 of PRDM2 gene in breast cancer cell lines. J Cell Physiol 227, 964–975. [DOI] [PubMed] [Google Scholar]
- 64.Sun W, Geyer CR & Yang J (2008) Cloning, expression, purification and crystallization of the PR domain of human retinoblastoma protein-binding zinc finger protein 1 (RIZ1). Int J Mol Sci 9, 943–950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Jiang G, Liu L, Buyse IM, Simon D & Huang S (1999) Decreased RIZ1 expression but not RIZ2 in hepatoma and suppression of hepatoma tumorigenicity by RIZ1. Int J Cancer 83, 541–546. [DOI] [PubMed] [Google Scholar]
- 66.Liu L, Shao G, Steele-Perkins G & Huang S (1997) The retinoblastoma interacting zinc finger gene RIZ produces a PR domain-lacking product through an internal promoter. J Biol Chem 272, 2984–2991. [DOI] [PubMed] [Google Scholar]
- 67.Tanadi C, Bambang A, Wendi IP, Sidharta VM, Hananta L & Sumarpo A (2020) The predictive value of PRDM2 in solid tumor: a systematic review and meta-analysis. PeerJ 8, e8826. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Sorrentino A, Rienzo M, Ciccodicola A, Casamassimi A & Abbondanza C (2018) Human PRDM2: Structure, function and pathophysiology. Biochim Biophys Acta Gene Regul Mech 1861, 657–671. [DOI] [PubMed] [Google Scholar]
- 69.Glass C, Wilson M, Gonzalez R, Zhang Y & Perkins AS (2014) The role of EVI1 in myeloid malignancies. Blood Cells Mol Dis 53, 67–76. [DOI] [PubMed] [Google Scholar]
- 70.Yuasa H, Oike Y, Iwama A, Nishikata I, Sugiyama D, Perkins A, Mucenski ML, Suda T & Morishita K (2005) Oncogenic transcription factor Evi1 regulates hematopoietic stem cell proliferation through GATA-2 expression. EMBO J 24, 1976–1987. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Morishita K, Suzukawa K, Taki T, Ihle JN & Yokota J (1995) EVI-1 zinc finger protein works as a transcriptional activator via binding to a consensus sequence of GACAAGATAAGATAAN1–28 CTCATCTTC. Oncogene 10, 1961–1967. [PubMed] [Google Scholar]
- 72.Bard-Chapeau EA, Jeyakani J, Kok CH, Muller J, Chua BQ, Gunaratne J, Batagov A, Jenjaroenpun P, Kuznetsov VA, Wei C-L et al. (2012) Ecotopic viral integration site 1 (EVI1) regulates multiple cellular processes important for cancer and is a synergistic partner for FOS protein in invasive tumors. Proc Natl Acad Sci USA 109, 2168–2173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 73.Yatsula B, Lin S, Read AJ, Poholek A, Yates K, Yue D, Hui P & Perkins AS (2005) Identification of binding sites of EVI1 in mammalian cells. J Biol Chem 280, 30712–30722. [DOI] [PubMed] [Google Scholar]
- 74.Funabiki T, Kreider BL & Ihle JN (1994) The carboxyl domain of zinc fingers of the Evi-1 myeloid transforming gene binds a consensus sequence of GAAGATGAG. Oncogene 9, 1575–1581. [PubMed] [Google Scholar]
- 75.Delwel R, Funabiki T, Kreider BL, Morishita K & Ihle JN (1993) Four of the seven zinc fingers of the Evi-1 myeloid-transforming gene are required for sequence-specific binding to GA(C/T)AAGA(T/C)AAGATAA. Mol Cell Biol 13, 4291–4300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Bogani D, Morgan MAJ, Nelson AC, Costello I, McGouran JF, Kessler BM, Robertson EJ & Bikoff EK (2013) The PR/SET domain zinc finger protein Prdm4 regulates gene expression in embryonic stem cells but plays a nonessential role in the developing mouse embryo. Mol Cell Biol 33, 3936–3950. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 77.Deng Q & Huang S (2004) PRDM5 is silenced in human cancers and has growth suppressive activities. Oncogene 23, 4903–4910. [DOI] [PubMed] [Google Scholar]
