

Abstract

A modular approach to prepare tri- and tetracyclic carbazoles by a sequential [3 + 2]heteroannulation is described. First, optimization of Pd-catalyzed Buchwald–Hartwig amination followed by C/N-arylation in a one-pot process is established. Second, mechanistic analyses identified the origins of chemo- and regioselective sequential control of both bond-forming steps. Finally, the substrate scope is demonstrated by the preparation of a range of tri- and tetracyclic carbazoles, including expedient access to several natural products and anti-cancer agents.
Introduction
Carbazoles are ubiquitous N-heterocycles used throughout medicinal chemistry and material sciences.1−3 From a pharmaceutical perspective, the carbazole core features extensively in drugs and natural products, many of which exhibit potent anti-proliferative activities (Figure 1a).4−9 The breadth of applications has inspired the development of numerous methodologies for their preparation.2,9,10 Pd-catalyzed processes in particular enable a [3 + 2]heteroannulation via Pd(0)-catalyzed Buchwald–Hartwig amination11 followed by Pd(II)-catalyzed C-arylation at a late stage in a synthetic workflow.12−25 However, a generalized set of guidelines that control the chemoselectivity of each C–N/C–C bond-forming reaction, and the factors that influence Pd(0)23 versus Pd(II)17 catalysis in a one-pot process have not been established. Furthermore, a mechanistic understanding of any regiocontrol underpinning the second C–H activation step for the formation of tetracyclic carbazoles is unknown.
Figure 1.

Generalized approach for the synthesis of tri–/tetracyclic carbazoles by Pd-catalyzed [3 + 2]heteroannulation.
In this manuscript, we establish reaction guidelines to prepare tri- and tetracyclic carbazoles by controlling the chemoselectivity of the first Pd(0)-catalyzed C–N bond-forming step and the regioselectivity of the second Pd(II)-catalyzed C/N-arylation (Figure 1b). Our rationale was to use o-chloroanilines (1) to define the A-ring of a carbazole core and heteroaryl bromides to form the C/D-rings of tricyclic and tetracyclic products. An initial version of this work was deposited in ChemRxiv on the 24th of November 2021.26
Results and Discussion
The motivation for this work was to develop a robust, one-pot synthetic framework that can access both tri- and tetracyclic carbazoles. The synthesis of tetracyclic carbazoles using a heteroaryl bromide substrate such as 3 has potentially two competing C–H activation sites. If the C-arylation proceeded via a Pd(II)-catalyzed intermediate, then we surmised that the more nucleophilic 5-position of an isoquinoline will result in preferential C–H bond activation at this site.27 However, the mechanistic determinants that control the regioselectivity of such Pd(II)-catalyzed C-arylation are not known with respect to a convergent [3 + 2]heteroannulation approach.28−31
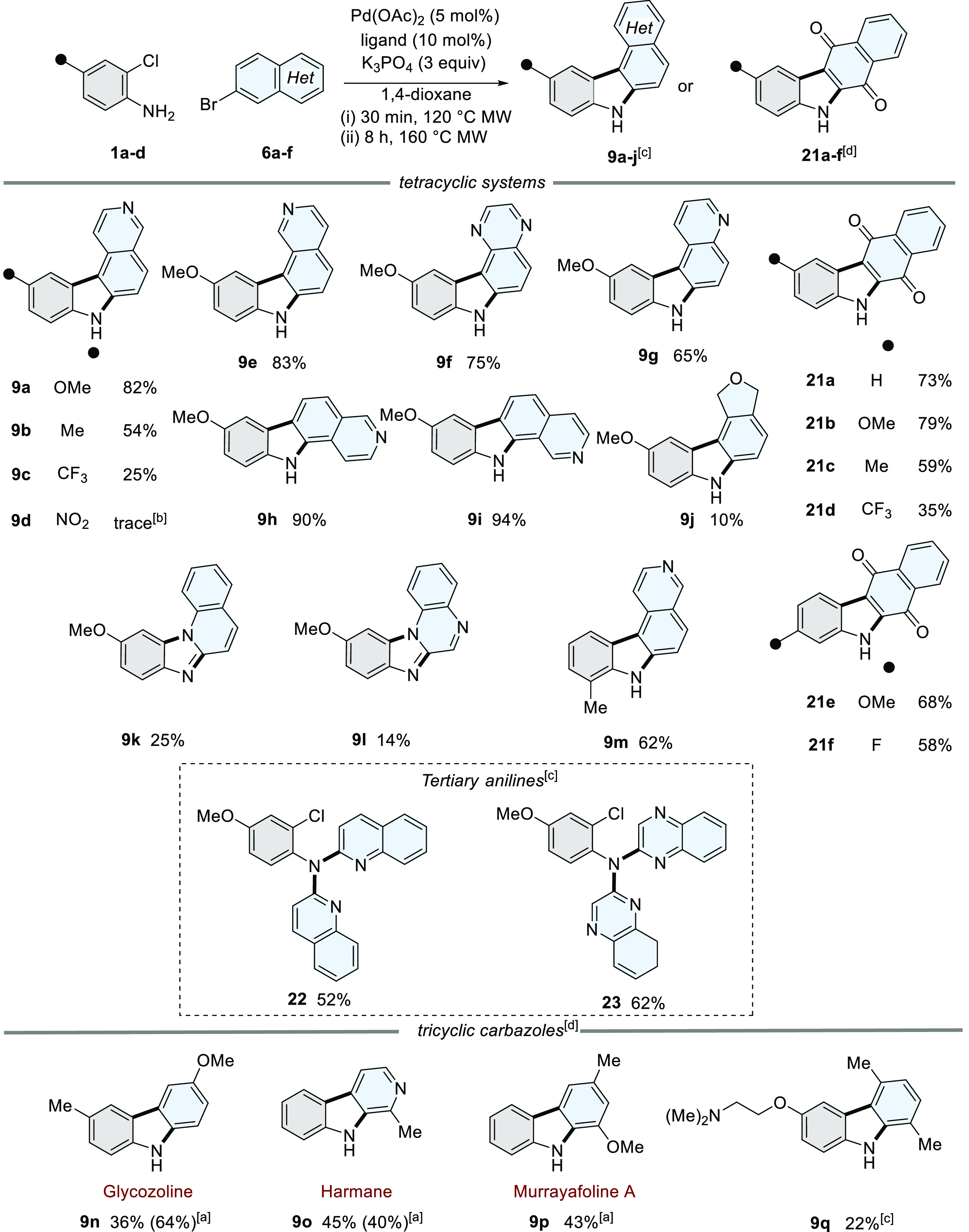
Our studies commenced with optimizing reaction conditions for the Pd(0)-catalyzed C–N bond-forming step using 4-methoxy-2-chloroaniline (1a) and 6-bromoisoquinoline (6a) as exemplar substrates (Scheme 1). An extensive screen (Table S1) identified DavePhos and K3PO4 as the optimal ligand/base pairing, which formed 7a in 87% yield.
Scheme 1. Scope (Isolated Yields) of Buchwald–Hartwig Amination.

Using these conditions, secondary anilines 7b–e (56–91%) were obtained using chloroanilines (1b–e) bearing electron-rich and electron-withdrawing substituents when reacted with 6-bromoisoquinoline (6a). The conditions tolerated a range of heteroaryl bromides to form 7g–j, except for 2-bromoquinoxaline (6d). In this case, a mixture of secondary (7i, 27%) and tertiary anilines (8, 25%) was formed. Using 5-bromo-1,3-dihydroisobenzofuran (6e) formed 7j in 18% yield. This process was also compatible with 2-bromonaphthalene-1,4-dione (6f) producing 1,4-napthoquinones32 (7k–p) in 59–82%. Whilst this reaction could conceivably proceed via Michael addition, the reaction afforded the secondary anilines 7k–p in 5–7% in the absence of a Pd catalyst.
With conditions for the Pd(0)-catalyzed C–N bond-forming step established, the optimization of the one-pot process was explored (Table S2). The optimal ligand/base pairing of HPCy3BF4 with K3PO4 was identified, which formed 9a from 1a and 6a in 82% yield (Scheme 2a). This highlights that the second C–H activation step is regioselective for the 7-position of 6a. To further understand how the nature of the chloroaniline and heteroaryl bromide substrates influenced the chemo- and regioselectivity of both bond-forming steps, a series of test reactions was undertaken. Exchanging 6-bromoisoquinoline (6a) for isoquinoline (10) formed dimethoxyphenazine (11) and the tertiary aniline (12) in 35 and 25% isolated yields, respectively (Scheme 2b). No dihydrophenazine was isolated from the reaction, which suggests that an in situ oxidation occurred.33 No reaction occurred when isoquinoline (10) was the coupling partner with 1-chloro-3-methoxybenzene (13, Scheme 2c). Only secondary aniline (15) was isolated when para-anisidine (14) was reacted with 6-bromoisoquinoline (6a, Scheme 2d). Taken collectively, these test reactions highlighted the following requirements for the preparation of the tetracyclic core: (i) aryl bromide is essential and prevents dimerization of the o-chloroaniline, (ii) whilst the absence of a bromo substituent in the heteroaryl substrate results in C–H activation at the same site, there is little regiocontrol, (iii) a chloro substituent is essential for C-arylation.
Scheme 2. Mechanistic Studies of the [3 + 2]Heteroannulation.

Reaction conditions: Pd(OAc)2 (5 mol %), HPCy3BF4 (10 mol %), K3PO4 (3 equiv), 1,4-dioxane; (i) 30 min, 120 °C MW; (ii) 8 h, 160 °C MW; wrt = with respect to.
The influence of the electron-donating aniline lone pair in the direct C-arylation step was then explored (Scheme 2e). We surmised that the acetylated substrate (16) would deactivate the C-ring and render the C-arylation less efficient. This indeed occurred as highlighted by the formation of deacetylated C-arylated regioisomers, 9a and 17, in 27% total yield (1.2:1.0, 9a:17). We assume that deacetylation occurs in situ because of the high temperatures and the presence of a base in the reaction mixture. An unexpected result was the formation of the linear tetracyclic carbazole 17, which arises from C–H activation of the isoquinoline 7-position of 16. The formation of 17 suggests that C–H activation of the 7-position is favored if the acetyl group is present prior to C-arylation. In contrast, if 7a is present, presumably formed by deactylation of 16, C–H activation at the 5-position occurs. DFT calculations confirmed that the 5-position is indeed the more electron-rich site (Scheme 2e and the Supporting Information Section 6). We speculate that the acetyl group (i.e., 16) directs C–H activation at this site via coordination of a Pd(II) species through the amide carbonyl.34−36
This series of reactions has guided us to propose that oxidative addition of the C–Cl bond in 7a occurs first and proceeds via a Pd(0) species to form 18 (Scheme 2f). Pd(II)-catalyzed C-arylation forms the palladacycle (20), which could proceed through a concerted metalation–deprotonation pathway via the formation of 19a.12
Alternatively, since the efficiency of the reaction is higher with the lone pair of aniline (18) donating into the ring, a Friedel–Crafts-like electrophilic aromatic substitution mechanism proceeding via imine (19b) followed by tautomerization might also be possible to form 20.37 Finally, reductive elimination of 20 produces the C-arylated product (9a). With mechanistic knowledge of the second C–C bond-forming step established, the substrate scope of the process was investigated (Scheme 3).
Scheme 3. Scope and Isolated Yields of the [3 + 2]Heteroannulation,,,
Addition of PivOH (30 mol %).
Formation of 7e in 79%; traces of 9d detected by LCMS.
Ligand = HPtBu3BF4.
Ligand = DavePhos.
The reaction conditions formed 7H-pyrido[3,4-c]carbazole analogues (9a–c). The presence of a nitro group resulted in only trace amounts of 9d formed, with the secondary aniline (7e) isolated in 79% yield. The reaction conditions also tolerated heteroatom changes and saturation in the D-ring of the heteroaryl bromide (9e–j).30 The [3 + 2]heteroannulation strategy was also compatible with the formation of carbazole-1,4-quinones (21a–f).28,38 Access to N-arylated products is also possible, forming a mixture of fused imidazoles (9k–l) via an N-arylation step, alongside tertiary anilines (22–23). Steric bulk ortho to the aniline substrate is also tolerated, forming 9m in 62%. However, when 3-methyl-2-chloroaniline is used as a substrate, a mixture of regioisomers was formed as an inseparable mixture (see Supporting Information).
The modularity of this strategy is also exemplified by the preparation of natural products glycozoline (9n),39 harmane (9o),40 and murrayafoline A (9p).41,42 In addition, preparation of 9q demonstrates that the reaction conditions tolerate functional groups bearing potential Pd-chelating sites and bulky substituents ortho to the corresponding aryl bromide.
Our [3 + 2]heteroannulation strategy was extended to the targeted synthesis of biologically active tetracyclic carbazoles. Carbazole-1,4-quinones (e.g., 21a–f) have established anti-cancer activity via topoisomerase inhibition or by the production of reactive oxygen species.38,43 We used 21b as a key intermediate for the targeted synthesis of a deaza analogue of the natural product 9-methoxyellipticine (Scheme 4a), producing 26 in three steps and in an overall yield of 20%.44
Scheme 4. Application of the [3 + 2]Heteroannulation Process for the Targeted Synthesis of Biologically Active Tetracyclic Carbazoles.
