

Abstract

A series of gold(I)–ethylene π-complexes containing a family of bulky phosphine ligands has been prepared. The use of these sterically congested ligands is crucial to stabilize the gold(I)–ethylene bond and prevent decomposition, boosting up their catalytic performance in the highly underexplored hydroamination of ethylene. The precatalysts bearing the most sterically demanding phosphines showed the best results reaching full conversion to the hydroaminated products under notably mild conditions (1 bar of ethylene pressure at 60 °C). Kinetic analysis together with density functional theory calculations revealed that the assistance of a second molecule of the nucleophile as a proton shuttle is preferred even when using an extremely congested cavity-shaped Au(I) complex. In addition, we have measured a strong primary kinetic isotopic effect that is consistent with the involvement of X–H bond-breaking events in the protodeauration turnover-limiting step.
Keywords: gold catalysis, π-complexes, ethylene functionalization, hydroamination, bulky phosphines
Introduction
For decades, gold has been considered too chemically inert to be used in catalysis.1 However, since the discovery of its ability to activate π-bonds toward nucleophilic addition, molecular gold complexes have played a prominent role in the catalytic transformation of unsaturated hydrocarbons.2 The number of reactions mediated by π-acid gold catalysis is extensive and includes hydrogenation, oxidation, diarylation, heteroarylation, or cycloadditions, among many others.3 A type of transformation that has been extensively studied as a versatile route to prepare nitrogen-containing compounds with optimal atom economy is hydroamination, that is, the addition of an N–H unit of nucleophilic amines (or related substrates) across a carbon–carbon multiple bond.4 Although these processes can be catalyzed by other transition metals5 and even through metal-free protocols,6 gold(I) complexes remain one of the most powerful hydroamination catalysts.3j,7,8 In fact, they can accomplish the intermolecular hydroamination of C≡C triple bonds9 and even the more challenging C=C double bonds,10,11 in some cases even for inactivated alkenes. For the latter, the Au(I)-catalyzed hydroamination of ethylene, the simplest alkene, has only been reported once.12
Coordination of a C–C multiple bond to form a gold π-complex is usually proposed as the initial step during π-acid-catalyzed reactions, including hydroamination. Thus, the isolation of gold π-complexes has gathered considerable interest associated with their catalytic relevance, because they serve as models for the transient gold π-complexes.13 Among those, cationic dicoordinate gold(I) π-complexes of substituted alkenes and alkynes have been isolated and characterized over the last decade using phosphine or N-heterocyclic carbene ligands.14 Chelating N- and P-based ligands have also proved useful to form tricoordinate gold π-complexes.15 However, despite the interest in developing efficient methods for ethylene functionalization, gold(I)–ethylene complexes are quite rare; only 10 examples can be found in the literature and mainly using chelating ligands.16,17 In fact, we have recently authenticated the first dicoordinate gold(I)–ethylene adduct using the extremely bulky tris-2-(4,4′-di-tert-butylbiphenylyl)phosphine (L1), previously reported by Straub,18 that kinetically stabilizes the coordination of ethylene.19 In contrast to related tricoordinate complexes, the bonding interactions are mainly electrostatic (i.e., ionic) with minimal Au → ethylene π-backdonation.
This strategy of kinetic stabilization using sterically demanding ligands to detect and isolate transient intermediates of relevance to catalytic processes has proved successful in the past. Our group has also committed to the task, capitalizing on the steric shrouding provided by terphenyl (C6H3-2,6-Ar2) phosphine ligands.20 For instance, these have been used to access unusual gold compounds, such as the first methyl-bridged cationic digold complexes21 and to study their relevance in C–C coupling processes,22 as well as to exploit gold species as frustrated Lewis pair constituents.23 In this study, we have selected a family of bulky phosphine ligands in an attempt to access rare Au(I)–ethylene adducts. More precisely, we have used both the commercial ligands trimesityl phosphine (L2) and tBuXPhos (L3), as well as a series of terphenyl phosphines (L4–L8) prepared by our group (Figure 1).24 We compare herein the stability of the resulting ethylene adducts with respect to the first of its class constructed around L1(19) and examine their catalytic competence for the underdeveloped Au(I)-catalyzed functionalization of ethylene through a model hydroamination reaction.
Figure 1.

Selected bulky phosphine ligands used in this study.
Results and Discussion
Synthesis of Gold(I)–Ethylene Complexes
The reaction of [AuCl(THT)] (THT = tetrahydrothiophene) with phosphine ligands L1–L8 in dichloromethane forms the air-stable, neutral phosphine chloride complexes 1–8. The steric bulkiness of the phosphine ligands was evaluated calculating the percent buried volume (%Vbur),25 which yielded notably large parameters (Table 1 and Figure S86). Nonetheless, there are clear differences in the steric shrouding imparted by the employed phosphines. Terphenyl phosphine ligands containing two small methyl groups bound to the phosphorus atom present lower %Vbur values, ranging from 38.2 in L5 to 46.2 in L4 after substituting the methyl groups on the flanking aryl rings of the terphenyl substituent by isopropyl termini. Similar %Vbur parameters were measured for L7 and the widely used trimesityl phosphine (L2). Introducing bulkier substituents bound to the phosphorus atom in L6 and L8 increased the %Vbur to around 53, comparable to that of the Buchwald-type phosphine L3. Albeit the former are considerably bulky, the tris biaryl tris-2-(4,4′-di-tert-butylbiphenylyl)phosphine phosphine (L1) clearly presents the highest %Vbur value of 67.0.19 As discussed in the following sections, the steric profile of the ligand seems to be crucial to impart stability to the aimed Au(I)–ethylene compounds, having a direct effect on catalytic performance.
Table 1. Selected Spectroscopic and Structural Data of Complexes 1–8·C2H4.
| compound | δ1H (ppm) | δ13C (ppm) | δ31P (ppm)a | %Vburb | d(C=C) (Å) |
|---|---|---|---|---|---|
| C2H4 | 5.43 | 116.8 | 1.313c | ||
| 1·C2H4 | 3.66; 3.79 | 110.2 | 13.1 (9.5) | 67.0 | 1.236(10)d |
| 2·C2H4 | 5.46 | 111.2 | 1.5 (−5.4) | 45.3 | |
| 3·C2H4 | 4.95 | 110.9 | 65.6 (58.6) | 55.5 | 1.353(15) |
| 4·C2H4 | 4.85 | 110.3 | 4,3 (−5.7) | 46.2 | |
| 5·C2H4 | 5.00 | 111.5 | 4.1 (−3.2) | 38.2 | |
| 6·C2H4 | 4.86 | 111.0 | 57.6 (53.3) | 53.5 | |
| 7·C2H4 | 5.16 | 111.8 | 9.4 (0.4) | 45.0 | |
| 8·C2H4 | 4.77 | 109.0 | 55.4 (48.8) | 53.7 | 1.384(10) |
Treatment of gold(I) chloride complexes 1–8 with AgSbF6 under an ethylene atmosphere at −30 °C caused instantaneous precipitation of AgCl and formation of the gold(I)–ethylene complexes 1–8·C2H4 (Scheme 1). Filtration of the aforementioned reaction mixtures through short pads of Celite followed by washing with pentane afforded the pure gold(I) π-complexes 1–8·C2H4 in good to excellent yields (53–93%). The reactions were conveniently monitored by 31P{1H} NMR spectroscopy, which revealed a systematic downfield shift in the range from 4.3 to 10.0 ppm compared to the corresponding gold(I) chloride complexes (Table 1). It is worth noting that attempts to prepare the related [(Ph3P)Au(C2H4)]+ complex led to immediate decomposition and formation of [(PPh3)2Au]+ and Au(0), likely because of the inability of the relatively small PPh3 ligand to kinetically stabilize the corresponding Au(I)–ethylene adduct.
Scheme 1. Synthesis of Gold(I)–Ethylene Complexes 1–8·C2H4 (L = L1–L8 from Figure 1).

Complexes 2–8·C2H4 were spectroscopically characterized in dichloromethane solution under an ethylene atmosphere to prevent decomposition, which accelerates upon removal of the gaseous substrate. In some cases and because of the chemical exchange between coordinated and free ethylene (vide infra), the two signals were undistinguishable. To unambiguously identify the resonances belonging to coordinated ethylene, 1H NMR spectroscopy and 13C{1H} NMR spectroscopy were also performed in the absence of ethylene, though in those cases signs of decomposition were evident from NMR spectroscopy results (see the SI for more details). Nonetheless, these studies permitted the unambiguous assignment of the targeted ethylene adducts; resonances associated with the coordinated olefin were found to differ from those of the free molecule (Table 1). Thus, coordination to gold(I) induces a noticeable upfield shift of the 1H NMR signals (∼0.5 ppm) with the exception of complex 2·C2H4, which is only slightly downfield shifted by 0.06 ppm. In turn, 13C{1H} NMR resonances are shifted in the same direction with upfield shifts about 7 ppm with respect to free ethylene (Table 1). These relatively small changes suggest little backdonation from Au to the ethylene π*(C=C) orbital, as noted earlier for 1·C2H4,19 and in contrast with the related tricoordinate gold(I)–ethylene complexes,13f in which the chemical shift differences can reach up to 3 and 55 ppm in 1H and 13C NMR spectra, respectively. As for the more sterically hindered complex 1·C2H4,19 the coordinated ethylene presented the largest shift in 1H NMR resonances, which appear as an AA′BB′ system at 3.79 and 3.66 ppm, contrasting with the rest of the compounds that led to a single broad peak due to four equivalent protons. We ascribed the shift in 1·C2H4 to ring-current effects due to the surrounding aryl rings, which could also hinder the rotation of bound ethylene giving rise to the observed AA′BB′ system. Chemical exchange between coordinated and free ethylene was observed in CD2Cl2 within the NMR timescale for all ethylene adducts; however, its rate could not be reliably quantified because of the rapid exchange even at low temperature and the close proximity of the respective NMR signals, which prevented accurate data analysis (see Figures S57–S61 in the Supporting Information).
Single crystals of complexes 3·C2H4 and 8·C2H4 suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into saturated dichloromethane solutions of the gold(I)–ethylene complexes at −30 °C. Both species adopt similar structures in the solid state, with the gold center in a linear environment and the ethylene molecule coordinated in an η2-fashion (Figure 2). It is worth noting that in contrast to complexes 1·C2H4 and 3·C2H4, the coordination of ethylene to gold in 8·C2H4 is highly nonsymmetric: the ethylene molecule is notably slipped, that is, whereas it presents similar Au–C bond distances, the P–Au–C angles of 173.55(19)° and 137.2(2)° are remarkably different. In complexes 3·C2H4 and 8·C2H4, the Au–C bond lengths (2.21–2.26 Å) are noticeably longer than those described for gold(I)–ethylene adducts bearing bidentate ligands (ca. 2.14–2.17 Å),15,16 but similar to 1·C2H4 (2.216(6) and 2.235(6) Å) and related cationic dicoordinate gold(I) π-complexes of other alkenes.14 The C=C double bond (3·C2H4, 1.353(15) Å; 8·C2H4, 1.384(10) Å) is slightly longer than that of free ethylene (1.313 Å)26 and complex 1·C2H4 (1.263(10) Å) and similar to those described for tricoordinate gold(I) ethylene compounds,16 despite the expected poor Au → ethylene π-backdonation.
Figure 2.

ORTEP representation of complexes 3·C2H4, 8·C2H4, and [6]∞. Thermal ellipsoids are set at 50% probability. Counteranions, solvent molecules, and hydrogen atoms are excluded for clarity, while iso-propyl and cyclohexyl groups are represented in wireframe format. Selected bond length (Å) and angles (°): compound 3·C2H4, (one of two independent molecules per asymmetric unit; selected parameters from the one not showing disorder in the ethylene ligand), P1–Au1, 2.289(2); Au1–C62, 2.237(9); Au1–C63, 2.261(9); C62–C63, 1.353(15); P1–Au1–C62, 149.2(3); P1–Au1–C63, 164.8(3); compound 8·C2H4, P1–Au1, 2.2977(16); Au1–C47, 2.210(7); Au1–C48, 2.227(7); C47–C48, 1.384(10); P1–Au1–C47, 173.54(19); P1–Au1–C48, 137.2(2); compound [6]∞, P1–Au1, 2.2633(15); Au1–C17, 2.301(6); Au1–C18, 2.403(6); C17–C18, 1.385(11); P1–Au1–C17, 169.2(2); P1–Au1–C18, 151.5(2).
It was mentioned above that gold(I)–ethylene complexes 2–8·C2H4 exhibit slow decomposition both in the solid state and in dichloromethane solution upon removal of the ethylene atmosphere, which contrasts with the remarkable stability of 1·C2H4 that we have attributed to the kinetic stabilization imparted by the cavity-shaped phosphine. For all other cases, monitoring the evolution of dichloromethane solutions of the ethylene adducts by 31P{1H} NMR spectroscopy revealed the presence of the corresponding [P–Au–P]+ decomposition products along with Au(0) nanoparticles as the major products.22,27,28 Nonetheless, the appearance of other broad 31P{1H} signals evinces the formation of additional species. For instance, after a few days in solution the decomposition spectrum of complex 6·C2H4 revealed the formation of a relatively broad 31P{1H} NMR signal at 50.3 ppm distinct to the one corresponding to [(PCyp2ArXyl2)2Au]+ (53.4 ppm). X-ray diffraction studies allowed us to ascertain the formation of a new gold(I) cationic species ([6]∞) with a highly unusual polymeric structure derived from ethylene release and subsequent η2-coordination of a side aryl ring of the terphenyl substituent of an adjacent cationic gold fragment (Figure 2). The η2-coordination of the xylyl ring is slightly slipped with different Au–C distances of 2.301(6) and 2.401(7) Å and notably different P–Au–C angles of 169.2(2)° and 151.5(2)°, respectively. This structure is reminiscent of π-arene complexes of gold formed in aromatic solvents, which have been reported in several occasions and whose geometric parameters are comparable to [6]∞.29 However, this seems to be the first polymeric structure of this kind in which the building blocks are solely units of [LAu]+ connected by π-coordination.
