

Summary
Tumor-associated macrophages (TAMs) promote metastasis and inhibit T cells, yet macrophages can be polarized to kill cancer cells. Macrophage polarization could thus be a strategy for controlling cancer. We show that macrophages from metastatic pleural effusions of breast cancer patients can be polarized to kill cancer cells with monophosphoryl lipid A (MPLA) and interferon γ (IFNγ). MPLA+IFNγ injected intratumorally or intraperitoneally reduces primary tumor growth and metastasis in breast cancer mouse models and suppresses metastasis and enhances chemotherapy response in an ovarian cancer model. Both macrophages and T cells are critical for the treatment’s anti-metastatic effects. MPLA+IFNγ stimulates type I IFN signaling, reprograms CD206+ TAMs to inducible NO synthase (iNOS)+ macrophages, and activates cytotoxic T cells through macrophage-secreted interleukin 12 (IL-12) and tumor necrosis factor α (TNFα). MPLA and IFNγ are used individually in clinical practice and together represent a previously unexplored approach for engaging a systemic anti-tumor immune response.
Keywords: tumor-associated macrophages, cytotoxic T cells, MPLA, IFNγ, metastasis treatment, anti-tumor immune response, breast cancer, ovarian cancer
Graphical Abstract

eTOC Blurb
Sun et al. combine monophosphoryl lipid A (MPLA) with interferon γ (IFN γ) to reprogram macrophages, activate cytotoxic T cells, suppress tumor growth and metastasis, and enhance chemotherapy response in mice. MPLA and IFNγ are used individually in clinical practice and together represents a previously unexplored approach for eliciting anti-metastatic responses.
Introduction
Cancer elicits an immune response that solid tumors can evade, in part by establishing an immunosuppressive microenvironment. The tumor microenvironment (TME) comprises tissue-resident cells (e.g., fibroblasts, endothelial cells), innate immune cells (e.g., macrophages), and adaptive immune cells (e.g., T cells) (Lim et al., 2018; Nakasone et al., 2012; Pogge von Strandmann et al., 2017). It was discovered over a century ago that intratumoral injection of dead bacteria, named “Coley’s toxin” after the physician who devised the treatment, led to durable anti-tumor responses in some patients (Wiemann and Starnes, 1994). These responses were likely caused by bacterial components activating immune cells, particularly macrophages. Yet, tumor-associated macrophages (TAMs) usually promote tumor growth and metastasis (Arwert et al., 2018; Kitamura et al., 2017; Yin et al., 2016). They also contribute to the immunosuppressive microenvironment, allowing tumors to evade intrinsic anti-tumor immune response and therapies (Cassetta and Kitamura, 2018; Peranzoni et al., 2018; Ruffell et al., 2014).
Macrophages exist on a continuum of phenotypes between classically and alternatively activated (Wynn et al., 2013). Classically activated macrophages are stimulated during acute inflammation by interferon γ (IFNγ) and toll-like receptor (TLR) ligands, including bacterially derived lipopolysaccharide (LPS). These macrophages secrete pro-inflammatory cytokines (tumor necrosis factor α [TNFα] and interleukins IL-1β, IL-2, IL-6, IL-12, and IL-23) and metabolize arginine to the “killer” molecule nitric oxide (NO) by upregulating inducible nitric oxide synthase (iNOS). Stimulating macrophages with IL-4/IL-13 leads to the alternatively activated phenotype and production of anti-inflammatory cytokines (IL-10, transforming growth factor [TGFβ]). TAMs usually exhibit traits of the alternatively activated macrophage subtype and have been clinically targeted by ablating strategies, such as anti-colony stimulating factor 1 receptor (CSF1R) therapeutics (Cannarile et al., 2017; Cassetta and Pollard, 2018). CSF1R inhibitors, e.g., PLX3397 (pexidartinib) and LY3022855, have been tested clinically. In patients with advanced tenosynovial giant cell tumor, PLX3397 is well tolerated and causes an anti-tumor response (Tap et al., 2015). However, PLX3397 is ineffective against recurrent glioblastoma (Butowski et al., 2015), and LY3022855 is ineffective against advanced, treatment-refractory breast or prostate cancer (Autio et al., 2020). Depending on context, macrophages can also eliminate cancer cells with NO, reactive oxygen species, or by phagocytosis, and they can stimulate cytotoxic T lymphocytes to kill tumors by presenting antigens and producing pro-inflammatory cytokines (Pozzi et al., 2005; Wynn et al., 2013). Since macrophages exhibit a high degree of plasticity, another strategy than macrophage ablation may be to “reprogram” TAMs to kill cancer cells and support cytotoxic T cells (Kowal et al., 2019; Pathria et al., 2019). Class IIa histone deacetylase (HDAC) inhibitors reprogram TAMs to be anti-tumor and increase chemotherapy and immunotherapy efficacy in breast cancer models (Guerriero et al., 2017). Similarly, inactivating macrophage phosphatidylinositol-3-Kinase γ (PI3Kγ) promotes an immunostimulatory transcriptional program, in turn restoring CD8+ T cell activation and increasing survival in mouse models of head and neck squamous cell carcinoma and breast carcinoma (Kaneda et al., 2016).
Reprogramming macrophages and/or directly stimulating adaptive immunity have been achieved using e.g., TLR agonists. TLRs are pattern recognition receptors that play fundamental roles in activating innate immunity (Adams, 2009). Nanoparticles loaded with the TLR7/8 agonist R848 accumulate in TAMs, polarize them to an inflammatory phenotype, and suppress colon cancer in a mouse model (Rodell et al., 2018). A TLR7/8 dual agonist conjugated to tumor-targeting antibodies elicits robust localized activation of macrophages and dendritic cells, resulting in tumor clearance and immunological memory in breast tumor models (Ackerman et al., 2021). Moreover, combining motolimod (a TLR8 agonist) with the epidermal growth factor receptor inhibitor cetuximab increases IL-6, CCL2, and CCL4 secretion and increases natural killer (NK) cell circulation and activation (Chow et al., 2017). Similarly, the TLR7 ligand imiquimod applied as a cream to breast cancer skin metastases shows anti-tumor activity and triggers a strong T-helper-1 (Th-1)/cytotoxic immune response in responding patients (Adams et al., 2012; Rozenblit et al., 2019). Yet, the therapeutic efficacy of motolimod and imiquimod is low: only 2 of 11 squamous cell carcinoma patients and 2 of 10 breast cancer patients achieved a partial response.
To improve the TLR agonists’ anti-tumor effects, we combined the TLR4 agonist monophosphoryl lipid A (MPLA) with IFNγ. MPLA is an LPS derivative; it can be chemically synthesized and displays reduced toxicity while maintaining most of LPS’s immunostimulatory activity (Mata-Haro et al., 2007). MPLA is used as an adjuvant in vaccines against cervical cancer and shingles (Lai et al., 2015; Monie et al., 2008). IFNγ is a cytokine essential for innate and adaptive immunity. It can promote macrophage activation, enhance antigen presentation, and regulate Th1/Th2 balance (Boehm et al., 1997; Schroder et al., 2004). Reprogramming TAMs in murine breast tumors depends on IFNγ (Clark et al., 2020). Furthermore, IFNγ 1b (a variant of IFNγ) is FDA-approved to treat chronic granulomatous disease and osteopetrosis (Key et al., 1992; Todd and Goa, 1992). Here, we tested if injecting MPLA with IFNγ (hereafter, “MPLA+IFNγ”) could provide a collaborative stimulus that would reprogram TAMs to become tumoricidal and activate T cells to elicit local and systemic immune responses to control metastatic breast cancer and ovarian cancer in mice.
Results
MPLA+IFNγ reprograms TAMs to be tumoricidal
We first evaluated whether TAMs could be reprogrammed to become tumoricidal. TAMs and tumor cells were isolated from the primary tumors of MMTV-PyMT mice (“PyMT” for short, a mouse model of luminal B breast cancer) (Figure S1A, S1B), co-cultured, and treated with a TLR agonist (LPS, MPLA, or polyI:C) alone or in combination with IFNγ. None of the TLR agonists alone or combined with IFNγ affected PyMT cancer cell viability in the absence of macrophages (Figure S1C). However, IFNγ together with either LPS or MPLA—both TLR4 agonists—activated TAMs, resulting in killing ~90% of cancer cells in 48 hours in the co-cultures (Figure 1A, 1B, Movies S1, S2). PolyI:C (a TLR3 agonist) combined with IFNγ, or LPS, MPLA, polyI:C, or IFNγ alone did not induce the tumoricidal activity of the macrophages (Figure 1A, 1B).
Figure 1. MPLA+IFNγ induces tumoricidal effects of breast TAMs from mice and patients.

(A) Macrophages and PyMT tumor cells were isolated from primary tumors of MMTV-PyMT (BL/6) mice, fluorescently labeled, and co-cultured for 48 hours as indicated. (B) Statistical analysis of relative PyMT cancer cell numbers for panel A. (C) The effect of macrophages on PyMT tumor cells were studied using: 1) CM: conditioned medium of vehicle- or MPLA+IFNγ-treated macrophages was added to the cancer cells; 2) Transwell: macrophages and cancer cells were seeded in upper or lower chambers, respectively; 3) Direct contact: macrophages and cancer cells were co-cultured in direct contact. B, C: Graph depicts the average results of three independent experiments. (D) Macrophages and EpCAM+ cancer cells were isolated from pleural effusions of breast cancer patients and co-cultured as indicated. EpCAM+ cancer cells did not proliferate during co-culture. For A–D, macrophages were stained with Deep Red Dye (falsely colored green); cancer cells were stained with CMFDA Dye. LPS, MPLA, polyI:C, mouse IFNγ, or human IFNγ was added at 0 hour. (E) Gene expressions in pleural effusion-derived macrophages (cultured without cancer cells) was determined by RT-qPCR 24 hours after indicated treatments were initiated. NOS2, CD40, TNF, and IL12B (p40 subunit of IL-12) are markers of tumoricidal macrophages. IL10 and MRC1 are markers of TAMs. The relative expression to ACTB was normalized to the relative expression in vehicle-treated samples, which was set to 1. D, E: Graph depicts the average results from three patients. One-way ANOVA was used for all panels. Data are represented as mean ± SD (standard deviation). See also Figure S1, Movies S1 and S2.
iNOS is required for macrophages’ tumoricidal activity (Dinapoli et al., 1996). Consistently, only the combination treatments—IFNγ + LPS or MPLA—induced iNOS expression (Figure S1D). Moreover, inhibiting iNOS activity with L-NIL blocked the tumoricidal effect of MPLA+IFNγ-activated macrophages (Figure S1E). Thus, TAMs can be reprogrammed to become tumoricidal using IFNγ with LPS or MPLA, and iNOS activity is required for their tumoricidal effects. iNOS metabolizes arginine to generate NO, which diffuse into adjacent cells and cause cell death (Kelm, 1999; Stuehr and Nathan, 1989). However, NO has a half-life of just 4-6 seconds (Kelm, 1999). Consistently, only when the MPLA+IFNγ-treated macrophages were directly cultured with cancer cells—not when the cells were separated by transwells (slowing diffusion) or when conditioned medium from MPLA+IFNγ-treated macrophages was added—could the macrophages kill the cancer cells (Figure 1C).
Nos2 (the gene coding for iNOS) expression is regulated by four transcription factors (NFκB, AP-1, STAT1, IRF1) (Gao et al., 1997; Martin et al., 1994; Salim et al., 2016; Xie et al., 1993). Signaling from TLR4, the MPLA receptor, can activate NFκB and AP-1 via MyD88 (Lu et al., 2008), and IFNγ activates the JAK/STAT1 pathway, leading to phosphorylated STAT1 dimer formation and IRF1 activation (Castro et al., 2018). Consistently, MPLA+IFNγ did not induce Nos2 expression or induce tumoricidal activity in Tlr4 knockout bone marrow-derived macrophages (BMDMs, Figure S1F–H). Furthermore, MPLA alone induced NFκB phosphorylation, while IFNγ alone induced STAT1 phosphorylation, and both transcription factors were phosphorylated by MPLA+IFNγ, as determined by Western blot of BMDMs (Figure S1I). Finally, inhibiting NFγB, STAT1/IRF1, or AP-1 was sufficient to prevent MPLA+IFNγ–induced Nos2 expression (Figure S1J). Together, these data suggest that MPLA and IFNγ induce separate, complementary signaling pathways that are required for maximal iNOS expression and thus tumoricidal activity.
