

Highlight
● IGSF1 may be the cause in patients with congenital hypogonadotropic hypogonadism and central hypothyroidism.
Introduction
Individuals with deletions and/or pathogenic variants of the Immunoglobulin superfamily 1 (IGSF1) gene may show congenital central hypothyroidism (CCH) (1,2,3). In addition, these individuals may have PRL deficiency and, in a small number of cases, GH deficiency. Furthermore, the onset of puberty tends to be delayed, and is often accompanied by giant testes. Despite the early replacement of thyroid hormone, CCH may be accompanied by developmental disorders and attention deficit hyperactivity syndrome (2, 3). However, asymptomatic cases have also been reported.
We report the identification of a novel nonsense variant (p.Arg1293Ter) of IGSF1 in a young male patient with congenital hypogonadotropic hypogonadism (CHH), CCH, and GH deficiency.
Case Report
Our patient was a 17-yr-old boy born after 36 wk of gestation by normal vaginal delivery (weight: 2150 g; length: 45 cm). Family history showed no indication of endocrine disorders. Cryptorchidism was noted at birth, but no follow up or endocrinological tests were done at that time.
During a health examination at 18 mo of age, a delay in psychomotor development was noted, and he was followed up at our neurodevelopmental department as an outpatient. At 5 yr of age, he was diagnosed with a pervasive developmental disorder.
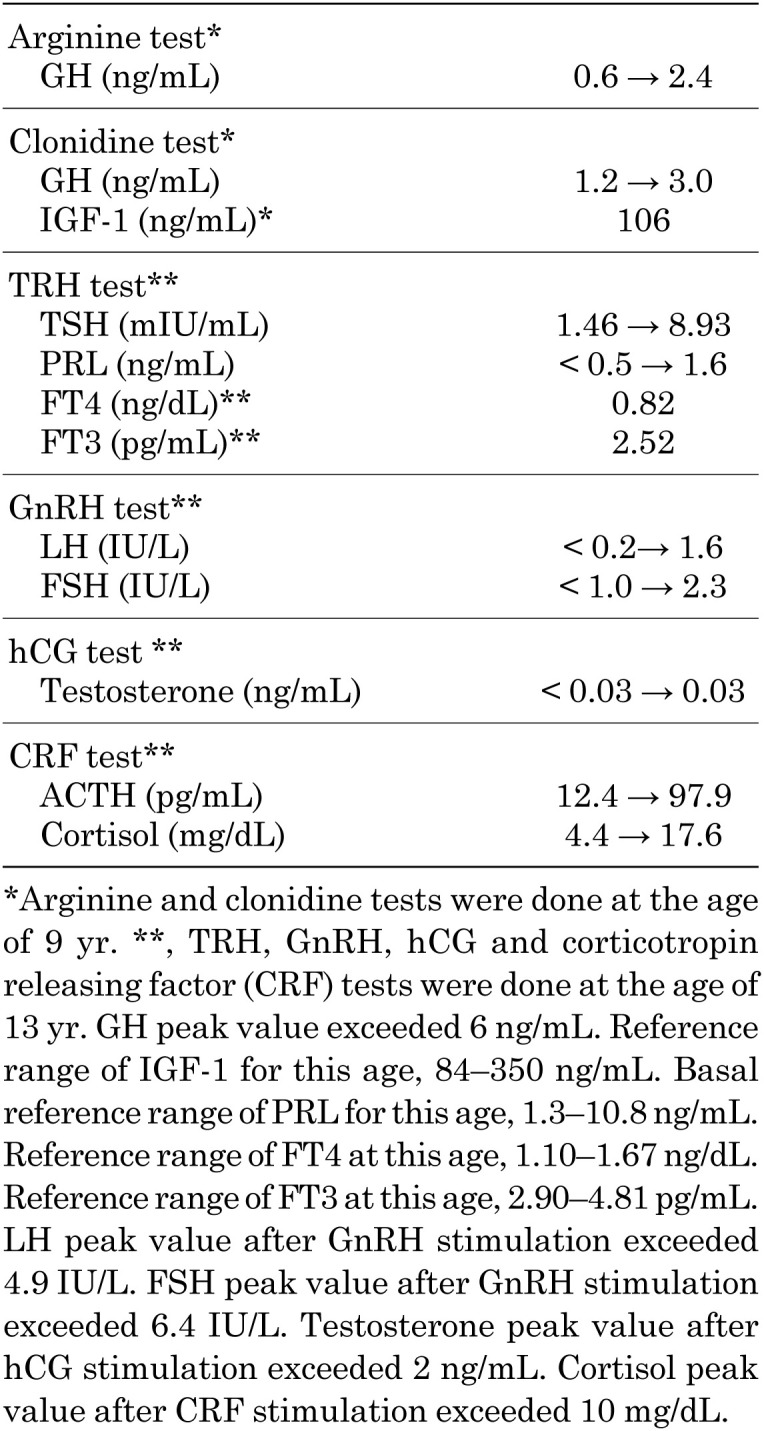
He was of a short stature at about 6 yr of age, and when 9, he was referred to endocrine outpatient department. At that time, he was 116.2 cm tall (–2.6 SD for a normal Japanese boy), and weighed 24 kg (–1 SD for normal Japanese boy) (Fig. 1A). Endocrinological examination showed IGF-1 in the normal range (106 ng/mL; normal range for his age, 84–350 ng/mL), while GH provocative tests of arginine and clonidine showed low responses of GH (0.6 to 2.4 ng/mL and 1.2 to 3.0 ng/mL, respectively) (Table 1). Thyroid function was as follows (normal range in parentheses): TSH 1.45 mU/L (0.24–3.50 mIU/L), FT3 2.45 pg/ml (2.45–4.6 pg/mL), and FT4 0.89 ng/dL (1.07–2.10 ng/dL). Based on these findings, the patient was diagnosed with GH deficiency, and GH supplementation was initiated, but CCH was not noted. At 13 yr of age, he had not developed secondary sexual characteristics, and showed a bilateral testicular volume of 2 mL and a micropenis (penile length, 2 cm). A detailed interview revealed that he had a reduced sense of smell. Endocrine tests are summarized in Table 1. Magnetic resonance imaging of the brain showed no olfactory bulb, but a normal pituitary gland and stalk (Fig. 1B, C). His testosterone level did not respond (< 0.03 to < 0.03 ng/mL) in the hCG loading test. Based on the above results, the patient was diagnosed with hypogonadotropic hypogonadism, central hypothyroidism, and PRL deficiency. Thyroid hormone replacement was initiated at this time, and testosterone replacement was started at 15 yr of age.
