

Abstract
A facile iridium/graphene-catalyzed methodology providing an efficient synthetic route for C–N bond formation is reported. This catalyst can directly promote the formation of C–N bonds, without pre-activation steps, and without solvents, alkalis and other additives. This protocol provides a direct N-alkylation of amines using a variety of primary and secondary alcohols with good selectivity and excellent yields. Charmingly, the use of diols resulted in intermolecular cyclization of amines, and such products are privileged structures in biologically active compounds. Two examples illustrate the advantages of this catalyst in organic synthesis: the tandem catalysis to synthesize hydroxyzine, and the intermolecular cyclization to synthesize cyclizine. Water is the only by-product, which makes this catalytic process sustainable and environmentally friendly.
The unprecedented iridium/graphene nanostructured catalyst directly promotes the formation of C–N bonds without the need for pre-activation steps, solvents, alkalis and other additives.
Introduction
Nitrogen-containing organic moieties are an essential structural unit, play a vital role in biologically active compounds, and are also of great significance in many fields of chemistry. The construction of the nitrogen-containing organic part is an important technology in synthetic organic chemistry, which is widely used in the synthesis of various medicines, agrochemicals, and fine chemicals.
Traditional synthetic routes for C–N bond formation are alkylation of amines with alkyl halides,1 which are usually fast but with tremendous disadvantages, for example, overalkylation reducing the yield of the desired product, side reactions bringing about tedious purification procedures, the use of toxic reagents harmful to human health, and stoichiometric amounts of waste unfriendly to the environment; therefore, many researchers have been devoted to this topic, including hydroamination,2 Buchwald–Hartwig coupling,3 and Ullmann reactions.4 Recently, a robust and sustainable method has been developed for this goal, which utilized a catalytic hydrogen autotransfer (HA) strategy to form a new C–N bond by employing less toxic and more readily available alcohols as the alkylating agents.5 This strategy has attracted a great deal of attention because the sole byproduct in this process is water, offering a sustainable method with high atom efficiency. Until now, various catalysts have been discovered for the N-alkylation of amines using alcohols as alkylating agents, in which most common catalysts were ruthenium6 and iridium7 complexes, but many other metals have also been explored.8 Most of these studies reported the scope of substrate development, but some used complicated catalytic systems, unstable and non-reusable catalysts, and most of them must use bases, solvents and other additives, which confines the development of these proposals for a true green process.
In this report, we introduce an iridium/graphene nanostructured catalyst for the N-alkylation of amines to prepare nitrogen-containing derivatives and heterocyclic compounds. This catalyst can directly promote the formation of C–N bonds, without pre-activation steps, and without solvents, alkalis and other additives. As a heterogeneous catalyst, it can be filtered of the reaction system, and reused. After many catalytic cycles, the structural characteristics, activity and reliability of the catalyst remain almost unchanged. In the reaction system, the only by-product is water, which can be used in tandem reactions or specific intermolecular cyclization reactions to construct biologically active drugs or other specific applications.
Results and discussion
Catalyst preparation and structural characterization
The catalyst, iridium–graphene nanostructured catalyst (GIrNC), was prepared by the reaction of graphene oxide (GO) with iridium(iii) chloride in a mixed solvent (ethoxylethanol : water = 1 : 1, v/v) at 110 °C under argon (Scheme 1),9 where the graphene oxide was synthesized by a modified Hummers' method.10 The infrared spectrum of graphene oxide (Fig. 1a) shows that some functional groups are formed on the surface of graphene oxide,11 including hydroxyl groups of carboxylic acid and alcohol (νO–H from 2800 to 3700 cm−1), carbonyl group (νC O at 1730 cm−1), C C stretching (νC C at 1616 cm−1) and C–O stretching (νC–O at 1027 and 968 cm−1), respectively. Fig. 1b and c show the infrared spectra of samples separated from the reaction mixture during the preparation of GIrNC at 5 and 24 hours reaction time, which shows that when GO reacts with iridium(iii) chloride, the νO–H intensity of GO decreases, and the absorption of νC O, νC C and νC–O still exists, implying that the protons of hydroxyl groups of carboxylic acid and alcohol gradually lost, and their conjugate base, carboxylate and alkoxide ions, trapped the iridium ions to form GIrNC.
Scheme 1. Illustration of the methods used for preparing iridium/graphene nanostructured catalyst.

Fig. 1. Infrared spectra of: (a) graphene oxide, (b) the sample separated from the reaction mixture during preparing GIrNC after reaction time of 5 hours, and (c) the sample separated from the reaction mixture during preparing GIrNC after reaction time of 24 hours.

The Ir 4f XPS of GIrNC (Fig. 2a) shows two characteristic peaks, the Ir 4f7/2 peak centered at 61.06 eV and the Ir 4f5/2 peak centered at 64.16 eV, which are obviously higher than those of Ir0 at 60.61, and 63.51 eV, respectively (Fig. 2b), which confirms that iridium should be ionic, and bonded to GO skeleton.
Fig. 2. Ir 4f XPS spectra of (a) GIrNC, and (b) Ir0.

To investigate the fine structure of GIrNC, the X-ray diffraction (XRD) pattern of GIrNC was acquired, and those of graphene, GO, and Ir0 were also obtained for comparing, which are shown in Fig. 3. The characteristic XRD peak of the graphene is at around 2θ = 26.8° corresponding to the (002) of carbon (Fig. 3a). The characteristic peak of the GO is at around 10.9° corresponding to the (001) of GO (Fig. 3b). For GIrNC (Fig. 3c), a broaden peak around 10.9° is observed, showing that parts of GO structure still remains in GIrNC. A peak around 2θ = 24.2° indicates that there are some graphene structures in GIrNC, which is attributed to the reconstruction of graphene structure, resulting from the decarboxylation, dehydration, and rearomatization of GO.
Fig. 3. XRD patterns of (a) graphene, (b) GO, (c) GIrNC, and (d) Ir0.

