

Summary
Human cytomegalovirus (HCMV) is a betaherpesvirus that can cause severe birth defects including vision and hearing loss, microcephaly, and seizures. Currently, no approved treatment options exist for in utero infections. Here, we aimed to determine the impact of HCMV infection on the transcriptome of developing neurons in an organoid model system. Cell populations isolated from organoids based on a marker for infection and transcriptomes were defined. We uncovered downregulation in key cortical, neurodevelopmental, and functional gene pathways which occurred regardless of the degree of infection. To test the contributions of specific HCMV immediate early proteins known to disrupt neural differentiation, we infected NPCs using a recombinant virus harboring a destabilization domain. Despite suppressing their expression, HCMV-mediated transcriptional downregulation still occurred. Together, our studies have revealed that HCMV infection causes a profound downregulation of neurodevelopmental genes and suggest a role for other viral factors in this process.
Subject areas: Biotechnology, Developmental neuroscience, Transcriptomics
Graphical abstract
Highlights
-
•
HCMV-infected organoids exhibit downregulation of key neurodevelopmental pathways
-
•
Significant transcriptional changes occur in cell populations with low HCMV viral load
-
•
Transcriptional downregulation is both IE1-dependent and -independent
Biotechnology; Developmental neuroscience; Transcriptomics
Introduction
Human cytomegalovirus (HCMV) is a betaherpesvirus with a 235-kbp double-stranded DNA (dsDNA) genome and the potential to express more than 700 proteins (Mocarski and Kemble, 1996; Mocarski, 2007; Stern-Ginossar et al., 2012). The virus infects most of the world’s population with seroprevalence of 40%–100% depending on age, socio-economic status, and geographic location (Sinzger et al., 2008; Griffiths et al., 2015). Infection by HCMV is lifelong and can result in a range of conditions. Vertical transmission can also occur causing congenital CMV (cCMV) infection (Mocarski and Kemble, 1996; Griffiths et al., 2015). A portion of infants born with cCMV infection will have long-term health problems including vision and hearing loss, microcephaly, and seizures. Neurological symptoms are likely the result of infection of neural progenitor cells (NPCs) (Luo et al., 2008; Sun et al., 2020). However, in vivo, fetal tissue studies have reported HCMV-positive cells throughout the cortex along with diffuse inflammatory infiltrate and widespread cellular effects even in areas lacking infiltrate (Gabrielli et al., 2009, 2012). These studies also highlight extensive laminar necrosis and infiltration of macrophages as well as activated microglia associated with regions of high HCMV positivity (Teissier et al., 2014). In vitro, NPCs derived from both induced pluripotent stem cells (iPSCs) and fetal stem cells are fully permissive for HCMV infection, though susceptibility may depend upon degree of NPC terminal differentiation (Odeberg et al., 2006; Luo et al., 2010; Pan et al., 2013; Gonzalez-Sanchez et al., 2015). In the fetal brain, NPCs are in the bilateral subventricular zone aligned with the developing ventricle, and express key transcription factors that are required for maintenance of the progenitor cell pool and subsequent differentiation. NPCs can differentiate into the multiple neuronal and glial lineages found in the central nervous system (CNS). HCMV infection of NPCs has been shown to alter differentiation and function (Luo et al., 2010; Liu et al., 2017a; Wu et al., 2018; Brown et al., 2019; Sison et al., 2019). However, the mechanisms controlling this effect remain to be fully established.
Upon infection, HCMV genes are expressed in three sequential steps known as immediate early (IE), early (E), and late (L). IE gene expression can be detected within hours of infection and along with early gene expression, leads to the onset of viral DNA synthesis. Viral particles are assembled and released within 72–96 hpi as has been shown in fibroblasts (Mocarski, 2007; Marcinowski et al., 2012; Griffiths et al., 2015). In general, IE proteins are responsible for inhibiting intrinsic and innate host cell responses and initiating the transcription of viral early genes (Adamson and Nevels, 2020). These genes regulate host cell function to allow for viral genome replication and packaging (Griffiths et al., 2015). Late viral genes are expressed following the onset of viral DNA synthesis and encode many structural proteins required for particle assembly and egress (Mocarski, 2007; Perng et al., 2011; Tirosh et al., 2015). Following primary infection, the virus may remain lytic, replicating and activating the immune system, or enter a latent stage in hematopoietic progenitor cells and monocytes during which the virus does not replicate (Griffiths et al., 2015). HCMV has broad cell tropism, being shown to replicate in fibroblasts, epithelial cells, monocytes, macrophages, and NPCs among others (Marcinowski et al., 2012; Pan et al., 2013; Gonzalez-Sanchez et al., 2015). In symptomatic children and adults, HCMV infection can be managed using (val)ganciclovir, cidofovir, and/or foscarnet, all of which inhibit viral DNA synthesis, or letermovir, which targets viral DNA packaging into nucleocapsids (Britt and Prichard, 2018; Pass and Arav-Boger, 2018; Altman et al., 2019; Steingruber and Marschall, 2020). Although clinical trials are being conducted for use of valganciclovir in infants and children with confirmed symptomatic and asymptomatic cCMV infections, there is no FDA-approved therapy for treating expecting mothers, making vaccine development a major public health priority.
Initial human microarray experiments using RNA isolated from HCMV-infected NPCs identified several downregulated neurodevelopmental genes such as NES, SOX2/4, and OLIG1 along with disruptions to differentiation (Luo et al., 2010; D'Aiuto et al., 2012). A subsequent mechanistic study in human NPCs demonstrated that expression of HCMV IE1 causes loss of SOX2 expression through sequestering of unphosphorylated STAT3 in the nucleus (Wu et al., 2018). A recent RNA-seq study conducted in HCMV-infected cerebral organoids support these findings and identify SOX2, OLIG1, ENO2, ALDOC, and FEZF2 among other downregulated neurodevelopmental genes (Sun et al., 2020). Additional work from us and others using the cerebral organoid model have found consistent disruptions to neuronal differentiation, cortical layering, calcium signaling, and electrical activity after HCMV infection (Brown et al., 2019; Sison et al., 2019; Sun et al., 2020). Disruption to these transcription factor networks do correspond with in vivo phenotypes like cerebral cortical necrosis, telencephalic leukoencephalopathy, and polymicrogyria observed in the fetal brain tissue of patients with cCMV (Gabrielli et al., 2009).
In the current study, we aim to identify whether specific viral proteins contribute to dysregulation of neurodevelopmental transcriptional networks that are altered in response to HCMV infection. We found extensive transcriptional downregulation that was not limited to cells expressing high levels of GFP, a surrogate marker of viral replication. Previous studies have suggested that IE1 viral protein expression can regulate host gene expression (Marcinowski et al., 2012; Pignoloni et al., 2016; Liu et al., 2017b), which we found to be the case for some cellular signaling-related genes. However, neurodevelopmental gene dysregulation appeared to be independent of IE1 and IE2 protein expression suggesting that other HCMV-related mechanisms are involved. Together, these data show that limiting IE1 and IE2 protein expression offers only minimal benefit to neurodevelopment and suggests that a more comprehensive therapeutic approach is needed to combat cCMV infection.
Results
Cells from infected neural organoids exhibit reduced transcriptional profiles despite varying levels of viral gene expression
Human iPSC-derived cerebral organoids were generated from a WT (4.2) independent healthy control iPSC line (Ebert et al., 2013; Sison et al., 2019) and infected after 30 days of development with a recombinant HCMV strain TB40/E expressing EGFP. An additional WT (21.8) independent healthy control iPSC line was used within the mock samples (see STAR Methods section for which line corresponds with what sample). Also, because tissues varied in size and likely cell numbers, we infected organoids using 500 infectious units per μg of tissue (IU/μg) and allowed infection to progress for two weeks. We detected GFP fluorescence by 4 days post infection (dpi). GFP signal increased by 8 dpi, and continued to be present at 12 dpi, indicating viral spread (Figure 1A). At 13–16 dpi, HCMV-infected and mock organoids (Figure 1B, one image from each organoid pool is shown) were dissociated into single cell suspensions, and cells were sorted based on levels of GFP fluorescence as an indirect measurement of infection. The gating parameters for live cells were set based on cells isolated from uninfected organoids (Figures S1 and S2). This approach is routinely used to investigate latency in HCMV-infected hematopoietic cells (Rak et al., 2018). Expression of the EGFP gene is driven by the SV40 promote and fluorescence is postulated to be proportional to transcriptionally competent HCMV genomes (Rak et al., 2018; Collins-McMillen et al., 2019). We sorted cells into groups exhibiting high, intermediate, and negligible to undetectable levels of fluorescence (Figure 1C). We defined these populations as GFP positive (+), GFP intermediate (Inter), and GFP (Low) based on gating strategies when compared to mock-infected organoids (Figure 1D). With this gating strategy, the gates defining GFP (Inter) are set broadly resulting in a mixed population of GFP (Low) and GFP (+) cells. These histograms also validate the absence of GFP (+) cells within mock-sorted organoids. The total number of live cells from a representative experiment was approximately 3.5 × 106 which consisted of 0.5 × 106 GFP (+) cells, 2 × 106 GFP (Inter) cells, and 1 × 106 GFP (Low) cells (Figure 1E). Infection did not cause large scale cell death as we observed that the number of live cells within each infected organoid group was comparable to the uninfected mock group (Figure 1F). High expression of GFP likely represents a population of cells exhibiting high viral genome copies and viral gene expression. In contrast, cells isolated based on the absence of fluorescence are anticipated to be uninfected or infected but with GFP fluorescence below the threshold of detection. To understand these differences, we quantified expression levels of HCMV gene classes, immediate early (IE; UL123, UL122), early-late (E-L; UL44), and late (L; UL99) with late expression dependent on viral genome synthesis (Chambers et al., 1999). Cell populations of GFP (+) and GFP (Inter) exhibited high levels of viral gene expression in all classes with differences consistent with their level of fluorescence (Figure 1G). In the GFP (Low) group, we did observe low but statistically significant levels of viral IE RNAs versus the limit of detection (Figure 1H) indicating that this population is a mixture of uninfected and infected cells within the tissue. This is further supported by the relative amounts of viral to host genomes in all infected organoid populations including a detectible number of genomes within the GFP (Low) population (Figure 1I).
Figure 1.

Three-dimensional cerebral organoid infection with HCMV
(A) On Day 30 of differentiation, organoids were weighed and infected at 500 IU/ug with HCMV strain TB40/E-GFP. Bright-field and fluorescent images of one representative infected organoid taken at 4, 8, and 12 dpi are shown.
(B) Images at 12 dpi of infected and uninfected organoids are displayed; one representative organoid from each pooled and sorted group is shown.
(C) Representative GFP intensity plot from FACs analysis of an infected organoid.
(D) Representative GFP intensity FACs from a Mock-infected organoid.
(E) Percentage of living cells in the isolated populations as determined by FACs; each infected organoid group was made up of three pooled organoids.
(F) Percentage of total live cells within whole infected and uninfected organoids as determined by FACs; each infected organoid group consisted of three pooled organoids.
(G) qPCR analysis of viral genes (UL122, UL123, UL44, and UL99) expression within Mock compared to GFP (+) and GFP (Inter) subpopulations.
(H) qPCR analysis of viral gene expression within Mock compared to the GFP (Low) subpopulation.
