

Abstract
Doxorubicin is an anthracycline widely used for the treatment of various cancers; however, the drug has a common deleterious side effect, namely a dose-dependent cardiotoxicity. Doxorubicin treatment increases the generation of reactive oxygen species, which leads to oxidative stress in the cardiac cells and ultimately DNA damage and cell death. The most common DNA lesion produced by oxidative stress is 7,8-dihydro-8-oxoguanine (8-oxoguanine), and the enzyme responsible for its repair is the 8-oxoguanine DNA glycosylase (OGG1), a base excision repair enzyme. Here, we show that the OGG1 deficiency has no major effect on cardiac function at baseline or with pressure overload; however, we found an exacerbation of cardiac dysfunction as well as a higher mortality in Ogg1 knockout mice treated with doxorubicin. Our transcriptomic analysis also showed a more extensive dysregulation of genes in the hearts of Ogg1 knockout mice with an enrichment of genes involved in inflammation. These results demonstrate that OGG1 attenuates doxorubicin-induced cardiotoxicity and thus plays a role in modulating drug-induced cardiomyopathy.
1. Introduction
Heart failure (HF) is a major cause of mortality and morbidity worldwide, posing a huge burden to public health systems in both developed and developing countries [1]. Despite the multifactorial nature of HF, oxidative stress is a common unifying feature, as the various pathologies that lead to HF are associated with increased generation of reactive oxygen species (ROS) [2]. ROS generation is particularly relevant in doxorubicin-induced cardiotoxicity [3], an anthracycline drug often used as a treatment of choice for various types of adult and paediatric cancers, including leukaemia, lymphomas, and solid tumours [3]. Despite the highly effective antineoplastic properties of doxorubicin, one major side effect that limits its clinical utility is the dose-dependent cardiotoxicity caused by the drug, which can lead to irreversible cardiomyopathy and subsequent HF [4, 5]. Indeed, it is estimated that up to 10% of patients on doxorubicin or its derivatives will develop delayed cardiac complications within ten years after completion of the therapy [3].
Doxorubicin disrupts protein complexes in the mitochondrial electron transport chain and also binds to the reductase domain of endothelial nitric oxide synthase, thereby increasing production of superoxide (O2−) in the cardiac cells [3]. Furthermore, doxorubicin has been shown to decrease the levels of the selenium-dependent glutathione peroxidase, an enzyme involved in detoxification of reactive oxygen metabolites with a subsequent accumulation of ROS [6]. The doxorubicin-induced increase in ROS in the cells leads to DNA damage, especially in mitochondrial DNA (mtDNA), lipid peroxidation of cellular and mitochondrial membrane, as well as impaired mitochondrial oxidative phosphorylation which ultimately leads to cell death by apoptosis and necrosis [7]. One of the most common DNA lesions produced by the increased ROS in the cells is the oxidation of guanine residues to 7,8-dihydro-8-oxoguanine (8-oxoguanine or 8-oxoG) [5]. Levels of 8-oxoG were found to be elevated in failing human hearts, suggesting that increased accumulation of DNA damage could be implicated in the pathophysiology of HF [2].
The primary enzyme responsible for the repair of 8-oxoG is the base excision repair (BER) enzyme, 8-oxoguanine DNA glycosylase (OGG1) and it excises 8-oxoG by cleaving the N-glycosidic bond of the lesion [2]. By preventing accumulation of DNA damage, especially in mtDNA, OGG1 is therefore critical in reducing mitochondrial fragmentation as well as improving mitochondrial function in muscle cells [8]. In addition, cardiac overexpression of human mitochondrial isoform of OGG1 (OGG1-2a) led to a reduction in mtDNA damage and cardiac fibrosis in a mouse pressure overload model of cardiac hypertrophy [9]. We therefore set out to study the role of OGG1 in pressure overload-induced hypertrophy and doxorubicin-induced cardiotoxicity, paying particular attention to the transcriptional changes induced by doxorubicin in both wild-type and Ogg1 knockout (KO) hearts.
2. Material and Methods
2.1. Generation of Ogg1 Knockout Mice
Ogg1+/- mice were previously established [10] and have been backcrossed to C57BL/6J for more than 20 generations. Age-matched male wild-type and Ogg1−/− (Ogg1-KO) mice obtained by mating Ogg1+/- mice were used for these studies. Six-week-old mice were used for the transverse aortic constriction (TAC) experiments, while 8-month-old mice were used for the doxorubicin study. Genotyping was performed on mouse ear clippings to confirm wild-type or Ogg1-KO status, as also previously described [10] using the following primers for Ogg1: mgO5-1, GTTAAGCTTCAAACGTGCCTC; mgO3-1, GAAGGACTGTCCAGAAGCTA; and mpII5′-3, AAAGTCTCTCATTAGTATCCC. A single band corresponding to 780 base pair reflects wild-type mice, whereas Ogg1-KO mice display a single band corresponding to 675 base pair. Heterozygous mice reveal dual bands of 675 base pairs and 780 base pairs.
2.2. Animal Experiments
Animal experiments were performed under a license approved by the IACUC (National University of Singapore). Pressure overload TAC surgery was performed on adult 6-week-old mice. Doxorubicin administration was carried out on 8-month-old mice. Echocardiography to monitor cardiac function and dimensions was performed on the mice 6 weeks after TAC surgery and 2 weeks after intraperitoneal injection with 15 mg/kg doxorubicin using the Vevo 2100 (Visualsonics) instrument. Purified adult mouse left ventricular cardiomyocytes (CMs) were isolated using a previously established protocol [11].
2.3. Mouse Cardiac Pressure Overload Surgery (TAC: Transverse Aortic Constriction)
TAC surgery was performed as described [12]. Briefly, anaesthesia was induced in mice in an induction chamber followed by partial thoracotomy for better visualisation of the transverse aortic arch. Ligation of the transverse aortic arch was done around the aorta distal between the innominate and left carotid arteries with a 7.0 Prolene suture overlying a 27.5 G needle. The sham-operated mice went through a similar surgical procedure without ligation of the transverse aortic arch. Enrofloxacin (0.1 mg/kg/each day for 3 days) and buprenorphine (0.03 mg/kg/each day for 3 days) were given as antimicrobial and analgesic, respectively, and were administered postsurgery.