Three new peaks were also observed on the pattern of GIrNC, located at around 37.7, 43.0, and 67.6°, respectively, which cannot be attributed to carbon and iridium elements, and should be attributed to the characteristic XRD peaks of the GIrNC. For comparison, the pattern of Ir0 is shown in Fig. 3d, three peaks at 40.6, 47.3, and 69.3° are observed, corresponding to (111), (200) and (220) of iridium, respectively.
Furthermore, transmission electron microscopy (TEM) is used to study the microstructure of GIrNC. The spherical-aberration corrected field emission TEM images of GIrNC are shown in Fig. 4a–c. The low magnification image (Fig. 4a) shows that some substructures are formed on the surface of graphene sheet. A representative high-resolution TEM (Fig. 4b) illustrates that the grain size of GIrNC ranges between 1 and 5 nm. Fig. 4c is an enlarged fragment taken from Fig. 4b (shown by the square symbol), where the hexagonal lattice of graphene and iridium structure can be observed, some of carbon atoms are replaced by iridium ions, and radii of iridium is about 1.25 Å belonging to the range of covalent bond of iridium. Fig. 4d–g show the EDS element mapping of the TEM GIrNC image. Carbon atom is evenly distributed in the marked area (Fig. 4e), iridium is present in the microparticle distribution area of the marked area (Fig. 4f), and oxygen mainly occurs where iridium exists (Fig. 4g), which means that the oxygen atom binds to iridium.
Fig. 4. Spherical-aberration corrected field emission TEM images of GIrNC and EDS elemental mappings from a TEM image. (a) A low-magnification TEM image, (b) a high-resolution TEM image, (c) an enlarged fragment taken from (b) (shown by the square symbol), (d) a marked area for EDS elemental mapping, (e–g) EDS elemental mappings from the marked area.

Initial catalytic studies
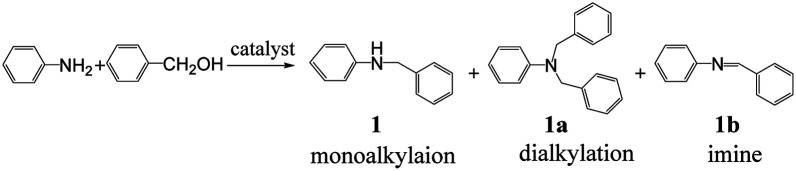
Our initial investigations using benzyl alcohol as an alkylating agent for N-alkylation of aniline showed that heterogeneous catalyst (GIrNC) exhibited 100% conversion of aniline at 110 °C after 24 h reaction (Table 1), in which, 60% of monoalkylation product (N-benzylaniline, 1), 36% of dialkylation product (N,N-dibenzylaniline, 1a) and ∼4% imine (1b) were obtained. In general, the alkylation of secondary amines is more difficult than primary amine; nevertheless, in the current situation, some of monoalkylated products are converted to dialkylated products before the imines are converted to monoalkylated products, which means that GIrNC has a very high catalytic activity for the N-alkylation of amines. However, it is possible to selectively obtain mono- or di-alkylated products by controlling the reaction conditions, which will be described in the second half of this article. In order to compare the catalytic ability of GIrNC, some other metal–graphene catalysts were also prepared, including copper–graphene catalyst (GCuNC, Table 1), nickel–graphene catalyst (GNiNC, Table 1) and cobalt–graphene catalyst (GCoNC, Table 1) and test the N-alkylation ability of aniline under similar reaction conditions. As shown in Table 1, other metal–graphene oxide catalysts have lower aniline conversion rates. When GNiNC was used as a catalyst, only 9% of aniline was converted into a mixture composed of 4% imine and 5% monoalkylation product (entry C). For GCuNC and GCoNC, the conversion rate of aniline is lower, and nearly 99% of aniline remains intact after 24 hours of reaction. Some previously reported iridium homogeneous catalysts are also listed in Table 1 for comparison of catalytic performance. For example, catalysts IrPNP,7f IrCl(BPEP–H),7g [Cp*Ir(Pro)Cl],7h and Tang's catalyst7i showed mild to good catalytic activity for N-alkylation with yields of 66, 90, 95 and 88%, respectively, but they should all use solvents or bases.
Catalytic activity comparison for N-alkylation of aniline.
| ||||
|---|---|---|---|---|
| Catalyst | Transformation of aniline (%) | Composition of reaction mixture (%) | ||
| 1 | 1a | 1b | ||
| GIrNCa | 100 | 60 | 36 | 4 |
| GCuNCa | 1 | 1 | 0 | 0 |
| GNiNCa | 9 | 5 | 0 | 4 |
| GCoNCa | 1 | 1 | 0 | 0 |
| IrPNPb,7f | — | 66 | — | — |
| IrCl(BPEP–H)b,7g | — | 90 | — | — |
| [Cp*Ir(Pro)Cl] b,7h | — | 95 | — | — |
| Tang's catalystb,7i | — | 88 | — | 9 |
Aniline (1 mmol), benzyl alcohol (2 mmol), catalyst (0.02 g) (the ratio of catalyst to aniline is 0.054 (g g−1)), without base and solvent, and reaction was carried out at 110 °C for 24 h. Composition of reaction mixture and conversion were determined by GC-MS.
Previous reports.
In order to optimize the catalytic conditions of the GIrNC, a series of catalytic reactions were carried out based on different catalyst loading ratios, and the effect of the catalyst loading on the selectivity of N-alkylation was studied. The ratio of catalyst to amine is in the range from 0.0025 to 0.185 (g mmol−1). The reaction was carried out in a Schlenk tube at 110 °C without base and solvent for 24 h. The composition of reaction mixture was determined by GC-MS and summarized in Fig. 5. For all the reactions, aniline has been completely converted. Under high catalyst loading ratio (0.185 g mmol−1), the dialkylated product (N,N-dibenzylaniline) is the main product (>95%) and the monoalkylated product (N-benzylaniline) is the minor product (∼4%), and trace amounts imine (<1%) was observed. In contrast, at a low loading ratio (0.005 g mmol−1), the monoalkylated product became the main product (99%), the imine concentration was low (1%), and no dialkylated product was detected; therefore, the selectivity of these reactions could be adjusted by adjusting the loading ratio of GIrNC.
Fig. 5. Composition distribution of nitrogen-containing products in the N-alkylation reaction of aniline and benzyl alcohol under various ratios of catalyst to aniline.