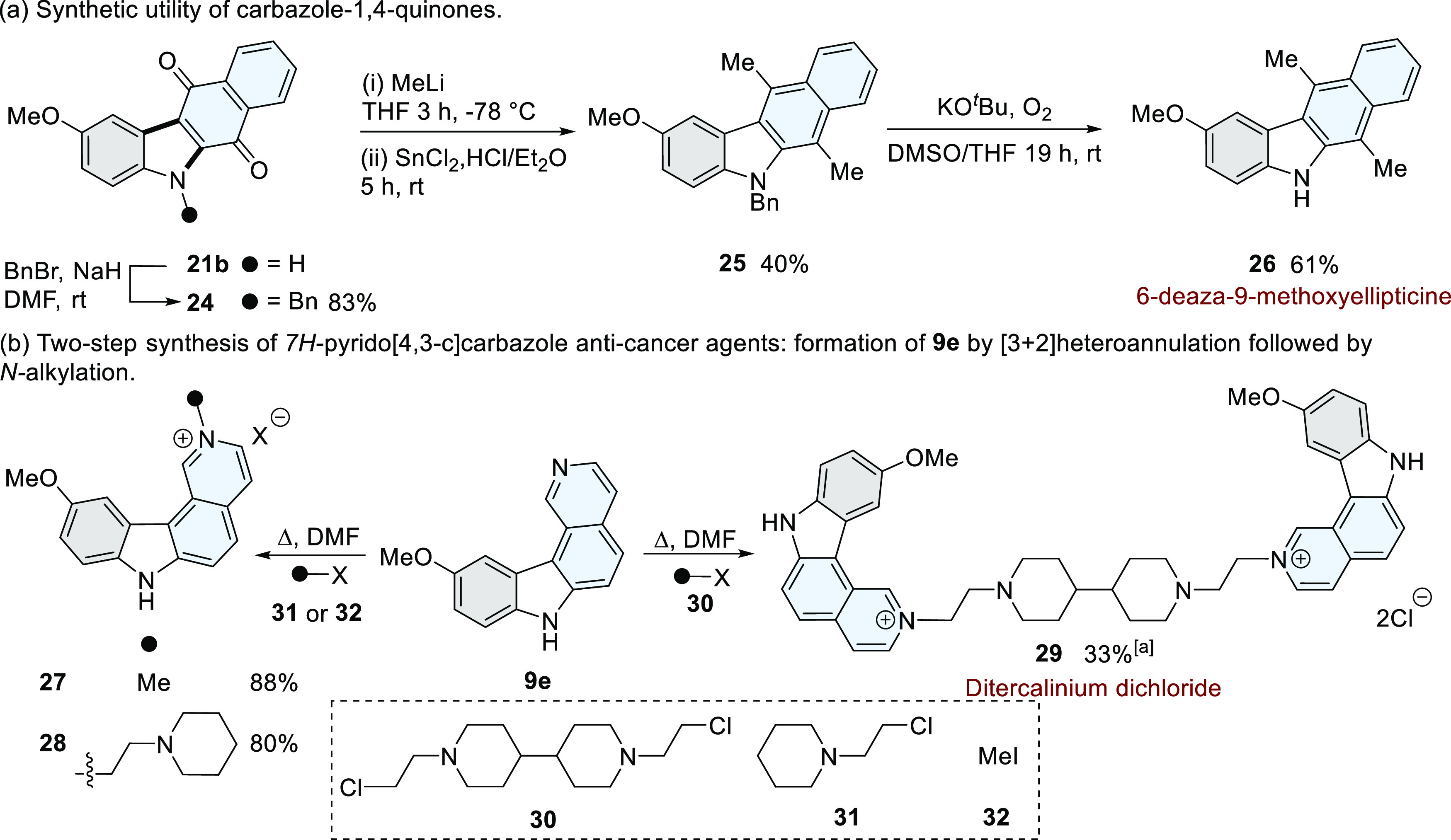
Yield calculated from the alkylation of 9e; X = iodide for 27 and chloride for 28.
Further exemplification of our strategy was demonstrated by the preparation of alkylated 7H-pyridocarbazoles (e.g., 9a–e), which have well-established anti-cancer activity.45,46 Previous preparative methods of this series of compounds have involved a six-step linear synthesis affording compound 9e in 28% overall yield.47 Our two-step convergent strategy accessed the 7H-pyrido[4,3-c]carbazole core 9e in a single step (83%), followed by alkylation (30–32) to produce mono-N-alkylated examples (27–28) and the potent anti-cancer agent ditercalinium dichloride (29, Scheme 4b).48
Conclusions
In summary, we have established a mechanistic framework for the preparation of fused tetracyclic carbazoles. The key to the modularity of this [3 + 2]heteroannulation approach is knowledge of the molecular hallmarks that underpin both the chemo- and regioselectivity of the process. The strategy is amenable for the diversification of tri- and tetracyclic carbazoles and is a scalable method for target-focused synthesis of tetracyclic carbazoles. We envisage that this convergent approach could find application in medicinal chemistry for structure–activity profiling and in broader synthetic applications that require step-efficient access to carbazole scaffolds.
Experimental Section
General Information
All reagents and solvents were obtained from commercial suppliers and were used without further purification unless otherwise stated. Purification was carried out according to standard laboratory methods. Starting materials were purchased from commercial suppliers and used without further purification unless otherwise stated. Dry solvents for reactions were purchased from Sigma-Aldrich and stored under nitrogen. Dichloromethane, chloroform, methanol, ethyl acetate, and petroleum ether (40–60 °C) for purification purposes were used as obtained from suppliers, without further purification. Reactions were carried out using conventional glassware for the preparation of starting materials. Microwave reactions were carried out in capped 2–5 mL microwave vials purchased from Biotage. Microwave reactions were carried out at elevated temperatures using a Biotage Initiator+ equipped with a Robot Eight microwave system. Thin-layer chromatography was carried out using Merck silica plates coated with a fluorescent indicator UV254, and they were analyzed under both 254 and 375 nm UV light or developed using potassium permanganate solution. Normal phase flash chromatography was carried out using 60 Å, 40–63 μm silica gel from Fluorochem. Semi-preparative reversed-phase HPLC purification was carried out on a Kinetex 5 μm, 150 × 21.2 mm XB C18 using a DIONEX 3000 series HPLC system equipped with a VWD3400 variable wavelength detector. Preparative purifications of small molecules were performed using a 30–90% gradient B (solvent A: 0.1% TFA in water, solvent B: 0.1% TFA in acetonitrile), with a flow rate of 12.0 mL/min. The absorbance of the UV-active material was detected at 254 nm. Analytical reversed-phase HPLC (RP-HPLC) was carried out on a Shimadzu Prominence instrument equipped with a PDA detector scanning from 190 to 600 nm using a Thermofisher Hypersil GOLD column 100 × 4.6 mm, with a particle size of 5 μm. The Fourier transform infrared (FTIR) spectra were obtained on a Shimadzu IR Affinity-1 instrument. Only major absorbance bands are reported. The 1H NMR, 13C NMR, and 19F NMR spectra were obtained on a Bruker AV 400 at 400, 101, and 376 MHz, respectively. Chemical shifts are reported in parts per million (ppm), and coupling constants are reported in hertz with DMSO-d6 referenced at 2.50 (1H) and 39.52 ppm (13C) and MeOD-d4 referenced at 3.31 (1H) and 49.0 ppm (13C). Assignment of 13C NMR signals is based on HSQC and HMBC experiments. The COSY and NOESY spectra were used to assign unequivocally atom connectivities. The high-resolution mass spectra were recorded on a Bruker microTOF II mass spectrometer at the SIRCAMs facility at the University of Edinburgh or on an LTQ Orbitrap xL at the EPSRC National Facility in Swansea.
General Experimental Procedure for Microwave-Assisted Buchwald–Hartwig Amination
Aryl bromide (0.50 mmol, 1.00 equiv), chloroaniline (0.60 mmol, 1.2 equiv), Pd(OAc)2 (5 mol %), DavePhos (10 mol %), and K3PO4 (1.50 mmol, 3 equiv) were added to a microwave vial (2–5 mL). 1,4-Dioxane (5 mL, 0.1 M) was added and the vial was capped, evacuated and purged with argon three times, and then heated at 120 °C for 30 min under microwave irradiation in a Biotage microwave. The reaction was allowed to cool to rt, diluted with ethyl acetate (50 mL), and the solid was filtered under vacuum. The organic phase was washed with water and brine, dried with Na2SO4, filtered, and the solvent was removed in vacuo. The crude compound was purified by silica column chromatography.
N-(2-Chloro-4-methoxyphenyl)isoquinolin-6-amine (7a)
The reaction was carried out according to the general procedure using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (7: 3), to obtain compound 7a in 87% yield (123 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.95 (s, 1H, H1), 8.29 (s, 1H, NH), 8.20 (d, 1H, J = 4.6 Hz, H3), 7.87 (d, 1H, J = 7.1 Hz, H8), 7.41–7.38 (m, 2H, H6′,4), 7.24 (dd, 1H, J = 7.1, 1.7 Hz, H7), 7.18 (d, 1H, J = 2.3 Hz, H3′), 7.00 (dd, 1H, J = 7.0, 2.3 Hz, H5′), 6.67 (d, 1H, J = 1.7 Hz, H5), 3.81 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 157.1 (C4′), 151.0 (C1), 147.9 (C6), 143.1 (C3), 137.3 (C4a), 130.6 (C2′), 130.3 (C1′), 128.8 (C8), 127.9 (C6′), 122.9 (C8a), 119.2 (C7), 118.6 (C4), 115.2 (C3′), 114.3 (C5′), 103.0 (C5), 55.7 (−OCH3).
IR v̅max (cm–1): 3230 (N–H stretch), 1618 (C=N stretch), 1476 (C=C stretch), 1284 (C–N stretch), 1212 (C–O stretch), 1039 (C–O stretch), 847 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H14O1N2Cl, 285.0789; found, 285.0787.
N-(2-Chloro-4-methylphenyl)isoquinolin-6-amine (7b)
The reaction was carried out according to the general procedure using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 4-methyl-2-chloroaniline (85 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (6:4), to obtain compound 7b in 91% yield (122 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.97 (s, 1H, H1), 8.35 (s, 1H, NH), 8.23 (d, 1H, J = 5.8 Hz, H3), 7.89 (d, 1H, J = 8.9 Hz, H8), 7.45 (d, 1H, J = 5.8 Hz, H4), 7.40 (d, 1H, J = 1.4 Hz, H3′), 7.38 (d, 1H, J = 8.1 Hz, H6′), 7.32 (dd, 1H, J = 8.9, 2.2 Hz, H7), 7.19 (dd, 1H, J = 8.1, 1.5 Hz, H5′), 6.90 (d, 1H, J = 2.1 Hz, H5), 2.32 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): 151.1 (C1), 146.8 (C6), 143.1 (C3), 137.1 (C4a), 135.3 (C2′), 135.1 (C1′), 130.5 (C3′), 128.8 (C8), 128.7 (C5′), 127.5 (C4′), 124.9 (C6′), 123.2 (C8a), 119.8 (C7), 118.8 (C4), 104.3 (C5), 20.1 (−CH3).
IR v̅max (cm–1): 3234 (N–H stretch), 3031 (C–H stretch), 1608 (C=N stretch), 1474 (C=C stretch), 821 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H14N2Cl, 269.0840; found, 269.0841.
N-(2-Chlorophenyl)isoquinolin-6-amine (7c)
The reaction was carried out according to the general procedure using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloroaniline (77 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (1:1), to obtain compound 7c in 85% yield (108 mg, green solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.00 (s, 1H, H1), 8.43 (s, 1H, NH), 8.26 (d, 1H, J = 5.8 Hz, H3), 7.93 (d, 1H, J = 8.9 Hz, H8), 7.55 (dd, 1H, J = 8.0, 1.4 Hz, H3′), 7.51 (dd, 1H, J = 8.0, 1.4 Hz, H7), 7.49–7.47 (m, 1H, H6′), 7.41–7.33 (m, 2H, H4,5′), 7.18–7.12 (m, 1H, H4′), 7.06 (d, 1H, J = 2.1 Hz, H5). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 151.1 (C1), 146.1 (C6), 143.1 (C3), 138.2 (C1′), 137.0 (C4a), 130.3 (C3′), 128.9 (C8), 128.1 (C5′), 126.7 (C2′), 124.8 (C4′), 123.7 (C7), 123.5 (C8a), 120.2 (C4), 118.9 (C6′), 105.5 (C5).
IR v̅max (cm–1): 3215 (N–H stretch), 3029 (C–H stretch), 1627 (C=N stretch), 1588 (C=C stretch), 1471 (C=C stretch), 737 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H12N2Cl, 255.0684; found, 255.0688.
N-(2-Chloro-4-(trifluoromethyl)phenyl)isoquinolin-6-amine (7d)
The reaction was carried out according to the general procedure using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-(trifluoromethyl)aniline (117 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (6:4), to obtain compound 7d in 78% yield (116 mg, yellow crystalline solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.12 (s, 1H, H1), 8.71 (s, 1H, NH), 8.35 (d, 1H, J = 5.7 Hz, H3), 8.03 (d, 1H, J = 8.9 Hz, H8), 7.88 (s, 1H, H5′), 7.64–7.61 (m, 3H, H4, 3′, 6′), 7.55 (dd, 1H, J = 8.8, 2.2 Hz, H7), 7.49 (d, 1H, J = 2.0 Hz, H5). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 151.4 (C1), 143.6 (C6), 143.3 (C3), 142.9 (C1′), 136.6 (C4a), 129.0 (C8), 127.2 (q, J = 3.7 Hz, C5′), 125.2 (q, J = 3.7 Hz, C3′), 124.4 (C8a), 123.7 (q, J = 272.9 Hz, −CF3), 123.6 (C2′), 122.4 (q, J = 33.2 Hz, C4′), 121.8 (C7), 119.4 (C4, 6′), 110.3 (C5). 19F NMR ((CD3)2SO, 471 MHz): δ −60.20.
IR v̅max (cm–1): 3220 (N–H stretch), 1612 (C=N stretch), 1487 (C=C stretch), 823 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H11N2ClF3, 323.0557; found, 323.0542.
N-(2-Chloro-4-nitrophenyl)isoquinolin-6-amine (7e)
The reaction was carried out following the general procedure using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 4-nitro-2-chloroaniline (104 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (1:1), to obtain compound 7e in 56% yield (83 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.20 (s, 1H, H1), 9.07 (s, 1H, NH), 8.42 (d, 1H, J = 5.8 Hz, H3), 8.34 (d, 1H, J = 2.7 Hz, H3′), 8.12 (d, 1H, J = 8.8 Hz, H8), 8.10 (dd, 1H, J = 9.2, 2.7 Hz, H5′), 7.73 (d, 1H, J = 2.0 Hz, H5), 7.71 (d, 1H, J = 5.8 Hz, H4), 7.64 (dd, 1H, J = 8.8, 2.0 Hz, H7), 7.48 (d, 1H, J = 9.2 Hz, H6′). 13C{1H} NMR (101 MHz, DMSO-d6): δ 151.7 (C1), 146.3 (C4′), 143.4 (C3), 142.1 (C6), 139.5 (C1′), 136.3 (C8a), 129.2 (C8), 125.9 (C3′), 125.2 (C4a), 124.3 (C5′), 123.2 (C7), 121.0 (C2′), 119.7 (C4), 115.7 (C6′), 114.4 (C5).
IR v̅max (cm–1): 3221 (N–H stretch), 1584 (C=N bend), 1508 (N–O stretch), 1474 (C=C stretch), 1333 (N–O stretch), 821 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H11O2N3Cl, 300.0534; found, 300.0538.