To formally test whether patient-derived TAMs could be activated to kill patient-derived cancer cells, we isolated macrophages and cancer cells from the metastatic pleural effusions of breast cancer patients (Figure S1K–M). MPLA+IFNγ could reprogram these macrophages, killing 80–90% of the cancer cells isolated from the corresponding patient in vitro (Figure 1D). We next used RT-qPCR to identify TAM reprogramming markers. MPLA+IFNγ increased the expression of tumoricidal macrophage markers (NOS2, CD40, TNF, IL12B) and reduced the expression of TAM markers (IL10, MRC1) in the patient-derived macrophages (Figure 1E). Tnf and Il12b upregulation and Il10 and CD206 downregulation were also observed in primary PyMT tumor-derived macrophages after MPLA+IFNγ treatment (Figure S1N, S1O).
The class IIa HDAC inhibitor TMP195 and PI3Kγ inhibitor IPI-549 can reprogram TAMs to control tumor growth and metastasis. Using TAMs from PyMT tumors and drug concentrations from prior publications (Guerriero et al., 2017; Kaneda et al., 2016), we found that TMP195 did not increase Nos2 or Il12b or inhibit Arg1, Mrc1, or Il10 expression, but did increase Cd86 and H2-Q2 expression. Similarly, IPI-549 only increased H2-Q2 and inhibited Il10 expression (Figure S1P). Furthermore, neither TMP195 nor IPI-549 activated TAMs to kill cancer cells (Figure S1Q). Thus, MPLA+IFNγ is specifically effective at inducing Nos2 expression and reprogramming TAMs to be tumoricidal in vitro.
MPLA+IFNγ suppresses breast tumor growth and lung metastasis
We next asked: Could MPLA+IFNγ control cancer cells in vivo, in the context of a complex TME? We used two metastatic breast cancer models: transplanted PyMT (luminal B subtype) or 4T1 (triple negative subtype) breast tumors. After determining optimal doses (1 μg MPLA + 1 μg IFNγ per PyMT tumor; 1 μg MPLA + 3 μg IFNγ per 4T1 tumor; Figure S2A, S2B), we found that intratumorally injected MPLA+IFNγ suppressed primary tumor growth in both models (Figure 2A, 2B). LPS can promote metastasis by inducing neutrophil extracellular traps (NETs) (Albrengues et al., 2018; Yang et al., 2020). Yet, MPLA+IFNγ did not increase NET levels in plasma of treated mice (Figure S2C, S2D). Importantly, MPLA+IFNγ did not enhance metastasis: it instead significantly inhibited lung metastasis. All vehicle-treated mice had detectable metastasis, while only 2 out of 5 PyMT and 4 out of 9 4T1 tumor-bearing mice had metastases after MPLA+IFNγ treatment (Figure 2C, 2D). IFNγ alone also inhibited tumor growth and lung metastasis, but less than the combination treatment, and MPLA alone had no effect. We also employed the parental MMTV-PyMT mouse model (on both the C57BL/6 and FVB/N genetic background). In this model, intratumoral injection of MPLA+IFNγ also suppressed primary tumor growth and lung metastasis (Figure 2E–J). Finally, since intratumoral injection is not the standard of care for any cancer type and not all tumors can be intratumorally injected, we tested intraperitoneal administration of MPLA+IFNγ: higher doses of MPLA (30 μg/mouse) and IFNγ (30 μg/mouse) were needed to reduce tumor growth (Figure S2E, S2F). Importantly, these higher doses did not change the body weight of C57BL/6 mice (Figure 2K), nor did it cause elevated serum levels of aspartate aminotransferase, alkaline phosphatase, albumin, or creatine kinase (markers of liver, heart, or kidney damage) (Figure 2L). Moreover, histological evaluation of liver, lung, kidney, bone marrow, heart, and spleen from the treated mice revealed no signs of toxicity (Figure 2M).
Figure 2. MPLA+IFNγ suppresses breast tumor growth and lung metastasis in mice.

(A, B) Tumor growth curve of (A) transplanted PyMT or (B) 4T1 tumors, arrows indicate time of treatments; 1 μg MPLA, 1 μg (A) or 3 μg (B) mouse IFNγ, or both (MPLA and IFNγ were mixed, then injected) per tumor. N=8 tumors/group. (C, D) Lung metastatic burden of (C) PyMT (N=4–6 mice/group) and (D) 4T1 tumor-bearing mice (N=5–9 mice/group) determined from histology. (E, H) Tumor growth curves, (F, I) representative H&E staining of lungs (scale bar: 4 mm), and (G, J) lung metastatic burden determined from histology of spontaneous C57BL/6-MMTV-PyMT (E–G, N=6-7 mice/group) and FVB/N-MMTV-PyMT (H–J, N=6-10 mice/group) mice (1 μg MPLA+1 μg IFNγ per tumor). Lungs were collected 3 days after last treatment. One-way ANOVA (2A–D) or t-test (2E, 2G, 2H, and 2J) was performed for tumor volume analysis at the end point and for lung metastasis analysis, and due to unequal group variances, Welch’s ANOVA was used for panels 2A, 2C, 2D, and Welch’s t-test was used for panels 2E, 2G, 2J. ***p<0.001, compared to vehicle. Data are represented as mean ± SD. (K) Body weight and (L) serum levels of AST (aspartate aminotransferase), ALP (alkaline phosphatase), albumin, and creatine kinase of tumor-free C57BL/6 mice treated with vehicle (N=9) or MPLA+IFNγ, treated as in Figure S2F (N=10). T-test was performed for panels 2K and 2L. Data are represented as mean ± SD. n.s.: not significant. (M) Representative H&E stained sections of liver, lung, kidney, bone marrow, heart, or spleen. Scale bar: 100 μm. Serum and tissues were collected 2 days after the 6th treatment. See also Figure S2.
We next benchmarked MPLA+IFNγ against other macrophage polarization approaches (TMP195 and IPI-549), as well as PLX3397 with anti-PD1 checkpoint immunotherapy (Wainberg et al., 2016). MPLA+IFNγ was more effective at reducing PyMT tumor growth and lung metastasis than the other treatments at previously published doses (DeNardo et al., 2011; Guerriero et al., 2017; Kaneda et al., 2016) (Figure S2G–I). Together, these results suggest that a safe and effective dose of MPLA+IFNγ could be used in future clinical trials.
MPLA+IFNγ stimulates chemokine secretion and type I IFN signaling
To explore how MPLA+IFNγ controls tumor growth and metastasis, we first focused on changes to the TME induced during early treatment phases. PyMT tumors were collected 24 hours after the 2nd intratumoral injection and digested. The cytokines released to the supernatant were analyzed by cytokine arrays (Figure 3A). We found that MPLA+IFNγ induced the secretion of a large number of chemokines responsible for immune cell recruitment, including CCL5 (leukocytes, including monocytes), CXCL9 (lymphocytes, particularly T cells), CXCL13 (B cells), CCL12 (monocytes), and CXCL5 (neutrophils) (Figure 3B). We also detected increased CD40 (a costimulatory protein on antigen-presenting cells), IL-12 (which enhances the cytotoxic activity of NK cells and CD8+ T cells), and BAFF (B-cell activating factor) (Figure 3B). Increased CXCL9 and CCL5 secretion after MPLA+IFNγ treatment was confirmed by enzyme-linked immunosorbent assay (ELISA) (Figure S3A, S3B). Thus, the combined MPLA+IFNγ treatment dramatically altered the cytokine microenvironment.
Figure 3. MPLA+IFNγ enhances chemokine secretion and stimulates the type I IFN signaling pathway in breast tumors.
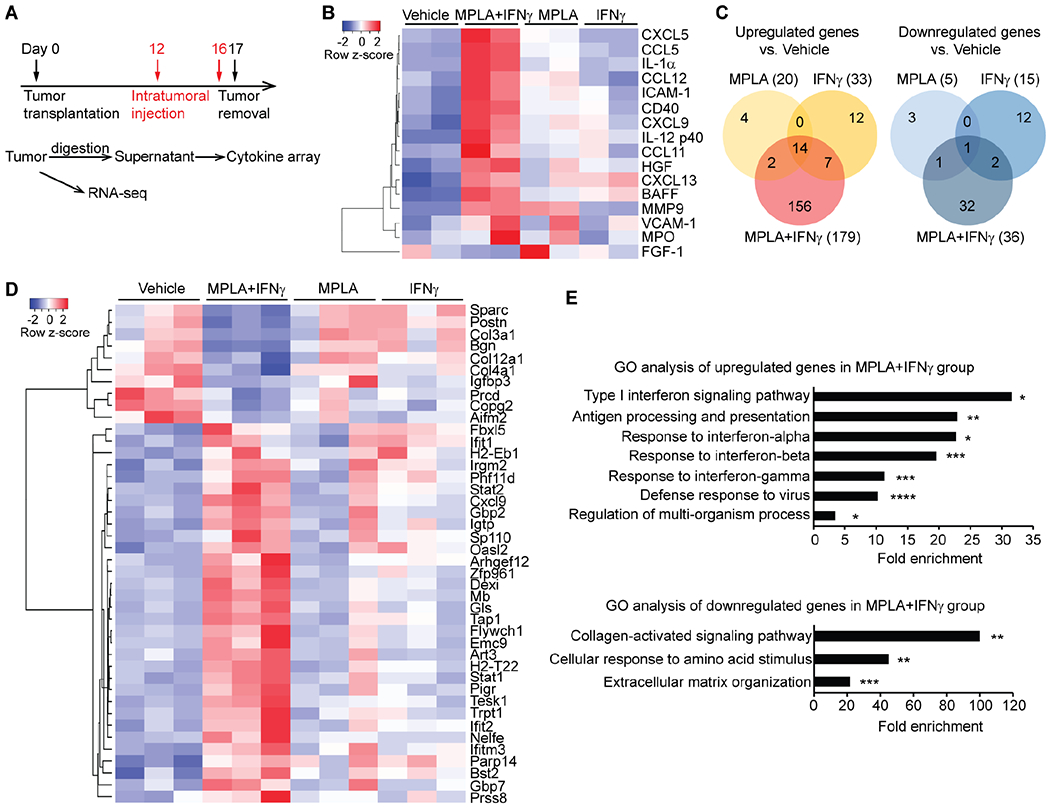
(A) Experimental design for obtaining cytokine array and RNA-seq data; 1 μg MPLA, 1 μg mouse IFNγ, or both were used per PyMT tumor. (B) Heatmap of the secreted cytokines that were changed by MPLA+IFNγ compared to vehicle. (C) Venn diagrams of upregulated and downregulated genes in tumors after the indicated treatments. MPLA+IFNγ uniquely upregulated 156 genes and downregulated 32 genes that did not show altered transcription when tumors were treated by MPLA or IFNγ alone. (D) Heatmap of the top genes that were uniquely changed in the MPLA+IFNγ group. (E) Gene ontology analysis of the genes that were uniquely up- or down-regulated in the MPLA+IFNγ group. *FDR<0.05, **FDR<0.01, ***FDR<0.001, ****FDR<0.0001, compared to vehicle. See also Figure S3 and Data file S1.
We next explored MPLA+IFNγ’s effect on gene regulation by RNA-seq of bulk tumors. There were only 20 upregulated and 5 downregulated genes after MPLA treatment alone; 33 upregulated and 15 downregulated genes after IFNγ treatment alone, but 179 upregulated and 36 downregulated genes after the combined MPLA+IFNγ treatment (Figure 3C, Data file S1). Both IFNγ alone and MPLA+IFNγ increased the expression of IFNγ-inducible genes (Data file S1). In addition, MPLA+IFNγ uniquely enhanced the expression of the type I IFN (IFNα/β)-inducible genes Stat1, Stat2, Irgm2, Gbp2, and Cxcl9 (Figure 3D). MPLA+IFNγ also uniquely decreased the expression of extracellular matrix (ECM)-associated genes (Col3a1, Col4a1, Col12a1, and Igfbp3) (Figure 3D). The altered transcription of select type I IFN-inducible and ECM-associated genes was verified in PyMT tumors, and was also shown to occur in MPLA+IFNγ-treated 4T1 tumors, by RT-qPCR (Figure S3C–F). These results indicate that MPLA+IFNγ activated immune cells and also had wider effects on the TME.