2.4. Doxorubicin Administration
Eight-month-old wild-type and Ogg1-KO mice were randomly assigned to either the control group or the doxorubicin treatment group. Doxorubicin HCl (Sigma-Aldrich, Cat#: D1515,) was dissolved in 0.9% saline and administered by intraperitoneal injection at a dose of 15 mg/kg of body weight [13]. The control mice received injections of saline of comparable volume.
2.5. Quantification of mtDNA Damage
Total DNA was isolated from adult mouse cardiomyocytes from the different treatment groups using the Purelink Genomic DNA extraction kit. 20 ng of extracted DNA was used for each reaction. mtDNA damage was detected by quantitative PCR as previously described [14]. Briefly, two fragments (117 bp and 10 kb) are amplified from the mtDNA with primers listed in Supplementary Table 1. As DNA lesions are likely distributed randomly, it is expected that the amplification of the 10 kb will be randomly inhibited. After 20 amplification cycles, which should still be during the logarithmic phase of the PCR, the PCR mixture was separated on an agarose gel stained with SYBR Safe DNA gel stain (Thermo Fisher Scientific, Cat# S33102) and imaged (Gel Logic 200 imaging systems). PCR products were quantified by densitometric analysis using the ImageJ (a free software from NIH). The linearity of the reaction was confirmed by including a control reaction containing 50% template DNA. The ratio between 10 kb and 117 bp was used to give an estimate of mtDNA damage [14].
2.6. Immunohistochemistry
Heart samples were fixed in 4% paraformaldehyde for 24 hours, then embedded in paraffin, and sectioned at 4 μm intervals. Paraffin samples were further treated with xylene (to remove paraffin) and rehydrated. Haematoxylin and eosin (HE) was applied to observe myocyte architecture and Masson trichrome (MT) to identify cardiac fibrosis. Quantification of fibrosis was calculated as the blue-stained areas relative to total ventricular area, using the ImageJ (NIH).
2.7. Cell Culture Experiments
Murine cardiomyocytes (CMs) and cardiac fibroblasts (CF) were obtained from 6-week-old wild-type mice and cultured as previously described [11]. Expression of Ogg1 mRNA was measured by reverse-transcription and quantitative real time PCR (RT-qPCR) in CMs exposed to oxidative stress (H2O2) and hypoxia (0.2% oxygen environment), as well as CFs in the heart exposed to profibrotic transforming growth factor beta 1 (TGF-β1) and proinflammatory interleukin-1β (IL-1β). TGF-β1 (10 ng/ml) and IL-1β (10 ng/ml) were purchased from PeproTech (NJ, USA). Experiments were conducted in serum-free conditions and treatments were applied for 24 hours.
2.8. RNA Extraction and RT-qPCR
Total RNA was prepared using TRIzol (Thermo Fisher Scientific, Cat#:15596018) according to the manufacturer's protocol. cDNA was synthesized using the qScript™ cDNA synthesis kit (Quanta Biosciences, Cat#:95049). QPCR analysis was performed using gene-specific primers, PerfeCTa SYBR® Green Fast Mix (Quanta Biosciences, Cat#95047), and Roche Lightcycler apparatus and software. Oligonucleotide primers were designed using PrimerBLAST (NCBI) and listed in Supplementary Table (ST) 1, 18 s was used as housekeeping.
2.9. RNA-Sequencing Analysis
Two sets of RNA-sequencing were performed. For the first set, total RNA was extracted from ventricular CMs from 2 biological replicates of 4-week-old mice, 2 Ogg1-KO and 2 wild type. For the second set, total RNAs were prepared from ventricular CMs from 2 biological replicates each of the following groups: wild-type mice injected with normal saline (WT saline), Ogg1-KO injected with normal saline (KO saline), wild-type mice treated with doxorubicin (WT DXB), and Ogg1-KO mice treated with doxorubicin (KO DXB). Paired-end libraries were made using TruSeq kits (Illumina). The libraries were sequenced on the HiSeq2000, generating 2 × 100-bp paired-end reads. Reads were mapped against the mouse genome using the TopHat version 2.0.11 with default parameters. Counting reads for each gene were computed using htseq-count [15]. Differential gene expression analysis was performed using edgeR [16]. A gene is considered differentially expressed between different groups if the fold change is greater than log2 0.5 and the P value is less than 0.05. Gene expression heat map was generated using R (version 3.1.2). Gene ontology analysis was performed by the AMIGO gene ontology [17].
2.10. Statistical Analysis
All animal in vivo experiments were performed with at least n = 3 biological replicates. Animal experimental groups were controlled for mouse age and sex. Student's t-test was performed to test for significance between the two groups. P value of less than 0.05 was considered significant.
3. Results
3.1. Expression of Ogg1 mRNA in the Mouse Heart
We first set out to evaluate Ogg1 mRNA levels in mouse CMs by performing RNA sequencing and RT-qPCR on freshly isolated CMs from 4-week-old wild-type and Ogg1-KO hearts. Our results revealed a relatively low abundance of Ogg1 mRNA in the wild-type CMs with fragments per kilobase of exon per million mapped reads (FPKM) value of ~2. In comparison, the FPKM level of cardiac-specific gene Actc1 was 8000. Ogg1 mRNA was however significantly depleted in the Ogg1-KO CMs,and also validated by RT-qPCR (Figures 1(a) and 1(b)). Ogg1-KO had very minor impact on the cardiac transcriptome at baseline as very few genes were significantly differentially expressed between wild-type and Ogg1-KO CMs (Figure 1(a), Supplementary Table 2). Next, we isolated adult CMs and CF from the wild-type heart, plated them in vitro, and exposed the cells to different stimuli to assess changes in Ogg1 mRNA under different conditions. Our results showed that though Ogg1 mRNA was slightly higher in the CF fraction, they were maintained relatively stable when exposed to different stimuli and only showed minimal downregulation in hypoxia in CM (Figure 1(c)).
