

Abstract
Classic MHC-I presentation relies on shuttling cytosolic peptides into the endoplasmic reticulum (ER) by the transporter associated with antigen processing (TAP). Viruses disable TAP to block MHC-I presentation and evade cytotoxic CD8+ T cells. Priming CD8+ T cells against these viruses is thought to rely solely on cross-presentation by uninfected TAP-functional dendritic cells (DCs). We found that protective CD8+ T cells could be mobilized during viral infection even when TAP was absent in all hematopoietic cells. TAP blockade depleted the endosomal recycling compartment (ERC) of MHC-I and as such impaired Toll-like receptor-regulated cross-presentation. Instead, MHC-I accumulated in ER-Golgi intermediate compartments (ERGIC), sequestered away from Toll-like receptor control, and coopted ER-SNARE Sec22b-mediated vesicular traffic to intersect with internalized antigen and rescue cross-presentation. Thus, when classic MHC-I presentation and ERC-dependent cross-presentation are impaired in DCs, cell-autonomous non-canonical cross-presentation relying on ERGIC-derived MHC-I counters TAP dysfunction to nevertheless mediate CD8+ T cell priming.
Keywords: MHC class I, cross-presentation, Toll-like receptor, CD8+ T cells, dendritic cells, TAP, virus, autoimmunity
Introduction
Major histocompatibility complex class I (MHC-I) molecules present peptides to CD8+ T cells1,2. TAP mediates cytosol-to-ER transfer of peptides generated by the proteasome and derived from cellular proteins or viruses and bacteria that infect the cells1,2. TAP is a central component of an ER-resident macromolecular peptide-loading complex (PLC) comprised of tapasin, calreticulin, and ERp573,4,5. Peptide-loaded MHC-I exit the ER to the ERGIC where they undergo quality control for conformational flexibility and folding1,6. In resting murine DCs, fully assembled MHC-I do not colocalize with calreticulin, TAP or the ERGIC-resident lectin ERGIC-53, consistent with successful export to the plasma membrane7,8,9. In DCs, MHC-I are also present in the endosomal recycling compartment (ERC)6,9,10.
The proteasome-TAP pathway is considered to be the conventional processing route for classic MHC-I presentation. Given the crucial role of TAP in translocating peptides to nascent MHC-I within the ER, clinically important human viruses such as Herpesviridae and Poxviridae have evolved to block TAP and evade CD8+ T cells4,5. When TAP is blocked, cells utilize alternative processing pathways to liberate peptides for MHC-I loading11,12,13. Cross-presentation also critically compensates for impairment in TAP-dependent MHC-I presentation14. Cross-presentation enables MHC-I presentation of peptides derived from extracellular sources including internalized viruses and infected dying cells6,14,15,16. It mobilizes CD8+ T cells against viruses that spare or functionally compromise antigen-presenting cells (APCs)14,17. Cross-presentation research has centered on pathways and sites of peptide processing14. We have investigated the regulation of cross-presentation by signals from Toll-like receptors (TLRs), which detect microbial structures and alert the immune system6,9. We identified a role for the ERC in supplying critical MHC-I numbers for cross-presentation during infection9. We delineated TLR-controlled vesicular ERC-to-phagosome trafficking that delivers ERC-resident MHC-I specifically to phagosomes carrying microbial ligands9. Our work showed control of MHC-I trafficking at the core of the regulation of cross-presentation.
The current paradigm holds that TAP is necessary for priming antiviral CD8+ T cells14. When tissues or hematopoietic APCs are infected, antiviral CD8+ T cells can be primed through cross-presentation by uninfected TAP-sufficient DCs that present peptides derived from phagocytosed infected dying cells17,18,19,20,21. However, CD8+ T cells primed to TAP-dependent peptides are mismatched to the viral peptides liberated by alternative TAP-independent processing and presented by MHC-I on infected tissues11,14. How does the immune system get around this problem? Here we report that a CD8+ T cell response could be generated during a respiratory viral infection in mice despite TAP deficiency in all hematopoietic cells. The TAP-independent CD8+ T cell response was even protective against a lethal viral infection. Mechanistically, blocking TAP in DCs altered sub-cellular MHC-I localization from the ERC –critical for TLR-regulated cross-presentation– to the ERGIC. Despite depletion of the ERC of MHC-I, TAP-deficient DCs nevertheless continued to conduct cross-presentation, but coopt ER-SNARE Sec22b-mediated vesicular traffic to deliver MHC-I from their new location in the ERGIC to phagosomes carrying internalized antigen. We refer to this cross-presentation as non-canonical because it relies on the ERGIC as the source of MHC-I molecules instead of the ERC. Although no longer regulated by TLR signaling, non-canonical cross-presentation rescued MHC-I presentation during viral TAP blockade and ensured CD8+ T cell priming.
Results
Altered MHC-I localization in DCs without functional TAP
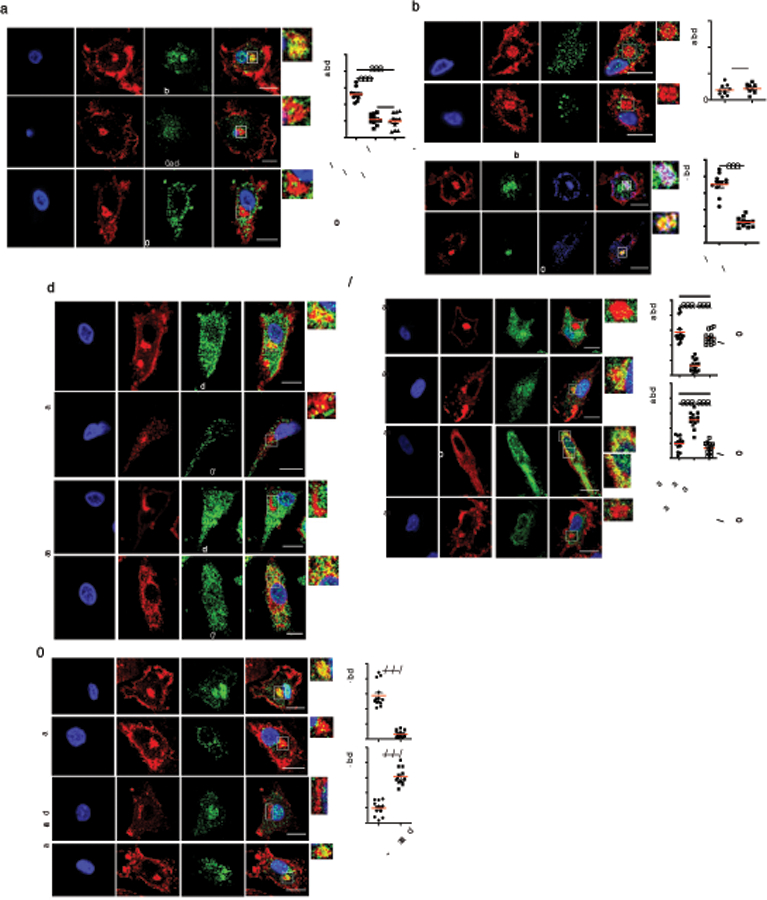
In TAP-deficient cell lines, thymic tissue and lymphocytes, MHC-I fail assembly and accumulate in the ERGIC22. Using an antibody, AF6–88.5 to detect mature peptide-loaded MHC-I molecules, we tested whether MHC-I would also be retained in the ERGIC in DCs derived from bone marrow progenitors from Tap1−/− mice. In wild-type (WT) DCs, MHC-I scarcely colocalized with the ERGIC marker, ERGIC-53, and as expected9 colocalized with two different ERC markers, cellubrevin/VAMP-3 and the small GTPase Rab11a (Fig. 1a). In Tap1−/− DCs, this pattern was reversed with MHC-I colocalizing with ERGIC-53 and only negligibly with VAMP-3 and Rab11a (Fig. 1a). ERGIC accumulation of MHC-I in Tap1−/− DCs at steady state was accompanied by reduced plasma membrane expression (Extended Data Fig. 1a,1b). Treatment of Wt DCs with Brefeldin A (BFA) phenocopied the ERGIC redistribution of MHC-I in Tap1−/− DCs (Fig. 1b), concordant with ‘disassembly’ of post-Golgi compartments and their merger with the ERGIC23. Rab11a also colocalized with MHC-I in the ERGIC upon BFA treatment (Fig. 1b).
Figure 1. MHC-I are retained in the ERGIC of murine DCs upon TAP deficiency.

a, Confocal micrographs of WT or Tap−/− resting bone marrow derived DCs (BMDCs) stained for H2-Kb (antibody clone AF6–88.5) and either ERGIC-53, VAMP-3 or Rab11a. b, Confocal micrographs of WT DCs stained for H-2Kb and either ERGIC-53 or Rab11a at resting state (n=9) or after treatment with 10µg/ml Brefeldin A (BFA). Scale bars represent 10µm. % colocalization shown for respective markers in boxed insets (Manders’ coefficient). Data represent mean ± SEM from three independent experiments, and each symbol represents a cell. ***P < 0.001; N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test.
We next reasoned that viruses interfering with TAP function to evade MHC-I presentation of endogenous viral peptides would consequently trigger a rearrangement in cellular MHC-I localization. We directly tested this hypothesis by infecting human monocyte-derived DCs (moDCs) with the clinically relevant human cytomegalovirus (HCMV) TB40/E strain, which encodes a TAP inhibitor US64. Here we made two important observations. First, concordant with our data in murine BM DCs9, human moDCs showed a large pool of intracellular MHC-I that colocalized with Rab11a and not ERGIC-resident ERGIC-53 and calreticulin (Fig. 2a and top two panels in Figs. 2d,f), or sorting endosome-resident ARF6 and EEA1 (Fig. 2b). Like in murine DCs9, the Rab11a+ pool of MHC-I in human moDCs colocalized with transferrin receptor (TfR), which can recycle through the ERC, and not with Golgi-resident giantin (Fig. 2c). Second, upon infection with HCMV, MHC-I no longer colocalized with Rab11a but with calreticulin instead (Fig. 2d, quantification in 2e), which like TAP and ERGIC-53 resides in the ERGIC9,24. Active infection of moDCs was necessary for ERGIC localization of MHC-I, as ultraviolet (UV)-inactivated HCMV did not disrupt steady state ERC localization of MHC-I (Fig. 2e). Nuclear detection of immediate early viral protein in infected and not uninfected or UV-inactivated virus treated moDCs confirmed active infection (Extended Data Fig. 2). Besides US6, HCMV encodes different effector proteins that interfere with several steps of the classic MHC-I pathway25,26. However, treatment of moDCs with a recombinant herpes simplex virus (HSV)-encoded TAP inhibitor protein ICP474 showed similar results demonstrating that TAP inhibition is specifically responsible for MHC-I accumulation in the ERGIC of HCMV-infected human moDCs (Fig. 2f). These results in murine and human DCs collectively indicate that in the absence of TAP expression or function, the plasma membrane and ERC localization of MHC-I is disrupted such that these molecules accumulate in the ERGIC instead.
