

Abstract
Stroke is the leading cause of serious long-term disability and the fifth leading cause of death in the United States. Treatment options for stroke are few in number and limited in efficacy. Neuroinflammation mediated by microglia and infiltrating peripheral immune cells is a major component of stroke pathophysiology. Interfering with the inflammation cascade after stroke holds the promise to modulate stroke outcome. The calcium activated potassium channel KCa3.1 is expressed selectively in the injured CNS by microglia. KCa3.1 function has been implicated in pro-inflammatory activation of microglia and there is recent literature suggesting that this channel is important in the pathophysiology of ischemia/reperfusion (stroke) related brain injury. Here we describe the potential of repurposing Senicapoc, a KCa3.1 inhibitor, to intervene in the inflammation cascade that follows ischemia/reperfusion.
Keywords: Drug repurposing, Microglia, Neuroinflammation, Stroke, Ischemia
Introduction
Stroke is the leading cause of serious long-term disability and the fifth leading cause of death in the United States [1]. Treatment options for stroke are few in number and limited in efficacy [2]. The cellular response to acute ischemic stroke, particularly the response of immune cells, has been studied extensively and has been recently reviewed in detail [3, 4]. Post-ischemic inflammation is characterized by a sequence of events involving the brain, its vessels, the circulating blood and lymphoid organs (Fig. 1). The responses begin in the intravascular compartment and includes release of inflammatory mediators such as cytokines, chemokines, proteases and small vasoactive compounds (including eicosanoids and endocannabinoids) that induce multiple changes in endothelial cell and leukocyte function. These changes result in blood–brain barrier (BBB) breakdown and leukocyte infiltration into the brain parenchyma. There is subsequent initiation of innate immune responses in the ischemic penumbra by microglia, macrophages and astrocytes through activation of danger recognition receptors such as the toll-like receptors (TLRs) or purinergic receptors (P2X, P2Y) by danger associated molecular patterns (DAMPs). T-cell based and adaptive immune responses are then initiated and can be broken down both temporally (acute vs. delayed phases) and functionally (detrimental vs. protective). Numerous active cellular processes and complex cellular interactions contribute to the resolution of post-ischemic inflammation (Fig. 1). Many of these processes also play a central role in preconditioning-mediated neuroprotection [5]. The kinetics of post-stroke immune reactions are critical in post-ischemic physiology and the concept of a biphasic or multi-phasic response to brain ischemia is now favored [6–8].
Fig. 1.
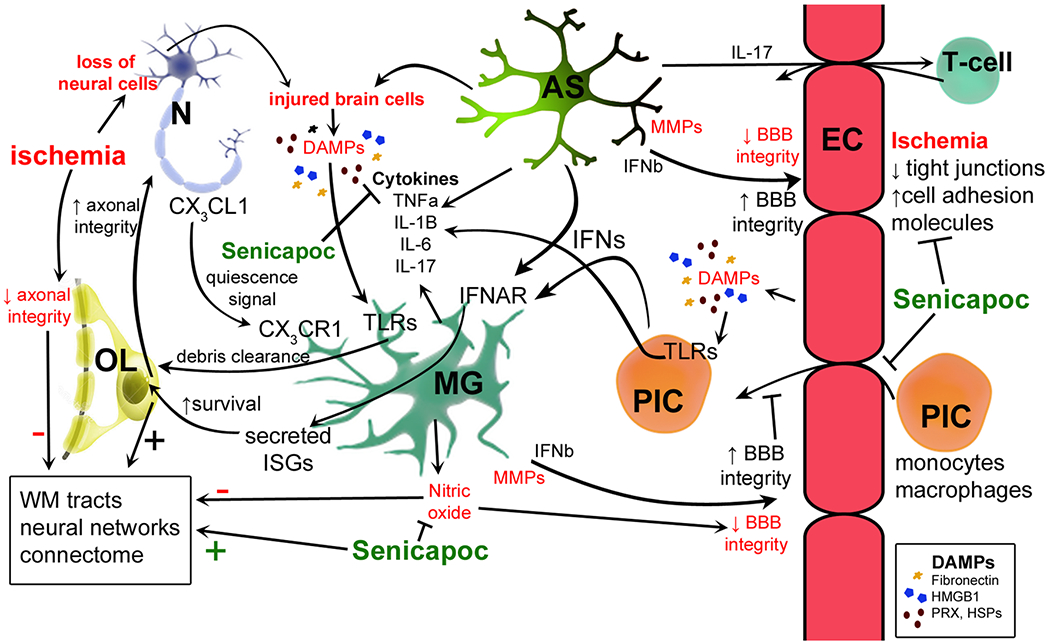
Summary of key neuroimmune pathways and interactions between cells of the CNS in ischemia. Astrocytes (AS) provide trophic support to neurons (N) through multiple mechanisms and secrete TGFβ, which is reparative to endothelial cells (EC). Both microglia (MG) and astrocytes secrete pro-inflammatory cytokines (TNFα, IL-1β, IL-6, IL-17) in response to ischemia. Neurons also signal via fractalkine (CX3CL1) to microglia which express cognate receptor CX3CR1. Both astrocytes and peripheral immune cells (PIC) are potential sources of type 1 inteferons (IFNα, β) that signal to microglia via IFNAR, triggering transcription of interferon stimulated genes (ISGs). ISG protein products may enhance oligodendrocyte (OL) viability in the setting of prolonged ischemia and in turn increase axonal integrity in white matter. The latter may limit long-term ischemia-induced injury to neural networks and protect the white matter-based connectome. ECs and other cells release danger associated molecular patterns (DAMPs), such as fibronectin, high mobility group box 1 (HMGB1), peroxiredoxin (PRX) and heat shock proteins (HSPs) that are endogenous ligands for numerous toll-like receptors (TLRs). PICs are capable of secreting many different cytokines, which have effects on multiple cell types. Senicapoc attenuates pro-inflammatory responses in microglia (reducing release of cytokines and nitric oxide) and in EC [attenuating ischemia-induced disruption of the blood–brain barrier (BBB)]. By modulating elements of the microglial and EC response to ischemia, Senicapoc may influence the neural environment indirectly in a number of ways (for example by enhancing white matter integrity as shown)