N-(2-Chloro-5-fluorophenyl)isoquinolin-6-amine (7f)
The reaction was carried out according to the general procedure using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-5-fluoroaniline (87 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (6:4), to obtain compound 7f in 74% yield (101 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.06 (s, 1H, H1), 8.51 (s, 1H, NH), 8.31 (d, 1H, J = 5.8 Hz, H3), 7.98 (d, 1H, J = 8.9 Hz, H8), 7.58 (d, 1H, J = 5.8 Hz, H4), 7.57 (dd, 1H, J = 9.0, 6.0 Hz, H3′), 7.47 (dd, 1H, J = 8.9, 2.2 Hz, H7), 7.33–7.27 (m, 2H, H5,6′), 6.95 (ddd, 1H, J = 8.8, 8.0, 3.0 Hz, H4′). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 161.2 (d, J = 244.7 Hz, C5′), 151.3 (C1), 144.7 (C4a), 143.2 (C3), 140.2 (d, J = 10.8 Hz, C1′) 136.9 (C8a), 131.5 (d, J = 10.0 Hz, C3′), 129.0 (C8), 124.0 (C6), 120.8 (C7), 120.5 (d, J = 3.3 Hz, C2′), 119.2 (C4), 110.5 (d, J = 20.9 Hz, C4′), 108.5 (d, J = 26.0 Hz, C6′), 107.7 (C5). 19F NMR ((CD3)2SO, 376 MHz): δ −113.1.
IR v̅max (cm–1): 3221 (N–H stretch), 1592 (C=N stretch), 1474 (C=C stretch), 852 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H11N2ClF, 273.0589; found, 273.0594.
N-(2-Chloro-4-methoxyphenyl)quinolin-6-amine (7g)
The reaction was carried out according to the general procedure using 6-bromoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (93 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel chromatography petroleum ether (40–60 °C):EtOAc (7:3), to obtain compound 7g in 59% yield (84 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.56 (dd, J = 4.2, 1.6 Hz, 1H, H2), 8.03–7.97 (m, 2H, NH, H4), 7.82 (d, J = 9.1 Hz, 1H, H8), 7.40 (dd, J = 9.0, 2.6 Hz, 1H, H7), 7.38 (d, J = 8.8 Hz, 1H, H6′), 7.31 (dd, J = 8.3, 4.2 Hz, 1H, H3), 7.16 (d, J = 2.8 Hz, 1H, H3′), 6.97 (dd, J = 8.8, 2.8 Hz, 1H, H5′), 6.85 (d, J = 2.5 Hz, 1H, H5), 3.80 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 156.4 (C4′), 146.3 (C2), 144.1 (C6), 143.0 (C8a), 133.7 (C4), 131.6 (C1), 129.8 (C4a), 129.3 (C8), 129.2 (C2′), 126.5 (C6′), 121.8 (C7), 121.5 (C3), 115.2 (C3′), 114.2 (C5′), 105.6 (C5), 55.7 (−OCH3).
IR v̅max (cm–1): 3215 (C–N stretch), 3016 (C–H stretch), 2846 (C–H stretch), 1631 (C=N stretch), 1498 (C=C stretch), 1214 (C–O stretch), 1050 (C–O stretch), 839 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H14O2N2Cl, 285.0789; found, 285.0793.
N-(2-Chloro-4-methoxyphenyl)quinoxalin-6-amine (7h)
The reaction was carried out according to the general procedure using 6-bromoquinoxaline (105 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (7:3) to obtain compound 7h in 87% yield (124 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.66 (d, 1H, J = 2.0 Hz, H3), 8.55 (d, 1H, J = 2.0 Hz, H2), 8.42 (s, 1H, NH), 7.87 (d, 1H, J = 9.1 Hz, H8), 7.46 (dd, 1H, J = 9.1, 2.6 Hz, H7), 7.42 (d, 1H, J = 8.8 Hz, H6′), 7.20 (d, 1H, J = 2.9 Hz, H3′), 7.01 (dd, 1H, J = 8.8, 2.9 Hz, H5′), 6.83 (d, 1H, J = 2.6 Hz, H5), 3.82 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 157.2 (C4′), 147.7 (C6), 145.3 (C3), 144.4 (C4a), 141.0 (C2), 137.4 (C8a), 130.5 (C1′), 130.2 (C8), 129.8 (C2′), 127.8 (C6′), 122.1 (C7), 115.3 (C3′), 114.3 (C5′), 105.3 (C5), 55.7 (−OCH3).
IR v̅max (cm–1): 3301 (N–H stretch), 3068 (C–H stretch), 2837 (C–H stretch), 1618 (C=N stretch), 1504 (C=C stretch), 1221 (C–O stretch), 1037 (C–O stretch), 852 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H13ON3Cl, 286.0742; found, 286.0740.
N-(2-Chloro-4-methoxyphenyl)quinoxalin-2-amine (7i)
The reaction was carried out according to the general procedure using 2-bromoquinoxaline (105 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (95 mg, 0.60, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (85:15), to obtain compound 7i in 27% yield (38 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.20 (s, 1H, NH), 8.64 (s, 1H, H3), 7.92 (d, 1H, J = 8.9 Hz, H6′), 7.84 (dd, 1H, J = 8.2, 1.2 Hz, H8), 7.62–7.53 (m, 2H, H5,6), 7.46–7.41 (m, 1H, H7), 7.15 (d, 1H, J = 2.9 Hz, H3′), 7.00 (dd, 1H, J = 8.9, 2.9 Hz, H5′), 3.81 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 156.6 (C4′), 150.5 (C2), 140.8 (C3), 139.8 (C8a), 137.1 (C4a), 129.9 (C5), 128.9 (C1′), 128.4 (C8), 128.0 (C2′), 126.9 (C6′), 126.2 (C6), 124.7 (C7), 114.6 (C3′), 113.6 (C5′), 55.7 (−OCH3).
IR v̅max (cm–1): 3340 (N–H stretch), 2934 (C–H stretch), 1590 (C=N stretch), 1536 (C=N stretch), 1502 (C=C stretch), 1214 (C–O stretch), 1040 (C–O stretch), 841 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H13ON3Cl, 286.0742; found, 286.0745.
N-(2-Chloro-4-methoxyphenyl)-N-(quinoxalin-2-yl)quinoxalin-2-amine (8)
The reaction was carried out according to the general procedure using 2-bromoquinoxaline (105 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (85:15) to obtained compound 8 in 25% yield (26 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.83 (s, 2H, 2 × H3), 8.06–8.00 (m, 2H, 2 × H5), 7.76–7.68 (m, 6H, 2 × H6/7/8), 7.58 (d, 1H, J = 8.8 Hz, H6′), 7.32 (d, 1H, J = 2.9 Hz, H3′), 7.12 (dd, 1H, J = 8.8, 2.9 Hz, H5′), 3.88 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 159.6 (C4′), 150.4 (2 × C2), 141.5 (2 × C3), 140.0 (2 × C8a), 138.8 (2 × C4a), 133.1 (C2′), 132.3 (C6′), 130.9 (C1′), 130.6 (2 × C6 or C7 or C8), 128.5 (2 × C5), 128.0 (2 × C6 or C7 or C8), 127.4 (2 × C6 or C7 or C8), 115.9 (C3′), 115.0 (C5′), 55.9 (−OCH3).
IR v̅max (cm–1): 1606 (C=N stretch), 1558 (C=N stretch), 1498 (C=C stretch), 1420 (C=C stretch), 1229 (C–O stretch), 1022 (C–O stretch), 761 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C23H17ON5Cl, 414.1116; found, 414.1115.
N-(2-Chloro-4-methoxyphenyl)-1,3-dihydroisobenzofuran-5-amine (7j)
The reaction was carried out according to the general procedure using 5-bromo-1,3-dihydroisobenzofuran (100 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (95 mg, 0.60, 1.2 equiv) as starting materials. The crude residue was purified by reversed-phase semi-preparative HPLC over a 5–95% gradient B, (solvent A: 0.1% TFA in water, solvent B: 0.1% TFA in acetonitrile) to obtain compound 7j in 18% yield (35 mg, white solid) as the TFA salt.
1H NMR ((CD3)2SO, 400 MHz): δ 7.49 (s, 1H, NH), 7.22 (d, 1H, J = 8.9 Hz, H6′), 7.08 (d, 1H, J = 2.8 Hz, H3′), 7.07 (d, 1H, J = 8.4 Hz, H7), 6.89 (dd, 1H, J = 8.9, 2.8 Hz, H5′), 6.71 (dd, 1H, J = 8.4, 2.0 Hz, H6), 6.66 (d, 1H, J = 2.0 Hz, H4), 4.88 (s, 4H, 2 × −CH2), 3.75 (s, 3H, −OCH3).
13C{1H} NMR ((CD3)2SO, 101 MHz): δ 155.4 (C4′), 144.9 (C4a), 140.0 (C3a), 132.8 (C1′), 128.9 (C7a), 127.7 (C2′), 124.7 (C6′), 121.4 (C7), 115.0 (C3′), 114.8 (C6), 114.0 (C5′), 107.3 (C4), 72.5 (C1 or C3), 72.3 (C1 or C3), 55.6 (−OCH3).
IR v̅max (cm–1): 3319 (N–H stretch), 2854 (C–H stretch), 1621 (C=C stretch), 1497 (C=C stretch), 1273 (C–O stretch), 1217 (C–O stretch), 1031 (C–O stretch), 821 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H15O2NCl, 276.0786; found, 276.0792.
2-((2-Chloro-4-methoxyphenyl)amino)naphthalene-1,4-dione (7k)
The reaction was carried out according to the general procedure using 2-bromo-1,4-napthoquinone (119 mg, 0.50 mmol, 1.0 equiv) and 4-methoxy-2-chloroaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc:NEt3 (100:8:1), to obtain compound 7k in 82% yield (129 mg, dark red solid). mp 155–158 °C.
1H NMR ((CD3)2SO, 400 MHz): δ 9.02 (s, 1H, NH), 8.06 (dd, 1H, J = 7.5, 1.0 Hz, H5), 7.94 (dd, 1H, J = 8.8, 1.2 Hz, H8), 7.86 (ddd, 1H, J = 7.5, 7.5, 1.4 Hz, H6), 7.79 (ddd, 1H, J = 7.5, 7.5, 1.4 Hz, H7), 7.36 (d, 1H, J = 8.8 Hz, H6′), 7.22 (d, 1H, J = 2.8 Hz, H3′), 7.04 (dd, 1H, J = 8.8, 2.8 Hz, H5′), 5.31 (s, 1H, H3), 3.82 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 182.1 (C1), 181.3 (C4), 158.7 (C4′), 147.7 (C2), 135.0 (C6), 132.6 (C7), 131.2 (C1′), 130.7 (C8a), 130.3 (C4a), 129.5 (C3′), 127.4 (C2′), 126.0 (C5), 125.4 (C8), 115.1 (C6′), 114.4 (C5′), 102.3 (C3), 55.8 (−OCH3).
IR v̅max (cm–1): 3331 (N–H stretch), 3021 (C–H stretch), 1677 (C=O stretch), 1577 (C=C stretch), 1489 (C=C stretch), 1216 (C–O stretch), 1031 (C–O stretch), 778 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C17H13O3NCl, 314.0578; found, 314.0577.
2-((2-Chloro-4-methylphenyl)amino)naphthalene-1,4-dione (7l)
The reaction was carried out according to the general procedure using 2-bromo-1,4-napthoquinone (119 mg, 0.50 mmol, 1.0 equiv) and 4-methyl-2-chloroaniline (85 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc:NEt3 (90:10:1), to obtain compound 7l in 72% yield (107 mg, red solid). mp 150–154 °C.
1H NMR ((CD3)2SO, 400 MHz): δ 9.01 (s, 1H, NH), 8.09–8.05 (m, 1H, H5), 7.96–7.92 (m, 1H, H8), 7.89–7.84 (m, 1H, H6), 7.82–7.77 (m, 1H, H7), 7.47 (d, 1H, J = 1.1 Hz, H3′), 7.35 (d, 1H, J = 8.1 Hz, H6′), 7.27 (dd, 1H, J = 8.1, 1.3 Hz, H5′), 5.41 (s, 1H, H3), 2.35 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 182.3 (C4), 181.2 (C1), 147.0 (C2), 138.5 (C1′), 135.0 (C6), 132.7 (C4a), 132.6 (C7), 132.1 (C2′), 130.5 (C8a), 130.3 (C3′), 129.6 (C4′), 129.0 (C5′), 127.9 (C6′), 126.1 (C5), 125.4 (C8), 102.7 (C3), 20.3 (−CH3).
IR v̅max (cm–1): 3342 (N–H stretch), 2927 (C–H stretch), 1675 (C=O stretch), 1599 (C=O stretch), 1526 (C=C stretch), 774 (C–Cl).
HRMS (ESI) m/z: [M + H]+ calcd for C17H13O2NCl, 298.0629; found, 298.0633.
2-((2-Chlorophenyl)amino)naphthalene-1,4-dione (7m)
The reaction was carried out according to the general procedure using 2-bromo-1,4-napthoquinone (119 mg, 0.50 mmol, 1.0 equiv) and 2-chloroaniline (77 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (9:1) to obtain compound 7m in 64% yield (90 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.08 (s, 1H, NH), 8.08 (dd, 1H, J = 7.7, 0.9 Hz, H5), 7.95 (dd, 1H, J = 7.5, 1.1 Hz, H8), 7.87 (ddd, 1H, J = 7.4, 7.4, 1.4 Hz, H6), 7.80 (ddd, 1H, J = 8.0, 6.5, 2.7 Hz, H7), 7.64 (dd, 1H, J = 7.5, 1.1 Hz, H3′), 7.51–7.44 (m, 2H, H4′,6′), 7.38 (ddd, 1H, J = 8.0, 6.5, 2.7 Hz, H5′), 5.47 (s, 1H, H3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 182.3 (C4), 181.2 (C1), 146.7 (C2), 135.0 (C6), 134.2 (C1′), 132.8 (C7), 132.5 (C4a), 130.3 (C3′), 129.7 (C8a), 128.4 (C2′), 128.3 (C5′), 128.0 (C4′, 6′), 126.1 (C5), 125.4 (C8), 102.9 (C3).
IR v̅max (cm–1): 3310 (N–H stretch), 3068 (C–H stretch), 2854 (C–H stretch), 1675 (C=O stretch), 1618 (C=O stretch), 1592 (C=C stretch), 1446 (C=C stretch), 741 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H11O2NCl, 284.0473; found, 284.0480.
