

Abstract
The synthesis of new chiral highly functionalized zwitterionic bicyclic lactams starting from acyclic β-enaminoesters derived from (R)-(−)-2-phenylglycinol is described. The key step involved an intramolecular non-classical Corey–Chaykovsky ring-closing reaction of the corresponding sulfonium salts derived from β-enaminoesters. This methodology permits the generation of two or three new stereogenic centers with high diastereoselectivity. The utility of these intermediates was demonstrated by the stereocontrolled total synthesis of cis-4-hydroxy-2-methyl piperidine and its corresponding pipecolic acid derivative.
The synthesis of new chiral highly functionalized zwitterionic bicyclic lactams starting from acyclic β-enaminoesters derived from (R)-(−)-2-phenylglycinol is described.
Stereocontrolled synthesis of polysubstituted piperidines is a very important field in organic synthesis due to the great variety of piperidine-moiety-containing drugs, making it the most prevalent nitrogen heterocycle in approved drugs;1 therefore, their synthesis has been the focus of considerable attention.
In this sense, chiral non-racemic bicyclic lactams derived from β-amino alcohols are commonly considered useful starting materials for preparing these substituted piperidine drugs.2 One of the most effective methodologies for the synthesis of these bicyclic lactams is via an aza-annulation of β-enaminoesters3 derived from suitable β-amino alcohols.4
We recently reported the preparation of new cyclic zwitterionic intermediates via an intramolecular non-classical Corey–Chaykovsky ring-closing reaction. Their utility was demonstrated by the synthesis of stereoisomers of σ1-receptor agonists trozamicol5 or the synthesis of (+)- and (−)-Geissman–Waiss lactone (Scheme 1).6
Scheme 1. Previous reports in the synthesis and utility of chiral cyclic zwitterionic intermediates.

Considering this precedent and highlighting the utility of cyclic zwitterionic intermediates, we present a new versatile methodology to access new chiral zwitterionic non-racemic bicyclic lactams starting from acyclic β-aminoesters derived from (R)-(−)-2-phenylglycinol and its utility in the diastereoselective synthesis of 2-substituted-4-hydroxy piperidines.
We commence our finding by synthesizing of a set of β-enaminoesters coming from (R)-(−)-2-phenylglycinol, which were prepared following the traditional methodologies, directly condensation of chiral amine with β-dicarbonyl compound7 or by addition to a corresponding alkyne.8 All enamino esters were obtained as an inseparable E : Z isomeric mixture and, these results are summarized below (Fig. 1).
Fig. 1. β-Enamino esters derived from (R)-(−)-2-phenylglycinol.

With a set of chiral β-enaminoesters in hand, next these compounds were condensed with bromo acetyl bromide,9 affording the desirable N-acyl oxazolidine with the formation of a new stereogenic center at the hemiaminal position in good to excellent diastereomeric ratio. The stereochemistry at the new stereogenic center of the major diastereoisomer was determined from its X-ray diffraction analysis of N-acyl oxazolidine 10 as (2R),10 therefore we assumed that this is the stereochemistry of the major diastereoisomer of each N-acyl oxazolidine (Scheme 2).
Scheme 2. Synthesis of N-acyl oxazolidines. a Only the major diastereoisomer is shown. bN-acyl oxazolidines 12 and 14 were probed to be unstable therefore were employed without purification for the next reaction.

Then, the diastereomeric mixture of 8 was selected as a model for preparing the desired zwitterionic chiral bicyclic lactam. To this end, 8 was treated with dimethyl sulfide in DCM,11 delivering the corresponding sulfonium salt which was immediately subjected to the intramolecular non-classical Corey–Chaykovsky ring-closing reaction to avoid its conversion to the undesired thioether derivative.
