

Abstract
We examined the mechanisms involved in protein kinase C (PKC)-dependent down-regulation of dopamine transporter (DAT) activity and cell surface expression by treating heterologously expressing cells with the clathrin-mediated endocytosis inhibitor concanavalin A (Con A) or the cholesterol depleter/membrane raft disrupter methyl-β-cyclodextrin (MβC) prior to treatment with the PKC activator phorbol 12-myristate, 13-acetate (PMA). Con A blocked PMA-induced surface reductions of DAT but only partially inhibited down-regulation, while MβC partially blocked down-regulation but did not inhibit loss of cell surface DAT, demonstrating that PKC-induced DAT down-regulation occurs by a combination of trafficking and non-trafficking processes. Using density-gradient centrifugation, we found that DATs are distributed approximately equally between Triton-insoluble, cholesterol-rich membrane rafts and Triton-soluble non-raft membranes. DATs in both populations are present at the cell surface and are active for dopamine and cocaine binding. PMA-induced loss of cell surface DAT occurred only from non-raft populations, demonstrating that non-raft DATs are regulated by trafficking events and indicating the likelihood that the cholesterol-dependent non-trafficking regulatory mechanism occurs in rafts. PMA did not affect the DAT raft-non-raft distribution but stimulated the phosphorylation of DAT to a substantially greater level in rafts than non-rafts. These findings reveal a previously unknown role for cholesterol in DAT function and demonstrate the presence of distinct subcellular DAT populations that possess multiple regulatory differences that may impact dopaminergic neurotransmission.
Keywords: cholesterol, dopamine, down-regulation, membrane rafts, trafficking, transport
The dopamine transporter (DAT) is a plasma membrane phosphoprotein that functions to regulate dopaminergic neurotransmission by clearing dopamine (DA) from the synaptic space (Giros et al. 1996). DAT is a major target for psychostimulants, such as cocaine, which blocks transmitter uptake, and amphetamines (AMPH), which are carried by the protein and induce substrate efflux through transport reversal (Eshleman et al. 1994; Sulzer et al. 1995). These and other drugs of abuse, environmental toxins, and innate factors are thought to contribute to dopaminergic disorders through interactions with DAT that lead to altered DA homeostasis (Miller et al. 1999).
Dopamine transport activity is tightly regulated by complex and poorly understood processes involving protein phosphorylation, substrate pre-treatments, and protein–protein interactions (Zahniser and Doolen 2001; Vaughan 2004) that are believed to rapidly modulate dopamine signaling in vivo (Gulley and Zahniser 2003; Mortensen and Amara 2003). Activation of protein kinase C (PKC) leads to reduced DA transport activity (down-regulation) accompanied by increased clathrin/dynamin-dependent endocytosis and reduced transporter recycling (Daniels and Amara 1999; Melikian and Buckley 1999; Loder and Melikian 2003), with reduced DAT cell surface expression largely assumed to represent the primary mechanism responsible for reduced DA uptake (Melikian 2004; Zahniser and Sorkin 2004). DAT down-regulation and decreased surface expression are also induced by the DAT substrates DA, AMPH, and methamphetamine via PKC- and dynamin-dependent processes, suggesting an activity-dependent feedback process that functions through related mechanisms (Saunders et al. 2000; Chi and Reith 2003; Cervinski et al. 2005; Gorentla and Vaughan 2005).
Protein kinase C and AMPHs also stimulate non-exocytotic DA efflux through transport reversal (Fischer and Cho 1979; Liang and Rutledge 1982; Kantor and Gnegy 1998), which is thought to influence dopaminergic neuronal activity and drug neurotoxicity (Falkenburger et al. 2001; Sulzer et al. 2005). PKC- and AMPH-stimulated phosphorylation of DAT on N-terminal serines (Foster et al. 2002; Cervinski et al. 2005) has been suggested to contribute to this mechanism by promoting a conformation of DAT favorable for reverse transport (Khoshbouei et al. 2004; Fog et al. 2006).
Although several studies have shown that PKC-induced internalization of DAT is blocked by inhibitors of clathrin-mediated endocytosis (Pristupa et al. 1998; Daniels and Amara 1999; Granas et al. 2003; Sorkina et al. 2003, 2005), less work has been done to characterize the effects of these compounds on DA transport down-regulation. We demonstrate in this report that a substantial fraction of phorbol 12 myristate 13 acetate (PMA)-induced DA transport down-regulation is retained after treatments that block DAT endocytosis, revealing the presence of a trafficking–independent mode of DAT regulation. In addition, total DA transport and transport down-regulation are significantly inhibited by agents that deplete cholesterol, demonstrating a previously unknown role for this lipid in dopaminergic functions. We also identify the presence of DAT in cholesterol-rich membrane raft microdomains, which may serve as a platform for regulating DAT activity, phosphorylation, and subcellular localization.
Materials and methods
Cell culture and transient transfections
Lewis Lung Carcinoma-Porcine Kidney (LLC-PK1) cells or LLC-PK1 cells stably expressing the rat (r) dopamine transporter (rDAT-LLCPK1) (Gu et al. 1994) were maintained in α-minimum essential medium supplemented with 10% fetal bovine serum, 2 mM L-glutamine, 200 μg/mL G418, and 1X penicillin/streptomycin in an incubation chamber gassed with 5% CO2 at 37°C. The cells were grown to ~ 70% confluence in various sized culture plates prior to experimentation.
For transient transfections, FuGENE 6 transfection reagent was used to co-transfect LLC-PK1 cells with cDNAs for WT rDAT and hemeagglutinin (HA) tagged human WT or K44A dynamin 1, or with cDNAs for WT rDAT and GFP-tagged human WT or Δ80–154 caveolin 1. Cells were grown in 12-well plates, transfected with 0.5 μg/well of the appropriate plasmid and maintained for 24 h prior to experimentation. Expression of proteins was verified by immunoblotting with DAT, HA, or GFP monoclonal antibodies.
Experimental pre-treatment
Concanavalin A (ConA), filipin, methyl–β–cyclodextrin (MβC), and sucrose were prepared in Krebs-Ringer-HEPES (KRH) buffer (25 mM HEPES, 125 mM NaCl, 4.8 mM KCl, 1.2 mM KH2PO4, 1.3 mM CaCl2, 1.2 mM MgSO4, 5.6 mM glucose, pH 7.4). rDAT-LLCPK1 cells were incubated with Con A (250 μg/mL), filipin (0.5 μg/mL), MβC (5 mM), or sucrose (0.45 M) for 30 min at 37°C followed by addition of vehicle (0.1% Me2SO4 final) or 200 nM PMA for an additional 30 min at 37°C prior to initiation of transport. Pre-treatment compounds were left on cells for all procedures with the exception of hypertonic sucrose, which was removed by washing immediately prior to transport assay. Concentrations of agents employed in pre-treatments were chosen based on published values shown to successfully modulate trafficking processes and empirically determined to minimize direct effects on DA uptake activity.
For quantification of down-regulation and comparison across multiple experiments, the transport activity in cells pre-treated with test compound followed by vehicle was normalized to 100%. The level of transport in the cells that received pre-treatment followed by PMA was converted to a fraction of that amount, and down-regulation is reported as the percent difference in transport activity between the vehicle and PMA treatments. Except for MβC and higher concentrations of filipin, pre-treatments did not affect transport activity. All experiments were repeated three or more times with similar results. Down-regulation values from multiple experiments were averaged for statistical analysis by Student’s t-test or ANOVA with Tukey’s post hoc analysis using Prism 3 software.
Dopamine and alanine uptake
rDAT-LLCPK1 cells grown in 12-well plates were rinsed twice with KRH buffer. Triplicate wells were treated with or without test compounds followed by vehicle or PMA as described above. Na+-dependent transport of DA and alanine were performed as previously described (Saunders et al. 2000; Chi and Reith 2003; Cervinski et al. 2005; Gorentla and Vaughan 2005) and initiated by the addition of 10 μL of a 100X solution of dopamine or alanine to obtain final concentrations of 3 μM plus 10 nM [3H]dopamine or [3H]alanine. Non-specific uptake was determined in the presence of 100 μM (−)-cocaine for dopamine and 1 mM serine for alanine (Kimmich et al. 1994). Uptake was allowed to proceed for 8 min and cells were rapidly washed three times with ice-cold KRH. Cells were solubilized with 1% sodium dodecylsulfate (SDS) and radioactivity was assessed by liquid scintillation counting at 62% efficiency.
Membrane raft isolation
rDAT-LLCPK1 cells grown in 100 mm plates and striatal tissue slices were lysed at 4°C in MES-buffered saline (MBS, 25 mM MES, 150 mM NaCl, 2 mM EDTA, pH 6.5; 0.5 mL per plate and 1 mL/50 mg original wet weight, respectively) containing 0.1% Triton X-100 and Complete Mini protease inhibitor using three up and down strokes of a tight fitting Dounce homogenizer. For 32PO4 metabolically labeled cells, the lysis buffer was supplemented with 125 mM Na2HPO4 and 50 mM NaF. The homogenates were brought to 40% sucrose by the addition of an equal volume of 80% sucrose in MBS and 1.8 mL was injected into the bottom of a Beckman SW41 centrifuge tube under a discontinuous sucrose gradient (4 mL 30% sucrose and 5 mL of 5% sucrose in MBS). The tubes were centrifuged at 152 000 g for 18 h at 4°C and nine 1.2 mL fractions were collected beginning at the top of each tube. Aliquots of each fraction (25 μL) were subjected to electrophoresis on 4–20% linear gradient sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels using Rainbow Molecular Mass markers as standards. Fractions were assayed for DAT and other proteins by immunoblotting with specific antibodies, for total protein using bicinchoninic acid protein assay reagent, for cholesterol using a Cholesterol E assay kit, and for ganglioside GM1 using dot blot analysis detected by horseradish peroxidase-conjugated cholera toxin B. For quantification of protein and lipid distributions, band or dot blot intensities were scanned and analyzed with LumiAnalyst 3.0 software (Boehringer, Indianapolis, IN, USA). Fractions 1 and 2 did not contain detectable levels of DAT or other marker proteins and are not shown. The sum of intensities in fractions 3–9 was set to 100% and the intensity of signal in each individual fraction is expressed as a percent of the sum total.
Detergent–free membrane raft isolation
Membrane rafts were isolated from cells lysed in the absence of detergent using the method of Macdonald and Pike (2005). Three 100 mm plates of rDAT-LLCPK1 cells at 4°C were washed and scraped into homogenization buffer containing 1 mM CaCl2 and 1 mM MgCl2 (HB). Cells were pelleted by centrifugation for 2 min at 1000 g, transferred with 1 mL of HB containing complete mini protease inhibitor to a Dounce homogenizer and lysed with 25 up and down strokes. The resulting lysate was centrifuged at 1000 g for 10 min and the post-nuclear supernatant was transferred to a separate tube. An equal volume of HB containing 50% OptiPrep (Axis-Shield, Norton, MA, USA) was added to this fraction, mixed well and placed on the bottom of a Beckman SW41 centrifuge tube at 4°C. An 8 mL gradient of 0–20% OptiPrep in HB was poured on top of the lysate that now contained 25% OptiPrep. The gradients were centrifuged at 4°C for 90 min at 52 000 g and 1.2 mL fractions were collected beginning at the top of each tube. Aliquots of each fraction were analyzed for DAT and protein markers as described above.
Cell surface protein biotinylation
rDAT-LLCPK1 cells subjected to various treatments were washed three times with ice-cold phosphate-buffered saline (PBS)/Ca-Mg (138 mM NaCl, 2.7 mM KCl, 1.5 mM KH2PO4, 9.6 mM Na2HPO4, 1 mM MgCl2, 0.1 mM CaCl2, pH 7.4) and incubated twice with 0.5 μg/mL of the membrane-impermeable reagent, sulfo-NHS-LC- biotin for 25 min at 4°C in PBS/Ca-Mg. The biotinylating reagent was removed and the reaction quenched by two sequential incubations with 100 mM glycine in PBS/Ca-Mg for 20 min at 4°C. Cells were washed with PBS/Ca-Mg and lysed at 4°C with 250 μL of radioimmunoprecipitation assay (RIPA) buffer (10 mM sodium phosphate, pH 7.3, 150 mM NaCl, 2 mM EDTA, 1% Triton X-100, 1% sodium deoxycholate, 0.1% SDS) containing one Complete Mini protease inhibitor tablet/10 mL or were prepared as described for lipid raft isolation. Total cell lysates (100 μg protein) or 125 μL of lipid raft fractions were incubated with NeutrAvidin beads for 2 h at ambient temperature. The beads were washed three times with RIPA buffer, and bound proteins were eluted with 40 μL of Laemmli buffer (62.5 mM Tris–HCl, pH 6.8, 20% glycerol, 2% SDS, 5% β-mercaptoethanol, and 0.01% bromophenyl blue), followed by immunoblotting with a DAT-specific monoclonal antibody (Gaffaney and Vaughan 2004). Band intensities were quantified using LumiAnalyst 3.0 software (Boehringer) and were normalized relative to the DAT content present in total extracts or fractions under each experimental condition. Surface localization of biotinylated DAT protein was validated by probing blots for intracellular protein using anti-protein phosphatase 1α (PP1α) antibody (1 : 1000).