Attempts to prepare other polymeric structures of this type by direct treatment of compounds 1–8 with equimolar amounts of AgSbF6 in dichloromethane were unsuccessful. In fact, while under an ethylene atmosphere instant precipitation of AgCl upon addition of the silver reagent was visually identified, this did not occur in the absence of the olefin, arguing in favor of the presence of silver within the resulting structure. This was not surprising considering our previous report on the reaction of complex 1 and AgSbF6, which resulted in the formation of a gold–silver trimetallic species without chloride abstraction. In the case of compounds 2 and [(Ph3P)AuCl], generation of the corresponding homoleptic [P–Au–P]+ complexes and Au(0) nanoparticles was exclusively observed. In contrast, complexes 3–8 bearing bulky biphenyl and terphenyl phosphine ligands do not lead to their corresponding [P–Au–P]+ complexes but form instead other species characterized by broad NMR resonances that we tentatively attribute to gold(I)–silver(I) multimetallic complexes by analogy with our prior studies on compound 1 (see Figures S64 and S65 in the Supporting Information).30 This notion is further supported by diffusion-ordered NMR experiments. For instance, 1H DOSY experimental data revealed a diffusion coefficient for the in situ equimolar reaction between complex 6 and AgSbF6, D equal to 9.13 × 10–10 m/s2 that accounts for only half of that for pure 6·C2H4 (D = 1.75 × 10–9 m/s2), (see Figures S63 and S64 for more details), indicating a larger structure attributable to a multimetallic species in the former case.
Catalytic Hydroamination of Ethylene
Having on hand the first examples of stable dicoordinate Au(I)–ethylene compounds, we next examined their catalytic potential in the hydroamination of ethylene. Initially, imidazolidine-2-one (9)12 was used as a model substrate to gauge the activity of all cationic gold(I)-ethylene species, obtained in situ from its corresponding neutral chloride precursors. Analogous to the conditions reported in Widenhoefer’s seminal investigations on intermolecular olefin hydroamination,12 solutions of compound 9 were pressurized with ethylene (4 bar) in the presence of 5 mol % of the gold(I) chloride complex 1–8 and 5 mol % of AgSbF6 as a halide scavenger in dioxane at 100 °C. Complexes 1, 3, 6, and 8 displayed great catalytic activity, reaching full conversion to the double hydroamination product 1,3-ethylimidazolidin-2-one (10) after 18 h (Table 2, entries 1, 4, 7, and 9), while formation of the monohydroaminated species was not detected. Interestingly, these complexes bear the bulkier phosphine ligands, with %Vbur values between 53.5 and 67.0. On the contrary, low or no conversion was obtained when employing complexes 2, 4, 5, 7, and [(Ph3P)AuCl] (Table 2, entries 3, 5, 6, 8, and 10), which present smaller phosphine ligands with %Vbur below 46.2. In addition, the previously isolated gold–ethylene complex 1·C2H4 was used as a catalyst reaching full conversion under our optimized conditions (Table 2, entry 26).
Table 2. Gold(I)-Catalyzed Hydroamination of Ethylene by Imidazolidine-2-onea.
| entry | catalyst | PC2H4 (bar) | conversion (%)b | 10:11 |
|---|---|---|---|---|
| 1 | 1 | 4 | >99 | 100:0 |
| 2 | 1·MeCNc | 4 | >99 | 100:0 |
| 3 | 2 | 4 | 0 | - |
| 4 | 3 | 4 | >99 | 100:0 |
| 5 | 4 | 4 | <5 | n.d.d |
| 6 | 5 | 4 | <5 | n.d.d |
| 7 | 6 | 4 | 95 | 100:0 |
| 8 | 7 | 4 | 20 | 15:85 |
| 9 | 8 | 4 | >99 | 100:0 |
| 10 | [(Ph3P)AuCl] | 4 | 0 | |
| 11 | 4 | 0 | ||
| 12 | L1 | 4 | 0 | |
| 13 | L3 | 4 | 0 | |
| 14 | 1 | 2 | >99 | 100:0 |
| 15 | 1·MeCNc | 2 | 98 | 100:0 |
| 16 | 3 | 2 | >99 | 100:0 |
| 17 | 6 | 2 | 50 | 35:65 |
| 18 | 8 | 2 | 56 | n.d.d |
| 19 | 1 | 1 | 98 | 100:0 |
| 20 | 1·MeCNc | 1 | 50 | 35:65 |
| 21 | 3 | 1 | 95 | 100:0 |
| 22 | 6 | 1 | 11 | 10:90 |
| 23 | 8 | 1 | 30 | 35:65 |
| 24 | 1e | 1 | 64 | 30:70 |
| 25 | 3e | 1 | 50 | 20:80 |
| 26 | 1·C2H4 | 1 | >99 | 100:0 |
| 27 | 1f | 1 | >99 | 100:0 |
| 28 | 1g | 1 | 96 | 66:33 |
| 29 | 3f | 1 | 70 | 31:69 |
| 30 | 3g | 1 | 64 | 37:63 |
| 29 | 13 | 1 | >99 | 100:0 |
Reaction was performed with imidazolidine-2-one (0.20 mmol) under the indicated ethylene pressure, gold catalyst (0.01 mmol), and AgSbF6 (0.01 mmol) as a chloride abstractor in 1,4-dioxane (1 mL) at 100 °C for 18 h.
Conversion was determined by 1H NMR spectroscopy with anisole as the internal standard.
In the absence of AgSbF6.
Not determined (n.d).
Catalyst loading at 2 mol %.
Reaction at 80 °C.
Reaction at 60 °C.
31P{1H} NMR spectroscopy analysis of the final catalytic mixtures after 18 h revealed the presence of the independently authenticated gold(I)–ethylene complexes in most cases, together with variable amounts of the corresponding free phosphine ligands. However, in the case of 2, 4, 5, and [(Ph3P)AuCl], the corresponding [P–Au–P]+ complexes were clearly observed as the major or sole gold-containing species. Formation of the latter under catalytic conditions is in agreement with our prior stability studies and can be understood as a deactivation pathway for the gold(I) complexes bearing the smaller phosphine ligands (Scheme 2), while more hindered phosphines prevent or slow down this unproductive route. Control experiments were also performed to investigate whether the presence of silver ions could have a direct influence on the catalytic outcome, as previously reported in other gold-catalyzed processes.31 No conversion was observed in the absence of the gold(I) complex (Table 2, entry 11) or in the presence of a combination of 5 mol % of L1 or L2 and AgSbF6 (Table 2, entries 12 and 13), indicating that under these conditions silver(I) is not capable of catalyzing the hydroamination of ethylene. In addition, the solvento complex 1·MeCN(19) was used in the absence of AgSbF6 achieving full conversion after 18 h, ruling out a direct silver-effect during gold catalysis (Table 2, entry 2).31
Scheme 2. Proposed Mechanism for the Assisted Hydroamination of Ethylene.

Deactivation of the gold(I) precatalyst by the formation of [P–Au–P]+ and Au(0) nanoparticles is indicated with a dashed gray arrow.
Complex 1·MeCN reaches full conversion with 2 bar of ethylene (Table 2, entry 15) but only 50% conversion under 1 bar of ethylene pressure (Table 2, entry 20). In this case, the presence of acetonitrile may compete with ethylene coordination at low pressure, as we have proved before,19 decreasing its catalytic activity. Complexes 6 and 8 only reach moderate conversions of ∼50% at 2 bar of ethylene pressure (Table 2, entries 17 and 18) and 30% and 11% (Table 2, entries 22 and 23) at 1 bar of ethylene pressure, respectively. For the latter two complexes, a mixture of the mono- (11) and dihydroaminated (10) products was detected by 1H NMR spectroscopy, suggesting that the double hydroamination process proceeds in a stepwise manner.
Considering the potential role of water as a proton shuttle in gold catalysis,32 and having in mind the existence of such proton rearrangements in this transformation (vide infra), we decided to investigate the addition of water and other additives. The results of these studies are collected in Table S1 in the Supporting Information. The presence of 10 mol % or 10 equiv of H2O or HFIP did not show any significant effect on the conversion. On the other hand, the presence of small amounts (or excess) of acids such as HOTf or CH3COOH has a detrimental effect on the catalytic transformation. Different bases such as tBuOK, Et3N, and DBU were also employed, which caused a complete shutdown of the catalytic activity.
Next, to compare better the reactivity of the most active catalysts, complexes 1 and 3, we investigated the hydroamination of ethylene under milder conditions. These two catalysts reach full conversion after 18 h when 2 and 1 bar of ethylene pressure was used (Table 2, entries 14, 16, 19, and 21). However, conversion drops to 64 and 50%, respectively, when the catalyst loading is lowered to 2 mol % at 1 bar of ethylene pressure at 100 °C (Table 2, entries 24 and 25). Remarkably, complex 1 also reaches full conversion even at 1 bar of ethylene pressure when the temperature is lowered to 80 °C and 96% conversion at only 60 °C (Table 1 entries 27 and 28). In contrast, conversion is reduced to 70 and 64% using complex 3 at 80 and 60 °C, respectively (entries 29 and 30). These results confirm the benefits of using the extremely bulky ligand L1, which slightly outperforms even the highly active catalyst based on L3 under particularly mild conditions.
Complex 1 is also able to successfully convert 1-methyl-imdazolidine-2-one (9′) into 1-methyl-2-ethylimidazolidin-2-one (10′) at 1 bar of ethylene pressure after 18 h at 60 °C, while only traces (<10%) of the hydroaminated product were observed when 2-oxazolidinone was used as a substrate even at 4 bar of ethylene pressure at 100 °C (see Supporting Information, Table S2, entries 1–4). In contrast, acyclic amide substrates could not be converted. Bulky amines, such as diisopropylamine or tert-butylamine, were also tested as substrates using gold(I) complexes 1 and 3, but no conversion was observed. In these cases, new signals were detected in the 31P{1H} NMR spectra of the final mixtures that differ from the corresponding gold(I) chloride and gold(I) π-ethylene complexes. For instance, from the reaction of complex 1 with diisopropylamine under catalytic conditions, a single crystal suitable for X-ray diffraction analysis was isolated and analyzed, confirming the coordination of the amine to the electrophilic Au(I) center (Figure S84) to form the corresponding [P–Au–NHiPr2]+ complex 12. Surprisingly, even more hindered amines like N-benzhydrylpropan-2-amine and tetramethylpiperidine are capable of displacing the ethylene molecule in gold(I) complex 1·C2H4 to yield gold adducts analogous to 12, as inferred from their corresponding 31P{1H} and 1H NMR spectra (Figure S66, see the SI for more details). Thus, the extreme steric profile of L1 does not seem to prevent amine coordination and as such the targeted nucleophilic attack of the amine toward the electrophilic carbon of the coordinated ethylene does not occur, preventing the initiation of the catalytic hydroamination process.
In view of these results, we decided to attempt the isolation of the Au(I)–imidazolidinone adduct that could act as an intermediate in the catalytic cycle. Complex 1 was reacted with AgSbF6 and imidazolidine-2-one 9 to form the corresponding complex 13, which was isolated as a stable solid under an inert atmosphere showing no decomposition at room temperature. Single crystals of complex 13 suitable for X-ray diffraction analysis were obtained by slow diffusion of pentane into a saturated dichloromethane solution of the gold(I) complex 13 at −30 °C. Complex 13 presents a solid-state structure with the gold center in a linear environment. In contrast to the isopropylamine ligand in complex 12, and despite the low oxophilicity of gold, imidazolidine-2-one coordinates at the metal through the oxygen atom (Figure 3). Complex 13 was utilized as a precatalyst affording full conversion toward hydroaminated ethylene within 18 h under the optimized conditions (Table 2, entry 31). To investigate the role of complex 13 in the mechanism, a solution of complex 13 in CD2Cl2 was charged with 1 bar of ethylene pressure showing full imidazolidine-2-one replacement by ethylene to exclusively form the gold–ethylene complex 1·C2H4, suggesting that under the catalytic conditions the presence of complex 13 is unlikely.
Figure 3.
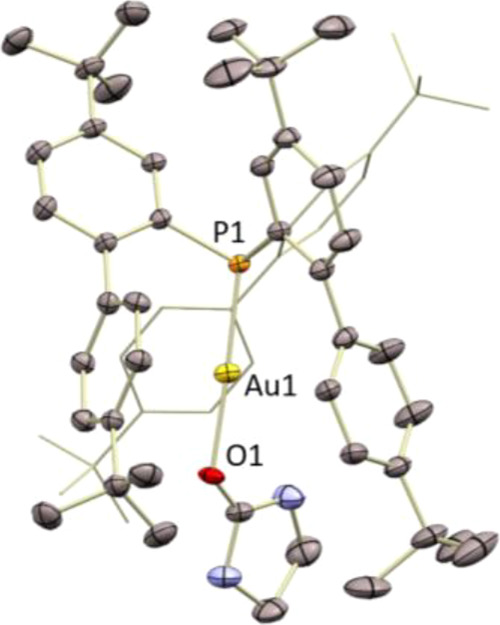
ORTEP representation of complex 13. Thermal ellipsoids are set at 50% probability. Counteranions and hydrogen atoms are excluded for clarity, while tert-butyl groups and one biaryl fragment are represented in wireframe format. Selected bond length (Å) and angles (°): P1–Au1, 2.2084(12); Au1–O1, 2.083(3); P1–Au1–O1, 177.54(12).
The catalytic activity of complex 1 toward the hydroamination of different 1-alkenes was also investigated (Table S2, see the Supporting Information for more details). Complex 1 converts imidazolidine-2-one to 1,3-diisopropylimidazolidin-2-one with complete Markovnikov regioselectivity at 6 bar of propene pressure after 18 h at 100 °C, while only 76% conversion is achieved when lowering the propene pressure to 4 bar. Complex 1 also fully converts 1-methyl-imdazolidine-2-one (9′) to 1-isopropyl-3-methylimidazolidin-2-one at 3 bar propene pressure after 18 h at 100 °C (87% conversion at 2 bar of propene pressure). The corresponding gold(I) π-propene complex 14 was also isolated and fully characterized, and X-ray diffraction analysis revealed a similar structure compared to the gold(I)–ethylene adduct 1·C2H4. The gold center is in a linear environment with the propene molecule coordinating gold in an η2 fashion (Figure S85). The Au–C bond lengths in the two independent molecules present in the asymmetric unit (2.22–2.27 Å) are similar to those in complex 1·C2H4 and the aforementioned ethylene adducts 3·C2H4 and 8·C2H4. The C=C double bond appears artificially shortened because of some degree of disorder on the olefin fragment and cannot be reliably determined. In addition, 1-alkenes with longer chains like 1-octene and cyclic alkenes such as cyclopentene and cyclohexene (Table S2, entries 12–17) are only moderately converted when using 1-methyl-imdazolidine-2-one (19–66%) at 100 °C even after longer reaction times (66 h). In contrast, 1-alkenes with bulkier substituents like 3,3-dimethyl-1-butene and styrene were not converted at all (<5% conversion, Table S2, entries 18 and 19), most likely because of the high steric profile of L1.