- 78.Tan SX, Hu RC, Xia Q, Tan YL, Liu JJ, Gan GX & Wang LL (2018) The methylation profiles of PRDM promoters in non-small cell lung cancer. Onco Targets Ther 11, 2991–3002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Meani N, Pezzimenti F, Deflorian G, Mione M & Alcalay M (2009) The tumor suppressor PRDM5 regulates Wnt signaling at early stages of zebrafish development. PLoS One 4, e4273. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Wang X, Chang H, Gao G, Su B, Deng Q, Zhou H, Wang Q, Lin Y & Ding Y (2020) Silencing of PRDM5 increases cell proliferation and inhibits cell apoptosis in glioma. Int J Neurosci 131, 144–153. [DOI] [PubMed] [Google Scholar]
- 81.Shu XS, Geng H, Li L, Ying J, Ma C, Wang Y, Poon FF, Wang X, Ying Y, Yeo W et al. (2011) The epigenetic modifier PRDM5 functions as a tumor suppressor through modulating WNT/beta-catenin signaling and is frequently silenced in multiple tumors. PLoS One 6, e27346. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Galli GG, Carrara M, Francavilla C, Honnens de Lichtenberg K, Olsen JV, Calogero RA & Lund AH (2013) Genomic and proteomic analyses of Prdm5 reveal interactions with insulator binding proteins in embryonic stem cells. Mol Cell Biol 33, 4504–4516. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Kinameri E, Inoue T, Aruga J, Imayoshi I, Kageyama R, Shimogori T & Moore AW (2008) Prdm proto-oncogene transcription factor family expression and interaction with the Notch-Hes pathway in mouse neurogenesis. PLoS One 3, e3859. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Komai T, Iwanari H, Mochizuki Y, Hamakubo T & Shinkai Y (2009) Expression of the mouse PR domain protein Prdm8 in the developing central nervous system. Gene Expr Patterns 9, 503–514. [DOI] [PubMed] [Google Scholar]
- 85.Cypris O, Eipel M, Franzen J, Rösseler C, Tharmapalan V, Kuo C-C, Vieri M, Nikolić M, Kirschner M, Brümmendorf TH et al. (2020) PRDM8 reveals aberrant DNA methylation in aging syndromes and is relevant for hematopoietic and neuronal differentiation. Clin Epigenetics 12, 125. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 86.Chen Z, Gao W, Pu L, Zhang L, Han G, Zuo X, Zhang Y, Li X, Shen H, Wu J et al. (2018) PRDM8 exhibits antitumor activities toward hepatocellular carcinoma by targeting NAP1L1. Hepatology 68, 994–1009. [DOI] [PubMed] [Google Scholar]
- 87.Orouji E, Peitsch WK, Orouji A, Houben R & Utikal J (2020) Unique role of histone methyltransferase PRDM8 in the tumorigenesis of virus-negative merkel cell carcinoma. Cancers 12, 1057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Altemose N, Noor N, Bitoun E, Tumian A, Imbeault M, Chapman JR, Aricescu AR & Myers SR (2017) A map of human PRDM9 binding provides evidence for novel behaviors of PRDM9 and other zinc-finger proteins in meiosis. Elife 6, e28383. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 89.Baudat F, Buard J, Grey C, Fledel-Alon A, Ober C, Przeworski M, Coop G & de Massy B (2010) PRDM9 is a major determinant of meiotic recombination hotspots in humans and mice. Science 327, 836–840. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 90.Myers S, Bowden R, Tumian A, Bontrop RE, Freeman C, MacFie TS, McVean G & Donnelly P (2010) Drive against hotspot motifs in primates implicates the PRDM9 gene in meiotic recombination. Science 327, 876–879. