

Abstract
Chimeric antigen receptor (CAR) T cell therapy has demonstrated remarkable outcomes in individuals with hematological malignancies, but its success has been hindered by barriers intrinsic to the tumor microenvironment (TME), particularly for solid tumors, where it has yet to make its mark. In this article, we provide an updated review and future perspectives on features of the TME that represent barriers to CART cell therapy efficacy, including competition for metabolic fuels, physical barriers to infiltration, and immunosuppressive factors. We then discuss novel and promising strategies to overcome these obstacles that are in preclinical development or under clinical investigation.
Keywords: tumor microenvironment, chimeric antigen receptor (CAR) T cell therapy, metabolic fuels, physical barriers, immunosuppression
Graphical abstract
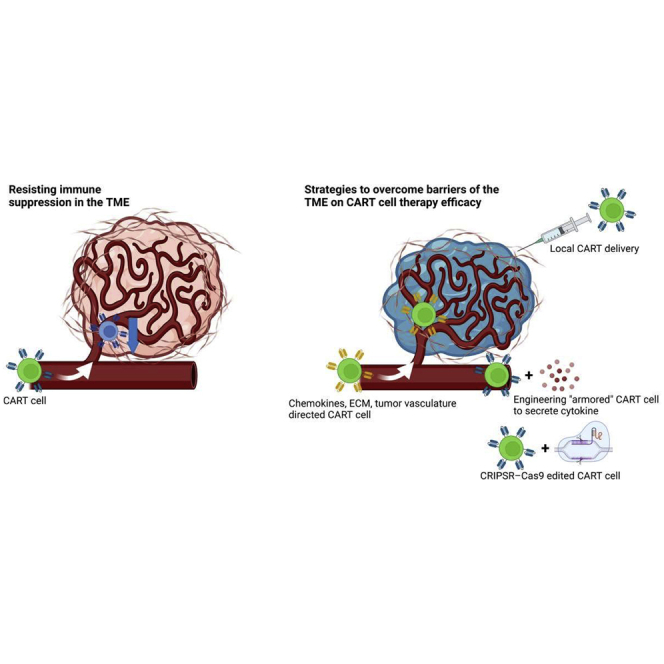
The success of chimeric antigen receptor (CAR) T cell therapy has been hindered by barriers intrinsic to the tumor microenvironment, including metabolic fuel competition, physical barriers to effective CAR T cell infiltration, and immunosuppressive cytokines and cell types. We review novel strategies and sophisticated CAR designs that may overcome these barriers.
Introduction
Chimeric antigen receptors (CARs) are recombinant receptors that redirect immune cells to recognize and target tumor cell surface molecules.1, 2, 3 Pivotal clinical trials of CAR T cells targeting CD19 (CART19) or B cell maturation antigen (CART-BCMA) have demonstrated unprecedented outcomes in individuals with relapsed/refractory B cell malignancies or multiple myeloma, respectively, which led to several recent US Food and Drug Administration (FDA) approvals.4, 5, 6, 7, 8, 9 However, the success of CAR T cell therapies has been largely limited to a subset of individuals with hematological malignancies who achieve durable remissions, and CAR T cells have had very limited activity in the solid tumor realm to date.10, 11, 12
The tumor microenvironment (TME) plays a critical role in the response to cancer treatment. The TME has the ability to dampen the efficacy of the infused CAR T cells, as evidenced by cellular and molecular profiles that conclusively indicate T cell dysfunction.13 Cancer cells take advantage of normal cellular homeostatic mechanisms to not only promote cancer survival and growth but also evade immune surveillance.14 Thus, bidirectional interaction between cancer cells and their microenvironment is crucial for cancer initiation and progression and has a great effect on treatment outcomes and disease prognosis.15,16 They have the ability to recruit stromal cells, immune cells, and endothelial cells by secreting cytokines and chemokines, shaping and remodeling the structure of a highly interconnected TME.16,17 A wide range of immune cells can infiltrate the tumor, and their phenotypes or composition within the TME have been strongly associated with clinical outcomes. Robust CD8+ T cell infiltration has been established as a good prognostic factor for individuals with solid tumors,18, 19, 20, 21 whereas the presence of M2-polarized macrophages22,23 or α-smooth muscle actin (SMA)-positive cancer-associated fibroblasts (CAFs) has been associated with less favorable outcomes.24,25 Therefore, the TME offers a wide range of potential therapeutic targets that can be leveraged to enhance CAR T cell efficacy.12,26,27
Here we present the overall effect of the TME on CAR T cell therapy efficacy with emphasis on competition for metabolic fuels, CAR T cell exhaustion mechanisms, immunosuppressive factors, and physical barriers (Figure 1). We then summarize promising strategies that allow CAR T cell therapy to overcome the challenges presented by the TME before providing our outlook on directions and potential applications (Table 1).
Figure 1.

TME factors limiting CAR T cell therapy efficacy
Barriers to chimeric antigen receptor (CAR) T cell infiltration include dysregulated tumor vasculature and dense fibrogenic extracellular matrix (ECM). Immunosuppressive cytokines (interleukin-10 [IL-10], IL-4, and transforming growth factor β [TGF-β]) and cells (myeloid-derived suppressor cells [MDSCs], tumor-associated macrophages [TAMs], and regulatory T [Treg] cells) hinder effective antitumor function of CAR T cells. Competition for metabolic fuels (glucose and O2) can suppress CAR T cell function. TME, tumor microenvironment.
Table 1.
Summary of select CAR T cell clinical trials targeting the TME
| Strategy/target | Treatment | Cancer type | ClinicalTrials.gov identifier |
|---|---|---|---|
| Enhancing CAR T cell infiltration | |||
| Chemokines | CXCR5-expressing EGFR-CAR T cells | non-small cell lung cancer | NCT04153799 |
| ECM | FAP-CAR T cells | mesothelioma | NCT01722149 |
| tumor vasculature | VEGF-R2-CAR T cells | metastatic melanoma renal cancer |
NCT01218867 |
| local CAR T cell delivery | intracranial Her2-CAR T cells | GBM glioma |
NCT02442297 |
| intratumoral Her2-CAR T cells | GBM glioma |
NCT03389230 | |
| intratumoral B7-H3-CAR T cells | GBM | NCT04077866 | |
| intraventricular NKG2D-CAR T cells | GBM | NCT04717999 | |
| intracranial IL13Ra2-CAR T cells | GBM glioma |
NCT02208362 | |
| intratumoral MG7-CAR T cells | liver metastases | NCT02862704 | |
| intrahepatic artery CEA- CAR T cells | liver metastases | NCT01373047 | |
| intratumoral GPC3-CAR T cells | hepatocellular carcinoma cholangiocarcinoma | NCT04951141 | |
| intratumoral GPC3-CAR T cells | hepatocellular carcinoma | NCT03130712 | |
| intrapleural mesothelin-CAR T cells | mesothelioma lung cancer breast cancer |
NCT02414269 | |
| intratumoral cMet-CAR T cells | breast cancer | NCT01837602 | |
| intratumoral T1E-CAR T cells | head and neck cancer | NCT01818323 | |
| Resisting immunosuppression | |||
| suppressive cytokines or inhibitory signals | TGF-β knockout EGFR-CAR T cells | advanced biliary tract cancer | NCT04976218 |
| TGF-β DNR PSMA-CAR T cells | prostate cancer | NCT04227275 | |
| TGF-β DNR PSMA-CAR T cells | prostate cancer | NCT03089203 | |
| PD1 DNR MSLN-CAR T cells | malignant mesothelioma | NCT04577326 | |
| pro-inflammatory cytokines | IL-12-secreting MUC16ecto- CAR T cells | ovarian cancer | NCT02498912 |
| IL-12-secreting EGFR-CAR T cells | colorectal cancer | NCT03542799 | |
| IL-7/CCL19-secreting CD19-CAR T cells | lymphoma | NCT04833504 | |
Effect of the TME on CAR T cell therapy efficacy
The TME is one of the greatest challenges affecting CAR T cell therapy efficacy because it hampers CART cell trafficking to the desired site of action, affects CART cell metabolic function, and creates an immunosuppressive environment leading to T cell exhaustion.28 In this section, we will summarize how various components of the TME are able to inhibit CAR T cells and review strategies utilized to overcome TME-induced CAR T cell inhibition.
Physical barriers
In addition to its role in liquid tumors, the TME in solid tumors also presents a physical barrier that excludes tumor-infiltrating effector T cells and CAR T cells through various mechanisms.