Mechanistic Considerations of the Gold(I)-Catalyzed Hydroamination of Ethylene
The gold(I)-catalyzed hydroamination of alkenes and related substrates has been recently studied computationally by Lledós and co-workers.10b,11c The proposed mechanism (Scheme 2) was described as a typical π-catalysis activation pathway, involving the coordination of the alkene to the gold(I) center (A) followed by the nucleophilic addition of the amide to the activated olefin (B). The next step involves the protodeauration process assisted by a proton shuttle (second amide molecule, C and D) to generate the hydroaminated product.
In this report, we have demonstrated that the use of sterically hindered phosphines is crucial to achieve good activities, which we attribute to the higher stability that they impart to the key π-ethylene intermediates, preventing (or slowing down) the formation of the corresponding [P–Au–P]+ complexes as the main deactivation route. In particular, the use of complex 1·C2H4, bearing an extremely bulky phosphine ligand, has given (along with 3·C2H4 to a slightly lesser extent) the best catalytic activities in this transformation. Therefore, we sought to gain mechanistic insight into this system, by means of kinetic experiments and density functional theory (DFT) calculations,33 to assess whether the previously proposed reaction mechanism11c was affected by the steric hindrance of phosphine L1 in the catalytic process.
As previously commented, chloride abstraction from the gold(I) chloride precatalyst generates the gold(I)–ethylene complex as the catalytically active species. Then, the nucleophilic addition of imidazolidine-2-one (9 = Nu) to the electrophilic carbon–carbon double bond constitutes the first step of the process. This transformation presents a barrier of 18.7 kcal/mol (TS1) relative to the independently computed reactants (Figure 4). This reaction is an endergonic process, yielding Int1 at 14.6 kcal/mol. From this step, we envisioned three possible mechanistic pathways, two in which the protodeauration step proceeds directly from the activated N-nucleophile 9 (intramolecular) and one assisted by a second molecule of imidazolidine-2-one (intermolecular) acting as a proton shuttle, as previously reported by Lledós and co-workers. These alternative intramolecular (paths A and B) and intermolecular (path C) routes are summarized in Scheme 3.
Figure 4.

Free energy profile for the Au(I)-catalyzed hydroamination of ethylene with imidazolidine-2-one (9 = Nu) assisted by a second molecule of imidazolidine-2-one acting as a proton shuttle (path C).
Scheme 3. Schematic Representation of the Three Possible Mechanistic Pathways (Paths A–C) for the Protodeauration Step.

We have computationally explored the three potential routes depicted in Scheme 3, with two alternative scenarios for path B (vide infra). For the first intramolecular protodeauration process (path A, Figure S88), a direct proton transfer from the nitrogen to the coordinated carbon atom is proposed. An initial rearrangement through a rotation of the coordinated nucleophile (TS2a, 19.7 kcal/mol) is followed by the proton transfer from the nitrogen to the coordinated carbon atom. The transition state for the proton transfer (TS3a), which leads to the hydroaminated product 11, was located at 45.9 kcal/mol (Figure S88). This barrier is too high to fit with our experimental observations. Alternatively, an intramolecular tautomerization through proton transfer from the nitrogen to the oxygen atom can be proposed, but the corresponding transition state (TS2b in Figure S90) is prohibitively high at 63.0 kcal/mol with respect to the separated reactants. Although the following proton transfer from the oxygen to the coordinated carbon atom (TS3b) drops to 23.8 kcal/mol (Figure S90), the overall kinetic barrier that accounts for 63.0 kcal/mol highly differs from our experimental results (experimentally we estimate an average ΔG373K of around 26.8 kcal/mol; see the Supporting Information for details).
Because the protodeauration step in path B seems indeed feasible, we examined an alternative way to access the required O–H intermediate. More precisely, we examined the intermolecular tautomerization process previously proposed by Lledós10g,11b and also related to the palladium intramolecular hydroamination of alkenes.34 The intermolecular proton transfer from the nitrogen atom to the oxygen atom of a second molecule of the imidazolidine-2-one occurs in a barrierless fashion, leading to Int2b′ at 18.6 kcal/mol with respect to the separated reactants.35 In contrast to the intramolecular scenario in path B (TS2b, 63.0 kcal/mol), the intermolecular tautomerization process through a proton transfer to a second molecule of 9 presents a negligible energy barrier (TS2b′, Figure S93). Then, the intramolecular proton transfer to the coordinated carbon atom (TS3b = TS3b′, at 23.8 kcal/mol) leads to the final hydroaminated product 11 (Figure S93, path B′).
Instead of mediating the above tautomerization, the protonated molecule of 9 may directly affect the intermolecular protodeauration as depicted in path C. We have also computed this route. The proton transfer from the protonated nucleophile (Nu–H) can occur directly to the coordinated carbon atom (TS2c, at 28.3 kcal/mol) leading to the final product 11 (Figure 4). This energy barrier (28.3 kcal/mol) is relatively higher than the one found for the intramolecular proton transfer from the oxygen to the coordinated carbon (TS3b′ in path B′), but it also fits reasonably well with the overall value measured experimentally (ΔG373K ≈ 26.8 kcal/mol). These studies indicate that the two intramolecular processes (paths A and B) present unfeasibly overall high energy barriers of 45.9 and 63.0 kcal/mol and that a second molecule of imidazolidine-2-one is required as a proton shuttle to mediate the subsequent intramolecular (path B′) or intermolecular (path C) protodeauration step. In any case, it is interesting to note that despite its extreme bulkiness, the flexibility of ligand L1 permits the accommodation of a second molecule of the amide in the cavity generated around the gold center, allowing the catalytic process.
To further support the intermolecular-assisted mechanism and to differentiate between the two aforementioned and close-in-energy potential routes, we studied experimentally the hydroamination of ethylene by 1-methyl-imidazolidine-2-one 9′ catalyzed by complex 13. It is worth noting that complex 13 was employed instead of complex 1 to avoid any potential complication associated with the presence of silver salts. In an initial experiment, we monitored by 1H NMR spectroscopy the conversion of 1-methyl-imidazolidine-2-one (0.3 M) under catalytic conditions of 10 mol % of complex 13 (0.03 M) at 100 °C in CDCl3 under 6 bar of ethylene pressure (0.8 M). A plot of 1/[9′] vs time was linear with a pseudo-second-order rate constant of 1.43 ± 0.03 × 10–3 M–1 s–1, indicating a pseudo-second-order dependence of the rate of the reaction on the concentration of 1-methyl-imdazolidine-2-one (Figure 5). This and subsequent kinetic experiments to be discussed were run in duplicate or triplicate in all cases. In addition, 1H and 31P NMR spectra of the catalytic reactions (Figures S78–S80) showed that the only gold species detected during the course of the catalysis, the one acting as the resting state, is the gold–ethylene complex 1·C2H4, in accordance with the potential profiles investigated by DFT (Figures 4 and S93). This indicates the key role of the gold(I)–ethylene adduct in the gold(I)-catalyzed hydroamination of ethylene, in agreement with the benefits derived from using ligands that stabilize this unusual resting state.
Figure 5.

Second-order kinetic representation of the consumption of 1-methyl-imidazolidine-2-one at 100 °C in CDCl3 under 6 bar of ethylene (kobs = 1.43 ± 0.03 × 10–3 M–1 s–1).
To determine the dependence of the rate of the hydroamination reaction on catalyst concentration, pseudo-second-order rate constants were determined for the gold(I)-catalyzed hydroamination of ethylene (6 bar) with 9′ (0.3 M) as a function of complex 13 from 0.055 to 0.450 M at 100 °C, which established a first-order dependence of the rate on catalyst concentration (Figure 6A). Likewise, to determine the dependence of the rate of hydroamination on ethylene pressure, pseudo-second-order rate constants were determined for the reaction of 9′ (0.2 M) with ethylene catalyzed by complex 13 (15 mol %, 0.03 M) as a function of ethylene pressure from 4 to 8 bar at 60 °C. A plot of the corresponding pseudo-second-order rate constants vs the ethylene pressure was almost flat, which establishes a zero-order dependence on ethylene concentration (Figure 6B). To differentiate between the two potential mechanisms proposed above, that is, the intramolecular (path B′, Figure S93) and the intermolecular (path C, Figure 4) protodeauration, we derived their corresponding differential equations (see Schemes S1 and S2 in the Supporting Information). While both alternative pathways agree with a zero-order dependence on ethylene and first-order dependence on the catalyst, only the route depicted in Figure 4 (path C) is in agreement with a second-order dependence on the nucleophile. Thus, we proposed the hydroamination of ethylene to proceed through that pathway.
Figure 6.

(A) Plot of pseudo-second-order rate constants vs catalyst concentration for the hydroamination of 9′ with ethylene (6 bar) catalyzed by complex 13 (0.0055–0.045 M) in CDCl3 at 100 °C. (B) Plot of pseudo-third-order (k = kobs/[13]) vs ethylene pressure for the hydroamination of 9′ with ethylene (5–8 bar) catalyzed by complex 13 (0.03 M) in CDCl3 at 60 °C.
To gain more insight into the proposed involvement of a proton transfer process in the turnover-limiting step during gold-catalyzed hydroamination, we evaluated the kinetic isotope effect (KIE) resulting from the deuteroamination of ethylene with the deuterated 1-methyl-imdazolidine-2-one (9′-d1). The corresponding plot of 1/[9′-d1] vs time was linear with a pseudo-second-order constant of 4.56 ± 0.02 × 10–4 M–1 s–1. Comparison of the pseudo-second-order constant determined for the hydroamination of ethylene with 1-methyl-imdazolidine-2-one gave a significant primary deuterium KIE of kH/kD = 3.14 (Figure S81), which corroborates the involvement of H-containing bond-breaking processes during the rate-limiting step of the catalytic reaction, which is attributed to intermolecular protodeauration.
Conclusions
In summary, we have synthesized and structurally characterized a family of highly unusual dicoordinate gold(I)–ethylene complexes bearing phosphine ligands with variable bulkiness. The use of bulky phosphines is crucial to stabilize the gold(I)–ethylene bond and prevent catalyst decomposition, two key aspects for catalytic performance. In fact, while there is no apparent decomposition for the more sterically hindered complex 1·C2H4, slow decomposition of complexes 2–8·C2H4 either in solution or in the solid state is detected. Interestingly, X-ray diffraction revealed a nonsymmetric coordination of ethylene at gold(I) with a slipped η2-coordination for complex 8·C2H4, further suggesting the lability of this type of coordination. Complexes 1–8 have been tested as precatalysts for the underdeveloped Au(I)-catalyzed hydroamination of ethylene. Precatalysts bearing the most sterically demanding phosphine 1 showed the best results achieving full conversion within 18 h under only 1 bar of ethylene pressure at 60 °C, highlighting the high catalytic potential of very sterically crowded catalysts. On the other hand, complexes with smaller phosphine ligands afforded little or no conversion in this transformation. Kinetic analysis together with DFT calculations shows that the preferred mechanistic pathway involves the assistance of a second molecule of the nucleophile even when using the more sterically congested cavity-shaped complex 1. In addition, a strong primary KIE has been observed, corroborating the involvement of H-containing bond-breaking processes in the rate-limiting step of the catalytic transformation that we attribute to intermolecular protodeauration.
Experimental Section
General Considerations
Unless otherwise stated, all reactions and manipulations were carried out under an atmosphere of dry argon or nitrogen using standard Schlenk techniques or in a nitrogen glovebox. Solvents were distilled under an inert atmosphere prior to use. Solution 1H, 13C, and 31P NMR spectra were recorded on Bruker AMX-300, DRX-400, and DRX-500 spectrometers at 298 K unless otherwise stated. Chemical shifts (δ) are expressed with a positive sign, in parts per million. 1H and 13C chemical shifts reported are referenced internally to residual protio (1H) or deutero (13C) solvent, while 31P chemical shifts are relative to 85% H3PO4. The following abbreviations and their combinations are used: br, broad; s, singlet; d, doublet; t, triplet; m, multiplet. The 1H and 13C resonance signals were attributed by means of 2D HSQC and HMBC experiments (Figure 7). For elemental analyses, a LECO TruSpec CHN elementary analyzer was utilized. [AuCl(THT)]36 (THT = tetrahydrothiophene) and all used phosphines (L1–L8)18,24 were prepared according to literature procedures. All other reagents were used as received from commercial suppliers.
Figure 7.

Labeling scheme used for 1H and 13C{1H} NMR assignments.
General Synthesis of Gold(I) Chloride Complexes
A solution of the corresponding phosphine (0.470 mmol) in toluene (10 mL) was added over a suspension of [AuCl(THT)] (150 mg, 0.470 mmol) in toluene (5 mL) at 0 °C. The initial white suspension was stirred for 12 h at rt until it became a clear solution. The solvent was removed under vacuum, and the resulting colorless solid was washed with pentane and dried to give the corresponding gold chloride complexes. Complexes 1–6 have been previously reported.19,21,23b
Compound 7
Complex 7 was prepared following the general procedure from L7 (221 mg, 63%). Crystals suitable for X-ray diffraction were grown by slow evaporation of pentane into a dichloromethane solution of complex 7 at −32 °C. 1H NMR (300 MHz, C6D6, 25 °C) δ: 7.61 (s, 2H, Hd), 7.58 (s, 4H, Hc), 7.19–7.02 (m, 3H, Ha + Hb), 1.43 (s, 36H, CH3(tBu)), 0.50 (d, 6H, 2JHP = 10.0 Hz, PCH3). 13C{1H} NMR (100 MHz, C6D6, 25 °C) δ: 152.3 (C4), 148.7 (d, 2JCP = 8 Hz, C3), 141.8 (s, C2), 131.3 (d, 3JPC = 7 Hz, CHa), 130.1 (s, CHb), 129.4 (d, 1JPC = 55 Hz, C1), 124.9 (s, CHc) 122.9 (s, CHd), 35.3 (s, C(tBu)), 31.8 (s, CH3(tBu)), 17.6 (d, 1JCP = 40 Hz, PCH3). 31P{1H} NMR (202 MHz, C6D6, 25 °C) δ: 0.4.