Metabolic phosphorylation and photoaffinity labeling of DAT
Phosphorylated DATs were prepared from rDAT-LLCPK1 cells metabolically labeled with 32PO4 as previously described (Cervinski et al. 2005). Briefly, cultured cells were incubated with phosphate-free medium for 1 h followed by exchange with fresh medium containing 0.5 mCi/mL of 32PO4. Cells were labeled for 2 h at 37°C, followed by application of vehicle or PMA for an additional 30 min at 37°C prior to lysis with 0.1% Triton X-100/MBS and density-gradient centrifugation.
Irreversible labeling of DATs with the cocaine photoaffinity analog [125I]RTI 82 was performed as previously described (Vaughan et al. 2005). Briefly, rDAT-LLCPK1 cells were incubated at 4°C for 1 h with KRH buffer containing 5 nM [125I]RTI 82. Cells were irradiated for 45 s with 245 nm ultraviolet light to attach the ligand to DAT, lysed with 0.1% Triton X-100/MBS, and subjected to density-gradient centrifugation.
Immunoprecipitation and immunoblotting
About 200 μL aliquots of sucrose density gradient fractions from 32PO4 and [125I]RTI 82 labeled samples were supplemented to 0.5% SDS, diluted with 800 μL of immunoprecipitation buffer (25 mM Tris–HCl, 0.1% Triton X-100, pH 8.0), and precipitated with rabbit polyclonal antiserum 16 generated against rDAT N-terminal amino acids 42–59 (Vaughan and Kuhar 1996). Aliquots of the same samples were immunoblotted with monoclonal antibody 16 (Gaffaney and Vaughan 2004) to determine DAT protein levels. Proteins were electrophoresed on 4–20% sodium dodecyl sulfate–polyacrylamide gel electrophoresis gels and autoradiographs were obtained using Hyperfilm MP (GE Healthcare Life Science, Piscataway, NJ, USA). Band densities were determined using an imaging densitometer and Molecular Analyst software (BioRad, Hercules, CA, USA). For quantification of immunoblotting and photoaffinity labeling, band densities from all fractions were summed and each fraction expressed as percent of the summed total. For basal phosphorylation, 32PO4 labeled band intensities in each fraction were normalized for amount of DAT protein and expressed as percent of the summed total. For stimulated phosphorylation, the basal phosphorylation levels in each fraction were defined as 100% and stimulated levels for that fraction were expressed as percent of basal level. Normalized phosphorylation values from multiple experiments were averaged for statistical analysis by Student’s t-test or ANOVA using Prism 3 software.
[3H]CFT binding
Whole cell membranes were prepared by scraping cells from plates in homogenization buffer (10 mM triethanolamine, 1 mM EDTA and 0.25 M sucrose), homogenizing with 10 up and down strokes in a Dounce homogenizer and collecting membranes by centrifugation at 12 000 g for 20 min after removal of nuclei and cell debris in an initial 1000 g centrifugation step. Membranes or sucrose density gradient fraction 5 were incubated with 5 nM [3H]CFT for 2 h at 4°C. For IC50 determinations, assay volumes contained 10−9 to 10−3 M DA or (−)-cocaine. Binding was terminated by rapid vacuum filtration using a Brandel tissue harvester over Whatman GF/B glass fiber filters soaked for 1 h in 0.1% bovine serum albumin. Filtered samples were assessed for radioactivity by liquid scintillation counting at 62% efficiency. IC50 values were determined using Prism 3 software. Data were analyzed using Student’s t-test on mean Log of IC50 values for normal distribution.
Materials
Carrier free 32PO4 was from ICN; [7,8-3H]DA (45 Ci/mmol), L-[2,3-3H]Alanine (54 Ci/mmol) and high range Rainbow molecular mass standards were from Amersham; [3H]CFT (87.0 Ci/mmol) was from Perkin-Elmer (Waltham, MA, USA) and DA was from Research Biochemicals International (Natwick, MA, USA); The amino precursor for [125I]RTI 82 was synthesized by Dr Joo Hwan Cha (Medicinal Chemistry Section, NIDA-IRP) using modifications (Xing et al. 2000; Varano et al. 2005) of the original procedure (Carroll et al. 1992), and radioiodinated as previously described (Lever et al. 1993). WT and K44A dynamin cDNA was obtained from Dr Sammanda Ramamoorthy (Medical University of South Carolina) via Dr Sandra Schmid (Scripps Research Institute, La Jolla, CA, USA); WT and Δ80–156 h-Caveolin cDNA was obtained from Dr. Brij Singh (University of North Dakota).
PMA and 1-oleoyl-2-acetyl-sn-glycerol (OAG) was from Calbiochem (San Diego, CA, USA); Monoclonal anti-protein phosphatase 1α antibody, MβC, filipin, Con A, (−)-cocaine, and other fine chemicals were from Sigma (St. Louis, MO, USA). Monoclonal anti-GFP antibody was from BD Bioscience (San Jose, CA, USA); Monoclonal anti-transferrin receptor antibody was from Zymed Laboratories (Carlsbad, CA, USA); Monoclonal anti-Na+/K+ ATPase and polyclonal anti-Lyn antibodies were from Santa Cruz Biotechnology (Santa Cruz, CA, USA). Monoclonal anti-HA-peroxidase conjugate antibody, FuGENE 6 transfection reagent and Complete Mini protease inhibitor tablets were from Roche Applied Bioscience (Indianapolis, IN, USA). Sulfo-NHS-LC-biotin, and bicinchoninic acid protein assay reagent were from Pierce; Cholesterol E assay kit was from Wako Chemicals and horseradish peroxidase-conjugated cholera toxin B was from Molecular Probes (Carlsbad, CA, USA). OptiPrep was from Axis-Shield (Norton, MA, USA).
Rats were purchased from Charles River Laboratories and were housed and treated in accordance with regulations established by the National Institutes of Health and approved by the University of North Dakota Institutional Animal Care and Use Committee.
Results
Clathrin-mediated endocytosis disrupters partially inhibit PMA-induced down-regulation of DA transport
To investigate the mechanisms underlying PKC-induced DAT down-regulation, we determined the [3H]DA transport capacity of rDAT-LLCPK1 cells pre-treated with several compounds well known to inhibit endocytosis. Because DAT has been thoroughly established to undergo clathrin- and dynamin-mediated endocytosis and intracellular sequestration in response to PKC activation (Daniels and Amara 1999; Granas et al. 2003; Sorkina et al. 2005) the discussion of results will be primarily oriented toward this process, although it is possible that PMA-binding proteins other than PKC and other trafficking events that impact DAT surface expression could also be affected by these treatments.
Cells were pre-treated for 30 min with vehicle or the clathrin-mediated endocytosis inhibitors Con A or hyperosmotic sucrose followed by 200 nM PMA for 30 min. In cells that received no pre-treatment, PMA reduced DA uptake by 34 ± 2% compared to control cells (Fig. 1a), a level of down-regulation comparable to that found in many previous studies (Vaughan 2000). In cells that received Con A or sucrose pre-treatment, PMA reduced DA uptake by 16 ± 2% and 18 ± 2%, respectively (both p < 0.001 relative to control down-regulation) (Fig. 1a), i.e., about half of the control level of down-regulation was lost while about half was retained.
Fig. 1.
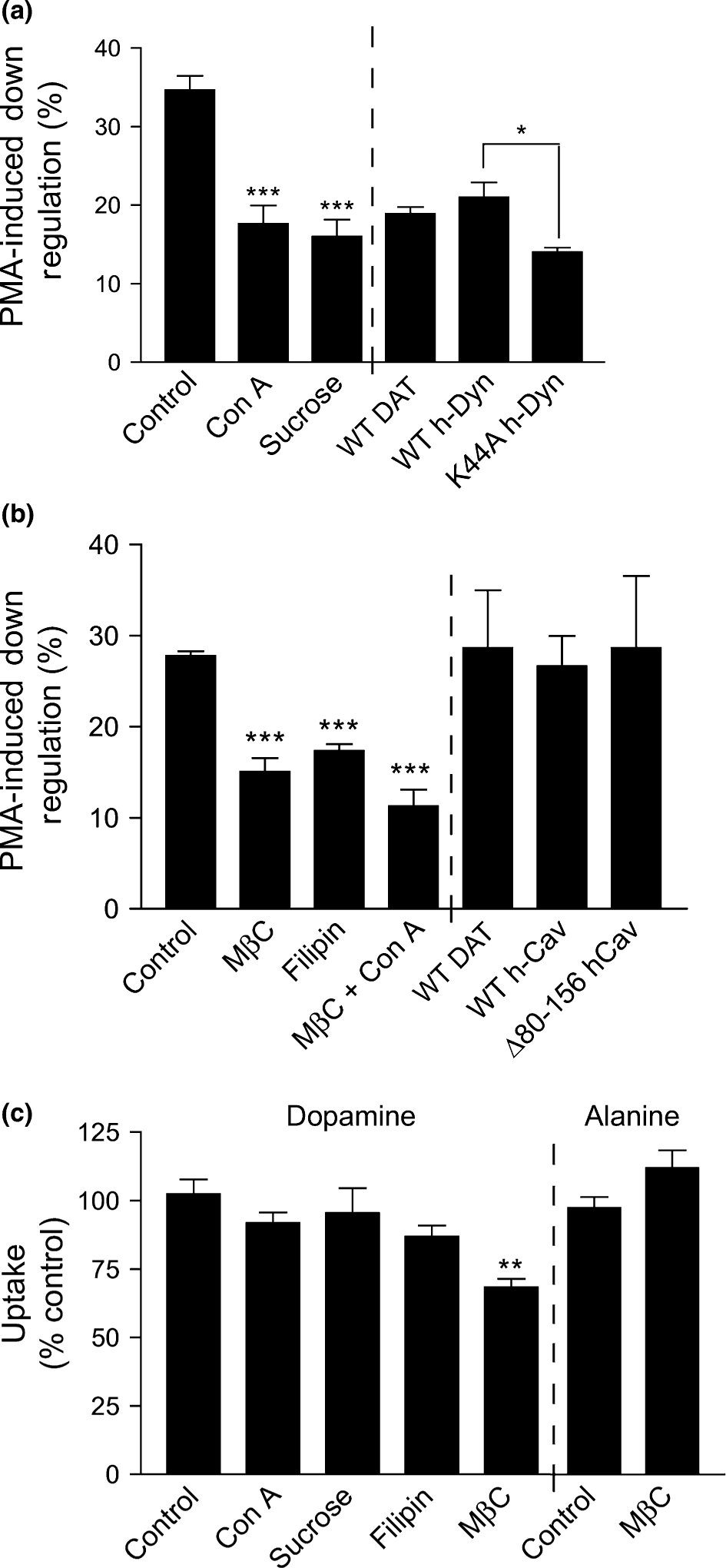
Effects of agents that inhibit clathrin-mediated endocytosis or deplete cholesterol on DAT down-regulation and DA transport. rDAT-LLCPK1 cells were treated with vehicle or the indicated inhibitors of clathrin-mediated endocytosis (a) or lipid rafts (b) followed by treatment with vehicle (control) or 200 nM β-PMA for 30 min at 37°C and assessment of [3H]DA uptake. Chemical inhibitors (left of dashed line) were applied for 30 min at 37°C prior to PMA. Final concentrations used were: Con A, 250 μg/mL; sucrose, 0.45 M; MβC, 5 mM; and filipin, 0.5 mg/mL. For transient transfections (right side of dashed line), LLC-PK1 cells were co-transfected with rDAT and WT or K44A h-dynamin-1 cDNA, or rDAT and WT or Δ80–154 caveolin-1 cDNA 24 h prior to assay for [3H]DA uptake. Down-regulation is presented as percent of DA uptake activity lost after PMA administration for each pre-treatment condition. (c) Total [3H]DA or [3H]alanine transport in rDAT-LLCPK1 cells after treatment with vehicle (control) or chemical endocytosis inhibitors. Data are presented as mean ± SE of three or more experiments performed in triplicate. *p < 0.05; **p < 0.01; ***p < 0.001 relative to controls (ANOVA).