Firstly, sulfonium salt was treated with KOH (2 equiv.) in a mixture of CH3CN : MeOH (9 : 1).6 Unfortunately, a mixture of expected zwitterion 22(a+b) and undesired pyridine-2-one 22c, was obtained. Therefore, screening experiments were performed. Gratifyingly, when the base was changed by K2CO3, the diastereomeric mixture of zwitterionic bicyclic lactams was exclusively obtained. 1H-NMR analysis of the crude reaction revealed the presence of a diastereomeric mixture of bicyclic zwitterionic intermediates in a 73 : 27 dr. Two hemiaminal protons were assigned as a doublet of doublets at 5.40 ppm and 5.12 ppm with coupling constants J = 9.8, 5.3 Hz for the minor diastereoisomer and J = 11.4, 4.2 Hz for the major diastereoisomer. The assignment was confirmed by a 1H–1H COSY experiment (Table 1).
Screening studies for nonclassical Corey–Chaykovsky ring-closing reactiona.
| ||
|---|---|---|
| Entry | Base | Global yield [ratio 22(a+b)b : 22c] |
| 1 | KOH (2.0 equiv.) | 95% [76 : 24] |
| 2 | KOH (1.0 equiv.) | 83% [82 : 18] |
| 3 | K2CO3 (1.2 equiv.) | 97% [100 : 0] |
All reactions were performed in CH3CN : MeOH (9 : 1), for 4 hours at room temperature.
22(a+b) was obtained in a 73 : 27 dr.
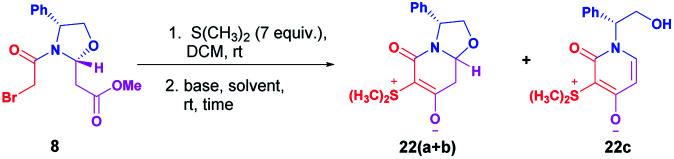
The absolute configuration at the hemiaminal position of the zwitterionic bicyclic diastereomeric mixture was determined by its X-ray diffraction analysis. A single crystal was found to include two independent molecules in the asymmetric unit of a triclinic cell (space group P1 with Z′ = 2), one of which presenting a disorder on the chiral center C8a. Combining these features results in a mixture of disatereoisomers with a ratio refined to 77.5/22.5%,12 close to that determined by NMR in solution, 73/27%. The determination of the absolute configuration in this crystal structure allowed the assignment for the major diastereoisomer 22a as (8aR) and the minor diastereoisomer 22b as (8aS) (Fig. 2).
Fig. 2. X-ray structure of the diastereomeric mixture of chiral zwitterionic bicyclic intermediates 22(a+b).
Once the optimal reaction conditions were established, the scope of our synthetic proposal was investigated. Chiral zwitterionic bicyclic intermediates containing alkyl or aryl substituents at the hemiaminal position were obtained in excellent chemical and stereochemical yields. Furthermore, the absolute configuration at the hemiaminal position was determined as (8aR) for the major diastereoisomer from the X-ray diffraction analysis of zwitterions 23a and 24a (Scheme 3).12 In the case of 24a, orthorhombic single crystals feature three independent molecules in the asymmetric unit (space group P212121, Z′ = 3), however, all display the same conformation and stereochemistry.
Scheme 3. Intramolecular non-classical Corey–Chaykovsky ring-closing reaction for the synthesis of chiral zwitterionic bicyclic intermediates.

To demonstrate the utility of these new chiral zwitterionic bicyclic compounds, the diastereoselective synthesis of 4-hydroxy piperidines was developed and 23a was selected as a model, therefore this compound was subjected to a desulfurization process to access the corresponding oxazolo[3,2-a]pyridine-5,7(6H)-dione 29. Catalytic hydrogenation was first attempted under various heterogeneous hydrogenation conditions. The use of Pd–C or RANEY®-Ni as a catalyst in acidic or neutral conditions led the oxazolo[3,2-a]pyridine-5,7(6H)-dione 29 in low chemical yield. The best result was obtained when reductive desulfurization was conducted using Zn (15 equiv.) in acetic acid, affording the compound 29 in 98% chemical yield (Table 2, entry 8). In addition, compound 29 provided suitable crystals for X-ray crystallographic analysis confirming the presence of an oxazolo[3,2-a]pyridine-5,7(6H)-dione core and enabling the determination of the absolute configuration at the hemiaminal position as (8aR),12 as in the starting material 23a.