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Parvanov ED, Petkov PM & Paigen K (2010) Prdm9 controls activation of mammalian recombination hotspots. Science 327, 835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Chen YC, Auer-Grumbach M, Matsukawa S, Zitzelsberger M, Themistocleous AC, Strom TM, Samara C, Moore AW, Cho LT-Y, Young GT et al. (2015) Transcriptional regulator PRDM12 is essential for human pain perception. Nat Genet 47, 803–808. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Bartesaghi L, Wang Y, Fontanet P, Wanderoy S, Berger F, Wu H, Akkuratova N, Bouçanova F, Médard J-J, Petitpré C et al. (2019) PRDM12 is required for initiation of the nociceptive neuron lineage during neurogenesis. Cell Rep 26, 3484–3492 e3484. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Chang JC, Meredith DM, Mayer PR, Borromeo MD, Lai HC, Ou Y-H & Johnson JE (2013) Prdm13 mediates the balance of inhibitory and excitatory neurons in somatosensory circuits. Dev Cell 25, 182–195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Watanabe S, Sanuki R, Sugita Y, Imai W, Yamazaki R, Kozuka T, Ohsuga M & Furukawa T (2015) Prdm13 regulates subtype specification of retinal amacrine interneurons and modulates visual sensitivity. J Neurosci 35, 8004–8020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 96.Mona B, Uruena A, Kollipara RK, Ma Z, Borromeo MD, Chang JC & Johnson JE (2017) Repression by PRDM13 is critical for generating precision in neuronal identity. Elife 6, e25787. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Yamaji M, Ueda J, Hayashi K, Ohta H, Yabuta Y, Kurimoto K, Nakato R, Yamada Y, Shirahige K & Saitou M (2013) PRDM14 ensures naive pluripotency through dual regulation of signaling and epigenetic pathways in mouse embryonic stem cells. Cell Stem Cell 12, 368–382. [DOI] [PubMed] [Google Scholar]
- 98.Yamamoto M, Suwa Y, Sugiyama K, Okashita N, Kawaguchi M, Tani N, Matsubara K, Nakamura A & Seki Y (2020) The PRDM14-CtBP1/2-PRC2 complex regulates transcriptional repression during the transition from primed to naive pluripotency. J Cell Sci 133, jcs240176. [DOI] [PubMed] [Google Scholar]
- 99.Okashita N, Kumaki Y, Ebi K, Nishi M, Okamoto Y, Nakayama M, Hashimoto S, Nakamura T, Sugasawa K, Kojima N et al. (2014) PRDM14 promotes active DNA demethylation through the ten-eleven translocation (TET)-mediated base excision repair pathway in embryonic stem cells. Development 141, 269–280. [DOI] [PubMed] [Google Scholar]
- 100.Okashita N, Suwa Y, Nishimura O, Sakashita N, Kadota M, Nagamatsu G, Kawaguchi M, Kashida H, Nakajima A, Tachibana M et al. (2016) PRDM14 drives OCT3/4 recruitment via active demethylation in the transition from primed to naive pluripotency. Stem Cell Reports 7, 1072–1086. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 101.Grabole N, Tischler J, Hackett JA, Kim S, Tang F, Leitch HG, Magnúsdóttir E & Surani MA (2013) Prdm14 promotes germline fate and naive pluripotency by repressing FGF signalling and DNA methylation. EMBO Rep 14, 629–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Ma Z, Swigut T, Valouev A, Rada-Iglesias A & Wysocka J (2011) Sequence-specific regulator Prdm14 safeguards mouse ESCs from entering extraembryonic endoderm fates. Nat Struct Mol Biol 18, 120–127. [DOI] [PubMed] [Google Scholar]