2-((2-Chloro-4-(trifluoromethyl)phenyl)amino)naphthalene-1,4-dione (7n)
The reaction was carried out according to the general procedure using 2-bromo-1,4-napthoquinone (119 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-(trifluoromethyl)aniline (117 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (9:1) to obtain compound 7n in 69% yield (122 mg, orange solid). mp 180–183 °C.
1H NMR ((CD3)2SO, 400 MHz): δ 9.10 (s, 1H, NH), 8.08 (dd, 1H, J = 7.5, 1.1 Hz, H8), 8.06 (d, 1H, J = 1.6 Hz, H3′), 7.96 (dd, 1H, J = 7.6, 1.2 Hz, H5), 7.88 (ddd, 1H, J = 7.5, 7.5, 1.5 Hz, H6), 7.85–7.78 (m, 2H, H5′, 7), 7.75 (d, 1H, J = 8.5 Hz, H6′), 5.77 (s, 1H, H3).
13C{1H} NMR ((CD3)2SO, 101 MHz): δ 182.7 (C4), 181.0 (C1), 146.0 (C2), 138.9 (C1′), 135.1 (C6), 133.0 (C7), 132.3 (C4a), 130.2 (C8a), 129.5 (C2′), 127.5 (q, J = 32.7 Hz, C4′), 127. 4 (q, J = 3.8 Hz, C3′), 127.2 (C6′), 126.2 (C8), 125.5 (C5), 125.4 (q, J = 3.8 Hz, C5′), 123.3 (q, J = 272.9 Hz, −CF3), 104.6 (C3). 19F NMR ((CD3)2SO, 376 MHz): δ −60.85.
IR v̅max (cm–1): 3338 (N–H stretch), 3055 (C–H stretch), 1677 (C=O stretch), 1642 (C=O stretch), 1580 (C=C stretch), 782 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C17H10O2NClF3, 352.0347; found, 352.0347.
2-((2-Chloro-5-methoxyphenyl)amino)naphthalene-1,4-dione (7o)
The reaction was carried out according to the general procedure using 2-bromo-1,4-napthoquinone (119 mg, 0.50 mmol, 1.0 equiv) and 5-methoxy-2-chloroaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (85:15), to obtain compound 7o in 59% yield (92 mg, red solid). mp 152–156 °C.
1H NMR ((CD3)2SO, 400 MHz): δ 9.05 (s, 1H, NH), 8.07 (dd, 1H, J = 7.6, 1.2 Hz, H8), 7.95 (dd, 1H, J = 7.6, 1.2 Hz, H5), 7.90–7.84 (m, 1H, H6), 7.83–7.77 (m, 1H, H7), 7.52 (d, 1H, J = 8.9 Hz, H3′), 7.03 (d, 1H, J = 3.0 Hz, H6′), 6.97 (dd, 1H, J = 8.9, 3.0 Hz, H4′), 5.48 (s, 1H, H3), 3.79 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 182.3 (C4=O), 181.2 (C1=O), 158.8 (C5′), 146.6 (C2), 135.8 (C1′), 135.0 (C6), 132.8 (C7), 132.6 (C4a), 130.7 (C3′), 130.3 (C8a), 126.1 (C8), 125.4 (C5), 120.9 (C2′), 114.0 (C4′), 113.3 (C6′), 103.3 (C3), 55.7 (−OCH3).
IR v̅max (cm–1): 3299 (N–H stretch), 2843 (C–H stretch), 1675 (C=O stretch), 1647 (C=O stretch), 1579 (C=C stretch), 1541 (C=C stretch), 1219 (C–O stretch), 1067 (C–O stretch), 776 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ cald for C17H13O3NCl, 314.0578; found, 314.0586.
2-((2-Chloro-5-fluorophenyl)amino)naphthalene-1,4-dione (7p)
The reaction was carried out according to the general procedure using 2-bromo-1,4-napthoquinone (119 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-5-fluoroaniline (87 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (9:1) to obtain compound 7p in 80% yield (120 mg, orange solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.08 (s, 1H, NH), 8.10–8.06 (m, 1H, H8), 7.98–7.94 (m, 1H, H5), 7.91–7.85 (m, 1H, H6), 7.85–7.79 (m, 1H, H7), 7.68 (dd, 1H, J = 9.0, 5.7 Hz, H3′), 7.43 (dd, 1H, J = 9.5, 3.0 Hz, H6′), 7.26 (ddd, 1H, J = 8.8, 8.1, 3.0 Hz, H4′), 5.60 (s, 1H, H3).
13C{1H} NMR ((CD3)2SO, 101 MHz): δ 182.6 (C4), 181.0 (C1), 160.9 (d, J = 246.8 Hz, C5′), 146.0 (C2), 136.4 (d, J = 10.8 Hz, C1′), 135.1 (C6), 132.9 (C7), 132.4 (C4a), 131.6 (d, J = 9.6 Hz, C3′), 130.2 (C8a), 126.2 (C8), 125.4 (C5), 125.1 (C2′), 115.2 (d, J = 22.8 Hz, C4′), 114.8 (d, J = 24.8 Hz, C6′), 103.9 (C3). 19F NMR ((CD3)2SO, 376 MHz): δ −112.74.
IR v̅max (cm–1): 3310 (N–H stretch), 3074 (C–H stretch), 2858 (C–H stretch), 1675 (C=O stretch), 1647 (C=O stretch), 1599 (C=C stretch), 1541 (C=C stretch), 722 (C–Cl stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H10O2NClF, 302.0379; found, 302.0380.
General Experimental Procedure A for Microwave-Assisted One-Pot Buchwald–Hartwig Amination/Direct Arylation
Aryl bromide (0.50 mmol, 1 equiv), chloroaniline (0.60 mmol, 1.2 equiv), Pd(OAc)2 (5 mol %), DavePhos (10 mol %), and K3PO4 (1.50 mmol, 3 equiv) were added to a microwave vial (2–5 mL). 1,4-Dioxane (5.0 mL, 0.1 M) was added and the vial was capped, evacuated and purged with argon three times, and heated at 120 °C for 30 min followed by 160 °C for 8 h under microwave irradiation in a Biotage microwave. The reaction was allowed to cool to rt, diluted with DCM (50 mL), and the solid was filtered under vacuum. The solvent was removed in vacuo, and the crude sample was dry-loaded onto silica gel without work up. The crude compound was purified by silica column chromatography.
General Experimental Procedure B for Microwave-Assisted One-Pot Buchwald–Hartwig Amination/Direct Arylation
Aryl bromide (0.50 mmol, 1 equiv), chloroaniline (0.60 mmol, 1.2 equiv), Pd(OAc)2 (5 mol %), P(Cy3), HBF4 (10 mol %), and K3PO4 (1.50 mmol, 3 equiv) were added to a microwave vial (2–5 mL). 1,4-Dioxane (5.0 mL, 0.1 M) was added, and the vial was capped, evacuated and purged with argon three times, and heated at 120 °C for 30 min followed by 160 °C for 8 h under microwave irradiation in a Biotage microwave. The reaction was allowed to cool to rt and diluted with DCM (50 mL). The solvent was removed in vacuo, and the crude sample dry-loaded onto silica gel without work up. The crude compound was purified by silica column chromatography.
10-Methoxy-7H-pyrido[3,4-c]carbazole (9a)
The reaction was carried out according to the general procedure B using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (1:1), to obtain compound 9a in 82% yield (102 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.94 (s, 1H, −NH), 9.30 (s, 1H, H4), 8.63 (d, 1H, J = 5.8 Hz, H2), 8.58 (d, J = 5.8 Hz, 1H, H1), 8.05–7.99 (m, 2H, H5,11), 7.85 (d, 1H, J = 8.8 Hz, H6), 7.60 (d, 1H, J = 8.8 Hz, H8), 7.13 (dd, 1H, J = 8.8, 2.4 Hz, H9), 3.96 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 154.1 (C10), 152.0 (C4), 144.1 (C2), 140.0 (C6a), 133.6 (C7a), 132.1 (C11c), 125.9 (C5), 123.6 (C11b), 123.0 (C11a), 116.4 (C1), 115.0 (C6), 114.3 (C9), 112.6 (C8), 112.3 (C4a), 103.9 (C11), 55.8 (−OCH3).
IR v̅max (cm–1): 2830 (C–H stretch), 1616 (C=N stretch), 1458 (C=C stretch), 1229 (C–O stretch), 1031 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13ON2, 249.1022; found, 249.1021.
10-Methyl-7H-pyrido[3,4-c]carbazole (9b)
The reaction was carried out according to the general procedure B using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methylaniline (85 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (1:1) to obtain compound 9b in 54% yield (63 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.97 (s, 1H, −NH), 9.31 (s, 1H, H4), 8.63 (d, 1H, J = 5.7 Hz, H2), 8.58 (d, 1H, J = 5.7 Hz, H1), 8.41 (s, 1H, H11), 8.04 (d, 1H, J = 8.8 Hz, H5), 7.86 (d, 1H, J = 8.8 Hz, H6), 7.58 (d, 1H, J = 8.3 Hz, H8), 7.30 (dd, 1H, J = 8.3, 1.2 Hz, H9), 2.58 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 152.0 (C4), 143.9 (C2), 139.7 (C6a), 137.0 (C7a), 132.0 (C11c), 129.0 (C10), 126.1 (C9), 125.8 (C5), 123.7 (C4a), 122.9 (C11a), 121.3 (C11), 116.4 (C1), 114.9 (C6), 112.2 (C11b), 111.6 (C8), 21.3 (−CH3).
IR v̅max (cm–1): 2917 (C–H stretch), 1616 (C=N stretch), 1474 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13N2, 233.1073; found, 233.1068.
10-(Trifluoromethyl)-7H-pyrido[3,4-c]carbazole (9c)
The reaction was carried out according to the general procedure B using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-(trifluoromethyl)aniline (117 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (7:2), to obtain compound 9c in 25% yield (35 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.53 (s, 1H, −NH), 9.38 (s, 1H, H4), 8.90 (s, 1H, H11), 8.74–8.63 (m, 2H, H1, 2), 8.17 (d, 1H, J = 8.8 Hz, H5), 7.95 (d, 1H, J = 8.8 Hz, H6), 7.88 (d, 1H, J = 8.6 Hz, H8), 7.78 (d, 1H, J = 8.6 Hz, H9). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 152.3 (C4), 144.6 (C2), 140.8 (C7a), 140.5 (C6a), 131.8 (C11c), 127.5 (C5), 125.4 (q, J = 272.6 Hz, −CF3), 124.0 (C4a), 122.2 (C11a), 121.2 (q, J = 4.0 Hz, C9), 120.8 (q, J = 30.8 Hz, C10), 118.8 (q, J = 4.0 Hz, C11), 116.6 (C1), 115.0 (C6), 112.6 (C8), 112.4 (C11b). 19F NMR (376 MHz, DMSO-d6): δ −58.1.
IR v̅max (cm–1): 1638 (C=N stretch), 1618 (C=N stretch), 1463 (C=C stretch), 1420 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H10N2F3, 287.0791; found, 287.0791.
10-Methoxy-7H-pyrido[4,3-c]carbazole (9e)
The reaction was carried out according to the general procedure B using 7-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (1:1), to obtain compound 9e in 83% yield (103 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.89 (s, 1H, −NH), 10.14 (s, 1H, H1), 8.53 (d, 1H, J = 5.5 Hz, H3), 8.07 (d, 1H, J = 2.4 Hz, H11), 7.99 (d, 1H, J = 8.8 Hz, H6), 7.96 (d, 1H, J = 5.5 Hz, H4), 7.91 (d, J = 8.8 Hz, 1H, H5), 7.61 (d, J = 8.8 Hz, 1H, H8), 7.14 (dd, J = 8.8, 2.4 Hz, 1H, H9), 3.98 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 154.0 (C10), 146.7 (C1), 140.5 (C3), 138.3 (C6a), 133.8 (C7a), 131.0 (C11b), 124.7 (C5, 11c), 122.1 (C11a), 121.6 (C4), 118.3 (C6), 115.0 (C9), 113.0 (C4a), 112.7 (C8), 104.2 (C11), 55.8 (−OCH3).
IR v̅max (cm–1): 2932 (C–H stretch), 1588 (C=N stretch), 1210 (C–O stretch), 1063 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13ON2, 249.1022; found, 249.1028.
10-Methoxy-7H-pyrazino[2,3-c]carbazole (9f)
The reaction was carried out according to the general procedure B using 6-bromoquinoxaline (105 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (7:2), to obtain compound 9f in 75% yield (93 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.96 (s, 1H, −NH), 9.04 (d, 1H, J = 2.0 Hz, H2), 8.86 (d, 1H, J = 2.0 Hz, H3), 8.29 (d, 1H, J = 2.6 Hz, H11), 8.05 (d, 1H, J = 9.1 Hz, H6), 7.98 (d, 1H, J = 9.1 Hz, H5), 7.61 (d, 1H, J = 8.8 Hz, H8), 7.13 (dd, 1H, J = 8.8, 2.6 Hz, H9), 3.91 (s, 3H, −OCH3).
13C{1H} NMR ((CD3)2SO, 101 MHz): δ 154.1 (C10), 144.2 (C2), 141.2 (C3), 140.3 (C11c), 139.5 (C6a), 138.7 (C4a), 133.8 (C7a), 126.5 (C5), 123.3 (C11a), 118.0 (C6), 115.0 (C9), 114.5 (C11b), 112.7 (C8), 104.4 (C11), 55.5 (−OCH3).
IR v̅max (cm–1): 3338 (N–H stretch), 2837 (C–H stretch), 1601 (C=N stretch), 1523 (C=C stretch), 1495 (C=C stretch), 1214 (C–O stretch), 1029 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H12ON3, 250.0975; found, 250.0970.
10-Methoxy-7H-pyrido[2,3-c]carbazole (9g)
The reaction was carried out according to the general procedure B using 6-bromoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (1:1), to obtain compound 9g in 65% yield (80 mg, off-white solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.77 (s, 1H, −NH), 9.17 (d, 1H, J = 8.3 Hz, H1), 8.82 (dd, 1H, J = 4.2, 1.4 Hz, H3), 8.02 (d, 1H, J = 2.3 Hz, H11), 7.99–7.92 (m, 2H, H5,6), 7.67 (dd, 1H, J = 8.3, 4.2 Hz, H2), 7.58 (d, 1H, J = 8.8 Hz, H8), 7.11 (dd, 1H, J = 8.8, 2.3 Hz, H9), 3.95 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 153.8 (C10), 146.1 (C3), 144.2 (C4a), 137.4 (C6a), 133.9 (C7a), 130.8 (C1), 127.4 (C5), 124.4 (C11c), 123.1 (C11a), 121.5 (C2), 116.9 (C6), 114.2 (C9), 113.3 (C11b), 112.6 (C8), 103.9 (C11), 55.8 (−OCH3).