Fig. 1.

(A) Growth chart of the patient. (B, C) Magnetic resonance imaging showing no olfactory bulb, but a normal pituitary gland and stalk. (D) Sanger sequencing showing the c.3877C>T (p. Arg1293Ter) (Red arrow).
Table 1. Endocrinological findings.
Genetic analysis was approved by the institutional review board of Jichi Medical University, and consents of the patient and his parents were obtained. A total of 25 genes (CHD7, FGF8, FGFR1, GNRH1, GNRHR, ANOS1, KISS1R, PROKR2, TACR3, IGSF1, LKISS1, PROK2, SOX10, TAC3, WDR11, HESX1, LHX3, LHX4, OTX2, POU1F1, PROKR2, PROP1, SOX2, SOX3, GLI2) were screened using a next-generation sequencer (Miseq system. Illumina). The identified variant of IGSF1 was sequenced by the Sanger method, to find a novel pathological variant of IGSF1, with c.3877C>T (p. Arg1293Ter) in a hemizygous state (Fig. 1D). No other pathogenic variant was identified.
Discussion
We identified a novel pathogenic nonsense variant of IGSF1 in a young male patient with CHH, olfactory bulb hypoplasia, CCH, GH deficiency, and PRL deficiency.
Characteristics of IGSF1 abnormality, CCH and PRL deficiencies are consistent in this patient. IGSF1 dysfunction may be accompanied by GH deficiency, which was alleviated by thyroid hormone replacement. However, in this 9-yr-old patient, IGSF1 dysfunction was coexistent with CCH. In addition, the patient was diagnosed with a pervasive developmental disorder. Patients with IGSF1 abnormalities are known to have developmental disorders and attention deficit hyperactivity syndrome. IGSF1 abnormalities have also been reported to cause giant testes and high birth weight, however, in this case, the testicular volume was only 2 mL and birth weight was low.
In our patient, CCH was accompanied by CHH. While pathogenic variant of IGSF1 may underlie CCH, the exact cause of CHH has not been determined. Many genes that cause CHH have been identified, but the CHH-related genes analyzed in the present study are limited. However, a report of CHH with analysis of IGSF1 had found 36 CHH-related genes in 130 patients with CHH from Brazil, and a rare variant of p.Pro237Ala of IGSF1 was identified in one of them, who had a normal sense of smell (4). Further analyses of a large number of CHH cases is required.
Asakura et al. (5) have reported a case with anterior pituitary hypoplasia, ectopic posterior lobe, and invisible stalk, accompanied by CCH and GH deficiency due to an N-terminal pathogenic variant of IGSF1; however, LH and FSH levels were found to be normal.
In conclusion, IGSF1 abnormalities should be considered in patients with CHH and central hypothyroidism.
Conflict of interests: The authors declare no conflict of interest.
Ethical consideration: Written informed consent for publication and genetic testing was obtained from the patient and patient’s parents.
Acknowledgments
This work was supported in part by the Japan Agency for Medical Research and Development (AMED) under grant number 20gk0110050h0001 (Chief Investigator: Go Tajima).
References
- 1.Nakamura A, Bak B, Silander TL, Lam J, Hotsubo T, Yorifuji T, et al. Three novel IGSF1 mutations in four Japanese patients with X-linked congenital central hypothyroidism. J Clin Endocrinol Metab 2013;98: E1682–91. doi: 10.1210/jc.2013-1224 [DOI] [PubMed] [Google Scholar]
- 2.Joustra SD, Heinen CA, Schoenmakers N, Bonomi M, Ballieux BE, Turgeon MO, et al. IGSF1 Clinical Care Group. IGSF1 Deficiency: Lessons from an extensive case series and recommendations for clinical management. J Clin Endocrinol Metab 2016;101: 1627–36. doi: 10.1210/jc.2015-3880 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Tajima T, Nakamura A, Oguma M, Yamazaki M. Recent advances in research on isolated congenital central hypothyroidism. Clin Pediatr Endocrinol 2019;28: 69–79. doi: 10.1297/cpe.28.69 [DOI] [PMC free article] [PubMed] [Google Scholar]
- 4.Amato LGL, Montenegro LR, Lerario AM, Jorge AAL, Guerra Junior G, Schnoll C, et al. New genetic findings in a large cohort of congenital hypogonadotropic hypogonadism. Eur J Endocrinol 2019;181: 103–19. doi: 10.1530/EJE-18-0764 [DOI] [PubMed] [Google Scholar]
- 5.Asakura Y, Abe K, Muroya K, Hanakawa J, Oto Y, Narumi S, et al. Combined growth hormone and thyroid-stimulating hormone deficiency in a Japanese patient with a novel frameshift mutation in IGSF1. Horm Res Paediatr 2015;84: 349–54. doi: 10.1159/000438672 [DOI] [PubMed] [Google Scholar]