To investigate the implications of the altered gene programs, we performed gene ontology (GO) analysis (Figure 3E), which revealed enhanced immunostimulatory responses (such as type I IFN signaling, antigen processing and presentation, and defense response to virus) and inhibited immunosuppressive responses (such as collagen-activated signaling and ECM organization). Overall, these data showed that only the combination MPLA+IFNγ treatment promoted the expression of genes involved in effector immune cell recruitment and stimulated the type I IFN signaling pathway, thus inducing changes associated with a strong anti-tumor response.
MPLA+IFNγ reprograms TAMs and activates cytotoxic T cells
We next elucidated the contributions of immune cells to the anti-tumor effects. By flow cytometry (Figure S4A–K), we found that as a percentage, there were almost three times as many immune (CD45+) cells in PyMT tumors after intratumoral injection of MPLA+IFNγ as in vehicle-treated tumors (Figure S4B). Among immune cells (Figure S4C–K), the proportion of B cells was decreased (Figure S4G), while the proportions of CD4+ T cells (Figure S4E) and inflammatory monocytes (Ly6C+Ly6G−, Figure 4A) were increased. Macrophage infiltration in PyMT tumors (F4/80+, Figures 4B, S4I) was also enhanced. Not only did the tumors contain more macrophages, but the proportion of macrophages that expressed iNOS (Figure 4C) or CD40 (Figure 4D) was increased, while the proportion of macrophages expressing CD206 was reduced (Figure 4E). MPLA+IFNγ also increased the numbers of iNOS+ or CD40+ cells, as determined by immunofluorescence (IF) staining (Figure 4F, 4G). Furthermore, IF staining showed STAT1 and NFκB p65 phosphorylation in macrophages (and other cells) when IFNγ was given with MPLA (Figure S4L), consistent with our in vitro data (Figure S1I, S1J). Together, these results suggest that MPLA+IFNγ had reprogrammed TAMs to tumoricidal macrophages in the mouse breast tumors.
Figure 4. MPLA+IFNγ reprograms TAMs to be tumoricidal in breast tumors.

(A) Flow cytometry of inflammatory monocytes in PyMT tumors. N=5 mice/group. (B) IF staining of macrophages (F4/80+) in PyMT tumors. In total, 6–14 fields of view (FOVs) were evaluated from 4 mice/group. Scale bar: 50 μm. (C–E) The percentages of tumoricidal macrophages (iNOS+ or CD40+) and TAMs (CD206+) of total macrophages were identified by flow cytometry. N=3–6 mice/group. (F, G) IF staining of iNOS+ (F) and CD40+ cells (G) in PyMT tumors. In total, 6–14 FOVs were evaluated from 4 mice/group. Scale bar: 50 μm. (H) Antigen-presenting activities of macrophages determined by the percentage of H-2Kb OVA+ cells in PyMT-chOVA tumors. N=5 mice/group. For all figure panels, the analysis was performed 2 days after the last (6th) intratumoral injection. One-way ANOVA was used for panels 4A, 4C–E, and 4H, and Welch’s ANOVA was used for panels 4B, 4F, and 4G. Data are represented as mean ± SD. See also Figure S4.
We next used a variant of the MMTV-PyMT model, the MMTV-PyMT-chOVA model (Engelhardt et al., 2012; Fein et al., 2020) to explore the effect of MPLA+IFNγ on MHC class I antigen-presentation. Cancer cells in the MMTV-PyMT-chOVA model express the ovalbumin (OVA) peptide SIINFEKL, allowing measurement of MHC class I presentation of OVA (H-2Kb OVA+). In the tumors of these mice, there was an almost twofold increase in the percentage of H-2Kb OVA+ macrophages (Figure 4H), H-2Kb OVA+ dendritic cells (Figure S4M), and CD103+CD11cHighCD11bHigh cells (Figure S4N) after MPLA+IFNγ treatment. In the PyMT tumors, MPLA+IFNγ significantly increased the infiltration of total (CD8a+, Figure 5A, S4D) and activated (CD107a+ or PD1+) (Figure 5B, 5C) cytotoxic T cells. Importantly, the combined treatment also promoted the production of effector memory T cells (CD44+CD62L−) in the tumors (Figure 5D). We also observed increased T cell accumulation in the lungs of tumor-bearing mice (which at the analyzed time point contain small metastasis) (Figure S5A), suggesting that disseminated cancer cells elicited a stronger T cell response in MPLA+IFNγ-treated mice. Together, these data suggest MPLA+IFNγ reverse immune suppression in the TME and activate lasting adaptive T cell immune responses to protect against future metastases.
Figure 5. MPLA+IFNγ can activate cytotoxic T cells through macrophage-secreted IL-12 and TNFα and antigen presentation.

(A) IF staining of CD8+ T cells in PyMT tumors. In total, 5–9 FOVs were evaluated from 4 mice/group. Scale bar: 50 μm. (B–D) The percentages of cytotoxic T cells (CD8+CD107a+, B), PD1+CD8+ T cells (C), and effector memory CD8+ T cells (CD44+CD62L−, D) in PyMT tumors were identified by flow cytometry. For panels A–D, the analysis was performed 2 days after the last (6th) intratumoral injection. (E–G) CD69 expression of CD8+ T cells in in vitro co-culture systems was identified by flow cytometry. (E) CD8+ T cells isolated from the spleens of PyMT tumor-bearing mice were cultured with or without macrophages derived from the bone marrow of the same mice, and MPLA or IFNγ or both were added to the cell cultures immediately after T cell seeding. (F) CD8+ T cells were cultured with macrophages similarly to (E), and neutralized antibodies against IL-12, TNFα, or CXCL9 were added 30 minutes before adding MPLA+IFNγ. (G) Macrophages derived from the bone marrow of PyMT tumor-bearing mice were activated with MPLA (or IFNγ or both) for 16 hours, then incubated with OVA-Q4H7 peptide (10−9 M) for 2 hours, washed with PBS and added to CD8+ T cells isolated from the spleens of OT1 mice. One-way ANOVA was used for panels 5C, 5E, and 5F, and Welch’s ANOVA was used for panels 5A, 5B, 5D, and 5G. Data are represented as mean ± SD. See also Figure S5.
Intratumoral injection of MPLA+IFNγ in tumors of the parental MMTV-PyMT mouse models also increased infiltration of immune cells, macrophages, CD8+ T cells; activated cytotoxic T cells (CD107a+ or PD1+); decreased the percentage of CD206+ macrophages; and increased the percentage of iNOS+ macrophages (Figure S5B). We also found more activated myeloid cells (CD40+) and cytotoxic, activated T cells (CD107a+ or PD1+) in the lungs (Figure S5C, S5D)—cells that may have been recruited and/or activated in response to the spontaneous dissemination of cancer cells. We next tested whether systemic MPLA+IFNγ treatment would also alter the PyMT tumor immune microenvironment. As we had found after intratumoral injection of MPLA+IFNγ, the total numbers of immune cells, macrophages, CD8+ T cells, activated CD107a+ CD8+ T cells, and PD1+ CD8+ T cells were increased after intraperitoneal injection, and there was a switch in macrophage marker expression, with decreased CD206 and increased iNOS expression (Figure S5E). Taken together, both local and systemic administration of MPLA+IFNγ can alter the immune microenvironment of primary tumors and lung metastases.
MPLA+IFNγ can activate cytotoxic T cells through macrophage-secreted cytokines and antigen presentation
Our in vivo data strongly suggested that MPLA+IFNγ activated cytotoxic T cells either directly or indirectly via macrophages. We incubated CD8+ T cells isolated from the spleens of PyMT tumor-bearing mice with MPLA+IFNγ in vitro, but found that the treatment did not directly stimulate T cell activation (Figure 5E). In contrast, when these CD8+ T cells were co-cultured with macrophages derived from the bone marrow of the same mice, MPLA+IFNγ increased the expression of CD69 on CD8+ T cells (Figure 5E). These results suggest that MPLA+IFNγ-activated macrophages stimulate cytotoxic T cell function. MPLA+IFNγ promoted the expression of IL-12, TNFα, and CXCL9 in macrophages and PyMT tumors (Figures 1E, S1N, 3B, S3A). Thus, we explored the possible roles of these three cytokines in macrophage-T cell crosstalk in vitro (Figure 5F). The increased expression of CD69 by T cells cultured with macrophages was partially reduced by incubation with the IL-12 or TNFα neutralizing antibody, indicating that MPLA+IFNγ activates cytotoxic T cells through the macrophage-secreted cytokines IL-12 and TNFα. Neutralizing CXCL9 had no effect on CD69 expression, consistent with a likely role in T cell recruitment rather than activation. Blocking IL-12 and TNFα together did not completely prevent the increased CD69 expression, suggesting that factors besides IL-12 and TNFα contribute to macrophage-T cell crosstalk.
We next challenged CD8+ T cells from OT1 mice with OVA peptide-incubated macrophages. OT1-derived T cells recognize cells with the MHC class I-bound OVA peptide. We found that the OT1 CD8+ T cells were activated, as determined indirectly by CD69 expression, by the combination of OVA and MPLA+IFNγ–treated macrophages (Figure 5G), while MPLA or IFNγ alone had no effect. Taken together, our in vitro results show that MPLA+IFNγ not only activates macrophages’ tumoricidal effects but also enhances their capacity to activate cytotoxic T cells.
Both macrophages and T cells are essential for MPLA+IFNα’s anti-tumor effects
We next performed in vivo antibody depletion experiments to test which immune cell populations are necessary for MPLA+IFNγ to eliminate metastatic breast cancer. Cell depletion was confirmed by flow cytometry (Figure S6A, S6B). In PyMT tumors, T cell depletion reduced the anti-tumor efficacy of MPLA+IFNγ (Figure S6C, S6D), but we were unable to determine the contribution of macrophages, as the anti-CSF1R antibody was ineffective in depleting them (Figure S6A). In 4T1 tumors, depleting either macrophages or T cells was effective (Figure S6B), and depleting either cell type abolished the suppressive effects of MPLA+IFNγ on both tumor growth and lung metastasis (Figure 6A, 6B). Of the two T cell populations, CD8+ T cells were required for MPLA+IFNγ to inhibit tumor growth and lung metastasis, while CD4+ T cells were required for tumor growth inhibition, but did not significantly affect lung metastasis (p=0.071). Thus, both the innate and adaptive immune systems are required for the anti-tumor effects of MPLA+IFNγ.
Figure 6. Both macrophages and T cells are essential for MPLA+IFNγ’s anti-tumor effects.

(A) Tumor growth curve of 4T1 tumors after T cell depletion (anti-CD4, anti-CD8, or anti-CD4/CD8) or macrophage depletion (anti-CSF1R). N=8 tumors/group. Immune cell-depleting antibodies were injected 1 day before intratumoral injection of MPLA+IFNγ. Welch’s ANOVA was performed for the tumor volume analysis at the end point. *p<0.05, **p<0.01, ***p<0.001, compared to MPLA+IFNγ+IgG. (B) Lung metastatic burden of 4T1 tumor-bearing mice was determined by H&E staining. Lung tissues were collected 3 days after the last MPLA+IFNγ treatment. N=5–7 mice/group. (C) Procedure for tumor re-challenge assay with or without T cell depletion (anti-CD4/CD8). i.t.: intratumoral; i.v.: intravenous. (D–E) Lung metastasis formed by newly injected breast cancer cells (ECFP+) without (D) or with (E) T cell depletion was determined by IF staining of lung sections for ECFP. N=4–5 mice/group. Welch’s ANOVA was used to analyze the data in panels 6B, 6D, and 6E. Data are represented as mean ± SD. See also Figure S6.
To test if MPLA+IFNγ induces a long-lasting immune response, tumor-bearing mice had their tumors surgically removed, recovered for 2 weeks, and were then re-challenged with intravenously injected cancer cells from MMTV-PyMT; ACTB-ECFP mice (Figure 6C), allowing us to identify the new cancer cells by their expression of enhanced cyan fluorescent protein (ECFP). Only mice treated with MPLA+IFNγ had significantly reduced ECFP+ metastatic burden, not mice treated with MPLA or IFNγ alone (Figure 6D). When T cells were depleted during MPLA+IFNγ treatment (Figure 6C), the ability of MPLA+IFNγ to inhibit new metastasis was abolished (Figure 6E). These data, together with enhanced memory T cell generation (Figure 5D), indicate that the combination treatment not only reverses immune suppression but also triggers anti-tumor memory, which may have a long-lasting effect on metastasis.