Screening experiments for desulfurization of zwitterion 23aa.
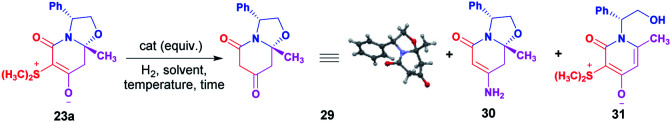
| ||||||
|---|---|---|---|---|---|---|
| Entry | Catalyst (mol%) | Solvent | Temp | Time | Product | Yield (%) |
| 1 | Pd/C (10% mol) | EtOH | rt | 16 h | 29 | Traces |
| 2 | Pd/C (10% mol) | EtOH | Reflux | 16 h | 29 | Traces |
| 3 | Pd/C (10% mol) | EtOH | Reflux | 4 h | 29 : 30 : 31 | 38 : 40 : 13 |
| 4 | RANEY®-Ni | THF : H2O (8 : 2) | rt | 8 h | 29 | 6 |
| 5 | RANEY®-Ni | Acetone | rt | 14 h | 29 | 19 |
| 6 | RANEY®-Ni | EtOH | −10 °C to rt | 8 h | 29 | 13 |
| 7 | RANEY®-Ni | Toluene | Reflux | 8 h | 31 | 38 |
| 8 | Zn (15 equiv.) | AcOH | rt | 7 days | 29 | 98 |
Ammonium formate was used as a hydrogen source.
Next, compound 29 was subjected to ketone reduction with NaBH4 (10 equiv.) in MeOH. The NMR spectrum of the crude reaction showed the presence of a diastereomeric mixture of 7-hydroxy oxazolo[3,2-a]pyridin-5-one 32(a+b) in an 87 : 13 diastereomeric ratio. Once the diastereomeric mixture was separated, the major diastereoisomer 32a crystallized, determining the new stereogenic center bearing the hydroxy group as (7R).12 The asymmetric unit of 32a contains two independent molecules (space group P21, Z′ = 2), both with the same geometry. The diastereoselectivity observed could be explained by the addition of the hydride reagent from the less hindered face, opposite to the methyl group (Scheme 4). After, reduction of the amide and hemiaminal functions of alcohol 32a was performed with BH3·DMS reagent, affording the 2,4-cis-2-methyl-4-hydroxy piperidine 33a as a major diastereoisomer for which the absolute configuration was unambiguously established by X-ray analysis.12 Indeed, the methyl and hydroxyl substituents are arranged cis in the piperidine ring.
Scheme 4. Synthesis of (2S,4S)-2-methylpiperidin-4-ol 35 and hydrochloride salt of (2R,4R,6S)-4-hydroxy-6-methylpiperidine-2-carboxylic acid 38.14.

Finally, complete synthesis of cis-4-hydroxy-2-methyl piperidine was accomplished by a simple debenzylation N-Boc protection reaction, followed by an acidic treatment to get 35 in quantitative yield. On the other hand, the synthesis of pipecolic acid derivative 38 was obtained through a 3-steps sequence reaction starting from intermediate 34, which involved silyl protection of the hydroxyl function followed by a diastereoselective deprotonation-electrophilic quenching endocyclic carbonylation process to access at the pipecolic acid analog 38. The absolute stereochemistry was assigned via the X-ray diffraction analysis of the corresponding hydrochloride salt. This compound crystallizes as a monohydrate, and the configuration (2R, 4R, 6S)12 is consistent with the refinement of a Flack parameter, x = 0.06(5)13 (Scheme 4).