(I) Quantification of relative viral vs. host genomes within each of the sorted infected organoid subpopulations (see STAR Methods for quantification procedure). Scale bar is 100 uM in panels A and B and error bars in E, F, G, and H represent mean ± SEM from 4 biological replicates. Also see Figures S1 and S2. Stars were assigned based on level of significance as determined by one-way ANOVA with Tukey post hoc test: ∗ = p ≤ 0.05, ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001, and ∗∗∗∗ = p ≤ 0.00001.
We quantified infection-induced changes to host cell gene expression using RNA sequencing on sorted cell populations from uninfected and infected organoids. These studies were completed using three biological replicate experiments across two sequencing runs. Three organoids were combined for each condition per biological replicate experiment. Mock and infected-sorted cell populations were mixed for each biological replicate resulting in the pooled sample. Libraries had at least 2.5 million aligned reads and the External RNA Controls Consortium (ERCC) spike-in was used to normalize variation in RNA expression across samples. After trimming, 80% of the bases had a quality score of 30, indicating the probability of an incorrect base call was 1 in 1000. One of the GFP (+) replicates was removed from our subsequent analysis due to a large drop off in total number of mapped reads likely related to technical error in the sequencing reaction. Initially, we performed principal component analysis (PCA) on the datasets which revealed two groups (Figure 2A). One group contained the Mock-infected samples, and the other contained samples isolated from infected organoids regardless of GFP signal strength (Figure 2A). We initially hypothesized that the subpopulations from infected organoids would cluster separately based on varying states of infection. However, as noted in Figure 1, we did detect low levels of viral transcripts and genomes in the GFP (Low) group despite negligible GFP expression indicating this population contains some infected cells.
Figure 2.

All infected organoid subpopulations cluster distinctly from mock samples with many genes being downregulated
(A) PCA with components being all mapped genes across samples split by axis determined by adj p value and compared between each sample.
(B) Hierarchical cluster analysis of all sequenced samples for the top 500 most variable genes according to read counts assigned. Hierarchical cluster analysis of only infected samples for the top 500 most variable genes according to read counts assigned.
(C) Volcano plot of the top 250 differentially expressed genes comparing GFP (+) vs. Mock and GFP (Low) vs. Mock as determined by fold change ±3 adj p value < 0.01 as determined by DESEQ2 analysis in R. Arrows denote genes downregulated in both groups.
(D) Venn diagram comparing the 2,500 most differentially expressed genes in GFP (Low) vs. Mock and GFP (+) vs. Mock.
(E) Volcano plot of the differentially expressed genes found from DESEQ2 analysis of GFP (+) vs. GFP (Low) using the same significance cutoff. Also see Table S1. Differential expression analysis conducted on GFP (+) vs. Mock in R, related to Figures 2 and 4, Table S2. Differential expression analysis conducted on GFP (Low) vs. Mock in R, related to Figures 2 and 4, Table S3. Differential expression and ontology analysis conducted on GFP (+) vs. GFP (Low) in R and using G-profiler, related to Figures 2 and 4.
We identified 1,222 cellular transcripts upregulated, 17,696 downregulated, and 2,882 not differentially expressed in GFP (+) cells compared to cells from mock-treated tissues based on differential expression analysis, using an adjusted p value < 0.05 and 3-fold cutoff. In the GFP (Low) population with substantially lower viral RNA and genomes, we observed 911 transcripts upregulated and 18,379 downregulated compared to mock. Tables S1 and S2 contain full gene lists from DESEQ2 analysis along with a gene list of those not differentially expressed for GFP (+) vs. Mock and GFP (Low) vs. Mock, respectively. Comparing GFP (+) vs. GFP (Low), we found 31 genes downregulated in GFP (+), with none significantly upregulated; Table S3 contains further details on the GFP (+) vs. GFP (Low) DESEQ2 analysis and gene ontology. Completion of hierarchical cluster analysis on the top 500 variable genes across samples showed distinct clustering between infected samples and mock samples (Figure 2B, left), and we did not observe any additional clustering based on GFP designation following the removal of the mock datasets (Figure 2B, right). The disproportional number of genes being downregulated is illustrated in the volcano plots for GFP (+) versus Mock and GFP (Low) versus Mock (Figure 2C). For both GFP (+) and GFP (Low) groups vs. Mock, over 90% of significantly differentially expressed genes were downregulated compared to cells isolated from Mock-infected tissue. Several genes like EEF2, TUBB, and HSP90AB1 are noted in Figure 2C and relate to altered pathways revealed by IPA analysis in Figure 4. A comparison of the top 2,500 differentially expressed genes in GFP (+) vs. Mock and GFP (Low) vs. Mock revealed an overlap of nearly 70% (Figure 2D). Differential expression analysis on GFP (+) vs. GFP (Low) revealed a small list of downregulated genes specific to GFP (+) that are plotted in Figure 2E, mostly involved in transcription factor activity and ion signaling (see Table S3 for full analysis). These observations demonstrate that HCMV infection profoundly impacts transcription in neural tissues and supports a hypothesis that infection induces tissue-wide changes that likely involve indirect effects on uninfected cells from the neighboring GFP (+)-infected cells.
Figure 4.

Pathway and ontology analysis conducted on GFP (+) vs. Mock and heatmaps displaying up- and downregulated genes
(A) Gene Ontology analysis of the top 3,000 differentially expressed genes found from DESEQ2 analysis in R of GFP (+) vs. Mock using cutoffs of fold change ± 3 and adj p value < 0.01. Terms listed were the five most significant across categories molecular function, biological process, and cellular components (for complete list see Table S5).
(B) Heatmaps showing 20 representative genes within the ontology classifications from (A); the values in the heatmap are the average raw read count number across GFP (+) or Mock samples.
(C) Most significantly impacted canonical pathways as determined by Ingenuity Pathway Analysis using the same gene list as in (A, see Table S6 for complete list).
(D) Heatmaps containing 20 of the most significantly upregulated genes in GFP (+) and GFP (Low) groups vs. Mock determined by adj p value and fold change. For ontology and IPA, significance is displayed as -log (adj p-value) using cutoffs stated above. Bolded genes within the heatmaps identify those used in subsequent Shield-1 experiments. Also see Figures S3, S4A–S4C, and S5 along with Tables S5 and S6.
Cell populations from HCMV-infected organoids display downregulation in critical neurodevelopmental and signaling genes
To determine which biological processes are disproportionately altered in response to infection, we began by analyzing the complete datasets using gene set enrichment analysis (GSEA) that defines significant differences between two biological states in hallmark genes defining specific biological processes. Full gene lists for each analyzed gene set and rank metrics can be found in Table S4. Compared to HCMV-infected GFP (+) cells, GSEA identified enrichment in genes upregulated in response to interferon alpha (Figure 3A). We have included a representative heatmap of 35 of the 97 hallmark genes in this category (Figure 3A). This interferon response is known to occur during HCMV infection (Boyle et al., 1999; Paulus et al., 2006). Although GFP (+) and GFP (Low) populations clustered together, the inflammatory response gene set was not significantly enriched in HCMV-infected GFP (Low) compared to Mock as it was in the GFP (+) cells, which was likely due to the lack of induction of CD47 and EIF2AK2 in the GFP (Low) samples (Figure 3A). In contrast, HCMV-infected GFP (+) cells exhibited negative correlation and dysregulation in several sets (Figure 3B). This included genes upregulated during the unfolded protein response (UPR), a subgroup of genes regulated by c-Myc, genes involved in the G2/M checkpoint, genes upregulated in response to TGFβ1, and genes upregulated by activation of the PI3K/Akt/mTOR pathway. Disruption to genes involved with c-Myc, TGFβ1, and Akt signaling in the GFP (+) population is likely related to HCMV-induced cell cycle dysregulation (Spector, 2015). We have included heatmaps of 20 genes within each hallmark set to further demonstrate that most genes were downregulated upon infection (Figure 3B). Others have demonstrated HCMV-mediated dysregulation of several of these processes in 2D cultures of primary human fibroblasts early post-infection (e.g. 4 dpi and earlier) with many of the activities dysregulated by HCMV immediate early genes (Michelson et al., 1994; Jault et al., 1995; Kudchodkar et al., 2006; Moorman et al., 2008). Our data show that HCMV-mediated disruption of c-Myc, TGFβ1, and Akt signaling is not limited to cultured fibroblasts, but also occurs in infected neural tissues and for prolonged periods of infection (14 dpi).
Figure 3.

Gene set enrichment analysis (GSEA) plots comparing infected organoid populations to mock
GSEA using the hallmark gene set on a list of all differentially expressed genes with fold change ± 3 and adj p value < 0.05.
(A) Hallmark interferon alpha response enrichment plots and corresponding heatmaps from GFP (+) vs. Mock and GFP (Low) vs. Mock, respectively.
(B) Additional enrichment plots and heatmaps (20 representative genes each) from GSEA comparing GFP (+) vs. Mock revealing significant de-enrichment in GFP (+) groups. A normalized enrichment score (NES) and normalized p value (NOM p value) for each plot are shown below; a cutoff of NOM p value < 0.05 was used to establish significance (for full GSEA analysis details see Table S4).
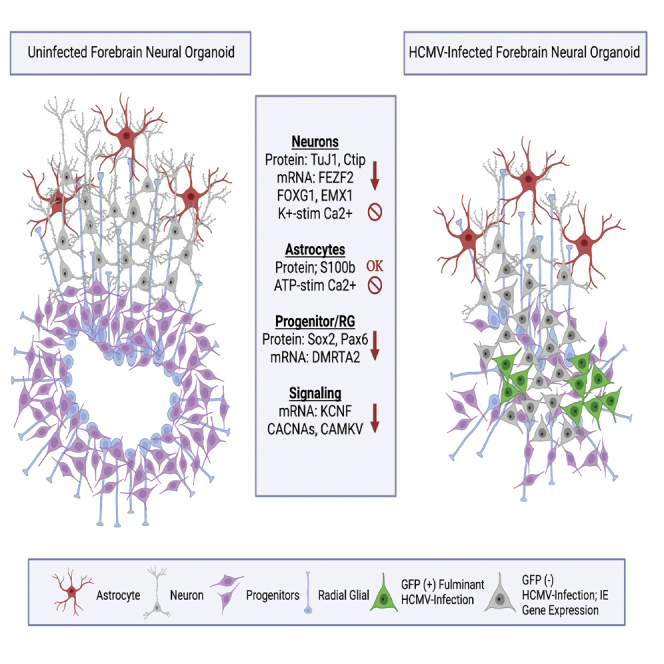
Next, we used gene ontology enrichment analysis to more precisely map specific processes that are impacted during infection. We analyzed the top 3,000 differentially expressed genes identified by DESEQ2 analysis, both increased and decreased, between cells isolated from HCMV-infected GFP (+) and Mock-infected organoids. As noted earlier, most expression was downregulated in infected populations. Using a p value cutoff of <0.01, gene ontology (GO) analysis identified significant changes in 804 terms related to biological processes, 95 molecular functions, and 201 cellular components. The top five statistically significant terms are shown in Figure 4A with full lists in Table S5 and additional enriched terms in Figure S3 (additional ontology analysis performed using DAVID is found in Figure S3). The GO terms ion signaling, cell-cell communication, and junction signaling were all significantly affected in HCMV GFP (+) cells (Figure 4B). For example, we observed a 10.9-fold reduction in expression of GJA1, 7.8-fold reduction in CACNA1C, 5.7-fold reduction in CACNA1G, 5.4-fold for CAMKV, and 5.8-fold for KCNF1 with fold changes determined by differential expression analysis of GFP (+) vs. Mock. We observed similar changes in the GFP (Low) group compared to Mock, with GJA1 reduced by 11.2-fold, CACNA1C by 4.7-fold, CACNA1G by 5.6-fold, and CAMKV by 7.1-fold (data found in Tables S1 and S2). KCNF1 was also downregulated but did not reach the level of significance in the GFP (Low) group. These data are consistent with previous studies demonstrating reduced expression of neuron-specific ion channel subunits and gap junctions (Luo et al., 2010; Khan et al., 2014) as well as our previous studies demonstrating reduced Ca2+ signaling in 2D NPC cultures and cells derived from HCMV-infected organoids (Sison et al., 2019).