The first major physical constraint is formation of a dense fibrogenic TME by stromal cells such as CAFs, which, under transforming growth factor β (TGF-β)-mediated activation, stimulate production of extracellular matrix (ECM) proteins that limit T cell motility and trafficking.29 TGF-β is a chemokine that can also limit T cell infiltration in solid tumors by directly acting on T cells through inhibition of expression of chemokine receptors such as CXCR3.30 It has been demonstrated that CAFs inhibit CAR T cell functions in preclinical models through complex multi-directional mechanisms that involve TGF-β as well contact cell-to-cell-mediated mechanisms.31
Another major physical constraint that leads to T cell exclusion from the TME is the aberrant vasculature of solid tumors that contributes to tissue hypoxia and limits T cell extravasation into the TME. Hypoxia promotes recruitment of immunosuppressive cells through secretion of various chemokines but also upregulates expression of CTLA4 or lymphocyte activation gene 3 protein (LAG3) on regulatory T (Treg) cells and programmed cell death 1 (PD-1) ligand 1 (PD-L1) on myeloid-derived suppressor cells (MDSCs), tumor-associated macrophages (TAMs), and tumor cells.32 The dysregulated vasculature also promotes downregulation of adhesion molecules necessary for T cell infiltration, such as vascular cell adhesion protein 1 (VCAM1) and intercellular adhesion molecule 1 (ICAM1), amplifying T cell exclusion.4,32,33
Immunosuppressive microenvironment
Other factors limiting CAR T cell efficacy include the presence of an immunosuppressive TME with a cytokine and chemokine profile that preferentially recruits immunosuppressive Treg cells, MDSCs, and TAMs. Treg cells specifically suppress cytotoxic T cell function through secretion of immunosuppressive cytokines, competitive consumption of interleukin-2 (IL-2), cytotoxic T lymphocyte antigen 4 (CTLA4)-mediated suppression of antigen-presenting cells (APCs), and prevention of T cell activation.34 MDSCs have been shown to have a detrimental effect on CAR T cells because of their potent immunosuppressive capabilities directly targeting effector T cells. MDSC-mediated CAR T cell suppression is profound, and low levels of MDSCs in individuals receiving CART19 have been associated with positive lymphoma and leukemia therapy response.35 TAMs are the most abundant immune-infiltrating cells in the TME and suppress T cell-mediated anti-tumor immunity through secretion of cytokines and amino-acid-depleting enzymes such as arginase 1 or indoleamine 2,3- dioxygenase (IDO) as well as increased recruitment of Treg cells.36, 37, 38
Some metabolites have also been shown to suppress effector T cell functions through various mechanisms. The enzyme IDO catalyzes conversion of tryptophan into kynurenine, suppressing effector T and natural killer (NK) cells while recruiting and activating immunosuppressive MDSCs.39 Elevated lactate levels produced by hypermetabolic tumor cells have been associated with nuclear factor of activated T cells (NFAT)-mediated dampening of T cell signaling,40 Treg cell expansion,41 and polarization of macrophages towards an immunosuppressive M2 phenotype.42 The interactions between tumor cells, the TME, and CAR T cells are complex. It has been suggested that tumor cells induce CAR T cell dysfunction through secretion of immunosuppressive extracellular vesicles.43
This vast array of TME metabolic and cellular features represents obstacles and targets to be exploited in the ongoing and future optimization of immunotherapeutic agents.
Competition for metabolic fuels
An effective CAR T cell antitumor response requires successful completion of a number of metabolically demanding tasks, including CAR T cell proliferation, cytokine secretion, and tumor cell killing. CAR T cells therefore face the challenge of competing with cancer cells for metabolic fuels in a nutrient-poor TME that supports oncogenic growth but limits effector T cell function.
Energy production in all cells is at the center of survival and metabolism, so the majority of cells rely primarily on the most energy-efficient mitochondrial oxidative phosphorylation pathway to generate adenosine 5′-trisphosphate (ATP). Cancer cells and rapidly proliferating T cells instead rely on aerobic glycolysis for less efficient ATP generation. The so-called “Warburg effect” is believed to facilitate less ATP production but greater nutrient uptake into the cancer cell to enhance proliferation and survival.44 Aerobic glycolysis works to alternatively ferment glucose into lactate rather than oxidize glucose through mitochondrial oxidative phosphorylation. T cell activation initiates glycolysis rapidly through T cell receptor (TCR)-induced pyruvate dehydrogenase kinase 1 (PDHK1), which metabolizes glucose into lactate and inhibits mitochondrial pyruvate import. This glycolytic process is necessary for effector T cell function and production of proinflammatory cytokines such as interferon gamma (IFNγ)45,46 Thus, proliferative cancer and T cells, including CAR T cells, rely on glycolysis as their primary means of energy metabolism. Unfortunately, the excessive glucose consumption by hyperactive cancer cells in the TME limits the glycolytic ability of CAR T cells, dampening TCR/CAR signaling and effector responses. These pressures ultimately decrease the efficacy of CAR T cell therapy by overriding the antitumor response as the tumor outcompetes the CAR T cells for nutrients.47, 48, 49
Exhausted CAR T cells
Besides the competition for fuels and limited glycolytic ability restricting CAR T cell function in the TME, other features of the TME can induce an exhausted T cell metabolic phenotype. This phenotype is characterized by loss of proliferative capacity and function while displaying upregulation of inhibitory receptors and dysregulated glycolysis enhanced beyond levels needed for effective T cell proliferation. Single-cell transcriptome analyses of intratumoral T cells isolated from mouse and human samples have revealed a substantial upregulation of glycolytic genes and decrease in transcription T cell factor 7 (TCF7) T cell self-renewal gene signatures in terminally exhausted T cells compared with progenitor or effector T cell subsets.50 The TME induces rapid exhaustion in these cells via repetitive TCR stimulation, leading to bioenergetic stress on glycolytic and oxidative metabolism. This stress results in transcriptional and enzymatic signature changes that characterize the dysfunctionally exhausted state of CAR T cells and associated compromised antitumor immunity.51,52
Strategies to overcome barriers of the TME on CAR T cell therapy efficacy
Enhancing CAR T cell infiltration
The TME poses several barriers to effective CAR T cell infiltration, including mismatched or downregulated chemokines and chemokine receptors that allow the tumor to circumvent immunosurveillance; physical barriers, such as the ECM, produced by tumor stromal cells; and abnormal tumor vasculature, which further limits the ability of CAR T cells to reach the target site. These unique challenges presented by the TME have been approached using a variety of strategies to engineer CAR T cells that are more effective at infiltrating the tumor site.
Localized delivery of CAR T cells
Most CAR T cell studies have employed intravenous administration. However, more localized routes of delivery may benefit the treatment of solid tumors by bypassing various obstacles to CAR T cell trafficking and limiting systemic toxicity. In a transgenic mouse model, repeated intratumoral administration of Her2-targeted CAR T cells eradicated spontaneous mammary tumors and showed trafficking to distal tumor sites and secondary lymphoid organs.53 Intrapleurally infused CAR T cells were superior to intravenously administered CAR T cells in an orthotopic model of pleural malignancy, requiring 30-fold fewer CAR T cells to induce a durable remission.54 In glioblastoma (GBM), CAR T cells have been delivered intravenously with the rationale that adoptively transferred T cells have been found in the brain or cerebrospinal fluid.55 In vivo, however, intraventricular administration of IL-13Ra2-targeted CAR T cells resulted in better antitumor efficacy than intravenously delivered CAR T cells.56 Several clinical studies have been performed with local delivery of CAR T cells for GBM. A phase I trial treated three individuals with intracranial CAR T cells to evaluate the safety profile of this approach.57 Another study in one individual with multiple lesions found that CAR T cells injected directly into one lesion eradicated disease only in that lesion, with recurrence and progression observed in uninjected sites. The individual responded well after CAR T cells were infused intraventricularly into a second tumor site, demonstrating a sustained antitumor response over several months of active therapy.58 CAR T cells have also been delivered regionally to hepatic metastases. Anti-Carcinoembryonic antigen (CEA) CAR T cells have been associated previously with severe colitis59 but were well tolerated with local delivery. CAR T cells delivered through the hepatic artery infiltrated into metastases in five of six individuals in this study, with some evidence of antitumor activity.60 A phase 0 trial of cMet-targeted CAR T cells injected intratumorally in individuals with metastatic breast cancer was well-tolerated and triggered immune responses at the tumor sites.61 Additional clinical studies of localized CAR T cell delivery methods are ongoing and will clarify the effect of route of administration on CAR T cell efficacy in solid tumors.