Figure 1.
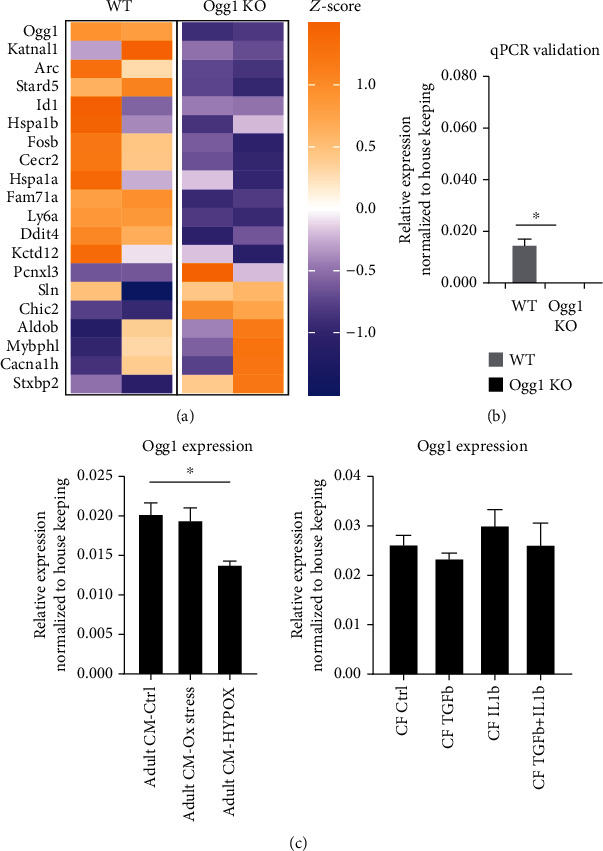
Expression of Ogg1 in cardiomyocytes and cardiac fibroblasts at baseline. (a) Heat map showing the transcriptomic differences in the cardiomyocytes (CM) of wild-type (WT) mice compared to Ogg1-KO mice at baseline identified through RNA-sequencing analysis with a few genes highlighted. Z-scores are computed on a gene-by-gene basis and obtained by subtracting the mean and dividing by standard deviation. (b) qPCR validation of Ogg1 mRNA expression in wild-type (WT) and Ogg1-KO CMs. All values are mean ± SE. n = 3∗P < 0.05. (c) Expression of Ogg1 mRNA evaluated by qPCR in cardiomyocytes (CM) subjected to oxidative stress (Ox stress) and hypoxia (HYPOX), as well as cardiac fibroblasts (CF) in the heart treated with different stimuli. All values are mean ± SE. n = 3∗P < 0.05.
3.2. Evaluation of Long-Term Effect of OGG1 Deficiency on Baseline Cardiac Function
OGG1 deficiency has been associated with the development of insulin resistance and obesity in mice with increasing age (12-15 months) [18]. We therefore assessed if OGG1 deficiency affects cardiac function over time. Using echocardiography, we monitored cardiac function by measuring the ejection fraction expressed in percentage (EF), left ventricular internal diameter (LVID), and left ventricular posterior wall (LVPW) in both Ogg1-KO and wild-type mice every month from the 7th to 12th months of age. No significant differences in the parameters were observed (Figures 2(a)–2(c)). Furthermore, there were no significant histopathological differences, comparing between Ogg1-KO and wild-type hearts (Figure 2(d)). OGG1deficiency therefore did not appear to result in any detectable difference in cardiac dysfunction with ageing. This lack of functional cardiac phenotype in Ogg1-KO mice is consistent with other studies that also reported no significant changes in cellular function over time in different cells of OGG1-deficient mice at baseline even up to 23 months [19, 20].
Figure 2.
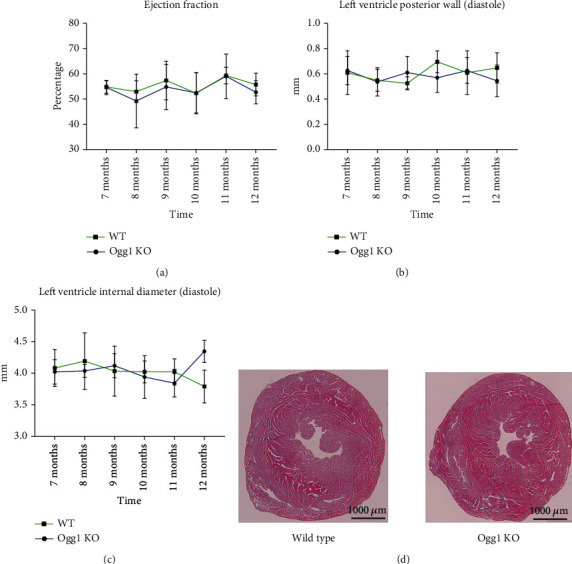
Heart morphology and function in aged WT and Ogg1-KO mice. (a) Ejection fraction, (b) left ventricular posterior wall, and (c) Left ventricle internal diameter measured monthly in both WT and Ogg1-KO mice from 7 months of age till 12 months. 3 mice per group. (d) Haematoxylin and Eosin staining of representative heart slices of WT and Ogg1-KO mice at 12 months of age.
3.3. Effect of OGG1 Deficiency on Cardiac Function in a Mouse Model of Cardiac Hypertrophy Induced by Pressure Overload
To assess the role of OGG1 on cardiac function under cardiac stress, we subjected both wild-type and Ogg1-KO mice to TAC surgery to induce pressure overload. Both groups of mice showed a reduction in EF and an increase in the thickness of the ventricular walls after the TAC surgery compared to the sham-operated mice. There were no significant differences in EF, LVID, and LVPW between wild-type and Ogg1-KO hearts with TAC surgery (Figures 3(a)–3(e)). Similarly, no significant differences in fibrosis were seen (Figure 3(f)). While this is the first study to perform TAC surgery in OGG1-deficient mice, an earlier study had analysed the effect of human mitochondrial OGG1 (OGG1-2a) overexpression on cardiac function with TAC surgery [9]. In the earlier study, there were no differences in EF and wall dimensions between wild-type and the OGG1 transgenic mice subjected to TAC surgery; however, there was a reduction in fibrosis in the OGG1 transgenic mice after TAC surgery. We however did not observe any differences in fibrosis in our OGG1-deficient mice.