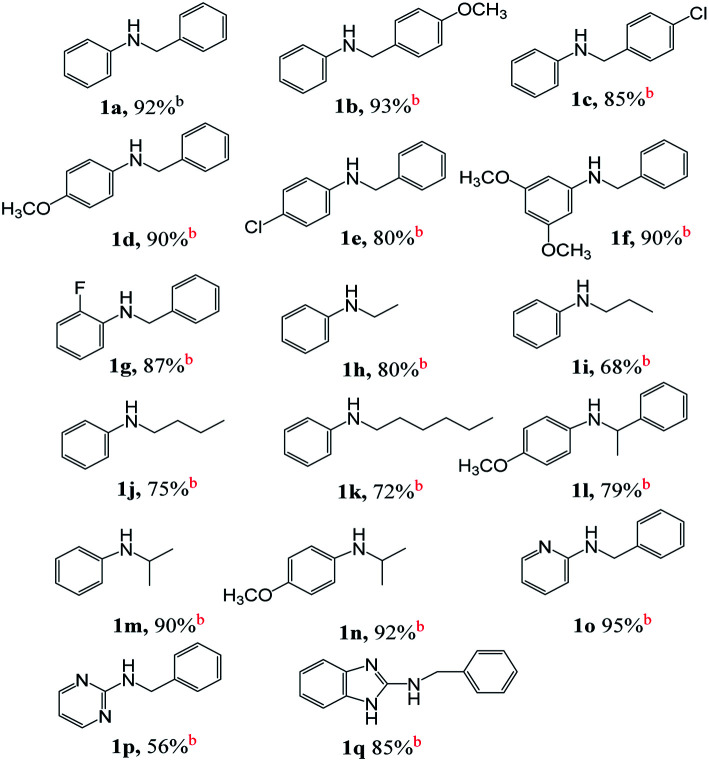
Substrate scope
On the basis of optimized conditions, we studied the substrate scope for the catalytic hydrogen autotransfer reaction of amine and alcohol coupling (Table 2). In the case of aniline alkylation, with benzyl alcohol, p-anisyl alcohol and 4-chlorobenzyl alcohol as alkylating agents, the isolated yields of secondary amine products 1a, 1b and 1c are 92%, 93% and 85% respectively. Substituted anilines, such as p-anisidine, 4-chloroaniline, and 5-aminoresorcinol dimethyl ether are also well alkylated by benzyl alcohol to obtain the corresponding secondary amines 1d, 1e, and 1f in high yields of 90%, 80% and 90%, respectively. Amines with ortho electron-withdrawing group can also be alkylated smoothly, for example, 2-fluoroaniline can be effectively converted to (2-fluorophenyl)benzylamine in great yield of 1g (87%).
GIrNC-catalyzed N-alkylation of amines using alcoholsa.
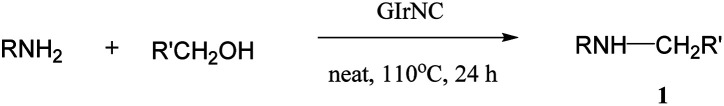
|
|---|
|
Amines (1 mmol), alcohols (2 mmol), catalyst (0.005 g), without base and solvent, and reaction was carried out at 110 °C for 24 h.
Isolated yields.
In order to evaluate whether our catalyst can expand the scope for N-alkylation of amines by aliphatic alcohols, we screened a variety of such substrates. Cheerly, aliphatic alcohols successfully alkylated amines with good selectivity and yields, for example, using ethanol, propanol, butanol and hexanol as alkylating agents for N-alkylation of aniline, the yields of secondary amine products 1h, 1i, 1j and 1k are 80%, 68%, 75% and 72%, respectively.
Because the formation of C–N bonds between nitrogen and secondary carbon is an important step in the construction of some natural products or drugs, for example, brucine, cinnarizine, hydroxyzine and ofloxacin, we tested the alkylation of amine by using secondary aliphatic alcohols as alkylating reagent to construct this kind of bonding. To our delight, both aromatic and aliphatic amines can be coupled with secondary aliphatic alcohols. Most of those reactions have a high conversion with good to excellent yield. For example, 4-methoxyaniline is alkylated with secondary benzylic alcohol (1-phenylethanol), the conversion rate is 90%, and the corresponding product 1l is obtained in yield of 79%. Secondary aliphatic alcohols also successfully alkylated aromatic amines, for example, aniline and 4-methoxyaniline were smoothly alkylated by iso-propanol to obtain the corresponding products 1m and 1n with yields of 90% and 92%, respectively.
We also found excellent scope and functional group tolerance. For example, the coupling yields of benzyl alcohol with aminopyridine, aminopyrimidine, and 2-aminobenzimidazole (products 1o–1q) are 95%, 56%, and 85%, respectively. The synthesis of 1o–1q shows efficient access to building blocks used in antihistamine and antitumor drugs, including tripelennamine, mepyramine, chloropyramine, and MSX-122.
Another feature of this reaction is that it is possible to use two different alcohols to achieve one-pot unsymmetrical N,N-dialkylation of amines with significant selectivity. For example, aniline was reacted with one equivalent of benzyl alcohol to provide monoalkylation. Subsequently, a different primary alcohol was introduced, which provides the unsymmetrical N,N-dialkylated products 2a, 2b, and 2c in high isolated yields of 82%, 80% and 86%, respectively (Scheme 2).
Scheme 2. GIrNC-catalyzed unsymmetrical N,N-dialkylation of aniline using alcohols.