Engineering CAR T cells to express chemokine receptors
Chemokines not only play a role in regulating tumor growth and progression but also shape the immune response within the TME.62 Mismatches between chemokine ligands and receptors or low expression of chemokine receptors allow the tumor to evade the immune response.63 Several groups have engineered CAR T cells to overexpress chemokine receptors with the goal of improving CAR T cell trafficking to the TME. Epidermal growth factor receptor (EGFR)-targeted CAR T cells that overexpressed CCR6 more effectively trafficked to adenocarcinoma cells in the lungs because of their abundant expression of CCL20, a ligand for CCR6.64 CXCR2-expressing CAR T cells have shown improved accumulation and antitumor effects in hepatocarcinoma sites65 and in mouse models of glioma, ovarian cancer, and pancreatic cancer.66 Anti-mesothelin CAR T cells that overexpressed CCR2b better infiltrated and eradicated mesothelin-positive tumors,67,68 and in an earlier study, GD2-targeted CAR T cells overexpressing CCR2b showed enhanced migration to CCL2-expressing neuroblastoma tumors.69 Determining relevant chemokine-chemokine receptor axes between CAR T cells and tumors can thus provide a basis to develop chemokine-overexpressing CAR T cells to better enhance solid tumor infiltration. In parallel to development of technologies aimed to enhance CAR T cell trafficking, strategies to enable real-time imaging of CAR T cells in vivo are being developed in preclinical and early-phase clinical trials.70,71 Such strategies would enable non-invasive assessment of CAR T cell trafficking into tumor cells and allow expedited testing of novel interventions to improve CAR T cell functions.
Engineering CAR T cells to normalize tumor vasculature
Abnormal tumor vasculature stems from an imbalance of pro- and anti-angiogenic factors, leading to structurally irregular blood vessels, impaired blood flow, hypoxic and necrotic regions, and higher interstitial fluid pressure.72,73 These factors make effective CAR T cell infiltration into the tumor difficult. Vascular endothelial growth factor receptor (VEGF-R) is upregulated in the TME to promote angiogenesis. Several studies have generated CAR T cells targeting VEGF-R to normalize tumor vasculature. In an early study, anti-VEGF-R2 CAR T cells were found to have increased tumor infiltration, which correlated with antitumor efficacy in five different syngeneic tumor models.74 A follow-up study showed that anti-VEFG-R2 CAR T cells combined with T cells targeting tumor antigens synergistically reduced tumor burden and allowed increased infiltration and expansion of adoptively transferred T cells in a syngeneic melanoma model, indicating that targeting TME- and tumor-associated surface markers is a viable strategy to boost T cell immunotherapy.75 Another group found that VEGF-R1-targeted CAR T cells demonstrated enhanced antitumor and antimetastatic activity when combined with IL-15-expressing T cells.76 Another study observed that GD2-targeted CAR T cells were only effective against a neuroblastoma xenograft model when combined with the anti-VEGF-A monoclonal antibody bevacizumab, which allowed CAR T cell infiltration into the tumor.77 A clinical trial of VEGF-R2-targeted CAR T cells in individuals with metastatic cancer was initiated; however, the study was terminated because no objective responses were observed (ClinicalTrials.gov: NCT01218867).
Engineering CAR T cells to reduce ECM density
Dense ECM with increased stiffness and cross-linking is often seen in tumors compared with normal tissue.78,79 CAFs play a pivotal role in remodeling the ECM in the TME. CAFs express fibroblast activation protein (FAP), which has been studied as a potential CAR T cell target. Although one study showed that FAP-targeted CAR T cells exerted limited antitumor activity and resulted in severe toxicities because of on-target, off-tumor effects,80 another study showed that FAP-targeted CAR T cells had minimal toxicity while promoting the endogenous T cell response and increasing numbers of tumor-infiltrating lymphocytes.81 Both studies used syngeneic models, but the latter study used a different single-chain fragment variable (scFv) that rendered the CAR T cells sensitive only to high levels of FAP. In a xenograft model, researchers found that combining FAP-targeted CAR T cells with EphA2-targeted CAR T cells synergistically reduced tumor growth.82 Alternatively, one group engineered CAR T cells to degrade ECM proteins more effectively. After finding that in-vitro-cultured T cells lack heparinase, a key enzyme in ECM degradation, they engineered CAR T cells to express heparinase and observed improved tumor infiltration and antitumor efficacy in xenograft models of neuroblastoma and melanoma.83 Recently, it was reported that SLAMF7 is expressed on CAFs and that targeting SLAMF7 in combination with targeting tumor cells is a potential strategy to reverse TME-induced CAR T cell inhibition.31
Resisting immune suppression in the TME
Innovative strategies to resist immunosuppression caused by anti-inflammatory cytokines and suppressive cells in the TME have been explored or are in development. These include engineering approaches to rewire suppressive cytokines and inhibitory signals, enhance secretion of pro-inflammatory cytokines, and deplete or redirect suppressive cell types.
Engineering synthetic receptors targeting suppressive cytokines and inhibitory signals
One strategy to overcome the immunosuppressive and tolerogenic state of the TME is to redirect or circumvent the endogenous response to suppressive cytokines and inhibitory signaling pathways. Suppressive cytokines commonly encountered in the TME include TGF-β, VEGF, IL-4, and IL-10, and they have the ability to not only directly inhibit T cell effector function but also recruit suppressor cell types. The field of CAR T cell therapy has witnessed a revolution in cell engineering because of advances in synthetic biology, cell engineering, and CRISPR tools.84 Synthetic receptors targeting these suppressive cytokines have been engineered, most notably against TGF-β and IL-4. The synthetic TGF-β dominant-negative receptor (DNR) is a truncated form of TGF-β receptor II (TGF-βRII) lacking the cytoplasmic signaling domain. TGF-β DNRs have been engineered to inhibit endogenous TGF-β signaling by forming signaling-incompetent ligand-receptor complexes, acting as a sink for TGF-β.85 T cells expressing this synthetic receptor can inhibit polarization of T cells into immunosuppressive Treg cells,86 augmenting anti-tumor immunity. CRIPSR-Cas9-mediated knockout of TGF-βRII in CAR T cells has demonstrated increased efficacy, as well as reduced exhaustion and polarization to Treg cells, when tested against solid tumor xenografts.87 When transduced as a second transgene in prostate-specific membrane antigen (PSMA)-targeted CAR, the synthetic TGF-β DNR has been shown to limit the immunosuppressive effects of TGF-β in the TME, and this construct is currently being investigated in a phase I clinical trial (ClinicalTrials.gov: NCT03089203).88 Although early reports suggest promising activity, there were two fatal cases of neurotoxicity associated with massive CAR T cell proliferation.89 This highlights the power of engineering CAR T cells and the need for extensive and careful evaluation of engineered therapy. Chimeric receptors have also been engineered to convert immunosuppressive signaling by fusing the ectodomain of an immunosuppressive cytokine with the endodomain of an immunostimulatory cytokine. Engineered chimeric IL-4 receptors have been combined with the intracellular signaling domains of immunostimulatory cytokines and have demonstrated potent in vitro T cell expansion and tumor cell killing when co-expressed with prostate stem cell antigen (PSCA)-targeted CARs.90,91 Leveraging this approach and aiming to simultaneously target multiple immunosuppressive cytokines, co-expression of an IL-4R-IL-7R chimera and TGF-βR–4-1BB chimera with a first-generation PSCA CAR resulted in enhanced potency and specificity of tumor cell killing in the TME with high TGF-β and IL-4 expression.92
Synthetic receptors have also been engineered to rewire inhibitory pathways such as the PD1-PDL1 and the FAS/Fas ligand (FASL) signaling axes.93,94 Co-expression of a PD1 DNR with mesothelin-targeted CAR enhanced CAR T cell function in xenograft models of high-PDL1-expressing pleural mesothelioma.93 Similarly, expression of a FAS DNR in CAR T cells has resulted in increased T cell longevity and enhanced tumor killing in syngeneic models because of inhibition of FASL-induced T cell apoptosis.94
Engineering “armored” CAR T cells to secrete pro-inflammatory cytokines
Another strategy to combat an immunosuppressive TME and increase CAR T cell function is secretion of pro-inflammatory soluble factors, which can reshape the TME for a favorable anti-tumor response. CAR T cells engineered to secrete IL-12 and IL-18 have been shown to stimulate recruitment of pro-inflammatory immune cells such as M1 macrophages and enhance the anti-tumor response through IFNγ secretion and Treg cell inhibition.95, 96, 97, 98 Overexpression of the IL-12β p40 subunit has also been shown to stimulate production of IL-23 by activated T cells, which promotes proliferation via STAT3 signalling.99 IL-7 and CCL19 are essential for formation of the T cell zone in lymphoid organs, and their overexpression in CAR T cells has been shown to promote CAR T cell survival and infiltration of pro-inflammatory dendritic cells and T cells in the TME in mouse models.100
Targeting suppressive cell types
Beyond a cytokine-driven approach to reshaping the immunosuppressive TME, direct inhibition of suppressor cell types within the TME has also proven to be an effective strategy in the quest to enhance CAR T cell efficacy.