Figure 3.
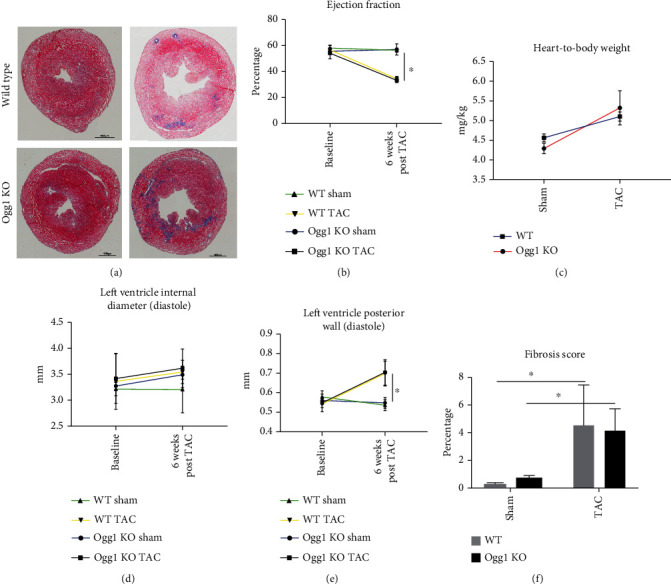
Cardiac function after transverse aortic constriction in WT and Ogg1-KO mice. (a) Representative heart sections stained with haematoxylin and eosin in WT and Ogg1-KO mice hearts to show left ventricle wall thickness and fibrosis after TAC surgery. (b) Ejection fraction, (c) heart weight-to-body weight, (d) left ventricle internal diameter, (e) left ventricle posterior wall, and (f) fibrosis score were assessed presurgery and 6 weeks post-TAC in WT and Ogg1-KO mice. All values are mean ± SD. n = 3 − 5∗P < 0.05.
3.4. Effect of OGG1 Deficiency on Cardiac Function and Mortality after Doxorubicin (DXB) Treatment
DXB induces oxidative stress in CMs with increased accumulation of 8-oxoG and mtDNA damage [5]; furthermore, DNA damage accumulates over time in different organs including the heart after DXB treatment [21]. We therefore examined the effect of DXB on the hearts of aged wild-type and Ogg1-KO mice (8-month-old mice). Mice received a single dose of 20 mg of DXB per kg of body weight and their cardiac function was analysed 2 weeks after the administration. First, we observed that about 50% (5 out of 10 mice) of Ogg1-KO mice died within 2 weeks after the DXB administration, while only 16% (1 out of 6 mice) of wild-type mice died within the 2 weeks (Figure 4(a)). Ogg1-KO mice that survived 2 weeks after the DXB administration exhibited a significantly greater EF reduction compared to wild-type mice, accompanied by significant LV dilatation (Figures 4(b) –4(d). DXB mediated LV dilatation is consistent with earlier reports [13], and this was more significant in Ogg1-KO mice. We next examined the level of mtDNA damage through PCR quantification of the ratio of long to short fragment. There was significantly greater mtDNA damage in the DXB-treated Ogg1-KO hearts compared to wild type (Figure 5(a)). Levels of mtDNA genes were however depleted equally in both wild-type and Ogg1-KO hearts upon treatment with DXB, an indication of mtDNA (Figure 5(b)) damage induced by DXB [22, 23].
Figure 4.
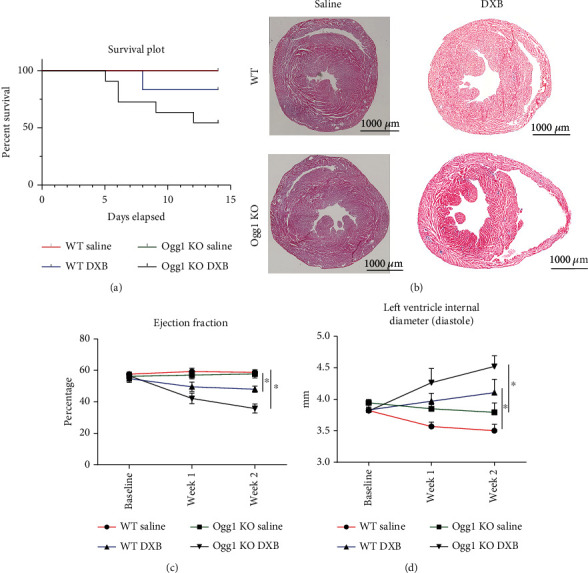
Cardiac function after doxorubicin treatment in WT and Ogg1-KO mice. (a) Survival plot 14 days after WT and KO mice were treated with 15 mg/kg doxorubicin. (b) Representative heart sections stained with haematoxylin and eosin 2 weeks after treatment with doxorubicin in both WT and Ogg1-KO mice. (c and d) Ejection fraction and left ventricle internal diameter measured at baseline, 1 and 2 weeks postdoxorubicin treatment. All values are mean ± SD. n = 3 − 7∗P < 0.05.
Figure 5.
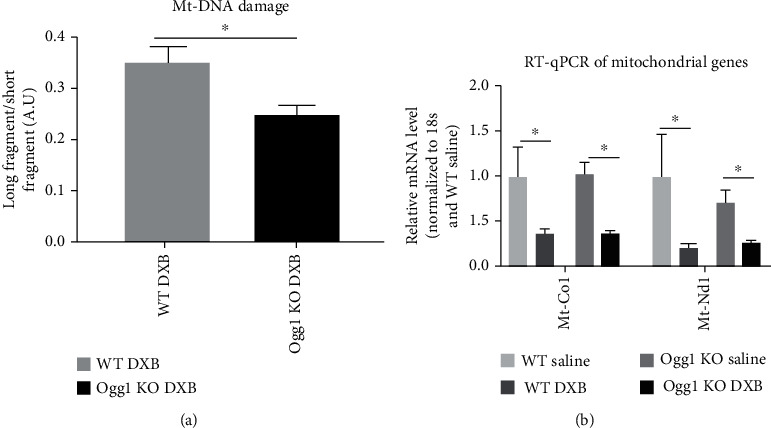
Mitochondrial DNA damage after doxorubicin treatment in WT and Ogg1-KO mice. (a) Mitochondrial DNA damage assessed after 2 weeks in doxorubicin-treated WT and Ogg1-KO mice. ∗P < 0.05. (b) qPCR of 2 mitochondrial genes quantified 2 weeks after treatment with doxorubicin. All values are mean ± SD. n = 3 − 7∗P < 0.05.