Figure 2. MHC-I are retained in the ERGIC of human DCs upon TAP blockade by virus or TAP-inhibitor.
a-f, Confocal micrographs of resting human monocyte-derived DCs stained for (a) MHC-I (clone W6/32) and either Rab11a, ERGIC-53 or Calreticulin, (b) ARF6 or EEA1, and (c) Transferrin receptor or Giantin, (d, e) MHC-I and either Rab11a or ERGIC-53 post infection with untreated or UV-irradiated HCMVTB40/E MOI=10, 48hr or treatment with ICP47 peptide 100mM, 24hr (g). Scale bars represent 10µm. In (a-f), % colocalization shown for markers in boxed insets (Manders’ coefficient). Both AF6–88.5 and W6/32 recognize folded MHC-I. Data represent mean ± SEM from three independent experiments, and each symbol represents a cell. ***P < 0.001; N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test.
Cross-presentation despite ERC depletion of MHC-I upon TAP blockade
In light of our previous findings that ERC-resident MHC-I are critical for the cross-presentation of phagocytosed antigens, we predicted a dramatic consequence of altered MHC-I localization on cross-presentation by TAP deficient cells9. TAP blockade is commonly used to assess involvement of the proteasome versus vacuolar hydrolytic enzymes in generating peptides for MHC-I presentation16. Potential altered MHC-I localization had not been taken into account. We first validated a TAP-independent readout for cross-presentation by transient treatment of DCs with US6 for 2 hours to block TAP translocation of proteasome-processed peptides without eliciting the effects of prolonged TAP dysfunction on ERC depletion of MHC-I. Transient treatment with active US6 did not disrupt ERC localization of MHC-I in resting DCs over the time frame of treatment, similar to treatment with inactive US6 (Fig. 3a). However, and as expected, only active and not inactive US6 effectively blocked TAP function by impairing the classic MHC-I presentation of viral infection-derived peptide to antigen-specific CD8+ T cells (Extended Data Fig. 3a). In contrast, neither cross-presentation of ovalbumin (OVA)-derived SIINFEKL peptide from phagocytosed recombinant (OVA)-expressing bacteria (E. coli-OVA) nor cross-presentation of IYSTVASSL peptide from recombinant influenza A virus hemagglutinin (HA)-coated microspheres were affected by transient treatment with active US6 (Fig. 3b). SIINFEKL cross-presentation by US6-treated DCs reflected the ability to generate the peptide from exogenous OVA-expressing phagocytic cargo by the vacuolar protease cathepsin S12,16,27, because transient US6 treatment of Cat S−/− DCs impaired cross-presentation (Fig. 3c). Although SIINFEKL can be generated by cathepsin S when TAP function is blocked, proteasomes described to be present within the phagosome lumen can also generate peptides through a vacuolar pathway independently of TAP13. Because transient US6 treatment did not redistribute MHC-I to the ERGIC, SIINFEKL cross-presentation under these conditions validated its use as a readout of TAP-independent cross-presentation27,28 in the absence of TAP function per se and without the effect of prolonged TAP dysfunction on subcellular MHC-I localization.
Figure 3. Intact cross-presentation independently of TAP function and despite ERC depletion of MHC-I.

a, Confocal micrographs of WT resting BMDCs treated with active or inactive 0.3mg/ml US6 for 2h and stained for MHC-I and either Rab11a or ERGIC-53. Scale bars represent 10µm. b, WT BMDC cross-presentation of SIINFEKL from recombinant ovalbumin (OVA)-expressing Escherichia coli (E. coli-OVA) to OT-I CD8+ T cells, and BALB/c BMDC cross-presentation of IYSTVASSL from recombinant influenza A virus hemagglutinin (HA) conjugated to beads with lipopolysaccharide (LPS) and presented to HA-specific CL4 CD8+ T cells. BMDCs incubated for 2h with active or inactive 0.3mg/ml US6. c-d, Cross-presentation of SIINFEKL by Cathepsin S−/− (Cat S−/−) BMDCs treated for 2h with inactive or active 0.3mg/ml US6 (c) or from E. coli-OVA by WT, Tap−/−, Cat S−/−, Cat S−/−Tap−/− BMDCs (d). Scale bars represent 10µm. All BMDCs from C57BL/6J except BALB/c BMDCs for (b). Data represent mean ± SEM from three independent experiments, and each symbol represents a cell (a) or an independent experimental replicate (b, c, d). ***P < 0.001; N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test.
We next investigated how altered MHC-I localization upon TAP blockade affected cross-presentation. We noted that cross-presentation of SIINFEKL from phagocytosed E. coli-OVA was intact in Tap1−/− DCs similar to WT DCs (Fig. 3d). This was irrespective of early or late time points (Extended Data Fig. 3b) and despite the distinct subcellular localization of MHC-I in Tap1−/− DCs. Cathepsin S and TAP played crucial roles in cross-presentation because similar to US6-treated Cat S−/− DCs (Fig. 3c), cathepsin S and TAP double deficient (Cat S−/−Tap1−/−) DCs were also impaired in the cross-presentation of SIINFEKL from E. coli-OVA (Fig. 3d). Cathepsin S deficiency alone in Cat S−/− DCs did not affect cross-presentation (Fig. 3d). As expected, classic MHC-I presentation of viral peptides by virus-infected Tap1−/− DCs was impaired (Extended Data Fig. 3c). Collectively, these results were consistent with previous studies reporting intact cross-presentation of phagocytic antigen by Tap1−/− DCs13,27,29,30,31. Our data indicate that phagocytic antigen cross-presentation in Tap1−/− DCs can proceed despite depletion of the ERC of MHC-I and the accumulation of MHC-I within the ERGIC.
Cross-presentation in the absence of TAP is not subject to TLR regulation
Because ERGIC delivery to phagosomes is TLR-independent9, we reasoned that ERGIC accumulation of MHC-I in the absence of TAP expression or function would enable phagosomal MHC-I recruitment bypassing the regulation imposed by TLRs on MHC-I trafficking. Hence, we tested cross-presentation by WT and Tap1−/− DCs with or without TLR ligands while providing exogenous co-stimulation in the form of anti-CD28 to negate differences in DC costimulatory molecule expression9. For phagocytic cargo, we used either apoptotic OVA-expressing B cells that had been cultured with or without LPS, or a GFP-SIINFEKL fusion protein (GFP-OT) conjugated to beads with or without LPS. As a readout for cross-presentation, we monitored proliferation of antigen-specific CD8+ T cells as the most sensitive readout for cognate peptide-MHC-I presentation by DCs (for a comparison of cross-presentation as a readout versus IL-2 production and 25D1 antibody-based detection of SIINFEKL:H2-Kb, see supplementary Figure S1A in reference 9). Notably, unlike the TLR-regulated cross-presentation by WT DCs9, Tap1−/− DCs cross-presented phagocytic antigen to similar levels regardless of the presence of TLR ligand (Fig. 4a). Concordant with these results, and in stark contrast to WT DCs that accumulated MHC-I specifically on phagosomes carrying TLR ligand9, MHC-I in Tap1−/− DCs were enriched around phagosomes regardless of their LPS content (Fig. 4b–d). TLR-regulated phagosomal ERC recruitment was maintained in Tap1−/− DCs as it continued to deliver Rab11a specifically to phagosomes carrying beads/LPS (Fig. 4b–4d), but the ERC did not deliver MHC-I in this case because it was devoid of MHC-I in Tap1−/− DCs. At this time point, all phagosomes acquired LAMP-1 (Fig. 4b,c), Sec22b and ERGIC-53 (Fig. 4d) to similar levels regardless of TAP expression or TLR signaling. These data demonstrated a loss of TLR-regulated MHC-I recruitment to phagosomes in Tap1−/− DCs and the uncoupling of their recruitment from that of Rab11a. Combined with the altered subcellular localization of MHC-I from the ERC to the ERGIC in Tap1−/− DCs, these results suggested TLR-independent phagosomal MHC-I recruitment from the ERGIC instead of the ERC in the absence of TAP.
Figure 4. Altered subcellular MHC-I localization in the absence of TAP bypasses TLR regulation of phagocytic antigen cross-presentation.

a, Cross-presentation of SIINFEKL by WT and Tap−/− BMDCs where SIINFEKL was derived from OVA+ or OVA+LPS+ apoptotic B cells or GFPOT 3μm beads +/− conjugation to LPS, all with 0.5μg/ml anti-CD28. b-d, WT or Tap1−/− BMDCs 3h post phagocytosis of 3μm beads alone or conjugated to LPS (b/LPS). Confocal micrographs show DIC, and representative staining for H2-Kb, Rab11a and LAMP-1 for a single confocal Z-stack and collapsed H2-Kb Z projections (b), quantification of % labeled phagosomes and results represented as mean ± SEM (c). Immunoblots on 15μg of phagosomal lysate/lane, cropped to show proteins as indicated (d). e-g, WT or Trif−/−Myd88−/− BMDCs were left untreated or treated with 10μg/ml Brefeldin A (BFA). Cross-presentation of SIINFEKL derived from E. coli- OVA with 0.5μg/ml anti-CD28 (e). BMDCs were stimulated for 3h post phagocytosis of 3μm beads conjugated to LPS (beads/LPS) (f, g). Confocal micrographs showing DIC and representative staining for H2-Kb, Rab11a and LAMP-1 for a single confocal Z-stack (f), and quantification of % labeled phagosomes and results represented as mean ± SEM (g). Results represented as mean ± SEM. ** p<0.01; *** p<0.001 ****P < 0.0001, N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test. Each symbol represents an independent experimental replicate (a, e) or a cell (c, g). Scale bars represent 10µm. Data represent at least three independent experiments.
Concordant with these findings, directing ERGIC accumulation of MHC-I in Trif−/−Myd88−/− DCs with BFA treatment enabled cross-presentation of peptide from phagocytic TLR ligand+ cargo as efficiently as their untreated and BFA-treated WT DC counterparts (Fig. 4e). Accordingly, MHC-I accumulated around beads/LPS carrying phagosomes in Trif−/−Myd88−/− DCs only upon treatment with BFA and to levels comparable to those in WT DCs (Fig. 4f,g). BFA-treated Trif−/−Myd88−/− DCs also accrued Rab11a (unlike Tap1−/− DCs) along with MHC-I around phagosomes containing beads/LPS (Fig. 4f,g) as a consequence to BFA-induced ERC disassembly and collapse into the ERGIC. These data demonstrate that segregation of MHC-I out of the ERC and into the ERGIC releases its trafficking from TLR control.
Delivery of ERC-resident MHC-I to phagosomes is controlled by IKK-2 dependent phosphorylation of SNAP23 upon TLR signaling, which serves to stabilize ERC-phagosome trans-SNARE complexes and mediates ERC-phagosome fusion9. When we treated DCs with the IKK-2 inhibitor TPCA-1 to block phosphorylation of SNAP239, we found impaired Rab11a delivery to beads/LPS carrying phagosomes in both WT and Tap1−/− DCs (Fig. 5a,b). However, we noted no effect of TPCA-1 on MHC-I delivery to TLR ligand+ phagosomes in Tap1−/− DCs (Fig. 5a,) (similar levels of MHC-I in collective Z projections) or on cross-presentation by these DCs (Fig. 5c), in sharp contrast to that in WT DCs and similar to that in WT DCs treated with a control ERK inhibitor PD184352 (Fig. 5a–d). Concordantly, whereas cross-presentation from TLR ligand+ apoptotic B cells was sensitive to IKK2 inhibition in WT DCs, cross-presentation of antigen from apoptotic B cells –regardless of LPS– was refractory to IKK2 inhibition in Tap1−/− DCs (Fig. 5d). These data are consistent with the TLR-independent phagosomal MHC-I delivery and cross-presentation in Tap1−/− DCs, and indicate constitutive TLR-independent cross-presentation by DCs upon TAP blockade.