Conclusions
In summary, we have developed a new concise high diastereoselective strategy route to access new zwitterionic bicyclic oxazolo intermediates starting from chiral β-enaminoesters derived from (R)-(−)-2-phenylglycinol. In addition, the utility of these new intermediates was also demonstrated in a short diastereoselective synthesis of (2S,4S)-2-methylpiperidin-4-ol and hydrochloride salt of (2R,4R,6S)-4-hydroxy-6-methylpiperidine-2-carboxylic acid. Applying this methodology to the synthesis of more complex pipecolic acids products is currently underway in our laboratory, and the results of these efforts will be reported in due course.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
We are grateful to CONACyT (Project A1-S-13280 and 268178) and VIEP-BUAP (Project 100148077-VIEP2021) for financial support. ERB thanks to CONACyT for the scholarship (658611). We thank A. L. García-Torres for HRMS spectrometry analyses.
Electronic supplementary information (ESI) available. CCDC 2125364–2125371. For ESI and crystallographic data in CIF or other electronic format see DOI: 10.1039/d1ra09298g
Notes and references
- Vitaku E. Smith D. T. Njardarson J. T. J. Med. Chem. 2014;57:10257. doi: 10.1021/jm501100b. [DOI] [PubMed] [Google Scholar]
- Roa L. F. Gnecco D. Galindo A. Terán J. L. Tetrahedron: Asymmetry. 2004;15:3393. doi: 10.1039/B200020M. [DOI] [Google Scholar]; Roa L. F. Gnecco D. Galindo A. Terán J. L. Bernès S. Tetrahedron: Asymmetry. 2004;15:847. doi: 10.1039/B200020M. [DOI] [Google Scholar]; Semak V. Escolano C. Arróniz C. Bosch J. Amat M. Tetrahedron: Asymmetry. 2010;21:2542. doi: 10.1039/B200020M. [DOI] [Google Scholar]; Amat M. Escolano C. Llor N. Huguet M. Pérez M. Bosch J. Tetrahedron: Asymmetry. 2003;14:1679. doi: 10.1039/B200020M. [DOI] [Google Scholar]; Amat M. Cantó M. Llor N. Bosch J. Chem. Commun. 2002:526. doi: 10.1039/B200020M. [DOI] [PubMed] [Google Scholar]; Groaning M. D. Meyers A. I. Tetrahedron. 2000;56:9843. doi: 10.1039/B200020M. [DOI] [Google Scholar]
- Cavé C. Daley V. d'Angelo J. Tetrahedron: Asymmetry. 1995;6:79. doi: 10.1016/0957-4166(94)00356-G. [DOI] [Google Scholar]; Ferraz H. M. C. Olveira E. O. Payret-Arrua M. E. Brandt C. A. J. Org. Chem. 1995;60:7357. doi: 10.1016/0957-4166(94)00356-G. [DOI] [Google Scholar]; Valduga C. J. Braibante H. S. Braibante E. F. J. J. Heterocycl. Chem. 1998;35:189. doi: 10.1016/0957-4166(94)00356-G. [DOI] [Google Scholar]; Amougay A. Letsh O. Pete J. P. Tetrahedron. 1996;52:2405. doi: 10.1016/0957-4166(94)00356-G. [DOI] [Google Scholar]; Cimarelli C. Palmieri G. J. Org. Chem. 1996;61:5557. doi: 10.1016/0957-4166(94)00356-G. [DOI] [Google Scholar]; Ali S. Khan A. T. Tetrahedron Lett. 2013;54:436. doi: 10.1016/0957-4166(94)00356-G. [DOI] [Google Scholar]