3.5. Cardiac Transcriptomic Changes Induced by OGG1 Deficiency and DXB Treatment
Finally, we examined transcriptomic changes induced by DXB treatment by performing RNA sequencing in the four groups: wild-type and Ogg1-KO CMs treated with normal saline as the control, or with DXB. First, we analysed for differences between Ogg1-KO and wild-type hearts treated with normal saline. Interestingly, despite absent visible phenotypic changes in Ogg1-KO hearts over time, there were 270 differentially expressed genes (Supplementary Table 3). Gene ontology of the biological processes in upregulated genes showed terms such as “MHC Protein complex assembly”, Cellular Response to interferon beta and gamma”, and “positive regulation of cell killing” (Supplementary Table 4). These terms point to an activation of immune system, consistent with previous studies showing that the DNA damage response induces interferon signalling and activates immune response [24–26]. Gene ontology terms in downregulated genes included processes such as monocarboxylic acid metabolic processes (Supplementary Table 5). Next, we analysed DXB-induced changes compared within each group (Ogg1-KO: DXB vs. saline; and wild type: DXB vs. saline). There were more dysregulated genes in Ogg1-KO (DXB vs. saline; 465 genes) compared to wild type (doxorubicin vs. saline; 284 genes) (Figures 6(a) and 6(b), Supplementary Tables 6 and 7), with an overlap of 70 genes, of which 38 were upregulated and 32 were downregulated (Supplementary Table 8). One of the most significantly upregulated gene in both conditions was the Sprr1a gene which is a gp130 pathway- and stress-inducible gene [27]. Gp130 acts as mediator for the IL-6 family of cytokines and is involved in the regulation of inflammation and cardiac stress [27]. Other shared upregulated genes included Ctgf, Lgals3, and Tnfrsf12a. These genes have been shown to be predictive of cardiac dysfunction and cell death [28]. Among downregulated genes were a number of mitochondrial genes, downregulated in both Ogg1-KO and wild-type mice treated with DXB, including Mt-Atp8, Mt-Nd2, Mt-Co3, Mt-Nd3, and Mt-Nd4. This observation is consistent with previously published studies on mtDNA damage induced by DXB [5]. Indeed, GO terms for commonly dysregulated genes included terms “ATP synthesis coupled electron transport”, “Reactive oxygen species metabolic process”, and “Cellular respiration” confirming the effect of DXB on mitochondrial respiration (Supplementary Table 8). To shed light on the added effect of OGG1 deficiency on the DXB-treated CMs, we first focused on the uniquely dysregulated genes in the Ogg1-KO CMs treated with DXB (KO-DXB). Similar to the results found in the saline-treated Ogg1-KO CMs, there was enrichment for terms such as response to interferon gamma and response to cytokines, further showing that the DNA damage due to the OGG1 deficiency may lead to increase in inflammatory cytokines (Supplementary Table 9). Finally, we compared the transcriptomic changes in the DXB-treated Ogg1-KO CMs (KO-DXB) to DXB-treated wild-type CMs (WT-DXB), where we observed 61 upregulated and 68 downregulated genes in the KO-DXB compared to WT-DXB (Supplementary Table 10). GO for dysregulated genes showed enrichment for Notch signalling (Supplementary Table 11). Indeed, there was upregulation of Notch ligands Jag1 and Dll4 as well as notch target gene Hey-1 [29]. These genes have also been found to be involved in the regulation of P53, a tumour suppressor often upregulated in response to DNA damage [30].
Figure 6.
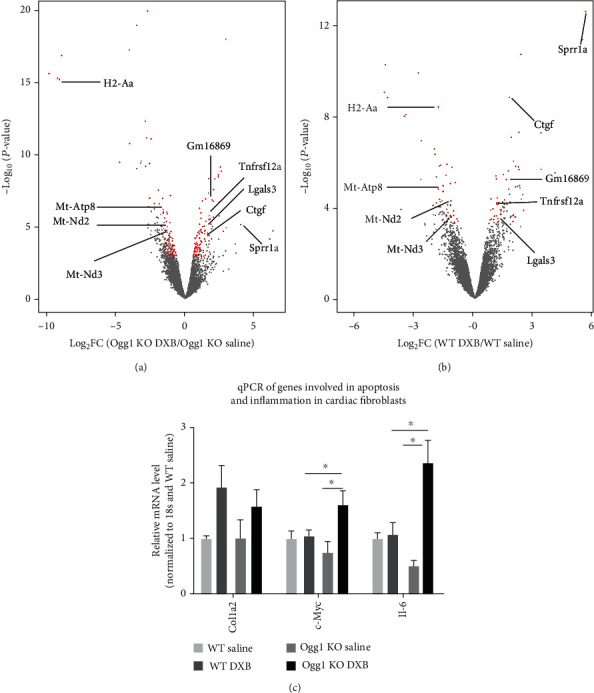
Transcriptomic profile after doxorubicin treatment in WT and Ogg1-KO mice. (a and b) Volcano plot showing genes that are changing after treatment with doxorubicin. The red dots are genes with fold change of log2 ≥ 0.5 and FDRvalue < 0.1. Some genes are highlighted. (c) qPCR of Col1a2, c-Myc, and Il-6 in cardiac fibroblasts in the heart after treatment with doxorubicin.