Figure 5. Non-canonical cross-presentation in the absence of TAP is independent of TLR signal-dependent phosphorylation of phagosomal SNAP-23.

a-d, WT or Tap−/− BMDCs +/− treatment with 10mM PD184352 ERK or TPCA-1 IKK-2 inhibitor. Confocal micrographs showing staining for H2-Kb, Rab11a and LAMP-1 for a single confocal Z-stack and collapsed H2-Kb Z projection (a), quantification of % labeled phagosomes and results represented as mean ± SEM (b), cross-presentation of SIINFEKL derived from E. coli-OVA by BMDCs (c), and cross-presentation of SIINFEKL from OVA+ or OVA+LPS+ apoptotic B cells (d). Results represented as mean ± SEM. ** p<0.01; *** p<0.001, N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test. Scale bars represent 10µm. Each symbol represents a cell (b) or an independent experimental replicate (c, d). Data represent at least three independent experiments.
The ERGIC and not ERC mediates cross-presentation in the absence of TAP
We next determined the relative contributions of the ERC and ERGIC to cross-presentation when subcellular MHC-I localization is altered upon TAP blockade. Disrupting the ERC by silencing Rab11a as previously described9, had no effect on phagosomal MHC-I delivery in Tap1−/− DCs, while interrupting ERGIC-to-phagosome trafficking by silencing Sec22b impaired phagosomal MHC-I delivery (Fig. 6a,b). Rab11a and Sec22b silencing had the opposite effect on WT DCs whereby MHC-I delivery to phagosomes was dependent on intact Rab11a+ ERC stores of MHC-I and unaffected by Sec22b silencing (Fig. 6a,b, Rab11a and Sec22b silencing validation by immunoblots shown in Extended Data Fig. 4a and Ref. 9). Accordingly, while cross-presentation by WT DCs was impaired by Rab11a silencing, it was unaffected in Tap1−/− DCs (Fig. 6c). Sec22b silencing, on the other hand, impaired cross-presentation by both WT and Tap1−/− DCs (Fig. 6c) due to impaired delivery of the PLC in both cases9,24, but also MHC-I from the ERGIC in Tap1−/− DCs (Fig. 6a). Together, these data demonstrate that the ERGIC accumulation of MHC-I upon TAP deficiency switches phagosomal recruitment of MHC-I from a TLR-regulated pSNAP23-dependent to a TLR-independent Sec22b-dependent event. Thus, in the absence of TAP, ERGIC-accumulated MHC-I is delivered to phagosomes via a TLR-independent, Sec22b-mediated ERGIC pathway. Because ERC-to-phagosomes and not ERGIC-to-phagosomes is normally the vesicular route that delivers MHC-I for cross-presentation, we have termed cross-presentation that relies on ERGIC-derived MHC-I molecules as non-canonical cross-presentation (Schematic summary, Supplementary Video 3).
Figure 6. Non-canonical cross-presentation is dependent on Sec22b-mediated trafficking of MHC-I to phagosomes.

a-c, WT or Tap−/− BMDC progenitors transduced with recombinant lentiviruses expressing scrambled, Rab11a or Sec22b specific shRNA. Confocal micrographs showing staining for indicated markers (a), quantification of % labeled phagosome and results represented as mean ± SEM (b), and DC cross-presentation of SIINFEKL from E. coli-OVA (c). Results represented as mean ± SEM. * p<0.05; ** p<0.01; *** p<0.001, N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test. Each symbol represents a cell (b) or an independent experimental replicate (c). Data represent at least three independent experiments.
Unregulated self-antigen cross-presentation upon TAP blockade
We reasoned that the recruitment of ERGIC-accumulated MHC-I to incoming cargo could serve to cell-autonomously counter TAP blockade in DCs and restore MHC-I presentation. To test this possibility, we generated influenza virus-infected Tap1−/− DCs as a model for a murine DC infected with a TAP-blocking virus, and challenged these infected DCs with a killed virus to engage cross-presentation. Although virus-infected Tap1−/− DCs could not activate CD8+ T cells specific to viral-derived antigen –consistent with blockade of classic MHC-I presentation– they could cross-present and stimulate CD8+ T cell proliferation upon internalization of inactivated, non-infectious SIINFEKL+ but not SIINFEKL– virus, and to levels similar to those by WT DCs (Fig. 7a). Consistent with the ERGIC accumulation of MHC-I in Tap1−/− DCs, CD8+ T cell proliferation to internalized viral antigen by infected Tap1−/− DCs was severely impaired by Sec22b silencing but not Rab11a silencing (Fig. 7b). Thus, the ERGIC pathway of MHC-I trafficking is responsible for cross-presentation in the absence of TAP. As controls, Sec22b and Rab11a silencing did not impair classic MHC-I presentation by WT DCs (Extended Data Fig. 4b). Thus, cross-presentation dependent on MHC-I traffic from the ERGIC represents a unique cell-autonomous immune response to viruses that block TAP and actively evade not only classic MHC-I presentation, but also TLR-regulated ERC-dependent cross-presentation when the ERC becomes depleted of MHC-I.
Figure 7. Non-canonical cross-presentation cell-autonomously counters TAP deficiency by presenting viral but also cellular derived antigens.

a, Classic MHC-I presentation of recombinant virus-derived SIINFEKL at 5h post-infection with recombinant SIINFEKL-expressing influenza A/PR8 virus (1º influenza-OTI, 2º No treatment) as a control for impaired classic MHC-I presentation upon TAP deficiency. SIINFEKL cross-presentation from heat inactivated influenza (2º HK influenza) or influenza-OTI (2º HK influenza-OTI). Heat-inactivated viruses cannot infect the DCs and were given to WT and Tap−/− BMDCs 3h post infection with influenza-OTI virus to engage cross-presentation of viral antigen, which was assessed 2h later. b, Same as (a) except Tap−/− BMDC progenitors were transduced with lentivirus expressing scrambled, Sec22b or Rab11a specific shRNA. c, WT and Tap−/− BMDCs were infected with influenza A/PR8 virus for 3h and then given OVA-expressing (OVA+) apoptotic cells to assess cross-presentation of SIINFEKL derived from cellular apoptotic cell OVA antigen by infected WT or Tap−/− DCs. Apoptotic cells not expressing OVA (OVA–) were controls. d, Expression of surface costimulatory molecules, CD40 and CD86 by WT and Tap−/− BMDCs at 12h post infection with influenza A virus. Results represented as mean ± SEM. ** p<0.01; *** p<0.001, N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test. Each symbol represents an independent experimental replicate. Data represent at least three independent experiments.
Because MHC-I trafficking from the ERGIC in Tap1−/− DCs is not subject to TLR regulation, we reasoned that its mobilization might come at the cost of indiscriminate cross-presentation of self and non-self antigens. We directly tested this possibility by infecting WT and Tap1−/− DCs with influenza A virus and gave these DCs an exogenous source of cellular self-antigen in the form of apoptotic cells. Interestingly, apoptotic cell antigen was not cross-presented efficiently by infected WT DCs when TLR ligands were absent within the apoptotic cell cargo (Fig. 7c). This was consistent with our previous findings that compartmentalized TLR signals from phagosomes determined antigen presentation regardless of the activation state of the DCs9,32. On the other hand, we observed apoptotic cell antigen cross-presentation selectively by infected Tap1−/− DCs from internalized OVA+ but not OVA− apoptotic cells and despite the absence of TLR ligand within apoptotic cell cargo (Fig. 7c). Apoptotic cell antigen cross-presentation occurred in a costimulatory context as Tap1−/− DCs upregulated T cell costimulatory molecule expression upon infection, and to levels similar to those expressed by WT DCs (Fig. 7d). These data indicate that while non-canonical cross-presentation counters viral TAP blockade and rescues the MHC-I presentation of viral antigens, it poses the risk of presenting self-antigens within the context of T cell co-stimulation.
CD8+ T cell immunity despite hematopoietic TAP deficiency
To test whether a CD8+ T cell response can be generated during viral infection in the absence of hematopoietic TAP expression, we reconstituted lethally irradiated WT recipient B6 mice with either TAP-sufficient or TAP-deficient bone marrow cells. Such chimeric mice had been used to demonstrate the critical role of cross-presentation by TAP-sufficient APCs to prime CD8+ T cells against tumors and against viruses lacking tropism to hematopoietic cells, precluding the presentation of viral peptides via the classic MHC-I presentation pathway in this model20,33. We infected our chimeric mice with a primary sublethal dose of influenza A/X31(H3N2) virus, and challenged 35 days later with a secondary lethal dose of influenza A/PR8(H1N1) virus in the presence or absence of CD8+ T cells (Fig. 8a). This standard heterotypic infection-challenge protocol excludes the role of surface viral hemagglutinin and neuraminidase specific antibodies in protection, and enables assessment of the role of CD8+ T cells against core viral polymerase acidic protein (PA) or nucleoprotein (NP) shared between X31 and PR8.
Figure 8. TAP deficiency in hematopoietic cells does not preclude the generation of a protective CD8+ T cell response to viral infection.

a, Schematic depicting generation of chimeric mice with a TAP-sufficient (WT→WT) or TAP-deficient (Tap−/−→WT) hematopoietic cell compartment, and their infection with a sublethal (10pfu) influenza A/X31(H3N2) followed by a heterotypic lethal (75pfu) influenza A/PR8 (H1N1) challenge on day 35 with or without depletion of CD8+ T cells by intranasal (i.n.) and intraperitoneal (i.p.) injection of control or anti-CD8α antibodies every other day. b, X31 titers in lungs of indicated mice at days 3 and 5 post X31 infection. c, % total CD8+ T cells in the blood at day 11 post X31. d-e, Representative FACS plots (d), and time course of influenza A virus-specific CD8+ T cell appearance in the blood post X31 and PR8 infection detected by H-2Dd tetramers loaded with either a polymerase acidic (PA) protein (SSLENFRAYV) or a nucleoprotein (NP) (ASNENMETM) epitopes (e). Representative FACS plot in (d) is at day 11 post X31. f, % initial body weight of mice after X31 infection. g, Confocal immunofluorescence micrographs on lung sections on days 3 and 5 post PR8 stained for CD8, CD11c, EpCAM and influenza A virus. White arrows, influenza virus-infected epithelial cells; yellow arrows, CD11c+ cells that engulfed infected epithelial cells. Scale bars represent 50μm. h, Quantification of CD8+ T cell and CD11c+ cell interactions in the lungs on days 3 and 5 post PR8 infection, upper panel and quantification of colocalization of EpCAM with influenza A virus staining in lungs on days 3 and 5 post PR8 infection, and in either control or anti-CD8α antibody treated mice, lower panel. i, % total CD8+ T cells in the blood at day 11 after PR8 infection. j, % total CD8+ T cells in the lungs at days 3 and 5 post PR8 infection. k, Representative FACS plot and % CD44+CD69+ and CD44+CD69− cells within the same CD8+ T cell gate in the lungs of infected mice day 3 post PR8 infection. l, % survival of (WT→WT) and TAP-deficient (Tap−/−→WT) mice post lethal PR8 challenge and treatment with antibody control (Ctl) or anti-CD8α antibody (anti-CD8). m-n, Percent of initial body weight post PR8 challenge. From day 8 when lethality began, % initial body weights are shown only for surviving mice representing control (WT→WT) and TAP-deficient (Tap−/−→WT) mice treated with antibody control (Ctl) (m), or as compared to those treated with anti-CD8α antibody (anti-CD8) (n). o, IFN-γ production by CD8+ T cells sorted from the lungs or the spleens of infected mice at day 10 post PR8 challenge and co-cultured for 4 days on WT or Tap−/− BMDCs infected or not with influenza A/PR8 (Flu). #: non-detected. N.S.= non statistically significant (P>0.05); *, P<0.05, **, P <0.01 and ***, P<0.001 using an unpaired two-tailed t-test (b, c, i, j, h, k, n), Log-rank (Mantel-Cox) test (l), and Mann-Whitney test (o). Results represented as mean ± SEM where each symbol represents a mouse (b, c, h, i, j, k, o), or the mean (e, f, m, n). All data represent at least 2 experiments (for a total n of 140 mice).