The growing appreciation of the suppressive role of the myeloid compartment of the TME has led to development of strategies targeting TAMs and MDSCs. NK cells express the cytotoxic receptor NKG2D, whose ligands are overexpressed on tumor-infiltrating MDSCs, providing an opportunity to reverse an MDSC-mediated suppressive TME. NK cells bearing an NKG2D-targeted CAR selectively targeted MDSCs and rescued impaired CAR T cell activity when combined with disialoganglioside (GD2)-targeted CART cells in a neuroblastoma xenograft model.101 The NKG2D CAR-NK cells secreted pro-inflammatory cytokines and chemokines in response to MDSCs present in the TME, resulting in tumor regression and prolonged survival compared with treatment with CAR T cells alone.101 Other studies have also shown that TAM depletion through colony-stimulating factor 1 receptor (CSF1R) inhibition102 and targeted PD-1 ablation on myeloid progenitor cells103 have resulted in more potent anti-tumor immunity and decreased tumor growth. Granulocyte macrophage-colony stimulating factor (GM-CSF) depletion resulted in enhancement of CAR T cell function through reversal of the immunosuppressive myeloid cell effect.38,104 These studies highlight the important role of the myeloid compartment in shaping an immune response and underscore the need to incorporate inhibition of myeloid cells in combinatorial approaches aiming to enhance CAR T cell efficacy.
Besides myeloid cells, Treg cells have also been shown to negatively affect the efficacy of CAR T cell therapy in the TME. In a pilot study evaluating the safety and feasibility of a single dose of peripherally infused EGFR class III variant (EGFRvIII)-targeting CAR T cells in individuals with EGFRvIII-expressing recurrent GBM, Treg cell density in tumor specimens was found to be increased after CAR T cell-EGFRvIII infusion compared with baseline, indicating a potential mechanism of adaptive resistance to CAR T cell therapy.105 To circumvent this Treg cell-mediated immunosuppression, the investigators developed a bicistronic construct to drive expression of an EGFRvIII-targeted CAR and a bispecific T cell engager (BiTE) against EGFR. Secretion of EGFR-specific BiTEs by these CART.BiTE cells allowed rewiring of Treg cells to become cytotoxic T cells.106
Conclusion and future perspectives
Despite the remarkable outcomes of CAR T cell therapy, which are rapidly shaping treatment paradigms for hematological malignancies, many challenges have to be overcome to achieve durable clinical benefits and maximize survival. Recent studies have uncovered many mechanisms in the TME that impede the efficacy of CAR T cell therapy, including competition for metabolic fuels, physical barriers to effective CAR T cell infiltration, and immunosuppressive cytokines and cell types. Improved understanding of therapeutic resistance intrinsic to the TME has informed novel strategies and sophisticated CAR designs. Such strategies include use of novel T cell engineering and gene-editing approaches to not only enhance CAR T cell delivery to the TME niche and rewire or counteract suppressive mechanisms but also effectively recruit the host immune response to augment antitumor immunity. Optimizing metabolic fuels in the TME to sustain the demands of CAR T cell function remains a critical area of ongoing investigation. Ultimately, the complicated task of overcoming the TME barriers to CAR T cell therapy will require a multidimensional approach involving synergistic combinations with other treatment modalities.
Acknowledgments
This work was partly supported by grants from the Mayo Clinic Center for Individualized Medicine (to S.S.K.) and the Passano Foundation (to R.S. and S.S.K.).
Author contributions
L.A.K.F., O.S., R.S., E.L.S., and S.S.K. wrote, edited, and approved the final version of the manuscript.
Declaration of interests
S.S.K. is an inventor on patents in the field of CAR immunotherapy that are licensed to Novartis (through an agreement between the Mayo Clinic, University of Pennsylvania, and Novartis), and Mettaforge (through the Mayo Clinic). R.S. and S.S.K. are inventors on patents in the field of CAR immunotherapy that are licensed to Humanigen (through the Mayo Clinic). S.S.K. receives research funding from Kite, Gilead, Juno, Celgene, Novartis, Humanigen, MorphoSys, Tolero, Sunesis, Viracta, and Lentigen. S.S.K. has participated in scientific advisory boards with Kite, Gilead, Juno, BMS, Novartis, and Humanigen. S.S.K. is on the DSMB for Humanigen.
References
- 1.Sadelain M., Rivière I., Brentjens R. Targeting tumours with genetically enhanced T lymphocytes. Nat. Rev. Cancer. 2003;3:35–45. doi: 10.1038/nrc971. [DOI] [PubMed] [Google Scholar]
- 2.Kenderian S.S., Ruella M., Gill S., Kalos M. Chimeric antigen receptor T-cell therapy to target hematologic malignancies. Cancer Res. 2014;74:6383–6389. doi: 10.1158/0008-5472.CAN-14-1530. [DOI] [PubMed] [Google Scholar]
- 3.Mohty M., Dulery R., Gauthier J., Malard F., Brissot E., Aljurf M., Bazarbachi A., Chabanon C., Kharfan-Dabaja M.A., Savani B.N., et al. CAR T-cell therapy for the management of refractory/relapsed high-grade B-cell lymphoma: a practical overview. Bone Marrow Transplant. 2020;55:1525–1532. doi: 10.1038/s41409-020-0892-7. [DOI] [PubMed] [Google Scholar]
- 4.Porter D.L., Hwang W.T., Frey N.V., Lacey S.F., Shaw P.A., Loren A.W., Bagg A., Marcucci K.T., Shen A., Gonzalez V., et al. Chimeric antigen receptor T cells persist and induce sustained remissions in relapsed refractory chronic lymphocytic leukemia. Sci. Transl. Med. 2015;7:303ra139. doi: 10.1126/scitranslmed.aac5415. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Bouchkouj N., Kasamon Y.L., de Claro R.A., George B., Lin X., Lee S., Blumenthal G.M., Bryan W., McKee A.E., Pazdur R. FDA approval summary: axicabtagene ciloleucel for relapsed or refractory large B-cell lymphoma. Clin. Cancer Res. 2019;25:1702–1708. doi: 10.1158/1078-0432.CCR-18-2743. [DOI] [PubMed] [Google Scholar]
- 6.Locke F.L., Neelapu S.S., Bartlett N.L., Siddiqi T., Chavez J.C., Hosing C.M., Ghobadi A., Budde L.E., Bot A., Rossi J.M., et al. Phase 1 results of ZUMA-1: a multicenter study of KTE-C19 anti-CD19 CAR T cell therapy in refractory aggressive lymphoma. Mol. Ther. 2017;25:285–295. doi: 10.1016/j.ymthe.2016.10.020. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Neelapu S.S., Locke F.L., Bartlett N.L., Lekakis L.J., Miklos D.B., Jacobson C.A., Braunschweig I., Oluwole O.O., Siddiqi T., Lin Y., et al. Axicabtagene ciloleucel CAR T-cell therapy in refractory large B-cell lymphoma. N. Engl. J. Med. 2017;377:2531–2544. doi: 10.1056/NEJMoa1707447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Munshi N.C., Anderson L.D., Jr., Shah N., Madduri D., Berdeja J., Lonial S., Raje N., Lin Y., Siegel D., Oriol A., et al. Idecabtagene vicleucel in relapsed and refractory multiple myeloma. N. Engl. J. Med. 2021;384:705–716. doi: 10.1056/NEJMoa2024850. [DOI] [PubMed] [Google Scholar]
- 9.Anagnostou T., Riaz I.B., Hashmi S.K., Murad M.H., Kenderian S.S. Anti-CD19 chimeric antigen receptor T-cell therapy in acute lymphocytic leukaemia: a systematic review and meta-analysis. Lancet Haematol. 2020;7:e816–e826. doi: 10.1016/S2352-3026(20)30277-5. [DOI] [PubMed] [Google Scholar]
- 10.Wagner J., Wickman E., DeRenzo C., Gottschalk S. CAR T cell therapy for solid tumors: bright future or dark reality? Mol. Ther. 2020;28:2320–2339. doi: 10.1016/j.ymthe.2020.09.015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Fucà G., Reppel L., Landoni E., Savoldo B., Dotti G. Enhancing chimeric antigen receptor T-cell efficacy in solid tumors. Clin. Cancer Res. 2020;26:2444–2451. doi: 10.1158/1078-0432.CCR-19-1835. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Sakemura R., Cox M.J., Hefazi M., Siegler E.L., Kenderian S.S. Resistance to CART cell therapy: lessons learned from the treatment of hematological malignancies. Leuk. Lymphoma. 2021;62:2052–2063. doi: 10.1080/10428194.2021.1894648. [DOI] [PubMed] [Google Scholar]