Compound 8
Complex 8 was prepared following the general procedure from L8 (295 mg, 71%). Crystals suitable for X-ray diffraction were grown by slow evaporation of a concentrated dichloromethane solution of complex 8. Anal. calcd for C46H67AuClP: C, 62.54; H, 7.64. Found: C, 62.36; H, 7.27. 1H NMR (300 MHz, CD2Cl2, 25 °C) δ: 7.56 (d, 2H, 4JHH = 1.7 Hz, CHd), 7.45 (td, 1H, 3JHH = 7.5 Hz, 5JHP = 1.6 Hz, CHb), 7.24 (dd, 2H, 3JHH = 7.5 Hz, 4JHP = 3.0 Hz, CHa), 7.06 (d, 4H, 4JHH = 1.8 Hz, CHc), 2.12–1.95 (m, 2H, CH(Cy)), 1.78–1.55 (m, 10H, CH2(Cy)), 1.52–1.45 (m, 2H, CH(Cy)), 1.40 (s, 36H, CH3(tBu)), 1.35–1.25 (m, 4H, CH2(Cy)), 1.19–1.11 (m, 2H, CH2(Cy)). 1.10–0.99 (m, 2H, CH2(Cy)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 151.3 (s, C4), 150.6 (s, C2), 142.9 (d, 3JPC = 5 Hz, C3), 133.4 (d, 3JPC = 7 Hz, CHa), 129.8 (s, CHb), 124.5 (s, CHc), 124.3 (d, 1JPC = 48 Hz, C1), 123.2 (CHc), 37.8 (d, 1JPC = 31 Hz, CH(Cy)), 35.7 (s, C(tBu)), 34.6 (d, 3JPC = 6 Hz, CH2(Cy)), 32.1 (s, CH3(tBu)), 31.7 (s, CH2(Cy)), 27.1 (d, 2JPC = 13 Hz, CH2(Cy)), 26.8 (d, 2JPC = 15 Hz, CH2(Cy)), 26.3 (s, CH2(Cy)). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 48.8.
General Synthesis of Gold(I)–Ethylene Complexes
In a glovebox, a Schlenk flask was charged with silver hexafluoroantimonate (8 mg, 0.022 mmol) in dichloromethane (1 mL). The corresponding gold(I) chloride complex (0.02 mmol) was transferred into a small glass vial and dissolved in dichloromethane (1 mL). The vial solution was loaded into a plastic syringe equipped with a stainless steel needle. Outside the glovebox, the Schlenk flask was cooled down to −30 °C. At this temperature, the solution of the gold(I) chloride complex was added to the AgSbF6 suspension while bubbling ethylene. The mixture was allowed to slowly warm up to room temperature and filtered through a short pad of Celite to remove the silver salts, and the solvent was removed under vacuum affording the corresponding gold(I)-ethylene complexes as colorless solids. Complex 1·C2H4 has been previously reported.19
Compound 2·C2H4
Complex 2·C2H4 was prepared following the general procedure from gold(I) chloride complex 2 (13 mg, 78%). Anal. calcd for C29H37AuF6PSb: C, 41.01; H, 4.39. Found: C, 41.08; H, 4.54. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.01 (bs, 6H, m-CH), 5.46 (bs, 4H, CH2(C2H4)), 2.66 (bs, 9H, o-CH3), 2.36 (s, 9H, p-CH3), 1.86 (bs, 9H, o-CH3). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 143.6 (s, p-C), 143.0 (bs, o-C), 132.9 (bs, m-CH), 123.3 (d, 1JCP = 56 Hz, C), 111.2 (d, 2JCP = 9 Hz, C(C2H4)), 24.4 (bs, o-CH3), 21.3 (s, p-CH3). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 1.5. 1H NMR (400 MHz, CD2Cl2, −30 °C) δ:7.06 (s, 3H, m-CH), 6.90 (s, 3H, m-CH), 5.43 (bs, 4H, CH2(C2H4)), 2.64 (s, 9H, o-CH3), 2.31 (s, 9H, p-CH3), 1.78 (s, 9H, o-CH3). 13C{1H} NMR (100 MHz, CD2Cl2, −30 °C) δ: 143.3 (d, 3JCP = 5 Hz, o-C), 143.0 (s, p-C), 142.2 (d, 3JCP = 16 Hz, o-C), 133.0 (d, 3JCP = 9 Hz, m-CH), 132.0 (d, 4JCP = 10 Hz, m-CH), 122.6 (bs, CH2(C2H4)), 122.5 (d, 1JCP = 56 Hz, C), 24.9 (d, 3JCP = 17 Hz, o-CH3), 24.0 (d, 3JCP = 5 Hz, o-CH3), 21.0 (s, p-CH3). 1H NMR (400 MHz, CD2Cl2, −70 °C) δ: 7.03 (d, 6H, 4JHP = 5.0 Hz, m-CH), 6.87 (s, 6H, m-CH), 5.67 (d, 4H, 3JHP = 2.9 Hz, CH2(C2H4)), 2.60 (s, 9H, o-CH3), 2.28 (s, 9H, p-CH3), 1.72 (s, 9H, o-CH3).
Compound 3·C2H4
Complex 3·C2H4 was prepared following the general procedure from gold(I) chloride complex 3 (16 mg, 89%). Crystals suitable for X-ray diffraction were grown by slow evaporation of pentane into a dichloromethane solution of complex 3·C2H4. Anal. calcd for C31H49AuF6PSb: C, 42.05; H, 5.58. Found: C, 41.83; H, 5.89. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.90 (m, 1H, CHd), 7.64 (m, 2H, CHb + CHc), 7.29 (s, 2H, CHe), 7.23 (m, 1H, CH), 4.95 (d, 4H, 3JHP = 2.6 Hz, CH2(C2H4)), 3.04 (hept, 1H, 3JHH = 6.9 Hz, CH(iPr)), 2.35 (hept, 2H, 3JHH = 6.9 Hz, CH(iPr)), 1.45 (d, 18H, 3JHP = 16.5 Hz, CH3(tBu)), 1.35 (d, 6H, 3JHH = 6.9 Hz, CH3(iPr)), 1.24 (d, 6H, 3JHH = 6.9 Hz, CH3(iPr)), 0.94 (d, 6H, 3JHH = 6.9 Hz, CH3(iPr)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 151.9 (s, C5), 149.0 (s, C4), 146.8 (d, 2JCP = 15 Hz, C2), 136.9 (d, 3JCP = 7 Hz, C3), 135.8 (d, 3JCP = 3 Hz, CHd), 135.4 (d, 2JCP = 8 Hz, CHa), 132.5 (d, 4JCP = 2 Hz, CHc), 128.7 (d, 3JCP = 7 Hz, CHb), 128.4 (d, 1JCP = 45 Hz, C1), 123.5 (s, CHe), 110.9 (d, 2JCP = 8 Hz, C(C2H4)), 39.9 (d, 1JCP = 24 Hz, C(tBu)), 34.7 (s, p-CH(iPr)), 31.6 (s, o-CH(iPr)), 31.6 (s, CH3(tBu)) 26.0 (s, CH3(iPr)), 24.6 (s, CH3(iPr)), 23.8 (s, CH3(iPr)). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 65.6.
Compound 4·C2H4
Complex 4·C2H4 was prepared following the general procedure from gold(I) chloride complex 4 (13 mg, 71%). Anal. calcd for C34H47AuF6PSb: C, 44.42; H, 5.15. Found: C, 44.48; H, 5.31. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.64 (td, 1H, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, CHb), 7.55 (t, 2H, 3JHH = 7.3 Hz, CHd), 7.40 (d, 4H, 3JHH = 7.3 Hz, CHc), 7.26 (dd, 2H, 3JHH = 7.6 Hz, 4JHP = 3.7 Hz, CHa), 4.85 (d, 4H, 3JHP = 3.0 Hz, CH2(C2H4)), 2.45 (hept, 3JHH = 6.8 Hz, 4H, CH(iPr)), 1.46 (d, 3JHP = 10.7 Hz, 6H, PCH3), 1.31 (d, 2H, 3JHH = 6.8 Hz, 12H, CH3(iPr)), 1.06 (d, 2H, 3JHH = 6.8 Hz, 12H, CH3(iPr)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 147.8 (s, C4), 146.2 (d, 2JPC = 12 Hz, C2), 138.2 (d, 3JPC = 6 Hz, C3), 133.7 (d, 3JPC = 8 Hz, CHa), 132.0 (s, CHb), 130.4 (s, CHd), 127.9 (d, 1JCP = 60 Hz C1), 124.6 (s, CHc), 110.3 (d, 2JCP = 9 Hz, CH2(C2H4)), 32.0 (s, CH(iPr)), 25.6 (s, CH3(iPr)), 23.2 (s, CH3(iPr)), 16.2 (d, 2JCP = 37 Hz, PCH3). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 4.3.
Compound 5·C2H4
Complex 5·C2H4 was prepared following the general procedure from gold(I) chloride complex 5 (9 mg, 53%). 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.72 (td, 1H, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, CHb), 7.36 (t, 2H, 3JHH = 7.3 Hz, CHd), 7.28 (d, 4H, 3JHH = 7.3 Hz, CHc), 7.15 (dd, 2H, 3JHH = 7.6 Hz, 4JHP = 3.6 Hz, CHa), 5.00 (bs, 4H, CH2(C2H4)), 2.04 (s, 12H, CH3(Xyl)), 1.49 (d, 2H, 2JHP = 10.4 Hz, PCH3). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 147.8 (d, 2JPC = 13 Hz, C2), 140.7 (s, C3), 137.3 (s, C4), 134.3 (s, CHb), 132.4 (d, 3JPC = 8 Hz, CHa), 129.3 (s, CHd), 129.0 (s, CHc), 125.6 (s, C1), 111.5 (s, CH2(C2H4)), 21.8 (s, CH3(Xyl)), 16.0 (d, 2JCP = 37 Hz, PCH3). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 4.1. 1H NMR (400 MHz, CD2Cl2, −30 °C) δ: 7.72 (td, 1H, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, CHb), 7.37 (t, 2H, 3JHH = 7.5 Hz, CHd), 7.28 (d, 4H, 3JHH = 7.5 Hz, CHc), 7.14 (dd, 2H, 3JHH = 7.6 Hz, 4JHP = 3.7 Hz, CHa), 5.36 (bs, 4H, CH2(C2H4)), 2.02 (s, 12H, CH3(Xyl)), 1.48 (d, 2H, 2JHP = 10.7 Hz, PCH3). 13C{1H} NMR (100 MHz, CD2Cl2, −30 °C) δ: 147.1 (d, 2JPC = 12 Hz, C2), 140.1 (d, 3JPC = 7 Hz, C3), 136.9 (s, C4), 134.0 (s, CHb), 131.8 (d, 3JPC = 7 Hz, CHa), 128.9 (s, CHd), 128.5 (s, CHc), 125.1 (d, 1JPC = 62 Hz, C1), 120.9 (bs, CH2(C2H4)), 21.6 (s, CH3(Xyl)), 15.6 (d, 2JCP = 37 Hz, PCH3).
Compound 6·C2H4
Complex 6·C2H4 was prepared following the general procedure from gold(I) chloride complex 6 (16 mg, 87%). Anal. calcd for C34H43AuF6PSb: C, 44.61; H, 4.73. Found: C, 44.68; H, 4.99. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.69 (td, 1H, 3JHH = 7.6 Hz, 5JHP = 1.8 Hz, CHb), 7.40–7.22 (m, 6H, CHd + CHc), 7.21–7.13 (m, 2H, CHa), 4.86 (d, 4H, 3JHP = 2.8 Hz, CH2(C2H4)), 2.45–2.28 (m, 2H, CH(Cy)), 2.08 (s, 12H, CH3(Xyl)), 1.93–1.85 (m, 2H, CH2(Cyp)), 1.76–1.57 (m, 6H, CH2(Cyp)), 1.56–1.43 (m, 4H, CH2(Cyp)), 1.40–1.19 (m, 4H, CH2(Cyp)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 148.2 (d, 2JPC = 10 Hz, C2), 140.9 (bs, C3), 137.8 (s, C4), 133.4 (s, CHa), 133.2 (s, CHb), 129.2 (s, CHc), 128.7 (s, CHd), 128.7 (d, 1JPC = 50 Hz, C1), 111.0 (d, 2JCP = 9 Hz, CH2(C2H4)), 38.3 (d, 1JPC = 31 Hz, CH(Cyp)), 36.3 (d, 3JPC = 8 Hz, CH2(Cyp)), 32.8 (d, 3JPC = 6 Hz, CH2(Cyp)), 25.8 (d, 2JPC = 12 Hz, CH2(Cyp)), 25.6 (d, 2JPC = 14 Hz, CH2(Cyp)), 21.9 (s, CH3(Xyl)). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 57.6. 1H NMR (400 MHz, CD2Cl2, −30 °C) δ: 7.71 (td, 1H, 3JHH = 7.6 Hz, 5JHP = 1.9 Hz, CHb), 7.49–7.38 (m, 2H, CHd), 7.34–7.23 (m, 2H, CHc), 7.23–7.17 (m, 2H, CHc), 7.11–7.04 (m, 2H, CHa), 4.86 (d, 4H, 3JHP = 2.8 Hz, CH2(C2H4)), 2.43–2.25 (m, 2H, CH(Cy)), 2.08 (s, 12H, CH3(Xyl)), 1.94–1.84 (m, 2H, CH2(Cyp)), 1.79–1.45 (m, 10H, CH2(Cyp)), 1.40–1.14 (m, 4H, CH2(Cyp)). 13C{1H} NMR (100 MHz, CD2Cl2, −30 °C) δ: 147.9 (s, C2), 147.3 (d, 2JPC = 19 Hz, C2), 141.8 (s, C3), 139.1 (s, C3), 137.5 (s, C4), 137.3 (s, C4), 133.2 (d, 4JPC = 7 Hz, CHa), 133.1 (s, CHb), 132.0 (d, 4JPC = 7 Hz, CHa), 129.5 (s, CHc), 129.2 (s, CHd), 128.8 (d, 1JPC = 46 Hz, C1), 127.7 (s, CHc), 111.0 (d, 2JCP = 9 Hz, CH2(C2H4)), 37.6 (d, 1JPC = 32 Hz, CH(Cyp)), 35.9 (d, 3JPC = 8 Hz, CH2(Cyp)), 32.3 (d, 3JPC = 6.5 Hz, CH2(Cyp)), 25.2 (d, 2JPC = 12 Hz, CH2(Cyp)), 25.1 (d, 2JPC = 14 Hz, CH2(Cyp)), 21.8 (s, CH3(Xyl)), 21.3 (s, CH3(Xyl)).