Lung viral titers in WT→WT and Tap−/−→WT chimeras were not significantly different post primary influenza infection (Fig. 8b), and the percentages of circulating CD8+ T cells in Tap−/− were statistically similar to those in WT chimeras (Fig. 8c). Influenza A virus-specific CD8+ T cell responses against two immunodominant TAP-dependent epitopes, from either PA or NP, peaked 10 days following primary infection in WT chimeras (Fig. 8d,e). As expected20, TAP-dependent PA and NP specific CD8+ T cells were absent in Tap−/− chimeras (Fig. 8d,e), and further demonstrated that lethal irradiation had been sufficient to eliminate recipient TAP-sufficient APCs. Despite the absence of a TAP-dependent CD8+ T cell response, weight loss in infected Tap−/− was similar to WT chimeras whereby both groups lost ≈10% of their initial body weight 7–8 days post primary infection, and gradually recovered over the next week without lethality and with statistically similar recovery kinetics (Fig. 8f). Therefore, hematopoietic TAP deficiency and the absence of an immunodominant TAP-dependent CD8+ T cell response did not render mice susceptible to sublethal infection with influenza virus.
Following lethal challenge with influenza A/PR8, we noted similar levels of viral infection of bronchiolar epithelial cells in WT and Tap−/− chimeras (Fig. 8g, white arrows, and quantification in Fig. 8h), CD11c+ cells labeled with influenza (Fig. 8g, yellow arrows, Supplementary Videos 2 and 3) indicating either direct infection or engulfment of dying infected epithelial cells, and similar numbers of CD8+ T cell interactions with CD11c+ cells (Fig. 8g and quantification in Fig. 8h). The percentages of total CD8+ T cells in the blood and lungs of Tap−/− chimeras were not significantly different from those in WT chimeras (Figs. 8i,j). Mirroring the CD8+ T cell response in the blood (Fig. 8d,e), only WT and not Tap−/− chimeras showed TAP-dependent virus-specific CD8+ T cells in the lungs following secondary viral infection (Extended Data Fig. 5a). After infection, the lungs of Tap−/− chimeras harbored an average 50% increase of activated CD44+ CD8+ T cells comparable to an average 56% increase in WT chimeras (60% in infected and 40% in naïve Tap−/− chimeras versus 78% in infected and 50% in naïve WT chimeras, Fig. 8k and Extended data Fig. 5b). Importantly, activated CD8+ T cells in Tap−/− chimeras did not label with TAP-dependent PA and NP H2-Db tetramers concordant with their TAP-independent generation (Extended Data Fig. 5a). Furthermore, the percentages of CD44+ CD8+ T cells expressing CD69, which prevents tissue egress, were similar in Tap−/− and WT chimeras (Fig. 8k). These data collectively showed an intact ability to mount CD8+ T cell responses during influenza infection despite hematopoietic TAP deficiency. Strikingly, despite lack of a TAP-dependent CD8+ T cell response (Fig. 8d,e), Tap−/− chimeras were not more susceptible than WT chimeras (82% and 83% survival, respectively, Fig. 8l, solid lines), whereby the majority of these mice lost ≈10% of their body weight 7–8 days post challenge and gradually began to recover thereafter with weight loss and recovery rates statistically similar to WT chimeras (Fig. 8m), and similar to weight loss following primary viral infection (Fig. 8f).
To determine the role of CD8+ T cells in protection against lethal viral challenge, we depleted CD8+ T cells from both the circulation and the lungs with an intranasal and intraperitoneal injection of anti-CD8α antibody (Fig. 8j and Extended Data Fig. 4c). Viral infection of bronchiolar epithelial cells (Fig. 8g and Extended Data Fig. 6a, quantification in Fig. 8h), lung viral titers (Extended Data Fig. 6b), and lung pathology scores and patchy inflammation (Extended Data Figs. 6c,d, respectively) were all similar in WT and Tap−/− chimeras post viral challenge irrespective of CD8+ T cell depletion. Notably, depletion of CD8+ T cells resulted in a statistically significant drop in the survival rates of both Tap−/− and WT chimeras with 69% and 67%, respectively, succumbing to lethal viral challenge in the absence of CD8+ T cells (Fig. 8l). Among surviving mice, CD8+ T cell depletion led to an additional 10% weight loss (≈20% on average around days 9–11 post viral challenge) in both WT and Tap−/− chimeras compared to CD8+ T cell-sufficient control antibody treated chimeras (≈10% on average) (Fig. 8n). These results demonstrated that protection of previously infected Tap−/− chimeras from lethal challenge with a heterotypic strain influenza virus was dependent on TAP-independent CD8+ T cells.
To probe the specificity of TAP-independent CD8+ T cells, we co-cultured CD8+ T cells isolated from the lungs and spleens of infected chimeras with either influenza A virus infected or uninfected DCs. We detected IFN-γ production when CD8+ T cells were re-activated by infected and not uninfected DCs showing that the CD8+ T cells generated after infection in both chimeras were specific to peptides presented by DCs only after viral infection (Fig. 8o). CD8+ T cells from infected WT chimeras produced IFN-γ when co-cultured with infected WT but not Tap−/− DCs in accordance with the expectation that the immunodominant CD8+ T cell response is directed against TAP-dependent epitopes (Fig. 8o). CD8+ T cells from Tap−/− chimeras produced IFN-γ when cocultured with infected Tap−/− DCs validating the specificity of these CD8+ T cells to TAP-independent epitopes presented by Tap−/− DCs upon infection (Fig. 8o). Notably, these TAP-independent CD8+ T cells also produced IFN-γ in response to infected WT DCs indicating the ability of primed TAP-independent effector CD8+ T cells to detect TAP-independent epitopes, whose presentation would be lower under TAP-sufficient conditions compared to TAP-dependent epitopes11. Altogether, these data show that the TAP-independent CD8+ T cell response contains CD8+ T cells specific to TAP-independent epitopes generated upon viral infection.
Discussion
The current paradigm holds that a productive CD8+ T cell response is severely impaired in the absence of TAP-sufficient DCs. We have discovered a novel cross-presentation pathway in DCs that we call non-canonical cross-presentation, which overcomes TAP blockade and primes TAP-independent CD8+ T cells. Our work reveals three new facets to MHC-I presentation upon TAP dysfunction. First, the absence of TAP function alters subcellular MHC-I localization in DCs from the plasma membrane and Rab11a+ ERC to the ERGIC. Second, by blocking TAP function, viruses severely compromise not only classic MHC-I presentation, but also ERC-dependent cross-presentation because of depletion of the ERC pools of MHC-I. Third, and contrary to expectation, TAP blockade in DCs does not abrogate their ability to prime CD8+ T cells. When both classic MHC-I presentation and ERC-dependent cross-presentation are impaired due to TAP blockade, DCs mobilize Sec22b-dependent MHC-I trafficking to phagosomes from the ERGIC instead of the ERC to rescue cross-presentation. Our findings imply that DCs infected with a virus that blocks TAP can also participate in priming CD8 T cells through non-canonical cross-presentation.
Despite the advantages to antiviral immunity, we show that cross-presentation during TAP blockade comes at the cost of bypassing the control that TLRs exert over cross-presentation. It is tempting to speculate whether the symptoms of chronic inflammation presented in individuals with TAP deficiency syndrome34, and perhaps cases of autoimmunity reported following HCMV infections35, might be triggered by unregulated cross-presentation of apoptotic cell antigens in the absence of functional TAP. Interestingly, downregulation of TAP transcripts has been observed in several MHC-I-linked autoimmune diseases including type I diabetes mellitus (T1DM), Sjögren’s syndrome, Graves’ disease, and Hashimoto’s disease36, and whether this disrupts the regulatory control of self versus non-self antigen cross-presentation is an important question for future investigations.
By generating bone marrow chimeric mice where all hematopoietic cells are deficient for TAP, we gained insight into the physiological relevance of CD8+ T cells primed by DCs lacking TAP function. TAP-deficient mice have greatly reduced numbers of CD8+ T cells because of the central role of MHC-I presentation in thymic CD8 T cell selection37. This is not the case in Tap−/− chimeric mice which retain TAP-sufficient radioresistant and non-hematopoietic thymic epithelial cells. Tap−/− chimeras were not particularly susceptible to a lethal infection with influenza A virus and recovered from infection similarly to WT chimeras, and despite their inability to mount the immunodominant TAP-dependent CD8+ T cell response. These findings were surprising in light of the fact that all DCs within these mice lacked TAP, including DCs that are infected directly with influenza virus as well as the bystander uninfected DCs reported to conduct cross-presentation because they retain TAP function17,18,19,20,21.
Despite the absence of TAP-dependent CD8+ T cells in Tap−/− chimeras, their protection against a lethal challenge of influenza virus was nevertheless dependent on CD8+ T cells. Indeed, systemic depletion of CD8+ T cells significantly compromised the ability of Tap−/− chimeras to survive the infection, and in effect phenocopied the previously reported susceptibility of Tap−/− mice, which lack CD8+ T cells38, to infection with the intracellular pathogen Mycobacterium tuberculosis39.
Our in vitro studies showed that virus-infected TAP-deficient DCs could retain the ability to cross-present exogenous viral-derived antigen through Sec22b-dependent and Rab11a-independent non-canonical cross-presentation. Sec22b-mediated vesicular traffic from the ERGIC normally delivers components of the MHC-I PLC, critical for cross-presentation, to antigen-containing phagosomes and endosomes9,40. The role of Sec22b in cross-presentation has been questioned41 because its importance was first demonstrated by shRNA-mediated knockdown of Sec22b in DCs24. Nevertheless, different contexts using both shRNA and DC-specific genetic deletion, and using multiple parallel cell biological and biochemical readouts of Sec22b-mediated ERGIC communication with phagosomes and its consequences, have supported initial shRNA-mediated knockdown results9,24,40. Future investigations will be important to address the role of Sec22b in mediating TAP-independent antiviral CD8+ T cell responses.