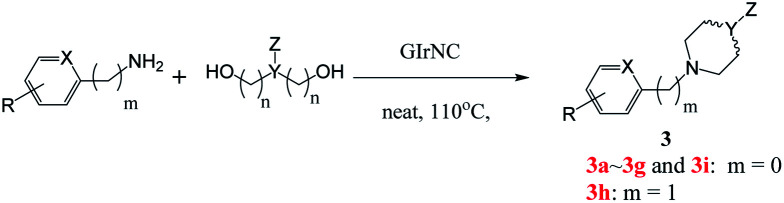
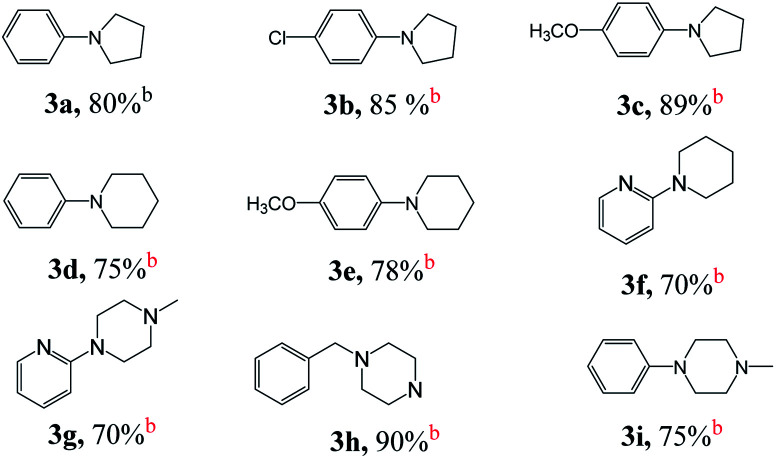
Next, we tested the intermolecular cyclization through the reaction of amine and diol. Aniline, 4-chloroaniline and 4-methoxyaniline react with 1,4-butanediol to form a cyclized five-membered cyclic amines 3a, 3b, and 3c in 80%, 85%, and 89% yields, respectively (Table 3). The reaction of 1,5-pentanediol with different amines gives six-membered cyclized products in good yields (3d–3e). Interestingly, by using diethanolamine or N-substituted diethanolamine, piperazine derivatives were obtained (3g–3i). Among them, pyridylpiperazine derivatives (3g) are known to be effective and selective α2-adrenergic receptor antagonists, and benzylpiperazine (BZP) (3h) is a recreational drug with euphoric properties. The phenylpiperazine derivative (3i) is characterized by the phenyl group attached to the piperazine ring. Many phenylpiperazine derivatives are medicines, such as antrafenine, bifeprunox, ciprofloxacin, dropropizine and elopiprazole.
GIrNC-catalyzed intermolecular cyclization of amines using diola.
|
|---|
|
Amines (1 mmol), alcohols (2 mmol), catalyst (0.005 g), without base and solvent, and reaction was carried out at 110 °C for 24 h.
Isolated yields.
The above results indicate that the GIrNC has good catalytic activity and C–N bond formation selectivity. The following two examples illustrate the advantages of this catalyst in organic synthesis.
Synthetic applications
First, cyclizine was successfully synthesized in a one-step reaction through intermolecular cyclization promoted by GIrNC catalysis. Cyclizine has been included in the World Health Organization Essential Medicines List for the treatment and prevention of nausea, vomiting and dizziness caused by motion sickness or dizziness.12Scheme 3a shows three previous reports used to prepare cyclizine. Both 3aI (ref. 13) and 3aII (ref. 14) routes require at least four steps, with total yields of 30% and 44%, respectively. Many harmful and expensive chemicals (such as formaldehyde, alkyl halide, and reducing agents) must be used, and toxic and ecologically unfavorable substances (such as hydrogen halides and solvents) are released into the environment. Recently, a more environmentally friendly method using a ruthenium complex catalyst has been found (3aIII),15 but the yield is very low (about 10%).
Scheme 3. Synthesis of cyclizine: (a) previous reports, (b) our case.

Our case (Scheme 3b) uses an intermolecular cyclization reaction to prepare cyclizine through one-step catalysis. In the reactor, N-methyldiethanolamine (MDEA) was mixed with diphenylamine and the catalyst GIrNC, and the reaction was carried out at 110 °C under nitrogen. After 24 hours, the reaction mixture was cooled, the catalyst was filtered off, and the filtrate was purified by column chromatography. The isolated yield of cyclizine is 60%, which is a very good yield compared with known methods.
Secondly, under the catalysis of GIrNC, hydroxyzine was also successfully obtained through a tandem reaction involving intermolecular cyclization and N-alkylation. Hydroxyzine has some C–N bonds, which are usually established through more complicated processes in traditional methods. Hydroxyzine is a first-generation antihistamine that belongs to the diphenylmethane and piperazine classes. Because it can bind to certain receptors in the brain, it is used as a powerful anti-anxiety drug and a mild anti-obsessive-compulsive disorder drug to treat mental anxiety and tension. Due to its antihistamine effect, it can also treat severe itching and hyperalgesia, and can even be used as a drug to relieve symptoms of opioid withdrawal; in addition, hydroxyzine derivatives are also research objects for the treatment of viral infections.16 So far, there are many methods for producing hydroxyzine, but most of them are stoichiometric reactions through a long production process. Scheme 4a is a patented process for industrial production of hydroxyzine, which includes at least three main steps. It requires the use of a variety of materials, including solvents, acids, bases, organic halides and other toxic chemicals. In addition, the harmful by-products produced during the manufacturing process require special equipment to handle, and multi-step synthesis pathways and side reactions lead to increased product costs.17
Scheme 4. Synthesis of hydroxyzine: (a) previous report, (b) our case.

Our case (Scheme 4b) provides a method for the preparation of hydroxyzine through a one-pot reaction in tandem catalysis. In a reactor, aminodiphenylmethane was mixed with diethanolamine (DEA) and the catalyst GIrNC, and the reaction was carried out at 110 °C under a nitrogen atmosphere for 24 hours. Then, diethylene glycol (DEG) was added to the above reaction mixture, and reacted again at 110 °C for 24 hours under nitrogen. Then, the reaction mixture was cooled, the catalyst was filtered off, and the filtrate was purified by column chromatography. The isolated yield of hydroxyzine obtained is 55%, which is a moderate yield for one-pot synthesis. The only by-product is water, no solvent, alkali or other additives are needed, and the catalyst can be recycled.
Reaction mechanism exploration
In order to gain insight into the N-alkylation process, we conducted a deuterium labelling experiment. The product distribution was analyzed by GCMS and 1H NMR spectroscopy, and is shown in Scheme 5. When the isotope-rich RND2 reacted with R′CH2OD, only one product labelled with deuterium on nitrogen (RNDCH2R′) was observed, while no deuterium label was found on the carbon. When RND2 reacts with R′CH2OH or RNH2 with R′CH2OD, about 50% of the products are labelled with deuterium on the nitrogen, about 50% of the products are not labelled with deuterium, and no products labelled with deuterium on the carbon are observed. The deuterium labelling experiments showed that N–H and O–H hydrogens were not transferred to the carbon atoms of the product, implying that only the carbon–hydrogen bonds of the alcohols are involved in autotransfer (HA) or hydrogen borrowing (HB) reactions.
Scheme 5. Deuterium labelling experiment and the product composition.

In addition, the progress of the N-alkylation reaction of aniline was also obtained to evaluate the change in the composition of the reaction mixture over time (Fig. 6), showing that the percentage of N-alkylation product increased sharply in the early stages, and intermediate imine was also found. After half an hour of reaction, the proportion of N-alkylated products still steadily increased, but the proportion of imines decreased significantly, indicating that the intermediate imine was rapidly converted to N-alkylated products. Which indicates that the intermediate imine accumulates at an early stage, but the reduction rate of imine is faster than the formation of imine, and also indicates that GIrNC has a higher catalytic activity for hydrogen autotransfer.
Fig. 6. Reaction progress of N-alkylation of aniline. Aniline (1 mmol) react with benzyl alcohol (2 mmol), and catalyst (0.005 g) without base and solvent; composition distribution of nitrogen-containing products were determined by GC-MS.