Modulating the neuroimmune response, and the microglial/macrophage phenotype in particular, is an attractive target in acute ischemic stroke therapy in part because this response evolves gradually over days to weeks, whereas many previously targeted physiological phenomena in stroke, such as glutamate-dependent excitotoxicity for example, tend to occur rapidly (minutes to hours after stroke onset) [9]. Thus, targeting the neuroimmune response in stroke offers a broader temporal therapeutic window and could translate to therapies beyond the current 3–6 h time window.
Microglia are CNS-resident immune cells [10, 11] derived from yolk sac macrophages that enter the CNS during early development and maintain themselves as a distinct population from circulating monocytes [12]. Microglia contribute to the maintenance of brain homeostasis by pruning synapses, clearing dead or dying cells as well as providing trophic support to other cells [13]. These functions suggest that microglia play a critical role in the normal physiology and development of the CNS [14]. Microglia play a significant role in the neuroinflammatory response to ischemia [15] (Fig. 1). The expression of TLRs and other pattern recognition receptors by microglia enables them to identify pathogens and upregulate a unique profile of innate and effector immune cytokines and chemokines in response to a wide range of stimuli [16]. Most abundantly expressed by microglia is TLR4, and both endogenous and exogenous TLR4 agonists potently activate classical pro-inflammatory responses in microglia [16, 17]. Although microglial activation has typically been considered a pro-inflammatory process, recent publications suggest that microglia could play a protective role in stroke [18, 19] through multiple mechanisms such as metabolic and physiological support of neurons [16], production of trophic factors [19], autophagy of damaged and repair of lesioned tissue [20]. Microglia are the first responders to ischemic injury, activating before peripheral monocytes/macrophages infiltrate the CNS [21]. Ischemia induces robust increases in microglia cell number [5, 22] and proliferation [22]. Pharmacologic or genetic ablation of microglia influences outcome in multiple rodent models of stroke [18, 23]. These findings have provided strong evidence to support a key role for innate immune signaling and microglia in both ischemia-induced injury and neuroprotection. Thus, pharmaco-therapeutics that can specifically modulate microglial or CNS-infiltrating monocyte gene expression and phenotype in the context of ischemia may be able to effectively skew the neuroimmune response in a direction that is more favorable to both neuronal survival and axonal/white matter integrity [15].
KCa3.1
KCa3.1 is a calcium activated potassium channel that is expressed in the injured CNS by microglia, infiltrating monocytes and cerebrovascular endothelial cells [24]. KCa3.1 leads to potassium efflux thereby increasing the driving force for Ca2+ entry, and subsequently affecting Ca2+ dependent immune mechanisms (Fig. 2). It has been shown that microglia in vitro express KCa3.1 and that its inhibition reduces production and release of nitric oxide and IL-1β from appropriately stimulated microglia [25]. Other studies have shown that inhibition of KCa3.1 reduces microglial synthesis of enzymes involved in production of eicosanoids (COX-2) and nitric oxide (iNOS) [26]. Several inhibitors of KCa3.1 have been reported [27, 28]. However, early inhibitors lacked potency and selectivity and were hampered by safety concerns [27, 29, 30].