We observed significant changes in the GO category nervous system development (Figure 4B). In the GFP (+) cells compared to Mock, we quantified a 9.7-fold reduction in SOX2, 8.3-fold in FOXG1, 8.4-fold in DMRTA2, 7.8-fold in EMX1, and 9.5-fold in FEZF2 (Table S1). In GFP (Low) cells, SOX2 decreased by 9.1-fold, FOXG1 by 7.5-fold, and FEZF2 by 8.5-fold while no significant changes occurred in DMRTA2 and EMX1 (Table S2). Other developmental genes that were significantly reduced in both GFP (+) and GFP (Low) include HES1, PAX6, and NEUROG1 and NEUROG2 (Figure 4B). Several of these transcription factors are decreased in in HCMV-infected NPCs (Luo et al., 2008, 2010; Li et al., 2015), and our data suggest similar effects are occurring within the context of a more complex 3D tissue model system. However, considering that these gene sets are still profoundly downregulated in the GFP (Low) populations, these data indicate that even low levels of viral transcript expression and/or microenvironment-mediated effects are sufficient to induce dramatic alterations in neurodevelopment.
The remaining most significantly enriched terms included cell cycle, cellular component organization, and RNA/protein binding (Figure 4A) with several genes spanning multiple categories. Examples of genes related to cell cycle included NPM1 and PP2R1A, to cellular organization included NEFL and DPYSL2, and to RNA-protein binding included EEF2 and RPL3 (Figure 4B). A complete list of GO terms enriched using the top 5,000 differentially expressed genes for GFP (+) and GFP (Low) vs. Mock as determined by adjusted p value of <0.01 and fold change of >5 are presented in Table S5. We observed conservation of enriched terms between GFP (+) and GFP (Low) populations including processes regulating protein expression, neuron differentiation, cellular component organization, and cell cycle.
Finally, we sought to define specific pathways using the Ingenuity Pathway Analysis tool. We analyzed the top 3,000 differentially expressed genes which defined 12 pathways as significantly altered during infection (Figure 4C). Most pathway components are downregulated and consistent with results from the GO analysis. Several of these pathways involve regulating protein translation and degradation including protein ubiquitination, signaling relating to BAG2, EIF2 signaling, regulation of EIF4 and p70S6K, and mTOR signaling (Figure S4A). Other dysregulated neurodevelopmental processes included synaptogenesis signaling pathway and Huntington disease signaling (Figure S4A). In contrast, RHOGDI signaling was one of a few pathways containing several upregulated components, and many of the unchanged genes between GFP (+) and Mock are related to GPCR signaling (Figure S4B). Finally, IPA’s core analysis tool generated a node diagram connecting regulators from the major affected canonical pathways (Figure S5). Several of these regulators fall into identified ontology classifications (TGFB1, RICTOR, NGF, and SP1) and have impacts on functional pathways that match our earlier analyses. Complete analyses from IPA comparing GFP (+) vs. Mock can be found in Table S6. We find it novel that these disruptions connect to specific neurodevelopmental processes like synaptogenesis and Huntington disease signaling that involve the UPR or require translation (Kalathur et al., 2015).
Impact of HCMV replication on cellular gene expression
The GFP (Low) cell populations isolated from infected neural organoids exhibit profound changes in host transcriptomes under conditions of significantly lower levels of HCMV IE gene expression than GFP (+). Previous studies have demonstrated that expression of HCMV IE1 disrupts SOX2 expression in a STAT3-dependent mechanism (Reitsma et al., 2013; Wu et al., 2018). Therefore, we hypothesized that expression of viral protein IE1 and/or IE2 might be necessary to downregulate other key neurodevelopmental genes identified by our RNA-seq experiments. To test their contributions, we infected cultures of NPCs using TB40r mGFP-IE-FKBP that expresses IE1 and IE2 with a destabilization domain (DD) tag in a shared exon (Glass et al., 2009). Addition of Shield-1 ligand to the culture media suppresses DD-dependent IE1 and IE2 degradation and increases virus steady-state levels (Glass et al., 2009). This strain also encodes GFP to allow for visualizing infected cells. We cultured neurosphere-derived NPCs for 3 and 7 days to mimic developmental states prior to infection. Cells were infected using TB40r mGFP-IE-FKBP at an MOI of 1.0 infectious unit per cell (IU/cell), based on titers from fibroblasts, and we added 1 μM Shield-1 ligand (Shield-1 (+)) or ethanol vehicle (Shield-1 (−)) starting at 2 hpi. We quantified changes in HCMV gene expression and observed a 85% reduction in UL123 (IE1), 80% reduction in UL122 (IE2), 60% reduction in UL44, and 75% reduction in UL99 expression in day 3 NPCs in Shield-1 (−) compared to Shield-1 (+) conditions (Figures 5A and 5B). Day 7 NPCs showed similar reductions (Figures 5C and 5D). Representative images from day 3 NPCs showed increased numbers of UL123 and GFP positive cells at 24 hpi in Shield-1 (+) versus Shield-1 (−) conditions, which increased at later time points in Shield-1 (+) (Figure 5E). We did observe several GFP and UL123-positive cells in the Shield-1 (−) condition (Figure 5E). These data indicate that in the absence of Shield-1, the onset of infection in NPCs and expression of IE1 and IE2 is substantially reduced but not eliminated. To further understand the viral proteins expressed in Shield-1 (+) and (−) conditions, we stained for viral tegument proteins pp65 and pp71 (Figures 6A and 6B). In both conditions, positive staining for both proteins are present; however, as expected, the expression is more abundant in the Shield-1 (+) group (Figures 6A and 6B). Staining for the neurodevelopmental transcription factor HES1, which has been shown to be downregulated by HCMV IE1 (Liu et al., 2017b), seemed to cause a loss of nuclear HES1 in Shield-1 (+) groups overtime. Moreover, we also observed cellular reorganization based on the presence of multinucleated cells and the formation of ring-like structures resembling replication zones in the Shield-1 (+) conditions (Figure 6B); these phenotypes were not detected in the Shield-1 (−) samples at the time points analyzed here. Together with data in Figure 5, infection in the absence of Shield-1 causes limited viral transcript and protein expression reducing the overall viral burden.
Figure 5.

Infection of NPCs with HCMV strain TB40r mGFP-IE-FKBP virus results in limited expression of viral proteins IE1 and IE2 (UL122/UL123) unless Shield-1 is administered
NPCs were infected at 3 or 7 days post plate down with TB40r mGFP-IE-FKBP at an MOI of 1. One group was then supplemented with 1 μM Shield-1 daily (Shield-1 +) while the other was given vehicle (Shield-1 -).
(A–D) Expression of HCMV viral genes (UL122 (IE), UL123 (IE), UL44 (E), and UL99 (L)) within NPCs infected 3- or 7- days post plate down at 24, 48, and 72 hours post infection (hpi) as determined by qPCR. A significant upregulation of viral transcripts was observed in (Shield-1 +) conditions compared to (Shield-1 -). Stars were assigned based on level of significance as determined by one-way ANOVA with Tukey post hoc test: ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001, and ∗∗∗∗ = p ≤ 0.00001.
(E) Immunocytochemical images at 24, 48, and 72 hpi from NPCs infected 3 days post plate down with HCMV-IE1/IE2-ddFKBP and probed for GFP (green), neurodevelopmental transcription factor SOX2 (red), UL123 (IE1) (purple), Hoescht (blue), and merge.
All images taken at 40X using Zeiss LSM980 and scale bar is 20 μm. qPCR data from 3 biological replicate experiments and error bars represent mean ± SEM.
Figure 6.

NPCs infected with TB40r mGFP-IE-FKBP and administered Shield-1 show positive staining for viral tegument proteins pp65 and pp71 by 24 hpi
NPCs were infected at 3 dpi with TB40r mGFP-IE-FKBP at an MOI of 1. One group was then supplemented with 1 μM Shield-1 daily (Shield-1 +) while the other was given vehicle (Shield-1 –), cells were fixed at noted times post infection for staining.
(A) Immunocytochemical images from the Shield-1 (−) group at 24, 48, and 72 hpi for viral tegument proteins pp65 or pp71 (purple), neurodevelopmental transcription factor HES1 (red), GFP (green), Hoechst (blue), and merge.
(B) Immunohistochemical images from the Shield (+) group using the same staining targets as (A). All images taken at 40X using Zeiss LSM980 with scale bar of 20 uM.
We next asked whether destabilizing IE1 and IE2 and limiting infection in the population might prevent downregulation of key developmental and cell signaling transcription factors observed in organoid populations. For these experiments, NPCs were infected at day 3 and analyzed for differences in cellular gene expression of nervous system developmental genes FEZF2, FOXG1, DMRTA2, and EMX1 in the Shield-1 (+) and Shield-1 (−) conditions (Figure 7). We observed significant reductions in FEZF2 and FOXG1 RNA levels by 24 hpi regardless of the presence or absence of Shield-1 (Figure 7A). Changes in DMRTA2 and EMX1 occurred later during infection at 48 or 72 hpi, respectively, and did not show a dependence on Shield-1 (Figure 7A). We next investigated the ion signaling, cell-cell communication, and junction organization genes KCNF1, CACNA1C, CAMKV, CACNA1G, and GJA1 as these genes were also reduced in our RNA-seq data (Figure 7B). We consistently observed decreases in KCNF1 and CACNA1C across time points independent of Shield addition; however, a trend toward a dependent effect was noted for CACNA1C at 48 hpi (Figure 7B). Decreases in CAMKV across time points were also observed independent of Shield-1 (Figure 7B). However, the Shield (−) group had significantly higher CAMKV expression than the Shield (+) condition at 24 and 48hpi, although this did not reach Mock levels (Figure 7B). When assessing CACNA1G and GJA1 expression in the presence and absence of Shield-1, we did identify a Shield-1-dependent effect at 24 and 48 hpi (Figure 7B). Together with our previous results (Sison et al., 2019), our data confirm that HCMV infection mediates a disruption of several key neurodevelopmental and functional genes. However, we further demonstrate that downregulation of key cortical developmental genes FEZF2, FOXG1, DMRTA2, and EMX1 is not dependent on robust expression of IE1 and IE2.
Figure 7.

Cellular gene targets are downregulated within NPCs infected with TB40r mGFP-IE-FKBP with varying dependence on Shield-1 administration
qPCRs were performed for several key neurodevelopmental transcription factors (FezF2, FOXG1, DMRTA2, and EMX1) and signaling, cell-cell communication, and junctional genes (CACNA1C, CACNA1G, CAMKV, KCNF1, and GJA1) within the HCMV-IE1/IE2-ddFKBP-infected (Shield +) and (Shield –) groups plus uninfected (Mock) NPCs at 3 days post plate down.
(A) No robust or consistent effect of Shield administration was observed for these targets; instead, genes were downregulated regardless of administration at all time points.