In previous reports, most of them provided a reaction mechanism to describe the hydrogen autotransfer or hydrogen borrowing reaction as Scheme 6.18 It consists of three steps: (a) dehydrogenation of alcohol to form carbonyl compound; (b) amine reacts with carbonyl compound to form imine; (c) reduction of imine to produce the N-alkylation product. However, this reaction mechanism cannot fully explain some experimental phenomena, such as the change in the composition of the reaction mixture during the reaction and how the reaction of the secondary amine proceeds.
Scheme 6. Reaction mechanism proposed in previous reports.

Fig. 7 shows the composition of reaction mixture when aniline reacts with benzyl alcohol (the molar ratio of amine to benzyl alcohol in the reactant is 1 : 2). Fig. 7a shows the early stage of the reaction. At this time, aniline and benzyl alcohol began to disappear, and imines and N-alkylated products were gradually formed. Only trace amounts (<0.1%) of benzaldehyde were observed. Fig. 7b shows the later stages of the reaction. The aniline disappeared completely, forming a large amount of N-alkylated products and a small amount (<1%) of imine. There was still an excess of benzyl alcohol and no benzaldehyde was observed. According to the previous reaction mechanism, accumulation of benzaldehyde should occur after aniline disappears, but benzaldehyde does not accumulate, and the residual amount of benzyl alcohol remains unchanged. Obviously, the formation of imines is controlled by the amount of aniline rather than the amount of benzaldehyde. We would rather say that imine is converted to benzaldehyde rather than benzaldehyde is converted to imine.
Fig. 7. The composition of the reaction mixture when aniline reacts with benzyl alcohol (the molar ratio of amine to benzyl alcohol in the reactant is 1 : 2): (a) at 1 h of reaction time; (b) at 24 h of reaction time.

For the reaction of secondary amines with carbonyl compounds, imines cannot be formed, but if the secondary amines contain α-hydrogens and react under harsh reaction conditions, enamines may be formed. However, for secondary amines without α-hydrogens, neither imine nor enamine can be formed. Fig. 8a shows the composition of the reaction mixture when the secondary amine piperazine reacts with benzyl alcohol (the molar ratio of piperazine to benzyl alcohol in the reactant is 1 : 3). During the reaction, no benzaldehyde and enamine were observed, only dibenzylpiperazine (DBZP) appears, and the remaining benzyl alcohol will not be converted to benzaldehyde.
Fig. 8. The composition of the reaction mixture: (a) piperazine reacts with benzyl alcohol (the molar ratio of piperazine to benzyl alcohol in the reactant is 1 : 3); (b) aniline reacts with 1,5-pentanediol (the molar ratio of aniline to 1,5-pentanediol in the reactant is 1 : 2).

Fig. 8b shows the intermolecular cyclization. An intermediate produced by the reaction of aniline and 1,5-pentanediol was observed (RT = 18.86). Cyclic amine (RT = 15.03) produced by intramolecular cyclization of intermediate was also observed. Theoretically, three aldehydes, one imine, and one enamine can be formed in the reaction mixture; however, none of them has been observed, which means that the imine or enamine tends to bind to the catalyst rather than release from the catalyst surface.
Fig. 9 shows the reaction of benzaldehyde with aniline, where 2 mmol of aldehyde is reacted with 1 mmol of aniline. At 1 min of reaction, trace amount of aniline was observed (Fig. 9a), the aniline was rapidly consumed and disappeared after 5 min of reaction. Excess aldehyde was still observed at 1 h (Fig. 9b) and 2 h (Fig. 9c) of the reaction, implying that the aldehyde is stable under the reaction conditions.
Fig. 9. Reaction of benzaldehyde and aniline (the molar ratio of benzaldehyde to aniline in the reactants is 2 : 1): (a) reaction for 1 minute; (b) reaction for 1 hour; (c) reaction for 2 hours.

Fig. 10 shows the reaction of benzyl alcohol with imine, where 1 mmol of aldehyde is reacted with 1 mmol of imine. Dissociation of the imine resulted in the formation of aldehyde (2.90%) and aniline (2.86%) at 1 hour of reaction (Fig. 10a). After 8 hours of reaction (Fig. 10b), the concentrations of aldehyde and aniline were 2.72% and 2.75%, respectively, indicating that the imine would dissociate and reach equilibrium under the reaction conditions.
Fig. 10. Reaction of benzyl alcohol and imine (the molar ratio of benzyl alcohol to imine in the reactants is 1 : 1): (a) reaction for 1 hour; (b) reaction for 8 hours.

Based on the previous reports15 and preliminary mechanistic studies, a plausible mechanism is proposed for the N-alkylation of amines using alcohols (Fig. 11). In the first step (step a) of the catalytic cycle, alcohol is absorbed by the GIrNC to form intermediate II. Then, the amine attacks the positively polarized carbon of the alcohol of the intermediate II, causing the hydride shift to form an iridium-hydride complex (III) (step b). If a primary amine is used, protons are transferred from nitrogen to oxygen (step c). Then the Lewis base (the lone pair of electrons of the amine or alcohol) captures the nitrogen protons of the intermediate IV and releases water to form the imine–iridium-hydride complex V (step d). Sometimes, imines may be released from complex V (step f), and the imines will be hydrolyzed to form aldehydes. In most of case, reductive elimination of complex V occurs, the catalyst GIrNC is regenerated and the N-alkylation product is obtained (step e). If a secondary amine is used, the lone pair of electrons on the nitrogen of complex VI will push the water away, forming an imine ion–iridium-hydride complex or enamine–iridium-hydride complex (VII) (step h). Finally, the reductive elimination of complex VII occurs, the catalyst GIrNC is regenerated and the N-alkylation product is obtained (step i).
Fig. 11. Proposed mechanism for the catalytic C–N bond formation using alcohols.

Reliability of GIrNC in catalytic cycle
Ideally, heterogeneous catalysts can be used in a continuous production process, and the product can easily be separated from the reaction mixtures. Therefore, we studied the catalyst reuse of GIrNC for the N-alkylation of aniline in five catalytic cycles without a regeneration step to evaluate the activity and stability of GIrNC in the long-term catalytic process. For each cycle, 1 mmol amine was mixture with 2 mmol alcohol and 0.005 g catalyst GIrNC. Reaction was carried out at 110 °C for 24 h, the recycled catalyst was isolated by centrifuged, and used for the next round. The yield of N-benzylaniline in each catalytic cycle is shown in Fig. 12. The yield of the first cycle is 95.9%. The yield of second cycle is 89.3%, decreased by 6.6%. Fortunately, after the second cycle, the yields remained between 89.4% and 89.7%, and the yield of the fifth cycle is 89.5%, indicating that the catalyst is quite stable under the reaction conditions. Because the catalytic ability of the catalyst remains almost unchanged after the second cycle, it can be seen that the catalyst will not encounter the problem of deactivation.
Fig. 12. Production yield of each catalytic cycle for the N-alkylation of aniline reacted with benzyl alcohol to produce N-benzylaniline.