MPLA+IFNγ suppresses metastatic ovarian cancer
Intraperitoneal MPLA+IFNγ’s effectiveness in our breast cancer model (Figure S2F, S5E) motivated us to explore this treatment for ovarian cancer, for which intraperitoneal chemotherapy is used and for which there are few effective treatments for late-stage disease. Furthermore, TAMs contribute to the immunosuppressive microenvironment in ovarian cancer (Rodriguez et al., 2018; Yigit et al., 2010). At 7 weeks after ID8-p53−/− cancer cell transplantation (Figure 7A), the mice were visibly sick (Figure S7A) and had bloody ascites in their abdominal cavity (Figure 7B). All mice had an elevated body weight (Figure S7B) and many metastases in the peritoneum, pancreas, mesentery, and diaphragm (Table S1). Yet, when MPLA+IFNγ was initiated 3 weeks after cancer cell transplantation (Figure 7A), the mice at the 7-week time point looked healthy (Figure S7A), and only 2 of 5 mice had visible metastases in the peritoneum (Table S1). Moreover, MPLA+IFNγ decreased the total cell number (Figure 7C), increased the percentage of CD45+ immune cells (Figure S7C), and reduced the proportion of EpCAM+ cancer cells in the ascites (Figure 7D). Strikingly, MPLA+IFNγ prolonged the median survival of nD8-p53−/− tumor-bearing mice from ~63 to ~100 days regardless of whether treatment was started 3 weeks (Figure 7E) or 6 weeks (Figure 7F) after tumor inoculation, when many mice already had a small amount of bloody ascites. Together, these findings indicate that MPLA+IFNγ suppressed the metastatic progression of ovarian cancer.
Figure 7. MPLA+IFNγ suppresses metastatic ovarian cancer in mice.

(A) Experimental design: three weeks after intraperitoneal ID8-p53−/− ovarian cancer cell inoculation, MPLA mixed with IFNγ was injected to the mice every 5 days, for six times. Two days after the last treatment, mice were euthanized and ascites/peritoneal lavage was collected for flow cytometry, i.p.: intraperitoneal. (B) Metastasis was evident from the appearance of the ascites/peritoneal lavage of the treated mice. (C, D) Metastasis was determined by the total cell number (C) and percentage of epithelial cancer cells (EpCAM+, D) in the ascites/peritoneal lavage. (E) Kaplan-Meier survival curves of DD8-p53−/− tumor-bearing mice treated with MPLA+IFNγ. Log-rank test, N=10 mice/group. Median survival times: Vehicle: 64 days; MPLA+IFNγ: 103 days. (F) Kaplan-Meier survival curves of DD8-p53−/− tumor-bearing mice treated with MPLA+IFNγ alone or in combination with cisplatin as indicated. Log-rank test, N=10 mice/group. Median survival times: Vehicle: 63 days; MPLA+IFNγ: 98.5 days; cisplatin: 105 days; and cisplatin followed by MPLA+IFNγ: undefined. (G–I) The proportions of monocytes (Ly6C+Ly6G−, G), tumoricidal macrophages (iNOS+, H), and TAMs (CD206+, I) in ascites/peritoneal lavage were determined by flow cytometry. N=5 mice/group. T-test was used for panels 7G and 7I, and Welch’s t-test was used for 7C, 7D, and 7H. Data are represented as mean ± SD. See also Figure S7 and Table S1.
Most patients with ovarian cancer are treated with chemotherapy, but the cancer will often recur (Herzog, 2006). We thus compared MPLA+IFNγ to cisplatin, a widely used chemotherapy for ovarian cancer. Both treatments increased the median survival of the mice, from 63 days for vehicle to 98.5 and 105 days for MPLA+IFNγ and cisplatin, respectively (Figure 7F). When MPLA+IFNγ was given after four cycles of cisplatin, the median survival time could not be determined (Figure 7F), as 70% of the mice were still alive at the endpoint of 150 days.
We next investigated the effect of MPLA+IFNγ on the ovarian cancer immune microenvironment. MPLA+IFNγ increased the percentage of monocytes in the ascites (Figure 7G), and similar to the breast cancer model, it altered the balance between iNOS+and CD206+ macrophages (Figure 7H, 7I): the percentage of iNOS+ macrophages increased, while that of CD206+ macrophages decreased. To determine if macrophages and T cells were both important for the anti-metastatic effects of MPLA+IFNγ, we depleted macrophages using an anti-CSF1 antibody (DeNardo et al., 2011; Guerriero et al., 2017) (Figure S7D). Macrophages were essential for the anti-metastatic effects of MPLA+IFNγ in the ovarian cancer model, measured by the total cell number in the ascites of the mice (Figure S7E) or the proportion of EpCAM+ cancer cells (Figure S7F). Furthermore, both CD4+ and CD8+ T cells contributed to inhibiting metastasis in this model (Figure S7E, S7F). The median survival of ID8-p53−/− tumor-bearing mice treated with MPLA+IFNγ (102 days) was longer than that of mice treated with TMP195 (51 days), IPI-549 (68 days), or PLX3397+anti-PD1 (70 days) (Figure S7G–I).
Altogether, our data show that MPLA+IFNγ dramatically suppresses metastatic breast and ovarian cancers by reprogramming the tumor-immunosuppressive microenvironment and stimulating the collaborative innate-adaptive anti-tumor immune response.
Discussion
Breast and ovarian tumors are infiltrated by large numbers of macrophages and T cells. Though these cells can kill cancer cells, they must be activated properly. With this study, we developed a strategy to reprogram the immune microenvironment of cancer. We revisited the approach used for Coley’s toxin and exploited the combined power of the TLR4 agonist MPLA with the cytokine IFNγ. The combination of MPLA and IFNγ reversed immune suppression in the TME and induced a systemic anti-tumor response. This treatment harnessed the power of macrophages and T cells, so that together, they eliminated metastatic breast and ovarian cancer. This result is in line with previous reports using other approaches to reprogram macrophages (Guerriero et al., 2017; Kaneda et al., 2016), strongly supporting the argument for engaging both innate and adaptive immunity for cancer immunotherapy.
MPLA+IFNγ stimulated innate immunity, but the adaptive immune response was also critical for the treatment’s in vivo effects. We found that MPLA+IFNγ did not stimulate T cells directly in vitro and that CD8+ T cell activation depended on macrophage-secreted cytokines, including IL-12 and TNFα. It is likely that MPLA+IFNγ in vivo also induces dendritic cells to produce IL-12 to activate cytotoxic T cell responses, as such effects have been reported (ten Brinke et al., 2010). Our data are consistent with reports that IL-12 bridges innate and adaptive immunity by enhancing the cytotoxic activity of CD8+ T cells (Trinchieri, 2003); that intratumor adoptive transfer of tumor-specific CD8+ T cells engineered with transiently expressed IL-12 mRNA eradicates tumors in melanoma mouse models (Etxeberria et al., 2019); and that genetically engineered myeloid cells expressing IL-12 activate antigen presentation and T cell response and reverse immune suppression to reduce metastasis in rhabdomyosarcoma and pancreatic tumor mouse models (Kaczanowska et al., 2021). TNFα can promote the activation and proliferation of naïve and effector T cells (Croft, 2009; Mehta et al., 2018). Macrophages from murine tumors and from malignant pleural effusions of breast cancer patients could be reprogrammed to be tumoricidal by MPLA+IFNγ ex vivo, suggesting that TAMs in the metastatic TME are sufficiently plastic to be reprogrammed to become tumoricidal. Moreover, the reprogrammed patient-derived macrophages expressed IL12B and TNF, suggesting that it will also be possible to activate both macrophage and T cell responses in cancer patients.
The collaborative therapeutic effect between MPLA and IFNγ in vivo is likely also related to MPLA+IFNγ’s induction of a robust type I IFN response in the tumors. Type I IFNs play critical roles in anticancer immunity [reviewed in (Parker et al., 2016; Vacchelli et al., 2015; Zitvogel et al., 2015)], and they can promote antigen presentation. This finding is consistent with our data showing increased antigen-presenting activities of macrophages and dendritic cells after combined MPLA+IFNγ treatment, but not by either treatment alone. Type I IFNs also skew the adaptive immune system to promote antigen-specific T cell responses and immunological memory. Indeed, MPLA+IFNγ significantly increased the abundance of activated cytotoxic T cells and effector memory T cells in tumors. In the metastasis re-challenge assay, MPLA+IFNγ–treated mice developed the fewest new metastatic lesions, and T cells were required for this effect, suggesting that MPLA+IFNγ triggers long-term anti-tumor T cell memory. Similar to our results, MPLA alone could not suppress murine melanoma or colon tumor growth, though MPLA does increase the anti-tumor responses of anti-PDL1 and anti-CTLA4 antibodies in the models (Jallad et al., 2020; Zhang et al., 2021). In situ anti-CD40+MPLA treatment activates local antigen-presenting cells, overcomes T cell exhaustion, and restores PD1 sensitivity in mouse models of melanoma, colorectal, hepatocellular, and bladder cancer (Khalil et al., 2019). Thus, MPLA combined with various other immunomodulatory drugs is a promising approach to improve anti-tumor immunity.
In our experiments, in situ injection of 1 μg MPLA (~50 μg/kg body weight) with 1–3 μg IFNγ (~50–150 μg/kg body weight) was sufficient to induce local immune modulation in breast tumors and to invoke a systemic anti-tumor immune response that protected against metastatic disease, either from spontaneously disseminated tumor cells or from cells injected as a re-challenge of the immune system. Intriguingly, under our experimental conditions, MPLA+IFNγ was more effective at suppressing metastasis in both breast and ovarian cancer models than other treatments that alter macrophage activation, namely TMP195, IPI-549, and PLX3397+anti-PD1. Along with our strategy, there are now several different approaches to activating the innate immune response to treat cancer. Potential drawbacks of invoking local immune modulation include the reliance on adequate immune cell infiltration available for reprogramming and the requirement of suitable injection sites in the tumor. As an alternative, we administrated MPLA+IFNγ intraperitoneally. Though this approach required higher doses (10–30 μg/ ~0.5–1.5 mg/kg body weight), we were reassured by the lack of overt toxicity.
MPLA and IFNγ are already FDA-approved biologies. If MPLA+IFNγ’s elimination of metastasis can be translated from our preclinical models to patients, then the approach may significantly improve the survival rates of late-stage breast and ovarian cancer patients.
STAR Methods
RESOURCE AVAILABILITY
Lead Contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the Lead Contact, Dr. Mikala Egeblad (egeblad@cshl.edu).
Materials Availability
This study did not generate new unique reagents.