(B) Downregulation of KCNF1, CACNA1C, CAMKV, CACNA1G, and GJA1was observed regardless of Shield administration; however, trends toward an effect of Shield-1 administration can be observed for CACNA1C at 48 hpi or CAMKV at 72 hpi. A significant Shield-1-dependent effect was noted for genes CACNA1G and GJA1 at both 48 and 72 hpi. Stars were assigned based on level of significance as determined by one-way ANOVA with Tukey post hoc test: ∗ = p ≤ 0.05, ∗∗ = p ≤ 0.01, ∗∗∗ = p ≤ 0.001, and ∗∗∗∗ = p ≤ 0.00001. Data from 3 biological replicate experiments and error bars represent mean ± SEM.
Discussion
Stem cell-derived organoids provide a unique model system in which to study the developing human brain. Their 3D structure more closely represents the developing brain compared to 2D monolayer culture system and allows for the generation of a layered cortical structure expressing a wide variety of the markers associated with the human brain (Lancaster et al., 2013; Xu et al., 2018; Sison et al., 2019). We and others have previously demonstrated that cerebral organoids can be infected by HCMV, which substantially alters organoid structure and function (Brown et al., 2019; Sison et al., 2019; Sun et al., 2020). However, a limitation of this model is the lack of inflammatory cells, namely microglia or macrophages, within the organoids. We do find a subtle but significant enrichment in the interferon alpha response when comparing GFP (+) vs. Mock populations that is not present in the GFP (Low) vs Mock comparisons using GSEA. Studies conducted using HCMV-infected fetal brain tissues have shown a major role for the inflammatory response and a particular correlation between the amount of inflammatory infiltrate produced and severity of damage (Gabrielli et al., 2009, 2012). However, our current data and our past work demonstrate a dramatic reduction in neurodevelopmental gene transcript expression and neural function even in the absence of immune cells (Sison et al., 2019) suggesting that multiple aspects of HCMV infection are at play. Thus, a more thorough examination of how the transcriptional landscape is altered upon HCMV infection and what role viral proteins or inflammatory infiltrate play in this process is needed to define a mechanism behind HCMV’s impact on neural development.
Neurodevelopment is a highly complex process dependent on both spatial and temporal cues that activate large transcription factor networks within the NPC population. Ultimately, these cues cause NPCs to develop into a variety of terminally differentiated CNS cell types. RNA-seq analysis of day 30 organoids sorted for GFP signal strength as a proxy for the level of infection revealed downregulation of developmental transcription factors DMRTA2, FEZF2, EMX1, and FOXG1 in all populations regardless of GFP levels. These transcription factors are involved in telencephalon development and regulation of early neuronal cell fate decision making. FEZF2 is expressed in progenitor cell populations and is critical for fate specification of subcerebral projection neurons through activation of downstream transcription factor networks (Chen et al., 2008; Wang et al., 2011; Guo et al., 2013). FOXG1 knockout mice experience severe microcephaly, which can also occur during cCMV infection (Martynoga et al., 2005; Kumamoto and Hanashima, 2017). In humans, FOXG1 syndrome, caused by mutations or deletions in the long (q) arm of chromosome 14, is characterized by impaired development and structural brain abnormalities (Tohyama et al., 2011; Chiola et al., 2019). Many of these abnormalities overlap with findings in brain tissue collected from congenitally infected fetuses, such as necrosis of the cerebral cortex, degeneration of radial glia populations, and telencephalic leukoencephalopathy (Gabrielli et al., 2009; Teissier et al., 2014). DMRTA2 and EMX1 are highly expressed in the developing dorsal telencephalon where research in mice reports reduced cortex size upon mutation (Shinozaki et al., 2004; Konno et al., 2012; Young et al., 2017). In addition to these roles, several of these transcription factors are involved in key signaling pathways with wide-ranging impacts. For example, DMRTA2 is cross regulated by the Wnt/β-catenin signaling pathway, FOXG1 is associated with the SHH pathway, and FEZF2 is an upstream regulator of cTIP2 and SATB2 regulating fate decisions (Chen et al., 2008; Kuwahara et al., 2010; Lui et al., 2011). Interestingly, despite no overt indication of active viral replication, the GFP (Low) population exhibits similar transcriptional effects as the GFP (+) and GFP (Inter) populations. This result suggests that viral infection has more widespread consequences that likely go beyond active viral replication. For example, the early downregulation of FEZF2 and FOXG1 transcript expression (Figure 7A) may directly or indirectly regulate expression of DMRTA2 and EMX1 such that their later downregulation (Figure 7A) is not related to the infection process but rather to misregulation of neurogenic networks. Neurodevelopmental literature indicates the importance of FEZF2 expression early in the neural differentiation process, as its expression identifies multipotent neural progenitors in humans, regulates telencephalic precursor differentiation in mouse embryonic stem cells, and controls multilineage neuronal differentiation in zebrafish through bHLH domains and homeodomain genes (Wang et al., 2011; Yang et al., 2012; Guo et al., 2013). Thus, it remains possible that HCMV-mediated downregulation to FEZF2 expression could have trickle down effects on transcription factors like FOXG1 and DMRTA2 that are responsible for specifying the dorsal and ventral telencephalic regions and generating the cerebral cortex, respectively (Martynoga et al., 2005; Konno et al., 2012). Although further studies are needed to elucidate the mechanisms underlying the global effect on the transcriptional profile, disruption of transcriptional interactions, cell-cell communication processes, spread of viral proteins, or distribution of tegument proteins that disrupt cellular differentiation and function could all be potential mechanisms.
These studies support the observations that NPCs exhibit impaired signaling, differentiation, and even apoptosis following HCMV infection (Luo et al., 2010; Brown et al., 2019; Sison et al., 2019; Sun et al., 2020). NPCs are the cell type most permissive to infection (Luo et al., 2008) and found abundantly within cultured cerebral organoids. They have also been found to comprise the largest proportion of HCMV-positive cells within infected human fetal brain tissue (Gabrielli et al., 2009). NPCs form early developmental structures and differentiate into a wide range of cell types in the human brain. Previous work has shown that IE1 sequesters unphosphorylated STAT3 in the nucleus and contributes to the reduction in SOX2 expression in HCMV-infected NPCs (Wu et al., 2018). However, little else is known about how HCMV downregulates other key neurodevelopmental targets involved in corticogenesis. Data from fibroblast cultures indicate the ability of HCMV infection to transcriptionally regulate host genes as well as degrade their protein products (Cohen and Stern-Ginossar, 2014; Khan et al., 2014). Further, overexpression of IE1 or IE2 alone in healthy fibroblasts infected with mutated virus unable to transcribe either major IE gene demonstrated rescue in gene targets previously downregulated by infection (Luo et al., 2010; Khan et al., 2014; Adamson and Nevels, 2020). Therefore, we used a conditional approach to assess the role of IE1 and IE2 on additional neurodevelopmental gene expression. Initially, infections were conducted at 7 days post plate down; however, we observed a lack of viral protein replication and low expression of viral proteins (Figures 5A and 5B). These results were not entirely unexpected as other groups have reported that NPC differentiation is inversely correlated to infectability (Odeberg et al., 2006; Gonzalez-Sanchez et al., 2015). While our experiments using HCMV TB40r mGFP-IE-FKBP support this idea, our data also show that infection at 7 days post plate down still results in transcript-level expression of several viral genes (Figures 5A and 5B). When focusing on the role of IE1 and IE2 expression in the 3-day NPCs, neurodevelopmental and signaling targets were clearly downregulated (Figure 7). These data suggest that transcriptional downregulation was largely independent of major IE protein expression, which further highlights the ability of other viral components and/or transcriptional networks to impact transcription and translation in infected NPCs. Staining for viral tegument proteins pp65 and pp71 (Figures 6A and 6B) did reveal expression in both Shield-1 (+) and (−) infection conditions similar to previous findings in fibroblasts (Pan et al., 2016). This finding is consistent with the role of IE1/IE2 in transactivating early and late viral gene expression and align with the small but detectable amount of IE1/IE2 transcripts present in the Shield-1 (−) condition (Figures 5A and 5B). Therefore, the presence of these transcripts and observable tegument proteins might be contributing to the gene target downregulation observed among in the Shield-1 (−) group.
Previous studies from us and others have shown that HCMV infection disrupts calcium and potassium signaling in infected NPCs and organoids (Brown et al., 2019; Sison et al., 2019; Sun et al., 2020). We therefore hypothesized that these functional deficits could be due to downregulation of channel encoding genes responsible for ion transport and maintaining membrane potential. Several such targets showed downregulation within the GFP (+) and GFP (Inter) groups (Figure 4D, Table S1). CACNA1C and CACNA1G are genes that encode unique voltage-gated calcium channel subtypes. CACNA1G expression gives rise to T-type calcium channels that belong to the low-voltage-activated subgroup. Recently, mutations in CACNA1G have been shown to be associated with early onset cerebellar ataxia and epilepsy (Calhoun et al., 2016; Barresi et al., 2020). The gene CACNA1C encodes a critical subunit leading to the formation of L-type calcium channels. L-type channels have a prominent role in controlling gene expression through coupling membrane depolarization with cAMP response element-binding protein (CREB) phosphorylation via local Ca2+/calmodulin-dependent protein kinase II (CAMKII) signaling; interestingly, both CAMKIIB and Calmodulin 1/3 (CALM1/3) are downregulated following infection (Zhang et al., 2005; Liang et al., 2016), thus limiting the ability of CREB to act as the transcriptional regulator of nearly 4,000 downstream gene targets (Wheeler et al., 2012; Chiola et al., 2019). In mice, it has been demonstrated that reduced expression of CACNA1C during development leads to a reduction in neurogenesis (Lee et al., 2016; Moon et al., 2018). These experiments demonstrate that altered expression of either CACNA1G or CACNA1C can cause phenotypes similar to those associated with cCMV infection. CAMKV, while not involved in the activation of CREB, is a Ca2+/calmodulin pseudo kinase that is required for the activity-dependent maintenance of dendritic spines (Saneyoshi et al., 2010; Liang et al., 2016). It is unsurprising that HCMV-infected organoids and NPCs that display decreased neuronal firing and ability to transport calcium have CAMKV deficits as many of these processes are cross regulated (Odeberg et al., 2006; Brown et al., 2019; Sison et al., 2019; Sun et al., 2020). Meanwhile, several potassium channel-encoding genes were downregulated in the GFP (+) and GFP (Inter) groups. These genes are important for potassium ion transport leading to neurotransmitter release and neural excitation. KCNF1, which encodes a member of the voltage-gated potassium channel subfamily F, was particularly interesting because previous studies have shown it to be downregulated upon HCMV infection in NPCs (Luo et al., 2010; Young et al., 2011; Brown et al., 2019), and mutations in it and other family members have been linked to seizures and epilepsy, phenotypes of cCMV infection (Kohling and Wolfart, 2016).
Finally, GJA1, which encodes the protein connexin-43, was downregulated in both GFP (+) and GFP (Inter) groups compared to mock (Figure 4B and Tables S1 and S2). In addition to ion channels and calcium sensors, NPCs rely on gap and tight junctions to communicate with each other through intercellular ion transport (Wei et al., 2004; Goodenough and Paul, 2009; Zhou and Jiang, 2014). This type of communication can inform neural cell fating by cuing NPCs to continue proliferating or to begin differentiating (Lemcke and Kuznetsov, 2013; Zhou and Jiang, 2014). There is some evidence to suggest that HCMV hijacks the existing junctional machinery to spread viral material (Silva et al., 2005; Cohen and Stern-Ginossar, 2014; Khan et al., 2014). In HCMV-infected fibroblasts, there is clear evidence that expression of IE genes specifically leads to decreased expression of GJA1 and GJC1 (Luo et al., 2008; Khan et al., 2014). Intriguingly, our data suggest that this is also the case in NPCs (Figure 7). As such, we postulate that remodeling of the cell-cell communication network could be a way by which the virus impacts both signaling and development in the host and promotes cell-to-cell viral spread early in infection.