The ability of APCs to present antigen independently of TAP function has previously been reported under conditions predicted to enable high levels of antigen presentation such as infection with high doses of influenza virus or lymphocytic choriomeningitis virus (LCMV), which replicates well in mice27,42. This contrasts with CD8+ T cell responses to other viruses that do not replicate as extensively in mice such as vaccinia and polio viruses, or to less immunogenic tumor-derived antigens where TAP expression by hematopoietic cells was deemed necessary for cross-priming20,33. Our data suggest that during a viral infection, a CD8+ T cell response could potentially be primed not only through cross-presentation by uninfected TAP-sufficient DCs but also cross-presentation by infected TAP-disabled DCs. CD8+ T cells from WT chimeras did not recognize infected TAP-deficient DCs upon recall in vitro, suggesting that TAP-independent responses are either not primed when TAP is functional or too small to be detected at this time point by the recall assay. On the other hand, once primed, effector TAP-independent CD8+ T cells could detect their cognate ligand on infected TAP-sufficient DCs demonstrating an underlying MHC-I presentation of TAP-independent alongside TAP-dependent epitopes during infection, and at levels sufficient for detection by effector TAP-independent CD8+ T cells but not for priming CD8+ T cells. Similarly, effector TAP-independent CD8+ T cells in Tap−/− chimeras confer protection against viral infection despite the absence of TAP-dependent CD8+ T cells, suggesting their ability to detect cognate TAP-independent epitopes on the infected TAP-sufficient lung epithelium, and potentially target these cells for removal. Precise assessment of TAP-independent CD8+ T cell responses alongside TAP-dependent ones awaits development of tools to understand how they differ from TAP-dependent responses. Interestingly, approximately 15% of HLA-B haplotypes can be highly expressed on the cell surface despite TAP deficiency43. It is tempting to speculate that the Sec22b-dependent MHC-I trafficking we describe here may prime virus-specific CD8+ T cells restricted to specific HLA haplotypes under TAP-inhibited conditions.
Our findings reveal a new facet of TAP in antigen cross-presentation and offer insight into TAP-independent MHC-I presentation in viral immunity and autoimmune sequelae of viral infections. Our findings may explain why individuals with congenital TAP deficiency syndrome do not have increased susceptibility to viral infections34,44,45. TAP-independent CD8+ T cells cross-primed by TAP-deficient DCs in these individuals may be well-matched to target the TAP-independent epitopes on the surface of their virally infected cells. In a similar scenario, many human cancers downmodulate TAP and present “T cell epitopes associated with impaired peptide processing” that can be targeted for cancer immunotherapy46,47,48. In hosts bearing such tumors, DCs are TAP sufficient and as such would prime TAP-dependent CD8+ T cells mismatched to the TAP-independent epitopes displayed on tumor cells. Our work provides new avenues for therapeutic strategies to mobilize CD8+ T cell responses against immune evasive infections and cancers.
Methods
Mice
C57BL/6J, BALB/c, OT-I, clone 4 (CL4), and Tap1−/− (referred to here as Tap−/−) mice were purchased from the Jackson Laboratories. Myd88−/− and Trif−/− mice were provided by S. Akira (Osaka University). Myd88−/− and Trif−/−mice were interbred to homozygosity to generate Trif−/−Myd88−/− mice, and were obtained from R. Medzhitov (Yale University). Actb-promoter-driven, membrane bound OVA (Actin-mOVA) transgenic mice were purchased from the Jackson Laboratories and bred to BALB/c mice for 8 generations. CatS−/−, CatS−/−xTap1−/−were provided by K. Rock (University of Massachusetts). We used male and female 6–10-week-old mice. All experiments were approved by the institutional animal care and use committee at Weill Cornell Medicine and the Icahn School of Medicine at Mount Sinai, and carried out in accordance with the ‘Guide for the Care and Use of Laboratory Animals’ (NIH publication 86–23, revised 1985).
Bone marrow chimeric mice
C57BL/6J female mice were injected with 200µg of anti-NK1.1 antibody (clone PK136, BioXcell) and the next day, were lethally irradiated twice with 2 doses of 600 Rads with a 4-h interval between irradiations. The day after irradiation, mice were reconstituted with 5×106 bone marrow cells from C57BL/6J or Tap1−/− mice. Reconstituted mice were used 8 weeks after.
Infection with Influenza A virus
Virus stocks of influenza A/X31(H3N2) and influenza A/PR/8 (H1N1) were produced in embryonated eggs and diluted in PBS. For virus infections, mice were anesthetized by intraperitoneal injection of a mixture of ketamine (100 mg/kg of body weight) and xylazine (5 mg/kg) before intranasal administration of either a sublethal dose at day 0 of influenza A virus/X31(H3N2) at 10 p.f.u. (LD50= 400 p.f.u.) or a lethal dose at day 35 of influenza A virus/PR/8 (H1N1) at 75 p.f.u. (LD50= 20 p.f.u.) in a volume of 50 μL. Sublethal dose was determined by lowest dose allowing mouse infection as determined by ≈10% weight loss and by viral lung titers. Lethal dose was determined as a dose of virus that kills 100% of naïve WT mice. Mice were monitored daily for clinical signs of illness, and body weights were recorded daily for 14 days. Upon reaching 75% of initial body weight, animals were humanely euthanized.
Anti-CD8α (clone 2.43, BioXcell) or control antibody (clone LTF-2, BioXcell) were injected into isoflurane anesthetized mice both intraperitoneally and intranasally every two days from day 33.
Pathology scoring
Mice were euthanized on days 3 or 5 post-challenge with influenza A/PR8. Lungs were perfused in situ with 1 mL of 10% buffered formalin, and sections stained with hematoxylin-eosin and evaluated by a comparative pathologist. Lung histopathology was assessed on a scale of 0 to 4: 0, no lesions; 1, mild changes with scattered cell necrosis/vacuolation and few/scattered inflammatory cells in the bronchiolar epithelium with minimal perivascular inflammation; 2, moderate multifocal vacuolation with peribronchiolar inflammation (<5 cell layers thick); 3, marked, multifocal/segmental necrosis, epithelial loss/effacement, and peribronchiolar inflammation (>5 cell layers thick); and 4, severe, coalescing areas of necrosis, parenchymal effacement with confluent areas of inflammation. Overall pathology scores/group were used for statistical analysis. Focal alveolar hyperplasia unrelated to the secondary influenza infection was noted in some mice signifying resolved inflammation likely due to either first infection with Influenza A/X31 or injection of the depleting antibody.
Plaque assay
On days 3 and 5 post-infection, mice were euthanized, and lungs collected and homogenized (BeadBlaster 24; Benchmark Scientific) in 1 mL of sterile phosphate-buffered saline (PBS). Lung homogenates were spun at 16,000×g for 10 min and supernatants were collected. Samples were stored at −80°C until titration was performed by standard plaque assay on MDCK cells.
Lung Preparation for flow cytometry
Lung tissues were minced with scissors in 1.5-mL tubes, and incubated in RPMI, 5% FBS, 1.6mg/ml collagenase D (Roche), 20μg/mL DNase (Roche) for 40 min on a rocker at 37°C before homogenization using a 20g syringe. Tissue suspensions were filtered through 70μm cell strainers (BD Falcon), pelleted, and stained for FACS with antibodies: anti-CD8α (clone 53–6.7, eBioscience), anti-CD8β (clone YTS156.7.7, BioLegend), anti-CD3ε (clone 17A2, BioLegend), anti-CD44 (clone IM7, BioLegend), and anti-CD69 (clone H1.2F3, BioLegend). MHC-I tetramers H-2Db loaded with the Influenza A epitope from nucleoprotein (NP, 366–374) ASNENMETM, or acid polymerase (PA, 224–233) SSLENFRAYV (both proteasome-TAP-dependent epitopes49,50). Tetramers were provided by NIH Tetramer Core. The gating strategy is available in the Supplemental Material to the manuscript.
Tissue Preparation for Confocal Microscopy
Tissues were processed as previously described51. Briefly, tissues were fixed in paraformaldehyde (PFA), lysine, and sodium periodate buffer (PLP, 0.05M phosphate buffer, 0.1M L-lysine, pH 7.4, 2 mg/mL NaIO4, and 10mg/mL PFA) overnight at 4°C followed by 30% sucrose overnight at 4oC and subsequent embedding in OCT media. Frozen tissues were sectioned at 20μm using Leica CM3050S cryostat, and FcR blocked with anti-CD16/32 Fc-block (Clone 93, BioLegend) diluted in PBS containing 2% donkey serum, 2% fetal bovine serum (FBS), and 0.1% Triton-X for 1h at 25oC. Sections were stained with anti-CD8α (Clone 53–6.7, BD Bioscience), anti-EpCAM (clone G8.8, eBioscience), anti-Influenza A virus (polyclonal, Abcam) and anti-CD11c (clone N418, BioLegend) diluted in PBS containing 2% goat serum, 2% FBS, 0.1% Triton-X, and 0.05% Fc block for 1h at 25oC. Images were acquired using a Zeiss LSM 880 confocal microscope (Carl Zeiss) with the Zen Black software. The imaging data were processed and analyzed using Imaris software version 9.0.1 (Bitplane, Oxford Instruments).
Cells
Bone marrow derived dendritic cells (BMDCs) were generated by culturing bone marrow progenitors for six days as previously described32 in RPMI 1640 medium supplemented with 10ng/ml granulocyte-macrophage colony-stimulating factor (GM-CSF) and 5% fetal bovine serum (FBS) (Hyclone), plus 100 units/mlLpenicillin, 100mg/ml streptomycin, 2mM L-glutamine, 10mM HEPES pH 7.2–7.5, 1nM sodium pyruvate, 1% MEM nonessential amino acids, and 55mM β-mercaptoethanol (all Sigma-Aldrich). Harvest of the BMDCs was performed by gentle pipetting to get the loosely attached cells from the wells.
CD8+ T cell proliferation and activation assays
All CD8+ T cell assays were conducted as described9 and all readouts were determined by FACS acquisition and analyses. 2×105 T cells were labeled with 5μM CFSE before co-culture with 1×105 GM-CSF BMDCs in 96-well U-bottom plates for 4 days. Percentage of proliferation are reported for the total dividing CD8+ T cell precursors in the well, as previously described52. All proliferation percentages reported were pre-gated on CD8+ T cells. The gating strategy is available in the Supplemental Material to the manuscript.
Cross-presentation of phagocytic antigen
E. coli-OVA were added to 1×106 BMDCs (1DC:25 E. coli) for 4.5h. Apoptotic cells were added to DC (1DC:2 apoptotic cells) for 18h. GFPOT or HA protein was coupled to 3μm streptavidin magnetic microspheres (Polysciences) and half of beads were additionally coupled to 100μg/ml biotinylated LPS (InvivoGen). Beads were added to DCs (1 DC:2 beads) for 5h. After stimulation, DCs were fixed, washed, and co-cultured with CD8+ T cells. DCs were fixed with 0.05% PFA for 5 minutes at 22ºCfollowed by a quenching step with 0.2 M glycine in RPMI medium. DCs were then extensively washed in PBS (at least 4 times) and were allowed to sit in RPMI for 20 minutes at 22ºC to allow release of excess PFA from the cells, followed by two more washes in PBS. Finally, DCs were counted and plated at 1×105 cells per well of a round-bottom 96-well plate. CD8+ T cells were isolated from spleens and lymph nodes of OT-I or CL4 TCR transgenic mice using CD8 Microbeads (Miltenyi). T cells were plated at 2×105 cells per well in the presence or absence of anti-CD28 (0.5μg/ml).