Compound 7·C2H4
Complex 7·C2H4 was prepared following the general procedure from gold(I) chloride complex 7 (12 mg, 63%). Anal. calcd for C38H55AuF6PSb: C, 46.79; H, 5.68. Found: C, 46.71; H, 5.94. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.77–7.65 (m, 1H, CHb), 7.64 (t, 2H, 3JHH = 1.7 Hz, CHd), 7.43 (dd, 2H, 3JHH = 7.6 Hz, 4JHP = 3.9 Hz, CHa), 7.25 (d, 4H, 4JHH = 1.7 Hz, CHc), 5.16 (s, 4H, CH2(C2H4)), 1.60 (d, 2H, 2JHP = 10.7 Hz, PCH3), 1.41 (s, 36H, CH3(tBu)).13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 152.6 (s, C4), 150.3 (s, 2JCP = 11 Hz, C3), 141.1 (d, 3JCP = 6 Hz, C2), 132.9 (d, 3JCP = 8 Hz, CHa), 132.1 (s, CHb), 125.5 (d, 1JPC = 60 Hz, C1), 125.0 (s, CHd), 123.5 (s, CHd), 111.8 (bs, CH2(C2H4)), 35.6 (s, C(tBu)), 31.9 (s, CH3(tBu)), 18.4 (d, 2JCP = 37 Hz, PCH3). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 9.4. 1H NMR (400 MHz, CD2Cl2, −30 °C) δ: 7.71 (td, 1H, 3JHH = 7.7 Hz, 5JHP = 1.7 Hz, CHb), 7.59 (t, 2H, 3JHH = 1.7 Hz, CHd), 7.43 (dd, 2H, 3JHH = 7.7 Hz, 4JHP = 3.8 Hz, CHa), 7.22 (d, 4H, 4JHH = 1.8 Hz, CHc), 5.35 (s, 4H, CH2(C2H4)), 1.56 (d, 2H, 2JHP = 10.4, PCH3), 1.37 (s, 36H, CH3(tBu)). 13C{1H} NMR (100 MHz, CD2Cl2, −30 °C) δ: 151.9 (s, C4), 149.9 (s, 2JCP = 11 Hz, C3), 140.6 (s, 3JCP = 7 Hz, C2), 132.3 (d, 3JCP = 7 Hz, CHa), 131.7 (s, CHb), 125.0 (d, 1JPC = 62 Hz, C1), 124.6 (s, CHd), 123.0 (s, CHd), 120.5 (s, CH2(C2H4)), 35.2 (s, C(tBu)), 31.4 (s, CH3(tBu)), 18.0 (d, 2JCP = 37 Hz, PCH3).
Compound 8·C2H4
Complex 8·C2H4 was prepared following the general procedure from gold(I) chloride complex 8 (21 mg, 93%). Crystals suitable for X-ray diffraction were grown by slow diffusion of pentane into a dichloromethane solution of complex 8·C2H4. Anal. calcd for C48H71AuF6PSb: C, 51.86; H, 6.44. Found: C, 51.92; H, 6.64. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.65 (s, 2H, CHd), 7.55 (td, 1H, 3JHH = 7.7 Hz, 5JHP = 1.8 Hz, CHb), 7.26 (bs, 2H, CHa), 7.13 (d, 4H, 4JHH = 1.8 Hz, CHc), 4.77 (s, 4H, CH2(C2H4)), 2.37–2.21 (m, 2H, CH(Cy)), 1.89–1.65 (m, 10H, CH2(Cy)), 1.42 (s, 36H, CH3(tBu)), 1.22–1.04 (m, 10H, CH2(Cy)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 152.8 (bs, C4), 149.9 (s, C3), 149.8 (s, C2), 133.9 (s, CHa), 131.4 (s, CHb), 123.9 (bs, CHc + CHd), 122.8 (d, 1JPC = 50 Hz, C1), 109.0 (s, CH2(C2H4)), 38.6 (d, 1JPC = 28 Hz, CH(Cy)), 35.7 (s, C(tBu)), 34.3 (d, 3JPC = 6 Hz, CH2(Cy)), 31.9 (s, CH3(tBu)), 31.9 (s, CH2(Cy)), 26.9 (d, 2JPC = 16 Hz, CH2(Cy)), 26.7 (d, 2JPC = 13 Hz, CH2(Cy)), 26.0 (s, CH2(Cy)). 31P{1H} NMR (162 MHz, CD2Cl2, 25 °C) δ: 55.4. 1H NMR (400 MHz, CD2Cl2, −30 °C) δ: 7.60 (s, 2H, CHd), 7.54 (td, 1H, 3JHH = 7.6 Hz, 5JHP = 1.8 Hz, CHb), 7.31 (d, 1H, 3JHH = 7.6 Hz, CHa), 7.19 (dd, 1H, 3JHH = 7.6 Hz, 4JHP = 4.3 Hz, CHa), 7.10 (s, 2H, CHc), 7.08 (s, 2H, CHc), 4.69 (d, 4H, 3JHP = 2.6 Hz, CH2(C2H4)), 2.26–2.07 (m, 2H, CH(Cy)), 1.84–1.58 (m, 10H, CH2(Cy)), 1.38 (s, 18H, CH3(tBu)), 1.37 (s, 18H, CH3(tBu)), 1.23–0.98 (m, 10H, CH2(Cy)). 13C{1H} NMR (100 MHz, CD2Cl2, −30 °C) δ: 152.3 (s, C4), 151.0 (s, C4), 149.1 (s, C3), 149.0 (s, C2), 142.7 (s, C3), 140.4 (s, C2), 133.6 (d, 3JPC = 6 Hz, CHa), 133.2 (d, 3JPC = 6 Hz, CHa), 131.0 (s, CHb), 124.3 (s, CHc),123.9 (s, CHd), 122.8 (s, CHc), 122.6 (d, 1JPC = 50 Hz, C1), 109.0 (d, 2JPC = 8 Hz, CH2(C2H4)), 37.2 (d, 1JPC = 29 Hz, CH(Cy)), 35.5 (s, C(tBu)), 35.2 (s, C(tBu)), 33.9 (d, 3JPC = 5 Hz, CH2(Cy)), 31.5 (s, CH3(tBu)), 31.3 (s, CH2(Cy)), 26.3 (d, 2JPC = 15 Hz, CH2(Cy)), 26.2 (d, 2JPC = 15 Hz, CH2(Cy)), 25.5 (s, CH2(Cy)).
Synthesis of Gold(I)–Amine Complex 12
A solution of complex 1 (32 mg, 0.02 mmol) in dichloromethane (1 mL) in the presence of diisopropylamine (5 mL, 0.03 mmol) was added to a suspension of silver hexafluoroantimonate (12 mg, 0.03 mmol) in dichloromethane (1 mL) at rt. The mixture was stirred for 30 min and filtered through a short pad of Celite to remove the silver salts, and the solvent was removed under vacuum affording complex 12 as a colorless solid (24 mg, 87%). Crystals suitable for X-ray diffraction were grown by slow diffusion of pentane into a dichloromethane solution of complex 12. 1H NMR (500 MHz, CD2Cl2, 25 °C) δ: 7.73 (d, 3H, 3JHH = 8.1 Hz, CHb), 7.51–7.35 (m, 12H, CHa + CHc), 7.23 (d, 6H, 3JHH = 8.0 Hz, CHe), 6.88 (d, 6H, 3JHH = 8.0 Hz, CHd), 2.73 (hept, 1H, 3JHH = 6.3 Hz, CH(iPr)), 2.16 (bs, 1H, CH(iPr)), 1.26 (s, 27H, CH3(tBu)), 1.20 (s, 27H, CH3(tBu)), 0.65 (d, 3H, 3JHH = 6.3 Hz, CH3(iPr)), 0.53 (d, 3H, 3JHH = 6.3 Hz, CH3(iPr)), 0.51 (d, 3H, 3JHH = 6.3 Hz, CH3(iPr)), 0.45 (d, 3H, 3JHH = 6.3 Hz, CH3(iPr)). 13C{1H} NMR (125 MHz, CD2Cl2, 25 °C) δ: 151.8 (d, 3JCP = 8 Hz, C2), 151.4 (s, C5), 143.6 (d, 2JCP = 15 Hz, C3), 138.7 (d, 3JCP = 6 Hz, C4), 134.9 (d, 3JCP = 10 Hz, CHa or CHc), 133.9 (d, 3JCP = 8 Hz, CHa or CHc), 130.2 (s, CHd), 129.6 (d, 4JCP = 3 Hz, CHb), 127.8 (d, 1JCP = 62 Hz, C1), 126.0 (s, CHe), 49.5 (s, CH(iPr)), 49.1 (s, CH(iPr)), 35.3 (s, C(tBu)), 35.1 (s, C(tBu)), 31.6 (s, CH3(tBu)), 31.3 (s, CH3(tBu)), 25.6 (s, CH3(iPr)), 23.8 (s, CH3(iPr)). 31P{1H} NMR (202 MHz, CD2Cl2, 25 °C) δ: 11.1.
Synthesis of Gold(I)–Imidazolidine-2-one Complex 13
A solution of complex 1 (106 mg, 0.10 mmol) in dichloromethane (1 mL) in the presence of imidazolidine-2-one (9 mg, 0.10 mmol) was added to a suspension of silver hexafluoroantimonate (38 mg, 0.11 mmol) in dichloromethane (1 mL) at rt. The mixture was stirred for 30 min and filtered through a short pad of Celite to remove the silver salts, and the solvent was removed under vacuum affording complex 13 as a white colorless solid (122 mg, 91%). Crystals suitable for X-ray diffraction were grown by slow diffusion of pentane into a dichloromethane solution of complex 13. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.70 (dd, 3H, 3JHH = 8.1 Hz, 4JHH = 1.8 Hz, CHb), 7.45–7.35 (m, 12H, CHa + CHc), 7.15 (d, 6H, 3JHH = 7.8 Hz, CHe), 6.68 (d, 6H, 3JHH = 7.8 Hz, CHd), 4.14 (bs, 2H, NH), 3.58 (s, 4H, CH2), 1.25 (s, 27H, CH3(tBu)), 1.24 (s, 27H, CH3(tBu)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 166.9 (CO), 151.9 (s, C5), 151.5 (d, 3JCP = 8 Hz, C2), 144.4 (d, 2JCP = 16 Hz, C3), 138.4 (d, 3JCP = 7 Hz, C4), 133.1 (d, 3JCP = 10 Hz, CHa or CHc), 133.0 (d, 3JCP = 8 Hz, CHa or CHc), 129.6 (s, CHd), 129.3 (s, CHb), 127.8 (d, 1JCP = 66 Hz, C1), 125.4 (s, CHe), 42.0 (s, CH2), 35.3 (s, C(tBu)), 35.0 (s, C(tBu)), 31.5 (s, CH3(tBu)), 31.4 (s, CH3(tBu)). 31P{1H} NMR (202 MHz, CD2Cl2, 25 °C) δ: 0.6.
Synthesis of Gold(I)–1-Propene Complex 14
In a glovebox, a Schlenk flask was charged with silver hexafluoroantimonate (8 mg, 0.022 mmol) in dichloromethane (1 mL). The corresponding gold(I) chloride complex (21 mg, 0.02 mmol) was transferred into a small glass vial and dissolved in dichloromethane (1 mL). The vial solution was loaded into a plastic syringe equipped with a stainless steel needle. Outside the glovebox, the solution of the gold(I) chloride complex was added to the AgSbF6 suspension while bubbling 1-propene and stirred for 10 min. The mixture was filtered through a short pad of Celite to remove the silver salts, and the solvent was removed under vacuum affording the complex 14 as colorless solid (23 mg, 87%). Crystals suitable for X-ray diffraction were grown by slow diffusion of pentane into a dichloromethane solution of complex 14. NMR analysis showed the presence of two isomers at rt with a 1:1 ratio that were analyzed altogether as 14 and 14′. 1H NMR (400 MHz, CD2Cl2, 25 °C) δ: 7.76 (d, 3H, 3JHH = 8.1 Hz, CHb + CHb′), 7.76 (d, 3H, 3JHH = 8.1 Hz, CHb′), 7.54 (d, 3H, 4JHH = 2.0 Hz, CHa), 7.51 (d, 3H, 4JHH = 2.0 Hz, CHa′), 7.44 (dd, 3H, 3JHH = 8.1 Hz, 4JHH = 2.0 Hz, CHc), 7.44 (dd, 3H, 3JHH = 8.1 Hz, 4JHH = 2.0 Hz, CHc’),7.26 (d, 6H, 3JHH = 8.6 Hz, CHe), 7.25 (d, 6H, 3JHH = 8.6 Hz, CHe′), 6.76 (d, 6H, 3JHH = 8.1 Hz, CHd), 6.76 (d, 6H, 3JHH = 8.1 Hz, CHd′), 4.64 (m, 1H, CHprop), 4.50 (m, 1H, CH(prop)’), 3.45 (m, 2H, CH2(prop)), 3.45 (m, 2H, CH2(prop)′),1.27 (s, 27H, CH3(tBu)), 1.23 (s, 3H, CH3(prop)), 1.23, (s, 3H, CH3(prop)′), 1.24 (s, 27H, CH3(tBu)). 13C{1H} NMR (100 MHz, CD2Cl2, 25 °C) δ: 152.3 (s, C5), 152.3 (s, C5′), 143.5 (d, 2JCP = 16 Hz, C3), 143.5 (d, 2JCP = 16 Hz, C3′), 138.6 (d, 3JCP = 7 Hz, C4), 138.5 (d, 3JCP = 7 Hz, C4’), 134.1 (d, 3JCP = 6 Hz, CHc), 134.0 (d, 3JCP = 6 Hz, CHc′), 133.7 (s, CHa), 133.7 (s, CHa′), 132.3 (s, CH(prop)), 132.2 (s, CH(prop)′), 130.0 (s, CHb), 130.0 (s, CHb), 129.9 (s, CHd), 129.9 (s, CHd′), 127.7 (d, 2JCP = 62 Hz, C1), 127.7 (d, 1JCP = 62 Hz, C1′), 126.1 (s, CHe + CHe′), 101.6 (d, 2JCP = 5 Hz, CH2(prop)), 101.1 (d, 2JCP = 5 Hz, CH2(prop)′), 35.4 (s, C(tBu)), 35.2 (s, C(tBu)), 22.9 (s, CH3(prop)), 21.9 (s, CH3(prop)′), 35.0 (s, C(tBu)), 31.6 (s, CH3(tBu)), 31.3 (s, CH3(tBu)). 31P{1H} NMR (202 MHz, CD2Cl2, 25 °C) δ: 13.8 and 13.6.