Despite the differences in viral strain, viral titer, and 2D vs. 3D culture models, the Shield-1 (−) NPCs and isolated GFP (Low) organoid cells have some interesting similarities. Previous studies show that the TB40r mGFP-IE-FKBP construct maintains a low level of viral transcript expression (Pan et al., 2016; Rak et al., 2018); however, our data suggest that this level of expression is likely not sufficient to cause full replication (Figure 5A and 5B). Similarly, IE viral transcripts were still significantly increased in the isolated GFP (Low) cells compared to mock conditions despite a lack of robust GFP expression (Figures 1C and 1D). It is possible that if the GFP (Low) cells were given more time in culture, GFP expression and E-L and L viral transcripts would be more evident. Nevertheless, significant downregulation of several gene targets was still observed in the absence of robust viral replication (Figures 2, 3, and 6) suggesting that the presence of some viral transcripts and/or viral material is sufficient to induce significant gene expression changes in neural tissues. These findings are supported by in vivo tissue studies in which infected fetal brains displayed necrosis of cerebral tissue, radial glia degeneration, and hypertrophic cell morphology even in brain regions with low HCMV positivity and immune infiltration (Teissier et al., 2014).
Taken together, we demonstrate that neurodevelopmental gene networks and critical neural signaling pathways can still be downregulated under conditions of HCMV infection where IE1 and IE2 protein levels are reduced indicating that other HCMV-related mechanisms are involved. Therefore, these data suggest that therapeutics designed to solely limit viral replication and/or viral gene and protein expression may be insufficient to impact the widespread neural differentiation and functional deficits induced by cCMV infection.
Limitations of the study
Owing to the unavailability of tissue samples from fetuses infected with HCMV and species-specific strain differences, we were limited to using iPSC models to model and test the impact of HCMV infection on human brain development. The cerebral organoid system, while having many advantages, does lack macrophages and microglial cell populations that have both been shown to exhibit major cellular responses to HCMV infection. As such, our results miss the contributions that these cells may make on the overall phenotype. We also would like to acknowledge that while the HCMV strain TB40r mGFP-IE-FKBP is a valuable tool to limit IE1 and IE2 expression, it does not fully eliminate expression. Thus, additional approaches are necessary to definitively elucidate the role of IE1 and IE2 protein expression in neural cell transcriptional downregulation. Although we identified multiple cellular pathways that were dysregulated upon HCMV infection in human neural cells, the downstream mechanisms and developmental implications as well as the role of specific viral genes need further study.
STAR★Methods
Key resources table
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Rabbit polyclonal anti HES1 | Invitrogen | PA5-28802 |
| Chicken polyclonal anti Tuj1 | GeneTex | GTX85469 |
| Mouse monoclonal anti UL44 | Virusys | CA006-100 |
| Mouse monoclonal anti IE1 | Shenk Lab | Clone 1B12 |
| Mouse monoclonal anti IE2 | Shenk Lab | Clone 3A9 |
| Mouse monoclonal anti pp28 | Shenk Lab | Clone 10B4-29 |
| Mouse monoclonal anti pp65 | Shenk Lab | Clone |
| Mouse monoclonal anti pp71 | Shenk Lab | Clone 2M10-9 |
| Rabbit polyclonal anti SOX2 | Millipore Sigma | AB5603 |
| Bacterial and virus strains | ||
| TB40/EeGFP | Goodrum Lab | |
| TB40r mGFP-IE-FKBP BAC | Borst Lab | |
| Chemicals, peptides, and recombinant proteins | ||
| Shield-1 | Aobious | AOB1848 |
| Human EGF | Miltenyi Biotec | 130-097-749 |
| Human bFGF | StemCell Technologies | 78003 |
| Heparin | Millipore Sigma | H5152 |
| Critical commercial assays | ||
| RNA Screentape | Agilent | 5067-5576 |
| NEBNext Poly(A) mRNA Magnetic Isolation Module | New England Biolabs | E7490L |
| NEBNext Library Quant Kit for Illumina | New England Biolabs | E7630L |
| DNA Screentape | Agilent | 5067-5582 |
| NextSeq PhiX Control Kit | Illumina | FC-110-3002 |
| NextSeq 500/550 High Output Kit v2.5 (75 Cycles) | Illumina | 20024906 |
| Reverse Transcription Kit | Promega | A3500 |
| Pierce BCA | Thermo Fisher Scientific | 23225 |
| STEMdiff Cerebral Organoid Kit | StemCell Technologies | 08570 |
| STEMdiff Cerebral Organoid Maturation Kit | StemCell Technologies | 08571 |
| DNeasy Blood and Tissues Kit | Qiagen | 69504 |
| Power SYBR Green PCR Master Mix | Thermo Fisher Scientific | 4367659 |
| Deposited data | ||
| Manuscript - BioRxiv | This paper | https://doi.org/10.1101/2021.08.01.45465 |
| Raw and Analyzed Data - GEO | This paper | ID# GSE193759 |
| Experimental models: Cell lines | ||
| Human iPSC WT 4.2 Line | ||
| Human iPSC WT 21.8 Line | ||
| Oligonucleotides | ||
| Primers for XX, see Table S7 | This paper | N/A |
| Software and algorithms | ||
| Ingenuity Pathway Analysis | Qiagen | https://qiagen.pathfactory.com |
| Basepair Tech | https://www.basepairtech.com | |
| Gene Set Enrichment Analysis | UC San Diego - Broad Institute | https://www.gsea-msigdb.org/gsea/index.jsp |
| Flowjo | https://www.flowjo.com | |
Resource availability
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Dr. Allison Ebert (aebert@mcw.edu).
Materials availability
This study did not generate new unique reagents.
Experimental model and subject details
Neural progenitor culture and infection
MRC-5 fibroblasts were cultured in Dulbecco’s modified Eagle medium (DMEM) (ThermoFisher Scientific) containing 7% fetal bovine serum (FBS) (Atlanta Biologicals) and 1% penicillin-streptomycin (ThermoFisher Scientific). Virus stocks were prepared by infecting MRC-5 fibroblasts (ATCC) with HCMV strain TB40/E encoding EGFP (Umashankar et al., 2011). Cell culture medium was collected and pelleted through a sorbitol cushion (20% sorbitol, 50 mM Tris-HCl, pH 7.2, 1 mM MgCl2) at 55,000 × g for 1 h in a Sorvall WX-90 ultracentrifuge and SureSpin 630 rotor (ThermoFisher Scientific). The TB40r mGFP-IE-FKBP BAC was kindly provided by E. Borst (unpublished). The virus was cultured in the presence of 1 μM Shield-1 (AOBIUS #AOB1848) and replaced every 24 h. Titers of viral stocks were determined by a limiting dilution assay with the 50% tissue culture infectious dose (TCID50) in MRC-5 cells in a 96-well dish. At 2 weeks post infection, HCMV IE1-positive cells were counted to determine viral titers, reported as the number of infectious units (IU) per milliliter. IE1-positive cells were determined using a mouse anti-HCMV IE1 (UL123) antibody (clone 1B12; generously provided by Tom Shenk, Princeton University, Princeton, NJ).
Two independent iPSC lines were used in organoid differentiations (4.2 and 21.5/8), meanwhile neural progenitor cells (NPC) were differentiated from the 4.2 line alone (Ebert et al., 2013). The iPSCs were grown and maintained in Essential 8 Media (ThermoFisher Scientific) and were cultured in feeder-free conditions on Matrigel (Corning). NPCs were differentiated and maintained as neurospheres (EZ spheres) in Stemline (Millipore Sigma) supplemented with 0.5% N-2 supplement (ThermoFisher Scientific), 100 ng/mL EGF (Miltenyi Biotech), 100 ng/mL fibroblast growth factor (FGF; Stem Cell Technologies), and 5 ug/mL heparin (Millipore Sigma) as described previously (Ebert et al., 2013). Plated NPCs were grown in Neurobasal medium (ThermoFisher Scientific) supplemented with 2% B-27 (ThermoFisher Scientific) and 1% antibiotic-antimycotic (ThermoFisher Scientific). Cells were plated at 1.0 × 104 cells per well of a 24-well plate onto Matrigel coated coverslips. Matrigel was diluted in DMEM, placed on coverslips for approximately 12 h, and aspirated off prior to plating cells.
Cerebral organoid culture and infection
Cerebral organoid cultures were differentiated from iPSCs according to the specification of the cerebral organoid kit from StemCell Technologies (#08570) that relies on an established protocol (Lancaster et al., 2013). These organoids were made from either of the two independent WT stem cell lines (4.2 and 21.5/8), Infected 1 was derived from the 4.2 line, Infected 2 and 3 were derived from the 4.2 line, Mock 1, 2, and 3 were derived from the 4.2 line, and Mock 4 was derived from the 21.8 line. (Ebert et al., 2013). Briefly, iPSCs were seeded at 9 × 103 cells per well onto 96-well ultralow attachment plates for embryoid body (EB) formation and grown in EB formation media (StemCell Technologies). At day 5, the induction of neural epithelium was initiated by moving the EBs into an ultralow attachment 24-well plate where they were then fed with induction media (StemCell Technologies). On day 7, neural tissues were embedded in Matrigel droplets and moved to ultralow attachment 6-well plates and fed expansion media (Stemcell Technologies). Then at day 10, the plate of developing organoids was transferred to a rocker which elicits the circulation of nutrients and prevents organoids from sticking to the dish. From this point on the organoids were fed maturation media (StemCell Technologies) every 3 days. At day 30, organoids were weighed and infected with 500 infectious units/ug HCMV strain TB40/E encoding GFP for 2 h then virus was removed, and fresh media added (Collins-McMillen et al., 2018; Rak et al., 2018; Collins-McMillen et al., 2019). Media was then changed every 3–4 days. At 14 days post infection, organoids were either fixed for cryosectioning, prepped for FACs sorting, or dissociated in accutase enzyme and lysed for protein or RNA isolation. Three organoids containing approximately 1-2 million cells each were combined prior to sorting to ensure sufficient cell numbers in each population for whole transcriptome analysis.
Method details
Flow cytometry, immunohistochemistry, and Western blot
Organoids were dissociated using the enzyme accutase at 37°C for 15–20 min, and then washed in PBS. Organoid sorting buffer (1% FBS and 2 mM EDTA in DPBS) was then added to the tube and gently blown at the organoid until it completely dissociated. This cell suspension was then filtered to remove any remaining cell clumps. Finally, the filtered suspension was placed on ice until ready for sorting. Fluorescence Activated Cell Sorting (FACs) was performed on BD FACS Diva 8.0.1. Live cell gating was established using the forward and side scatter plots from an uninfected organoid. Infected organoids were then further sorted based on GFP fluorescence signal into GFP (+), GFP (Inter), or GFP (Low) that were consistent across replicates. Gating analysis and plots were generated using Flowjo.