For detailed instructions on preparation of bacteria, apoptotic cells or beads, please see supplemental methods.
Bacteria:
Recombinant E. coli expressing flagellin-ovalbumin fusion protein (E. coli-OVA), previously described32, were prepared by growing E. coli in Luria-Betrani (LB) broth to an optical density at 600 nm (OD600) of 0.6 and adding 0.2 mM Isopropyl-beta-D-thiogalactopyranoside (IPTG) (Gold biotechonology, St. Louis, MO) (for inducing ovalbumin expression) for an additional 6h. Bacteria were diluted in PBS to 0.6 OD600, and killed by heating at 60°C for 60 minutes. Heat killed bacteria were stored up to 18h at 4°C prior to adding to DCs. On day 6 of DC culture, based on an estimation of 1×106 DCs/well of a tissue culture treated 24 well plate, 25×106 heat killed E. coli-OVA were added per well and plates centrifuged at 2,000 rpm for 2 minutes.
Apoptotic cells:
B cells from the spleen of Actin-mOVA BALB/c mice were sorted using CD19 Microbeads (Miltenyi). OVA+LPS+ B cells were prepared from B cells stimulated with 25μg/mL LPS for 3 days as previously described32. OVA+ B cells were prepared by stimulation of B cells with 10μg/mL anti-mouse Ig (IgM+IgG+IgA, H+L) (Southern Biotech). All B cells were induced to undergo apoptosis by UV irradiation at 2.5mJ/cm2 for 18h. Apoptotic cells were then added to BMDCs (1DC: 2 apoptotic cells). DCs were harvested 18h post phagocytosis, and kept on ice while counted.
Beads:
3μm streptavidin magnetic microspheres (Polysciences) were andincubated with biotinylated GFPOT (gift from S. Trombetta, as previously described53) or biotinylated hemagglutinin (HA) for 1h, with constant shaking at 22ºC. Recombinant Influenza A Virus HA (A/Puerto Rico/8/1934 (H1N1)) was purchased from Sino Biological Inc. (China) and biotinylated using EZ-link Sulfo-NHS-LC-Biotin kit (Pierce).
To allow for LPS binding, ≈62 ng of GFPOT or HA protein was conjugated to 1×106 microspheres. Beads were washed four times in PBS-0.5% FCS and 100μg/ml biotinylated LPS (InvivoGen) added to half the microsphere preparation in PBS for 1h at 22ºC while gently mixing. Beads were extensively washed prior to adding to DC cultures (1DC: 2 beads) in 24 well plates.
Viral infection of BMDCs and MHC-I presentation of viral antigen
To assess classic MHC-I antigen presentation, DCs were infected with Influenza virus strain A/PR/8/1934 (H1N1) (PR8) expressing the SIINFEKL epitope of OVA antigen54 at a multiplicity of infection (MOI) = 2.5 for 5h prior to fixation and co-culture with OT-I T cells. Alternatively, WT DCs were treated with active or inactive 0.3 mg/ml US6 at the time of infection. US6 was kept in the culture for duration of infection. In indicated experiments, DCs were infected with PR8-OTI (MOI=2.5) for 3h and pulsed with heat inactivated PR8-OTI (1DC:100 viral particles) for an additional 2h. Viral particles were heat inactivated by incubating at 60°C for 40 minutes.
Re-activation ex-vivo of CD8 T cells for Interferon-γ (IFN-γ) production
WT or Tap1−/− chimeric mice were infected with influenza A/X31 and challenged at day 35 with influenza A/PR8. Spleen and lung single-cell suspensions were prepared at day 10 post challenge and CD8+ T cells were sorted with magnetic separation (Miltenyi Biotec). BMDCs from WT and Tap1−/− mice and infected or not with influenza A/PR8 for 5h at 37ºC, fixed and extensively washed as above. Sorted CD8+ T cells and BMDCs were co-cultured for 4 days and supernatants collected for IFN-γ measurements by ELISA (BioLegend).
Human DC related experiments
Generation
Human monocyte-derived DCs were obtained from healthy human blood donors (New York Blood Center), following a standard protocol as previously described55. Briefly, after Ficoll-Hypaque gradient centrifugation, CD14+ cells were isolated from the mononuclear fraction using a MACS CD14 isolation kit (Milteny Biotec). CD14+ cells were differentiated for 6 days in DC medium (RPMI supplemented with 100 units/mLpenicillin, 100μg/mL streptomycin, 2mM L-glutamine, 10mM HEPES pH 7.2–7.5, 1nM sodium pyruvate, 1% MEM nonessential amino acids and 55μM β-mercaptoethanol (all Sigma-Aldrich) in presence of 500 U/ml human granulocyte-macrophage colony-stimulated factor (GM-CSF) (R&D), 1,000 U/mL human Interleukin 4 (IL-4) (R&D) and 10% fetal bovine serum.
TAP inhibition
CMV TB40/E virus was propagated on fibroblasts and purified by density gradient centrifugation56. Infectious virus yield was assayed by 50% tissue culture infectious dose (TCID50) assay. moDCs were infected with TB40/E at a multiplicity of infection (MOI) of 10 PFU per cell in DMEM (10% FBS) for 2hat 37°C with shaking every 15 minutes and then washed twice with PBS prior to culturing for 48h. Staining for viral nuclear immediate early (IE) protein confirmed infection. For UV inactivation, virus inoculum was UV irradiated at 360mJ/cm2. DCs were treated with 100μM ICP47 peptide (kind gift from Peter Cresswell) for 24hr.
Immunofluorescence and confocal microscopy
DCs were seeded onto Alcian blue-treated glass coverslips and stimulated or not with phagocytic cargo. Streptavidin beads with or without conjugation to biotinylated LPS were added to DCs (1DC:2beads) for 3h. Coverslips were fixed and permeabilized prior to staining. Images were acquired on a Leica SP5 DM microscope with a 633/1.4 NA oil immersion objective.
Antibodies
The following antibodies (catalog numbers c#) indicated for those antibodies without clone numbers) were used for immunofluorescence and immunoblot analysis: purified rabbit anti-ERGIC-53/p58 (Sigma-Aldrich) (c# E1031), rabbit polyclonal anti-Sec22b (c# 186 003) (Synaptic Systems), goat polyclonal anti-Giantin (N-18) (Santa Cruz), mouse polyclonal anti-H2-Kb (c# ab93364), rabbit polyclonal anti-Calreticulin (EPR3924), rabbit polyclonal anti-Rab11 (c# ab3612), rabbit polyclonal anti-VAMP3/cellubrevin (c# ab43080) and rabbit polyclonal anti-ARF6 (c# ab77581) (Abcam), rabbit anti-TAP2 (#706, gift from S. Amigorena), mouse monoclonal anti-transferrin receptor (Invitrogen), mouse monoclonal anti-H2-Kb (AF6–88.5) (BD PharMingen), rat anti-mouse LAMP1 (eBio1D4B) (eBioscience), Alexa 488-labeled cholera toxin B subunit (Molecular Probes) and rabbit polyclonal anti-EEA1 (Affinity Bioreagents). Mouse monoclonal anti-human HLA-ABC antibody (W6/32) that recognizes folded MHC class I molecules was purified from hybridoma supernatants57. MAB810X clone 8B1.2 conjugated to Alexa Fluor 488 was against CMV viral nuclear immediate early (IE) (Millipore). Secondary antibodies were anti-species conjugated to Alexa 488, 594 or 647 (Molecular Probes) or peroxidase (Jackson Laboratories).
Antibodies for FACS included anti-mouse antibodies: CD8α (53–6.7), CD86 (GL-1), CD11c (N418) and CD40 (1C10) (BioLegend), H2-Kb (AF6–88.5) (BD PharMingen), CD69 (H1.2F3) and CD44 (IM7) (eBioscience).
Colocalization coefficients
Percentage of colocalization of MHC-I with organelle markers (Manders’ coefficient) in boxed insets was calculated using the JACoP plugin on ImageJ software9,58. Thresholds were fixed for each experiment. For quantification, at least 10–20 resting cells were analyzed to generate individual coefficients. Bar graphs depict M1 coefficients for MHC-I colocalization to different organelle markers.
Phagosome isolation and immunoblot analyses
Phagosomes were isolated and analyzed by immunoblot as previously described32. Briefly, DCs were pulsed with equal numbers of magnetic streptavidin microspheres either conjugated or not to LPS (100 μg/mL) (1DC:2beads) for 3h. DCs were dounce-homogenized and phagosomes containing magnetic beads isolated with magnet and resuspended in lysis buffer for immunoblots.
Lentiviral knockdown of Sec22b and Rab11a
Sec22b and Rab11a were silenced in DCs as described previously9,24. Briefly, VSV-pseudo-typed lentiviruses encoding shRNAs against Rab11a and Sec22b were generated by calcium phosphate transfection of 293T cells with third generation split packaging plasmids. Plasmids encoding these lentiviral vectors were purchased from Sigma. Recombinant lentiviruses were produced encoding the following target sequences for:
Sec22b shRNA
(5′-CCGGCCCTATTCCTTCATCGAGTTTCTCGAGAAACTCGATGAAGGAA TAGGGTTTTTG-3′)
Rab11a shRNA
(5′-CCGGAGTAGGTGCCTTATTGGTTTACTCGAGTAAACCAATAAGGCACCTACTTTTTTG-3′)
as well as a scrambled control
(5′-CCTAAGGTTAAGTCGCCCTCGCTCGAGCGAGGGCGACTTAACCTTAGG-3′)
Validation of Sec22b and Rab11a specific shRNAs were described previously9,24. Validation included (1) verification of knockdowns of Sec22b and Rab11a (but not closely related Rab11b) transcripts in DCs derived from progenitors that had been lentivirally transduced with those shRNAs, (2) functional validation that the shRNAs were associated with predicted outcomes such as the disruption of the IFN-β response in DCs after E. coli phagocytosis, which relies on ERC stores of TLR4 being recruited to phagosomes, as previously described59, and (3) dissolution of the ERC upon knockdown of Rab11a (ERC formation relies on Rab11a activity), and impaired cross-presentation upon knockdown of Sec22b due to impaired delivery of MHC-I peptide loading complex components, as described24.
DC progenitors were transduced with recombinant lentiviruses to silence Rab11a and Sec22b and were subsequently differentiated into DCs.
Immunoblots
BMDCs were harvested, washed in cold PBS, and lysed in 50mM Tris-HCl (pH 7.9), 300mM NaCl, 1% Triton X-100, supplemented with protease inhibitor and phosphatase inhibitor cocktails (respectively Complete Protease and Phostop, Roche). Whole cell extracts were cleared by centrifugation at 14,000 rpm for 10 min at 4°C. Protein concentrations were determined using the Bradford method. Samples were denatured in Laemmli buffer for SDS-PAGE resolution. Proteins were transferred onto a PVDF membrane (Millipore). Membranes were blocked with 7% evaporated milk in PBS 0.2% Tween and were incubated with primary antibodies and peroxidase-conjugated secondary antibodies (all diluted in PBS 7% milk 0.2% Tween). Bound antibodies visualized using Amersham™ ECL or Pierce® ECL2, and imaged using developing machine or Amersham™ Imager 600 (GE Healthcare). Anti-Sec22b was from Santa Cruz (sc-101267), anti-β-Actin was from Cell Signaling (3700).