- 103.Chia NY, Chan Y-S, Feng B, Lu X, Orlov YL, Moreau D, Kumar P, Yang L, Jiang J, Lau M-S et al. (2010) A genome-wide RNAi screen reveals determinants of human embryonic stem cell identity. Nature 468, 316–320. [DOI] [PubMed] [Google Scholar]
- 104.Sybirna A, Tang WWC, Pierson Smela M, Dietmann S, Gruhn WH, Brosh R & Surani MA (2020) A critical role of PRDM14 in human primordial germ cell fate revealed by inducible degrons. Nat Commun 11, 1282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 105.Mzoughi S, Zhang J, Hequet D, Teo SX, Fang H, Xing QR, Bezzi M, Seah MKY, Ong SLM, Shin EM et al. (2017) PRDM15 safeguards naive pluripotency by transcriptionally regulating WNT and MAPK-ERK signaling. Nat Genet 49, 1354–1363. [DOI] [PubMed] [Google Scholar]
- 106.Mzoughi S, Di Tullio F, Low DHP, Motofeanu C-M, Ong SLM, Wollmann H, Wun CM, Kruszka P, Muenke M, Hildebrandt F et al. (2020) PRDM15 loss of function links NOTCH and WNT/PCP signaling to patterning defects in holoprosencephaly. Sci Adv 6, eaax9852. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 107.Mann N, Mzoughi S, Schneider R, Kühl SJ, Schanze D, Klämbt V, Lovric S, Mao Y, Shi S, Tan W et al. (2021) Mutations in PRDM15 are a novel cause of Galloway-Mowat syndrome. J Am Soc Nephrol 32, 580–596. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 108.Nishikata I, Sasaki H, Iga M, Tateno Y, Imayoshi S, Asou N, Nakamura T & Morishita K (2003) A novel EVI1 gene family, MEL1, lacking a PR domain (MEL1S) is expressed mainly in t(1;3)(p36;q21)-positive AML and blocks G-CSF-induced myeloid differentiation. Blood 102, 3323–3332. [DOI] [PubMed] [Google Scholar]
- 109.Harms MJ, Lim H-W, Ho Y, Shapira SN, Ishibashi J, Rajakumari S, Steger DJ, Lazar MA, Won K-J & Seale P (2015) PRDM16 binds MED1 and controls chromatin architecture to determine a brown fat transcriptional program. Genes Dev 29, 298–307. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 110.Baizabal JM, Mistry M, García MT, Gómez N, Olukoya O, Tran D, Johnson MB, Walsh CA & Harwell CC (2018) The epigenetic state of PRDM16-regulated enhancers in radial glia controls cortical neuron position. Neuron 98, 945–962.e948. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 111.Buyse IM, Takahashi EI & Huang S (1996) Physical mapping of the retinoblastoma interacting zinc finger gene RIZ to D1S228 on chromosome 1p36. Genomics 34, 119–121. [DOI] [PubMed] [Google Scholar]
- 112.Yang XH & Huang S (1999) PFM1 (PRDM4), a new member of the PR-domain family, maps to a tumor suppressor locus on human chromosome 12q23-q24.1. Genomics 61, 319–325. [DOI] [PubMed] [Google Scholar]
- 113.Casamassimi A, Rienzo M, Di Zazzo E, Sorrentino A, Fiore D, Proto MC, Moncharmont B, Gazzerro P, Bifulco M & Abbondanza C (2020) Multifaceted role of PRDM proteins in human cancer. Int J Mol Sci 21, 2648. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 114.Paigen K & Petkov PM (2018) PRDM9 and its role in genetic recombination. Trends Genet 34, 291–300. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 115.Koh-Stenta X, Poulsen A, Li R, Wee JLK, Kwek PZ, Chew SY, Peng J, Wu L, Guccione E, Joy J et al. (2017) Discovery and characterisation of the automethylation properties of PRDM9. Biochem J 474, 971–982. [DOI] [PubMed] [Google Scholar]
- 116.Juretic A, Spagnoli GC, Schultz-Thater E & Sarcevic B (2003) Cancer/testis tumour-associated antigens: immunohistochemical detection with monoclonal antibodies. Lancet Oncol 4, 104–109. [DOI] [PubMed] [Google Scholar]