Neural progenitor cells were fixed on coverslips with 4% paraformaldehyde (PFA) for 20 min at 4°C, washed with phosphate-buffered saline (PBS), and placed in PBS. For immunohistochemistry, coverslips were blocked with 5% normal donkey serum (S30; Sigma) and 0.1% Triton in PBS for 30 min, incubated in primary antibodies overnight at 4°C, and incubated in secondary antibodies for 1 h at room temperature. The nuclear stain Hoechst was used to label nuclei. Primary antibodies used were HES1 (rabbit, PA5-28802; Thermo Fisher), IE1 (UL123) (mouse, 1:1000, clone 1B12, Shenk Lab), SOX2 (rabbit, AB5603; Millipore Sigma), pp65 (mouse, 1:500, clone, Shenk Lab), pp71 (mouse, 1:500, clone 2M10-9, Shenk Lab) and Tuj1 (chicken, GTX85469; GeneTex). Species-appropriate fluorescent secondary antibodies were used. An upright TS100 Nikon fluorescence microscope and NIS Elements were used for imaging and analysis.
RNA sequencing, qRT-PCR, and viral DNA quantification
Total RNA was isolated from organoid samples following FACs sorting for live cells and GFP expression. To insure a high enough RNA concentration, 3 infected organoids were combined for each N. After isolation RNA was analyzed on the tape station using screen tape from Agilent (#5067-5576) for quantity and quality. External RNA Controls Consortium (ERCC) spike-in control from Invitrogen (#4456740) was added to each sample to minimize external sources of variability. cDNA libraries were then generated for each sample using the NEBNext Poly(A) mRNA Magnetic Isolation Module (NEB #E7490). Libraries were then analyzed by qPCR generating CT values using NEBNEXT Library Quant Kit (#E7630) and by tape station with D1000 screen tape from Agilent (#5067-5582) to calculate fragment base pair size. These measures were then analyzed by the NEB bio-calculator tool to get an estimated library concentration. Libraries were then diluted to 4 nM and pooled. Another qPCR was run with the pooled library to confirm concentration and a new dilution was performed if necessary. The diluted pooled library (25 pM) was then combined with diluted NEXTSeq PhiX control (#FC-110-3002). The PhiX control and library were then combined and loaded into the cartridge. Sequencing was performed using the NextSeq 500/550 High Output Kit v2.5 (#20024906) from Illumina. Preliminary analysis was then performed using the online platform Basepair Technologies.
Total RNA was isolated, and reverse transcribed into cDNA using the Promega RT Kit (#A3500). q-RT-PCR was performed using specific primer sequences as outlined in (Table S7). The resulting CT values for each technical replicate were averaged and the CT average of the gene of interest was subtracted from the average CT of the GAPDH calculating the delta CT. A reference sample was then set (the uninfected group was used for this purpose except when assessing viral transcripts in which case the sample hypothesized to express the highest level of viral transcripts was selected). This reference/control delta CT value once calculated was used to calculate the delta delta CT value for the rest of the samples via subtraction. Following calculation of the delta delta CT value, the value was multiplied by a common normalization factor and plotted as a fold change.
DNA was isolated from organoid samples following FACs sorting for live cells and GFP expression using Qiagen DNeasy Blood and Tissue Kit (69504). DNA yield from three independent organoids was then combined to boost concentration as with the RNA isolation. After DNA isolation qPCR was run for housekeeping gene GAPDH and HCMV gene UL123 to estimate total viral genomes within our GFP (Low), GFP (Inter), and GFP (+) sorted populations. Relative expression was calculated using a five-point standard curve made from dilutions of our GFP (+) cells (group with highest viral gene expression). Once the relative expression values were determined for each sample, we divided the GAPDH by UL123 and plotted this ratio as relative host/viral genomes.
Quantification and statistical analysis
Statistical analysis
All statistical analysis was performed using Graph Pad Version 9. Data were collected from 2 independent organoid differentiations and 1 set of infections for bulk RNA-seq experiments. For experiments conducted using NPCs, 3 biological replicates were included for all qPCRs and data was analyzed by one-way ANOVA or Student’s T-test as appropriate with Tukey post hoc test for qPCR. p < 0.05 was considered significant (details on qPCR quantification method can be found above).
Bioinformatic analyses
Fastq files were exported from Illumina sequence hub following sequencing then optical duplicates were removed and reference genome created using the raw fasta sequences for hg19. The raw RNA Seq reads are mapped and quantified to combined reference genome using Salmon v1.4.0. The read count matrix for all transcripts were utilized in R to make PCA plots and to run differential expression analysis. DESeq2 in R was used to perform the comparisons against mock samples to get significant up regulated and down regulated genes at adjusted p value < 0.05 and fold change of 3. Basepair Tech software and expression count analysis of the trimmed reads was conducted using STAR. Differentially expressed genes were identified by DESeq2 analysis using cutoffs of adjusted p-value < 0.05 and log2 fold change of ± 3. Gene ontology analysis was subsequently performed using G-profiler and DAVID. Additional RNA seq analysis was performed using ingenuity pathway analysis from Qiagen and Gene Set Enrichment Analysis tool from UC San Diego and the Broad Institute. For both G-profiler and IPA analysis a list of the 3,000 most differentially expressed genes across samples analyzed were used. All these genes met the threshold of adj p value < 0.01 and log2 fold change ±9 as determined by DESeq2 analysis. For GSEA analysis the entire list of differentially expressed genes identified by DESeq2 meeting the adj p value < 0.05 and log2 fold change of ±3 cutoffs were used.
Gene set enrichment analysis was performed using GSEA version 3.0 software with default parameters, classic enrichment, and phenotype permutations at 1000 times. GSEA was performed on differentially expressed genes determined from DESeq2 analysis of GFP (+) vs. Mock and GFP (Low) vs. Mock samples using genes that met the cutoff of adj p value of <0.05 and log2 fold change ±3. The enrichment plots displayed in Figure 3 were generated from analysis of GFP (+) vs. Mock or GFP (Low) vs. Mock using the software’s hallmark gene set.
Acknowledgments
The authors would like to thank Tom Shenk for providing antibodies against HCMV proteins and Eva Borst for providing the TB40r mGFP-IE-FKBP virus. We thank Benedetta Bonacci in the Versiti-Blood Research Institute Flow Cytometry Core for technical assistance and Emma Thomas for the graphical abstract. Creation of the graphical abstract was supported by the VI4-ArtLab Artist-in-Residence program. Finally, we thank members of the Terhune, Hudson, Rao, and Ebert laboratories for helpful discussions and input on the project. Research reported in this publication was supported by the National Institute of Allergy and Infectious Diseases division of the National Institutes of Health under award number R01AI132414 (S.S.T. and A.D.E.) and the National Cancer Institute division of the National Institutes of Health under award number R01CA2042031 (S.R.). The content is solely the responsibility of the authors and does not necessarily represent the official views of the National Institutes of Health.
Author contributions
B.S.O., R.L.M., S.S.T., and A.D.E. were responsible for study design. B.S.O., R.L.M., M.L.S., K.P., S.R., S.S.T., and A.D.E. conducted experiments, analyzed data, and/or interpreted data. S.R., S.S.T., and A.D.E. provided resources. B.S.O., R.L.M., and K.P. contributed to writing the manuscript. B.S.O., R.L.M., M.L.S., S.S.T., and A.D.E. wrote and edited the final manuscript.
Declaration of interests
All authors declare no conflicts of interests related to the manuscript.
Published: April 15, 2022
Footnotes
Supplemental information can be found online at https://doi.org/10.1016/j.isci.2022.104098.
Supplemental information
Total of 19897 significant DE genes were found, of which 1733 were upregulated (9%) and the remaining 18164 were downregulated (91%). 2882 genes were unchanged, adj p-value > 1. Additional analysis details found in S1 or methods.
20271 significant DE genes were found with 1384 upregulated (7%) and 18887 downregulated (93%). 1687 genes were unchanged, adj p-value > 1. Additional analysis details found in S2 or methods.
31 significant DE genes were found, all of which were downregulated. 5,000 genes remained unchanged; adj p-value > 1. Additional analysis details found in S3 or methods.
All comparisons were made relative to Mock, and each list contain all significant differentially expressed genes as revealed by DESeq2. Additional analysis details found in S4 or methods and reference Figure 3.
Results based on list of most significantly differentially expressed genes as determined by adj p-value for GFP (+) vs. Mock in reference to Figure 4. Additional analysis detail in S5 and methods.
A complete list of canonical pathways, upstream regulators, and related disease or functional outcomes identified by IPA analysis of GFP (+) vs. Mock shown in Figure 4. Additional analysis details found in S6 or methods.