We also examined the contribution of the clathrin pathway to DAT down-regulation using the K44A dynamin dominant negative mutant, which blocks clathrin-mediated endocytosis (Song et al. 2004). Although dynamins can also participate in non-clathrin mechanisms, DAT internalization has been shown to be clathrin-dependent (Daniels and Amara 1999; Granas et al. 2003; Sorkina et al. 2005), and to date other endocytotic trafficking pathways have not been identified for DAT. For these experiments, rDAT was transiently co-expressed with WT or K44A dynamin prior to PMA treatment. Expression of DAT and equal expression of the dynamin forms was verified by western blotting (not shown). In these experiments, cells transiently transfected with DAT alone or in combination with WT dynamin showed DA transport down-regulation levels of 18.9 ± 0.8% and 21.0 ± 1.9%, respectively, which were not statistically different. However, cells co-transfected with DAT and K44A dynamin showed transport down-regulation of 14.0 ± 0.6% (p < 0.05 relative to WT dynamin) (Fig. 1a), a partial loss of down-regulation that is similar to that produced by chemical inhibitors of clathrin-mediated endocytosis. Collectively, these results demonstrate that in rDAT-LLCPK1 cells clathrin-dependent endocytosis accounts for only about one-third to one-half of the DA transport down-regulation induced by PMA, and the retention of the remaining portion of down-regulation suggests the presence of an additional mechanism in this process.
Membrane raft disrupters partially inhibit PMA-induced down-regulation of DA transport
To investigate the role of other mechanisms in DA transport down-regulation we treated rDAT-LLCPK1 cells with methyl-β-cyclodextrin (MβC), which desorbs cholesterol from membranes, or filipin, which complexes with and functionally depletes cholesterol (Schnitzer et al. 1994; Ravichandran and Divakar 1998). These treatments are known to disrupt the structure of cholesterol-rich membrane rafts (Pike 2006).
Cells were pre-treated with vehicle, MβC or filipin for 30 min followed by treatment with PMA for an additional 30 min. In these experiments, control down-regulation averaged 28 ± 1%, while the MβC- and filipin-treated cells displayed down-regulation levels of 15 ± 2% and 17 ± 1%, respectively (both p < 0.001 relative to control down-regulation) (Fig. 1b). These treatments thus also induce the loss of approximately one-third to one-half of the control level of down-regulation.
Cholesterol is also required for caveolae-mediated endocytosis (Roy et al. 1999; Nichols 2003). To examine the involvement of caveolae in DA transport down-regulation we transiently co-transfected LLC-PK1 cells with WT rDAT and WT h-caveolin or the Δ80–156 h-caveolin-1 dominant negative truncation mutant that interferes with caveolae-mediated endocytosis (Brazer et al. 2003; Jayanthi et al. 2004). Expression of DAT and equal expression of both caveolin forms were verified by western blotting (not shown). The levels of PMA-induced down-regulation of DA transport in the WT and Δ80–156 h-caveolin expressing cells (27 ± 3% and 29 ± 8%, respectively) were not statistically different from each other or from control cells transfected with DAT alone (29 ± 6%, p > 0.05, Fig. 1b). These results, as well as others shown below, indicate that in these cells DAT down-regulation does not occur via caveolae-mediated endocytosis.
Together these findings suggest that in rDAT-LLCPK1 cells PMA-dependent down-regulation of DA transport occurs by a combination of clathrin-dependent and cholesterol-dependent processes. To determine if these mechanisms were additive for down-regulation we treated rDAT-LLCPK1 cells with MβC and Con A together prior to addition of PMA. The level of down-regulation remaining after treatment with both compounds (11 ± 2%) (Fig. 1b), was slightly less than but not statistically different from that obtained by individual treatments. The overall transport activity remained significantly reduced relative to that from non-PMA treated cells (p < 0.05), demonstrating that down-regulation was not completely blocked. The retention of this fraction of down-regulation and the lack of additivity of MβC and Con A effects may indicate an incomplete blockade of cholesterol-dependent events, the involvement of additional processes in DAT down-regulation, or the presence of mechanisms that oppose complete inhibition of down-regulation.
Inhibition of DA transport activity by MβC
In addition to affecting DAT down-regulation, the concentration of MβC that we used (5 mM) decreased total DA transport activity by 32 ± 3% relative to control (p < 0.01, Fig. 1c). Surface biotinylation showed that MβC also caused a 16 ± 3% decrease in DAT surface levels (Fig. 2a and b), indicating that some internalization or sequestration of DAT occurred in response to the treatment, but by an amount that may not fully account for the loss of transport activity, suggesting the possibility that optimal DA transport requires cholesterol. We also found that [3H]DA transport activity in rat striatal synaptosomes was reduced by 12 ± 1% and 31 ± 2% with 3.75 and 5 mM MβC, respectively, while 2.5 mM MβC was without effect, indicating that similar cholesterol effects on DAT are occurring in neurons.
Fig. 2.

Con A but not MβC blocks PMA-induced reduction of DAT cell surface levels. rDAT-LLCPK1 cells were treated with vehicle, 250 μg/mL Con A, or 5 mM MβC at 37°C prior to treatments with vehicle or 200 nM PMA for 30 min at 37°C. Cells were cooled to 4°C and subjected to surface biotinylation and lysates were chromatographed on NeutrAvidin beads. (a) Representative DAT immunoblot of column eluates. (b and c) Quantification of DAT biotinylation intensities from three independent experiments performed in duplicate. Data are presented as mean ± SE of band intensities relative to the non-treated sample (b) or relative to the non-PMA treated sample for each treatment group (c). *, p < 0.05***, p < 0.001 relative to control (ANOVA).
Because the extraction of cholesterol by MβC and filipin can permeabilize membranes, this reduction in transport activity could be due to loss of ion gradients required to drive substrate translocation. However, we found that Na+-dependent [3H]alanine transport (Kimmich et al. 1994) was not decreased by 5 mM MβC (Fig. 1c), suggesting that the reduction in DA transport is not the result of disruption of the electrochemical gradient. The concentration of filipin that suppressed PMA-induced down-regulation (0.5 μg/mL) did not affect overall DA uptake (Fig. 1c), but higher concentrations (1–5 μg/mL) dose-dependently reduced transport by close to 100% (not shown). In this case, the magnitude of loss suggests an effect on membrane permeability that may mask other cholesterol effects. Pre-treatments with Con A (250 μg/mL) and 0.45 M sucrose had no effect on overall DA transport (Fig. 1c).
Effect of Con A and MβC on DAT cell surface levels
Phorbol 12-myristate, 13-acetate-induced changes in surface DAT levels after treatments with clathrin pathway and cholesterol disrupters were assessed using cell surface biotinylation (Fig. 2). In non-PMA treated cells surface levels of DAT were not affected by Con A and were decreased by 16 ± 3% by MβC (Fig. 2a and b). To quantify effects of PMA on DAT surface expression under each condition, each of these levels was normalized to 100% (Fig. 2c). In control rDAT-LLCPK1 cells, surface DAT levels were decreased by 18 ± 3% after treatment with PMA (p < 0.001), reflective of increased endocytosis and/or decreased recycling, while in Con A pre-treated cells, surface levels of DAT remained at 98 ± 3% of control after PMA treatment (p > 0.05). This demonstrates that PMA-induced loss of cell surface DAT is essentially completely inhibited by clathrin pathway blockers, as has been shown using clathrin siRNA (Sorkina et al. 2005). Although surface DAT levels were reduced by MβC alone, after application of PMA DAT levels were further reduced by 18 ± 4% (Fig. 2c; p < 0.001), demonstrating that MβC treatment does not block PMA-induced DAT endocytosis/sequestration. The findings that Con A and MβC each inhibit about half the total level of PMA-induced down-regulation, while PMA-induced loss of cell surface DAT is blocked by Con A but is unaffected by MβC, indicates that the MβC-sensitive portion of DA transport down-regulation occurs independently of DAT trafficking. The retention of DAT endocytosis/trafficking after cholesterol depletion also argues against the involvement of caveolae-dependent endocytosis in these processes.
Association of DAT with membrane rafts
The effects of cholesterol disrupters on DA transport down-regulation suggested the involvement of DAT with cholesterol-rich membrane raft microdomains (Foster et al. 2006). To examine this possibility we determined the distribution of DAT in fractions obtained from discontinuous sucrose density centrifugation of Triton X-100 lysates. In this procedure, samples are loaded at the bottom of the gradient and buoyant low-density membrane rafts float upward and accumulate at the interface of the 5 and 30% sucrose steps while detergent solubilized membrane proteins remain in the 40% sucrose (Pike 2003).
For these experiments, rDAT-LLCPK1 cells were homogenized in 0.1% Triton X-100 and centrifuged as described. The density gradient was divided into nine equal fractions that were immunoblotted for DAT and other proteins. The majority of DAT immunoreactivity was present in fractions 5–9 spanning the 30% and 40% sucrose steps, and only negligible amounts were found in fractions 1–4 from the 5% sucrose layer (Fig. 3, fractions 1 and 2 not shown). In 10 independent experiments we obtained a highly reproducible distribution of DAT between membrane raft fraction 5 at the 5–30% sucrose interface and non-raft fractions 8 and 9 in the 40% sucrose step, with average DAT intensities for fractions 5, 8, and 9 of 28 ± 2%, 24 ± 1%, and 23 ± 2%, respectively (Fig. 3a and d). Most of the remainder of DAT (~ 25%) was recovered in fractions 6 and 7 from the 30% sucrose step, which may contain rafts with a slightly higher density or different lipid content than those in fraction 5 (Anderson and Jacobson 2002). While many membrane raft constituents are stable in 1% Triton (Pike 2004), lysis of rDAT-LLCPK1 cells with 1% Triton X-100 led to essentially complete extraction of DAT from fraction 5 and recovery of DAT immunoreactivity in fractions 8 and 9 (not shown), indicating that higher detergent concentrations overcome the forces that maintain DATs in rafts.
Fig. 3.

Dopamine transporter (DAT) is distributed between membrane raft and non-raft populations. rDAT-LLCPK1 cells were treated with vehicle (a) or 200 nM PMA (b) for 30 min at 37°C followed by solubilization with 0.1% Triton X-100 and sucrose gradient centrifugation. Aliquots of gradient fractions 3–9 and total homogenate (T) were analyzed by immunoblotting using antibodies against the indicated proteins. GM1 content was assessed by dot-blot analysis using horseradish peroxidase-linked cholera toxin subunit B. Blots shown are representative of at least three independent experiments. Sucrose concentrations and designations of raft-non-raft are indicated at the top. (c) Total protein (△) and cholesterol (•) content of gradient fractions (means ± SE of three independent experiments). Error bars are contained within the symbols. Cholesterol and protein values from PMA treated cells were essentially identical to control values and are omitted for clarity. (d) Quantification of DAT protein distribution after vehicle (gray bars) or PMA pre-treatment (black bars) (means ± SE of 10 independent experiments). Striatal tissue slices were treated with vehicle (e) or 10 μM OAG (f) for 30 min at 30°C followed by solubilization with 0.1% Triton X-100 and sucrose gradient centrifugation. Aliquots of gradient fractions 3–9 and total homogenate (T) were analyzed by immunoblotting using antibodies against DAT. GM1 content was assessed by dot-blot analysis using horseradish peroxidase-linked cholera toxin subunit B.