Given that OGG1 deficiency in the Ogg1-KO mice was global, we also assessed the level of other injury-associated genes in CFs. In particular, Il6 and Myc were upregulated in the DXB-treated CFs harvested from Ogg1-KO mice (KO-DXB), compared to those from wild-type mice (WT-DXB) (Figure 6(c)). Il6, has been shown to be upregulated by DXB and is highly correlated with HF severity and cardiac remodelling [31]. Similarly, proapoptotic c-Myc was also shown before to be upregulated by DXB [32]. These results suggest that the impact of the OGG1 deficiency on cardiac homeostasis is mediated by both CFs and CMs.
4. Discussion
The role of OGG1-mediated base excision repair (BER) in the pathophysiology of oxidative stress-induced damage in different tissues has been the subject of many foregoing studies [19, 20]. In the cardiovascular system, the function of OGG1 has been analysed in ischemia reperfusion injury [14], TAC [9], and in atherosclerosis [33, 34], using gene knockout or overexpression approaches. Furthermore, OGG1 has been implicated in the development of DXB-induced cardiomyopathy in Sirt3-KO mouse model [5], in the prevention of mtDNA damage after abdominal aortic constriction [35], in the diabetic cardiomyopathy in mice [36], and in the end-stage cardiomyopathy in humans [2], implying that it may have an overall role in cardiac homeostasis. For all studies involving CMs, OGG1 levels correlate with levels of DNA damage but not always with cardiac function. Indeed a study found that despite increased DNA damage in Ogg1-KO mice, there was no evidence of mitochondrial dysfunction or cardiac dysfunction in Ogg1-KO mice [20]. In our study, cardiac EF in Ogg1-KO mice was not significantly perturbed up to 12 months of age. Similarly, there were no significant differences in cardiac function between the Ogg1-KO and wild-type mice after TAC surgery. These results suggest therefore that OGG1-mediated BER may not be critical for proper stress response of CMs. Indeed, expression of Ogg1 mRNA in adult CMs is relatively low with FPKM values ranging from 1 to 2 in RNA sequencing data and its expression is not altered in TAC [12].
Nevertheless, systemic administration of DXB led to a more pronounced reduction in EF and increase in chamber dilatation in the DXB-treated Ogg1-KO mice, compared to wild type. There was also higher mortality in Ogg1-KO mice and larger number of dysregulated genes in the DXB-treated Ogg1-KO mouse hearts, suggesting that OGG1-mediated BER may ameliorate the DXB-induced cardiotoxicity. Our RNA sequencing data revealed an activation of the inflammatory pathway in Ogg1-KO mice. This is consistent with an earlier report that demonstrated that OGG1 deficiency led to increased inflammatory response in the lungs of mice exposed to hyperoxia [37]. They observed that the bronchioalveolar lavage fluid from the lungs of Ogg1-KO mice contained higher levels of inflammatory cytokines such as IL-6 and IFN-γ [37].
As DXB administration in our study was systemic, and the OGG1 deficiency was global in Ogg1-KO mice, we cannot rule out that the striking phenotypic and transcriptomic changes observed may have arisen in part due to signalling from other OGG1-deficient cells and tissues. Indeed, OGG1 deficiency has been shown to lead to increased NLRP3 inflammasome activation in macrophages, increased IL-1β production, and increased apoptosis in atherosclerotic plaques [34]. Nevertheless, our study provides the first report of DXB treatment in Ogg1-KO mice and also provides a transcriptomic analysis of both the impact of DXB on CMs as well as the potentiating effect of OGG1 deficiency on the cardiac injury produced by the drug. Further studies will be aimed at unravelling the mechanism through which OGG1 deficiency exacerbates cardiac dilatation in the DXB-treated hearts as well as the role of OGG1 in CFs.
Acknowledgments
This work was funded by the Biomedical Research Council (BMRC), Agency for Science, Technology and Research (A∗STaR), Special Positioning Fund (SPF2014/004), Individual Research Grants and a Clinician Scientist Award from National Medical Research Council (NMRC) of Singapore (R.S.Y.F.), and performed partly in the Cooperative Research Project Program of the Medical Institute of Bioregulation, Kyushu University (R.S.Y.F, Y.N.).
Contributor Information
Chukwuemeka George Anene-Nzelu, Email: mdccgoa@nus.edu.sg.
Roger Sik-Yin Foo, Email: roger.foo@nus.edu.sg.
Yusaku Nakabeppu, Email: yusaku@bioreg.kyushu-u.ac.jp.
Data Availability
All supporting data to be made available upon request.
Conflicts of Interest
The authors declare no conflicts of interest.
Supplementary Materials
Supplementary Table 1: primer list.
Supplementary Table 2: differentially expressed genes between 4-week-old WT and KO CMs.
Supplementary Table 3: differentially expressed genes between KO saline-treated and WT saline-treated CMs.
Supplementary Table 4: gene ontology terms in upregulated genes in the KO saline compared to WT Saline.
Supplementary Table 5: gene ontology terms in downregulated genes in the KO saline compared to WT Saline.
Supplementary Table 6: differentially expressed genes between KO doxorubicin-treated and KO saline-treated CMs.
Supplementary Table 7: differentially expressed genes between WT doxorubicin-treated and WT saline-treated CMs.
Supplementary Table 8: commonly dysregulated genes and gene ontology terms in KO DXB- and WT DXB-treated cells.
Supplementary Table 9: gene ontology terms in the genes only dysregulated in the KO DXB-treated CMs.
Supplementary Table 10: differentially expressed genes between KO doxorubicin-treated and WT doxorubicin-treated CMs.
Supplementary Table 11: gene ontology terms in upregulated genes in the KO doxorubicin-treated CMs compared to WT doxorubicin-treated CMs.