General Procedure for the Gold(I)-Catalyzed Hydroamination of Ethylene
A mixture of amide (0.20 mmol), gold chloride complex (0.01 mmol), and silver hexafluoroantimoniate (4 mg, 0.01 mmol) in dioxane (1 mL) was placed in a Fischer Porter tube together with a magnetic stirring bar under a nitrogen atmosphere. The tube was freeze-pumped to remove the nitrogen gas, filled with the indicated ethylene pressure, and stirred at 100 °C for 18 h. After this time, the mixture was cooled down to rt and diluted in CH2Cl2 (5 mL), and anisole (22 mL, 0.20 mmol) was added as the internal standard. The mixture was then filtered through a short pad of Celite, the solvents were removed under reduced pressure, and the sample was analyzed by NMR spectroscopy in CDCl3.
Acknowledgments
This work was supported by the European Research Council (ERC Starting Grant, CoopCat, Project 756575) and the Spanish Ministry of Science and Innovation (Project PID2019-110856GA-I00). M.N. acknowledges the Spanish Ministry of Science and Innovation and Junta de Andalucía for postdoctoral programs (FJC2018-035514-I and DOC_00149). The use of computational facilities at the Supercomputing Center of Galicia (CESGA) is acknowledged. Dr. Juan J. Moreno is gratefully acknowledged for his advice and help in the DFT calculation analysis.
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acscatal.1c05823.
Accession Codes
CCDC 2129167–2129172 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
The authors declare no competing financial interest.
Supplementary Material
References
- a Hammer B.; Norskov J. K. Why gold is the noblest of all metals. Nature 1995, 376, 238–240. 10.1038/376238a0. [DOI] [Google Scholar]; b Schmidbaur H. Gold-Chemie: ein Eldorado. Naturwiss. Rundsch. 1995, 48, 443–451. [Google Scholar]
- a Teles J. H.; Brode S.; Chabanas M. Cationic Gold(I) Complexes: Highly Efficient Catalysts for the Addition of Alcohols to Alkynes. Angew. Chem., Int. Ed. 1998, 37, 1415–1418. . [DOI] [PubMed] [Google Scholar]; b Hashmi A. S. K.; Schwarz L.; Choi J.-H.; Frost T. M. A New Gold-Catalyzed C–C Bond Formation. Angew. Chem., Int. Ed. 2000, 39, 2285–2288. . [DOI] [PubMed] [Google Scholar]; c Hashmi A. S. K.; Frost T. M.; Bats J. W. Highly Selective Gold-Catalyzed Arene Synthesis. J. Am. Chem. Soc. 2000, 122, 11553–11554. 10.1021/ja005570d. [DOI] [Google Scholar]; d Hashmi A. S. K. Homogeneous catalysis by gold. Gold Bull. 2004, 37, 51–65. 10.1007/BF03215517. [DOI] [Google Scholar]; e Hashmi A. S. K.; Toste F. D.. Modern Gold Catalyzed Synthesis; Wiley-VCH, 2012. [Google Scholar]; f Toste F. D.; Michelet V.. Gold Catalysis: An Homogeneous Approach; Imperial College Press, 2014. [Google Scholar]; g Slaughter L. M.Homogeneous Gold Catalysis; Springer, 2015. [Google Scholar]; h Hashmi A. S. K. Introduction: Gold Chemistry. Chem. Rev. 2021, 121, 8309–8310. 10.1021/acs.chemrev.1c00393. [DOI] [PubMed] [Google Scholar]; i Wang T.; Hashmi A. S. K. 1,2-Migration onto Gold Carbene Centers. Chem. Rev. 2021, 121, 8948–8978. 10.1021/acs.chemrev.0c00811. [DOI] [PubMed] [Google Scholar]; j Campeau D.; Rayo D. F. K.; Mansour A.; Murativ K.; Gagosz F. Gold-Catalyzed Reactions of Specially Activated Alkynes, Allenes, and Alkenes. Chem. Rev. 2021, 121, 8756–8867. 10.1021/acs.chemrev.0c00788. [DOI] [PubMed] [Google Scholar]
- a Fürstner A. Gold and platinum catalysis–a convenient tool for generating molecular complexity. Chem. Soc. Rev. 2009, 38, 3208–3221. 10.1039/b816696j. [DOI] [PubMed] [Google Scholar]; b Krause N.; Winter C. Gold-Catalyzed Nucleophilic Cyclization of Functionalized Allenes: A Powerful Access to Carbo and Heterocycles. Chem. Rev. 2011, 111, 1994–2009. 10.1021/cr1004088. [DOI] [PubMed] [Google Scholar]; c Corma A.; Leyva-Pérez A.; Sabater M. J. Gold-Catalyzed Carbon-heteroatom Bond-Forming Reactions. Chem. Rev. 2011, 111, 1657–1712. 10.1021/cr100414u. [DOI] [PubMed] [Google Scholar]; d Rudolph M.; Hashmi A. S. K. Gold catalysis in total synthesis–an update. Chem. Soc. Rev. 2012, 41, 2448–2462. 10.1039/C1CS15279C. [DOI] [PubMed] [Google Scholar]; e Chiarucci M.; Bandini M. New developments in gold-catalyzed manipulation of inactivated alkenes. Beilstein J. Org. Chem. 2013, 9, 2586–2614. 10.3762/bjoc.9.294. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Braun I.; Asiri A. M.; Hashmi A. S. K. Gold Catalysis 2.0. ACS Catal. 2013, 3, 1902–1907. 10.1021/cs400437s. [DOI] [Google Scholar]; g Friend C. M.; Hashmi A. S. K. Gold Catalysis. Acc. Chem. Res. 2014, 47, 729–730. 10.1021/ar5000506. [DOI] [PubMed] [Google Scholar]; h Soriano E.; Fernández I. Allenes and computational chemistry: from bonding situations to reaction mechanisms. Chem. Soc. Rev. 2014, 43, 3041–3105. 10.1039/c3cs60457h. [DOI] [PubMed] [Google Scholar]; i Obradors C.; Echavarren A. M. Intriguin mechanistic labyrinths in gold(I) catalysis. Chem. Commun. 2014, 50, 16–28. 10.1039/C3CC45518A. [DOI] [PMC free article] [PubMed] [Google Scholar]; j Dorel R.; Echavarren A. M. Gold(I)-Catalyzed Activation of Alkynes for the Construction of Molecular Complexity. Chem. Rev. 2015, 115, 9028–9072. 10.1021/cr500691k. [DOI] [PMC free article] [PubMed] [Google Scholar]; k Halliday C. J. V.; Lynam M. Gold-alkynyls in catalysis: alkyne activation, gold cumulenes and nuclearity. Dalton Trans. 2016, 45, 12611–12626. 10.1039/C6DT01641C. [DOI] [PubMed] [Google Scholar]; l Mascareñas J. L.; Varela I.; López F. Allenes and Derivatives in Gold(I)- and Platinum(II)-Catalyzed Formal Cycloadditions. Acc. Chem. Res. 2019, 52, 465–479. 10.1021/acs.accounts.8b00567. [DOI] [PMC free article] [PubMed] [Google Scholar]; m Zucarello G.; Zanini M.; Echavarren A. M. Buchwald-Type Ligands on Gold(I) Catalysis. Isr. J. Chem. 2020, 60, 360–372. 10.1002/ijch.201900179. [DOI] [Google Scholar]
- a Müller T. E.; Hultzsch K. C.; Yus M.; Foubelo F.; Tada M. Hydroamination: Direct Addition of Amines to Alkenes and Alkynes. Chem. Rev. 2008, 108, 3795–3892. 10.1021/cr0306788. [DOI] [PubMed] [Google Scholar]; b Nishina N.; Yamamoto Y. Late Transition Metal-Catalyzed Hydroamination. Top. Organomet. Chem. 2013, 43, 115–144. [Google Scholar]; c Huang K.; Arndt M.; Gooßen K.; Heydt H.; Gooßen L. J. Late Transition Metal-Catalyzed Hydroamination and Hydroamidation. Chem. Rev. 2015, 115, 2596–2697. 10.1021/cr300389u. [DOI] [PubMed] [Google Scholar]; d Patel M.; Saunthwal R. K.; Verma A. K. Base-Mediated Hydroamination of Alkynes. Acc. Chem. Res. 2017, 50, 240–254. 10.1021/acs.accounts.6b00449. [DOI] [PubMed] [Google Scholar]; e Streiff S.; Jérome F. Hydroamination of non-activated alkenes with ammonia: a holy grail in catalysis. Chem. Soc. Rev. 2021, 50, 1512–1521. 10.1039/C9CS00873J. [DOI] [PubMed] [Google Scholar]
- For selected examples of transition metal catalyzed hydroamination of olefins via nucleophilic attack of the amine nitrogen on the olefin see:; a Yang S.; Li Q.-Z.; Xu C.; Xu Q.; Shi M. Rhodium-catalyzed asymmetric hydroamination and hydroindolation of keto-vinylidenecyclopropanes. Chem. Sci. 2018, 9, 5074–5081. 10.1039/C8SC01595C. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Lepori C.; Bernoud E.; Guillot R.; Tobisch S.; Hannedouche J. Experimental and Computational Mechanistic Studies of the β-Diketaminatoiron(II)-Catalyzed Hydroamination of Primary Aminoalkanes. Chem. – Eur. J. 2019, 25, 835–844. 10.1002/chem.201804681. [DOI] [PubMed] [Google Scholar]; c Foster D.; Gao P.; Zhang Z.; Sipos G.; Sobolev A. N.; Nealon G.; Falviene L.; Cavallo L.; Dorta R. Design, scope and mechanism of highly active and selective chiral NHC-iridium catalysts for the intramolecular hydroamination of a variety of unactivated aminoalkenes. Chem. Sci. 2021, 12, 3751–3767. 10.1039/D0SC05884J. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Bernoud E.; Lepori C.; Mellah M.; Schulz E.; Hannedouche J. Recent advances in metal free- and late transition metal-catalysed hydroamination of unactivated alkenes. Catal. Sci. Technol. 2015, 5, 2017–2037. 10.1039/C4CY01716A. [DOI] [Google Scholar]; b Peng X.; Kaga A.; Hirao H.; Chiba S. Hydroamination of alkenyl N-arylhydrazones mediated by t-BuOK for the synthesis of nitrogen heterocycles. Org. Chem. Front. 2016, 3, 609–613. 10.1039/C6QO00053C. [DOI] [Google Scholar]; c Gentry E. C.; Knowles R. R. Synthetic Applications of Proton-Coupled Electron Transfer. Acc. Chem. Res. 2016, 49, 1546–1556. 10.1021/acs.accounts.6b00272. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Margrey K. A.; Nicewicz D. A. A General Approach to Catalytic Alkene Anti-markovnikov Hydrofunctionalization Reactions via Acridinium Photoredox Catalysis. Acc. Chem. Res. 2016, 49, 1997–2006. 10.1021/acs.accounts.6b00304. [DOI] [PubMed] [Google Scholar]; e Trowbridge A.; Walton S. M.; Gaunt M. J. New Strategies for the Transition-Metal Catalyzed Synthesis of Aliphatic Amines. Chem. Rev. 2020, 120, 2613–2692. 10.1021/acs.chemrev.9b00462. [DOI] [PubMed] [Google Scholar]; f Yu Z.-L.; Cheng Y.-F.; Jiang N.-C.; Wang J.; Fan L.-W.; Yuan Y.; Li Z.-L.; Gu Q.-S.; Liu X.-Y. Desymmetrization of unactivated bis-alkenes via chiral Bronsted acid-catalysed hydroamination. Chem. Sci. 2020, 11, 5987–5993. 10.1039/D0SC00001A. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Kim K.; Park S.; Lee Y. KOt-Bu-Catalyzed Chemo- and Regioselective Hydroamiantion of Allylic Sulfones with Indoles. Eur. J. Org. Chem. 2021, 2021, 125–137. 10.1002/ejoc.202001394. [DOI] [Google Scholar]
- a Widenhoefer R. A.; Han X. Gold-Catalyzed Hydroamination of C–C Multiple Bonds. Eur. J. Org. Chem. 2006, 2006, 4555–4563. 10.1002/ejoc.200600399. [DOI] [Google Scholar]; b Leung C. H.; Baron M.; Biffis A. Gold-Catalyzed Intermolecular Alkyne Hydrofunctionalization–Mechanistic Insights. Catalysts 2020, 10 (10), 1210. 10.3390/catal10101210. [DOI] [Google Scholar]
- a Zhang J.; Yang C.-G.; He C. Homogeneous Gold-Catalyzed Oxidative Carboheterofunctionalization of Alkenes. J. Am. Chem. Soc. 2006, 128, 1798–1799. 10.1021/ja053864z. [DOI] [PubMed] [Google Scholar]; b Liu X.-Y.; Li C.-H.; Che C.-M. Phosphine Gold(I)-Catalyzed Hydroamination of Alkenes under Thermal and Microwave-Assisted Conditions. Org. Lett. 2006, 8, 2707–2710. 10.1021/ol060719x. [DOI] [PubMed] [Google Scholar]; c Giner X.; Nájera C.; Kovács G.; Lledós A.; Ujaque G. Gold versus Silver-Catalyzed Intermolecular Hydroamination of Alkenes and Dienes. Adv. Synth. Catal. 2011, 353, 3451–3466. 10.1002/adsc.201100478. [DOI] [Google Scholar]; d Timmerman J. C.; Widenhoefer R. A. Gold-Catalyzed Intermolecular ant-Markovnikov Hydroamination of Methylenecyclopropanes with 2-Pyridones. Adv. Synth. Catal. 2015, 357, 3703–3706. 10.1002/adsc.201500866. [DOI] [Google Scholar]; e Timmerman J. C.; Robertson B. D.; Widenhoefer R. A. Gold-Catalyzed Intermolecular Anti-Markovnikov Hydroamination of Alkylidenecyclopropanes. Angew. Chem., Int. Ed. 2015, 54, 2251–2254. 10.1002/anie.201410871. [DOI] [PubMed] [Google Scholar]; f Wang C.; Ren X.-R.; Qi C.-Z.; Yu H.-Z. Mechanistic Study on Gold-Catlyzed Highly Selective Hydroamiantion of Alkylidenecyclopropanes. J. Org. Chem. 2016, 81, 7326–7335. 10.1021/acs.joc.6b00726. [DOI] [PubMed] [Google Scholar]; g Couce-Rios A.; Lledós A.; Fernández I.; Ujaque G. Origin of the Anti-Markovnikov Hydroamination of Alkenes Catalyzed by L–Au(I) Complexes: Coordination Mode Determines Regioselectivity. ACS Catal. 2019, 9, 848–858. 10.1021/acscatal.8b03843. [DOI] [Google Scholar]