To check the structural stability of catalyst during catalysis, Raman spectra of original and reused catalysts were acquired for inspecting the structural characteristics of catalyst. The Raman spectrum of original catalyst shows a broad D band peak (the vibration of carbon atoms with sp3 electronic configuration) at 1343 cm−1 and a G band peak (in-plane vibration of sp2-bonded carbon atoms) at 1583 cm−1 (Fig. 13). During five catalytic cycles, the location of D band and G band peaks of the catalyst remain unchanged. The integrated peak area ratio of the D to G band (ID/IG) of the original catalyst is 1.15, and the ID/IG value of catalyst after one run is 1.08, showing the ID/IG value changes slightly after one run. The ID/IG value of catalyst after the 3rd run is the same as the catalyst after the 5th run (1.07) and there is no significant difference between the catalyst after the first run. The changes in the full width at half maximum (FWHM) of the D band of original catalyst, and the catalysts after the 1st, 3rd, and 5th runs are 93, 93, 92 and 92 cm−1, respectively, showing that there is no significant change in the FWHM of the D band during the catalysis process. Through the Raman spectroscopy analysis of the original catalyst and the reused catalyst, we can see that although the catalyst has a slight structural change after the first run, the structural characteristics of the catalyst remain almost unchanged since then.
Fig. 13. The Raman spectra of original and reused catalysts: (a) original catalyst; (b) the catalysts of the first cycle; (c) the catalysts of the third cycle; (d) the catalysts of the fifth cycle.

The Ir 4f XPS of original catalyst shows two characteristic peaks, the Ir 4f7/2 peak is centered at 61.06 eV and the Ir 4f5/2 peak is centered at 64.16 eV (Fig. 14a). For the reused catalyst (Fig. 14b–d), the position, relative intensity and peak shape of the Ir 4f7/2 and Ir 4f5/2 peaks are the same as the original catalyst, showing the oxidation state of iridium remains unchanged, and the bonding mode of iridium on graphene is stable. We also checked the atomic percentage of iridium in the catalyst. The newly prepared catalyst has an iridium atomic ratio of 3.2%. The atomic ratio of iridium in the catalysts of the first, third and fifth cycles are 2.9%. 2.8% and 2.8% respectively, which can explain why the yield of N-alkylation products decreased by 6.6% from the first cycle to the second cycle, but remained almost unchanged after the second cycle. We can infer that GIrNC is stable, but a few iridium atoms are loosely bound and may leave the catalyst surface in the first run; however, most of the catalytically active sites are firmly fixed on the graphene surface and remain active for a long time.
Fig. 14. Ir 4f XPS spectra of catalyst: (a) original catalyst; (b) the catalysts of the first cycle; (c) the catalysts of the third cycle; (d) the catalysts of the fifth cycle.