Treatment with inhibitors
DCs were pretreated for 1h at 37°C with the following inhibitors (all resuspended in DMSO): ERK inhibitor PD184352 (10 μM), IKK-2 inhibitor TPCA-1 (10 μM) as well as piceatannol (25 μM). For soluble TAP inhibitor US6, DCs were treated with 0.3 mg/ml of inactive or active US6 at the time of addition of the antigen.
Preparation of US6
The active soluble domain of US6 (amino acids 20–146) and the further truncated, inactive US6 (amino acids 20–125) were recombinantly produced in E. coli grown in LB broth. Inclusion bodies were collected and solubilized in 8M urea, 20mM TRIS-HCl pH 7.5, 150mM NaCl. The protein solution was subjected to Ni-NTA-agarose (Qiagen), washing with 8M urea, 20mM TRIS-HCl pH 7.5, 150mM NaCl, 20mM imidazole and eluted with 6M urea, 20mM TRIS-HCl pH 7.5, 150mM NaCl, 200mM imidazole. The eluate was dialyzed in 20mM sodium phosphate buffer, pH 6.5, leading to precipitation of protein. Precipitate was then collected and stored in 20mM sodium phosphate buffer, pH 6.5, on ice. Precipitates were then resolubilized in 8M urea, 20mM TRIS-HCl pH 7.5, 150mM NaCl.
Statistical analyses
Statistical significance was determined by the two-sided unpaired Student’s t test (except when indicated) and analysis of variance. *p < 0.05, **p < 0.01, ***p < 0.001, N.S (not statistically significant) p > 0.05, asterisk symbols denote not detected.
Data and materials availability
Source Data and uncropped immunoblot images are included in the Supplemental Material to the manuscript. All other data supporting the findings of the paper are available from the corresponding author upon reasonable request.
Extended Data
Extended Data Fig. 1. Steady state residual expression of MHC-I at the plasma membrane of Tap–/– DC.

a, Confocal micrographs of WT or Tap−/− resting DCs stained for cholera toxin B subunit (CTB), H2-Kb and ERGIC-53. b, FACS analyses of WT or Tap−/− DCs for surface H2-Kb. Scale bars represent 10 µm. Data represent at least three independent experiments.
Extended Data Fig. 2. HCMV protein expression is detected only in DC treated with live and not UV-inactivated virus.
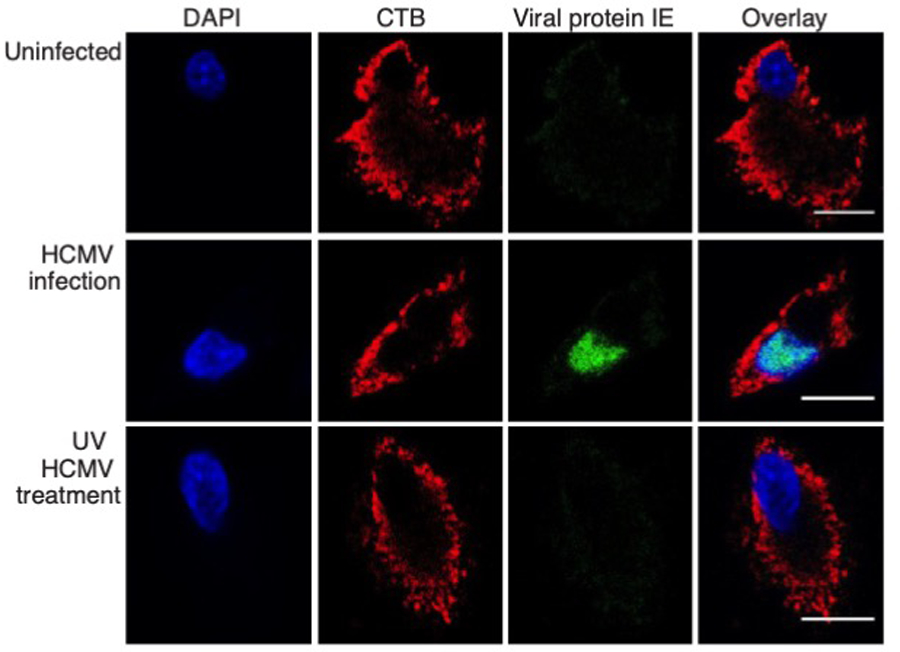
Confocal micrographs of human DCs stained for immediate early (IE) viral protein along with CTB, 48h post infection with either HCMVTB40/E or its UV-irradiated counterpart at a MOI=10. Scale bars represent 10 µm. Data represent at least three independent experiments.
Extended Data Fig. 3. Impaired TAP function blocks classic MHC-I presentation but not cross-presentation.

a, Classic MHC-I presentation by WT DCs of SIINFEKL derived from influenza PR/8-OTI virus to OVA-specific CD8+ T cells. DCs were infected with influenza PR/8-OT-I virus for 5h and incubated in the presence of active or inactive US6. b, c, WT and Tap−/− DC cross-presentation of SIINFEKL from E. coli-OVA at several time points (b), or at 4.5h post-phagocytosis of either E. coli-OVA or 5h post-infection with influenza PR/8-OTI virus (c). Data represent at least three independent experiments except for (b) depicting two independent experiments. The mean ± SEM are presented and each symbol represents biological replicate. **P < 0.01; N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test.
Extended Data Fig. 4. Classic MHC-I presentation of viral antigen by Wt DC is not affected by Sec22b or Rab11a inhibition.

a, Immunoblots for the expression of β-actin, Sec22b and Rab11a on whole cell extracts prepared from DCs seven days post-differentiation of bone marrow progenitors that had been transduced with lentiviruses encoding for control shRNA or shRNA targeting either Rab11a or Sec22b. Immunoblots were cropped to show indicated proteins. b, WT DC progenitors were transduced with recombinant lentiviruses expressing scrambled, Sec22b or Rab11a specific shRNA. Classic MHC-I presentation of SIINFEKL at 5h after infection with recombinant SIINFEKL-expressing Influenza-OTI (PR/8-OTI) (left panels, no secondary treatment) or cross-presentation of SIINFEKL from heat-inactivated Influenza-OTI given to DCs at 3h following infection by Influenza-OTI virus. Cross-presentation was assessed 2h later. Data represent at least three independent experiments. The mean ± SEM are presented and each symbol represents biological replicate. N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test.
Extended Data Fig. 5. Lung CD8 T cells during lethal challenge with influenza A virus.

a, Percent of influenza A specific CD8+ T cells in the lungs of WT→WT and Tap−/−→WT mice at days 3 and 5 post lethal challenge with 75 p.f.u. influenza A PR8 virus. Data show two different H-2Dd tetramers loaded with either a polymerase acidic protein epitope (SSLENFRAYV) or a nucleoprotein epitope (ASNENMETM). b, Representative FACS plot and % CD8+CD44+ cells in the lung of naïve chimeric mice. c, Flow cytometry plots showing CD8+ T cells in the lungs or spleens of WT mice on days 1 and 2 following injection with anti-CD8α antibody (clone 2.43) intranasally (i.n., 200µg), intraperitoneally (i.p., 100µg), or via both routes (i.p.+i.n.). Percent of CD3+CD8β+ T cells remaining are indicated in the blue gates. The mean ± SEM are presented and each symbol represents a mouse. N.S.= non statistically significant (P>0.05) using an unpaired two-tailed t-test.
Extended Data Fig. 6. CD8 T cells mediate protection of Tap⎯/⎯ chimeric mice against lethal challenge with influenza A virus.

a, Confocal micrographs at 20X magnification of lung sections from indicated anti-CD8α-treated mice stained for CD8α, CD11c, EpCAM and influenza A virus. Legend to the right shows the color code for each antibody. Scale bar represents 50 µm. b, Lung PR8 viral titers at days 3 and 5 in WT→WT and Tap−/−→WT chimeric mice. Each symbol is one mouse. c, Pathology scores of infected mice at different time points post lethal PR8 challenge in WT→WT and Tap−/−→WT mice. Slides were scored by a blinded pathologist for perivascular, bronchiolar or alveolar inflammation, epithelial degeneration or necrosis, and intraluminal debris or hemorrhage. Each symbol is one mouse. d, Hematoxylin-and-eosin staining of lung sections from WT→WT and Tap−/−→WT mice on days 3 or 5 post PR8 challenge. Scale bar represents 100 μm. The mean ± SEM are presented and each symbol represents a mouse. Data represent 2 experiments (for a total n of 140 mice).
Supplementary Material
Acknowledgments:
We are grateful to Virginia Gillespie for expert pathology on mouse lung tissues. We thank S. Trombetta (Boehringer Ingelheim), P. Cresswell (Yale University), Wenjing Li, T.M. Moran (Icahn School of Medicine at Mount Sinai), Dabeiba Bernal Rubiov, and Ana Fernandez-Sesma (Icahn School of Medicine at Mount Sinai) for reagents and technical advice. We are grateful to current Blander lab members and to H. Gupta, M.A. Blander and S.J. Blander for discussions and support.
Funding:
ISMMS-Microscopy Shared Resource Facility was supported by grants NIH 5R24 CA095823–04, S10 RR0 9145–01, and NSF DBI-9724504. This work was supported by NIH grants AI073899 and AI123284 to J.M.B., NIH/NIAID Center for Research on Influenza Pathogenesis contract as part of the CEIRS Network HHSN266200700010C to A.G.-S, AI101820 and AI112318 to D.T., and AI143861 to K.M.K., German Research Foundation grants SFB 807–Membrane Transport and Communication and TA157/7, and European Research Council (ERC Advanced Grant 789121) to R.T. J.M.B was also supported by NIH grants DK111862 and AI127658, the Burroughs Wellcome Fund, and the Leukemia and Lymphoma Society. G.B. was supported by a fellowship and currently career development award from the Crohn’s and Colitis Foundation. T.G. was supported by an American Heart Association pre-doctoral fellowship and NIH F32CA224438.
Footnotes
Competing interests: Authors declare no competing interests.