IR v̅max (cm–1): 3146 (N–H stretch), 2997 (C–H stretch), 1571 (C=N stretch), 1534 (C=C stretch), 1493 (C=C stretch), 1227 (C–O stretch), 1035 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13ON2, 249.1022; found, 249.1026.
8-Methoxy-11H-pyrido[4,3-a]carbazole (9h)
The reaction was carried out according to the general procedure B using 5-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, hexane:EtOAc (3:7), to obtain compound 9h in 90% yield (111 mg, gray solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.30 (s, 1H, −NH), 9.36 (s, 1H, H4), 8.62 (d, 1H, J = 5.7 Hz, H2), 8.38 (d, 1H, J = 8.5 Hz, H6), 8.32 (d, J = 5.7 Hz, 1H, H1), 7.81 (d, 1H, J = 2.3 Hz, H7), 7.75 (d, 1H, J = 8.5 Hz, H5), 7.60 (d, 1H, J = 8.8 Hz, H10), 7.13 (dd, 1H, J = 8.8, 2.3, H9), 3.89 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 153.7 (C8), 152.0 (C4), 142.8 (C2), 134.1 (C10a), 133.7 (C4a), 126.6 (C11a), 124.1 (C11b), 123.0 (C6b), 121.2 (C6), 120.3 (C6a), 117.4 (C5), 115.7 (C9), 114.9 (C1), 112.5 (C10), 102.3 (C7), 55.6 (−OCH3).
IR v̅max (cm–1): 3149 (N–H stretch), 1619 (C=N stretch), 1480 (C=C stretch), 1439 (C=C stretch), 1215 (C–O stretch), 1027 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13ON2, 249.1022; found, 249.1020.
8-Methoxy-11H-pyrido[3,4-a]carbazole (9i)
The reaction was carried out according to the general procedure B using 8-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, hexane:EtOAc (3:7), to obtain 9i in 94% yield (104 mg, gray solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.39 (s, 1H, −NH), 9.86 (s, 1H, H1), 8.55 (d, 1H, J = 5.6 Hz, H3), 8.48 (d, 1H, J = 8.5 Hz, H6), 7.91 (d, 1H, J = 5.6 Hz, H4), 7.79 (d, 1H, J = 2.5 Hz, H7), 7.61 (d, 1H, J = 8.5 Hz, H5), 7.59 (d, 1H, J = 8.8 Hz, H10), 7.10 (dd, 1H, J = 8.8, 2.5, H9), 3.89 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 153.8 (C8), 146.4 (C1), 142.6 (C3), 134.7 (C4a), 134.4 (C11a), 133.7 (C10a), 124.6 (C6), 123.2 (C6b), 121.1 (C4), 118.8 (C6a), 116.8 (C11b), 116.6 (C5), 114.9 (C9), 112.3 (C10), 102.2 (C7), 55.6 (−OCH3).
IR v̅max (cm–1): 3074 (N–H stretch), 2962 (C–H stretch), 1627 (C=N stretch), 1474 (C=C stretch), 1437 (C=C stretch), 1217 (C–O stretch), 1027 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13ON2, 249.1022; found, 249.1032.
9-Methoxy-3,6-dihydro-1H-furo[3,4-c]carbazole (9j)
The reaction was carried out according to the general procedure A using 5-bromo-1,3-dihydroisobenzofuran (99 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by reversed-phase semi-preparative HPLC over a 5–95% gradient B, (solvent A: 0.1% TFA in water, solvent B: 0.1% TFA in acetonitrile) to obtain compound 9j in 10% yield (35 mg, white solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.15 (s, 1H, −NH), 7.41 (d, 1H, J = 8.8 Hz, H7), 7.38 (d, 1H, J = 8.3 Hz, H5), 7.30 (d, 1H, J = 2.6 Hz, H10), 7.29 (d, 1H, J = 8.3 Hz, H4), 7.06 (dd, 1H, J = 8.8, 2.6 Hz, H8), 5.52 (t, 2H, J = 2.2 Hz, 2 × H1), 5.14 (t, 2H, J = 2.2 Hz, 2 × H3), 3.84 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 153.1 (C9), 140.0 (C5a), 134.8 (C6a), 131.5 (C10c), 128.2 (C3a), 121.6 (C10a), 118.0 (C4), 116.0 (C10b), 114.6 (C8), 111.6 (C7), 110.0 (C5), 104.2 (C10), 72.8 (C3), 72.4 (C1), 55.7 (−OCH3).
IR v̅max (cm–1): 3246 (N–H stretch), 2867 (C–H stretch), 1478 (C=C stretch), 1439 (C=C stretch), 1214 (C–O stretch), 1014 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H14O2N, 240.1019; found, 240.1030.
10-Methoxybenzo[4,5]imidazo[1,2-a]quinolone (9k)
The reaction was carried out according to the general procedure B using 2-bromoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, hexane:EtOAc (3:7), to obtain compound 9k in 25% yield (31 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.78 (d, J = 8.5 Hz, 1H, H1), 8.08 (d, 1H, J = 2.3 Hz, H11), 8.03 (dd, 1H, J = 7.8, 1.5 Hz, H4), 7.88–7.82 (m, 3H, H2,5,8), 7.59 (d, 1H, J = 9.5 Hz, H6), 7.59–7.54 (m, 1H, H3), 7.21 (dd, 1H, J = 8.9, 2.3 Hz, H9), 4.00 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 156.0 (C10), 147.1 (C6a), 138.8 (C7a), 134.8 (C12a), 130.7 (C11a), 130.4 (C5), 130.0 (C2), 129.5 (C4), 124.4 (C3), 123.0 (C4a), 120.3 (C8), 117.5 (C6), 115.6 (C1), 113.8 (C9), 98.3 (C11), 56.1 (−OCH3).
IR v̅max (cm–1): 1636 (C=N stretch), 1608 (C=N stretch), 1487 (C=C stretch), 1448 (C=C stretch), 1219 (C–O stretch), 1026 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13ON2, 249.1022; found, 249.1026.
N-(2-Chloro-4-methoxyphenyl)-N-(quinolin-2-yl)quinolin-2-amine (22)
The reaction was carried out according to the general procedure B using 2-bromoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, hexane:EtOAc (3:7) to obtain compound 22 in 26% yield (54 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.22 (d, 2H, J = 9.0 Hz, 2 × H4), 7.88 (d, 2H, J = 8.0 Hz, 2 × H5), 7.66–7.59 (m, 4H, 2 × H7,8), 7.45 (ddd, 2H, J = 8.1, 5.5, 2.8 Hz, 2 × H6), 7.38 (d, 1H, J = 8.8 Hz, H6′), 7.28 (d, 2H, J = 8.9 Hz, 2 × H3), 7.24 (d, 1H, J = 2.9 Hz, H3′), 7.07 (dd, 1H, J = 8.8, 2.9 Hz, H5′), 3.87 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 158.9 (C4′), 155.3 (2 × C2), 146.4 (2 × C8a), 137.3 (2 × C4), 133.5 (C1′, 2′), 132.6 (C6′), 129.7 (2 × C7), 127.6 (2 × C5), 127.2 (2 × C8), 125.2 (2 × C4a), 124.7 (2 × C6), 116.6 (2 × C3), 115.5 (C3′), 114.6 (C5′), 55.8 (−OCH3).
IR v̅max (cm–1): 1597 (C=N stretch), 1575 (C=N stretch), 1502 (C=C stretch), 1428 (C=C stretch), 1230 (C–O stretch), 1040 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C25H19ON3Cl, 412.1211; found, 412.1230.
10-Methoxybenzo[4,5]imidazo[1,2-a]quinoxaline (9l)
The reaction was carried out according to the general procedure B using 2-bromoquinoxaline (105 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, hexane:EtOAc (3:7), to obtain compound 9l in 14% yield (17 mg, white solid).
1H NMR ((CD3)2SO, 400 MHz): δ 9.23 (s, 1H, H6), 8.81 (d, 1H, J = 8.3 Hz, H1), 8.14 (dd, 1H, J = 9.0, 1.4 Hz, H4), 8.10 (d, 1H, J = 2.3 Hz, H11), 8.00 (d, 1H, J = 9.0 Hz, H8), 7.87 (ddd, 1H, J = 8.3, 7.6, 1.4 Hz, H2), 7.69 (ddd, 1H, J = 8.0, 7.6, 1.0 Hz, H3), 7.33 (dd, 1H, J = 9.0, 2.3 Hz, H9), 4.04 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 157.6 (C10), 146.1 (C6), 140.5 (C6a), 138.5 (C7a), 135.2 (C4a), 130.2 (C4), 130.0 (C2, 11a), 129.1 (C12a), 125.7 (C3), 122.1 (C8), 116.3 (C9), 115.9 (C1), 97.4 (C11), 56.2 (−OCH3).
IR v̅max (cm–1): 2843 (C–H stretch), 1580 (C=N stretch), 1590 (C=N stretch), 1463 (C=C stretch), 1225 (C–O stretch), 1026 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H12ON3, 250.0975; found, 250.0982.
8-Methyl-7H-pyrido[3,4-c]carbazole (9m)
The reaction was carried out according to the general procedure B using 6-bromoisoquinoline (104 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-6-methylaniline (85 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, DCM:MeOH (0 to 2% MeOH), to obtain compound 9m in 62% yield (72 mg, white solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.01 (s, 1H, −NH), 9.32 (s, 1H, H4), 8.63 (d, 1H, J = 5.8 Hz, H2), 8.56 (d, 1H, J = 5.7 Hz, H1), 8.42 (dd, 1H, J = 7.2, 2.4 H11), 8.06 (d, 1H, J = 8.9 Hz, H5), 7.91 (d, 1H, J = 8.9 Hz, H6), 7.31–7.24 (m, 2H, H9,H10), 2.65 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 152.2 (C4), 144.1 (C2), 139.6 (C6a), 138.1 (C7a), 132.0 (C11c), 125.9 (C9), 125.2 (C5), 123.9 (C4a), 122.4 (C11a), 121.1 (C11), 120.3 (C8), 119.1 (C10), 116.3 (C1), 114.9 (C6), 112.8 (C11b), 17.5 (−CH3).
HRMS (ESI) m/z: [M + H]+ calcd for C16H13N2, 233.1073; found, 233.1070.
Glycozoline (9n)1
Preparation 1
The reaction was carried out according to the general procedure A using 4-bromotoluene (85 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (100:6), to obtain compound 9m in 36% yield (38 mg, brown crystalline solid).
Preparation 2
The reaction was carried out according to the general procedure A using 4-bromotoluene (85 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials with the addition of PivOH (15.3 mg, 0.15 mmol, 0.3 equiv). The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (100:6) to obtain compound 9n in 64% yield (66 mg, brown crystalline solid).
1H NMR ((CD3)2SO, 400 MHz): δ 10.85 (s, 1H, −NH), 7.87 (d, 1H, J = 1.0 Hz, H5), 7.61 (d, 1H, J = 2.5 Hz, H4), 7.34 (d, 1H, J = 8.8 Hz, H1), 7.32 (d, 1H, J = 8.3 Hz, H8), 7.16 (dd, 1H, J = 8.3, 1.0 Hz, H7), 6.98 (dd, 1H, J = 8.8, 2.5 Hz, H2), 3.83 (s, 3H, −OCH3), 2.45 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 152.8 (C3), 138.6 (C8a), 134.8 (C9a), 126.7 (C6, 7), 126.5 (C4a), 122.5 (C4b), 119.9 (C5), 114.5 (C2), 111.5 (C1), 110.6 (C8), 102.8 (C4), 55.5 (−OCH3), 21.0 (−CH3).
IR v̅max (cm–1): 3401 (N–H stretch), 3003 (C–H stretch), 2835 (C–H stretch), 1474 (C=C stretch), 1458 (C=C stretch), 1212 (C–O stretch), 1037 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C14H14ON, 212.1070; found, 212.1079.
Harmane (9o)48
The reaction was carried out according to the general procedure A using 3-bromo-2-methylpyridine (86 mg, 0.50 mmol, 1.0 equiv) and 2-chloroaniline (77 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, EtOAc (100%), to obtain compound 9o in 45% yield (40 mg, purple solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.53 (s, 1H, NH), 8.20 (d, 1H, J = 5.3 Hz, H3), 8.18 (d, 1H, J = 8.0 Hz, H5), 7.92 (d, 1H, J = 5.3 Hz, H4), 7.61–7.57 (m, 1H, H8), 7.52 (ddd, 1H, J = 8.3, 7.0, 1.2 Hz, H7), 7.22 (ddd, 1H, J = 8.0, 7.0, 1.2 Hz, H6), 2.76 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 142.1 (C1), 140.3 (C8a), 137.5 (C3), 134.4 (C9a), 127.7 (C7), 126.8 (C4a), 121.7 (C5), 121.0 (C4b), 119.1 (C6), 112.6 (C4), 111.9 (C8), 20.4 (−CH3).
IR v̅max (cm–1): 3062 (N–H stretch), 2954 (C–H stretch), 1627 (C=N stretch), 1571 (C=C stretch), 1506 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C12H11N2, 183.0917; found, 183.0927.
Murrayafoline A (9p)
The reaction was carried out according to the general procedure A using 2-bromo-5-methylanisole (101 mg, 0.50 mmol, 1.0 equiv) and 2-chloroaniline (77 mg, 0.60 mmol, 1.2 equiv) as starting materials with the addition of PivOH (15.3 mg, 0.15 mmol, 0.3 equiv). The crude residue was purified by silica gel column chromatography, hexane:EtOAc (7:3), to obtain compound 9p in 43% yield (45 mg, brown crystalline solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.12 (s, 1H, −NH), 8.00 (d, 1H, J = 8.0 Hz, H5), 7.49–7.46 (m, 1H, H4), 7.45–7.41 (m, 1H, H8), 7.32 (ddd, 1H, J = 8.3, 7.1, 1.3 Hz, H7), 7.09 (ddd, 1H, J = 8.0, 7.1, 1.1 Hz, H6), 6.82 (d, 1H, J = 1.1 Hz, H2), 3.96 (s, 3H, −OCH3), 2.46 (s, 3H, −CH3).
13C{1H} NMR ((CD3)2SO, 101 MHz): δ 145.3 (C1), 139.7 (C8a), 128.0 (C9a), 127.8 (C3), 125.0 (C7), 123.4 (C4a/4b), 122.4 (C4a/4b), 120.0 (C5), 118.2 (C6), 112.2 (C4), 111.2 (C8), 107.7 (C2), 55.2 (−OCH3), 21.5 (−CH3).