- Hickmott P. W. Sheppard G. J. Chem. Soc. C. 1971:1358. doi: 10.1039/J39710001358. [DOI] [Google Scholar]; Hickmott P. W. Sheppard G. J. Chem. Soc. C. 1971:2112. doi: 10.1039/J39710001358. [DOI] [Google Scholar]; Agami C. Dechoux L. Hebbe S. Tetrahedron Lett. 2003;44:5311. doi: 10.1039/J39710001358. [DOI] [Google Scholar]; Agami C. Dechoux L. Ménard C. Hebbe S. J. Org. Chem. 2002;67:7573. doi: 10.1039/J39710001358. [DOI] [PubMed] [Google Scholar]; Agami C. Dechoux L. Hebbe S. Ménard C. Tetrahedron. 2004;60:5433. doi: 10.1039/J39710001358. [DOI] [Google Scholar]
- López-González R. Zarate A. Aparicio D. M. Mendoza A. Gnecco D. Juárez J. R. Romero-Ceronio N. Orea L. Terán J. L. Tetrahedron Lett. 2016;57:1683. doi: 10.1016/j.tetlet.2016.03.029. [DOI] [Google Scholar]
- López-González R. Gnecco D. Juárez J. R. Gómez-Calvario V. Bernès S. Aparicio D. M. Terán J. L. Tetrahedron Lett. 2020;61:151697. doi: 10.1016/j.tetlet.2020.151697. [DOI] [Google Scholar]
- Martin D. F. Janusonis G. A. Martín B. B. J. Am. Chem. Soc. 1961;83:73. doi: 10.1039/B210165C. [DOI] [Google Scholar]; Rechsteiner B. Teixier-Boullet F. Hamelin J. Tetrahedron Lett. 1993;34:5071. doi: 10.1039/B210165C. [DOI] [Google Scholar]; Arcadi A. Bianchi G. Di Giuseppe S. Marinelli F. Green Chem. 2003;5:64. doi: 10.1039/B210165C. [DOI] [PubMed] [Google Scholar]; Khosropour A. R. Khodaei M. Kookhazadeh M. Tetrahedron Lett. 2004;45:1725. doi: 10.1039/B210165C. [DOI] [Google Scholar]; Zhang Z. H. Yin L. Want Y. M. Adv. Synth. Catal. 2006;348:184. doi: 10.1039/B210165C. [DOI] [Google Scholar]; Dalpozzo R. Deino A. Nardi M. Russo B. Procopio A. Synthesis. 2006:1127. doi: 10.1039/B210165C. [DOI] [Google Scholar]; Lin J. Shang L. F. Monatsh. Chem. 2007;138:77. doi: 10.1039/B210165C. [DOI] [Google Scholar]; Epofano F. Genovese S. Curini M. Tetrahedron Lett. 2007;48:2717. doi: 10.1039/B210165C. [DOI] [Google Scholar]
- Turunen B. J. Georg G. I. J. Am. Chem. Soc. 2006;128:8702. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]; Panda N. Mothkuri R. J. Org. Chem. 2012;77:9407. doi: 10.1021/ja0609046. [DOI] [PubMed] [Google Scholar]; Pilotzi H. Gnecco D. Orea M. L. Juárez J. R. Aparicio D. M. Terán J. L. Heterocycles. 2018;96:895. doi: 10.1021/ja0609046. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Aparicio D. M. Gnecco D. Juárez J. R. Orea M. L. Waksman N. Salazar R. Flores-Alamo M. Terán J. L. Tetrahedron. 2012;68:10252. doi: 10.1016/j.tet.2012.09.047. [DOI] [Google Scholar]
- CCDC: 2125364 (10), contain the ESI crystallographic data for this paper.†
- Aparicio D. M. Terán J. L. Gnecco D. Galindo A. Juárez J. R. Orea M. L. Mendoza A. Tetrahedron: Asymmetry. 2009;20:2764. doi: 10.1016/j.tetasy.2009.12.004. [DOI] [Google Scholar]
- CCDC: 2125365 (22(a+b)), 2125366 (23a), 2125367 (24a), 2125368 (29), 2125369 (32a), 2125370 (33a), 2125371 (38) contain the ESI†
- Flack H. D. Bernardinelli G. Acta Crystallogr. Sect. A. 1999;55:908. doi: 10.1107/S0108767399004262. [DOI] [PubMed] [Google Scholar]
- In this article, Authors reported the corresponding pipecolic acid enantiomer: ; Fowler L. S. Thomas L. H. Ellis D. Sutherland A. Chem. Commun. 2011;47:6569. doi: 10.1039/C1CC11916H. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.