Experimental
Materials and methods
Iridium chloride (IrCl3, anhydrous) was obtained from the Seedchem Co. Crystalline graphite was purchased from SHOWA Co. All other chemicals including ethanol were purchased from Acros and used as received. Aqueous solutions were prepared with double-distilled water from a Millipore system (>18 MΩ cm). For thin layer chromatography (TLC), silica aluminium foils with fluorescent indicator 254 nm (from Merck) were used. Column chromatography was performed using SD Fine silica gel 60–120 mesh using a gradient of dichloromethane and hexane as mobile phase. High-resolution mass spectra were recorded on a Waters QTOF mass spectrometer. UV-vis spectra were obtained using a Hitachi U-3900 spectrophotometer. The infrared spectra were recorded on Agilent Technologies Model Cary 630 FTIR instruments. Mass spectra were taken with a Finnigan/Thermo Quest MAT 95XL instrument with electron impact ionization for organic compounds. Transmission electron microscopy (TEM) images were carried out on a JEOL JEM-ARM200FTH microscopy operated at 80 kV with cold field emission gun (CFEG), spherical-aberration corrector, and high angle annual dark field detector. For the TEM resolution: point image resolution is 0.19 nm, lattice image resolution is 0.10 nm, information limit is 0.10 nm, bright-field lattice image resolution is 0.136 nm, and dark-field lattice image resolution is 0.08 nm, respectively. Energy-dispersive X-ray spectroscopy (EDS) was also performed on the TEM. Raman spectra (RAMaker Raman spectrometer) with an excitation laser of 532 nm were also used to characterize the samples. 1H NMR and 13C NMR spectra were recorded on a 600 MHz Agilent Technologies DD2 FT-NMR spectrometer. NMR shifts are reported as delta (δ (ppm)=) units in parts per million (ppm) and coupling constants (J) are reported in hertz (Hz).
Preparation of catalysts GIrNC, GCuNC, GNiNC, and GCoNC
A solution prepared by dissolving 3 mmol of metal salts (IrCl3, CuCl2, NiCl2, or CoCl2) in 250 ml of mixed solvent (ethyl ethanol : water = 1 : 1, v/v) was added to the 500 ml of GO solution (2 mg ml−1). The mixture solution was stirred at room temperature for 0.5 h and ultrasonicated for 0.5 h, and then the mixture was refluxed for 48 hours under argon. The obtained graphene oxide–metal complex dispersion was purified by filtration and washing with DI water and ethanol.
General procedure for N-alkylation reaction
(a) General procedure for variety of aromatic amines and primary alcohol, 1 mmol of amine was mixed with 2 mmol of alcohol in the presence of catalyst GINC (0.005 g for primary amines, 0.03 g for secondary amines and for heteroaromatic amines) in a Schlenk tube without solvent or base, and the reaction was heated to 110 °C for 24 h, and monitored by GCMS spectroscopy. The residue was purified by column chromatography using dichloromethane/n-hexane (20–100%) or methanol/dichloromethane (3–10%) as eluent to afford pure products. The desired coupling products were fully characterized by 1H, 13C NMR, and MS spectroscopies. (b) General procedure for variety of aliphatic amines and primary alcohol, 1 mmol of amine was mixed with 2 mmol of alcohol in the presence of catalyst GINC (0.005 g for primary amines, 0.03 g for secondary amines) in a Schlenk tube without solvent or base, and the reaction was heated to 110 °C for 24 h, and monitored by GCMS spectroscopy. The residue was purified by column chromatography using dichloromethane/n-hexane (5–80%) as eluent to afford pure products. The desired coupling products were fully characterized by 1H, 13C NMR, and MS spectroscopies. (c) General procedure for variety of amines and secondary alcohols, 1 mmol of amine was mixed with 2 mmol of alcohol and 0.05 g of catalyst GINC in a Schlenk tube without solvent or base, and the reaction was heated to 110 °C for 24 h, and monitored by GCMS spectroscopy. The residue was purified by column chromatography using dichloromethane/n-hexane (10–100%) as eluent to afford pure products. The desired coupling products were fully characterized by 1H, 13C NMR, and MS spectroscopies.
Catalyst reuse studies
1 mmol aniline was mixture with 2 mmol benzyl alcohol and 0.005 g catalyst GINC in a Schlenk tube. Reaction was carried out at 110 °C for 24 h, after cooling, 5 ml dichloromethane added to the reaction mixture and the mixture was centrifuged to separate out the catalyst, the clean supernatant was analyzed by GC-MS to identify the product composition. And then, 1 mmol aniline and 2 mmol benzyl alcohol was added to a Schlenk tube containing the recovered GINC for the next catalytic cycle.
Intermolecular cyclyzation to synthesize cyclizine
In a Schlenk tube, 1 mmol of 2-diphenylamine was mixed with 2 mmol of N-methyldiethanolamine (MDEA) and 0.05 g of catalyst GIrNC, and the reaction was carried at 110 °C for 48 h under nitrogen. Then, the reaction mixture was cooled down, the residue was purified by column chromatography using triethylamine/n-hexane/dichloromethane (1 : 40 : 60, volume ratio) and triethylamine/methanol/dichloromethane (1 : 10 : 90, volume ratio) as eluent to afford pure products; 60% isolated yield of cyclizine was obtained. The desired products were fully characterized by 1H, 13C NMR, and MS spectroscopies.
Tandem catalysis to synthesize hydroxyzine
In a Schlenk tube, 1 mmol of aminodiphenylmethane was mixed with 2 mmol of diethanolamine and 0.05 g of catalyst GIrNC, and the reaction was carried at 110 °C for 48 h under nitrogen. Then, 2 mmol of diethylene glycol was added to the above reaction mixture, the reaction was performed again at 110 °C for 48 h under nitrogen. Then, the reaction mixture was cooled down, the residue was purified by column chromatography using dichloromethane/n-hexane (50%) and methanol/dichloromethane (10%) as eluent to afford pure products; 60% isolated yield of hydroxyzine was obtained (calculated based on aminodiphenylmethane). The desired products were fully characterized by 1H, 13C NMR, and MS spectroscopies.
Conclusions
In conclusion, we have successfully demonstrated an unprecedented iridium/graphene nanostructured catalyst for the effective and direct coupling of amines and alcohols to form CN bonds. The reactions can be carried out under neat conditions, without pre-activation steps, and without solvents, alkalis and other additives. Both aromatic and aliphatic amines can be alkylated, and both aromatic and aliphatic alcohols can be used as alkylating agents with excellent selectivity and yield. The asymmetric N,N-dialkylation of amines using alcohols is achieved in a one-pot synthesis. Advantageously, various functional groups such as halides, alkoxy, hydroxyl, and heteroaromatic groups can be tolerated in this catalytic protocol. Interestingly, the use of diols resulted in the formation of five- and six-membered cyclic amines and piperazine derivatives, which are analogous to various drugs and biologically active molecules. This catalytic method also proved its feasibility in drug synthesis, for example, under the catalysis of GIrNC, cyclizine was prepared through a one-step reaction of intermolecular cyclyzation, and hydroxyzine was obtained through a tandem reaction. This synthetic strategy can inspire the development of new C–N bond formation reactions and sustainable transformations.