Fig. 2.
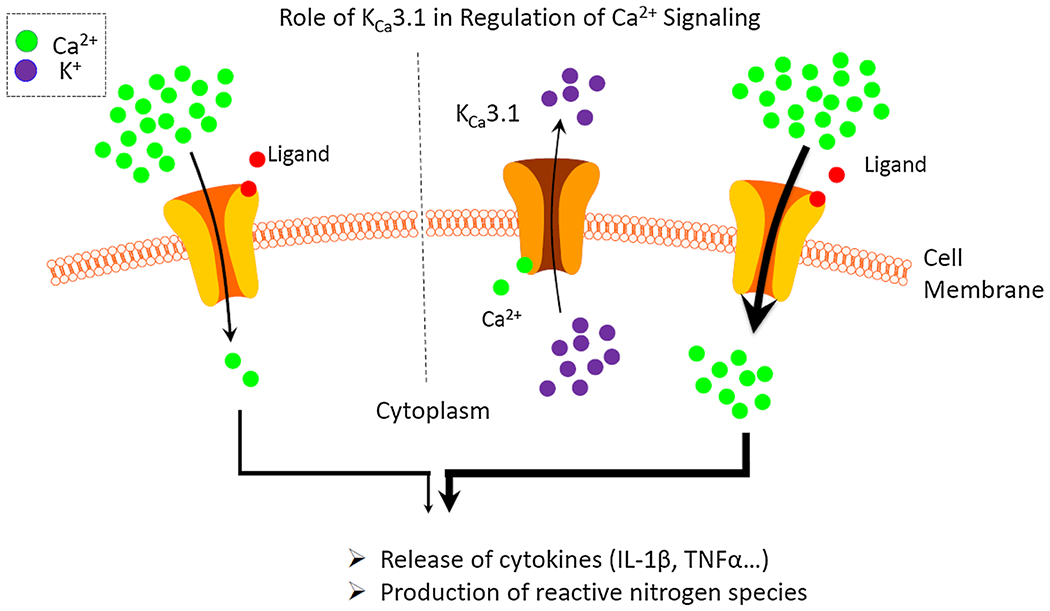
Role of KCa3.1 in regulation of calcium signaling. Upon activation of a calcium permeable cell membrane receptor increasing intracellular calcium concentrations lead to activation of the KCa3.1. The resulting potassium efflux leads to depolarization of the cell thereby increasing the driving force for calcium influx through calcium permeable plasma channels. The augmented calcium influx in turn increases calcium-regulated processes such as cytokine release or production of reactive oxygen species
KCa3.1 Inhibitor TRAM-34
TRAM-34 was described as a more selective inhibitor of KCa3.1 with better potency and good CNS penetration. TRAM-34 potently inhibits KCa3.1 channels with an IC50 of 20 nM in recombinant cell lines and was (incorrectly) reported to no effect cytochrome P450-dependent enzymes [31]. It has been used to investigate the physiology of KCa3.1 channels in immune cells and the involvement of KCa3.1 channels in several CNS disorders, including multiple sclerosis [32], optic nerve transection [33], spinal cord injury [34], ischemic stroke [24, 35], and glioblastoma multiforme [36]. Wulff and colleagues evaluated TRAM-34 in a rat model of ischemic stroke [35]. After administration of TRAM-34 at 40 mg/kg intraperitoneal (i.p.), plasma and brain concentrations reached ~1 μmol/L at 8 h, dropping to 0.4 μmol/L by 12 h. Free plasma concentrations were determined to be approximately 2%. From these data, it is estimated that the plasma and brain concentrations are 20 and 8 nM, respectively, at 12 h (before the second dose). Thus, when given i.p. the TRAM-34 concentrations are at or near the IC50 values for KCa3.1 inhibition. The high doses needed to achieve concentrations above IC50 values suggested that bioavailability for TRAM-34 is a significant issue. Unbound CNS levels of the TRAM-34 are not much higher than the IC50 for KCa3.1 inhibition in microglia in vitro and the t½ suggests that CNS KCa3.1 inhibition is only achieved for a few hours after administration. While it is always challenging to develop a CNS penetrant drug able to provide 24 h coverage even with a multiple dosing paradigm, there is significant room for improvement in drug levels achieved as well as t½. Nevertheless, administration of TRAM-34 at both 10 and 40 mg/kg i.p. significantly attenuated post stroke infarct volume and neuronal loss. TRAM-34 also improved the neurological deficit score and significantly reduced the extent of microglial ED1 staining. Especially promising was the finding that TRAM-34 improved the outcome in this model of stroke even when given 12 h after the ischemic insult. Current pharmacologic treatments for acute ischemic stroke need to be given within 3–4.5 h [2], a temporal challenge that severely limits the reach of currently available therapies.