To further support the nature of the density gradient contents, we assayed the fractions for several raft and non-raft markers. Fraction 5 had the highest content of ganglioside GM1 (~ 53% of total) and cholesterol (~ 43% of total), but was low (~ 6% of total) in protein (Fig. 3a and c), all of which are characteristics of membrane rafts (Edidin 2003; Pike 2004). Fractions 6 and 7 were also enriched in GM1 and cholesterol and low in protein relative to non-raft fractions, but to lesser extents than fraction 5. Fractions 8 and 9 were enriched in transferrin receptor (TfR) (Fig. 3a), which is found primarily in non-raft membranes under Triton X-100 extraction conditions (Jayanthi et al. 2004; Dalskov et al. 2005; Macdonald and Pike 2005), and were low in cholesterol and GM1, and high in protein (Fig. 3c). These fractions are thus likely to contain solubilized non-raft plasma membrane and endosomal proteins (Pike 2004; Shaikh and Edidin 2006). The plasma membrane marker Na+/K+ ATPase was found primarily in non-raft fractions 8 and 9 although some was detected in fraction 5, indicating that both raft and non-raft fractions contained plasma membrane proteins.
To determine if PKC affected the raft-non-raft distribution of DATs, Triton lysates were prepared from cells treated for 30 min with PMA. This caused no alteration in the density gradient distribution of DAT (Fig. 3b and d), TfR, Na+/K+ ATPase, or GM1 (Fig. 3b), or total protein or cholesterol (not shown). Transition of DAT into or out of rafts thus does not accompany PMA-induced DAT down-regulation. However, we did find that PMA induced the translocation of PKCα into fraction 5 (Fig. 3b), as previously characterized for membrane rafts (Anderson 1998; Monastyrskaya et al. 2005), further supporting the identity of this fraction.
Extraction of rat striatal tissue with 0.1% Triton X-100 also led to the recovery of DAT in the same low and high density gradient fractions characterized as raft and non-raft by GM1 content (Fig. 3e), indicating that a similar distribution occurs in neurons. In this case, the distribution of DAT was quantified as 35 ± 6% in raft membranes and 65 ± 8% in non-raft membranes. Treatment of striatal slices with the PKC activator 1-oleoyl-2-acetyl-sn-glycerol (OAG), which is more effective than PMA in inducing DAT down-regulation and phosphorylation in rat striatal tissue (Vaughan et al. 1997; Foster et al. 2002), did not lead to any change in the DAT raft-non-raft distribution (Fig. 3f), similar to results obtained with heterologous cells. Identification of Triton-extracted expressed and native DATs in both raft and non-raft density gradient fractions has also been independently reported recently (Adkins et al. 2007).
To confirm the in vivo association of DATs with membrane rafts independently of potential detergent effects, we lysed rDAT-LLCPK1 cells with a detergent-free method and fractionated the lysates on 0–20% continuous OptiPrep density gradients with a 25% cushion (Macdonald and Pike 2005). In these preparations, DAT immunoreactivity was distributed throughout the 0–20% gradient (Fig. 4). The tyrosine kinase Lyn, a well-known membrane raft-associated protein was also present across the fractions with a distribution that significantly overlapped that of DAT, while TfR was found primarily in higher density fractions 5–9 with clear separation from DAT (Fig. 4b). Based on the natural breaks in the distributions of these markers we designated fractions 1–4 as rafts and fractions 5–9 as non-rafts. Using this criterion, DAT raft-non-raft distribution was 64.6 ± 0.1% and 35.4 ± 0.1%; Lyn was 34 ± 2% and 66 ± 2%, and TfR was 5 ± 1% and 95 ± 1%. The Na+/K+ ATPase was found in a bimodal distribution that overlapped with the DAT/Lyn, and TfR peaks, again indicating that both raft and non-raft populations contained plasma membrane proteins. The close agreement of these results with those obtained from Triton extracted lysates strongly supports the endogenous association of DATs with membrane raft domains.
Fig. 4.

Dopamine transporter (DAT) is distributed between membrane raft and non-raft populations in detergent-free preparations. rDAT-LLCPK1 cells were disrupted with a Dounce homogenizer and lysates were subjected to continuous OptiPrep density-gradient centrifugation. (a) Aliquots of gradient fractions 1–9 and total homogenate (T) were analyzed by immunoblotting using antibodies against the indicated proteins. Blots shown are representative of two or more independent experiments. OptiPrep concentrations of the fractions are indicated at the top, and raft-non-raft designations are based on the breaks observed between peaks of raft and non-raft markers. (b) Quantification of the distribution of DAT, Lyn and TfR across the gradient fractions. Data are presented as mean ± SE of band intensities as a percent of the sum total for the protein within the gradient fractions. Where absent, error bars lie within the symbols.
Effect of MβC on DAT raft distribution
To test the effects of cholesterol depletion on DAT presence in rafts we treated cells with MβC for 30 min prior to 0.1% Triton X-100 lysis and density gradient fractionation. This treatment reduced the cholesterol content of fraction 5 by 52 ± 7% (control, 211 ± 3 μg/mg protein; MβC, 102 ± 15 μg/mg protein, p < 0.01), but did not affect the distribution of DAT across the gradient (Fig. 5a–c). However, the distribution of caveolin, a cholesterol binding raft-associated protein, was altered (Fig 5a, b and d), with levels in fraction 5 reduced by 32 ± 11% (p < 0.01) and a trend toward higher levels apparent in fractions 7, 8 and 9 (Fig. 5d). Thus although raft composition is altered under these conditions, the reduction of DA transport down-regulation induced by MβC is not associated with translocation of DAT into or out of rafts.
Fig. 5.
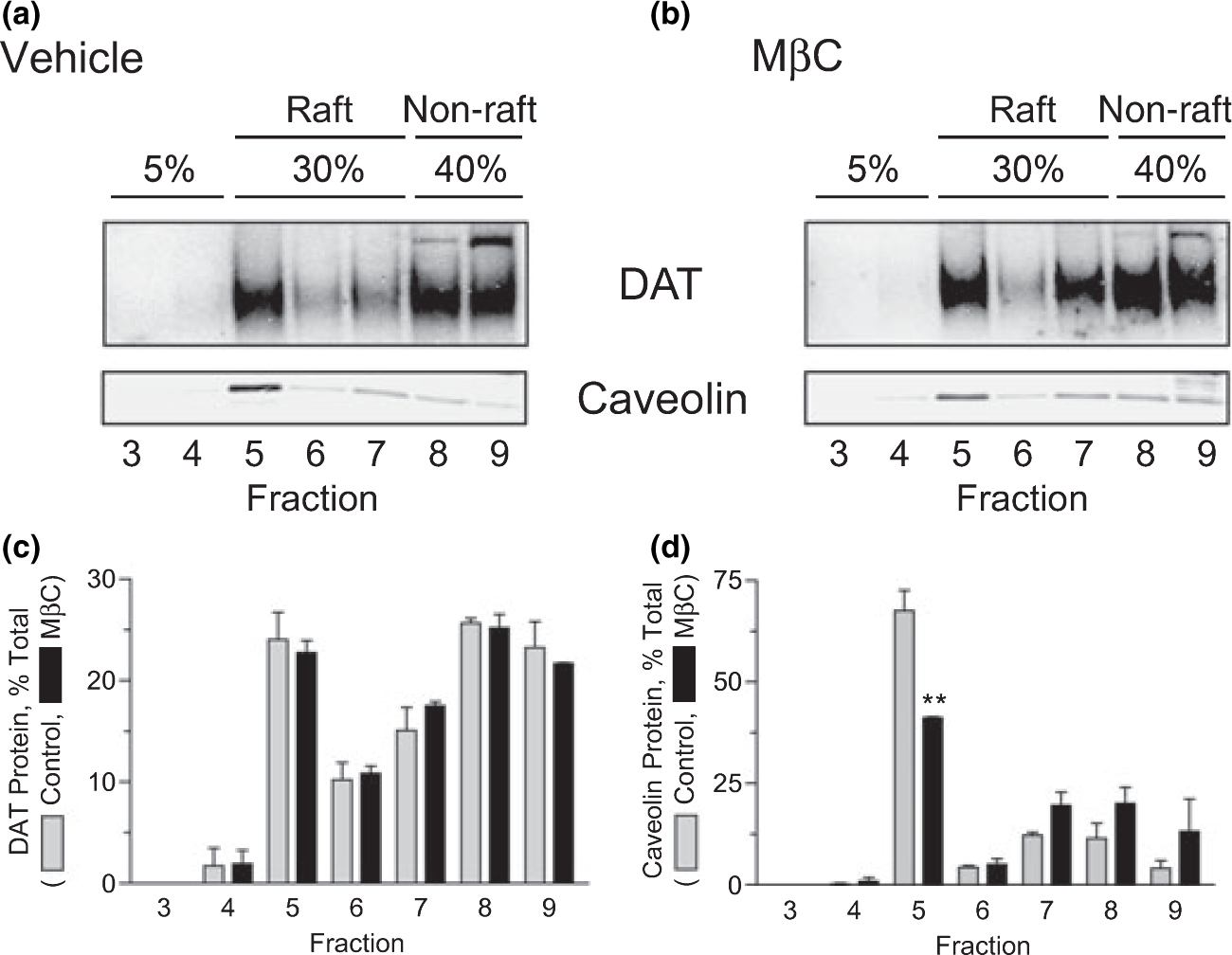
Cholesterol depletion does not alter DAT membrane raft association. rDAT-LLCPK1 cells were treated with vehicle (a) or 5 mM MβC (b) for 30 min at 37°C. Cells were solubilized with 0.1% Triton X-100 followed by sucrose gradient centrifugation and fractions were immunoblotted for DAT or Caveolin. (A and B) Representative immunoblots with sucrose concentrations indicated at the top. (C and D) Quantification of DAT (c) and caveolin (d) protein distribution after vehicle (gray bars) or MβC (black bars) treatment (means ± SE of two or more independent experiments, **, p < 0.01).
Cell surface DAT levels in raft and non-raft fractions
To determine if DATs in raft and non-raft fractions were present at the cell surface and/or underwent PMA-induced changes in trafficking, we subjected control and PMA-treated cells to surface biotinylation followed by Triton extraction and density gradient centrifugation. Biotinylated and non-biotinylated proteins in total lysates and fractions 5, 8, and 9 were separated by NeutrAvidin-Sepharose chromatography and DAT was detected by immunoblotting (Fig. 6a). The specificity of the cell surface biotinylating reagent was verified by immunoblotting for the cytosolic enzyme protein phosphatase 1α (PP1α), which was detected only in non-biotinylated fractions (Fig. 6c).
Fig. 6.
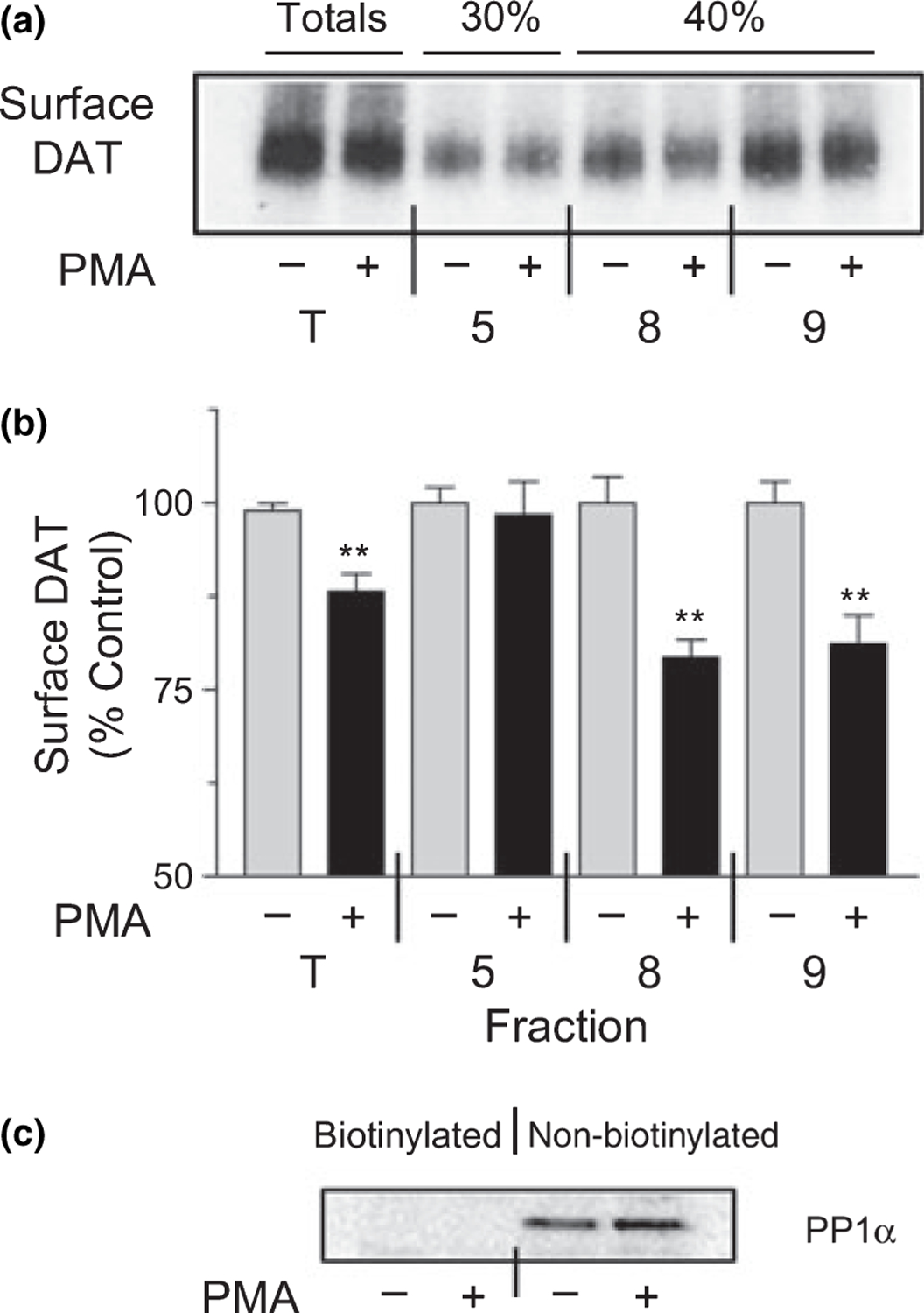
Membrane raft-associated DATs do not undergo PMA-induced endocytosis. rDAT-LLCPK1 cells were treated with vehicle or 200 nM PMA for 30 min at 37°C followed by cell surface protein biotinylation at 4°C. Cells were solubilized with 0.1% Triton X-100 followed by sucrose gradient centrifugation. (a) Aliquots of gradient fractions 5, 8, 9 and total homogenate (T) were incubated with NeutrAvidin beads and eluates of bound samples were immunoblotted for DAT. (b) Histogram indicating intensities of biotinylated bands from each fraction (means ± SE of three independent experiments). **, p < 0.01 relative to the corresponding vehicle treated control (Student’s t-test). (c) Immunoblot of PP1α performed on aliquots of biotinylated and non-biotinylated fractions of total homogenates.