IR v̅max (cm–1): 3418 (N–H stretch), 2921 (C–H stretch), 1590 (C=C stretch), 1454 (C=C stretch), 1232 (C–O stretch), 1040 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C14H14ON, 212.1070; found, 212.1070.
2-((5,8-Dimethyl-9H-carbazol-3-yl)oxy)-N,N-dimethylethan-1-amine (9p)
The reaction was carried out according to the general procedure B using 2-bromo-p-xylene (93 mg, 0.50 mmol, 1.0 equiv) and 2-chloro-4-(2-(dimethylamino)ethoxy)aniline (128 mg, 0.60 mmol, 1.2 equiv) as starting materials and HPtBu3BF4 (14.5 mg, 10 mol %). The crude residue was purified by silica gel column chromatography, DCM:MeOH (100:3) to obtain compound 9p in 22% yield (31 mg, brown solid).
1H NMR ((CD3)2SO, 400 MHz): δ 11.04 (s, −NH), 7.67 (d, 1H, J = 2.4 Hz, H4), 7.46 (d, 1H, J = 8.7 Hz, H1), 7.12 (dd, 1H, J = 8.7, 2.4 Hz, H2), 7.06 (d, 1H, J = 7.2 Hz, H7), 6.82 (d, 1H, J = 7.2 Hz, H6), 4.40 (t, 2H, J = 5.1 Hz, H2′), 3.44 (t, 2H, J = 5.1 Hz, H1′), 2.82 (s, 6H, −N(CH3)2), 2.77 (s, 3H, C5–CH3), 2.50 (s, 3H, C8–CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 151.1 (C3), 139.8 (C8a), 135.2 (C9a), 129.6 (C5), 125.8 (C7), 123.6 (C4a), 120.3 (C4b), 119.6 (C6), 117.5 (C8), 114.1 (C2), 111.4 (C1), 107.0 (C4), 63.7 (C2′), 56.0 (C1′), 41.2 ((−NCH3)2), 20.2 (C5–CH3), 16.7 (C8–CH3).
IR v̅max (cm–1): 3857 (N–H stretch), 2925 (C–H stretch), 1523 (C=C stretch), 1465 (C–H stretch), 1057 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C18H23ON2, 283.1805; found, 283.1812.
5H-Benzo[b]carbazole-6,11-dione (21a)
The reaction was carried out according to the general procedure A using 2-bromonaphthalene-1,4-dione (119 mg, 0.50 mmol, 1.0 equiv) and 2-chloroaniline (77 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, CHCl3 (100%), to obtain compound 21a in 73% yield (90 mg, orange solid).
1H NMR ((CD3)2SO, 400 MHz): δ 13.08 (s, 1H, −NH), 8.22 (d, 2H, J = 8.0 Hz, H1), 8.12 (ddd, 2H, J = 7.4, 7.4, 1.5 Hz, H7, 10), 7.87 (ddd, 1H, J = 7.4, 7.4, 1.5 Hz, H9), 7.82 (ddd, 1H, J = 7.4, 7.4, 1.5 Hz, H8), 7.61 (d, 1H, J = 8.3 Hz, H4), 7.46 (ddd, 1H, J = 8.3, 7.0, 1.2 Hz, H3), 7.33–7.41 (m, 1H, H2). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 180.4 (C11), 177.6 (C6), 138.2 (C4a), 137.2 (C5a), 134.3 (C9), 134.1 (C10a), 133.2 (C8), 132.6 (C6a), 127.0 (C3), 126.1 (C10), 126.0 (C7), 124.0 (C11b), 123.9 (C2), 122.4 (C1), 117.4 (C11a), 113.9 (C4).
IR v̅max (cm–1): 3263 (N–H stretch), 1649 (C=O stretch), 1590 (C=C stretch), 1474 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H10O2N, 248.0706; found, 248.0703.
2-Methoxy-5H-benzo[b]carbazole-6,11-dione (21b)
The reaction was carried out according to the general procedure A using 2-bromonaphthalene-1,4-dione (119 mg, 0.50 mmol, 1 equiv) and 2-chloro-4-methoxyaniline (94 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc:NEt3 (70:30:1), to obtain compound 21b in 79% yield (93 mg, orange solid).
1H NMR ((CD3)2SO, 400 MHz): δ 13.02 (s, 1H, −NH), 8.15–8.08 (m, 2H, H7,10), 7.86 (dd, 1H, J = 7.5, 1.5 Hz, H9), 7.81 (dd, 1H, J = 7.4, 1.7 Hz, H8), 7.63 (d, 1H, J = 2.5 Hz, H1), 7.51 (d, 1H, J = 9.1 Hz, H4), 7.10 (dd, 1H, J = 9.0, 2.6 Hz, H3), 3.86 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 180.2 (C11), 177.2 (C6), 157.0 (C2), 137.0 (C5a), 134.2 (C9, 10a), 133.4 (C4a), 133.1 (C8), 132.7 (C6a), 126.0 (C7, 10), 124.8 (C11b), 118.4 (C3), 117.0 (C11a), 115.0 (C4), 102.1 (C1), 55.4 (−OCH3).
IR v̅max (cm–1): 3195 (N–H stretch), 1673 (C=O stretch), 1644 (C=O stretch), 1592 (C=C stretch), 1530 (C=O stretch), 1223 (C–O stretch), 1020 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C15H12ON3, 278.0812; found, 278.0812.
2-Methyl-5H-benzo[b]carbazole-6,11-dione (21c)
The reaction was carried out according to the general procedure A using 2-bromonaphthalene-1,4-dione (119 mg, 0.50 mmol, 1.0 equiv) and 4-methyl-2-chloroaniline (85 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, CHCl3 (100%), to obtain compound 21c in 59% yield (77 mg, orange solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.98 (s, 1H, −NH), 8.15–8.06 (m, 2H, H7,10), 8.01 (s, 1H, H1), 7.89–7.84 (m, 1H, H9), 7.84–7.78 (m, 1H, H8), 7.50 (d, 1H, J = 8.5 Hz, H4), 7.29 (d, 1H, J = 8.5 Hz, H3), 2.46 (s, 3H, −CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 180.3 (C11), 177.5 (C6), 137.1 (C5a), 136.7 (C4a), 134.2 (C10a), 134.1 (C9), 133.4 (C2), 133.2 (C8), 132.7 (C6a), 128.9 (C3), 126.1 (C10), 126.0 (C7), 124.2 (C11b), 121.6 (C1), 116.9 (C11a), 113.5 (C4), 21.3 (−CH3).
IR v̅max (cm–1): 3174 (N–H stretch), 1670 (C=O stretch), 1642 (C=O stretch), 1595 (C=C stretch), 1538 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C17H12O2N, 262.0863; found, 262.0861.
2-(Trifluoromethyl)-5H-benzo[b]carbazole-6,11-dione (21d)
The reaction was carried out according to the general procedure A using 2-bromonaphthalene-1,4-dione (119 mg, 0.50 mmol, 1.0 equiv) and 4-trifluoromethyl-2-chloroaniline (117 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc:NEt3 (100:20:4), to obtain compound 21d in 35% yield (54 mg, orange solid).
1H NMR ((CD3)2SO, 400 MHz): δ 13.44 (s, 1H, −NH), 8.49–8.45 (m, 1H, H1), 8.16–8.08 (m, 2H, H7, 10), 7.91–7.81 (m, 2H, H8, 9), 7.80–7.71 (m, 2H, H3, 4). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 180.3 (C11), 177.5 (C6), 139.5 (C4a), 139.0 (C5a), 134.5 (C8), 133.8 (C9), 133.5 (C7a), 132.5 (C10a), 126.2 (C7, 10), 124.7 (q, J = 272.9 Hz, −CF3), 124.3 (q, J = 31.8 Hz, C2), 123.1 (C11b), 123.0 (q, J = 3.4 Hz, C3), 119.6 (q, J = 4.3 Hz, C1), 117.6 (C11a), 115.1 (C4). 19F NMR ((CD3)2SO, 376 MHz): δ −59.7.
IR v̅max (cm–1): 3234 (N–H stretch), 1655 (C=O stretch), 1631 (C=O stretch), 1586 (C=C stretch), 1541 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C17H9O2NF3, 316.0580; found, 316.0585.
3-Methoxy-5H-benzo[b]carbazole-6,11-dione (21e)
The reaction was carried out according to the general procedure A using 2-bromonaphthalene-1,4-dione (119 mg, 0.5 mmol, 1.0 equiv) and 2-chloro-5-methoxyaniline (95 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc (9:1), to obtain compound 21e in 68% yield (94 mg, red solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.89 (s, 1H, −NH), 8.11–8.03 (m, 3H, H1, 7, 10), 7.87–7.76 (m, 2H, H8, 9), 7.00 (dd, 1H, J = 8.8, 2.0 Hz, H2), 6.98 (d, 1H, J = 2.0 Hz, H4), 3.85 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 180.5 (C11), 176.8 (C6), 159.4 (C3), 139.8 (C4a), 136.4 (C5a), 134.0 (C6a), 133.9 (C8), 133.2 (C9), 132.7 (C10a), 126.0 (C7/10), 125.9 (C7/10), 123.2 (C1), 118.0 (C11a/11b), 117.9 (C11a/11b), 115.5 (C2), 95.1 (C4), 55.4 (−OCH3).
IR v̅max (cm–1): 3189 (N–H stretch), 2927 (C–H stretch), 1664 (C=O stretch), 1629 (C=O stretch), 1532 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C17H12O3N, 278.0812; found, 278.0809.
3-Fluoro-5H-benzo[b]carbazole-6,11-dione (21f)
The reaction was carried out according to the general procedure A using 2-bromonaphthalene-1,4-dione (119 mg, 0.50 mmol, 1.0 equiv) and 5-fluoro-2-chloroaniline (87 mg, 0.60 mmol, 1.2 equiv) as starting materials. The crude residue was purified by silica gel column chromatography, petroleum ether (40–60 °C):EtOAc:NEt3 (100:20:4) to obtain compound 21f in 58% yield (76 mg, orange solid).
1H NMR ((CD3)2SO, 400 MHz): δ 13.12 (s, 1H, −NH), 8.19 (dd, 1H, J = 8.8, 5.5 Hz, H1), 8.11–8.05 (m, 2H, H7, 10), 7.87 (m, 2H, H8, 9), 7.29 (dd, 1H, J = 9.4, 2.2 Hz, H4), 7.27–7.20 (m, 1H, H2). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 180.3 (C11), 177.1 (C6), 161.5 (d, J = 243.5 Hz, C3), 138.6 (d, J = 13.2 Hz, C4a), 138.0 (C5a), 134.2 (C8), 133.8 (C6a), 133.3 (C9), 132.5 (C10a), 126.0 (C7, 10), 124.1 (d, J = 10.5 Hz, C1), 120.6 (C11a/11b), 117.4 (C11a/11b), 113.1 (d, J = 25.2 Hz, C2), 99.6 (d, J = 26.1 Hz, C4). 19F NMR ((CD3)2SO, 376 MHz): δ −113.2.
IR v̅max (cm–1): 3230 (N–H stretch), 2928 (C–H stretch), 1647 (C=O stretch), 1588 (C=C stretch), 1472 (C=C stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C16H9O2NF, 266.0612; found, 266.0609.
2-Methoxy-5-(4-methoxybenzyl)-5H-benzo[b]carbazole-6,11-dione (24)
2-Methoxy-5H-benzo[b]carbazole-6,11-dione (21b) (74 mg, 0.27 mmol, 1.0 equiv) was stirred in anhydrous DMF (8 mL) and cooled to 0 °C in an ice bath. NaH (16 mg, 0.66 mmol, 2.4 equiv) was added and stirred for 1 h. Next, benzyl bromide (0.078 mL, 0.66 mmol, 2.4 equiv) was added dropwise and the reaction was stirred for 2 h at rt. The reaction mixture was diluted with EtOAc, washed with 5% w/v aqueous solution of LiCl followed by water and brine, dried over Na2SO4, and filtered. The filtrate was then concentrated in vacuo and purified by silica gel flash column chromatography (petroleum ether (40–60 °C):EtOAc (10:1)) to afford the desired compound 24 in 83% yield (81 mg, orange solid).
1H NMR ((CD3)2SO, 500 MHz): δ 8.13 (dd, 1H, J = 7.5, 1.1 Hz, H7 or H10), 8.10 (dd, 1H, J = 7.5, 1.2 Hz, H7 or H10), 7.90–7.85 (m, 1H, H9), 7.85–7.80 (m, 1H, H8), 7.75 (d, 1H, J = 2.5 Hz, H1), 7.71 (d, 1H, J = 9.2 Hz, H4), 7.33–7.28 (m, 2H, 2 × H2′), 7.27–7.20 (m, 3H, 2 × H3′and H4′), 7.15 (dd, 1H, J = 9.2, 2.6 Hz, H3), 6.04 (s, 2H, −CH2), 3.88 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 125 MHz): δ 180.3 (C11), 177.9 (C6), 158.6 (C4′), 157.5 (C2), 134.6 (C4a), 134.4 (C5a), 134.2 (C9), 133.5 (C8), 133.3 (C10a), 133.2 (C6a), 128.6 (2 × C2′), 127.5 (C4′), 126.6 (2 × C3′), 126.3 (C7 or C10), 125.7 (C7 or C10), 124.2 (C11b), 118.8 (C3), 117.7 (C11a), 113.8 (C4), 102.3 (C1), 55.5 (Ar–OCH3), 47.8 (−CH2).
IR v̅max (cm–1): 2926 (C–H stretch), 1647 (C=O stretch), 1489 (C–H bend), 1234 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C24H18O3N, 368.1281; found, 368.1284.
5-Benzyl-2-methoxy-6,11-dimethyl-5H-benzo[b]carbazole (25)
5-Benzyl-2-methoxy-5H-benzo[b]carbazole-6,11-dione (71 mg, 0.19 mmol, 1.0 equiv) was dissolved in anhydrous THF (7 mL) under an argon atmosphere. The reaction was cooled to −80 °C, and a 1.6 M MeLi in THF solution (0.55 mL, 0.87 mmol) was added dropwise over 5 min. The reaction mixture was stirred at −80 °C for 3 h. Next, the reaction was quenched with water and extracted with CHCl3. The combined organic phases were washed with water and brine, dried over Na2SO4, and concentrated in vacuo. The isolated crude solid was dissolved in anhydrous THF (5 mL) and added to a solution of SnCl2 (0.37 g, 1.94 mmol) in a mixture of 37% HCl (4.8 mL) and Et2O (4.8 mL). The reaction mixture was stirred vigorously for 5 h and then extracted with CHCl3. The combined organic phases were washed with water and brine, dried over Na2SO4, and the filtrate was concentrated in vacuo. The crude was purified by silica gel flash column chromatography, petroleum ether (40–60 °C):EtOAc (7:3), to afford the desired compound 25 in 40% yield (28 mg, cream solid).