References
- 1.Ponikowski P., Anker S. D., AlHabib K. F., et al. Heart failure: preventing disease and death worldwide. ESC Heart Fail . 2014;1(1):4–25. doi: 10.1002/ehf2.12005. [DOI] [PubMed] [Google Scholar]
- 2.Siggens L., Figg N., Bennett M., Foo R. Nutrient deprivation regulates DNA damage repair in cardiomyocytes via loss of the base-excision repair enzyme OGG1. The FASEB Journal . 2012;26(5):2117–2124. doi: 10.1096/fj.11-197525. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Octavia Y., Tocchetti C. G., Gabrielson K. L., Janssens S., Crijns H. J., Moens A. L. Doxorubicin-induced cardiomyopathy: from molecular mechanisms to therapeutic strategies. Journal of Molecular and Cellular Cardiology . 2012;52(6):1213–1225. doi: 10.1016/j.yjmcc.2012.03.006. [DOI] [PubMed] [Google Scholar]
- 4.Minotti G., Menna P., Salvatorelli E., Cairo G., Gianni L. Anthracyclines: molecular advances and pharmacologic developments in antitumor activity and cardiotoxicity. Pharmacological Reviews . 2004;56(2):185–229. doi: 10.1124/pr.56.2.6. [DOI] [PubMed] [Google Scholar]
- 5.Pillai V. B., Bindu S., Sharp W., et al. Sirt3 protects mitochondrial DNA damage and blocks the development of doxorubicin-induced cardiomyopathy in mice. American Journal of Physiology. Heart and Circulatory Physiology . 2016;310(8):H962–H972. doi: 10.1152/ajpheart.00832.2015. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Doroshow J. H., Locker G. Y., Myers C. E. Enzymatic defenses of the mouse heart against reactive oxygen metabolites: alterations produced by doxorubicin. The Journal of Clinical Investigation . 1980;65(1):128–135. doi: 10.1172/JCI109642. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Shi J., Abdelwahid E., Wei L. Apoptosis in anthracycline cardiomyopathy. Current Pediatric Reviews . 2011;7(4):329–336. doi: 10.2174/157339611796892265. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Torres-Gonzalez M., Gawlowski T., Kocalis H., Scott B. T., Dillmann W. H. Mitochondrial 8-oxoguanine glycosylase decreases mitochondrial fragmentation and improves mitochondrial function in H9C2 cells under oxidative stress conditions. American Journal of Physiology. Cell Physiology . 2014;306(3):C221–C229. doi: 10.1152/ajpcell.00140.2013. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Wang J., Wang Q., Watson L. J., Jones S. P., Epstein P. N. Cardiac overexpression of 8-oxoguanine DNA glycosylase 1 protects mitochondrial DNA and reduces cardiac fibrosis following transaortic constriction. American Journal of Physiology. Heart and Circulatory Physiology . 2011;301(5):H2073–H2080. doi: 10.1152/ajpheart.00157.2011. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Sakumi K., Tominaga Y., Furuichi M., et al. Ogg1 knockout-associated lung tumorigenesis and its suppression by Mth1 gene disruption. Cancer Research . 2003;63(5):902–905. [PubMed] [Google Scholar]
- 11.Ackers-Johnson M., Li P. Y., Holmes A. P., O’Brien S. M., Pavlovic D., Foo R. S. A simplified, Langendorff-free method for concomitant isolation of viable cardiac myocytes and nonmyocytes from the adult mouse heart. Circulation Research . 2016;119(8):909–920. doi: 10.1161/CIRCRESAHA.116.309202. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Lee D. P., Tan W. L. W., Anene-Nzelu C. G., et al. Robust CTCF-based chromatin architecture underpins epigenetic changes in the heart failure stress-gene response. Circulation . 2019;139(16):1937–1956. doi: 10.1161/CIRCULATIONAHA.118.036726. [DOI] [PubMed] [Google Scholar]
- 13.Nozaki N., Shishido T., Takeishi Y., Kubota I. Modulation of doxorubicin-induced cardiac dysfunction in toll-like receptor-2-knockout mice. Circulation . 2004;110(18):2869–2874. doi: 10.1161/01.CIR.0000146889.46519.27. [DOI] [PubMed] [Google Scholar]
- 14.Bliksoen M., Baysa A., Eide L., et al. Mitochondrial DNA damage and repair during ischemia-reperfusion injury of the heart. Journal of Molecular and Cellular Cardiology . 2015;78:9–22. doi: 10.1016/j.yjmcc.2014.11.010. [DOI] [PubMed] [Google Scholar]
- 15.Anders S., Pyl P. T., Huber W. HTSeq--a python framework to work with high-throughput sequencing data. Bioinformatics . 2015;31(2):166–169. doi: 10.1093/bioinformatics/btu638. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 16.Robinson M. D., McCarthy D. J., Smyth G. K. edgeR: a Bioconductor package for differential expression analysis of digital gene expression data. Bioinformatics . 2010;26(1):139–140. doi: 10.1093/bioinformatics/btp616. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Carbon S., Ireland A., Mungall C. J., et al. AmiGO: online access to ontology and annotation data. Bioinformatics . 2009;25(2):288–289. doi: 10.1093/bioinformatics/btn615. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Sampath H., Vartanian V., Rollins M. R., Sakumi K., Nakabeppu Y., Lloyd R. S. 8-Oxoguanine DNA glycosylase (OGG1) deficiency increases susceptibility to obesity and metabolic dysfunction. PLoS One . 2012;7(12, article e51697) doi: 10.1371/journal.pone.0051697. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Osterod M., Hollenbach S., Hengstler J. G., Barnes D. E., Lindahl T., Epe B. Age-related and tissue-specific accumulation of oxidative DNA base damage in 7,8-dihydro-8-oxoguanine-DNA glycosylase (Ogg1) deficient mice. Carcinogenesis . 2001;22(9):1459–1463. doi: 10.1093/carcin/22.9.1459. [DOI] [PubMed] [Google Scholar]
- 20.Stuart J. A., Bourque B. M., de Souza-Pinto N. C., Bohr V. A. No evidence of mitochondrial respiratory dysfunction in OGG1-null mice deficient in removal of 8-oxodeoxyguanine from mitochondrial DNA. Free Radical Biology & Medicine . 2005;38(6):737–745. doi: 10.1016/j.freeradbiomed.2004.12.003. [DOI] [PubMed] [Google Scholar]