- a Mizushima E.; Hayashi T.; Tanaka M. Au(I)-Catalyzed Highly Efficient Intermolecular Hydroamination of Alknes. Org. Lett. 2003, 5, 3349–3352. 10.1021/ol0353159. [DOI] [PubMed] [Google Scholar]; b Kramer S.; Dooleweerdt K.; Lindhardt A. T.; Rottländer M.; Skrydstrup T. Highly Regioselective Au(I)-Catalyzed Hydroamination of Ynamides and Propiolic Acid Derivatives with Anilines. Org. Lett. 2009, 11, 4208–4211. 10.1021/ol901565p. [DOI] [PubMed] [Google Scholar]; c Patil N. T.; Lakshmi P. G. V. V.; Singh V. Au(I)-Catalyzed Direct Hydroamination/Hydroarylation and Double Hydroamination of Terminal Alkynes. Eur. J. Org. Chem. 2010, 2010, 4719–4731. 10.1002/ejoc.201000389. [DOI] [Google Scholar]; d Alvarado E.; Badaj A. C.; Larocque T. G.; Lavoie G. G. N-Heterocylic Carbnes and Imidazole-2-thiones as Ligands for the Gold(I)-Catalysed Hydroamination of Phenylacetylene. Chem. – Eur. J. 2012, 18, 12112–12121. 10.1002/chem.201201448. [DOI] [PubMed] [Google Scholar]; e Siewert J.-E.; Schumann A.; Fischer M.; Schmidt C.; Taeufer T.; Hering-Junghans C. Terphenyl(bisamino)phosphines: electron-rich ligands for gold-catalysis. Dalton Trans. 2020, 49, 12354–12364. 10.1039/D0DT02435J. [DOI] [PubMed] [Google Scholar]; f Jia T.; Fan S.; Li F.; Ye X.; Zhang W.; Song Z.; Shi X. Achieving Aliphatic Amine Addition to Arylalkynes via the Lewis Acid Assisted Triazole-Gold (TA-Au) Catalyst System. Org. Lett. 2021, 23, 6019–6023. 10.1021/acs.orglett.1c02098. [DOI] [PMC free article] [PubMed] [Google Scholar]
- For examples of Au(I)-catalyzed intermolecular hydroamination of allenes see:; a LaLonde R. L.; Sherry B. D.; Kang E. J.; Toste F. D. Gold(I)-Catalyzed Enantioselective Intramolecular Hydroamination of Allenes. J. Am. Chem. Soc. 2007, 129, 2452–2453. 10.1021/ja068819l. [DOI] [PubMed] [Google Scholar]; b Kinder R. E.; Zhang Z.; Widenhoefer R. A. Intermolecular Hydroamination of Allenes with N-Unsubstituted Carbamates Catalyzed by a Gold(I) N-Heterocyclic Carbene Complex. Org. Lett. 2008, 10, 3157–3159. 10.1021/ol8010858. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Duncan A. N.; Widenhoefer R. A. Gold(I)-Catalyzed Intermolecular Hydroamination of Allenes with Arylamines. Synlett 2010, 419–422. 10.1055/s-0029-1218555. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Wang Z. J.; Benitez D.; Tkatchouk E.; Goddard W. A. III; Toste F. D. Mechanistic Study of Gold(I)-Catalyzed Intermolecular Hydroamination of Allenes. J. Am. Chem. Soc. 2010, 132, 13064–13071. 10.1021/ja105530q. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Kinjo R.; Donnadieu B.; Bertrand G. Gold-Catalyzed Hydroamination of Alkynes and Allenes with Parent Hydrazine. Angew. Chem., Int. Ed. 2011, 50, 5560–5563. 10.1002/anie.201100740. [DOI] [PubMed] [Google Scholar]; f Butler K. L.; Tragni M.; Widenhoefer R. A. Gold(I)-Catalyzed Stereoconvergent, Intermolecular Enantioselective Hydroamination of Allenes. Angew. Chem., Int. Ed. 2012, 51, 5175–5178. 10.1002/anie.201201584. [DOI] [PMC free article] [PubMed] [Google Scholar]; g Couce-Rios A.; Kovács G.; Ujaque G.; Lledós A. Hydroamination of C–C Multiple Bonds with Hydrazine Catalyzed by N-Heterocylic Carbene–Gold(I) Complexes: Substrate and Ligands Effects. ACS Catal. 2015, 5, 815–829. 10.1021/cs501705b. [DOI] [Google Scholar]; h Harris R. J.; Nakafuku K.; Duncan A. N.; Carden R. G.; Timmerman J. C.; Widenhoefer R. A. Kinetics and Mechanism of the Gold-Catalyzed Hydroamination of 1,1-Dimethylallene with N-Methylaniline. Chem. – Eur. J. 2021, 27, 10377–10386. 10.1002/chem.202100741. [DOI] [PubMed] [Google Scholar]
- For examples of intermolecular Au(I)-catalyzed hydroamination of dienes see:; a Brouwer C.; He C. Efficient Gold-Catalyzed Hydroamination of 1,3-dienes. Angew. Chem., Int. Ed. 2006, 45, 1744–1747. 10.1002/anie.200504495. [DOI] [PubMed] [Google Scholar]; b Kovács G.; Ujaque G.; Lledós A. The Reaction Mechanism of the Hydroamination of Alkenes Catalyzed by Gold(I)–Phosphine The Role of the Counterion and the N-Nucleophile Substituents in the Proton-Transfer Step. J. Am. Chem. Soc. 2008, 130, 853–864. 10.1021/ja073578i. [DOI] [PubMed] [Google Scholar]; c Giner X.; Nájera C. (Triphenyl phosphite)gold(I)-Catalyzed Intermolecular Hydroamination of Alkenes and 1,3-Dienes. Org. Lett. 2008, 10, 2919–2922. 10.1021/ol801104w. [DOI] [PubMed] [Google Scholar]
- Zhang Z.; Lee S. D.; Widenhoefer R. A. Intermolecular Hydroamiantion of Ethylene and 1-Alkenes with Cyclic Ureas Catalyzed by Achiral and Chiral Gold(I) Complexes. J. Am. Chem. Soc. 2009, 131, 5372–5373. 10.1021/ja9001162. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dias H. V. R.; Fianchini M.; Cundari T. R.; Campana C. F. Synthesis and characterization of the gold(I) tris(ethylene) complex [Au(C2H4)3][SbF6]. Angew. Chem., Int. Ed. 2008, 47, 556–559. 10.1002/anie.200703515. [DOI] [PubMed] [Google Scholar]; b Schmidbaur H.; Schier A. Gold η2-Coordination of Unsaturated and Aromatic Hydrocarbons: The Key Step in Gold-Catalyzed Organic Transformations. Organometallics 2010, 29, 2–23. 10.1021/om900900u. [DOI] [Google Scholar]; c Cinellu M. A. In Modern Gold Catalyzed Synthesis; Wiley-VCH: Weinheim, 2012, pp 175–179. [Google Scholar]; d Brooner R. E. M.; Widenhoefer R. A. Carionic, Two-Coordinate Gold π Complexes. Angew. Chem., Int. Ed. 2013, 52, 11714–11724. 10.1002/anie.201303468. [DOI] [PubMed] [Google Scholar]; e Jones A. C. Gold π-Complexes as Model Intermediates in Gold Catalysis. Top. Curr. Chem. 2015, 357, 133–166. 10.1007/128_2014_593. [DOI] [PubMed] [Google Scholar]; f Navarro M.; Bourissou D. π-Alkene/Alkyne and Carbene Complexes of Gold(I) Stabilized by Chelating Ligands. Adv. Organomet. Chem. 2021, 76, 101–144. 10.1016/bs.adomc.2021.02.001. [DOI] [Google Scholar]
- a Shapiro N. D.; Toste F. D. Synthesis and structural characterization of isolable phosphine coinage metal π-complexes. Proc. Natl. Acad. Sci. U. S. A 2008, 105, 2779–2782. 10.1073/pnas.0710500105. [DOI] [Google Scholar]; b Brown T. J.; Dickens M. G.; Widenhoefer R. A. Synthesis, X-ray Crystal Structures, and Solution Behavior of Monomeric, Cationic, Two-Coordinate Gold(I) π-Alkene Complexes. J. Am. Chem. Soc. 2009, 131, 6350–6351. 10.1021/ja9015827. [DOI] [PMC free article] [PubMed] [Google Scholar]; c Zuccaccia D.; Belpassi L.; Tarantelli F.; Macchioni A. Ion Pairing in Cationic Olefin–Gold(I) Complexes. J. Am. Chem. Soc. 2009, 131, 3170–3171. 10.1021/ja809998y. [DOI] [PubMed] [Google Scholar]; d de Frémont P.; Marion N.; Nolan S. P. Carionic NHC–gold(I) complexes: Synthesis, Isolation, and catalytic reactivity. J. Organomet. Chem. 2009, 694, 551–560. 10.1016/j.jorganchem.2008.10.047. [DOI] [Google Scholar]; e Brown T. J.; Dickens M. G.; Widenhoefer R. A. Synthesis and X-ray crystal structure of cationic, two-coordnate gold(I) π-alkene complexes that contain a sterically hindered o-biphenylphosphine ligands. Chem. Commun. 2009, 6451–6453. 10.1039/b914632f. [DOI] [PubMed] [Google Scholar]; f Hooper T. N.; Green M.; McGrady J. E.; Patel J. R.; Russell C. A. Synthesis and structural characterization of stable cationic gold(I) alkene complexes. Chem. Commun. 2009, 3877–3879. 10.1039/b908109g. [DOI] [PubMed] [Google Scholar]; g Moltoch P.; Blahut J.; Cisarova I.; Roithová J. X-ray characterization of triphenylphosphine-gold(I) olefin π-complexes and the revision of their stability in solution. J. Organomet. Chem. 2017, 848, 114–117. 10.1016/j.jorganchem.2017.07.011. [DOI] [Google Scholar]; h Griebel C.; Hodges D.; Yager B. R.; Liu F. L.; Zhow W.; Makaravage K. J.; Zhu Y.; Norman S. G.; Lan R.; Day C. S.; Jones A. C. Bisphenyl Phosphines. Structure and Synthesis of Gold(I) Alkene π-Complexes with Variable Phosphine Donicity and Enhanced Stability. Organometallics 2020, 39, 2665–2671. 10.1021/acs.organomet.0c00278. [DOI] [Google Scholar]
- a Cinellu M. A.; Minghetti G.; Stoccoro S.; Zucca A.; Manassero M. Reduction of gold(III) oxo complexes with alkenes. Synthesis of unprecedented gold alkene complexes,, [Au(N,N)(alkene)][PF6]. Crytsal structure of [Au(bipyip)(η2-CH2=CHPh)][PF6] (bipyip = 6-isopropyl-2,2′-bipyridine). Chem. Commun. 2004, 1618–1619. 10.1039/B404890C. [DOI] [PubMed] [Google Scholar]; b Cinellu M. A.; Minghetti G.; Cocco F.; Stoccoro S.; Zucca A.; Manassero M.; Arca M. Synthesis and properties of gold alkene complexes. Crystal structure of [Au(bipyoXyl)(η2-CH2=CHPh)](PF6) and DFT calculations on the model cation [Au(bipy)(η2-CH2=CH2)]+. Dalton Trans. 2006, 5703–5716. 10.1039/B610657A. [DOI] [PubMed] [Google Scholar]; c Flores J. A.; Dias H. V. R. Monomeric Copper(I), Silver(I) and Gold(I) Alkyne complexes and the Coinage Metal Family Group Trends. Inorg. Chem. 2008, 47, 4448–4450. 10.1021/ic800373u. [DOI] [PubMed] [Google Scholar]; d Dias H. V. R.; Wu J. Structurally Similar, thermally Stable Copper(I), Silver(I), and Gold(I) Ethylene Complexes Supported by a Fluorinated Scorpionate. Organometallics 2012, 31, 1511–1517. 10.1021/om201185v. [DOI] [Google Scholar]; e Klimovica K.; Krischbaum K.; Daugulis O. Synthesis and Properties of “Sandwich” Diimine-Coinage Metal Ethylene Complexes. Organometallics 2016, 35, 2938–2943. 10.1021/acs.organomet.6b00487. [DOI] [PMC free article] [PubMed] [Google Scholar]; f Navarro M.; Toledo A.; Joost M.; Amgounne A.; Mallet-Ladeira S.; Bourissou D. π Complexes of P^P and P^N chelated cold(I). Chem. Commun. 2019, 55, 7974–7977. 10.1039/C9CC04266K. [DOI] [PubMed] [Google Scholar]; g Navarro M.; Toledo A.; Mallet-Ladeira S.; Sosa-Carrizo E. D.; Miqueu K.; Bourissou D. Versatility and adaptative behaviour of the P^N chelating ligand MeDalphos within gold(I) π complexes. Chem. Sci. 2020, 11, 2750–2758. 10.1039/C9SC06398F. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Dias H. V. R.; Wu J. Thermally Stable Gold(I) Ehtylene Adducts: [(HB{3,5-(CF3)2Pz}3)Au(CH2=CH2)] and [(HB{3-(CF3),5-(Ph)Pz}3)(CH2=CH2)]. Angew. Chem., Int. Ed. 2007, 46, 7814–7816. 10.1002/anie.200703328. [DOI] [PubMed] [Google Scholar]; b Ridlen S. G.; Wu J.; Kulkarni N. V.; Dias H. V. R. Isolable Ethylene Complexes of Copper(I), Silver(I), and Gold(I) Supported by Fluorinated Scorpionates [HB{3-(CF3),5-(CH3)Pz}3] and [(HB{3-(CF3),5-(Ph)Pz}3)]. Eur. J. Inorg. Chem. 2016, 2016, 2573–2580. 10.1002/ejic.201501365. [DOI] [Google Scholar]; c Harper M. J.; Arthur C. J.; Crosby J.; Emmet E. J.; Falconer R. L.; Fensham-Smith A. J.; Gates P. J.; Leman T.; McGrady J. E.; Bower J.; Russell C. A. Oxidative Addition, Transmetallation, and Reductive Elimination at a 2,2’-Bipyridyl-Ligated Gold Center. J. Am. Chem. Soc. 2018, 140, 4440–4445. 10.1021/jacs.8b01411. [DOI] [PubMed] [Google Scholar]; d Yang Y.; Antoni P.; Zimmer M.; Sekine K.; Mulks F. F.; Hu L.; Zhang L.; Rudolph M.; Rominger F.; Hashmi A. S. K. Dual Gold/Silver Catalysis Involving Alkynylgold(III) Intermediates Formed by Oxidative Addition and Silver(I)-Catalyzed C–H Activation for the Direct Alkynylation of Cyclopropenes. Angew. Chem., Int. Ed. 2019, 58, 5129–5133. 10.1002/anie.201812577. [DOI] [PubMed] [Google Scholar]; e Wu J.; Noonikara-Poyil A.; Muñoz-Castro A.; Dias H. V. R. Gold(I) ethylene complexes supported by electron-rich scorpionates. Chem. Commun. 2021, 57, 978–981. 10.1039/D0CC07717H. [DOI] [PubMed] [Google Scholar]