References
- 1.Blum JS, Wearsch PA & Cresswell P Pathways of antigen processing. Annual review of immunology 31, 443–473 (2013). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 2.Neefjes J, Jongsma ML, Paul P & Bakke O Towards a systems understanding of MHC class I and MHC class II antigen presentation. Nature reviews. Immunology 11, 823–836 (2011). [DOI] [PubMed] [Google Scholar]
- 3.Blees A et al. Structure of the human MHC-I peptide-loading complex. Nature 551, 525–528 (2017). [DOI] [PubMed] [Google Scholar]
- 4.Mayerhofer PU & Tampe R Antigen translocation machineries in adaptive immunity and viral immune evasion. Journal of molecular biology 427, 1102–1118 (2015). [DOI] [PubMed] [Google Scholar]
- 5.Praest P, Liaci AM, Forster F & Wiertz E New insights into the structure of the MHC class I peptide-loading complex and mechanisms of TAP inhibition by viral immune evasion proteins. Molecular immunology 113, 103–114 (2019). [DOI] [PubMed] [Google Scholar]
- 6.Blander JM Regulation of the Cell Biology of Antigen Cross-Presentation. Annual review of immunology 36, 717–753 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Ackerman AL, Kyritsis C, Tampe R & Cresswell P Early phagosomes in dendritic cells form a cellular compartment sufficient for cross presentation of exogenous antigens. Proceedings of the National Academy of Sciences of the United States of America 100, 12889–12894 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Delamarre L, Holcombe H & Mellman I Presentation of exogenous antigens on major histocompatibility complex (MHC) class I and MHC class II molecules is differentially regulated during dendritic cell maturation. The Journal of experimental medicine 198, 111–122 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Nair-Gupta P et al. TLR Signals Induce Phagosomal MHC-I Delivery from the Endosomal Recycling Compartment to Allow Cross-Presentation. Cell 158, 506–521 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Montealegre S, Abramova A, Manceau V, de Kanter AF & van Endert P The role of MHC class I recycling and Arf6 in cross-presentation by murine dendritic cells. Life Sci Alliance 2 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Oliveira CC & van Hall T Alternative Antigen Processing for MHC Class I: Multiple Roads Lead to Rome. Frontiers in immunology 6, 298 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 12.Rock KL & Shen L Cross-presentation: underlying mechanisms and role in immune surveillance. Immunological reviews 207, 166–183 (2005). [DOI] [PubMed] [Google Scholar]
- 13.Sengupta D, Graham M, Liu X & Cresswell P Proteasomal degradation within endocytic organelles mediates antigen cross-presentation. The EMBO journal 38, e99266 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 14.Cruz FM, Colbert JD, Merino E, Kriegsman BA & Rock KL The Biology and Underlying Mechanisms of Cross-Presentation of Exogenous Antigens on MHC-I Molecules. Annual review of immunology 35, 149–176 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 15.Heath WR & Carbone FR Cross-presentation in viral immunity and self-tolerance. Nature reviews. Immunology 1, 126–134 (2001). [DOI] [PubMed] [Google Scholar]
- 16.Joffre OP, Segura E, Savina A & Amigorena S Cross-presentation by dendritic cells. Nature reviews. Immunology 12, 557–569 (2012). [DOI] [PubMed] [Google Scholar]
- 17.Gutierrez-Martinez E et al. Cross-Presentation of Cell-Associated Antigens by MHC Class I in Dendritic Cell Subsets. Frontiers in immunology 6, 363 (2015). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 18.Helft J et al. Cross-presenting CD103+ dendritic cells are protected from influenza virus infection. The Journal of clinical investigation 122, 4037–4047 (2012). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Silvin A et al. Constitutive resistance to viral infection in human CD141(+) dendritic cells. Sci Immunol 2 (13):eaai8071 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Sigal LJ, Crotty S, Andino R & Rock KL Cytotoxic T-cell immunity to virus-infected non-haematopoietic cells requires presentation of exogenous antigen. Nature 398, 77–80 (1999). [DOI] [PubMed] [Google Scholar]
- 21.Whitney PG et al. Effective Priming of Herpes Simplex Virus-Specific CD8(+) T Cells In Vivo Does Not Require Infected Dendritic Cells. Journal of virology 92 (3):e01508–17 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Donaldson JG & Williams DB Intracellular assembly and trafficking of MHC class I molecules. Traffic 10, 1745–1752 (2009). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Nebenfuhr A, Ritzenthaler C & Robinson DG Brefeldin A: deciphering an enigmatic inhibitor of secretion. Plant physiology 130, 1102–1108 (2002). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Cebrian I et al. Sec22b regulates phagosomal maturation and antigen crosspresentation by dendritic cells. Cell 147, 1355–1368 (2011). [DOI] [PubMed] [Google Scholar]
- 25.Hewitt EW The MHC class I antigen presentation pathway: strategies for viral immune evasion. Immunology 110, 163–169 (2003). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Noriega V, Redmann V, Gardner T & Tortorella D Diverse immune evasion strategies by human cytomegalovirus. Immunologic research 54, 140–151 (2012). [DOI] [PubMed] [Google Scholar]
- 27.Shen L, Sigal LJ, Boes M & Rock KL Important role of cathepsin S in generating peptides for TAP-independent MHC class I crosspresentation in vivo. Immunity 21, 155–165 (2004). [DOI] [PubMed] [Google Scholar]
- 28.Tiwari N et al. A transporter associated with antigen-processing independent vacuolar pathway for the MHC class I-mediated presentation of endogenous transmembrane proteins. Journal of immunology 178, 7932–7942 (2007). [DOI] [PubMed] [Google Scholar]
- 29.Bertholet S et al. Leishmania antigens are presented to CD8+ T cells by a transporter associated with antigen processing-independent pathway in vitro and in vivo. Journal of immunology 177, 3525–3533 (2006). [DOI] [PubMed] [Google Scholar]
- 30.Merzougui N, Kratzer R, Saveanu L & van Endert P A proteasome-dependent, TAP-independent pathway for cross-presentation of phagocytosed antigen. EMBO reports 12, 1257–1264 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 31.Lawand M, Abramova A, Manceau V, Springer S & van Endert P TAP-Dependent and -Independent Peptide Import into Dendritic Cell Phagosomes. Journal of immunology 197, 3454–3463 (2016). [DOI] [PubMed] [Google Scholar]
- 32.Blander JM & Medzhitov R Toll-dependent selection of microbial antigens for presentation by dendritic cells. Nature 440, 808–812 (2006). [DOI] [PubMed] [Google Scholar]
- 33.Huang AY, Bruce AT, Pardoll DM & Levitsky HI In vivo cross-priming of MHC class I-restricted antigens requires the TAP transporter. Immunity 4, 349–355 (1996). [DOI] [PubMed] [Google Scholar]
- 34.Lankat-Buttgereit B & Tampe R The transporter associated with antigen processing: function and implications in human diseases. Physiological reviews 82, 187–204 (2002). [DOI] [PubMed] [Google Scholar]
- 35.Halenius A & Hengel H Human cytomegalovirus and autoimmune disease. BioMed research international 2014, 472978 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Fu Y, Yan G, Shi L & Faustman D Antigen processing and autoimmunity. Evaluation of mRNA abundance and function of HLA-linked genes. Annals of the New York Academy of Sciences 842, 138–155 (1998). [DOI] [PubMed] [Google Scholar]
- 37.Aldrich CJ et al. Positive selection of self- and alloreactive CD8+ T cells in Tap-1 mutant mice. Proceedings of the National Academy of Sciences of the United States of America 91, 6525–6528 (1994). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Van Kaer L, Ashton-Rickardt PG, Ploegh HL & Tonegawa S TAP1 mutant mice are deficient in antigen presentation, surface class I molecules, and CD4–8+ T cells. Cell 71, 1205–1214 (1992). [DOI] [PubMed] [Google Scholar]
- 39.Behar SM, Dascher CC, Grusby MJ, Wang CR & Brenner MB Susceptibility of mice deficient in CD1D or TAP1 to infection with Mycobacterium tuberculosis. The Journal of experimental medicine 189, 1973–1980 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Alloatti A et al. Critical role for Sec22b-dependent antigen cross-presentation in antitumor immunity. The Journal of experimental medicine 214, 2231–2241 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Wu SJ et al. A Critical Analysis of the Role of SNARE Protein SEC22B in Antigen Cross-Presentation. Cell Rep 19, 2645–2656 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Sigal LJ & Rock KL Bone marrow-derived antigen-presenting cells are required for the generation of cytotoxic T lymphocyte responses to viruses and use transporter associated with antigen presentation (TAP)-dependent and -independent pathways of antigen presentation. The Journal of experimental medicine 192, 1143–1150 (2000). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Geng J, Zaitouna AJ & Raghavan M Selected HLA-B allotypes are resistant to inhibition or deficiency of the transporter associated with antigen processing (TAP). PLoS Pathog 14, e1007171 (2018). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Cerundolo V & de la Salle H Description of HLA class I- and CD8-deficient patients: Insights into the function of cytotoxic T lymphocytes and NK cells in host defense. Seminars in immunology 18, 330–336 (2006). [DOI] [PubMed] [Google Scholar]
- 45.de la Salle H et al. HLA class I deficiencies due to mutations in subunit 1 of the peptide transporter TAP1. The Journal of clinical investigation 103, R9–R13 (1999). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Garrido G et al. Tumor-targeted silencing of the peptide transporter TAP induces potent antitumor immunity. Nature communications 10, 3773 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Marijt KA, Van Der Burg SH & van Hall T TEIPP peptides: exploration of unTAPped cancer antigens. Oncoimmunology 8, 1599639 (2019). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Marijt KA & van Hall T To TAP or not to TAP: alternative peptides for immunotherapy of cancer. Curr Opin Immunol 64, 15–19 (2020). [DOI] [PubMed] [Google Scholar]
Methods-only References
- 49.Uger RA & Barber BH Presentation of an influenza nucleoprotein epitope incorporated into the H-2Db signal sequence requires the transporter-associated with antigen presentation. Journal of immunology 158, 685–692 (1997). [PubMed] [Google Scholar]
- 50.Zhong W, Reche PA, Lai CC, Reinhold B & Reinherz EL Genome-wide characterization of a viral cytotoxic T lymphocyte epitope repertoire. The Journal of biological chemistry 278, 45135–45144 (2003). [DOI] [PubMed] [Google Scholar]
- 51.Perez OA et al. CD169(+) macrophages orchestrate innate immune responses by regulating bacterial localization in the spleen. Sci Immunol 2 (2017). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Suchin EJ et al. Quantifying the frequency of alloreactive T cells in vivo: new answers to an old question. Journal of immunology 166, 973–981 (2001). [DOI] [PubMed] [Google Scholar]
- 53.Drutman SB & Trombetta ES Dendritic cells continue to capture and present antigens after maturation in vivo. Journal of immunology 185, 2140–2146 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.Moltedo B, Li W, Yount JS & Moran TM Unique type I interferon responses determine the functional fate of migratory lung dendritic cells during influenza virus infection. PLoS Pathog 7, e1002345 (2011). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Rodriguez-Madoz JR et al. Inhibition of the type I interferon response in human dendritic cells by dengue virus infection requires a catalytically active NS2B3 complex. Journal of virology 84, 9760–9774 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Noriega VM et al. Human cytomegalovirus modulates monocyte-mediated innate immune responses during short-term experimental latency in vitro. Journal of virology 88, 9391–9405 (2014). [DOI] [PMC free article] [PubMed] [Google Scholar]
- 57.Parham P, Barnstable CJ & Bodmer WF Use of a monoclonal antibody (W6/32) in structural studies of HLA-A,B,C, antigens. Journal of immunology 123, 342–349 (1979). [PubMed] [Google Scholar]
- 58.Bolte S & Cordelieres FP A guided tour into subcellular colocalization analysis in light microscopy. J Microsc 224, 213–232 (2006). [DOI] [PubMed] [Google Scholar]
- 59.Husebye H et al. The Rab11a GTPase controls Toll-like receptor 4-induced activation of interferon regulatory factor-3 on phagosomes. Immunity 33, 583–596 (2010). [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.