While TRAM-34 shows selectivity for KCa3.1 over other calcium-activated potassium channels [31], it may inhibit additional targets confounding the interpretation of any results [37]. Schilling and Eder have demonstrated that TRAM-34 blocks non-selective cation current in primary microglia stimulated with lysophosphatidylcholine (LPC) with an IC50 that was similar to its IC50 for KCa3.1 channels [37]. Furthermore, another presumed KCa3.1 blocker, charybdotoxin, had no effect on LPC signals [37]. Hence, TRAM-34 may modulate immune cell function by a mechanism unrelated to KCa3.1 inhibition. Furthermore, it has recently been demonstrated that TRAM-34 inhibits some cytochrome P450 isoforms, namely human CYP2B6, CYP2C19 and CYP3A4 with IC50 values in the low micromolar range [38]. In addition, TRAM-34 shows metabolic instability and has a short half-life (~2 h in rats and primates) potentially complicating chronic dosing [39]. Thus, although TRAM-34 is a valuable experimental and potentially effective therapeutic agent, it has issues that may confound interpretation of mechanism in pre-clinical models and may limit its clinical utility.
KCa3.1 Inhibitor Senicapoc: Pharmacokinetics and Selectivity
The potent and selective KCa3.1 inhibitor Senicapoc (ICA-17043, IC50 of 11 nM) was initially developed for the treatment of sickle cell anemia [40–43]. The drug was well tolerated in Phase 1 clinical trials in both healthy volunteers and in patients with sickle cell disease [40, 41]. In a double blind placebo controlled Phase 2 study, Senicapoc (at 10 mg/day) reduced hemolysis and significantly increased hematocrit and hemoglobin levels in patients with sickle cell disease [42]. In a subsequent Phase 3 trial, Senicapoc was tested for its effects on vaso-occlusive pain crisis [41]. However, despite properly engaging erythrocyte KCa3.1, reducing hemolysis and increasing hemoglobin and hematocrit levels, Senicapoc had no effect on pain outcome measures and the trial was terminated [41]. While this was disapointing, it is important to point out, that the drug did what it was supposed to do on a molecular and cellular level. The clinical trial failed because the outcome measure chosen, which was distal to the proposed mode of action.
While the peripheral pharmacokinetics of Senicapoc have been described in detail [44], its ability to cross the blood–brain barrier has only recently been reported [45]. After 10 mg/kg oral dosing in rats, Senicapoc achieved free plasma concentrations of 17 and 65 nM and free brain concentrations of 37 and 136 nM at 1 and 4 h post-dose, respectively. Cerebrospinal fluid (CSF) concentrations were determined to be 25 and 121 nM at 1 and 4 h post-dosing which are in-line with the free brain concentrations. These data suggest that Senicapoc achieves CNS concentrations greater than its IC50 value for KCa3.1 channels (11 nM) and thus should be sufficient to inhibit it [44]. Furthermore, Senicapoc achieves free brain concentrations several fold higher than TRAM-34.
In the same study, Senicapoc’s selectivity was assessed in a screen of ~70 additional neuronal drug targets (50 neuronal receptors, 8 enzymes, 5 transporters and 7 ion channels) [45]. None of the targets tested was inhibited by Senicapoc at 1 μM, providing additional evidence that Senicapoc is selective for KCa3.1 channels. In vivo, Senicapoc was tested in the chronic constriction injury model of neuropathic pain [46]. Senicapoc dose dependently (10, 30 and 100 mg/kg p.o.) attenuated the mechanical hypersensitivity induced by the peripheral nerve injury, although only the highest dose was significant [45]. Furthermore, in contrast to reported locomotor effects in kcnn4−/− mice [47] that have no functional KCa3.1, the authors did not observe any significant impact of Senicapoc on locomotor activity [45]. While the study does not shed light on the cell types in the CNS that express KCa3.1, it clearly demonstrates that Senicapoc was efficacious in ameliorating pain behaviors in rats with peripheral nerve injury and these conclusions were supported by the free drug concentrations attained in plasma, brain and CSF.