- 13.Hou A.J., Chen L.C., Chen Y.Y. Navigating CAR-T cells through the solid-tumour microenvironment. Nat. Rev. Drug Discov. 2021;20:531–550. doi: 10.1038/s41573-021-00189-2. [DOI] [PubMed] [Google Scholar]
- 14.Quail D.F., Joyce J.A. Microenvironmental regulation of tumor progression and metastasis. Nat. Med. 2013;19:1423–1437. doi: 10.1038/nm.3394. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Joyce J.A., Pollard J.W. Microenvironmental regulation of metastasis. Nat. Rev. Cancer. 2009;9:239–252. doi: 10.1038/nrc2618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Fridman W.H., Pagès F., Sautès-Fridman C., Galon J. The immune contexture in human tumours: impact on clinical outcome. Nat. Rev. Cancer. 2012;12:298–306. doi: 10.1038/nrc3245. [DOI] [PubMed] [Google Scholar]
- 17.Gabrilovich D.I., Ostrand-Rosenberg S., Bronte V. Coordinated regulation of myeloid cells by tumours. Nat. Rev. Immunol. 2012;12:253–268. doi: 10.1038/nri3175. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Cheng L., Huang Z., Zhou W., Wu Q., Donnola S., Liu J.K., Fang X., Sloan A.E., Mao Y., Lathia J.D., et al. Glioblastoma stem cells generate vascular pericytes to support vessel function and tumor growth. Cell. 2013;153:139–152. doi: 10.1016/j.cell.2013.02.021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Galon J., Costes A., Sanchez-Cabo F., Kirilovsky A., Mlecnik B., Lagorce-Pagès C., Tosolini M., Camus M., Berger A., Wind P., et al. Type, density, and location of immune cells within human colorectal tumors predict clinical outcome. Science. 2006;313:1960–1964. doi: 10.1126/science.1129139. [DOI] [PubMed] [Google Scholar]
- 20.Pagès F., Mlecnik B., Marliot F., Bindea G., Ou F.S., Bifulco C., Lugli A., Zlobec I., Rau T.T., Berger M.D., et al. International validation of the consensus immunoscore for the classification of colon cancer: a prognostic and accuracy study. Lancet. 2018;391:2128–2139. doi: 10.1016/S0140-6736(18)30789-X. [DOI] [PubMed] [Google Scholar]
- 21.Mahmoud S.M., Paish E.C., Powe D.G., Macmillan R.D., Grainge M.J., Lee A.H., Ellis I.O., Green A.R. Tumor-infiltrating CD8+ lymphocytes predict clinical outcome in breast cancer. J. Clin. Oncol. 2011;29:1949–1955. doi: 10.1200/JCO.2010.30.5037. [DOI] [PubMed] [Google Scholar]
- 22.Sugimoto M., Mitsunaga S., Yoshikawa K., Kato Y., Gotohda N., Takahashi S., Konishi M., Ikeda M., Kojima M., Ochiai A., et al. Prognostic impact of M2 macrophages at neural invasion in patients with invasive ductal carcinoma of the pancreas. Eur. J. Cancer. 2014;50:1900–1908. doi: 10.1016/j.ejca.2014.04.010. [DOI] [PubMed] [Google Scholar]
- 23.Jensen T.O., Schmidt H., Møller H.J., Høyer M., Maniecki M.B., Sjoegren P., Christensen I.J., Steiniche T. Macrophage markers in serum and tumor have prognostic impact in American Joint Committee on Cancer stage I/II melanoma. J. Clin. Oncol. 2009;27:3330–3337. doi: 10.1200/JCO.2008.19.9919. [DOI] [PubMed] [Google Scholar]
- 24.Marsh D., Suchak K., Moutasim K.A., Vallath S., Hopper C., Jerjes W., Upile T., Kalavrezos N., Violette S.M., Weinreb P.H., et al. Stromal features are predictive of disease mortality in oral cancer patients. J. Pathol. 2011;223:470–481. doi: 10.1002/path.2830. [DOI] [PubMed] [Google Scholar]
- 25.Neuzillet C., Tijeras-Raballand A., Ragulan C., Cros J., Patil Y., Martinet M., Erkan M., Kleeff J., Wilson J., Apte M., et al. Inter- and intra-tumoural heterogeneity in cancer-associated fibroblasts of human pancreatic ductal adenocarcinoma. J. Pathol. 2019;248:51–65. doi: 10.1002/path.5224. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Petitprez F., Vano Y.A., Becht E., Giraldo N.A., de Reyniès A., Sautès-Fridman C., Fridman W.H. Transcriptomic analysis of the tumor microenvironment to guide prognosis and immunotherapies. Cancer Immunol. Immunother. 2018;67:981–988. doi: 10.1007/s00262-017-2058-z. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Fridman W.H., Miller I., Sautès-Fridman C., Byrne A.T. Therapeutic targeting of the colorectal tumor stroma. Gastroenterology. 2020;158:303–321. doi: 10.1053/j.gastro.2019.09.045. [DOI] [PubMed] [Google Scholar]
- 28.Srivastava S., Riddell S.R. Chimeric antigen receptor T cell therapy: challenges to bench-to-bedside efficacy. J. Immunol. 2018;200:459–468. doi: 10.4049/jimmunol.1701155. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Valkenburg K.C., de Groot A.E., Pienta K.J. Targeting the tumour stroma to improve cancer therapy. Nat. Rev. Clin. Oncol. 2018;15:366–381. doi: 10.1038/s41571-018-0007-1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 30.Gunderson A.J., Yamazaki T., McCarty K., Fox N., Phillips M., Alice A., Blair T., Whiteford M., O’Brien D., Ahmad R., et al. TGFβ suppresses CD8+ T cell expression of CXCR3 and tumor trafficking. Nat. Commun. 2020;11:1749. doi: 10.1038/s41467-020-15404-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Sakemura R., Hefazi M., Siegler E.L., Cox M.J., Larson D.P., Hansen M.J., Roman C.M., Schick K.J., Can I., Tapper E.E., et al. Targeting cancer-associated fibroblasts in the bone marrow prevents resistance to CART-cell therapy in multiple myeloma. Blood. 2022 doi: 10.1182/blood.2021012811. blood.2021012811. [DOI] [PubMed] [Google Scholar]
- 32.Huang Y., Kim B.Y.S., Chan C.K., Hahn S.M., Weissman I.L., Jiang W. Improving immune-vascular crosstalk for cancer immunotherapy. Nat. Rev. Immunol. 2018;18:195–203. doi: 10.1038/nri.2017.145. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Lanitis E., Irving M., Coukos G. Targeting the tumor vasculature to enhance T cell activity. Curr. Opin. Immunol. 2015;33:55–63. doi: 10.1016/j.coi.2015.01.011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Togashi Y., Shitara K., Nishikawa H. Regulatory T cells in cancer immunosuppression—implications for anticancer therapy. Nat. Rev. Clin. Oncol. 2019;16:356–371. doi: 10.1038/s41571-019-0175-7. [DOI] [PubMed] [Google Scholar]
- 35.Enblad G., Karlsson H., Gammelgard G., Wenthe J., Lovgren T., Amini R.M., Wikstrom K.I., Essand M., Savoldo B., Hallbook H., et al. A phase I/IIa trial using CD19-targeted third-generation CAR T cells for lymphoma and leukemia. Clin. Cancer Res. 2018;24:6185–6194. doi: 10.1158/1078-0432.CCR-18-0426. [DOI] [PubMed] [Google Scholar]
- 36.Rodriguez-Garcia A., Palazon A., Noguera-Ortega E., Powell D.J., Jr., Guedan S. CAR-T cells hit the tumor microenvironment: strategies to overcome tumor escape. Front. Immunol. 2020;11:1109. doi: 10.3389/fimmu.2020.01109. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Sterner R.M., Kenderian S.S. Myeloid cell and cytokine interactions with chimeric antigen receptor-T-cell therapy: implication for future therapies. Curr. Opin. Hematol. 2020;27:41–48. doi: 10.1097/MOH.0000000000000559. [DOI] [PubMed] [Google Scholar]
- 38.Sterner R.M., Sakemura R., Cox M.J., Yang N., Khadka R.H., Forsman C.L., Hansen M.J., Jin F., Ayasoufi K., Hefazi M., et al. GM-CSF inhibition reduces cytokine release syndrome and neuroinflammation but enhances CAR-T cell function in xenografts. Blood. 2019;133:697–709. doi: 10.1182/blood-2018-10-881722. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Mellor A.L., Munn D.H. Ido expression by dendritic cells: tolerance and tryptophan catabolism. Nat. Rev. Immunol. 2004;4:762–774. doi: 10.1038/nri1457. [DOI] [PubMed] [Google Scholar]
- 40.Brand A., Singer K., Koehl G.E., Kolitzus M., Schoenhammer G., Thiel A., Matos C., Bruss C., Klobuch S., Peter K., et al. LDHA- associated lactic acid production blunts tumor immunosurveillance by T and NK cells. Cell Metab. 2016;24:657–671. doi: 10.1016/j.cmet.2016.08.011. [DOI] [PubMed] [Google Scholar]