- 117.Liu J, Lichtenberg T, Hoadley KA, Poisson LM, Lazar AJ, Cherniack AD, Kovatich AJ, Benz CC, Levine DA, Lee AV et al. (2018) An integrated TCGA pan-cancer clinical data resource to drive high-quality survival outcome analytics. Cell 173, 400–416.e411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 118.Houle AA, Gibling H, Lamaze FC, Edgington HA, Soave D, Fave MJ, Agbessi M, Bruat V, Stein LD, Awadalla P et al. (2018) Aberrant PRDM9 expression impacts the pan-cancer genomic landscape. Genome Res 28, 1611–1620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 119.Feichtinger J, Aldeailej I, Anderson R, Almutairi M,Almatrafi A, Alsiwiehri N, Griffiths K, Stuart N, Wakeman JA, Larcombe L et al. (2012) Meta-analysis of clinical data using human meiotic genes identifies a novel cohort of highly restricted cancer-specific marker genes. Oncotarget 3, 843–853. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 120.Hussin J, Sinnett D, Casals F, Idaghdour Y, Bruat V, Saillour V, Healy J, Grenier J-C, de Malliard T, Busche S et al. (2013) Rare allelic forms of PRDM9 associated with childhood leukemogenesis. Genome Res 23, 419–430. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 121.Dettman EJ & Justice MJ (2008) The zinc finger SET domain gene Prdm14 is overexpressed in lymphoblastic lymphomas with retroviral insertions at Evi32. PLoS One 3, e3823. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 122.Dettman EJ, Simko SJ, Ayanga B, Carofino BL, Margolin JF, Morse HC & Justice MJ (2011) Prdm14 initiates lymphoblastic leukemia after expanding a population of cells resembling common lymphoid progenitors. Oncogene 30, 2859–2873. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 123.Nishikawa N, Toyota M, Suzuki H, Honma T, Fujikane T, Ohmura T, Nishidate T, Ohe-Toyota M, Maruyama R, Sonoda T et al. (2007) Gene amplification and overexpression of PRDM14 in breast cancers. Cancer Res 67, 9649–9657. [DOI] [PubMed] [Google Scholar]
- 124.Nandy SB, Orozco A, Lopez-Valdez R, Roberts R, Subramani R, Arumugam A, Dwivedi AK, Stewart V, Prabhakar G, Jones S et al. (2017) Glucose insult elicits hyperactivation of cancer stem cells through miR-424-cdc42-prdm14 signalling axis. Br J Cancer 117, 1665–1675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 125.Carofino BL, Ayanga B, Tracey LJ, Brooke-Bisschop T & Justice MJ (2016) PRDM14 promotes RAG-dependent Notch1 driver mutations in mouse T-ALL. Biol Open 5, 645–653. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 126.Carofino BL, Ayanga B & Justice MJ (2013) A mouse model for inducible overexpression of Prdm14 results in rapid-onset and highly penetrant T-cell acute lymphoblastic leukemia (T-ALL). Dis Model Mech 6, 1494–1506. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 127.Bi HX, Shi HB, Zhang T & Cui G (2015) PRDM14 promotes the migration of human non-small cell lung cancer through extracellular matrix degradation in vitro. Chin Med J 128, 373–377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 128.Shibuya K, Kudoh J, Okui M & Shimizu N (2005) Identification of a novel zinc finger protein gene (ZNF298) in the GAP2 of human chromosome 21q. Biochem Biophys Res Commun 332, 557–568. [DOI] [PubMed] [Google Scholar]