Unlike TRAM-34, Senicapoc has no known off target effects at concentrations that block KCa3.1 [45]. It also does not suffer from metabolic instability or effects on cytochrome P450. Most importantly, Senicapoc has been tested in humans in clinical trials without any significant side effects. The finding that Senicapoc is also CNS penetrant opens up its use for CNS indications. With these significant advantages of Senicapoc over TRAM-34 the lingering question is whether Senicapoc also ameliorates the sequelae of ischemia/reperfusion in rodent models of stroke, similar to TRAM-34?
Senicapoc: Lessons from Rodent Trial for Neuropathic Pain
Senicapoc ameliorated pain behaviors in a model of neuropathic pain [45]. Since experimental surgery-related inflammation is resolved 7 days after the animals are tested, it supported the hypothesis that the efficacy was mediated by inhibition of KCa3.1 on microglia in the spinal cord or brain rather than peripheral immune cells. In addition to the prior studies in rats, we report here the ability of Senicapoc to penetrate the CNS in mice. The data were similar to those in rats (see Table 1) with Senicapoc reaching higher levels in brain than plasma and showing a similar t½ demonstrating that Senicapoc readily crossed the blood brain barrier and achieved concentrations well above the IC50. Whether or not Senicapoc is CNS penetrant in humans, specifically stroke patients, is not known. However, based on the rat and mouse pharmacokinetic data CNS penetrance in man seems likely.
Table 1.
Pharmacokinetics of the KCa3.1 blockers TRAM-34, NS6180 and Senicapoc
| TRAM-34 | NS1680 | Senicapoc | Senicapoc | |
|---|---|---|---|---|
| Molecular weight (g/mol) | 345 | 323 | 323 | |
| Species | Rat | Mouse | ||
| Dose (mg/kg) | 10 | 10 | 10 | 10 |
| T½ (hours) | ~2 | 3.8 | 1 | 3.1 |
| Tmax (hours) | 0.5–1 | 4 | 0.33 | |
| In vitro IC50 (nM)a | 20 [31] | 9.4 hu | 12 hu | |
| Cmax total (nM) | ||||
| Plasma | ~2500 | 186 | 2400 [45] | 709 |
| Brain | ~2500 | 17,000 [45] | ||
| % unbound | ||||
| Plasma | 2 | 2.7 [45] | 1.5 | |
| Cmax unbound (nM) | ||||
| Brain | 0.8 [45] | 0.8 | ||
| Plasma | 50 | 65 | ||
| Cmax unbound (nM) | ||||
| Brain | 50 | 136 | ||
| CSF | 121 | |||
| Reference | [35] | [48] | [44] | Unpublished data |
IC50 reported are for human KCa3.1 expressed in recombinant cells. All data are for compounds dosed per os (p.o.)
To address in vivo side effects of Senicapoc, the most relevant being sedation in pain models, the authors tested effect of the drug on rat locomotor activity [45]. Senicapoc did not alter activity at doses required for efficacy in the chronic constriction injury model of neuropathic pain. While the data suggest that KCa3.1 inhibition has few adverse effects, it should be kept in mind that the locomotor activity test should by no means be the extent of side effect testing pre-clinically. In the case of Senicapoc, however, the significance of these pre-clinical findings is enhanced by the human clinical trials that demonstrated that Senicapoc is safe and has a low incidence of side effects.
Based on the animal studies, the major drawback to both TRAM-34 and Senicapoc is the short half-life (see Table 1). In contrast to the preclinical studies in rodents, clinical trials showed an unexpected t½ of 23 days. This raises the question whether Senicapoc covalently binds to plasma proteins whose t½ is approximately 21 days which is significantly longer than that of the unbound drug. It is important to note that the potential covalent protein binding, should not impact the ability of Senicapoc to penetrate the CNS, although it would make dose titration more complex.
To date, the only CNS disease model in which Senicapoc has been evaluated is the chronic constriction injury model of neuropathic pain [45]. While many devastating neurological and perhaps psychiatric diseases could be potentially treated by Senicapoc, the studies with TRAM-34 lay a foundation for efficacy of this mechanism in stroke. Finding a treatment for stroke that can be given beyond the narrow therapeutic window of current treatments would be a major advance. The data on efficacy of the KCa3.1 inhibitor, TRAM-34, outside of this narrow therapeutic window suggests that inhibition of KCa3.1 could become a promising treatment strategy in acute stroke. Senicapoc, having been in clinical trials and found to be safe, is uniquely positioned to be repurposed for the treatment of stroke and potentially, to be a groundbreaking treatment which is so desperately needed by patients and care providers.
Acknowledgements
The study was supported by Lundbeck Research USA.