- 41.Angelin A., Gil-de-Gómez L., Dahiya S., Jiao J., Guo L., Levine M.H., Wang Z., Quinn W.J., III, Kopinski P.K., Wang L., et al. Foxp3 reprograms T cell metabolism to function in low- glucose, high- lactate environments. Cell Metab. 2017;25:1282–1293.e7. doi: 10.1016/j.cmet.2016.12.018. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Colegio O.R., Chu N.Q., Szabo A.L., Chu T., Rhebergen A.M., Jairam V., Cyrus N., Brokowski C.E., Eisenbarth S.C., Phillips G.M., et al. Functional polarization of tumour-associated macrophages by tumour- derived lactic acid. Nature. 2014;513:559–563. doi: 10.1038/nature13490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Cox M.J., Lucien F., Sakemura R., Boysen J.C., Kim Y., Horvei P., Roman C.M., Hansen M.J., Tapper E.E., Siegler E.L., et al. Leukemic extracellular vesicles induce chimeric antigen receptor T cell dysfunction in chronic lymphocytic leukemia. Mol. Ther. 2021;29:1529–1540. doi: 10.1016/j.ymthe.2020.12.033. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Vander Heiden M.G., Cantley L.C., Thompson C.B. Understanding the Warburg effect: the metabolic requirements of cell proliferation. Science. 2009;324:1029–1033. doi: 10.1126/science.1160809. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Menk A.V., Scharping N.E., Moreci R.S., Zeng X., Guy C., Salvatore S., Bae H., Xie J., Young H.A., Wendell S.G., et al. Early TCR signaling induces rapid aerobic glycolysis enabling distinct acute T cell effector functions. Cell Rep. 2018;22:1509–1521. doi: 10.1016/j.celrep.2018.01.040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Chang C.H., Curtis J.D., Maggi L.B., Jr., Faubert B., Villarino A.V., O'Sullivan D., Huang S.C., van der Windt G.J., Blagih J., Qiu J., et al. Posttranscriptional control of T cell effector function by aerobic glycolysis. Cell. 2013;153:1239–1251. doi: 10.1016/j.cell.2013.05.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Chang C.H., Qiu J., O'Sullivan D., Buck M.D., Noguchi T., Curtis J.D., Chen Q., Gindin M., Gubin M.M., van der Windt G.J., et al. Metabolic competition in the tumor microenvironment is a driver of cancer progression. Cell. 2015;162:1229–1241. doi: 10.1016/j.cell.2015.08.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Ho P.C., Bihuniak J.D., Macintyre A.N., Staron M., Liu X., Amezquita R., Tsui Y.C., Cui G., Micevic G., Perales J.C., et al. Phosphoenolpyruvate is a metabolic checkpoint of anti-tumor T cell responses. Cell. 2015;162:1217–1228. doi: 10.1016/j.cell.2015.08.012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu J., Villa M., Sanin D.E., Buck M.D., O'Sullivan D., Ching R., Matsushita M., Grzes K.M., Winkler F., Chang C.H., et al. Acetate promotes T cell effector function during glucose restriction. Cell Rep. 2019;27:2063–2074.e5. doi: 10.1016/j.celrep.2019.04.022. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vardhana S.A., Hwee M.A., Berisa M., Wells D.K., Yost K.E., King B., Smith M., Herrera P.S., Chang H.Y., Satpathy A.T., et al. Impaired mitochondrial oxidative phosphorylation limits the self-renewal of T cells exposed to persistent antigen. Nat. Immunol. 2020;21:1022–1033. doi: 10.1038/s41590-020-0725-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Bengsch B., Johnson A.L., Kurachi M., Odorizzi P.M., Pauken K.E., Attanasio J., Stelekati E., McLane L.M., Paley M.A., Delgoffe G.M., et al. Bioenergetic insufficiencies due to metabolic alterations regulated by the inhibitory receptor PD-1 are an early driver of CD8(+) T cell exhaustion. Immunity. 2016;45:358–373. doi: 10.1016/j.immuni.2016.07.008. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Gemta L.F., Siska P.J., Nelson M.E., Gao X., Liu X., Locasale J.W., Yagita H., Slingluff C.L., Jr., Hoehn K.L., Rathmell J.C., et al. Impaired enolase 1 glycolytic activity restrains effector functions of tumor-infiltrating CD8(+) T cells. Sci. Immunol. 2019;4:eaap9520. doi: 10.1126/sciimmunol.aap9520. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Globerson-Levin A., Waks T., Eshhar Z. Elimination of progressive mammary cancer by repeated administrations of chimeric antigen receptor-modified T cells. Mol. Ther. 2014;22:1029–1038. doi: 10.1038/mt.2014.28. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Adusumilli P.S., Cherkassky L., Villena-Vargas J., Colovos C., Servais E., Plotkin J., Jones D.R., Sadelain M. Regional delivery of mesothelin-targeted CAR T cell therapy generates potent and long-lasting CD4-dependent tumor immunity. Sci. Transl. Med. 2014;6:261ra151. doi: 10.1126/scitranslmed.3010162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Maude S.L., Frey N., Shaw P.A., Aplenc R., Barrett D.M., Bunin N.J., Chew A., Gonzalez V.E., Zheng Z., Lacey S.F., et al. Chimeric antigen receptor T cells for sustained remissions in leukemia. N. Engl. J. Med. 2014;371:1507–1517. doi: 10.1056/NEJMoa1407222. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Brown C.E., Aguilar B., Starr R., Yang X., Chang W.C., Weng L., Chang B., Sarkissian A., Brito A., Sanchez J.F., et al. Optimization of IL13Ralpha2-targeted chimeric antigen receptor T cells for improved anti-tumor efficacy against glioblastoma. Mol. Ther. 2018;26:31–44. doi: 10.1016/j.ymthe.2017.10.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Brown C.E., Badie B., Barish M.E., Weng L., Ostberg J.R., Chang W.C., Naranjo A., Starr R., Wagner J., Wright C., et al. Bioactivity and safety of IL13Ralpha2-redirected chimeric antigen receptor CD8+ T cells in patients with recurrent glioblastoma. Clin. Cancer Res. 2015;21:4062–4072. doi: 10.1158/1078-0432.CCR-15-0428. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Brown C.E., Alizadeh D., Starr R., Weng L., Wagner J.R., Naranjo A., Ostberg J.R., Blanchard M.S., Kilpatrick J., Simpson J., et al. Regression of glioblastoma after chimeric antigen receptor T-cell therapy. N. Engl. J. Med. 2016;375:2561–2569. doi: 10.1056/NEJMoa1610497. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 59.Parkhurst M.R., Yang J.C., Langan R.C., Dudley M.E., Nathan D.A., Feldman S.A., Davis J.L., Morgan R.A., Merino M.J., Sherry R.M., et al. T cells targeting carcinoembryonic antigen can mediate regression of metastatic colorectal cancer but induce severe transient colitis. Mol. Ther. 2011;19:620–626. doi: 10.1038/mt.2010.272. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Katz S.C., Burga R.A., McCormack E., Wang L.J., Mooring W., Point G.R., Khare P.D., Thorn M., Ma Q., Stainken B.F., et al. Phase I hepatic immunotherapy for metastases study of intra-arterial chimeric antigen receptor-modified T-cell therapy for CEA+ liver metastases. Clin. Cancer Res. 2015;21:3149–3159. doi: 10.1158/1078-0432.CCR-14-1421. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.Tchou J., Zhao Y., Levine B.L., Zhang P.J., Davis M.M., Melenhorst J.J., Kulikovskaya I., Brennan A.L., Liu X., Lacey S.F., et al. Safety and efficacy of intratumoral injections of chimeric antigen receptor (CAR) T cells in metastatic breast cancer. Cancer Immunol. Res. 2017;5:1152–1161. doi: 10.1158/2326-6066.CIR-17-0189. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Nagarsheth N., Wicha M.S., Zou W. Chemokines in the cancer microenvironment and their relevance in cancer immunotherapy. Nat. Rev. Immunol. 2017;17:559–572. doi: 10.1038/nri.2017.49. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Martinez M., Moon E.K. CAR T cells for solid tumors: new strategies for finding, infiltrating, and surviving in the tumor microenvironment. Front. Immunol. 2019;10:128. doi: 10.3389/fimmu.2019.00128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Jin L., Cao L., Zhu Y., Cao J., Li X., Zhou J., Liu B., Zhao T., et al. Enhance anti-lung tumor efficacy of chimeric antigen receptor-T cells by ectopic expression of C–C motif chemokine receptor 6. Sci. Bull. 2020;66:803–812. doi: 10.1016/j.scib.2020.12.027. [DOI] [PubMed] [Google Scholar]