Data and Code Availability
RNA-seq data have been deposited in the Sequence Read Archive (SRA) with the accession number GSE146211 (also listed in the Key Resources Table).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-mouse iNOS | BD Biosciences | Cat #610431 |
| Goat polyclonal anti-mouse CD206 | R&D Systems | Cat #AF2535 |
| Mouse monoclonal anti-β-actin | Santa Cruz Biotechnology | Cat #sc-47778 |
| Rabbit monoclonal anti-NFκB p65 | Cell Signaling Technology | Cat #8242 |
| Rabbit monoclonal anti-Phospho-NFκB p65 (Ser536) (93H1) | Cell Signaling Technology | Cat #3033 |
| Rabbit monoclonal anti-STAT1 | Cell Signaling Technology | Cat #14994 |
| Rabbit monoclonal anti-Phospho-STAT1 (Tyr701) (58D6) | Cell Signaling Technology | Cat #9167 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | Cat #sc-32233 |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR | Cat #926-32210 |
| IRDye 800CW Donkey anti-Goat IgG Secondary Antibody | LI-COR | Cat #926-32214 |
| Goat polyclonal anti-Neutrophil Elastase | Santa Cruz Biotechnology | Cat #sc-9521 |
| BV785™ anti-mouse CD45 Antibody | BioLegend | Cat #103149 |
| Alexa Fluor® 647 anti-mouse CD3 Antibody | BioLegend | Cat #100209 |
| APC/Cy7 anti-mouse CD3 Antibody | BioLegend | Cat #100221 |
| APC anti-mouse/human CD11b Antibody | BioLegend | Cat #101212 |
| FITC anti-mouse CD4 Antibody | BioLegend | Cat #100510 |
| PE anti-mouse CD8a Antibody | BioLegend | Cat #100708 |
| APC/Cy7 anti-mouse CD8a Antibody | BioLegend | Cat #100714 |
| PE/Cy5 anti-mouse CD19 Antibody | BioLegend | Cat #115509 |
| PE/Cy7 anti-mouse CD11c Antibody | BioLegend | Cat #117318 |
| APC/Cy7 anti-mouse CD11c Antibody | BioLegend | Cat #117324 |
| APC/Cy7 anti-mouse Ly6C Antibody | BioLegend | Cat #128026 |
| FITC anti-mouse Ly6G Antibody | BioLegend | Cat #127605 |
| PerCP/Cy5.5 anti-mouse F4/80 Antibody | BioLegend | Cat #123127 |
| Alexa Fluor® 488 anti-mouse F4/80 Antibody | BioLegend | Cat #123120 |
| APC anti-mouse CD326 (EpCAM) Antibody | BioLegend | Cat #118214 |
| PE/Cy7 anti-mouse CD40 Antibody | BioLegend | Cat #124621 |
| FITC anti-mouse CD206 (MMR) Antibody | BioLegend | Cat #141704 |
| FITC anti-mouse CD69 Antibody | BioLegend | Cat #104505 |
| PE anti-mouse CD107a (LAMP-1) Antibody | BioLegend | Cat #121612 |
| FITC anti-mouse CD279 (PD-1) Antibody | BioLegend | Cat #135213 |
| PerCP/Cy5.5 anti-mouse/human CD44 Antibody | BioLegend | Cat #103032 |
| APC anti-mouse CD62L Antibody | BioLegend | Cat #104412 |
| APC anti-mouse H-2Kb bound to SIINFEKL Antibody | BioLegend | Cat #141605 |
| Alexa Fluor® 647 anti-human CD326 (EpCAM) Antibody | BioLegend | Cat #324212 |
| BV510™ anti-human CD45 Antibody | BioLegend | Cat #368525 |
| APC anti-human CD68 Antibody | BioLegend | Cat #333810 |
| PE Rat Anti-CD11b Clone M1/70 (RUO) | BD Biosciences | Cat #557397 |
| CD335 (NKp46) Monoclonal Antibody (29A1.4), eFluor 450 | Invitrogen | Cat #48335180 |
| iNOS Monoclonal Antibody (CXNFT), PE | Invitrogen | Cat #12592082 |
| CD103 (Integrin alpha E) Monoclonal Antibody (2E7), FITC | Invitrogen | Cat #11103182 |
| PE anti-mouse CD40 Antibody | BioLegend | Cat #124610 |
| FITC anti-mouse CD8a Antibody | BioLegend | Cat #100706 |
| Rat monoclonal anti-PyMT antigen | Abcam | Cat #ab15085 |
| Alexa Fluor 488 goat anti-Rat IgG secondary antibody | Invitrogen | Cat #A11006 |
| Alexa Fluor® 647 anti-mouse CD4 Antibody | BioLegend | Cat #100424 |
| Rat monoclonal anti-Mouse F4/80 | Bio-Rad | Cat #MCA497R |
| Rabbit anti-Phospho-NFkB p65 (Ser276) Antibody | Invitrogen | Cat #PA5-37718 |
| Rabbit polyclonal anti-CFP | Bio-Rad | Cat #AHP2986 |
| Alexa Fluor 488 goat anti-rabbit IgG secondary antibody | Invitrogen | Cat #A11070 |
| Goat polyclonal anti-Mouse IL-12 | R&D Systems | Cat #AF-419-NA |
| Rat monoclonal anti-Mouse TNF-alpha | R&D Systems | Cat #MAB4101 |
| Goat polyclonal anti-Mouse CXCL9/MIG | R&D Systems | Cat #AF-492-NA |
| InVivoMAb anti-mouse CD8α | BioXCell | Cat #BE0004-1 |
| InVivoMAb anti-mouse CD4 | BioXCell | Cat #BE0003-1 |
| InVivoMAb anti-mouse CSF1R (CD115) | BioXCell | Cat #BE0213 |
| InVivoMAb anti-mouse CSF1 | BioXCell | Cat #BE0204 |
| InVivoPlus anti-mouse PD1 | BioXCell | Cat #BP0273 |
| Biological Samples | ||
| Metastatic pleural effusions from breast cancer patients | New York University | IRB #i17-01382 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Matrigel | Corning | Cat #356231 |
| Human M-CSF | ProSpec | Cat #CYT-308 |
| L-glutamine | Gibco | Cat #25030081 |
| Sodium pyruvate | Gibco | Cat #11360070 |
| 2-mercaptoethanol | Gibco | Cat #31350010 |
| B27 supplement | Gibco | Cat #17504044 |
| Human EGF | ProSpec | Cat #CYT-217 |
| Insulin | Gibco | Cat #12585014 |
| Collagenase/hyaluronidase | STEMCELL Technologies | Cat #07912 |
| DNase I | Roche | Cat #4716728001 |
| Dispase | STEMCELL Technologies | Cat #07913 |
| Collagenase D | Sigma | Cat #11088866001 |
| DNase I | Sigma | Cat #045362820 |
| Liberase DL | Sigma | Cat #05466202001 |
| TrypLE Express Enzyme | Gibco | Cat #12605010 |
| Mouse M-CSF | ProSpec | Cat #CYT-439 |
| Fibronectin | Sigma Aldrich | Cat #F1141 |
| CellTracker™ Deep Red Dye | Invitrogen | Cat #C34565 |
| CellTracker™ Green CMFDA Dye | Invitrogen | Cat #C7025 |
| LPS | Sigma Aldrich | Cat #L4391 |
| MPLA for in vitro experiments | InvivoGen | Cat #tlrl-mpls |
| MPLA for in vivo experiments | InvivoGen | Cat #vac-mpls |
| PolyI:C | InvivoGen | Cat #tlrl-picw |
| Mouse IFNγ | R&D Systems | Cat #485-MI/CF |
| Human IFNγ | R&D Systems | Cat #285-IF/CF |
| TMP195 | MedChemExpress | Cat #HY-18361 |
| IPI-549 | MedChemExpress | Cat #HY-100716 |
| PLX3397 (Pexidartinib) | MedChemExpress | Cat #HY-16749 |
| L-NIL (N6-(1-Iminoethyl)-L-lysine hydrochloride) | TOCRIS | Cat #1139 |
| Creatine | Selleckchem | Cat #S5588 |
| JSH-23 | Selleckchem | Cat #S7351 |
| SR11302 | TOCRIS | Cat #2476 |
| CellTiter 96® AQueous One Solution reagent | Promega | Cat #G3582 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems | Cat #A25780 |
| Taqman™ Universal Master Mix II, no UNG | Applied Biosystems | Cat #4440040 |
| RIPA lysis buffer | Thermo Fisher Scientific | Cat #89990 |
| Protease and phosphatase inhibitor | Thermo Fisher Scientific | Cat #78440 |
| Cisplatin | MBL | Cat #JM-1550-1000 |
| Sodium citrate solution | Sigma | Cat #C3821 |
| ABTS Substrate Solution | Thermo Fisher Scientific | Cat #37615 |
| Fc receptor blocker | Innovex | Cat #NB309 |
| Goat serum | Dako | Cat #X0907 |
| Mounting media | Electron Microscopy Sciences | Cat #17985-16 |
| OVA-Q4H7 peptide | AnaSpec | Cat #AS-64405 |
| Critical Commercial Assays | ||
| RNeasy Mini kit | Qiagen | Cat #74104 |
| TaqMan™ Reverse Transcription Reagents | Invitrogen | Cat #N8080234 |
| Cell Death Detection ELISA Kit | Roche | Cat #11774425001 |
| Proteome Profiler Mouse XL Cytokine Array Kit | R&D Systems | Cat #ARY028 |
| Mouse CXCL9/MIG DuoSet ELISA | R&D Systems | Cat #DY492 |
| Mouse CCL5/RANTES DuoSet ELISA | R&D Systems | Cat #DY478 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R&D Systems | Cat #DY008 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat #20020594 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat #554714 |
| Comprehensive Chemistry test of serum | IDEXX BioAnalytics | Cat #6006 |
| CD8a+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat #130-104-075 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE146211 |
| Experimental Models: Cell Lines | ||
| Mouse: 4T1 cells | ATCC | Cat #CRL-2539 |
| Mouse: ID8-p53−/− cells | Walton et al., 2016 | N/A |
| Experimental Models: Organisms/strains | ||
| MMTV-PyMT mice (on C57BL/6 background) | Jackson Laboratory | Cat #022974 |
| MMTV-PyMT mice (on FVB/N background) | Jackson Laboratory | Cat #002374 |
| MMTV-PyMT-chOVA mice (on C57BL/6 background) | Engelhardt et al., 2012 | N/A |
| ACTB-ECFP mice | Jackson Laboratory | Cat #004218 |
| OT1 mice | Jackson Laboratory | Cat #003831 |
| Tlr4 KO mice (on C57BL/6 background) | Jackson Laboratory | Cat #029015 |
| MMTV-PyMT; ACTB-ECFP mice (on C57BL/6 background) | This paper | N/A |
| Oligonucleotides | ||
| Primers for human CD40, ACTB, NOS2, TNF, MRC1, IL12B, IL10, mouse Stat1, Stat2, Ifit1, Irgm2, Gbp2, Cxcl9, Igfbp3, Col4a1, Actb, Tnf, Il12b, Il10, Nos2, Mrc1, Arg1, see Table S2 | This paper | N/A |
| Taqman Probe for mouse Tbp | Thermo Fisher Scientific | Mm00446973-m1 |
| Taqman Probe for mouse Cd86 | Thermo Fisher Scientific | Mm00444543-m1 |
| Taqman Probe for mouse H2-Q2 | Thermo Fisher Scientific | Mm04213368-g1 |
| Software and Algorithms | ||
| STAR | Dobin et al., 2012 | N/A |
| FeatureCounts | Liao et al., 2013 | N/A |
| DESeq2 | Love et al., 2014 | N/A |
| Other | ||
| CD326 (EpCAM) microbeads, human | Miltenyi Biotec | Cat #130-061-101 |
| CD11b MicroBeads, human and mouse | Miltenyi Biotec | Cat #130-049-601 |
| 24-well glass bottom plate | MatTek | Cat #P24G-1.5-13-F |
| PVDF membrane | Bio-Rad | Cat #1620177 |
| Relfex 7 mm wound clips | CellPoint Scientific | Cat #203-1000 |
| 96-well Enzyme ImmunoAssay/Radio Immuno-Assay (EIA/RIA) plates | Costar | Cat #3590 |
| Not TC-treated culture dishes | Corning | Cat #430597 |
| Cell culture insert for 24-well Plate with 1.0 μm Transparent PET Membrane | Corning | Cat #353104 |
| Rodent Chow 5053 With PLX3397 Diet | Research Diets, Inc. | N/A |
| Rodent Chow 5053 With IPI-549 Diet | Research Diets, Inc. | N/A |
EXPERIMENTAL MODEL AND SUBJECT DETAIL
Patient samples
Metastatic pleural effusions from three female breast cancer patients were collected during a standard therapeutic procedure used to palliate symptoms. Patient 1 was 34 years old, and the tumor was estrogen-receptor (ER) positive (+), human epidermal growth factor receptor (HER)-2 negative (−); patient 2 was 35 years old, and the tumor was ER+/HER2+; and patient 3 was 53 years old, and the tumor was ER+/HER2−. All research was performed in accordance with New York University and Cold Spring Harbor Laboratory Institutional Review Board guidelines and regulations, and written informed consent was obtained from all patients.
Animals
OT1 mice and TLR4 KO (knockout) mice (on C57BL/6 [referred to hereafter as BL/6] background) were purchased from Jackson Laboratory. MMTV-PyMT mice (on BL/6 or FVB/N background) were bred at CSHL. To obtain MMTV-PyMT; ACTB-ECFP mice, ACTB-ECFP mice (on BL/6 background, originally obtained from Jackson Laboratory) were intercrossed with MMTV-PyMT mice. MMTV-PyMT-chOVA mice (Engelhardt et al., 2012) were a kind gift from Dr. Mathew Krummel (University of California, San Francisco).
Six- to 8-week-old female BL/6 and BALB/c host mice were purchased from Jackson Laboratory and acclimated to the animal housing facility for one week prior to performing experiments. Animals were housed All animal experiments were conducted in accordance with procedures approved by the Institutional Animal Care and Use Committee at CSHL and the National Institutes of Health (NIH) Guide for the Care and Use of Laboratory Animals.