References
- 1.Mozaffarian D, Benjamin EJ, Go AS, Arnett DK, Blaha MJ, Cushman M, de Ferranti S, Despres JP, Fullerton HJ, Howard VJ, Huffman MD, Judd SE, Kissela BM, Lackland DT, Lichtman JH, Lisabeth LD, Liu S, Mackey RH, Matchar DB, McGuire DK, Mohler ER 3rd, Moy CS, Muntner P, Mussolino ME, Nasir K, Neumar RW, Nichol G, Palaniappan L, Pandey DK, Reeves MJ, Rodriguez CJ, Sorlie PD, Stein J, Towfighi A, Turan TN, Virani SS, Willey JZ, Woo D, Yeh RW, Turner MB, American Heart Association Statistics C, Stroke Statistics S (2015) Heart disease and stroke statistics—2015 update: a report from the American Heart Association. Circulation 131:e29–322 [DOI] [PubMed] [Google Scholar]
- 2.Prabhakaran S, Ruff I, Bernstein RA (2015) Acute stroke intervention: a systematic review. JAMA 313:1451–1462 [DOI] [PubMed] [Google Scholar]
- 3.Iadecola C, Anrather J (2011) The immunology of stroke: from mechanisms to translation. Nat Med 17:796–808 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Macrez R, Ali C, Toutirais O, Le Mauff B, Defer G, Dirnagl U, Vivien D (2011) Stroke and the immune system: from pathophysiology to new therapeutic strategies. Lancet Neurol 10:471–480 [DOI] [PubMed] [Google Scholar]
- 5.McDonough A, Weinstein JR (2016) Neuroimmune response in ischemic preconditioning. Neurother 13:748–761 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Hayakawa K, Nakano T, Irie K, Higuchi S, Fujioka M, Orito K, Iwasaki K, Jin G, Lo EH, Mishima K, Fujiwara M (2010) Inhibition of reactive astrocytes with fluorocitrate retards neurovascular remodeling and recovery after focal cerebral ischemia in mice. J Cereb Blood Flow Metab 30:871–882 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Lo EH (2008) A new penumbra: transitioning from injury into repair after stroke. Nat Med 14:497–500 [DOI] [PubMed] [Google Scholar]
- 8.Maki T, Hayakawa K, Pham LD, Xing C, Lo EH, Arai K (2013) Biphasic mechanisms of neurovascular unit injury and protection in CNS diseases. CNS Neurol Disord Drug Targets 12:302–315 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Dirnagl U (2012) Pathobiology of injury after stroke: the neurovascular unit and beyond. Ann N Y Acad Sci 1268:21–25 [DOI] [PubMed] [Google Scholar]
- 10.Benarroch EE (2013) Microglia: multiple roles in surveillance, circuit shaping, and response to injury. Neurology 81:1079–1088 [DOI] [PubMed] [Google Scholar]
- 11.Michell-Robinson MA, Touil H, Healy LM, Owen DR, Durafourt BA, Bar-Or A, Antel JP, Moore CS (2015) Roles of microglia in brain development, tissue maintenance and repair. Brain 138:1138–1159 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Ginhoux F, Greter M, Leboeuf M, Nandi S, See P, Gokhan S, Mehler MF, Conway SJ, Ng LG, Stanley ER, Samokhvalov IM, Merad M (2010) Fate mapping analysis reveals that adult microglia derive from primitive macrophages. Science 330:841–845 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zuchero JB, Barres BA (2015) Glia in mammalian development and disease. Development 142:3805–3809 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Jha MK, Lee WH, Suk K (2015) Functional polarization of neuroglia: implications in neuroinflammation and neurological disorders. Biochem Pharmacol 103:1–16 [DOI] [PubMed] [Google Scholar]
- 15.Weinstein JR, Koerner IP, Möller T (2010) Microglia in ischemic brain injury. Future Neurol 5:227–246 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.van Rossum D, Hanisch UK (2004) Microglia. Metab Brain Dis 19:393–411 [DOI] [PubMed] [Google Scholar]
- 17.Dave KR, Saul I, Prado R, Busto R, Perez-Pinzon MA (2006) Remote organ ischemic preconditioning protect brain from ischemic damage following asphyxial cardiac arrest. Neurosci Lett 404:170–175 [DOI] [PubMed] [Google Scholar]
- 18.Lalancette-Hebert M, Gowing G, Simard A, Weng YC, Kriz J (2007) Selective ablation of proliferating microglial cells exacerbates ischemic injury in the brain. J Neurosci 27:2596–2605 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Nedergaard M, Dirnagl U (2005) Role of glial cells in cerebral ischemia. Glia 50:281–286 [DOI] [PubMed] [Google Scholar]