Data and code availability
Bulk RNA-seq data have been deposited at GEO (GEO: GSE193759) and the preprint manuscript has been made publicly available on BioRXiv as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
References
- Adamson C.S., Nevels M.M. Bright and early: inhibiting human cytomegalovirus by targeting major immediate-early gene expression or protein function. Viruses. 2020;12:110. doi: 10.3390/v12010110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Altman A.M., Mahmud J., Nikolovska-Coleska Z., Chan G. HCMV modulation of cellular PI3K/AKT/mTOR signaling: new opportunities for therapeutic intervention? Antivir. Res. 2019;163:82–90. doi: 10.1016/j.antiviral.2019.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barresi S., Dentici M.L., Manzoni F., Bellacchio E., Agolini E., Pizzi S., Ciolfi A., Tarnopolsky M., Brady L., Garone G., et al. Infantile-onset syndromic cerebellar ataxia and CACNA1G mutations. Pediatr. Neurol. 2020;104:40–45. doi: 10.1016/j.pediatrneurol.2019.09.005. [DOI] [PubMed] [Google Scholar]
- Boyle K.A., Pietropaolo R.L., Compton T. Engagement of the cellular receptor for glycoprotein B of human cytomegalovirus activates the interferon-responsive pathway. Mol. Cell. Biol. 1999;19:3607–3613. doi: 10.1128/mcb.19.5.3607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Britt W.J., Prichard M.N. New therapies for human cytomegalovirus infections. Antivir. Res. 2018;159:153–174. doi: 10.1016/j.antiviral.2018.09.003. [DOI] [PubMed] [Google Scholar]
- Brown R.M., Rana P., Jaeger H.K., O'Dowd J.M., Balemba O.B., Fortunato E.A. Human cytomegalovirus compromises development of cerebral organoids. J. Virol. 2019;93:e00957-19. doi: 10.1128/JVI.00957-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Calhoun J.D., Hawkins N.A., Zachwieja N.J., Kearney J.A. Cacna1g is a genetic modifier of epilepsy caused by mutation of voltage-gated sodium channel Scn2a. Epilepsia. 2016;57:E103–E107. doi: 10.1111/epi.13390. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chambers J., Angulo A., Amaratunga D., Guo H., Jiang Y., Wan J.S., Bittner A., Frueh K., Jackson M.R., Peterson P.A., et al. DNA microarrays of the complex human cytomegalovirus genome: profiling kinetic class with drug sensitivity of viral gene expression. J. Virol. 1999;73:5757–5766. doi: 10.1128/jvi.73.7.5757-5766.1999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen B., Wang S.S., Hattox A.M., Rayburn H., Nelson S.B., McConnell S.K. The Fezf2-Ctip2 genetic pathway regulates the fate choice of subcortical projection neurons in the developing cerebral cortex. Proc. Natl. Acad. Sci. U S A. 2008;105:11382–11387. doi: 10.1073/pnas.0804918105. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chiola S., Do M.D., Centrone L., Mallamaci A. Foxg1 overexpression in neocortical pyramids stimulates dendrite elongation via Hes1 and pCreb1 upregulation. Cereb. Cortex. 2019;29:1006–1019. doi: 10.1093/cercor/bhy007. [DOI] [PubMed] [Google Scholar]
- Cohen Y., Stern-Ginossar N. Manipulation of host pathways by human cytomegalovirus: insights from genome-wide studies. Semin. Immunopathol. 2014;36:651–658. doi: 10.1007/s00281-014-0443-7. [DOI] [PubMed] [Google Scholar]
- Collins-McMillen D., Buehler J., Peppenelli M., Goodrum F. Molecular determinants and the regulation of human cytomegalovirus latency and reactivation. Viruses. 2018;10:444. doi: 10.3390/v10080444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Collins-McMillen D., Rak M., Buehler J.C., Igarashi-Hayes S., Kamil J.P., Moorman N.J., Goodrum F. Alternative promoters drive human cytomegalovirus reactivation from latency. Proc. Natl. Acad. Sci. U S A. 2019;116:17492–17497. doi: 10.1073/pnas.1900783116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- D'Aiuto L., Di Maio R., Heath B., Raimondi G., Milosevic J., Watson A.M., Bamne M., Parks W.T., Yang L., Lin B., et al. Human induced pluripotent stem cell-derived models to investigate human cytomegalovirus infection in neural cells. PLoS One. 2012;7:e49700. doi: 10.1371/journal.pone.0049700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ebert A.D., Shelley B.C., Hurley A.M., Onorati M., Castiglioni V., Patitucci T.N., Svendsen S.P., Mattis V.B., McGivern J.V., Schwab A.J., et al. EZ spheres: a stable and expandable culture system for the generation of pre-rosette multipotent stem cells from human ESCs and iPSCs. Stem Cell Res. 2013;10:417–427. doi: 10.1016/j.scr.2013.01.009. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gabrielli L., Bonasoni M.P., Lazzarotto T., Lega S., Santini D., Foschini M.P., Guerra B., Baccolini F., Piccirilli G., Chiereghin A., et al. Histological findings in foetuses congenitally infected by cytomegalovirus. J. Clin. Virol. 2009;46:S16–S21. doi: 10.1016/j.jcv.2009.09.026. [DOI] [PubMed] [Google Scholar]
- Gabrielli L., Bonasoni M.P., Santini D., Piccirilli G., Chiereghin A., Petrisli E., Dolcetti R., Guerra B., Piccioli M., Lanari M., et al. Congenital cytomegalovirus infection: patterns of fetal brain damage. Clin. Microbiol. Infect. 2012;18:E419–E427. doi: 10.1111/j.1469-0691.2012.03983.x. [DOI] [PubMed] [Google Scholar]
- Glass M., Busche A., Wagner K., Messerle M., Borst E.M. Conditional and reversible disruption of essential herpesvirus proteins. Nat. Methods. 2009;6:577–579. doi: 10.1038/nmeth.1346. [DOI] [PubMed] [Google Scholar]
- Gonzalez-Sanchez H.M., Monsivais-Urenda A., Salazar-Aldrete C.A., Hernandez-Salinas A., Noyola D.E., Jimenez-Capdeville M.E., Martinez-Serrano A., Castillo C.G. Effects of cytomegalovirus infection in human neural precursor cells depend on their differentiation state. J. Neurovirol. 2015;21:346–357. doi: 10.1007/s13365-015-0315-5. [DOI] [PubMed] [Google Scholar]
- Goodenough D.A., Paul D.L. Gap junctions. Cold Spring Harb. Perspect. Biol. 2009;1:a002576. doi: 10.1101/cshperspect.a002576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Griffiths P., Baraniak I., Reeves M. The pathogenesis of human cytomegalovirus. J. Pathol. 2015;235:288–297. doi: 10.1002/path.4437. [DOI] [PubMed] [Google Scholar]
- Guo C., Eckler M.J., McKenna W.L., McKinsey G.L., Rubenstein J.L.R., Chen B. Fezf2 expression identifies a multipotent progenitor for neocortical projection neurons, astrocytes, and oligodendrocytes. Neuron. 2013;80:1167–1174. doi: 10.1016/j.neuron.2013.09.037. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jault F.M., Jault J.M., Ruchti F., Fortunato E.A., Clark C., Corbeil J., Richman D.D., Spector D.H. Cytomegalovirus infection induces high levels of cyclins, phosphorylated Rb, and p53, leading to cell cycle arrest. J. Virol. 1995;69:6697–6704. doi: 10.1128/jvi.69.11.6697-6704.1995. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kalathur R.K., Giner-Lamia J., Machado S., Barata T., Ayasolla K.R., Futschik M.E. The unfolded protein response and its potential role in Huntington's disease elucidated by a systems biology approach. F1000Research. 2015;4:103. doi: 10.12688/f1000research.6358.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Khan Z., Yaiw K.C., Wilhelmi V., Lam H., Rahbar A., Stragliotto G., Soderberg-Naucler C. Human cytomegalovirus immediate early proteins promote degradation of connexin 43 and disrupt gap junction communication: implications for a role in gliomagenesis. Carcinogenesis. 2014;35:145–154. doi: 10.1093/carcin/bgt292. [DOI] [PubMed] [Google Scholar]
- Kohling R., Wolfart J. Potassium channels in epilepsy. Cold Spring Harb. Perspect. Med. 2016;6:a02287. doi: 10.1101/cshperspect.a022871. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Konno D., Iwashita M., Satoh Y., Momiyama A., Abe T., Kiyonari H., Matsuzaki F. The mammalian DM domain transcription factor Dmrta2 is required for early embryonic development of the cerebral cortex. PLoS One. 2012;7:e46577. doi: 10.1371/journal.pone.0046577. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kudchodkar S.B., Yu Y., Maguire T.G., Alwine J.C. Human cytomegalovirus infection alters the substrate specificities and rapamycin sensitivities of raptor- and rictor-containing complexes. Proc. Natl. Acad. Sci. U S A. 2006;103:14182–14187. doi: 10.1073/pnas.0605825103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kumamoto T., Hanashima C. Evolutionary conservation and conversion of Foxg1 function in brain development. Dev. Growth Differ. 2017;59:258–269. doi: 10.1111/dgd.12367. [DOI] [PubMed] [Google Scholar]
- Kuwahara A., Hirabayashi Y., Knoepfler P.S., Taketo M.M., Sakai J., Kodama T., Gotoh Y. Wnt signaling and its downstream target N-myc regulate basal progenitors in the developing neocortex. Development. 2010;137:1035–1044. doi: 10.1242/dev.046417. [DOI] [PubMed] [Google Scholar]
- Lancaster M.A., Renner M., Martin C.A., Wenzel D., Bicknell L.S., Hurles M.E., Homfray T., Penninger J.M., Jackson A.P., Knoblich J.A. Cerebral organoids model human brain development and microcephaly. Nature. 2013;501:373–379. doi: 10.1038/nature12517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee A.S., De Jesus-Cortes H., Kabir Z.D., Knobbe W., Orr M., Burgdorf C., Huntington P., McDaniel L., Britt J.K., Hoffmann F., et al. The neuropsychiatric disease-associated gene cacna1c mediates survival of Young hippocampal neurons. eNeuro. 2016;3 doi: 10.1523/ENEURO.0006-16.2016. ENEURO.0006-16.2016. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lemcke H., Kuznetsov S.A. Involvement of connexin43 in the EGF/EGFR signalling during self-renewal and differentiation of neural progenitor cells. Cell. Signal. 2013;25:2676–2684. doi: 10.1016/j.cellsig.2013.08.030. [DOI] [PubMed] [Google Scholar]
- Li X.J., Liu X.J., Yang B., Fu Y.R., Zhao F., Shen Z.Z., Miao L.F., Rayner S., Chavanas S., Zhu H., et al. Human cytomegalovirus infection dysregulates the localization and stability of NICD1 and Jag1 in neural progenitor cells. J. Virol. 2015;89:6792–6804. doi: 10.1128/JVI.00351-15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liang Z.Y., Zhan Y., Shen Y., Wong C.C.L., Yates J.R., Plattner F., Lai K.O., Ip N.Y. The pseudokinase CaMKv is required for the activity-dependent maintenance of dendritic spines. Nat. Commun. 2016;7:13282. doi: 10.1038/ncomms13282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.J., Jiang X., Huang S.N., Sun J.Y., Zhao F., Zeng W.B., Luo M.H. Human cytomegalovirus infection dysregulates neural progenitor cell fate by disrupting Hes1 rhythm and down-regulating its expression. Virol. Sin. 2017;32:188–198. doi: 10.1007/s12250-017-3956-0. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X.J., Yang B., Huang S.N., Wu C.C., Li X.J., Cheng S., Jiang X., Hu F., Ming Y.Z., Nevels M., et al. Human cytomegalovirus IE1 downregulates Hes1 in neural progenitor cells as a potential E3 ubiquitin ligase. PLoS Pathog. 2017;13:e1006542. doi: 10.1371/journal.ppat.1006542. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lui J.H., Hansen D.V., Kriegstein A.R. Development and evolution of the human neocortex. Cell. 2011;146:18–36. doi: 10.1016/j.cell.2011.06.030. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M.H., Schwartz P.H., Fortunato E.A. Neonatal neural progenitor cells and their neuronal and glial cell derivatives are fully permissive for human cytomegalovirus infection. J. Virol. 2008;82:9994–10007. doi: 10.1128/JVI.00943-08. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Luo M.H., Hannemann H., Kulkarni A.S., Schwartz P.H., O'Dowd J.M., Fortunato E.A. Human cytomegalovirus infection causes premature and abnormal differentiation of human neural progenitor cells. J. Virol. 2010;84:3528–3541. doi: 10.1128/JVI.02161-09. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marcinowski L., Lidschreiber M., Windhager L., Rieder M., Bosse J.B., Radle B., Bonfert T., Gyory I., de Graaf M., da Costa O.P., et al. Real-time transcriptional profiling of cellular and viral gene expression during lytic cytomegalovirus infection. PLoS Pathog. 2012;8:e1002908. doi: 10.1371/journal.ppat.1002908. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martynoga B., Morrison H., Price D.J., Mason J.O. Foxg1 is required for specification of ventral telencephalon and region-specific regulation of dorsal telencephalic precursor proliferation and apoptosis. Dev. Biol. 2005;283:113–127. doi: 10.1016/j.ydbio.2005.04.005. [DOI] [PubMed] [Google Scholar]