- a Lang S. M.; Bernhardt T. M.; Barnett R. N.; Landman U. Methane Activation and Catalytic Ethylene Formation on Free Au2+. Angew. Chem., Int. Ed. 2010, 49, 980–983. 10.1002/anie.200905643. [DOI] [PubMed] [Google Scholar]; b Lang S. M.; Bernhardt T. M.; Bakker M.; Yoon B.; Landman U. The interaction of ethylene with free gold cluster cations: infrared photodissociation spectroscopy combined with electronic and vibrational structure calculations. J. Phys.: Condens. Matter 2018, 30, 504001. 10.1088/1361-648X/aaeafd. [DOI] [PubMed] [Google Scholar]; c Metz R. B.; Altinay G.; Kostko O.; Ahmed M. Probing Reactivity of Gold Atoms with Acetylene and Ethylene with VUV Photoionization Mass Spectrometry and Ab Initio Studies. J. Phys. Chem. A 2019, 123, 2194–2202. 10.1021/acs.jpca.8b12560. [DOI] [PubMed] [Google Scholar]
- Keller J.; Schlierf C.; Nolte P.; Straub B. F. One Pot Syntheses of Sterically Shielding Phosphorous Ligands by Selective Stepwise Nucleophilic Substitution at Triphenyl Phosphite. Synthesis 2006, 2, 354–365. 10.1055/s-2005-921758. [DOI] [Google Scholar]
- Navarro M.; Miranda-Pizarro J.; Moreno J. J.; Navarro-Gilabart C.; Fernández I.; Campos J. A Dicoordinate Gold(I)–Ethylene Complex. Chem. Commun. 2021, 57, 9280–9283. 10.1039/D1CC02769G. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Moreno J. J.; Espada M. F.; Campos J.; López-Serrano J.; Macgregor S. A.; Carmona E. Base Promoted, Remote C–H Activation of (η5-C5Me5)Ir(III) Center Involving Reversible C–C Bond Formation of Bound C5Me5. J. Am. Chem. Soc. 2019, 141, 2205–2210. 10.1021/jacs.8b11752. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Ortega-Moreno L.; Peloso R.; López-Serrano J.; Iglesias-Sigüenza J.; Maya C.; Carmona E. A Cationic Unsaturated Platinum(II) COmplex that promotes the Tautomerization of Acetylene to Vinylidene. Angew. Chem., Int. Ed. 2017, 56, 2772–2775. 10.1002/anie.201700087. [DOI] [PubMed] [Google Scholar]; c Marin M.; Moreno J. J.; Alcaide M. M.; Álvarez E.; López-Serrano J.; Campos J.; Nicasio M. C.; Carmona E. Evaluating stereoelectronic properties of bulky dialkylterphenyl phosphine ligands. J. Organomet. Chem. 2019, 896, 120–128. 10.1016/j.jorganchem.2019.06.003. [DOI] [Google Scholar]
- Espada M. F.; Campos J.; López-Serrano J.; Poveda M. L.; Carmona E. Methyl-, Ethenyl-, and Ethynyl-Bridged Cationic Digold Complexes Stabilized by Coordination to a Bulky Terphenylphosphine Ligand. Angew. Chem., Int. Ed. 2015, 54, 15379–15384. 10.1002/anie.201508931. [DOI] [PubMed] [Google Scholar]
- Miranda-Pizarro J.; Luo Z.; Moreno J. J.; Dickie D. A.; Campos J.; Gunnoe B. Reductive C–C Coupling from Molecular Au(I) Hydrocarbyl Complexes: A Mechanistic Study. J. Am. Chem. Soc. 2021, 143, 2509–2522. 10.1021/jacs.0c11296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Campos J. Dihydrogen and Acetylene Activation by a Gold(I)/Platinum(0) Transition Metal Only Frustrated Lewis Pair. J. Am. Chem. Soc. 2017, 139, 2944–2947. 10.1021/jacs.7b00491. [DOI] [PubMed] [Google Scholar]; b Arndt S.; Rudolph M.; Hahsmi A. S. K. Gold-based frustrated Lewis acid/base pairs (FLPs). Gold Bull. 2017, 50, 267–282. 10.1007/s13404-017-0219-7. [DOI] [Google Scholar]; c Hidalgo N.; Moreno J. J.; Pérez-Jiménez M.; Maya C.; López-Serrano J.; Campos J. Evidence for Genuine Bimetallic Frustrated Lewis Pair Activation of Dihydrogen with Gold(I)/Platinum(0) Systems. Chem. – Eur. J. 2020, 26, 5982–5993. 10.1002/chem.201905793. [DOI] [PubMed] [Google Scholar]; d Hidalgo N.; Moreno J. J.; Pérez-Jiménez M.; Maya C.; López-Serrano J.; Campos J. Tuning Activity and Selectivity during Alkyne Activation by Gold(I)/Platinum(0) Frustrated Lewis Pairs. Organometallics 2020, 39, 2534–2544. 10.1021/acs.organomet.0c00330. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Alférez M. G.; Moreno J. J.; Hidalgo N.; Campos J. Reversible Hydride Migration from C5Me5 to RhI Revealed by a Cooperative Bimetallic Approach. Angew. Chem., Int. Ed. 2020, 59, 20863–20867. 10.1002/anie.202008442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Marín M.; Moreno J. J.; Navarro-Gilabert C.; Álvarez E.; Maya C.; Peloso R.; Nicasio M. C.; Carmona E. Synthesis, Structure, and Nickel Carbonyl Complexes of Dialkylterphenyl Phosphines. Chem. – Eur. J. 2019, 25, 260–272. 10.1002/chem.201803598. [DOI] [PubMed] [Google Scholar]
- Clavier H.; Nolan S. P. Percent buried volume for phosphine and N-heterocyclic carbene ligands: steric properties in organometallic chemistry. Chem. Commun. 2010, 46, 841–861. 10.1039/b922984a. [DOI] [PubMed] [Google Scholar]
- Nes G. J. H.; Vos A. Single-crystal structures and electron density distributions of ethane, ethylene and acetylene. III. Single-crystal X-ray structure determination of ethylene at 85 K. Acta Crystallogr., Sect. B: Struct. Crystallogr. Cryst. Chem. 1979, B35, 2593–2601. 10.1107/S0567740879009961. [DOI] [Google Scholar]
- Bayler A.; Bowmaker G. A.; Schmidbaur H. Propeller Isomerism in Bis(trimesitylphosphine)gold(I), -silver(I), and -copper(I) Tetrafluoroborates. Inorg. Chem. 1996, 35, 5959–5960. 10.1021/ic960421a. [DOI] [Google Scholar]
- Zhdanko A.; Ströbele M.; Maier M. E. Coordination Chemistry of Gold Catalysts in Solution: A Detailed NMR Study. Chem. – Eur. J. 2012, 18, 14732–14744. 10.1002/chem.201201215. [DOI] [PubMed] [Google Scholar]
- a Li Q.-S.; Wan C.-Q.; Zou R.-Y.; Xu F.-B.; Song H.-B.; Wan X.-J.; Zhang Z.-Z. Gold(I) η2-Arene Complexes. Inorg. Chem. 2006, 45, 1888–1890. 10.1021/ic051869r. [DOI] [PubMed] [Google Scholar]; b Xu F.-B.; Li Q.-S.; Wu L.-Z.; Leng X.-B.; Li Z.-C.; Zeng X.-S.; Chow Y. L.; Zhang Z.-Z. Formation of Group 11 Metal(I)-Arene Complexes: Bonding Mode and Molecule-Responsive Spectral Variations. Organometallics 2003, 22, 633–640. 10.1021/om020668h. [DOI] [Google Scholar]; c Lavallo V.; Frey G. D.; Kousar S.; Donnadieu B.; Bertrand G. Allene formation by gold catalyzed cross-coupling of masked carbenes with vinylidenes. Proc. Natl. Acad. Sci. U. S. A. 2007, 104, 13569–13573. 10.1073/pnas.0705809104. [DOI] [PMC free article] [PubMed] [Google Scholar]; d Jiménez-Núñez E.; Claverie C. K.; Nieto-Oberhuber C.; Echavarren A. M. Prins cyclizations inAu-catalyzed reactions of enynes. Angew. Chem., Int. Ed. 2006, 45, 5452–5455. 10.1002/anie.200601575. [DOI] [PubMed] [Google Scholar]; e Weber S. G.; Rominger F.; Straub B. F. Isolated Silver Intermediate of Gold Precatalyst Activation. Eur. J. Inorg. Chem. 2012, 2012, 2863–3867. 10.1002/ejic.201200327. [DOI] [Google Scholar]; f Parvin N.; Mishra B.; George A.; Neralkar M.; Hossain H.; Parameswaran P.; Hotha S.; Kahn S. N-Heterocyclic silylene/germylene ligands in Au(I) catalysis. Chem. Commun. 2020, 56, 7625–7628. 10.1039/D0CC03156A. [DOI] [PubMed] [Google Scholar]
- a Zhu Y.; Day C. S.; Zhang L.; Hauser K. J.; Jones A. C. A Unique Au–Ag–Au Triangular Motif in a Trimetallic Halonium Dication: Silver Incroporation in a Gold(I) Catalyst. Chem. – Eur. J. 2013, 19, 12264–12271. 10.1002/chem.201302152. [DOI] [PubMed] [Google Scholar]; b Homs A.; Escofet I.; Echavarren A. M. On the Silver Effect and the Formation of Chloride-Bridged Digold Complexes. Org. Lett. 2013, 15, 5782–5785. 10.1021/ol402825v. [DOI] [PMC free article] [PubMed] [Google Scholar]
- a Weber D.; Gagné M. R. Dinuclear Gold-Silver Resting States May Explain Silver Effects in Gold(I) Catalysis. Org. Lett. 2009, 11, 4962–4965. 10.1021/ol902116b. [DOI] [PMC free article] [PubMed] [Google Scholar]; b Wang D.; Cai R.; Sharma S.; Jirak J.; Thummanapelli S. K.; Akhmedov N. G.; Zhang H.; Liu X.; Petersen J. L.; Shi X. “Silver Effect” in Gold(I) Catalysis: An Overlooked Important Factor. J. Am. Chem. Soc. 2012, 134, 9012–9019. 10.1021/ja303862z. [DOI] [PubMed] [Google Scholar]; c Franchino A.; Montesinos-Magraner M.; Echavarren A. M. Silver-Free Catalysis with Gold(I) Chloride Complexes. Bull. Chem. Soc. Jpn. 2021, 94, 1099–1117. 10.1246/bcsj.20200358. [DOI] [Google Scholar]; d Hu L.; Dietl M. C.; Han C.; Rudolph M.; Rominger K.; Hashmi A. S. K. Au–Ag Bimetallic Catalysis: 3-Alkynyl Benzofurans from Phenols via Tandem C–H Alkynylation/Oxy-Alkynylation. Angew. Chem., Int. Ed. 2021, 60, 10637–10642. 10.1002/anie.202016595. [DOI] [PMC free article] [PubMed] [Google Scholar]; e Han C.; Liu Y.; Tian X.; Rominger F.; Hashmi A. S. K. Dual Gold/Silver Catalysis: Indolizines from 2-Substituted Pyridine Derivatives via a Tandem C(sp3)–H Alkynylation/Iminoauration. Org. Lett. 2021, 23, 9480–9484. 10.1021/acs.orglett.1c03667. [DOI] [PubMed] [Google Scholar]
- Krauter C. M.; Hashmi A. S. K.; Pernpointner M. A New Insight into Gold(I)-Catalyzed Hydration of Alkynes: Proton Transfer. ChemCatChem 2010, 2, 1226–1230. 10.1002/cctc.201000136. [DOI] [Google Scholar]
- Calculations were performed with the Gaussian 09 program employing the hybrid functional PBE0. Geometry optimizations were carried out without geometry constraints and included solvent (dichloromethane) and dispersion effects (Grimme’s D3 parameter set). The Goodvibes program was used to implement concentration, temperature and Truhlar’s quasiharmonic free energy corrections.
- Cochran B. M.; Michael F. E. Mechanistic Studies of a Palladium-Catalyzed Intramolecular Hydroamination of Unactivated Alkenes: Protonolysis of a Stable Palladium Alkyl Complex Is the Turnover-Limiting Step. J. Am. Chem. Soc. 2008, 130, 2786–2792. 10.1021/ja0734997. [DOI] [PubMed] [Google Scholar]
- The reaction coordinate was thoroughly mapped by means of relaxed potential energy scans, which confirmed its barrierless nature.
- Uson A.; Laguna M.; Briggs D. A.; Murray H. H.; Fackler J. P. (Twreahydrothiophene) gold(I) or gold(III) complexes. Inorg. Synth. 2007, 26, 85–91. 10.1002/9780470132579.ch17. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.