- 20.Cunningham LA, Wetzel M, Rosenberg GA (2005) Multiple roles for MMPs and TIMPs in cerebral ischemia. Glia 50:329–339 [DOI] [PubMed] [Google Scholar]
- 21.Umekawa T, Osman AM, Han W, Ikeda T, Blomgren K (2015) Resident microglia, rather than blood-derived macrophages, contribute to the earlier and more pronounced inflammatory reaction in the immature compared with the adult hippocampus after hypoxia-ischemia. Glia 63:2220–2230 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Denes A, Vidyasagar R, Feng J, Narvainen J, McColl BW, Kauppinen RA, Allan SM (2007) Proliferating resident microglia after focal cerebral ischaemia in mice. J Cereb Blood Flow Metab 27:1941–1953 [DOI] [PubMed] [Google Scholar]
- 23.Szalay G, Martinecz B, Lenart N, Kornyei Z, Orsolits B, Judak L, Csaszar E, Fekete R, West BL, Katona G, Rozsa B, Denes A (2016) Microglia protect against brain injury and their selective elimination dysregulates neuronal network activity after stroke. Nat Commun 7:11499. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Chen YJ, Wallace BK, Yuen N, Jenkins DP, Wulff H, O’Donnell ME (2015) Blood-brain barrier KCa3.1 channels: evidence for a role in brain Na uptake and edema in ischemic stroke. Stroke 46:237–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Dale E, Staal RG, Eder C, Möller T (2016) KCa 3.1-a microglial target ready for drug repurposing? Glia 64:1733–1741 [DOI] [PubMed] [Google Scholar]
- 26.Nguyen HM, Grossinger EM, Horiuchi M, Davis KW, Jin LW, Maezawa I, Wulff H (2016) Differential Kv1.3, KCa3.1, and Kir2.1 expression in “classically” and “alternatively” activated microglia. Glia 65:106–121 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Wulff H, Castle NA (2010) Therapeutic potential of KCa3.1 blockers: recent advances and promising trends. Expert Rev Clin Pharmacol 3:385–396 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Wulff H, Kolski-Andreaco A, Sankaranarayanan A, Sabatier JM, Shakkottai V (2007) Modulators of small- and intermediate-conductance calcium-activated potassium channels and their therapeutic indications. Curr Med Chem 14:1437–1457 [DOI] [PubMed] [Google Scholar]
- 29.Suzuki S, Kurata N, Nishimura Y, Yasuhara H, Satoh T (2000) Effects of imidazole antimycotics on the liver microsomal cytochrome P450 isoforms in rats: comparison of in vitro and ex vivo studies. Eur J Drug Metab Pharmacokinet 25:121–126 [DOI] [PubMed] [Google Scholar]
- 30.Zhang W, Ramamoorthy Y, Kilicarslan T, Nolte H, Tyndale RF, Sellers EM (2002) Inhibition of cytochromes P450 by antifungal imidazole derivatives. Drug Metab Dispos 30:314–318 [DOI] [PubMed] [Google Scholar]
- 31.Wulff H, Miller MJ, Hansel W, Grissmer S, Cahalan MD, Chandy KG (2000) Design of a potent and selective inhibitor of the intermediate-conductance Ca2+-activated K+ channel, IKCa1: a potential immunosuppressant. Proc Natl Acad Sci USA 97:8151–8156 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Reich EP, Cui L, Yang L, Pugliese-Sivo C, Golovko A, Petro M, Vassileva G, Chu I, Nomeir AA, Zhang LK, Liang X, Kozlowski JA, Narula SK, Zavodny PJ, Chou CC (2005) Blocking ion channel KCNN4 alleviates the symptoms of experimental autoimmune encephalomyelitis in mice. Eur J Immunol 35:1027–1036 [DOI] [PubMed] [Google Scholar]