Biotinylated DAT was found in total lysates (T) and in fractions 5, 8, and 9 (Fig. 6a), demonstrating the presence of surface DATs in all of these populations. In cells that received PMA treatment, surface levels of DAT in the unfractionated lysates and in fractions 8 and 9 were reduced by 12 ± 2%, 21 ± 2%, and 19 ± 4%, respectively (all p < 0.01 relative to vehicle control) (Fig. 6b). However, in three separate experiments, the DATs in fraction 5 consistently showed no change in surface levels in response to PMA (98 ± 5% of control, p > 0.05) (Fig. 6b). These results demonstrate that PMA-induced loss of cell surface DAT occurs from non-raft but not from raft populations.
Phosphorylation of DAT in raft and non-raft fractions
Dopamine transporter is known to undergo constitutive levels of phosphorylation and dephosphorylation in untreated cells and to display increased phosphorylation upon activation of PKC (Vaughan et al. 1997; Vaughan 2004). To determine if these properties are related to the DAT raft-non-raft distribution, cells were metabolically labeled with 32PO4, treated with or without PMA, and Triton lysates were subjected to density gradient centrifugation. Fractions were immunoprecipitated and immunoblotted for DAT, and phosphorylation and protein levels were assessed by densitometry.
Basal and PMA-stimulated DAT phosphorylation were evident in both raft and non-raft fractions (Fig. 7a). To quantify the levels of DAT phosphorylation in each fraction, the densitometric phosphorylation values were normalized for the amount of immunoblotted DAT protein in each fraction (levels equivalent to those in Figs 3a, b, and 5a). Basal values are expressed as a percent of the summed total of all fractions and PMA-stimulated phosphorylation for each fraction is expressed as a percent of the basal level.
Fig. 7.
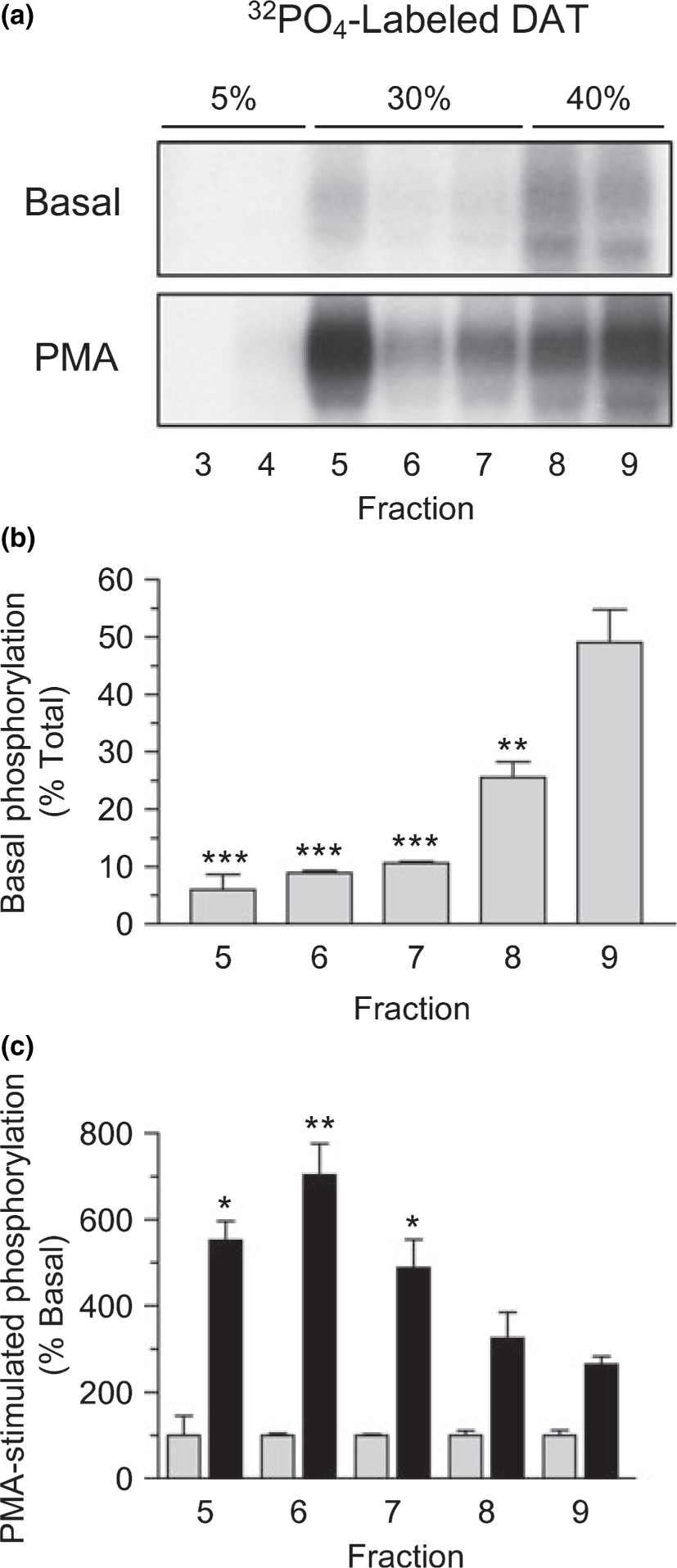
DAT phosphorylation in raft and non-raft membranes. rDAT-LLCPK1 cells were metabolically labeled with 32PO4 and treated with vehicle (basal) or 200 nM PMA for 30 min at 37°C. Cells were solubilized in 0.1% Triton X-100 followed by sucrose gradient centrifugation. (a) Aliquots of fractions 3–9 were immunoprecipitated for DAT and subjected to SDS-PAGE and autoradiography. (b) Quantification of DAT basal phosphorylation intensities normalized to DAT protein level in each fraction and expressed as a percent of the summed total for fractions 3–9 (means ± SE from three independent experiments). (c) Quantification of PMA-stimulated DAT phosphorylation intensities in each fraction (black bars), expressed as a percent of the basal phosphorylation for that fraction (gray bars, 100%). *, p < 0.05; **, p < 0.01; ***, p < 0.001 relative to fraction 9 DAT phosphorylation level for each condition (Student’s t-test).
Basal DAT phosphorylation was detected in fractions 5–9, and when corrected for DAT protein levels, was significantly lower in fractions 5, 6, and 7 than in fractions 8 and 9 (Fig. 7b). Of the total normalized basal DAT phosphorylation, fraction 9 contained 49 ± 6%, while fractions 5, 6, and 7 contained 6 ± 3%, 9 ± 0.4%, and 11 ± 0.3%, respectively (all p < 0.001 relative to fraction 9). The basal phosphorylation level in fraction 8 (26 ± 3%) was also less than in fraction 9 (p < 0.01). This difference may suggest that fractions 8 and 9 contain distinct DAT populations (e.g., plasma membranes vs. endosomes) that differ in phosphorylation characteristics, although alternative subcellular fractionation methods are needed to verify this.
Following PMA treatment, the DATs in all gradient fractions showed stimulated phosphorylation (Fig. 7a), but the increases were not equivalent in all fractions (Fig. 7c). Relative to the basal phosphorylation level, DATs in fractions 8 and 9 showed 2–3-fold phosphorylation increases, while those in fractions 5, 6, and 7 showed 5–7-fold increases (p < 0.05 and p < 0.01 relative to fraction 9), indicating that membrane rafts represent preferential sites of PKC-stimulated DAT phosphorylation.
Cocaine and dopamine binding to raft and non-raft DATs
Because rafts are prepared from solubilized cells, the density gradient fractions cannot be analyzed for DA transport. However, we analyzed the binding properties of raft and non-raft DATs using the irreversible photoaffinity cocaine analog [125I]RTI 82 (Vaughan and Kuhar 1996) to assess the ability of the DATs in each fraction to bind cocaine prior to cell lysis. Whole cells were labeled with [125I]RTI 82 followed by 0.1% Triton lysis and density gradient centrifugation. Immunoblots of each fraction were performed to determine total DAT levels, and photoaffinity labeled protein was extracted by immunoprecipitation. The photolabeling profile (Fig. 8) paralleled that of DAT protein levels assessed by immunoblotting (Figs 3a, d, and 5a). The presence of photoaffinity labeled DAT at levels proportional to that of DAT protein in each fraction indicates that the DATs in both populations are similarly active for binding the ligand prior to cell lysis. Because 10 μM DA displaces > 99% of [125I]RTI 82 DAT labeling in total cell lysates, DATs in both rafts and non-rafts must also be functional for DA binding suggesting that under transport permissive conditions DATs in both populations may be active for DA transport.
Fig. 8.

Irreversible cocaine binding to DATs in raft and non-raft membranes. rDAT-LLCPK1 cells were irreversibly labeled with the cocaine photoaffinity analog [125I]RTI 82 at 4°C and cells were solubilized with 0.1% Triton X-100 followed by sucrose gradient centrifugation. (a) Autoradiograph of DATs immunoprecipitated from each fraction. (b) Quantification of [125I]RTI 82 photolabeled DAT intensities. Results are representative of two independent experiments.
To further assess the functional capacity of DATs residing in density gradient fractions we performed reversible binding assays with the cocaine analog [3H]CFT using whole cell membranes prepared from parallel plates of cells as positive controls. Fractions 8 and 9 derived by detergent extraction were inactive for binding, demonstrating that solubilization of DAT from membranes with Triton destroys binding activity. Fraction 5, however, displayed easily detectable [3H]CFT binding, presumably because the retention of the protein in membrane raft structure preserved its native conformation.
To obtain an estimate of the affinity of DATs in fraction 5 for cocaine and DA we determined the IC50 values for these compounds to displace [3H]CFT binding (Fig. 9). The cocaine IC50 values for fraction 5 and whole cell membranes were not statistically different (451 ± 62 and 318 ± 64 nM, respectively; Fig. 9a). However, in fraction 5 the IC50 for DA displacement of [3H]CFT binding was 2-fold higher than for whole cell membranes (15.3 ± 1.6 vs. 6.9 ± 0.2 μM, p < 0.05, Fig. 9b). Further work will be required to determine if this is an endogenous characteristic of fraction 5 DATs or is induced exogenously by the low level (0.03%) of remaining Triton in the fraction or the exposure to 0.1% Triton during the extraction. The unchanged cocaine potency of fraction 5, however, suggests that major structural alterations did not occur to DATs during the detergent extraction. If the DATs located in rafts posses an innately lower potency for DA compared to the DAT population as a whole, this could result in kinetically distinct populations of transporters.