Tumor mouse models
PyMT cancer cells for orthotopic transplantation were isolated from the primary tumors of MMTV-PyMT mice or MMTV-PyMT-chOVA mice (BL/6 background). The cancer cells were isolated and were injected immediately into the 4th mammary fat pads of BL/6 mice (2×105 cells/fat pad in 20 μl 1:1 PBS/Matrigel, 2 tumors per mouse) to obtain the transplanted PyMT tumor model. For 4T1 cells, 5×104 cells/fat pad were transplanted into the 4th mammary fat pads of BALB/c mice (2 tumors per mouse). ID8-p53−/− ovarian cancer cells were cultured as previously described (Walton et al., 2016). A total of 5×106 ID8-p53−/− cells (in 200 μl PBS) were injected intraperitoneally into BL/6 mice.
METHOD DETAILS
Isolation of macrophages and tumor cells from pleural effusions of breast cancer patients
Pleural effusions were shipped on ice to CSHL, and cells were isolated less than 24 hours after sample collection, as follows: the effusions were spun down at 450 x g for 10 minutes to remove the liquid supernatant, and the cell pellet was washed with Dulbecco’s Phosphate-Buffered Saline (DPBS) twice, then resuspended in Ammonium-Chloride-Potassium (ACK) lysis buffer on ice for 5–10 minutes; an additional spin down at 300 x g for 10 minutes was performed to eliminate the lysed red blood cells. Then, CD326 (EpCAM) microbeads were applied to enrich for EpCAM+ and EpCAM− cells, according to the manufacturer’s instructions. Macrophages were obtained from the EpCAM− cells after 2 hours of adherence to the tissue culture dish and were cultured with 50 ng/ml human macrophage colony-stimulating factor (M-CSF) in RPMI-1640 containing 20% fetal bovine serum (FBS), 200 μM L-glutamine, 100 μM sodium pyruvate, and 50 μM 2-mercaptoethanol. EpCAM+ cells were cultured with Dulbecco’s Modified Eagle Medium (DMEM)-F12 containing 20% FBS, 2% B27, 20 ng/ml human EGF, and 4 μg/ml insulin.
Isolation and culturing of macrophages and cancer cells from primary breast tumors of mice
The primary tumors (6–8 mm in diameter) of MMTV-PyMT mice (on BL/6 background) were mechanically dissociated and digested with commercial collagenase/hyaluronidase solution containing DNase I (4 U/ml) at 37 °C for 45 minutes. After pulse centrifugation (450 x g), the supernatant (mostly immune cells) and cell pellets (mostly cancer cells) were collected. CD11b MicroBeads were used to enrich CD11b+ cells from the supernatant, and macrophages were obtained by allowing the CD11b+ cells to adhere to the tissue culture dish for 2 hours (non-adherent cells were removed by washing with PBS). Cancer cells were isolated from the tumor cell pellets and dissociated into single tumor cells with TrypLE Express Enzyme containing DNase I (4 U/ml) at 37 °C for 10 minutes.
Mouse macrophages were cultured with RPMI-1640 containing 20% FBS, 200 μM L-glutamine, 100 μM sodium pyruvate, 50 μM 2-mercaptoethanol, and 10 ng/ml mouse M-CSF. Cancer cells were cultured with DMEM + 10% FBS.
Isolation of bone marrow-derived macrophages
Macrophages were differentiated from the bone marrow of wild-type (WT) BL/6 mice and TLR4 KO mice. The bone marrow was cultured for 6 days in the mouse macrophage culture medium on Not TC-treated culture dishes. The attached cells at the end of this culture period are unpolarized macrophages (Marim et al., 2010).
In vitro macrophage and tumor cell co-culture assay
A 24-well glass bottom plate was coated with fibronectin (1:100). For the direct contact condition, macrophages were seeded (1.5×105/well) to the plate and 2 hours later, labeled with CellTracker™ Deep Red Dye (1 μM). Tumor cells were labeled with CellTracker™ Green CMFDA (5-chloromethylfluorescein diacetate) Dye (1 μM) and seeded (5×104/well) to the plate. From that point forward, the culture medium was changed from a macrophage culture medium to cancer cell medium. Two hours later, LPS (100 ng/ml), MPLA (a low-toxicity derivative of LPS; 100 ng/ml; this product is produced by chemical synthesis and contains 6 fatty acyl groups), polyI:C (1 μg/ml), mouse IFNγ (33 ng/ml) or human IFNγ (50 ng/ml), TMP195 (a class IIa HDAC inhibitor, 500nM), or IPI-549 (a PI3Kγ inhibitor, 1 μM) was added to the medium (time point: 0 hour), alone or in indicated combinations, and live cell imaging was performed with a Yokagawa spinning disk confocal microscope (Solamere Technology Group) and LiveCell Stage Top Incubation System (Pathology Devices) for 48 hours. To inhibit iNOS activity, L-NIL (N6-(1-Iminoethyl)-L-lysine hydrochloride, 200 μM) was added to the medium before MPLA+IFNγ. For the conditioned medium (CM) condition, macrophages were treated with MPLA+IFNγ for 24 hours before collection of the CM. Tumor cells were fluorescently labeled and seeded to the plate in DMEM + 10% FBS. Two hours later, the medium was replaced with the CM from MPLA+IFNγ-treated macrophages (time point: 0 hour). For the transwell condition, macrophages were seeded to the cell culture insert (pore size: 1 μm), and tumor cells were seeded to the lower glass bottom plate. Two hours later, MPLA+IFNγ was added to the medium of macrophages (time point: 0 hour). In all the conditions, cancer cell numbers at 0 hour and 48 hours were determined by counting the cells in 5 fields of view, and cell numbers at 48 hours relative to those at 0 hour were then calculated and reported.
MTS assay
PyMT tumor cells were seeded to a 96-well plate (5×103/well) and treated with LPS (100 ng/ml), MPLA (100 ng/ml), or polyI:C (1 μg/ml) with or without mouse IFNγ (33 ng/ml) for 48 hours. Next, 20 μl CellTiter 96® AQueous One Solution reagent was added to 100 μl medium and incubated for 2 hours at 37 °C. Absorbance at 490 nm was measured using a SpectraMax MiniMax 300 Imaging Cytometer (Molecular Devices).
RNA extraction and quantitative real-time PCR
Total RNA from macrophages (cultured alone) or primary tumors was extracted using an RNeasy Mini kit, following the manufacturer’s instructions. Reverse transcription was performed with TaqMan™ Reverse Transcription Reagents. Quantitative real-time PCR was performed in a 384-well format on a QuantStudio 6-flex Instrument (Applied Biosystems) using PowerUp™ SYBR™ Green Master Mix or Taqman™ Universal Master Mix II. Relative quantitation was performed with the 2(−ΔΔCT) method using ACTB, Actb, or Tbp expression for normalization. The PCR primers are listed in the Key Resources Table and Table S2.
Western blot
Macrophages were lysed by RIPA lysis buffer with protease and phosphatase inhibitors. Protein extracts were resolved through 8% SDS-PAGE, transferred to a polyvinylidene difluoride (PVDF) membrane, probed with antibodies against iNOS (1:1000), CD206 (1:1000), STAT1 (1:1000), p-STAT1 (Tyr701, 1:1000), NFκB p65 (1:1000), p-p65 (Ser536, 1:1000), or β-actin (1:2000), GAPDH (1:2000), and then with fluorescent secondary antibody (1:2000). Binding was visualized by an Odyssey® Classic Imaging System.
In vivo tumor experiments
For PyMT and 4T1 tumors, tumor size was measured by caliper, and tumor volume was calculated as volume = (length×width2)/2. Treatment was initiated when tumors reached 5–6 mm in size and was given twice a week: 1 μg MPLA/tumor and indicated doses of mouse IFNγ were injected into the tumors to optimize the doses by comparing their inhibitory effects on tumor growth. Because the growth of PyMT tumors treated with MPLA+0.33, 1, or 3 μg IFNγ was almost equally suppressed compared to controls (Figure S2A) and the growth of 4T1 tumors treated with MPLA+3 or 9 μg IFNγ was inhibited similarly (Figure S2B), we chose the following doses: 1 μg MPLA with 1 μg IFNγ per tumor (or 1 μg MPLA alone or 1 μg IFNγ alone) was used for the intratumoral injection of PyMT tumors, and 1 μg MPLA with 3 μg IFNγ per tumor (or 1 μg MPLA alone or 3 μg IFNγ alone) was used for the intratumoral injection of 4T1 tumors. For the spontaneous MMTV-PyMT tumors (BL/6 background and FVB/N background), treatment was initiated when the total tumor volume of each mouse was approximately 150 mm3, and tumors that had reached 5–6 mm were intratumorally injected with 1 μg MPLA+1 μg IFNγ twice a week. Total volumes of all tumors were calculated for each mouse and reported. For intraperitoneal administration to mice with PyMT tumors, 30 μg MPLA with 30 μg IFNγ per mouse were used. At the indicated time points (tumor size<2 cm), mice were euthanized in a CO2 chamber before performing a cardiac perfusion with PBS, and tumors and lungs were removed for analysis.
For the toxicology analysis experiments (shown in Figure 2K–M), tumor-free C57BL/6 mice were intraperitoneally injected with 30 μg MPLA+30 μg IFNγ every 3 or 4 days, for six times total. The body weight of the mice was measured before each injection. Two days after the last treatment, the mice were euthanized. The serum was collected for Comprehensive Chemistry Test, and liver, lung, kidney, heart, spleen, and femur were collected for hematoxylin and eosin (H&E) staining.
For the tumor re-challenge experiments (shown in Figure 6C), transplanted PyMT tumors were treated with MPLA+IFNγ (intratumoral injection) four times. Two days after the last treatment, breast tumors were surgically removed by making a small incision at the skin near the site of the tumor. The tumor along with 4th mammary fat pad was excised and the wound was closed using Relfex 7 mm wound clips. Two weeks after tumor removal, ECFP+ PyMT cancer cells were injected into the tail vein of the mice. The ECFP+ PyMT cancer cells were isolated from the primary tumors of MMTV-PyMT; ACTB-ECFP mice as described above, and 5×105 cells (in 100 μl PBS) were injected into the tail vein of the mice whose tumors had been surgically removed. Seventeen days later, the lungs were removed for analysis.
At indicated times, 10 μg MPLA with 20 μg mouse IFNγ was administered intraperitoneally to the ID8-p53−/− tumor-bearing mice (Figure 7A–D, every 5 days), for six times total. At the end of the experiment (2 days after last treatment), the mice were weighed and intraperitoneally injected with PBS, and the ascites/peritoneal lavage was collected (~12 ml collected per mouse) for cell counting and flow cytometry. Ascites/peritoneal lavage was spun down at 300 x g for 10 minutes, and the cell pellet was resuspended in ACK lysis buffer on ice for 5–10 minutes, then spun down at 300 x g for an additional 10 minutes to eliminate the red blood cells. The total cell numbers were determined by a CellDrop cell counter. For the survival experiments, cisplatin (10 μg/g body weight) was administrated intraperitoneally to the mice every week, for a total of four times. MPLA (10 μg) with mouse IFNγ (20 μg) was administered as indicated (Figure 7E, every 5 days; Figure 7F, every 3–4 days). Mice were euthanized when the presence of ascites was evident and affected the mobility of the mouse.
For the comparison experiments of different approaches (shown in Figure S2G–I and Figure S7G–I), MPLA+IFNγ or anti-PD 1 antibody (200 μg/mouse, intraperitoneal injection every 3 days, three times in total) was administered on days indicated by arrows in figures showing the experimental design (Figure S2G and Figure S7G). During the indicated period, TMP195 (50 mg/kg body weight, in 10% DMSO + 90% sesame oil) was administrated intraperitoneally to the mice every day; IPI-549 and PLX3397 (pexidartinib, a CSF1R inhibitor) were administered in the mouse chow. The chow was custom-made. To obtain estimated daily doses (IPI-549: 15 mg/kg body weight/day and PLX3399: 40 mg/kg body weight/day), we assumed a female mouse body weight of 20 g and a daily chow intake of 3.5 g.