Conflicts of interest
There are no conflicts to declare.
Supplementary Material
Acknowledgments
This work was supported by Ministry of Science and Technology (110-2113-M-153 -002). The authors would like to thank Ms Yin-Mei Chang (Instrument Center, NTHU) for TEM analysis and thank the Instrument Center of National Chung Hsing University for help with measurements of Nuclear Magnetic Resonance and LC-MS/MS. We are grateful to thank Ms Swee-Lan Cheah (Instrument Center, NTHU) for HRXPS analysis.
Electronic supplementary information (ESI) available: Details of characterization of products, gas chromatogram of purified compounds, 1H NMR spectra, mass spectra and high-MS data. See DOI: 10.1039/d1ra09052f
References
- Yang Y.-S. Shen Z.-L. Loh T.-P. Org. Lett. 2009;11:1209–1212. doi: 10.1021/ol8027362. [DOI] [PubMed] [Google Scholar]
- Blieck R. Bahri J. Taillefer M. Monnier F. Org. Lett. 2016;18:1482–1485. doi: 10.1021/acs.orglett.6b00452. [DOI] [PubMed] [Google Scholar]
- Lee D.-H. Taher A. Hossain S. Jin M.-J. Org. Lett. 2011;13:5540–5543. doi: 10.1021/ol202177k. [DOI] [PubMed] [Google Scholar]
- Zhao X. She Y. Fang K. Li G. J. Org. Chem. 2017;82:1024–1033. doi: 10.1021/acs.joc.6b02595. [DOI] [PubMed] [Google Scholar]
- (a) Zhang G. Yin Z. Zheng S. Org. Lett. 2016;18:300–303. doi: 10.1021/acs.orglett.5b03461. [DOI] [PubMed] [Google Scholar]; (b) Subaramanian M. Midya S. P. Ramar P. M. Balaraman E. Org. Lett. 2019;21:8899–8903. doi: 10.1021/acs.orglett.9b02990. [DOI] [PubMed] [Google Scholar]
- (a) Huang M. Li Y. Lan X.-B. Liu J. Zhao C. Liu Y. Ke Z. Org. Biomol. Chem. 2021;19:3451–3461. doi: 10.1039/D1OB00362C. [DOI] [PubMed] [Google Scholar]; (b) Piehl P. Amuso R. Spannenberg A. Gabriele B. Neumann H. Beller M. Catal. Sci. Technol. 2021;11:2512–2517. doi: 10.1039/D0CY02210A. [DOI] [Google Scholar]; (c) Shan S. P. Xiaoke X. Gnanaprakasam B. Dang T. T. Ramalingam B. Huynh H. V. Seayad A. M. RSC Adv. 2015;5:4434–4442. doi: 10.1039/C4RA15398G. [DOI] [Google Scholar]
- (a) Feng X. Huang M. Polyhedron. 2021;205:115289. doi: 10.1016/j.poly.2021.115289. [DOI] [Google Scholar]; (b) Luo N. Zhong Y. Shui H. Luo R. J. Org. Chem. 2021;86(21):15509–15521. doi: 10.1021/acs.joc.1c01930. [DOI] [PubMed] [Google Scholar]; (c) Yuan K. Jiang F. Sahli Z. Achard M. Roisnel T. Bruneau C. Angew. Chem., Int. Ed. 2012;51:8876–8880. doi: 10.1002/anie.201204582. [DOI] [PubMed] [Google Scholar]; (d) Zou Q. Wang C. Smith J. Xue D. Xiao J. Chem.–Eur. J. 2015;21:9656–9661. doi: 10.1002/chem.201501109. [DOI] [PubMed] [Google Scholar]; (e) Kawahara R. Fujita K. Yamaguchi R. J. Am. Chem. Soc. 2010;132:15108–15111. doi: 10.1021/ja107274w. [DOI] [PubMed] [Google Scholar]; (f) Vellakkaran M. Singh K. Banerjee D. ACS Catal. 2017;7:8152–8158. doi: 10.1021/acscatal.7b02817. [DOI] [Google Scholar]; (g) Chang Y.-H. Nakajima Y. Ozawa F. Organometallics. 2013;32:2210–2215. doi: 10.1021/om4000743. [DOI] [Google Scholar]; (h) Wetzel A. W€ockel S. Schelwies M. Brinks M. K. Rominger F. Hofmann P. Limbach M. Org. Lett. 2013;15:266–269. doi: 10.1021/ol303075h. [DOI] [PubMed] [Google Scholar]; (i) Luo N.-H. Zhong Y.-H. Wen H.-L. Luo R. ACS Omega. 2020;5:27723–27732. doi: 10.1021/acsomega.0c04192. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Prabha D. Pachisia S. Gupta R. Inorg. Chem. Front. 2021;8:1599–1609. doi: 10.1039/D0QI01374A. [DOI] [Google Scholar]; (b) Wu D. Bu Q. Guo C. Dai B. Mol. Catal. 2021;503:111415. doi: 10.1016/j.mcat.2021.111415. [DOI] [Google Scholar]; (c) Quintard A. Rodriguez J. ChemSusChem. 2016;9:28–30. doi: 10.1002/cssc.201501460. [DOI] [PubMed] [Google Scholar]; (d) Santoro F. Psaro R. Ravasio N. Zaccheria F. RSC Adv. 2014;4:2596–2600. doi: 10.1039/C3RA44364G. [DOI] [Google Scholar]; (e) Likhar P. R. Arundhathi R. Kantam M. L. Prathima P. S. Eur. J. Org. Chem. 2009;31:5383–5389. doi: 10.1002/ejoc.200900628. [DOI] [Google Scholar]; (f) Yan T. Feringa B. L. Barta K. ACS Catal. 2016;6:381–388. doi: 10.1021/acscatal.5b02160. [DOI] [Google Scholar]
- (a) Chen T.-R. Wang Y.-X. Lee W.-J. Chen K. H.-C. Chen J.-D. Nanotechnology. 2020;31:285705. doi: 10.1088/1361-6528/ab814d. [DOI] [PubMed] [Google Scholar]; (b) Chen T.-R. Lin Y.-S. Wang Y.-X. Lee W.-J. Chen K. H.-C. Chen J.-D. RSC Adv. 2020;10:4436–4445. doi: 10.1039/C9RA10294A. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (a) Zhang Q. Zheng H. Geng Z. Jiang S. Ge J. Fan K. Duan S. Chen Y. Wang X. Luo Y. J. Am. Chem. Soc. 2013;13:12468–12469. doi: 10.1021/ja407110r. [DOI] [PubMed] [Google Scholar]; (b) Zhang H. Bhat V. V. Gallego N. C. Contescu C. I. ACS Appl. Mater. Interfaces. 2012;4:3239–3243. doi: 10.1021/am300593k. [DOI] [PubMed] [Google Scholar]
- (a) Chen X. Chen B. Environ. Sci. Technol. 2015;49:6181–6189. doi: 10.1021/es5054946. [DOI] [PubMed] [Google Scholar]; (b) Lee M. Yang S. Kim K.-J. Kim S. Lee H. J. Phys. Chem. C. 2014;118:1142–1147. doi: 10.1021/jp411682b. [DOI] [Google Scholar]
- Clubiey M. Henson T. Peck A. W. Riddington C. Br. J. Clin. Pharmacol. 1977;4:652. doi: 10.1111/j.1365-2125.1977.tb00824.x. [DOI] [PubMed] [Google Scholar]
- Afanasyev O. I. Kuchuk E. Usanov D. L. Chusov D. Chem. Rev. 2019;119:11857–11911. doi: 10.1021/acs.chemrev.9b00383. [DOI] [PubMed] [Google Scholar]
- Brown A. R. Rees D. C. Rankovic Z. Morphy J. R. J. Am. Chem. Soc. 1997;119:3288–3295. doi: 10.1021/ja963829f. [DOI] [Google Scholar]
- Abbenhuis R. A. T. M. Boersma J. van Koten G. J. Org. Chem. 1998;63:4282–4290. doi: 10.1021/jo972260d. [DOI] [Google Scholar]
- (a) He S. Xiao J. Dulcey A. E. Lin B. Rolt A. Hu Z. Hu X. Wang A. Q. Xu X. Southall N. Ferrer M. Zheng W. Liang T. J. Marugan J. J. J. Med. Chem. 2016;59:841–853. doi: 10.1021/acs.jmedchem.5b00752. [DOI] [PMC free article] [PubMed] [Google Scholar]; (b) Baker J. D. Uhrich R. L. Strovas T. J. Saxton A. D. Kraemer B. C. ACS Chem. Neurosci. 2020;11:2277–2285. doi: 10.1021/acschemneuro.0c00214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Morren H. and Brussels F., US Pat., 2,899,436, Aug. 11, 1959
- (a) Dobereiner G. E. Crabtree R. H. Chem. Rev. 2010;110:681–703. doi: 10.1021/cr900202j. [DOI] [PubMed] [Google Scholar]; (b) Crabtree R. H. Organometallics. 2011;30:17–19. doi: 10.1021/om1009439. [DOI] [Google Scholar]; (c) Hamid M. H. S. A. Slatford P. A. Williams J. M. J. Adv. Synth. Catal. 2007;349:1555–1575. doi: 10.1002/adsc.200600638. [DOI] [Google Scholar]; (d) Nixon T. D. Whittlesey M. K. Williams J. M. J. Dalton Trans. 2009:753–762. doi: 10.1039/B813383B. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.