- 129.Mzoughi S, Fong JY, Papadopoli D, Koh CM, Hulea L, Pigini P, Di Tullio F, Andreacchio G, Hoppe MM, Wollmann H et al. (2020) PRDM15 is a key regulator of metabolism critical to sustain B-cell lymphomagenesis. Nat Commun 11, 3520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 130.Northcott PA, Buchhalter I, Morrissy AS, Hovestadt V, Weischenfeldt J, Ehrenberger T, Gröbner S, Segura-Wang M, Zichner T, Rudneva VA et al. (2017) The whole-genome landscape of medulloblastoma subtypes. Nature 547, 311–317. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 131.Hofvander J, Tayebwa J, Nilsson J, Magnusson L, Brosjö O, Larsson O, Vult von Steyern F, Mandahl N, Fletcher CD & Mertens F (2015) Recurrent PRDM10 gene fusions in undifferentiated pleomorphic sarcoma. Clin Cancer Res 21, 864–869. [DOI] [PubMed] [Google Scholar]
- 132.Hofvander J, Puls F, Pillay N, Steele CD, Flanagan AM, Magnusson L, Nilsson J & Mertens F (2019) Undifferentiated pleomorphic sarcomas with PRDM10 fusions have a distinct gene expression profile. J Pathol 249, 425–434. [DOI] [PubMed] [Google Scholar]
- 133.Parvanov ED, Tian H, Billings T, Saxl RL, Spruce C, Aithal R, Krejci L, Paigen K & Petkov PM (2017) PRDM9 interactions with other proteins provide a link between recombination hotspots and the chromosomal axis in meiosis. Mol Biol Cell 28, 488–499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 134.Nady N, Gupta A, Ma Z, Swigut T, Koide A, Koide S & Wysocka J (2015) ETO family protein Mtgr1 mediates Prdm14 functions in stem cell maintenance and primordial germ cell formation. Elife 4, e10150. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 135.Davis JN, McGhee L & Meyers S (2003) The ETO (MTG8) gene family. Gene 303, 1–10. [DOI] [PubMed] [Google Scholar]
- 136.Ivanochko D, Halabelian L, Henderson E, Savitsky P, Jain H, Marcon E, Duan S, Hutchinson A, Seitova A, Barsyte-Lovejoy D et al. (2019) Direct interaction between the PRDM3 and PRDM16 tumor suppressors and the NuRD chromatin remodeling complex. Nucleic Acids Res 47, 1225–1238. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 137.Reibnegger G, Fuchs D, Hausen A, Werner ER, Werner-Felmayer G & Wachter H (1990) Unconjugated pteridines and the activation of macrophages by interferon gamma. Cancer Chemother Pharmacol 25, 308–309. [DOI] [PubMed] [Google Scholar]
- 138.Roche AE, Bassett BJ, Samant SA, Hong W, Blobel GA & Svensson EC (2008) The zinc finger and C-terminal domains of MTA proteins are required for FOG-2-mediated transcriptional repression via the NuRD complex. J Mol Cell Cardiol 44, 352–360. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 139.Abbehausen C (2019) Zinc finger domains as therapeutic targets for metal-based compounds - an update. Metallomics 11, 15–28. [DOI] [PubMed] [Google Scholar]
- 140.Rea S, Eisenhaber F, O’Carroll D, Strahl BD, Sun ZW, Schmid M, Opravil S, Mechtler K, Ponting CP, Allis CD et al. (2000) Regulation of chromatin structure by site-specific histone H3 methyltransferases. Nature 406, 593–599. [DOI] [PubMed] [Google Scholar]
- 141.Sun X, Gao H, Yang Y, He M, Wu Y, Song Y, Tong Y & Rao Y (2019) PROTACs: great opportunities for academia and industry. Signal Transduct Target Ther 4, 64. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 142.Rinaldi C & Wood MJA (2018) Antisense oligonucleotides: the next frontier for treatment of neurological disorders. Nat Rev Neurol 14, 9–21. [DOI] [PubMed] [Google Scholar]
- 143.Jimenez JJ, Chale RS, Abad AC & Schally AV (2020) Acute promyelocytic leukemia (APL): a review of the literature. Oncotarget 11, 992–1003. [DOI] [PMC free article] [PubMed] [Google Scholar]