- 65.Liu G., Rui W., Zheng H., Huang D., Yu F., Zhang Y., Dong J., Zhao X., Lin X. CXCR2-modified CAR-T cells have enhanced trafficking ability that improves treatment of hepatocellular carcinoma. Eur. J. Immunol. 2020;50:712–724. doi: 10.1002/eji.201948457. [DOI] [PubMed] [Google Scholar]
- 66.Jin L., Tao H., Karachi A., Long Y., Hou A.Y., Na M., Dyson K.A., Grippin A.J., Deleyrolle L.P., Zhang W., et al. CXCR1- or CXCR2-modified CAR T cells co-opt IL-8 for maximal antitumor efficacy in solid tumors. Nat. Commun. 2019;10:4016. doi: 10.1038/s41467-019-11869-4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 67.Moon E.K., Carpenito C., Sun J., Wang L.C., Kapoor V., Predina J., Powell D.J., Riley J.L., June C.H., Albelda S.M., et al. Expression of a functional CCR2 receptor enhances tumor localization and tumor eradication by retargeted human T cells expressing a mesothelin-specific chimeric antibody receptor. Clin. Cancer Res. 2011;17:4719–4730. doi: 10.1158/1078-0432.CCR-11-0351. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 68.Wang Y., Wang J., Yang X., Yang J., Lu P., Zhao L., Li B., Pan H., Jiang Z., Shen X., et al. Chemokine receptor CCR2b enhanced anti-tumor function of chimeric antigen receptor T cells targeting mesothelin in a non-small-cell lung carcinoma model. Front. Immunol. 2021;12:628906. doi: 10.3389/fimmu.2021.628906. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Craddock J.A., Lu A., Bear A., Pule M., Brenner M.K., Rooney C.M., Foster A.E. Enhanced tumor trafficking of GD2 chimeric antigen receptor T cells by expression of the chemokine receptor CCR2b. J. Immunother. 2010;33:780–788. doi: 10.1097/CJI.0b013e3181ee6675. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Sakemura R., Bansal A., Siegler E.L., Hefazi M., Yang N., Khadka R.H., Newsom A.N., Hansen M.J., Cox M.J., Roman C.M., et al. Development of a clinically relevant reporter for chimeric antigen receptor T-cell expansion, trafficking, and toxicity. Cancer Immunol. Res. 2021;9:1035–1046. doi: 10.1158/2326-6066.CIR-20-0901. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Sakemura R., Can I., Siegler E.L., Kenderian S.S. In vivo CART cell imaging: paving the way for success in CART cell therapy. Mol. Ther. Oncolytics. 2021;20:625–633. doi: 10.1016/j.omto.2021.03.003. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Khawar I.A., Kim J.H., Kuh H.J. Improving drug delivery to solid tumors: priming the tumor microenvironment. J. Control. Release. 2015;201:78–89. doi: 10.1016/j.jconrel.2014.12.018. [DOI] [PubMed] [Google Scholar]
- 73.Upreti M., Jyoti A., Sethi P. Tumor microenvironment and nanotherapeutics. Transl. Cancer Res. 2013;2:309–319. doi: 10.3978/j.issn.2218-676X.2013.08.11. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Chinnasamy D., Yu Z., Theoret M.R., Zhao Y., Shrimali R.K., Morgan R.A., Feldman S.A., Restifo N.P., Rosenberg S.A. Gene therapy using genetically modified lymphocytes targeting VEGFR-2 inhibits the growth of vascularized syngenic tumors in mice. J. Clin. Invest. 2010;120:3953–3968. doi: 10.1172/JCI43490. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 75.Chinnasamy D., Tran E., Yu Z., Morgan R.A., Restifo N.P., Rosenberg S.A. Simultaneous targeting of tumor antigens and the tumor vasculature using T lymphocyte transfer synergize to induce regression of established tumors in mice. Cancer Res. 2013;73:3371–3380. doi: 10.1158/0008-5472.CAN-12-3913. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Wang W., Ma Y., Li J., Shi H.S., Wang L.Q., Guo F.C., Zhang J., Li D., Mo B.H., Wen F., et al. Specificity redirection by CAR with human VEGFR-1 affinity endows T lymphocytes with tumor-killing ability and anti-angiogenic potency. Gene Ther. 2013;20:970–978. doi: 10.1038/gt.2013.19. [DOI] [PubMed] [Google Scholar]
- 77.Bocca P., Di Carlo E., Caruana I., Emionite L., Cilli M., De Angelis B., Quintarelli C., Pezzolo A., Raffaghello L., Morandi F., et al. Bevacizumab-mediated tumor vasculature remodelling improves tumor infiltration and antitumor efficacy of GD2-CAR T cells in a human neuroblastoma preclinical model. Oncoimmunology. 2017;7:e1378843. doi: 10.1080/2162402X.2017.1378843. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Klemm F., Joyce J.A. Microenvironmental regulation of therapeutic response in cancer. Trends Cell Biol. 2015;25:198–213. doi: 10.1016/j.tcb.2014.11.006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Kanapathipillai M., Mammoto A., Mammoto T., Kang J.H., Jiang E., Ghosh K., Korin N., Gibbs A., Mannix R., Ingber D.E., et al. Inhibition of mammary tumor growth using lysyl oxidase-targeting nanoparticles to modify extracellular matrix. Nano Lett. 2012;12:3213–3217. doi: 10.1021/nl301206p. [DOI] [PubMed] [Google Scholar]
- 80.Tran E., Chinnasamy D., Yu Z., Morgan R.A., Lee C.C., Restifo N.P., Rosenberg S.A. Immune targeting of fibroblast activation protein triggers recognition of multipotent bone marrow stromal cells and cachexia. J. Exp. Med. 2013;210:1125–1135. doi: 10.1084/jem.20130110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.Wang L.C., Lo A., Scholler J., Sun J., Majumdar R.S., Kapoor V., Antzis M., Cotner C.E., Johnson L.A., Durham A.C., et al. Targeting fibroblast activation protein in tumor stroma with chimeric antigen receptor T cells can inhibit tumor growth and augment host immunity without severe toxicity. Cancer Immunol. Res. 2014;2:154–166. doi: 10.1158/2326-6066.CIR-13-0027. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Kakarla S., Chow K.K., Mata M., Shaffer D.R., Song X.T., Wu M.F., Liu H., Wang L.L., Rowley D.R., Pfizenmaier K., et al. Antitumor effects of chimeric receptor engineered human T cells directed to tumor stroma. Mol. Ther. 2013;21:1611–1620. doi: 10.1038/mt.2013.110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Caruana I., Savoldo B., Hoyos V., Weber G., Liu H., Kim E.S., Ittmann M.M., Marchetti D., Dotti G. Heparanase promotes tumor infiltration and antitumor activity of CAR-redirected T lymphocytes. Nat. Med. 2015;21:524–529. doi: 10.1038/nm.3833. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 84.Manriquez-Roman C., Siegler E.L., Kenderian S.S. CRISPR takes the front seat in CART-cell development. BioDrugs. 2021;35:113–124. doi: 10.1007/s40259-021-00473-y. [DOI] [PubMed] [Google Scholar]
- 85.Bollard C.M., Rössig C., Calonge M.J., Huls M.H., Wagner H.J., Massague J., Brenner M.K., Heslop H.E., Rooney C.M. Adapting a transforming growth factor β- related tumor protection strategy to enhance antitumor immunity. Blood. 2002;99:3179–3187. doi: 10.1182/blood.v99.9.3179. [DOI] [PubMed] [Google Scholar]
- 86.Hou A.J., Chang Z.L., Lorenzini M.H., Zah E., Chen Y.Y. TGF- β–responsive CAR- T cells promote anti- tumor immune function. Bioeng. Transl. Med. 2018;3:75–86. doi: 10.1002/btm2.10097. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 87.Tang N., Cheng C., Zhang X., Qiao M., Li N., Mu W., Wei X.F., Han W., Wang H. TGF-β inhibition via CRISPR promotes the long-term efficacy of CAR T cells against solid tumors. JCI Insight. 2020;5:e133977. doi: 10.1172/jci.insight.133977. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 88.Narayan V., Gladney W., Plesa G., Vapiwala N., Carpenter E., Maude S.L., Lal P., Lacey S.F., Melenhorst J.J., Sebro R., et al. A phase I clinical trial of PSMA-directed/TGFβ-insensitive CAR-T cells in metastatic castration-resistant prostate cancer. J. Clin. Oncol. 2019 doi: 10.1200/JCO.2019.37.7_suppl.TPS347. [DOI] [Google Scholar]
- 89.Exclusive: Carl June's Tmunity encounters a lethal roadblock as 2 patient deaths derail lead trial, raise red flag forcing a rethink of CAR-T for solid tumors. https://endpts.com/exclusive-carl-junes-tmunity-encounters-a-lethal-roadblock-as-2-patient-deaths-derail-lead-trial-raise-red-flag-forcing-a-rethink-of-car-t-for-solid-tumors/.