Fig. 9.
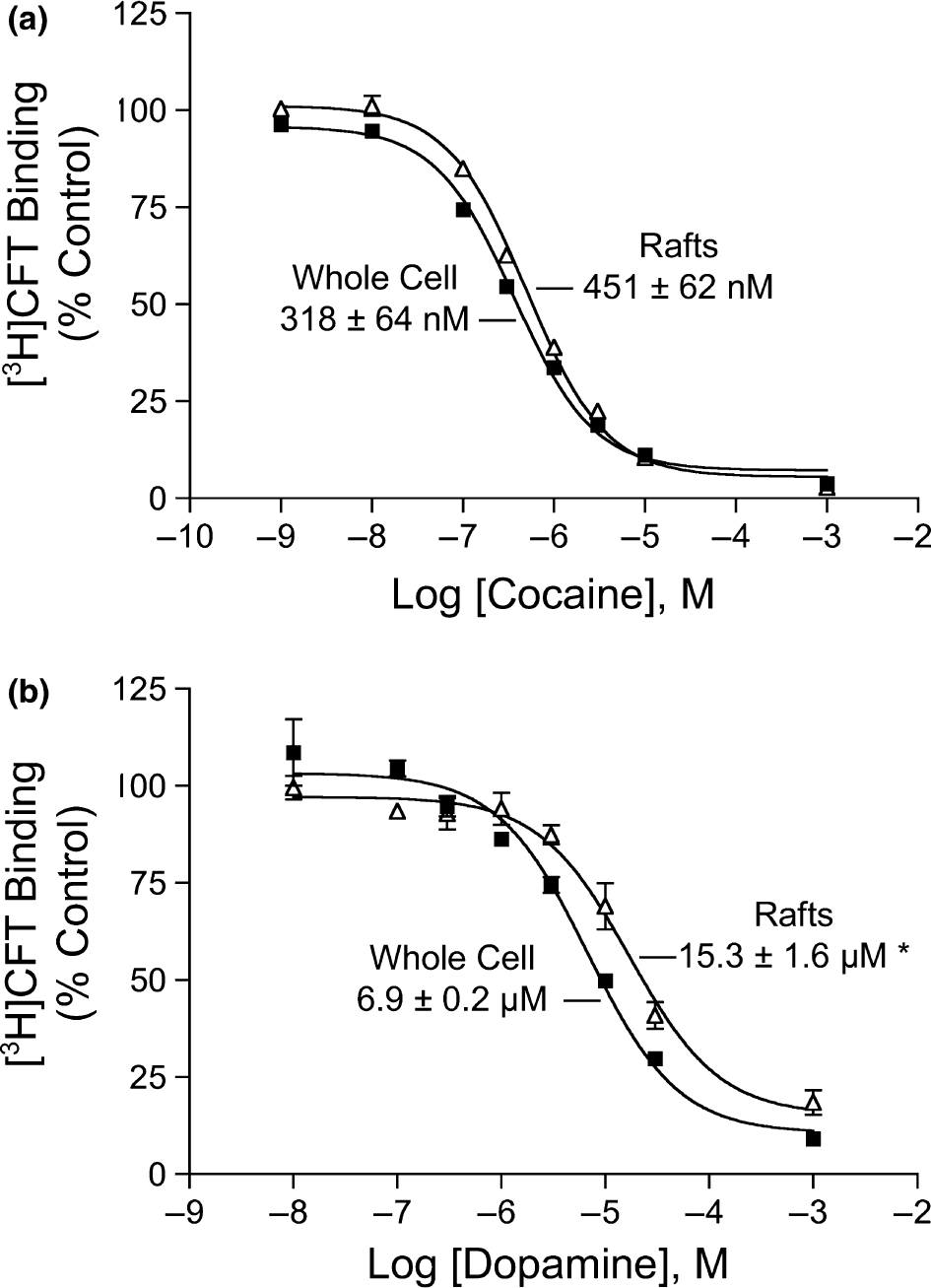
Dopamine (DA) and cocaine displacement of [3H]CFT binding in raft and whole cell membranes. Membrane rafts (△) and whole cell membranes (■) prepared from rDAT-LLCPK1 cells were assayed for reversible binding with 5 nM [3H]CFT in the presence or absence of 10−9–10−3 M cocaine (a) or 10−8–10−3 M dopamine (b). Results shown are representative of three independent experiments, and where absent, error bars are contained within the symbols. IC50 values were calculated using nonlinear regression analysis and are presented as means ± SE *, p < 0.05 relative to whole cell membranes.
Discussion
Membrane rafts are heterogeneous, dynamic membrane microdomains enriched in cholesterol and glycosphingolipids such as GM1 (Brown and London 2000; Pike 2005, 2006). The tight interaction between these lipids results in highly ordered membrane structures that are resistant to solubilization by detergents (Brown and London 2000; Pike 2003) and are wider than non-raft regions (Munro 2003). Membrane rafts contain selective subsets of proteins that serve to compartmentalize cellular processes and may represent focal points for signaling pathways (Pike 2006). In some (Parton and Richards 2003), but not all (Pike 2003, 2004) cases, rafts are associated with internalization and endocytotic cargo delivery. The results presented in this paper demonstrate that DATs are partitioned between raft and non-raft microdomains in both heterologous cells and rat striatal tissue and that multiple biochemical and regulatory properties that have the potential to affect dopaminergic neuronal activity are differentially associated with the two populations and/or with dependence on cholesterol.
We found in LLC-PK1-rDAT cells that PKC-mediated down-regulation of DA transport occurs by a combination of at least two mechanisms that segregate via raft-non-raft partitioning (Fig. 10). DATs present in non-raft membranes undergo a Con A blockable decrease in cell surface levels in response to PMA, and the partial blockade of PMA-induced transport down-regulation by Con A and other clathrin pathway inhibitors is consistent with regulation of these DATs primarily by control of surface expression. Thus, when PKC is activated but DAT endocytosis is blocked, transport down-regulation is only partially lost because the non-raft DATs remain at the surface and are active for transport, while the non-endocytotic cholesterol-dependent component of down-regulation is not affected.
Fig. 10.

Model of DAT regulation in raft and non-raft membranes. DATs are shown in raft (left) and non-raft (right) pools, with both populations subject to PKC-induced down-regulation. Membrane sphingolipids and cholesterol are depicted in red and yellow, respectively, with raft membranes displaying increased width and higher cholesterol and sphinolipid concentrations than non-raft membranes. Basal and stimulated phosphorylation levels of DATs in each pool are schematically indicated by labeled symbols (P) on the N-terminal tail that roughly correspond to the relative phosphorylation levels shown in Fig. 7. Right: PKC-induced down-regulation of non-raft DATs occurs by endocytosis and is not affected by cholesterol depletion. Clathrin and dynamin inhibitors block this process without affecting down-regulation of the raft DATs. Left: PKC-induced down-regulation of raft-associated DATs does not involve endocytosis but is reduced by cholesterol disrupters. Here we hypothesize that this is achieved through PKC induction of a cholesterol-sensitive association of DAT with a binding partner (BP, green) that suppresses transport (indicated by the size of inwardly directed arrows). In this model, cholesterol depletion prevents the DAT-BP association, leading to the loss of this portion of down-regulation without affecting down-regulation of non-raft DATs.
Conversely, the finding that MβC did not inhibit PMA-induced reduction of cell surface DAT indicates that the partial blockade of down-regulation by MβC and filipin occurs because only the trafficking-independent regulatory processes are suppressed by these treatments while trafficking-mediated down-regulation is retained. The lack of PMA-induced reduction of cell surface DAT in raft fractions, in conjunction with the high cholesterol levels in the raft compared to non-raft fractions indicates the likelihood that the cholesterol-dependent portion of down-regulation occurs in rafts, although this remains to be more directly established.
These results demonstrate that DAT is subject to a previously unknown cholesterol-dependent mode of regulation in response to PMA that occurs in the absence of trafficking in a heterologous system. Whether these processes occur in neurons and how these findings relate to other DAT regulatory processes remains to be determined. Non-trafficking regulation has been reported for hDAT variant V382A (Mazei-Robison and Blakely 2005), and trafficking-independent regulation of DAT and the serotonin transporter (SERT) have also been suggested based on kinetic differences between PMA-induced transport reduction and transporter endocytosis (Loder and Melikian 2003; Jayanthi et al. 2005), but it is not known if these finding are related to raft mechanisms. In addition, DATs that undergo down-regulation in response to AMPH treatment are proposed to remain active until internalized (Kahlig et al. 2004), suggesting that substrate-induced down-regulation may operate primarily through trafficking, and highlighting the many aspects of DAT regulation that are not understood.
In addition to inhibiting PMA-induced DAT down-regulation, MβC also decreased total DA transport activity under conditions in which Na+-dependent transport of alanine was not affected. This may be due at least in part to a reduction in cell surface DAT levels, but whether an additional mechanism is involved is not known. Other studies have shown loss of DA and serotonin transport by MβC (Magnani et al. 2004; Adkins et al. 2007), and membrane cholesterol is required for stabilization of the structure of SERT and optimal in vitro reconstitution of the GABA transporter (Shouffani and Kanner 1990; Scanlon et al. 2001), which together suggest that cholesterol may have a direct effect on intrinsic activity of neurotransmitter transporters, but additional study is needed to support this more conclusively.
Although both PMA and MβC induce reduced DAT surface expression, they do not affect the partitioning of DAT between raft-non-raft populations in rDAT-LLC-PK1 cells. This demonstrates that association of DAT with membrane rafts is PKC-independent and insensitive to a certain degree of cholesterol depletion, and that transport down-regulation induced by these treatments does not involve movement between these populations. While many proteins translocate into or out of rafts in response to PMA and MβC, others show little or no sensitivity to these treatments (Pike 2003), comparable to our observation with DAT. These results, however, do not exclude the possibility that other signals or a greater degree of cholesterol depletion could induce DAT movement between these compartments. Interestingly, SERT and the norepinephrine transporter (NET), which have also been found in membrane rafts, display raft to non-raft redistribution following PMA treatment (Jayanthi et al. 2004; Samuvel et al. 2005). These differences from DAT may suggest differences among the monoamine transporters in their targeting and regulatory mechanisms or could be due to the different cellular systems employed.
The molecular basis of the cholesterol-dependent component of DAT regulation is unknown, but because it occurs in the absence of intracellular trafficking or translocation into or out of rafts, it is possible that the process occurs at the cell surface and within the raft structure. One possibility is that PKC induces a cholesterol-dependent interaction between DAT and a binding partner (BP) that suppresses transport. The cholesterol dependence of the process is consistent with the interaction occurring predominantly in rafts, and cholesterol depletion could disrupt down-regulation if it induces translocation of the BP out of rafts or if the DAT-BP interaction directly requires cholesterol. This concept is depicted schematically in Fig. 10.
In many cell types membrane rafts display polarized subcellular distributions linked to interactions with actin cytoskeleton (Caroni 2001; Foger et al. 2001; Itoh et al. 2002; Hering et al. 2003), and in neurons raft localization and protein composition are subject to dynamic modulation implicated in synaptic plasticity (Golub et al. 2004). DATs are localized primarily to dendrites, axons, and synaptic terminals of nigrostriatal neurons (Nirenberg et al. 1996), and are sorted to apical membrane domains in cultured Madin-Darby canine kidney (MDCK) epithelial cells (Gu et al. 1996). The mechanisms responsible for these polar distributions have not been identified but rafts are potential candidates to serve in this capacity. A recent study showing that cholesterol depletion increases DAT lateral mobility suggests that rafts stabilize DAT subcellular location through cytoskeletal contacts (Adkins et al. 2007), supporting this idea and consistent with raft-related regulation of DAT occurring through raft-specific protein or lipid interactions (Fig. 10).