- 33.Kaushal V, Koeberle PD, Wang Y, Schlichter LC (2007) The Ca2+-activated K+ channel KCNN4/KCa3.1 contributes to microglia activation and nitric oxide-dependent neurodegeneration. J Neurosci 27:234–244 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Bouhy D, Ghasemlou N, Lively S, Redensek A, Rathore KI, Schlichter LC, David S (2011) Inhibition of the Ca(2) (+)-dependent K(+) channel, KCNN4/KCa3.1, improves tissue protection and locomotor recovery after spinal cord injury. J Neurosci 31:16298–16308 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Chen YJ, Raman G, Bodendiek S, O’Donnell ME, Wulff H (2011) The KCa3.1 blocker TRAM-34 reduces infarction and neurological deficit in a rat model of ischemia/reperfusion stroke. J Cereb Blood Flow Metab 31:2363–2374 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.D’Alessandro G, Catalano M, Sciaccaluga M, Chece G, Cipriani R, Rosito M, Grimaldi A, Lauro C, Cantore G, Santoro A, Fioretti B, Franciolini F, Wulff H, Limatola C (2013) KCa3.1 channels are involved in the infiltrative behavior of glioblastoma in vivo. Cell Death Dis 4:e773. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Schilling T, Eder C (2007) TRAM-34 inhibits nonselective cation channels. Pflugers Arch 454:559–563 [DOI] [PubMed] [Google Scholar]
- 38.Agarwal JJ, Zhu Y, Zhang QY, Mongin AA, Hough LB (2013) TRAM-34, a putatively selective blocker of intermediate-conductance, calcium-activated potassium channels, inhibits cytochrome P450 activity. PLoS ONE 8:e63028. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Maezawa I, Jenkins DP, Jin BE, Wulff H (2012) Microglial KCa3.1 channels as a potential therapeutic target for Alzheimer’s disease. Int J Alzheimers Dis 2012:868972. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Ataga KI, Orringer EP, Styles L, Vichinsky EP, Swerdlow P, Davis GA, Desimone PA, Stocker JW (2006) Dose-escalation study of ICA-17043 in patients with sickle cell disease. Pharmacotherapy 26:1557–1564 [DOI] [PubMed] [Google Scholar]
- 41.Ataga KI, Reid M, Ballas SK, Yasin Z, Bigelow C, James LS, Smith WR, Galacteros F, Kutlar A, Hull JH, Stocker JW, Investigators ICAS (2011) Improvements in haemolysis and indicators of erythrocyte survival do not correlate with acute vaso-occlusive crises in patients with sickle cell disease: a phase III randomized, placebo-controlled, double-blind study of the Gardos channel blocker senicapoc (ICA-17043). Br J Haematol 153:92–104 [DOI] [PubMed] [Google Scholar]
- 42.Ataga KI, Smith WR, De Castro LM, Swerdlow P, Saunthararajah Y, Castro O, Vichinsky E, Kutlar A, Orringer EP, Rigdon GC, Stocker JW, Investigators ICA (2008) Efficacy and safety of the Gardos channel blocker, senicapoc (ICA-17043), in patients with sickle cell anemia. Blood 111:3991–3997 [DOI] [PubMed] [Google Scholar]
- 43.Ataga KI, Stocker J (2009) Senicapoc (ICA-17043): a potential therapy for the prevention and treatment of hemolysis-associated complications in sickle cell anemia. Expert Opin Investig Drugs 18:231–239 [DOI] [PubMed] [Google Scholar]
- 44.McNaughton-Smith GA, Burns JF, Stocker JW, Rigdon GC, Creech C, Arrington S, Shelton T, de Franceschi L (2008) Novel inhibitors of the Gardos channel for the treatment of sickle cell disease. J Med Chem 51:976–982 [DOI] [PubMed] [Google Scholar]
- 45.Staal RG, Khayrullina T, Zhang H, Davis S, Fallon SM, Cajina M, Nattini ME, Hu A, Zhou H, Poda SB, Zorn S, Chandrasena G, Dale E, Cambpell B, Biilmann Ronn LC, Munro G, Möller T (2017) Inhibition of the potassium channel KCa3.1 by senicapoc reverses tactile allodynia in rats with peripheral nerve injury. Eur J Pharmacol 795:1–7 [DOI] [PubMed] [Google Scholar]
- 46.Bennett GJ, Xie YK (1988) A peripheral mononeuropathy in rat that produces disorders of pain sensation like those seen in man. Pain 33:87–107 [DOI] [PubMed] [Google Scholar]
- 47.Lambertsen KL, Gramsbergen JB, Sivasaravanaparan M, Ditzel N, Sevelsted-Moller LM, Olivan-Viguera A, Rabjerg M, Wulff G, Kohler R (2012) Genetic KCa3.1-deficiency produces locomotor hyperactivity and alterations in cerebral monoamine levels. PLoS ONE 7:e47744. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Strobaek D, Brown DT, Jenkins DP, Chen YJ, Coleman N, Ando Y, Chiu P, Jorgensen S, Demnitz J, Wulff H, Christophersen P (2013) NS6180, a new K(Ca) 3.1 channel inhibitor prevents T-cell activation and inflammation in a rat model of inflammatory bowel disease. Br J Pharmacol 168:432–444 [DOI] [PMC free article] [PubMed] [Google Scholar]