- 21.Moller P., Lohr M., Folkmann J. K., Mikkelsen L., Loft S. Aging and oxidatively damaged nuclear DNA in animal organs. Free Radical Biology & Medicine . 2010;48(10):1275–1285. doi: 10.1016/j.freeradbiomed.2010.02.003. [DOI] [PubMed] [Google Scholar]
- 22.Li J., Wang P. Y., Long N. A., et al. p53 prevents doxorubicin cardiotoxicity independently of its prototypical tumor suppressor activities. Proceedings of the National Academy of Sciences of the United States of America . 2019;116(39):19626–19634. doi: 10.1073/pnas.1904979116. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Wallace K. B., Sardao V. A., Oliveira P. J. Mitochondrial determinants of doxorubicin-induced cardiomyopathy. Circulation Research . 2020;126(7):926–941. doi: 10.1161/CIRCRESAHA.119.314681. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Brzostek-Racine S., Gordon C., Van Scoy S., Reich N. C. The DNA damage response induces IFN. Journal of Immunology . 2011;187(10):5336–5345. doi: 10.4049/jimmunol.1100040. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Tang M. L., Khan M. K., Croxford J. L., Tan K. W., Angeli V., Gasser S. The DNA damage response induces antigen presenting cell-like functions in fibroblasts. European Journal of Immunology . 2014;44(4):1108–1118. doi: 10.1002/eji.201343781. [DOI] [PubMed] [Google Scholar]
- 26.Tretina K., Park E. S., Maminska A., MacMicking J. D. Interferon-induced guanylate-binding proteins: guardians of host defense in health and disease. The Journal of Experimental Medicine . 2019;216(3):482–500. doi: 10.1084/jem.20182031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Pradervand S., Yasukawa H., Muller O. G., et al. Small proline-rich protein 1A is a gp130 pathway- and stress-inducible cardioprotective protein. The EMBO Journal . 2004;23(22):4517–4525. doi: 10.1038/sj.emboj.7600454. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Henninger C., Huelsenbeck S., Wenzel P., et al. Chronic heart damage following doxorubicin treatment is alleviated by lovastatin. Pharmacological Research . 2015;91:47–56. doi: 10.1016/j.phrs.2014.11.003. [DOI] [PubMed] [Google Scholar]
- 29.Kratsios P., Catela C., Salimova E., et al. Distinct roles for cell-autonomous Notch signaling in cardiomyocytes of the embryonic and adult heart. Circulation Research . 2010;106(3):559–572. doi: 10.1161/CIRCRESAHA.109.203034. [DOI] [PubMed] [Google Scholar]
- 30.Sasaki Y., Ishida S., Morimoto I., et al. The _p53_ family member genes are involved in the Notch signal pathway∗. The Journal of Biological Chemistry . 2002;277(1):719–724. doi: 10.1074/jbc.M108080200. [DOI] [PubMed] [Google Scholar]
- 31.Narikawa M., Umemura M., Tanaka R., et al. Doxorubicin induces trans-differentiation and MMP1 expression in cardiac fibroblasts via cell death-independent pathways. PLoS One . 2019;14(9, article e0221940) doi: 10.1371/journal.pone.0221940. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Zhang Y. W., Shi J., Li Y. J., Wei L. Cardiomyocyte death in doxorubicin-induced cardiotoxicity. Archivum Immunologiae et Therapiae Experimentalis (Warsz) . 2009;57(6):435–445. doi: 10.1007/s00005-009-0051-8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 33.Shah A., Gray K., Figg N., Finigan A., Starks L., Bennett M. Defective Base excision repair of oxidative DNA damage in vascular smooth muscle cells promotes atherosclerosis. Circulation . 2018;138(14):1446–1462. doi: 10.1161/CIRCULATIONAHA.117.033249. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Tumurkhuu G., Shimada K., Dagvadorj J., et al. Ogg1-dependent DNA repair regulates NLRP3 inflammasome and prevents atherosclerosis. Circulation Research . 2016;119(6):e76–e90. doi: 10.1161/CIRCRESAHA.116.308362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Vela-Guajardo J. E., Perez-Trevino P., Rivera-Alvarez I., Gonzalez-Mondellini F. A., Altamirano J., Garcia N. The 8-oxo-deoxyguanosine glycosylase increases its migration to mitochondria in compensated cardiac hypertrophy. Journal of the American Society of Hypertension . 2017;11(10):660–672. doi: 10.1016/j.jash.2017.08.004. [DOI] [PubMed] [Google Scholar]
- 36.Cividini F., Scott B. T., Dai A., et al. _O_ -GlcNAcylation of 8-oxoguanine DNA glycosylase (Ogg1) impairs oxidative mitochondrial DNA lesion repair in diabetic hearts. The Journal of Biological Chemistry . 2016;291(51):26515–26528. doi: 10.1074/jbc.M116.754481. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Ye Y., Lin P., Zhang W., et al. DNA repair interacts with autophagy to regulate inflammatory responses to pulmonary hyperoxia. Journal of Immunology . 2017;198(7):2844–2853. doi: 10.4049/jimmunol.1601001. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Supplementary Table 1: primer list.
Supplementary Table 2: differentially expressed genes between 4-week-old WT and KO CMs.
Supplementary Table 3: differentially expressed genes between KO saline-treated and WT saline-treated CMs.
Supplementary Table 4: gene ontology terms in upregulated genes in the KO saline compared to WT Saline.
Supplementary Table 5: gene ontology terms in downregulated genes in the KO saline compared to WT Saline.
Supplementary Table 6: differentially expressed genes between KO doxorubicin-treated and KO saline-treated CMs.
Supplementary Table 7: differentially expressed genes between WT doxorubicin-treated and WT saline-treated CMs.
Supplementary Table 8: commonly dysregulated genes and gene ontology terms in KO DXB- and WT DXB-treated cells.
Supplementary Table 9: gene ontology terms in the genes only dysregulated in the KO DXB-treated CMs.
Supplementary Table 10: differentially expressed genes between KO doxorubicin-treated and WT doxorubicin-treated CMs.
Supplementary Table 11: gene ontology terms in upregulated genes in the KO doxorubicin-treated CMs compared to WT doxorubicin-treated CMs.
Data Availability Statement
All supporting data to be made available upon request.