The different regulatory and kinetic transport characteristics we identified in raft and non-raft fractions also suggest the potential for functionally distinct populations of DAT that could be spatially and temporally regulated via raft processes. The lower DA binding potency found in raft DATs could result in regions of reduced DA clearance and prolonged dopaminergic transmission, whereas regions containing the higher DA potency non-raft DATs would exhibit relatively greater transport activity and more rapid DA clearance. The distinct down-regulation mechanisms associated with DAT raft and non-raft populations might also serve different functions in fine-tuning DA uptake, through currently unknown characteristics such as time courses of onset and reversal, and induction by specific physiological signals.
Raft and non-raft DATs also display differences in phosphorylation properties. Basal phosphorylation was lower in rafts than in non-rafts, suggesting that rafts represent sites of relatively lower constitutive phosphorylation and/or higher dephosphorylation. Whether this is related to raft-non-raft compartmentalization of the DAT kinase or phosphatase is not known. Based on inhibitor studies, the basal DAT kinase does not appear to be PKC (Cervinski et al. 2005), and its identity is currently unknown. DAT dephosphorylation is mediated in vivo and in vitro by PP1 and PP2A (Foster et al. 2003), and although DAT forms co-immunoprecipitation complexes with PP2A (Bauman et al. 2000), we were not able to detect either of these phosphatases in DAT-containing raft fractions by immunoblotting (not shown). Conversely, the highest level of DAT phosphorylation after PMA treatment was found in rafts, suggesting that these structures represent preferred sites of PKC-stimulated DAT phosphorylation. Because DAT does not move into or out of raft fractions with PMA treatment this suggests that a higher DAT phosphorylation stoichiometry occurs in rafts compared to non-rafts, either through increased molar phosphoryl incorporation or through increased numbers of phosphorylated transporters. Several PKC isoforms are known to associate with membrane rafts (Anderson 1998; Bi et al. 2001; Dalskov et al. 2005), and we observed significant levels of PKCα translocation into membrane rafts after PMA stimulation. Although it is not known if PKC directly phosphorylates DAT in vivo, the potential for this to occur is supported by our finding that a recombinant DAT N-terminal tail sequence is phosphorylated in vitro by several PKC isoforms (Gorentla and Vaughan 2006). The most parsimonious explanation for our results is thus that activated PKC translocates to membrane rafts where it phosphorylates the raft-resident DATs. As the only known function for DAT phosphorylation is to promote PKC- and AMPH-stimulated DA efflux (Khoshbouei et al. 2004), membrane rafts could represent neuronal sites specialized for non-exocytotic DA release. Because efflux is related to the dendrodendritic autoinhibition of DA neuron firing (Falkenburger et al. 2001), this underscores the significance of DAT raft-non-raft distribution and any associated cellular localization.
Collectively, the results presented in this study demonstrate that DATs are partitioned between membrane raft and non-raft populations where they are subject to distinct regulatory controls that could provide fine modulation of dopamine clearance and efflux. Malfunctions in the molecular mechanisms responsible for DAT raft-non-raft targeting could thus lead to inappropriate DAT localization or regulation that could adversely impact neurotransmission related to dopaminergic diseases or drug abuse. These findings also suggest the potential for dopaminergic processes to be impacted in cholesterol-related disorders.
Acknowledgements
This work was supported by grants R01 DA13147 and DA15175 to RAV from the National Institute on Drug Abuse, ND EPSCoR IIG to RAV and JDF, and P20 RR016741 to the University of North Dakota from the INBRE program of the National Center for Research Resources. We thank Dr Sammanda Ramamoorthy (Medical University of South Carolina) and Dr Sandra Schmid (Scripps Research Institute) for WT and K44A dynamin cDNA and for helpful discussion, Dr Brij Singh (University of North Dakota) for WT and Δ80–156 h-caveolin-1 cDNA, Dr Joo Hwan Cha and Dr Amy Newman (Medicinal Chemistry Section, NIDA-IRP) for synthesizing the RTI 82 precursor, and Dr Siegfried Detke (University of North Dakota) for advice on transient transfections.
Abbreviations used:
- AMPH
amphetamine
- ANOVA
analysis of variance
- BP
binding partner
- Con A
concanavalin A
- DA
dopamine
- DAT
dopamine transporter
- HA
hemeagglutinin
- LLC-PK1
Lewis lung carcinoma – porcine kidney
- MBS
MES-buffered saline
- MES
MES; 2-(4-morpholino)-ethane sulfonic acid
- MβC
methyl-β-cyclodextrin
- NET
norepinephrine transporter
- OAG
1-oleoyl-2-acetyl-sn-glycerol
- PBS
phosphate-buffered saline
- PKC
protein kinase C
- PMA
phorbol 12 myristate 13 acetate
- SERT
serotonin transporter
- TfR
transferrin receptor
References
- Adkins EM, Samuvel DJ, Fog JU, Eriksen J, Jayanthi LD, Vaegter CB, Ramamoorthy S and Gether U (2007) Membrane Mobility and Microdomain Association of the Dopamine Transporter Studied with Fluorescence Correlation Spectroscopy and Fluorescence Recovery after Photobleaching. Biochemistry 46, 10484–10497. [DOI] [PubMed] [Google Scholar]
- Anderson RG (1998) The caveolae membrane system. Annu. Rev. Biochem. 67, 199–225. [DOI] [PubMed] [Google Scholar]
- Anderson RG and Jacobson K (2002) A role for lipid shells in targeting proteins to caveolae, rafts, and other lipid domains. Science 296, 1821–1825. [DOI] [PubMed] [Google Scholar]
- Bauman AL, Apparsundaram S, Ramamoorthy S, Wadzinski BE, Vaughan RA and Blakely RD (2000) Cocaine and antidepressant-sensitive biogenic amine transporters exist in regulated complexes with protein phosphatase 2A. J. Neurosci. 20, 7571–7578. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Bi K, Tanaka Y, Coudronniere N, Sugie K, Hong S, van Stipdonk MJ and Altman A (2001) Antigen-induced translocation of PKC-theta to membrane rafts is required for T cell activation. Nat. Immunol. 2, 556–563. [DOI] [PubMed] [Google Scholar]
- Brazer SC, Singh BB, Liu X, Swaim W and Ambudkar IS (2003) Caveolin-1 contributes to assembly of store-operated Ca2+ influx channels by regulating plasma membrane localization of TRPC1. J. Biol. Chem. 278, 27208–27215. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Brown DA and London E (2000) Structure and function of sphingolipid- and cholesterol-rich membrane rafts. J. Biol. Chem. 275, 17221–17224. [DOI] [PubMed] [Google Scholar]
- Caroni P (2001) New EMBO members’ review: actin cytoskeleton regulation through modulation of PI(4,5)P(2) rafts. EMBO J. 20, 4332–4336. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Carroll FI, Gao Y, Abraham P, Lewin AH, Lew R, Patel A, Boja JW and Kuhar MJ (1992) Probes for the cocaine receptor. Potentially irreversible ligands for the dopamine transporter. J. Med. Chem. 35, 1813–1817. [DOI] [PubMed] [Google Scholar]
- Cervinski MA, Foster JD and Vaughan RA (2005) Psychoactive substrates stimulate dopamine transporter phosphorylation and down-regulation by cocaine-sensitive and protein kinase C-dependent mechanisms. J. Biol. Chem. 280, 40442–40449. [DOI] [PubMed] [Google Scholar]
- Chi L and Reith ME (2003) Substrate-induced trafficking of the dopamine transporter in heterologously expressing cells and in rat striatal synaptosomal preparations. J. Pharmacol. Exp. Ther. 307, 729–736. [DOI] [PubMed] [Google Scholar]
- Dalskov SM, Immerdal L, Niels-Christiansen LL, Hansen GH, Schousboe A and Danielsen EM (2005) Lipid raft localization of GABA A receptor and Na+, K+-ATPase in discrete microdomain clusters in rat cerebellar granule cells. Neurochem. Int. 46, 489–499. [DOI] [PubMed] [Google Scholar]
- Daniels GM and Amara SG (1999) Regulated trafficking of the human dopamine transporter. Clathrin- mediated internalization and lysosomal degradation in response to phorbol esters. J. Biol. Chem. 274, 35794–35801. [DOI] [PubMed] [Google Scholar]
- Edidin M (2003) The state of lipid rafts: from model membranes to cells. Annu. Rev. Biophys. Biomol. Struct. 32, 257–283. [DOI] [PubMed] [Google Scholar]
- Eshleman AJ, Henningsen RA, Neve KA and Janowsky A (1994) Release of dopamine via the human transporter. Mol. Pharmacol. 45, 312–316. [PubMed] [Google Scholar]
- Falkenburger BH, Barstow KL and Mintz IM (2001) Dendrodendritic inhibition through reversal of dopamine transport. Science 293, 2465–2470. [DOI] [PubMed] [Google Scholar]
- Fischer JF and Cho AK (1979) Chemical release of dopamine from striatal homogenates: evidence for an exchange diffusion model. J. Pharmacol. Exp. Ther. 208, 203–209. [PubMed] [Google Scholar]
- Fog JU, Khoshbouei H, Holy M et al. (2006) Calmodulin kinase II interacts with the dopamine transporter C terminus to regulate amphetamine-induced reverse transport. Neuron 51, 417–429. [DOI] [PubMed] [Google Scholar]
- Foger N, Marhaba R and Zoller M (2001) Involvement of CD44 in cytoskeleton rearrangement and raft reorganization in T cells. J. Cell Sci. 114, 1169–1178. [DOI] [PubMed] [Google Scholar]
- Foster JD, Pananusorn B and Vaughan RA (2002) Dopamine transporters are phosphorylated on N-terminal serines in rat striatum. J. Biol. Chem. 277, 25178–25186. [DOI] [PubMed] [Google Scholar]
- Foster JD, Pananusorn B, Cervinski MA, Holden HE and Vaughan RA (2003) Dopamine transporters are dephosphorylated in striatal homogenates and in vitro by protein phosphatase 1. Brain Res. Mol. Brain Res. 110, 100–108. [DOI] [PubMed] [Google Scholar]
- Foster JD, Adkins SA and Vaughan RA (2006) Membrane raft association and trafficking-independent regulation of the dopamine transporter. Program No. 39.13, Abstract Viewer/Itinerary Planner. Society for Neuroscience, Washington, DC, Online. [Google Scholar]
- Gaffaney JD and Vaughan RA (2004) Uptake inhibitors but not substrates induce protease resistance in extracellular loop two of the dopamine transporter. Mol. Pharmacol. 65, 692–701. [DOI] [PubMed] [Google Scholar]
- Giros B, Jaber M, Jones SR, Wightman RM and Caron MG (1996) Hyperlocomotion and indifference to cocaine and amphetamine in mice lacking the dopamine transporter. Nature 379, 606–612. [DOI] [PubMed] [Google Scholar]
- Golub T, Wacha S and Caroni P (2004) Spatial and temporal control of signaling through lipid rafts. Curr. Opin. Neurobiol. 14, 542–550. [DOI] [PubMed] [Google Scholar]
- Gorentla BK and Vaughan RA (2005) Differential effects of dopamine and psychoactive drugs on dopamine transporter phosphorylation and regulation. Neuropharmacology 49, 759–768. [DOI] [PubMed] [Google Scholar]
- Gorentla BK and Vaughan RA (2006) Multiple protein kinases phosphorylate the N-terminal tail of DAT in vitro. Program No. 35.16, Abstract Viewer/Itinerary Planner. Society for Neuroscience, Washington, DC, Online. [Google Scholar]
- Granas C, Ferrer J, Loland CJ, Javitch JA and Gether U (2003) N-terminal truncation of the dopamine transporter abolishes phorbol ester- and substance P receptor-stimulated phosphorylation without impairing transporter internalization. J. Biol. Chem. 278, 4990–5000. [DOI] [PubMed] [Google Scholar]
- Gu H, Wall SC and Rudnick G (1994) Stable expression of biogenic amine transporters reveals differences in inhibitor sensitivity, kinetics, and ion dependence. J. Biol. Chem. 269, 7124–7130. [PubMed] [Google Scholar]