- 90.Wilkie S., Burbridge S.E., Chiapero-Stanke L., Pereira A.C., Cleary S., van der Stegen S.J., Spicer J.F., Davies D.M., Maher J. Selective expansion of chimeric antigen receptor- targeted T- cells with potent effector function using interleukin-4. J. Biol. Chem. 2010;285:25538–25544. doi: 10.1074/jbc.M110.127951. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 91.Mohammed S., Sukumaran S., Bajgain P., Watanabe N., Heslop H.E., Rooney C.M., Brenner M.K., Fisher W.E., Leen A.M., Vera J.F. Improving chimeric antigen receptor- modified T cell function by reversing the immunosuppressive tumor microenvironment of pancreatic cancer. Mol. Ther. 2017;25:249–258. doi: 10.1016/j.ymthe.2016.10.016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 92.Sukumaran S., Watanabe N., Bajgain P., Raja K., Mohammed S., Fisher W.E., Brenner M.K., Leen A.M., Vera J.F. Enhancing the potency and specificity of engineered T cells for cancer treatment. Cancer Discov. 2018;8:972–987. doi: 10.1158/2159-8290.CD-17-1298. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 93.Cherkassky L., Morello A., Villena-Vargas J., Feng Y., Dimitrov D.S., Jones D.R., Sadelain M., Adusumilli P.S., et al. Human CAR T cells with cell-intrinsic PD-1 checkpoint blockade resist tumor-mediated inhibition. J. Clin. Invest. 2016;126:3130–3144. doi: 10.1172/JCI83092. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 94.Yamamoto T.N., Lee P.H., Vodnala S.K., Gurusamy D., Kishton R.J., Yu Z., Eidizadeh A., Eil R., Fioravanti J., Gattinoni L., et al. T cells genetically engineered to overcome death signaling enhance adoptive cancer immunotherapy. J. Clin. Invest. 2019;129:1551–1565. doi: 10.1172/JCI121491. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 95.Chmielewski M., Kopecky C., Hombach A.A., Abken H. IL-12 release by engineered T cells expressing chimeric antigen receptors can effectively muster an antigen- independent macrophage response on tumor cells that have shut down tumor antigen expression. Cancer Res. 2011;71:5697–5706. doi: 10.1158/0008-5472.CAN-11-0103. [DOI] [PubMed] [Google Scholar]
- 96.Pegram H.J., Lee J.C., Hayman E.G., Imperato G.H., Tedder T.F., Sadelain M., Brentjens R.J. Tumor- targeted T cells modified to secrete IL-12 eradicate systemic tumors without need for prior conditioning. Blood. 2012;119:4133–4141. doi: 10.1182/blood-2011-12-400044. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 97.Hu B., Ren J., Luo Y., Keith B., Young R.M., Scholler J., Zhao Y., June C.H. Augmentation of antitumor immunity by human and mouse CAR T cells secreting IL-18. Cell Rep. 2017;20:3025–3033. doi: 10.1016/j.celrep.2017.09.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 98.Avanzi M.P., Yeku O., Li X., Wijewarnasuriya D.P., van Leeuwen D.G., Cheung K., Park H., Purdon T.J., Daniyan A.F., Spitzer M.H., et al. Engineered tumor- targeted T cells mediate enhanced anti- tumor efficacy both directly and through activation of the endogenous immune system. Cell Rep. 2018;23:2130–2141. doi: 10.1016/j.celrep.2018.04.051. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 99.Ma X., Shou P., Smith C., Chen Y., Du H., Sun C., Porterfield Kren N., Michaud D., Ahn S., Vincent B., et al. Interleukin-23 engineering improves CART cell function in solid tumors. Nat. Biotechnol. 2020;38:448–459. doi: 10.1038/s41587-019-0398-2. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 100.Adachi K., Kano Y., Nagai T., Okuyama N., Sakoda Y., Tamada K. IL-7 and CCL19 expression in CAR-T cells improves immune cell infiltration and CAR-T cell survival in the tumor. Nat. Biotechnol. 2018;36:346–351. doi: 10.1038/nbt.4086. [DOI] [PubMed] [Google Scholar]
- 101.Parihar R., Rivas C., Huynh M., Omer B., Lapteva N., Metelitsa L.S., Gottschalk S.M., Rooney C.M. NK cells expressing a chimeric activating receptor eliminate MDSCs and rescue impaired CAR- T cell activity against solid tumors. Cancer Immunol. Res. 2019;7:363–375. doi: 10.1158/2326-6066.CIR-18-0572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 102.Cassetta L., Kitamura T. Targeting tumor- associated macrophages as a potential strategy to enhance the response to immune checkpoint inhibitors. Front. Cell Dev. Biol. 2018;6:38. doi: 10.3389/fcell.2018.00038. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 103.Strauss L., Mahmoud M.A., Weaver J.D., Tijaro-Ovalle N.M., Christofides A., Wang Q., Pal R., Yuan M., Asara J., Patsoukis N., et al. Targeted deletion of PD-1 in myeloid cells induces antitumor immunity. Sci. Immunol. 2020;5:eaay1863. doi: 10.1126/sciimmunol.aay1863. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 104.Sterner R.M., Cox M.J., Sakemura R., Kenderian S.S. Using CRISPR/Cas9 to knock out GM-CSF in CAR-T cells. J. Vis. Exp. 2019;149:e59629. doi: 10.3791/59629. [DOI] [PubMed] [Google Scholar]
- 105.O’Rourke D.M., Nasrallah M.P., Desai A., Melenhorst J.J., Mansfield K., Morrissette J.J., Martinez-Lage M., Brem S., Maloney E., Shen A., et al. A single dose of peripherally infused EGFRvIII- directed CAR T cells mediates antigen loss and induces adaptive resistance in patients with recurrent glioblastoma. Sci. Transl. Med. 2017;9:eaaa0984. doi: 10.1126/scitranslmed.aaa0984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 106.Choi B.D., Yu X., Castano A.P., Bouffard A.A., Schmidts A., Larson R.C., Bailey S.R., Boroughs A.C., Frigault M.J., Leick M.B., et al. CAR-T cells secreting BiTEs circumvent antigen escape without detectable toxicity. Nat. Biotechnol. 2019;37:1049–1058. doi: 10.1038/s41587-019-0192-1. [DOI] [PubMed] [Google Scholar]