Lung metastasis analysis
Lungs were placed in PBS in a vacuum desiccator for 2 hours, then fixed in 4% paraformaldehyde (PFA) for 4 hours, infiltrated with 30% sucrose in PBS for 48 hours, and embedded in OCT compound. The blocks were then placed in a custom-made cutting chamber with razor blade inserts every 2 mm, and the block was cut into 2 mm sections. These sectioned lung pieces were then re-embedded in fresh OCT and sent to the Histology Core at CSHL for cross-sectional cuts from the entire lung tissue and for H&E staining. The metastatic burden was calculated as the percentage of metastasis/lung area, determined using Aperio eSlide Capture Devices software (Leica Biosystems).
ELISA for NETs
Plasma samples were obtained by cardiac blood collection using a syringe with a 25 G needle containing 25 μl sodium citrate solution. Whole blood was centrifuged at 1,300 x g for 10 minutes at 4 °C, and the top plasma layer was collected. As in a previous study (Albrengues et al., 2018), 96-well Enzyme ImmunoAssay/Radio Immuno-Assay (EIA/RIA) plates were coated overnight at 4 °C with an anti-elastase antibody (1:250) in 15 mM Na2CO3 and 35 mM NaHCO3 at pH 9.6. The next day, the wells were washed three times with PBS, blocked in 5% bovine serum albumin (BSA) for 2 hours at room temperature, and washed three times with PBS. Then, 50 μl plasma samples were added to the wells and incubated for 2 hours at room temperature on a shaker, and the plates were washed three times with a wash buffer (1% BSA, 0.05% Tween 20 in PBS). Next, anti-DNA-peroxidase conjugated antibody (1:50, part of Cell Death Detection ELISA Kit) in 1% BSA in PBS was added to the wells for 2 hours at room temperature, and the wells were washed five times with wash buffer before the addition of 2,2’-azino-bis(3-ethylbenzothiazoline-6-sulphonic acid) (ABTS). Optical density was read 40 minutes later at 405 nm using a SpectraMax MiniMax 300 Imaging Cytometer (Molecular Devices).
Cytokine array
Twenty-four hours after the second intratumoral injection, transplanted PyMT tumors were collected, weighed, and digested with commercial collagenase/hyaluronidase (25 mg tumor/1 ml enzyme solution) containing DNase I at 37 °C for 30–45 minutes. The supernatant that contained secreted cytokines was collected and spun down to remove cells and cell debris (12,000 x g, 30 minutes). Supernatant samples (1 ml) and a Proteome Profiler Mouse XL Cytokine Array Kit were used according to the manufacturer’s instructions. Films were scanned, and spots were analyzed using ImageJ software. The heatmap was generated with http://www2.heatmapper.ca/expression/.
ELISA of cytokines
The samples were collected in the same manner as for the cytokine array analysis. CXCL9 and CCL5 secreted by tumors were assayed using a DuoSet ELISA and ancillary reagent kit according to the manufacturer’s guidelines. Absorbance at 450 nm was measured using a SpectraMax MiniMax 300 Imaging Cytometer (Molecular Devices).
Sample and library preparation for RNA-seq
Twenty-four hours after the second intratumoral injection, transplanted PyMT tumors were collected and total RNA was extracted using an RNeasy Mini Kit. RNA-seq libraries were prepared using a TruSeq Stranded mRNA Library Prep Kit and sequenced using aNextSeq500 High Output Illumina platform with 76 base single end reads at the CSHL Next Generation Sequencing Core Facility. This procedure yielded an average of 33.3 million filtered reads per sample.
Flow cytometry
Two days after the last intratumoral or intraperitoneal injection, tumors were harvested and processed as previously described (Fein et al., 2020). Briefly, tumors were mechanically dissociated and digested with collagenase/hyaluronidase containing DNase I (4 U/ml) at 37 °C for 30–45 minutes, lysed with ACK lysis buffer, and filtered with a 40 μm cell strainer. Lungs were digested with Dispase (2.5 U/ml)/Collagenase D (0.1 mg/ml)/Liberase DL (0.2 mg/ml) containing DNase I (25 U/ml) at 37 °C for 30 minutes. For flow cytometry staining, cells (1×106) were incubated with mouse Fc receptor blocker at 4 °C for 10 minutes, then stained with the appropriate antibodies to surface markers at 4 °C for 30 minutes in the dark, and/or fixed/permeabilized (Fixation/Permeabilization Solution Kit) and stained with intracellular antibodies at 4 °C for 30 minutes. The stained populations were analyzed using a Fortessa flow cytometer (BD Biosciences) and FlowJo software (BD Biosciences). The antibodies used are listed in detail in the Key Resources Table. The PyMT tumors in our model are derived from transplanted primary cells, and the percentages of CD8/CD4 and F4/80 out of CD45+ cells varied between different experiments, likely due to the diversity of the PyMT tumors used to generate donor cells in individual experiments. We compared treatments only between groups of mice transplanted with the same primary cell population.
Immunofluorescence staining
Tumors were fixed in 4% PFA, infiltrated with 30% sucrose in PBS, and embedded in OCT compound, except the samples for staining p-STAT1 and p-p65, which were embedded in paraffin. The lungs were embedded in OCT compound. Tissues were cryosectioned at the Histology Core at CSHL. OCT compound-embedded tissue sections were post-fixed with ice-cold methanol and acetone (1:1 ratio), rinsed with PBS, blocked with Fc receptor blocker, and incubated with 1 × blocking buffer (5% goat serum, 2.5% BSA, 0.1% Triton X-100 in PBS) (Albrengues et al., 2018). Tumor sections were then incubated with fluorochrome-conjugated antibody (1:100, Alexa Fluor 488 anti-mouse F4/80; or PE anti-mouse CD40; or PE anti-iNOS; or FITC anti-mouse CD8a) in 0.5 × blocking buffer overnight at 4 °C. Lung sections were incubated with anti-PyMT antigen antibody (1:100) overnight. The second day, tissue sections were counterstained with DAPI and mounted with mounting media, while lung sections were incubated with Alexa Fluor 488 goat anti-Rat secondary antibody (1:1500) for 45 minutes, then incubated with Alexa Fluor 647 anti-mouse CD4 (1:100) and PE anti-mouse CD8a (1:100) overnight. The third day, lung sections were counterstained with DAPI and mounted with mounting media. Images were obtained with a Leica SP8 confocal microscope. The percentage of F4/80, CD8a, CD40, or iNOS positive cells in tumor sections was determined using ImageJ.
For the staining of F4/80 with p-STAT1 or p-p65, paraffin sections were deparaffinized and rehydrated and antigen retrieval was carried out by boiling slides in Tris EDTA buffer (10 mM Tris Base, 1 mM EDTA, pH 9.0) for 10 min in a pressure cooker. The slides were blocked and incubated with anti-F4/80 antibody (1:100), and anti-p-STAT1 antibody (1:100) or anti-p-p65 antibody (1:50) overnight at 4°C. The second day, the sections were incubated with Alexa Fluor 488 goat anti-Rat secondary antibody (1:1500) and Alexa Fluor 568 goat anti-Rabbit secondary antibody (1:1500), and were counterstained with DAPT. The numbers of p-STAT1+F4/80+ cells and the numbers of p-p65+F4/80+ cells per field of view were determined.
For the tumor re-challenge assay, lung sections were stained with anti-ECFP antibody (1:100) and Alexa Fluor 488 goat anti-rabbit secondary antibody (1:1500). Images were scanned using an Aperio ScanScope® FL System (Leica Biosystems), and ECFP+ lung metastasis was determined using Aperio eSlide Capture Devices software (Leica Biosystems).
In vitro T cell activation experiments
CD8+ T cells were isolated from the spleens of transplanted PyMT tumor-bearing mice with a CD8a+ T Cell Isolation Kit, according to the manufacturer’s instructions. Macrophages were differentiated from the bone marrow of these mice. CD8+ T cells were cultured with or without macrophages under different experimental conditions, as indicated below and in the figure legends, in RPMI-1640 with 20% FBS, 200 μM L-glutamine, 100 μM sodium pyruvate, and 50 μM 2-mercaptoethanol. Macrophages were seeded 2 hours before T cells to allow for their attachment, and MPLA (100 ng/ml), mouse IFNγ (33 ng/ml), or the two together were added to the culture system immediately after the seeding of T cells. T cell activation was identified by CD69 expression using flow cytometry, similar to the experiments described above using cells isolated from tumors. To block the cytokines that activate CD8+ T cells, neutralizing antibodies against IL-12 (1 μg/ml), TNFα (5 μg/ml), or CXCL9 (6 μg/ml) were added to the co-culture system 30 minutes before MPLA+IFNγ.
For the antigen-presenting assay, CD8+ T cells were isolated from the spleens of OT1 mice with a CD8a+ T Cell Isolation Kit. Macrophages were derived from the bone marrow of PyMT tumor-bearing mice as above and treated with MPLA (100 ng/ml), IFNγ (33 ng/ml), or the two together for 16 hours, then incubated with OVA-Q4H7 peptide (10−9 M) for 2 hours. The pre-treated macrophages were washed with PBS and finally added to CD8+ T cells. CD69 expression of CD8+ T cells was identified by flow cytometry.
Immune cell depletion/neutralization experiments
Anti-CD4, anti-CD8, anti-CSFIR, or anti-CSF1 antibodies were injected intraperitoneally one day before treatment with MPLA+IFNγ. For the first depletion, 0.5 mg of anti-CD4, 0.5 mg of anti-CD8, 1 mg of anti-CSF1R, or 1 mg of anti-CSF1 were injected per mouse. For the subsequent depletions, 0.2 mg of anti-CD4, 0.2 mg of anti-CD8, 0.5 mg of anti-CSF1R, or 0.5 mg of anti-CSF1 were injected per mouse.
QUANTIFICATION AND STATISTICAL ANALYSIS
Statistical methods
Prism software (version 8.4.1, GraphPad) was used to analyze the data. In survival experiments, Kaplan-Meier survival curves were plotted and a log rank test was performed. In all other experiments with only two groups, the unpaired t-test (equal variances) or unpaired Welch’s t-test (unequal variances) was used. One-way ANOVA (with equal variances) or Welch’s ANOVA (unequal variances) was performed for experiments with more than two groups. Bartlett’s test or Brown-Forsythe test was used to assess equality of variances among groups. When the ANOVA was significant, group means were compared as indicated in the figures, and p-values adjusted for multiple comparisons were obtained using the Holm-šidák procedure (equal variances) or Dunnett T3 procedure (unequal variances). All graphs present the mean, the error bars represent the standard deviation (SD), and p-values are presented in the figures. All p-values<0.05 were considered statistically significant. In all in vitro experiments, the average results were taken from at least three independent experiments or three patients. In all in vivo experiments, the results are representative of at least two independent experiments (the numbers of mice and statistical tests are listed in the figure legends). Mice were randomized before the administration of therapy.
Analysis of RNA-seq data
FASTQ sequencing files were processed and analyzed using the Galaxy server hosted by the CSHL Bioinformatics Core Facility. Reads were aligned to the GRCm38/mm10 reference mouse genome using STAR (Dobin et al., 2012). Gene read counts were determined using FeatureCounts (Liao et al., 2013). Differential gene expression analysis was performed using DESeq2 (Love et al., 2014), with a 0.1 false discovery rate (FDR)-adjusted p-value cutoff for significance. DESeq2-normalized read counts of the top genes that were uniquely changed in the MPLA+IFNγ group were visualized as a heatmap using http://www2.heatmapper.ca/expression/. Genes that were uniquely upregulated or downregulated in the MPLA+IFNγ group were used as the input for biological process gene ontology analysis (http://geneontology.org/).
Supplementary Material
Data file S1. List of upregulated and downregulated genes in the MPLA+IFNγ, MPLA, and IFNγ treatment groups, related to Figure 3. RNA-seq data were collected following the experimental design in Figure 3A. After the combined MPLA+INFγ treatment, 179 genes were upregulated and 36 genes were downregulated in PyMT tumors, compared to vehicle. After treatment with MPLA alone, 20 genes were upregulated and 5 genes were downregulated. After treatment with INFγ alone, 33 genes were upregulated and 15 genes were downregulated.
Movie S1. Un-activated breast tumor-derived macrophages do not kill cancer cells, related to Figure 1. Macrophages and PyMT tumor cells were isolated from the primary tumors of MMTV-PyMT mice, and macrophages were stained with CellTracker™ Deep Red Dye, while tumor cells were stained with CellTracker™ CMFDA Dye. Deep Red dye is falsely colored green for clarity. Time indicated is time after imaging was initiated.