- Michelson S., Alcami J., Kim S.J., Danielpour D., Bachelerie F., Picard L., Bessia C., Paya C., Virelizier J.L. Human cytomegalovirus infection induces transcription and secretion of transforming growth factor beta 1. J. Virol. 1994;68:5730–5737. doi: 10.1128/jvi.68.9.5730-5737.1994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mocarski E.S. Human Herpesviruses: Biology, Therapy, and Immunoprophylaxis. Cambridge University Press; 2007. Betaherpes viral genes and their functions; pp. 204–230. [PubMed] [Google Scholar]
- Mocarski E.S., Jr., Kemble G.W. Recombinant cytomegaloviruses for study of replication and pathogenesis. Intervirology. 1996;39:320–330. doi: 10.1159/000150503. [DOI] [PubMed] [Google Scholar]
- Moon A.L., Haan N., Wilkinson L.S., Thomas K.L., Hall J. CACNA1C: association with psychiatric disorders, behavior, and neurogenesis. Schizophr. Bull. 2018;44:958–965. doi: 10.1093/schbul/sby096. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moorman N.J., Cristea I.M., Terhune S.S., Rout M.P., Chait B.T., Shenk T. Human cytomegalovirus protein UL38 inhibits host cell stress responses by antagonizing the tuberous sclerosis protein complex. Cell Host Microbe. 2008;3:253–262. doi: 10.1016/j.chom.2008.03.002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Odeberg J., Wolmer N., Falci S., Westgren M., Seiger A., Soderberg-Naucler C. Human cytomegalovirus inhibits neuronal differentiation and induces apoptosis in human neural precursor cells. J. Virol. 2006;80:8929–8939. doi: 10.1128/JVI.00676-06. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan D., Xuan B.Q., Sun Y.M., Huang S.W., Xie M.R., Bai Y.D., Xu W.J., Qian Z.K. An intein-mediated modulation of protein stability system and its application to study human cytomegalovirus essential gene function. Sci. Rep. 2016;6:26167. doi: 10.1038/srep26167. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pan X., Li X.J., Liu X.J., Yuan H., Li J.F., Duan Y.L., Ye H.Q., Fu Y.R., Qiao G.H., Wu C.C., et al. Later passages of neural progenitor cells from neonatal brain are more permissive for human cytomegalovirus infection. J. Virol. 2013;87:10968–10979. doi: 10.1128/JVI.01120-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pass R.F., Arav-Boger R. Maternal and fetal cytomegalovirus infection: diagnosis, management, and prevention. F1000Research. 2018;7:255. doi: 10.12688/f1000research.12517.1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Paulus C., Krauss S., Nevels M. A human cytomegalovirus antagonist of type I IFN-dependent signal transducer and activator of transcription signaling. Proc. Natl. Acad. Sci. U S A. 2006;103:3840–3845. doi: 10.1073/pnas.0600007103. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perng Y.C., Qian Z.K., Fehr A.R., Xuan B.Q., Yu D. The human cytomegalovirus gene UL79 is required for the accumulation of late viral transcripts. J. Virol. 2011;85:4841–4852. doi: 10.1128/JVI.02344-10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pignoloni B., Fionda C., Dell'Oste V., Luganini A., Cippitelli M., Zingoni A., Landolfo S., Gribaudo G., Santoni A., Cerboni C. Distinct roles for human cytomegalovirus immediate early proteins IE1 and IE2 in the transcriptional regulation of MICA and PVR/CD155 expression. J. Immunol. 2016;197:4066–4078. doi: 10.4049/jimmunol.1502527. [DOI] [PubMed] [Google Scholar]
- Rak M.A., Buehler J., Zeltzer S., Reitsma J., Molina B., Terhune S., Goodrum F. Human cytomegalovirus UL135 interacts with host adaptor proteins to regulate epidermal growth factor receptor and reactivation from latency. J. Virol. 2018;92:e00919-18. doi: 10.1128/JVI.00919-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reitsma J.M., Sato H., Nevels M., Terhune S.S., Paulus C. Human cytomegalovirus IE1 protein disrupts interleukin-6 signaling by sequestering STAT3 in the nucleus. J. Virol. 2013;87:10763–10776. doi: 10.1128/JVI.01197-13. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saneyoshi T., Fortin D.A., Soderling T.R. Regulation of spine and synapse formation by activity-dependent intracellular signaling pathways. Curr. Opin. Neurobiol. 2010;20:108–115. doi: 10.1016/j.conb.2009.09.013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shinozaki K., Yoshida M., Nakamura M., Aizawa S., Suda Y. Emx1 and Emx2 cooperate in initial phase of archipallium development. Mech. Dev. 2004;121:475–489. doi: 10.1016/j.mod.2004.03.013. [DOI] [PubMed] [Google Scholar]
- Silva M.C., Schroer J., Shenk T. Human cytomegalovirus cell-to-cell spread in the absence of an essential assembly protein. Proc. Natl. Acad. Sci. U S A. 2005;102:2081–2086. doi: 10.1073/pnas.0409597102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sinzger C., Digel M., Jahn G. Cytomegalovirus cell tropism. Curr. Top. Microbiol. Immunol. 2008;325:63–83. doi: 10.1007/978-3-540-77349-8_4. [DOI] [PubMed] [Google Scholar]
- Sison S.L., O'Brien B.S., Johnson A.J., Seminary E.R., Terhune S.S., Ebert A.D. Human cytomegalovirus disruption of calcium signaling in neural progenitor cells and organoids. J. Virol. 2019;93:e00954-19. doi: 10.1128/JVI.00954-19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Spector D.H. Human cytomegalovirus riding the cell cycle. Med. Microbiol. Immunol. 2015;204:409–419. doi: 10.1007/s00430-015-0396-z. [DOI] [PubMed] [Google Scholar]
- Steingruber M., Marschall M. The cytomegalovirus protein kinase pUL97: host interactions, regulatory mechanisms and antiviral drug targeting. Microorganisms. 2020;8:515. doi: 10.3390/microorganisms8040515. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stern-Ginossar N., Weisburd B., Michalski A., Le V.T., Hein M.Y., Huang S.X., Ma M., Shen B., Qian S.B., Hengel H., et al. Decoding human cytomegalovirus. Science. 2012;338:1088–1093. doi: 10.1126/science.1227919. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun G.Q., Chiuppesi F., Chen X.W., Wang C., Tian E., Nguyen J., Kha M., Trinh D., Zhang H.N., Marchetto M.C., et al. Modeling human cytomegalovirus-induced microcephaly in human iPSC-derived brain organoids. Cell Rep. Med. 2020;1:100002. doi: 10.1016/j.xcrm.2020.100002. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Teissier N., Fallet-Bianco C., Delezoide A.L., Laquerriere A., Marcorelles P., Khung-Savatovsky S., Nardelli J., Cipriani S., Csaba Z., Picone O., et al. Cytomegalovirus-induced brain malformations in fetuses. J. Neuropathol. Exp. Neurol. 2014;73:143–158. doi: 10.1097/NEN.0000000000000038. [DOI] [PubMed] [Google Scholar]
- Tirosh O., Cohen Y., Shitrit A., Shani O., Le-Trilling V.T.K., Trilling M., Friedlander G., Tanenbaum M., Stern-Ginossar N. The transcription and translation landscapes during human cytomegalovirus infection reveal novel host-pathogen interactions. PLoS Pathog. 2015;11:e1005288. doi: 10.1371/journal.ppat.1005288. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tohyama J., Yamamoto T., Hosoki K., Nagasaki K., Akasaka N., Ohashi T., Kobayashi Y., Saitoh S. West syndrome associated with mosaic duplication of FOXG1 in a patient with maternal uniparental disomy of chromosome 14. Am. J. Med. Genet. A. 2011;155a:2584–2588. doi: 10.1002/ajmg.a.34224. [DOI] [PubMed] [Google Scholar]
- Umashankar M., Petrucelli A., Cicchini L., Caposio P., Kreklywich C.N., Rak M., Bughio F., Goldman D.C., Hamlin K.L., Nelson J.A., et al. A novel human cytomegalovirus locus modulates cell type-specific outcomes of infection. PLoS Pathog. 2011;7:e1002444. doi: 10.1371/journal.ppat.1002444. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang Z.B., Boisvert E., Zhang X., Guo M., Fashoyin A., Du Z.W., Zhang S.C., Li X.J. Fezf2 regulates telencephalic precursor differentiation from mouse embryonic stem cells. Cereb. Cortex. 2011;21:2177–2186. doi: 10.1093/cercor/bhr006. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wei C.J., Xu X., Lo C.W. Connexins and cell signaling in development and disease. Annu. Rev. Cell Dev. Biol. 2004;20:811–838. doi: 10.1146/annurev.cellbio.19.111301.144309. [DOI] [PubMed] [Google Scholar]
- Wheeler D.G., Groth R.D., Ma H., Barrett C.F., Owen S.F., Safa P., Tsien R.W. Ca(v)1 and Ca(v)2 channels engage distinct modes of Ca2+ signaling to control CREB-dependent gene expression. Cell. 2012;149:1112–1124. doi: 10.1016/j.cell.2012.03.041. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wu C.C., Jiang X., Wang X.Z., Liu X.J., Li X.J., Yang B., Ye H.Q., Harwardt T., Jiang M., Xia H.M., et al. Human cytomegalovirus immediate early 1 protein causes loss of SOX2 from neural progenitor cells by trapping unphosphorylated STAT3 in the nucleus. J. Virol. 2018;92:e00340-18. doi: 10.1128/JVI.00340-18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu H., Jiao Y., Qin S., Zhao W., Chu Q., Wu K. Organoid technology in disease modelling, drug development, personalized treatment and regeneration medicine. Exp. Hematol. Oncol. 2018;7:30. doi: 10.1186/s40164-018-0122-9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang N., Dong Z.Q., Guo S. Fezf2 regulates multilineage neuronal differentiation through activating basic helix-loop-helix and homeodomain genes in the zebrafish ventral forebrain. J. Neurosci. 2012;32:10940–10948. doi: 10.1523/JNEUROSCI.2216-12.2012. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young A., Machacek D.W., Dhara S.K., Macleish P.R., Benveniste M., Dodla M.C., Sturkie C.D., Stice S.L. Ion channels and ionotropic receptors in human embryonic stem cell derived neural progenitors. Neuroscience. 2011;192:793–805. doi: 10.1016/j.neuroscience.2011.04.039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Young F.I., Keruzore M., Nan X.S., Gennet N., Bellefroid E.J., Li M. The doublesex-related Dmrta2 safeguards neural progenitor maintenance involving transcriptional regulation of Hes1. Proc. Natl. Acad. Sci. U S A. 2017;114:E5599–E5607. doi: 10.1073/pnas.1705186114. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang X.M., Odom D.T., Koo S.H., Conkright M.D., Canettieri G., Best J., Chen H.M., Jenner R., Herbolsheimer E., Jacobsen E., et al. Genome-wide analysis of cAMP-response element binding protein occupancy, phosphorylation, and target gene activation in human tissues. Proc. Natl. Acad. Sci. U S A. 2005;102:4459–4464. doi: 10.1073/pnas.0501076102. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou J.Z., Jiang J.X. Gap junction and hemichannel-independent actions of connexins on cell and tissue functions--an update. FEBS Lett. 2014;588:1186–1192. doi: 10.1016/j.febslet.2014.01.001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Total of 19897 significant DE genes were found, of which 1733 were upregulated (9%) and the remaining 18164 were downregulated (91%). 2882 genes were unchanged, adj p-value > 1. Additional analysis details found in S1 or methods.
20271 significant DE genes were found with 1384 upregulated (7%) and 18887 downregulated (93%). 1687 genes were unchanged, adj p-value > 1. Additional analysis details found in S2 or methods.
31 significant DE genes were found, all of which were downregulated. 5,000 genes remained unchanged; adj p-value > 1. Additional analysis details found in S3 or methods.
All comparisons were made relative to Mock, and each list contain all significant differentially expressed genes as revealed by DESeq2. Additional analysis details found in S4 or methods and reference Figure 3.
Results based on list of most significantly differentially expressed genes as determined by adj p-value for GFP (+) vs. Mock in reference to Figure 4. Additional analysis detail in S5 and methods.
A complete list of canonical pathways, upstream regulators, and related disease or functional outcomes identified by IPA analysis of GFP (+) vs. Mock shown in Figure 4. Additional analysis details found in S6 or methods.
Data Availability Statement
Bulk RNA-seq data have been deposited at GEO (GEO: GSE193759) and the preprint manuscript has been made publicly available on BioRXiv as of the date of publication. Accession numbers are listed in the key resources table. Microscopy data reported in this paper will be shared by the lead contact upon request. Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.