1H NMR ((CD3)2SO, 400 MHz): δ 8.36 (dd, 1H, J = 8.8, 0.8 Hz, H10), 8.15 (dd, 1H, J = 8.9, 0.8 Hz, H7), 7.92 (d, 1H, J = 2.5 Hz, H1), 7.55–7.48 (m, 1H, H8), 7.48–7.42 (m, 1H, H9), 7.39 (d, 1H, J = 8.8 Hz, H4), 7.29 (m, 2H, 2 × H3′), 7.22 (m, 1H, H4′), 7.15 (dd, 1H, J = 8.8, 2.5 Hz, H3), 7.08 (m, 2H, 2 × H2′), 5.83 (s, 2H, −CH2), 3.90 (s, 3H, −OCH3), 3.20 (s, 3H, C11–CH3), 2.84 (s, 3H, C6–CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 153.4 (C2), 139.5 (C4a), 139.2 (C5a), 139.1 (C1′), 131.8 (C6a), 128.7 (2 × C3′), 126.9 (C4′,11a), 126.7 (C11), 125.6 (2 × C2′), 124.8 (C8), 124.5 (C10), 123.7 (C10a), 123.4 (C11b), 123.3 (C7), 122.2 (C9), 114.4 (C3), 109.8 (C4), 109.6 (C6), 108.2 (C1), 55.8 (−OCH3), 49.0 (−CH2), 15.2 (C11–CH3), 13.6 (C6–CH3).
IR v̅max (cm–1): 2922 (C–H stretch), 1599 (C=C stretch), 1437 (C–H bend), 1204 (C–O stretch).
HRMS (ESI) m/z: [M + H]+ calcd for C27H25O2N, 366.1852; found, 366.1854.
2-Methoxy-6,11-dimethyl-5H-benzo[b]carbazole (26)
The N-debenzylation protocol was based on a literature procedure.49 5-Benzyl-2-methoxy-6,11-dimethyl-5H-benzo[b]carbazole (25) (10 mg, 0.027 mmol, 1 equiv) was dissolved in DMSO (0.5 mL) and added to a flame-dried vial. While stirring the solution at room temperature, KOtBu (21 mg, 0.19 mmol, 7 equiv) in THF (0.5 mL) was added. An oxygen balloon was then bubbled into the solution for 30 min. After stirring at room temperature for 5 h, the reaction was quenched with a saturated ammonium chloride aqueous solution. The product was extracted three times with EtOAc. The combined organic phases were dried over Na2SO4, filtered, and concentrated in vacuo. The crude was purified by silica gel column chromatography using hexane:EtOAc (7:3) to afford the desired compound 26 in 61% yield (4.5 mg, yellow solid). Characterization of 26 is in agreement with that previously described in the literature.5
1H NMR ((CD3)2SO, 400 MHz): δ 10.83 (s, 1H, NH), 8.32 (dd, 1H, J = 8.7, 0.7 Hz, H10), 8.10 (d, 1H, J = 8.7, 0.7 Hz, H7), 7.86 (d, 1H, J = 2.5 Hz, H1), 7.51 (ddd, 1H, J = 8.7, 6.5, 1.3 Hz, H8), 7.45–7.39 (m, 2H, H9,4), 7.14 (dd, 1H, J = 8.7, 2.5 Hz, H3), 3.89 (s, 3H, Ar–OCH3), 3.16 (s, 3H, C11–CH3), 2.80 (s, 3H, C6–CH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 152.6 (C2), 138.9 (C5a), 137.5 (C4a), 130.6 (C6a), 126.4 (C10a), 125.8 (C11), 124.5 (C10), 124.3 (C8), 123.7 (C11b), 123.1 (C7), 122.7 (C11a), 121.5 (C9), 114.6 (C3), 110.7 (C4), 108.8 (C1), 107.7 (C6), 55.8 (Ar–OCH3), 15.1 (C11–CH3), 12.6 (C6–CH3).
LC-MS (ESI +ve mode): m/z = 276.1. Retention time = 8.19 min.
10-Methoxy-2-methyl-7H-pyrido[4,3-c]carbazol-2-ium (27)
To a solution of 10-methoxy-7H-pyrido[4,3-c]carbazole 9e (80 mg, 0.32 mmol, 1.0 equiv) in DMF (1.3 mL) was added MeI (230 mg, 1.61 mmol, 5.3 equiv). The reaction was stirred for 19 h at 50 °C in a DrySyn heating block. The resulting yellow precipitate was filtered and washed with cold Et2O. The resulting solid was recrystallized from hot EtOH twice to yield compound 27 in 88% yield (111 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.50 (s, 1H, NH), 10.28 (s, 1H, H1), 8.71 (dd, 1H, J = 6.7, 1.2 Hz, H3), 8.67 (d, 1H, J = 6.7 Hz, H4), 8.44 (d, 1H, J = 8.8 Hz, H6), 8.30 (d, 1H, J = 2.3 Hz, H11), 8.24 (d, 1H, J = 8.8 Hz, H5), 7.75 (d, 1H, J = 8.9 Hz, H8), 7.32 (dd, 1H, J = 8.9, 2.4 Hz, H9), 4.67 (s, 3H, −NCH3), 4.00 (s, 3H, −OCH3). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 154.7 (C10), 143.3 (C1), 139.4 (C6a), 134.4 (C7a), 133.5 (C3), 133.1 (C4a), 126.0 (C4), 124.3 (C5), 124.2 (C6), 123.4 (C11c), 121.3 (C11a), 116.1 (C9), 113.7 (C11b), 113.4 (C8), 105.5 (C11), 56.3 (−OCH3), 48.0 (−NCH3).
IR v̅max (cm–1): 3129 (N–H stretch), 1631 (C=N stretch), 1562 (C=C stretch), 1497 (C=C stretch), 1029 (C–O stretch).
HRMS (ESI) m/z: [M]+ calcd for C17H15ON2, 263.1179; found, 263.1179.
10-Methoxy-2-(2-(piperidin-1-yl)ethyl)-7H-pyrido[4,3-c]carbazol-2-ium (28)45
To a solution of 10-methoxy-7H-pyrido[4,3-c]carbazole 9e (60 mg, 0.24 mmol, 1.0 equiv) in DMF (0.5 mL) was added a solution of 1-(2-chloroethyl)piperidine hydrochloride (178 mg, 1.0 mmol, 4.0 equiv) in a 1:1 mixture of DMF:water (1 mL). The reaction was stirred for 19 h at 80 °C in a DrySyn heating block. The resulting yellow precipitate was filtered and washed with cold Et2O. The resulting solid was recrystallized from hot EtOH three times to yield compound 28 in 80% yield (77 mg, yellow solid).
1H NMR (400 MHz, DMSO-d6): δ 12.61 (s, 1H, NH), 11.00 (s, 1H, NH), 10.46 (s, 1H, H1) 8.86 (d, 1H, J = 6.7 Hz, H3), 8.76 (d, 1H, J = 6.7 Hz, H4), 8.50 (d, 1H, J = 8.9 Hz, H6), 8.41–8.34 (m, 1H, H11), 8.26 (d, 1H, J = 8.9 Hz, H5), 7.76 (d, 1H, J = 8.9 Hz, H8), 7.34 (dd, 1H, J = 8.9, 1.9 Hz, H9), 5.56–5.42 (m, 2H, 2 × H1″), 4.04 (s, 3H, −OCH3), 3.95–3.82 (m, 2H, 2 × H2″), 3.67–3.54 (m, 2H, 2 × H2′ or H6′), 3.11–2.94 (m, 2H, 2 × H2′ or H6′), 1.90–1.77 (m, 4H, 2 × H3′,5′), 1.76–1.70 (m, 1H, H4′), 1.49–1.40 (m, 1H, H4′). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 154.7 (C10), 143.8 (C1), 139.4 (C6a), 134.5 (C7a), 133.7 (C4a), 132.5 (C3), 126.4 (C4), 124.8 (C6), 124.3 (C5), 123.6 (C11c), 121.3 (C11a), 116.1 (C9), 114.1 (C11b), 113.5 (C8), 105.9 (C11), 56.4 (−OCH3), 55.0 (C5), 54.2 (C2″), 52.6 (2 × C2′, 6′), 22.2 (2 × C3′, 5′), 21.2 (C4′).
IR v̅max (cm–1): 3390 (N–H stretch), 2958 (C–H stretch), 1634 (C=N stretch), 1219 (C–O stretch), 1029 (C–O stretch).
HRMS (ESI) m/z: [M]+ calcd for C23H26ON3, 360.2070; found, 360.2081.
Ditercalinium (29)47
10-Methoxy-7H-pyrido[4,3-c]carbazole 9e (100 mg, 0.40 mmol, 1.0 equiv) and 1,1′-bis(2-chloroethyl)-4,4′-bipiperidine (89 mg, 0.24 mmol, 0.6 equiv) were charged to a 2–5 mL microwave vial that was evacuated and purged with argon twice. DMF (4 mL) was added, and the vial was heated to 80 °C in a DrySyn heating block for 22 h. H2O (0.1 mL) was added to further solubilize the starting materials, and the reaction was heated for a further 24 h at 80 °C. DMF (1 mL) was added to redissolve the formation of a yellow precipitate, and the reaction was heated to 120 °C for a further 24 h. The reaction was cooled, and the precipitate was isolated by filtration. The precipitate was washed with cold Et2O and then recrystallized from hot EtOH five times to isolate 29 in 33% yield (52 mg, yellow solid).
1H NMR ((CD3)2SO, 400 MHz): δ 12.69 (s, 2H, NH), 11.63 (s, 2H, NH), 10.51 (s (br), 2H, H1), 9.00–8.78 (s (br), 2H, H3), 8.77–8.67 (d, 2H, J = 5.8 Hz, H4), 8.49 (d, 2H, J = 8.8 Hz, H6), 8.45–8.29 (s (br), 2H, H11), 8.25 (d, 2H, J = 8.9 Hz, H5), 7.75 (d, 2H, J = 8.9 Hz, H8), 7.31 (dd, 2H, J = 9.0, 2.2 Hz, H9), 5.71–5.26 (m, 4H, H1″), 4.03, 4.00–3.77 (m, 4H, H2″), 3.73–3.50 (m, 4H, 4 × H2′eq or 4 × H2′ax), 3.11–2.91 (m, 4H, 4 × H2′eq or 4 × H2′ax), 2.06–1.51 (m, 8H, 4 × H3′eq and 4 × H3′ax), 1.48–1.29 (m, 2H, H4′). 13C{1H} NMR ((CD3)2SO, 101 MHz): δ 154.7 (C10), 143.7 (C1), 139.4 (C6a), 134.4 (C7a), 133.6 (C4a), 132.6 (C3), 126.4 (C4), 124.8 (C6), 124.3 (C5), 123.6 (C11c), 121.3 (C11a), 116.3 (C9), 114.1 (C11b), 113.4 (C8), 105.7 (C11), 56.4 (−OCH3), 55.3 (C5), 54.3 (C2″), 52.6 (2 × C2′), 37.3 (2×C4′), 25.8 (2 × C3′).
IR v̅max (cm–1): 3342 (N–H stretch), 2927 (C–H stretch), 1616 (C=N stretch), 1472 (C=C stretch), 1402 (C=C stretch), 1214 (C–O stretch), 1031 (C–O stretch).
HRMS (ESI) m/z: [M]2+ calcd for C46H50O2N6, 359.1992; found, 359.1991.
Acknowledgments
E.C. thanks the University of Strathclyde for a PhD studentship. O.I.P. thanks GSK and the Engineering and Physical Sciences Research Council (EPSRC) for an industrial CASE studentship. A.T.S. and G.A.B. thank the Biotechnology and Biological Research Council for funding (BB/R006857/1).
Supporting Information Available
The Supporting Information is available free of charge at https://pubs.acs.org/doi/10.1021/acs.joc.1c02943.
Experimental procedures, characterization data, the 1H and 13C NMR spectra for new compounds, DFT calculations, and crystallographic data (PDF)
Accession Codes
CCDC 2102051–2102053 contain the supplementary crystallographic data for this paper. These data can be obtained free of charge via www.ccdc.cam.ac.uk/data_request/cif, or by emailing data_request@ccdc.cam.ac.uk, or by contacting The Cambridge Crystallographic Data Centre, 12 Union Road, Cambridge CB2 1EZ, UK; fax: +44 1223 336033.
Author Contributions
§ E.C. and A.T.S. contributed equally. Conceptualization was done by G.A.B., E.C., A.T.S. and O.I.P.; methodology was performed by E.C., O.I.P., A.T.S, and J.B.; validation was done by E.C. and A.T.S.; formal analysis was done by E.C. and A.T.S.; and manuscript preparation was done by E.C., A.T.S., and G.A.B.
The authors declare no competing financial interest.
Notes
All data underpinning this publication are openly available from the University of Strathclyde KnowledgeBase at https://doi.org/10.15129/f45b6f45-b8f7-435a-b5a3-097e3ff7397c.