- Gu HH, Ahn J, Caplan MJ, Blakely RD, Levey AI and Rudnick G (1996) Cell-specific sorting of biogenic amine transporters expressed in epithelial cells. J. Biol. Chem. 271, 18100–18106. [DOI] [PubMed] [Google Scholar]
- Gulley JM and Zahniser NR (2003) Rapid regulation of dopamine transporter function by substrates, blockers and presynaptic receptor ligands. Eur. J. Pharmacol. 479, 139–152. [DOI] [PubMed] [Google Scholar]
- Hering H, Lin CC and Sheng M (2003) Lipid rafts in the maintenance of synapses, dendritic spines, and surface AMPA receptor stability. J. Neurosci. 23, 3262–3271. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Itoh K, Sakakibara M, Yamasaki S, Takeuchi A, Arase H, Miyazaki M, Nakajima N, Okada M and Saito T (2002) Cutting edge: negative regulation of immune synapse formation by anchoring lipid raft to cytoskeleton through Cbp-EBP50-ERM assembly. J. Immunol. 168, 541–544. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Samuvel DJ and Ramamoorthy S (2004) Regulated internalization and phosphorylation of the native norepinephrine transporter in response to phorbol esters. Evidence for localization in lipid rafts and lipid raft-mediated internalization. J. Biol. Chem. 279, 19315–19326. [DOI] [PubMed] [Google Scholar]
- Jayanthi LD, Samuvel DJ, Blakely RD and Ramamoorthy S (2005) Evidence for biphasic effects of protein kinase C on serotonin transporter function, endocytosis, and phosphorylation. Mol. Pharmacol. 67, 2077–2087. [DOI] [PubMed] [Google Scholar]
- Kahlig KM, Javitch JA and Galli A (2004) Amphetamine regulation of dopamine transport. Combined measurements of transporter currents and transporter imaging support the endocytosis of an active carrier. J. Biol. Chem. 279, 8966–8975. [DOI] [PubMed] [Google Scholar]
- Kantor L and Gnegy ME (1998) Protein kinase C inhibitors block amphetamine-mediated dopamine release in rat striatal slices. J. Pharmacol. Exp. Ther. 284, 592–598. [PubMed] [Google Scholar]
- Khoshbouei H, Sen N, Guptaroy B, Johnson L, Lund D, Gnegy ME, Galli A and Javitch JA (2004) N-terminal phosphorylation of the dopamine transporter is required for amphetamine-induced efflux. PLoS Biol. 2, E78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kimmich GA, Randles J and Wilson J (1994) Na(+)-coupled alanine transport in LLC-PK1 cells. Am. J. Physiol. 267, C1119–1129. [DOI] [PubMed] [Google Scholar]
- Lever JR, Carroll FI, Patel A, Abraham P, Boja JW, Lewin AH and Lew R (1993) Radiosynthesis of a photoaffinity probe for the cocaine receptor of the dopamine transporter: 3-(p-chlorophenyl) tropan-2-carboxylic acid m-([1251]-iodo)-p-azidophenethyl ester ([1251]-RTI-82). J. Labelled Compds. Radiopharm. 33, 1131–1137. [Google Scholar]
- Liang NY and Rutledge CO (1982) Evidence for carrier-mediated efflux of dopamine from corpus striatum. Biochem. Pharmacol. 31, 2479–2484. [DOI] [PubMed] [Google Scholar]
- Loder MK and Melikian HE (2003) The dopamine transporter constitutively internalizes and recycles in a protein kinase C-regulated manner in stably transfected PC12 cell lines. J. Biol. Chem. 278, 22168–22174. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Macdonald JL and Pike LJ (2005) A simplified method for the preparation of detergent-free lipid rafts. J. Lipid Res. 46, 1061–1067. [DOI] [PubMed] [Google Scholar]
- Magnani F, Tate CG, Wynne S, Williams C and Haase J (2004) Partitioning of the serotonin transporter into lipid microdomains modulates transport of serotonin. J. Biol. Chem. 279, 38770–38778. [DOI] [PubMed] [Google Scholar]
- Mazei-Robison MS and Blakely RD (2005) Expression studies of naturally occurring human dopamine transporter variants identifies a novel state of transporter inactivation associated with Val382Ala. Neuropharmacology 49, 737–749. [DOI] [PubMed] [Google Scholar]
- Melikian HE (2004) Neurotransmitter transporter trafficking: endocytosis, recycling, and regulation. Pharmacol. Ther. 104, 17–27. [DOI] [PubMed] [Google Scholar]
- Melikian HE and Buckley KM (1999) Membrane trafficking regulates the activity of the human dopamine transporter. J. Neurosci. 19, 7699–7710. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Miller GW, Gainetdinov RR, Levey AI and Caron MG (1999) Dopamine transporters and neuronal injury. Trends Pharmacol. Sci. 20, 424–429. [DOI] [PubMed] [Google Scholar]
- Monastyrskaya K, Hostettler A, Buergi S and Draeger A (2005) The NK1 receptor localizes to the plasma membrane microdomains, and its activation is dependent on lipid raft integrity. J. Biol. Chem. 280, 7135–7146. [DOI] [PubMed] [Google Scholar]
- Mortensen OV and Amara SG (2003) Dynamic regulation of the dopamine transporter. Eur. J. Pharmacol. 479, 159–170. [DOI] [PubMed] [Google Scholar]
- Munro S (2003) Lipid rafts: elusive or illusive? Cell 115, 377–388. [DOI] [PubMed] [Google Scholar]
- Nichols B (2003) Caveosomes and endocytosis of lipid rafts. J. Cell Sci. 116, 4707–4714. [DOI] [PubMed] [Google Scholar]
- Nirenberg MJ, Vaughan RA, Uhl GR, Kuhar MJ and Pickel VM (1996) The dopamine transporter is localized to dendritic and axonal plasma membranes of nigrostriatal dopaminergic neurons. J. Neurosci. 16, 436–447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parton RG and Richards AA (2003) Lipid rafts and caveolae as portals for endocytosis: new insights and common mechanisms. Traffic 4, 724–738. [DOI] [PubMed] [Google Scholar]
- Pike LJ (2003) Lipid rafts: bringing order to chaos. J. Lipid Res. 44, 655–667. [DOI] [PubMed] [Google Scholar]
- Pike LJ (2004) Lipid rafts: heterogeneity on the high seas. Biochem. J. 378, 281–292. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pike LJ (2005) Growth factor receptors, lipid rafts and caveolae: an evolving story. Biochim. Biophys. Acta 1746, 260–273. [DOI] [PubMed] [Google Scholar]
- Pike LJ (2006) Rafts defined: a report on the Keystone Symposium on Lipid Rafts and Cell Function. J. Lipid Res. 47, 1597–1598. [DOI] [PubMed] [Google Scholar]
- Pristupa ZB, McConkey F, Liu F, Man HY, Lee FJ, Wang YT and Niznik HB (1998) Protein kinase-mediated bidirectional trafficking and functional regulation of the human dopamine transporter. Synapse 30, 79–87. [DOI] [PubMed] [Google Scholar]
- Ravichandran R and Divakar S (1998) Inclusion of ring A of cholesterol inside the β-cyclodextrin cavity: evidence from oxidation reactions and structural studies. J Incl Phenom Mol Recognit Chem 30, 253–270. [Google Scholar]
- Roy S, Luetterforst R, Harding A, Apolloni A, Etheridge M, Stang E, Rolls B, Hancock JF and Parton RG (1999) Dominant-negative caveolin inhibits H-Ras function by disrupting cholesterol-rich plasma membrane domains. Nat. Cell Biol. 1, 98–105. [DOI] [PubMed] [Google Scholar]
- Samuvel DJ, Jayanthi LD, Bhat NR and Ramamoorthy S (2005) A role for p38 mitogen-activated protein kinase in the regulation of the serotonin transporter: evidence for distinct cellular mechanisms involved in transporter surface expression. J. Neurosci. 25, 29–41. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Saunders C, Ferrer JV, Shi L, Chen J, Merrill G, Lamb ME, Leeb-Lundberg LM, Carvelli L, Javitch JA and Galli A (2000) Amphetamine-induced loss of human dopamine transporter activity: an internalization-dependent and cocaine-sensitive mechanism. Proc. Natl Acad. Sci. USA 97, 6850–6855. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Scanlon SM, Williams DC and Schloss P (2001) Membrane cholesterol modulates serotonin transporter activity. Biochemistry 40, 10507–10513. [DOI] [PubMed] [Google Scholar]
- Schnitzer JE, Oh P, Pinney E and Allard J (1994) Filipin-sensitive caveolae-mediated transport in endothelium: reduced transcytosis, scavenger endocytosis, and capillary permeability of select macromolecules. J. Cell Biol. 127, 1217–1232. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shaikh SR and Edidin MA (2006) Membranes are not just rafts. Chem. Phys. Lipids 144, 1–3. [DOI] [PubMed] [Google Scholar]
- Shouffani A and Kanner BI (1990) Cholesterol is required for the reconstruction of the sodium- and chloride-coupled, gamma-aminobutyric acid transporter from rat brain. J. Biol. Chem. 265, 6002–6008. [PubMed] [Google Scholar]
- Song BD, Leonard M and Schmid SL (2004) Dynamin GTPase domain mutants that differentially affect GTP binding, GTP hydrolysis, and clathrin-mediated endocytosis. J. Biol. Chem. 279, 40431–40436. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Doolen S, Galperin E, Zahniser NR and Sorkin A (2003) Oligomerization of dopamine transporters visualized in living cells by fluorescence resonance energy transfer microscopy. J. Biol. Chem. 278, 28274–28283. [DOI] [PubMed] [Google Scholar]
- Sorkina T, Hoover BR, Zahniser NR and Sorkin A (2005) Constitutive and protein kinase C-induced internalization of the dopamine transporter is mediated by a clathrin-dependent mechanism. Traffic 6, 157–170. [DOI] [PubMed] [Google Scholar]
- Sulzer D, Chen TK, Lau YY, Kristensen H, Rayport S and Ewing A (1995) Amphetamine redistributes dopamine from synaptic vesicles to the cytosol and promotes reverse transport. J. Neurosci. 15, 4102–4108. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sulzer D, Sonders MS, Poulsen NW and Galli A (2005) Mechanisms of neurotransmitter release by amphetamines: A review. Prog. Neurobiol. 75, 406–433. [DOI] [PubMed] [Google Scholar]
- Varano F, Catarzi D, Colotta V, Calabri FR, Lenzi O, Filacchioni G, Galli A, Costagli C, Deflorian F and Moro S (2005) 1-Substituted pyrazolo[1,5-c]quinazolines as novel Gly/NMDA receptor antagonists: synthesis, biological evaluation, and molecular modeling study. Bioorg. Med. Chem. 13, 5536–5549. [DOI] [PubMed] [Google Scholar]
- Vaughan RA (2000) Regulation of dopamine transporters by phosphorylation and impact on cocaine action, in Cerebral Signal Transduction: From First to Fourth Messengers (Reith MEA, ed.), pp. 375–400. Humana Press Inc, Totowa, NJ. [Google Scholar]
- Vaughan RA (2004) Phosphorylation and regulation of psychostimulant-sensitive neurotransmitter transporters. J. Pharmacol. Exp. Ther. 310, 1–7. [DOI] [PubMed] [Google Scholar]
- Vaughan RA and Kuhar MJ (1996) Dopamine transporter ligand binding domains. Structural and functional properties revealed by limited proteolysis. J. Biol. Chem. 271, 21672–21680. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Huff RA, Uhl GR and Kuhar MJ (1997) Protein kinase C-mediated phosphorylation and functional regulation of dopamine transporters in striatal synaptosomes. J. Biol. Chem. 272, 15541–15546. [DOI] [PubMed] [Google Scholar]
- Vaughan RA, Parnas ML, Gaffaney JD, Lowe MJ, Wirtz S, Pham A, Reed B, Dutta SM, Murray KK and Justice JB (2005) Affinity labeling the dopamine transporter ligand binding site. J. Neurosci. Methods 143, 33–40. [DOI] [PubMed] [Google Scholar]
- Xing D, Chen P, Keil R et al. (2000) Synthesis, biodistribution, and primate imaging of fluorine-18 labeled 2beta-carbo-1′-fluoro-2-propoxy-3beta-(4-chlorophenyl)tr opanes. Ligands for the imaging of dopamine transporters by positron emission tomography. J. Med. Chem. 43, 639–648. [DOI] [PubMed] [Google Scholar]
- Zahniser NR and Doolen S (2001) Chronic and acute regulation of Na+/Cl− -dependent neurotransmitter transporters: drugs, substrates, presynaptic receptors, and signaling systems. Pharmacol. Ther. 92, 21–55. [DOI] [PubMed] [Google Scholar]
- Zahniser NR and Sorkin A (2004) Rapid regulation of the dopamine transporter: role in stimulant addiction? Neuropharmacology 47(Suppl. 1), 80–91. [DOI] [PubMed] [Google Scholar]