Supplementary Material
References
- Cui L.-S.; Nomura H.; Geng Y.; Kim J. U.; Nakanotani H.; Adachi C. Controlling Singlet–Triplet Energy Splitting for Deep-Blue Thermally Activated Delayed Fluorescence Emitters. Angew. Chem., Int. Ed. 2017, 56, 1571. 10.1002/anie.201609459. [DOI] [PubMed] [Google Scholar]
- Schmidt A. W.; Reddy K. R.; Knölker H.-J. Occurrence, Biogenesis, and Synthesis of Biologically Active Carbazole Alkaloids. Chem. Rev. 2012, 112, 3193. 10.1021/cr200447s. [DOI] [PubMed] [Google Scholar]
- Caruso A.; Ceramella J.; Iacopetta D.; Saturnino C.; Mauro M. V.; Bruno R.; Aquaro S.; Sinicropi M. S. Carbazole Derivatives as Antiviral Agents: An Overview. Molecules 2019, 24, 1912. 10.3390/molecules24101912. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller C. M.; McCarthy F. O. Isolation, biological activity and synthesis of the natural product ellipticine and related pyridocarbazoles. RSC Adv. 2012, 2, 8883. 10.1039/c2ra20584j. [DOI] [Google Scholar]
- Andrews W. J.; Panova T.; Normand C.; Gadal O.; Tikhonova I. G.; Panov K. I. Old Drug, New Target: Ellipticine Selectively inhibit RNA Polymerase I Transcription. J. Biol. Chem. 2013, 288, 4567. 10.1074/jbc.M112.411611. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tian E.; Landowski T. H.; Stephens O. W.; Yaccoby S.; Barlogie B.; Shaughnessy J. D. Jr. Ellipticine derivative NSC 338258 represents a potential new antineoplastic agent for the treatment of multiple myeloma. Mol. Cancer Ther. 2008, 7, 500. 10.1158/1535-7163.MCT-07-0524. [DOI] [PubMed] [Google Scholar]
- Fellous R.; Coulaud D.; El Abed I.; Roques B. P.; Le Pecq J.-B.; Delain E.; Gouyette A. Cytoplasmic Accumulation of Ditercalinium in Rat Hepatocytes and Induction of Mitochondrial Damage. Cancer Res. 1988, 48, 6542. [PubMed] [Google Scholar]
- Conchon E.; Anizon F.; Aboab B.; Prudhomme M. Synthesis and Biological Activities of New Checkpoint Kinase 1 Inhibitors Structurally Related to Granulatimide. J. Med. Chem. 2007, 50, 4669. 10.1021/jm070664k. [DOI] [PubMed] [Google Scholar]
- Issa S.; Prandina A.; Bedel N.; Rongved P.; Yous S.; Le Borgne M.; Bouaziz Z. Carbazole scaffolds in cancer therapy: a review from 2012 to 2018. J. Enzyme Inhib. Med. Chem. 2019, 34, 1321. 10.1080/14756366.2019.1640692. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Georgiades S. N.; Nicolaou P. G.Recent advances in carbazole syntheses. In Advances Heterocyclic Chemistry; Scriven E. F. V.; Ramsden C. A., Eds.; Academic Press: 2019, Vol. 129, p 1. [Google Scholar]
- Ruiz-Castillo P.; Buchwald S. L. Applications of Palladium-Catalyzed C–N Cross-Coupling Reactions. Chem. Rev. 2016, 116, 12564. 10.1021/acs.chemrev.6b00512. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gorelsky S. I.; Lapointe D.; Fagnou K. Analysis of the Palladium-Catalyzed (Aromatic)C–H Bond Metalation-Deprotonation Mechanism Spanning the Entire Spectrum of Arenes. J. Org. Chem. 2012, 77, 658. 10.1021/jo202342q. [DOI] [PubMed] [Google Scholar]
- Lafrance M.; Fagnou K. Palladium-Catalyzed Benzene Arylation: Incorporation of Catalytic Pivalic Acid as a Proton Shuttle and a Key Element in Catalyst Design. J. Am. Chem. Soc. 2006, 128, 16496. 10.1021/ja067144j. [DOI] [PubMed] [Google Scholar]
- Hong B.; Luo T.; Lei X. Late-Stage Diversification of Natural Products. ACS Cent. Sci. 2020, 6, 622. 10.1021/acscentsci.9b00916. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bedford R. B.; Betham M.; Charmant J. P. H.; Weeks A. L. Intramolecular direct arylation in the synthesis of fluorinated carbazoles. Tetrahedron 2008, 64, 6038. 10.1016/j.tet.2008.01.143. [DOI] [Google Scholar]
- Bedford R. B.; Betham M. N–H Carbazole Synthesis from 2-Chloroanilines via Consecutive Amination and C–H Activation. J. Org. Chem. 2006, 71, 9403. 10.1021/jo061749g. [DOI] [PubMed] [Google Scholar]
- Tsang W. C. P.; Munday R. H.; Brasche G.; Zheng N.; Buchwald S. L. Palladium-Catalyzed Method for the Synthesis of Carbazoles via Tandem C–H Functionalization and C–N Bond Formation. J. Org. Chem. 2008, 73, 7603. 10.1021/jo801273q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Watanabe T.; Oishi S.; Fujii N.; Ohno H. Palladium-Catalyzed Direct Synthesis of Carbazoles via One-Pot N-Arylation and Oxidative Biaryl Coupling: Synthesis and Mechanistic Study. J. Org. Chem. 2009, 74, 4720. 10.1021/jo9003376. [DOI] [PubMed] [Google Scholar]
- Watanabe T.; Ueda S.; Inuki S.; Oishi S.; Fujii N.; Ohno H. One-pot synthesis of carbazoles by palladium-catalyzed N-arylation and oxidative coupling. Chem. Commun. 2007, 4516. 10.1039/b707899d. [DOI] [PubMed] [Google Scholar]
- Börger C.; Kataeva O.; Knölker H.-J. Novel approach to biscarbazole alkaloids via Ullmann coupling – synthesis of murrastifoline-A and bismurrayafoline-A. Org. Biomol. Chem. 2012, 10, 7269. 10.1039/c2ob26229k. [DOI] [PubMed] [Google Scholar]
- Hostyn S.; Van Baelen G.; Lemière G. L. F.; Maes B. U. W. Synthesis of α-Carbolines Starting from 2,3-Dichloropyridines and Substituted Anilines. Adv. Synth. Catal. 2008, 350, 2653. 10.1002/adsc.200800077. [DOI] [Google Scholar]
- Laha J. K.; Petrou P.; Cuny G. D. One-Pot Synthesis of α-Carbolines via Sequential Palladium-Catalyzed Aryl Amination and Intramolecular Arylation. J. Org. Chem. 2009, 74, 3152. 10.1021/jo802776m. [DOI] [PubMed] [Google Scholar]
- Wienhold M.; Molloy J. J.; Daniliuc C. G.; Gilmour R. Coumarins by Direct Annulation: β-Borylacrylates as Ambiphilic C3-Synthons. Angew. Chem., Int. Ed. 2021, 60, 685. 10.1002/anie.202012099. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jordan-Hore J. A.; Johansson C. C. C.; Gulias M.; Beck E. M.; Gaunt M. J. Oxidative Pd(II)-Catalyzed C–H Bond Amination to Carbazole at Ambient Temperature. J. Am. Chem. Soc. 2008, 130, 16184. 10.1021/ja806543s. [DOI] [PubMed] [Google Scholar]
- Alberico D.; Scott M. E.; Lautens M. Aryl–Aryl Bond Formation by Transition-Metal-Catalyzed Direct Arylation. Chem. Rev. 2007, 107, 174. 10.1021/cr0509760. [DOI] [PubMed] [Google Scholar]
- Burley G. A. C. E.; Taladriz-Sender A.; Paisley O. I.; Kennedy A. R.; Bush J. T.. A Chemo- and Regioselective Tandem [3 2]Heteroannulation Strategy for Carbazole Synthesis: Synergizing Two Mechanistically Distinct Bond-Forming Processes. ChemRxiv 2021. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen P.; Norris D.; Haslow K. D.; Murali Dhar T. G.; Pitts W. J.; Watterson S. H.; Cheney D. L.; Bassolino D. A.; Fleener C. A.; Rouleau K. A.; Hollenbaugh D. L.; Townsend R. M.; Barrish J. C.; Iwanowicz E. J. Identification of novel and potent isoquinoline aminooxazole-based IMPDH inhibitors. Bioorg. Med. Chem. Lett. 2003, 13, 1345. 10.1016/S0960-894X(03)00107-0. [DOI] [PubMed] [Google Scholar]
- Ramkumar N.; Nagarajan R. Total Synthesis of Ellipticine Quinones, Olivacine, and Calothrixin B. J. Org. Chem. 2014, 79, 736. 10.1021/jo402593w. [DOI] [PubMed] [Google Scholar]
- Kim D.; Choi G.; Kim W.; Kim D.; Kang Y. K.; Hong S. H. The site-selectivity and mechanism of Pd-catalyzed C(sp2)–H arylation of simple arenes. Chem. Sci. 2021, 12, 363. 10.1039/D0SC05414C. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Campeau L.-C.; Parisien M.; Jean A.; Fagnou K. Catalytic Direct Arylation with Aryl Chlorides, Bromides, and Iodides: Intramolecular Studies Leading to New Intermolecular Reactions. J. Am. Chem. Soc. 2006, 128, 581. 10.1021/ja055819x. [DOI] [PubMed] [Google Scholar]
- Campeau L.-C.; Thansandote P.; Fagnou K. High-Yielding Intramolecular Direct Arylation Reactions with Aryl Chlorides. Org. Lett. 2005, 7, 1857. 10.1021/ol050501v. [DOI] [PubMed] [Google Scholar]
- Vaidya S. D.; Toenjes S. T.; Yamamoto N.; Maddox S. M.; Gustafson J. L. Catalytic Atroposelective Synthesis of N-Aryl Quinoid Compounds. J. Am. Chem. Soc. 2020, 142, 2198. 10.1021/jacs.9b12994. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Winkler J. D.; Twenter B. M.; Gendrineau T. Synthesis of substituted phenazines via palladium-catalyzed aryl ligation. Heterocycles 2012, 84, 1345. 10.3987/COM-11-S(P)76. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sambiagio C.; Schönbauer D.; Blieck R.; Dao-Huy T.; Pototschnig G.; Schaaf P.; Wiesinger T.; Zia M. F.; Wencel-Delord J.; Besset T.; Maes B. U. W.; Schnürch M. A comprehensive overview of directing groups applied in metal-catalysed C–H functionalisation chemistry. Chem. Soc. Rev. 2018, 47, 6603. 10.1039/C8CS00201K. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang Z.; Qiu F.-C.; Gao J.; Li Z.-W.; Guan B.-T. Palladium-Catalyzed Oxidative Arylation of Tertiary Benzamides: Para-Selectivity of Monosubstituted Arenes. Org. Lett. 2015, 17, 4316. 10.1021/acs.orglett.5b02135. [DOI] [PubMed] [Google Scholar]
- Zhu R.-Y.; Farmer M. E.; Chen Y.-Q.; Yu J.-Q. A Simple and Versatile Amide Directing Group for C–H Functionalizations. Angew. Chem., Int. Ed. 2016, 55, 10578. 10.1002/anie.201600791. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rousseaux S.; García-Fortanet J.; Del Aguila Sanchez M. A.; Buchwald S. L. Palladium(0)-Catalyzed Arylative Dearomatization of Phenols. J. Am. Chem. Soc. 2011, 133, 9282. 10.1021/ja203644q. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Suematsu N.; Ninomiya M.; Sugiyama H.; Udagawa T.; Tanaka K.; Koketsu M. Synthesis of carbazoloquinone derivatives and their antileukemic activity via modulating cellular reactive oxygen species. Bioorg. Med. Chem. Lett. 2019, 29, 2243. 10.1016/j.bmcl.2019.06.038. [DOI] [PubMed] [Google Scholar]
- Yan Q.; Gin E.; Wasinska-Kalwa M.; Banwell M. G.; Carr P. D. A Palladium-Catalyzed Ullmann Cross-Coupling/Reductive Cyclization Route to the Carbazole Natural Products 3-Methyl-9H-carbazole, Glycoborine, Glycozoline, Clauszoline K, Mukonine, and Karapinchamine A. J. Org. Chem. 2017, 82, 4148. 10.1021/acs.joc.7b00044. [DOI] [PubMed] [Google Scholar]
- Ding S.; Shi Z.; Jiao N. Pd(II)-Catalyzed Synthesis of Carbolines by Iminoannulation of Internal Alkynes via Direct C–H Bond Cleavage Using Dioxygen as Oxidant. Org. Lett. 2010, 12, 1540. 10.1021/ol100272s. [DOI] [PubMed] [Google Scholar]
- Humne V.; Dangat Y.; Vanka K.; Lokhande P. Iodine-catalyzed aromatization of tetrahydrocarbazoles and its utility in the synthesis of glycozoline and murrayafoline A: a combined experimental and computational investigation. Org. Biomol. Chem. 2014, 12, 4832. 10.1039/C4OB00635F. [DOI] [PubMed] [Google Scholar]
- Szabó T.; Volk B.; Milen M. Recent Advances in the Synthesis of β-Carboline Alkaloids. Molecules 2021, 26, 663. 10.3390/molecules26030663. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang C.; Li S.; Ji L.; Liu S.; Li Z.; Li S.; Meng X. Design, synthesis and antitumor activity of non-camptothecin topoisomerase I inhibitors. Bioorg. Med. Chem. Lett. 2015, 25, 4693. 10.1016/j.bmcl.2015.06.042. [DOI] [PubMed] [Google Scholar]
- Boogaard A. T.; Pandit U. K.; Koomen G.-J. Ring D modifications of ellipticine. Part 2. Chlorination of ellipticine via its N–Hxide and synthesis and selective acetylation of 5,6,11-trimethyl-5H-benzo[b]carbazole. Tetrahedron 1994, 50, 4811. 10.1016/S0040-4020(01)85018-3. [DOI] [Google Scholar]
- Pelaprat D.; Oberlin R.; Guen I. L.; Le Pecq J. B.; Roques B. P. DNA intercalating compounds as potential antitumor agents. 1. Preparation and properties of 7H-pyridocarbazoles. J. Med. Chem. 1980, 23, 1330. 10.1021/jm00186a009. [DOI] [PubMed] [Google Scholar]
- Leon P.; Garbay-Jaureguiberry C.; Barsi M. C.; Le Pecq J. B.; Roques B. P. Modulation of the antitumor activity by methyl substitutions in the series of 7H-pyridocarbazole monomers and dimers. J. Med. Chem. 1987, 30, 2074. 10.1021/jm00394a024. [DOI] [PubMed] [Google Scholar]
- Leon P.; Garbay-Jaureguiberry C.; Lambert B.; Le Pecq J. B.; Roques B. P. Asymmetrical bisintercalators as potential antitumor agents. J. Med. Chem. 1988, 31, 1021. 10.1021/jm00400a023. [DOI] [PubMed] [Google Scholar]
- Zhao Z.; Sun Y.; Wang L.; Chen X.; Sun Y.; Lin L.; Tang Y.; Li F.; Chen D. Organic base-promoted efficient dehydrogenative/decarboxylative aromatization of tetrahydro-β-carbolines into β-carbolines under air. Tetrahedron Lett. 2019, 60, 800. 10.1016/j.tetlet.2019.02.020. [DOI] [Google Scholar]
- Haddach A. A.; Kelleman A.; Deaton-Rewolinski M. V. An efficient method for the N-debenzylation of aromatic heterocycles. Tetrahedron Lett. 2002, 43, 399. 10.1016/S0040-4039(01)02192-X. [DOI] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.