Movie S2. MPLA+IFNγ activates breast tumor-derived macrophages to kill cancer cells, related to Figure 1. Macrophages and PyMT tumor cells were isolated from the primary tumors of MMTV-PyMT mice, and macrophages were stained with CellTracker™ Deep Red Dye, while tumor cells were stained with CellTracker™ CMFDA Dye. Deep Red dye is falsely colored green for clarity. MPLA with mouse IFNγ was added before imaging was initiated. Time indicated is time after imaging was initiated.
Supplemental Figures and Tables
Highlights.
MPLA+IFNγ repolarizes TAMs from mice and patients to tumoricidal macrophages.
MPLA+IFNγ activates cytotoxic T cells through macrophage-secreted IL-12 and TNFα.
MPLA+IFNγ suppresses tumor growth & metastasis in breast and ovarian cancer models.
MPLA+IFNγ enhances the response to chemotherapy in ovarian cancer.
Acknowledgments
We thank the patients and the staff of the Clinical Trials Office of New York University (NYU) Langone’s Perlmutter Cancer Center and the Center for Biospecimen Research and Development at NYU Langone Health. We thank J. Erby Wilkinson (University of Michigan) for the histological analysis of tissues. This work was supported by grants from the New York State Department of Health to M.E. and S.A. (DOH01-C31850GG-3450000; the opinions, results, findings, and/or interpretations of data contained therein do not necessarily represent the opinions, interpretations, or policy of the State); the Katie Oppo Research Fund and Swim Across America funds to M.E.; METAvivor Research and Support, Inc. to L.S.; NCI 5P01CA013106-Project 3 to D.L.S.; Ovarian Cancer Action (OCARC18-22) to I.A.M.; and by funds from the CSHL Cancer Center (P30-CA045508) and the NYU Perlmutter Cancer Center (P30CA016087, Center for Biospecimen Research and Development at NYU Langone Health). The graphic abstract was drawn using pictures from Servier Medical Art (http://smart.servier.com/), licensed under a Creative Commons Attribution 3.0 Unported License (https://creativecommons.org/licenses/by/3.0/).
Declaration of interests:
M.E. is a member of the research advisory board for brensocatib for Insmed; a member of the scientific advisory board for Vividion Therapeutics; a consultant for Protalix; and holds shares in Agios. I.A.M. is on the Advisory Boards for Clovis Oncology, AstraZeneca, GSK, Epsila Bio, Roche and Scancell; has institutional funding from AstraZeneca; and is a Trustee of Worldwide Cancer Research. S.A. has uncompensated consulting or advisory roles with Bristol Meyers Squibb, Genentech, and Merck; and research funding to her institution from Amgen, Bristol Meyers Squibb, Celgene, Genentech, Merck and Novartis. T.K. is currently an employee of Novartis. The other authors declare that they have no conflicts of interest.
Footnotes
Publisher's Disclaimer: This is a PDF file of an unedited manuscript that has been accepted for publication. As a service to our customers we are providing this early version of the manuscript. The manuscript will undergo copyediting, typesetting, and review of the resulting proof before it is published in its final form. Please note that during the production process errors may be discovered which could affect the content, and all legal disclaimers that apply to the journal pertain.
References
- Ackerman SE, Pearson CI, Gregorio JD, Gonzalez JC, Kenkel JA, Hartmann FJ, Luo A, Ho PY, LeBlanc H, Blum LK, et al. (2021). Immune-stimulating antibody conjugates elicit robust myeloid activation and durable antitumor immunity. Nature Cancer 2, 18–33. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S (2009). Toll-like receptor agonists in cancer therapy. Immunotherapy 1, 949–964. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Adams S, Kozhaya L, Martiniuk F, Meng TC, Chiriboga L, Liebes L, Hochman T, Shuman N, Axelrod D, Speyer J, et al. (2012). Topical TLR7 agonist imiquimod can induce immune-mediated rejection of skin metastases in patients with breast cancer. Clin. Cancer Res 18, 6748–6757. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Albrengues J, Shields MA, Ng D, Park CG, Ambrico A, Poindexter ME, Upadhyay P, Uyeminami DL, Pommier A, Kuttner V, et al. (2018). Neutrophil extracellular traps produced during inflammation awaken dormant cancer cells in mice. Science 361, eaao4227. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, and Condeelis JS (2018). A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Rep. 23, 1239–1248. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Autio KA, Klebanoff CA, Schaer D, Kauh JSW, Slovin SF, Adamow M, Blinder VS, Brahmachary M, Carlsen M, Comen E, et al. (2020). Immunomodulatory Activity of a Colony-stimulating Factor-1 Receptor Inhibitor in Patients with Advanced Refractory Breast or Prostate Cancer: A Phase I Study. Clin. Cancer Res 26, 5609–5620. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Boehm U, Klamp T, Groot M, and Howard JC (1997). CELLULAR RESPONSES TO INTERFERON-γ. Annu. Rev. Immunol 15, 749–795. [DOI] [PubMed] [Google Scholar]
- Butowski N, Colman H, De Groot JF, Omuro AM, Nayak L, Wen PY, Cloughesy TF, Marimuthu A, Haidar S, Perry A, et al. (2015). Orally administered colony stimulating factor 1 receptor inhibitor PLX3397 in recurrent glioblastoma: an Ivy Foundation Early Phase Clinical Trials Consortium phase II study. Neuro Oncol. 18, 557–564. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cannarile MA, Weisser M, Jacob W, Jegg A-M, Ries CH, and Rüttinger D (2017). Colony-stimulating factor 1 receptor (CSF1R) inhibitors in cancer therapy. J. Immunother. Cancer 5, 53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, and Kitamura T (2018). Targeting Tumor-Associated Macrophages as a Potential Strategy to Enhance the Response to Immune Checkpoint Inhibitors. Front. Cell Dev. Biol 6, 38. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cassetta L, and Pollard JW (2018). Targeting macrophages: therapeutic approaches in cancer. Nat. Rev. Drug Discov 17, 887–904. [DOI] [PubMed] [Google Scholar]
- Castro F, Cardoso AP, Gonçalves RM, Serre K, and Oliveira MJ (2018). Interferon-Gamma at the Crossroads of Tumor Immune Surveillance or Evasion. Front. Immunol 9, 847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chow LQM, Morishima C, Eaton KD, Baik CS, Goulart BH, Anderson LN, Manjarrez KL, Dietsch GN, Bryan JK, Hershberg RM, et al. (2017). Phase Ib Trial of the Toll-like Receptor 8 Agonist, Motolimod (VTX-2337), Combined with Cetuximab in Patients with Recurrent or Metastatic SCCHN. Clin. Cancer Res 23, 2442–2450. [DOI] [PubMed] [Google Scholar]
- Clark NM, Martinez LM, Murdock S, deLigio JT, Olex AL, Effi C, Dozmorov MG, and Bos PD (2020). Regulatory T Cells Support Breast Cancer Progression by Opposing IFN-γ-Dependent Functional Reprogramming of Myeloid Cells. Cell Rep. 33, 108482. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Croft M (2009). The role of TNF superfamily members in T-cell function and diseases. Nat. Rev. Immunol 9, 271–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, Gallagher WM, Wadhwani N, Keil SD, Junaid SA, et al. (2011). Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 1, 54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dinapoli MR, Calderon CL, and Lopez DM (1996). The altered tumoricidal capacity of macrophages isolated from tumor-bearing mice is related to reduce expression of the inducible nitric oxide synthase gene. J. Exp. Med 183, 1323–1329. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, Batut P, Chaisson M, and Gingeras TR (2012). STAR: ultrafast universal RNA-seq aligner. Bioinformatics 29, 15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, Egeblad M, and Krummel MF (2012). Marginating dendritic cells of the tumor microenvironment cross-present tumor antigens and stably engage tumor-specific T cells. Cancer cell 21, 402–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Etxeberria T, Bolanos E, Quetglas JI, Gros A, Villanueva A, Palomero J, Sanchez-Paulete AR, Piulats JM, Matias-Guiu X, Olivera I, et al. (2019). Intratumor Adoptive Transfer of IL-12 mRNA Transiently Engineered Antitumor CD8+ T Cells. Cancer Cell 36, 613–629. [DOI] [PubMed] [Google Scholar]
- Fein MR, He X-Y, Almeida AS, Bružas E, Pommier A, Yan R, Eberhardt A, Fearon DT, Van Aelst L, Wilkinson JE, et al. (2020). Cancer cell CCR2 orchestrates suppression of the adaptive immune response. J. Exp. Med 217, e20181551. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gao J, Morrison DC, Parmely TJ, Russell SW, and Murphy WJ (1997). An Interferon-γ-activated Site (GAS) Is Necessary for Full Expression of the Mouse iNOS Gene in Response to Interferon-γ and Lipopolysaccharide. J. Biol. Chem 272, 1226–1230. [DOI] [PubMed] [Google Scholar]
- Guerriero JL, Sotayo A, Ponichtera HE, Castrillon JA, Pourzia AL, Schad S, Johnson SF, Carrasco RD, Lazo S, Bronson RT, et al. (2017). Class IIa HDAC inhibition reduces breast tumours and metastases through anti-tumour macrophages. Nature 543, 428–432. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Herzog TJ (2006). The current treatment of recurrent ovarian cancer. Curr. Oncol. Rep 8, 448–454. [DOI] [PubMed] [Google Scholar]
- Jallad M-AN, Jurjus AR, Rahal EA, and Abdelnoor AM (2020). Triple Immunotherapy Overcomes Immune Evasion by Tumor in a Melanoma Mouse Model. Front. Oncol 10, 839. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaczanowska S, Beury DW, Gopalan V, Tycko AK, Qin H, Clements ME, Drake J, Nwanze C, Murgai M, Rae Z, et al. (2021). Genetically engineered myeloid cells rebalance the core immune suppression program in metastasis. Cell 184, 2033–2052. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kaneda MM, Messer KS, Ralainirina N, Li H, Leem CJ, Gorjestani S, Woo G, Nguyen AV, Figueiredo CC, Foubert P, et al. (2016). PI3Kγ is a molecular switch that controls immune suppression. Nature 539, 437–442. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kelm M (1999). Nitric oxide metabolism and breakdown. Biochim. Biophy. Acta Bioenerg 1411, 273–289. [DOI] [PubMed] [Google Scholar]
- Key LL Jr., Ries WL, Rodriguiz RM, and Hatcher HC (1992). Recombinant human interferon gamma therapy for osteopetrosis. J. Pediatr 121, 119–124. [DOI] [PubMed] [Google Scholar]
- Khalil DN, Suek N, Campesato LF, Budhu S, Redmond D, Samstein RM, Krishna C, Panageas KS, Capanu M, Houghton S, et al. (2019). In situ vaccination with defined factors overcomes T cell exhaustion in distant tumors. J. Clin. Invest 129, 3435–3447. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kitamura T, Doughty-Shenton D, Cassetta L, Fragkogianni S, Brownlie D, Kato Y, Carragher N, and Pollard JW (2017). Monocytes Differentiate to Immune Suppressive Precursors of Metastasis-Associated Macrophages in Mouse Models of Metastatic Breast Cancer. Front. Immunol 8, 2004. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kowal J, Kornete M, and Joyce JA (2019). Re-education of macrophages as a therapeutic strategy in cancer. Immunotherapy 11, 677–689. [DOI] [PubMed] [Google Scholar]
- Lal H, Cunningham AL, Godeaux O, Chlibek R, Diez-Domingo J, Hwang SJ, Levin MJ, McElhaney JE, Poder A, Puig-Barbera J, et al. (2015). Efficacy of an adjuvanted herpes zoster subunit vaccine in older adults. N. Engl. J. Med 372, 2087–2096. [DOI] [PubMed] [Google Scholar]
- Liao Y, Smyth GK, and Shi W (2013). featureCounts: an efficient general purpose program for assigning sequence reads to genomic features. Bioinformatics 30, 923–930. [DOI] [PubMed] [Google Scholar]
- Lim B, Woodward WA, Wang X, Reuben JM, and Ueno NT (2018). Inflammatory breast cancer biology: the tumour microenvironment is key. Nat. Rev. Cancer 18, 485–499. [DOI] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lu Y-C, Yeh W-C, and Ohashi PS (2008). LPS/TLR4 signal transduction pathway. Cytokine 42, 145–151. [DOI] [PubMed] [Google Scholar]
- Marim FM, Silveira TN, Lima DS Jr., and Zamboni DS (2010). A method for generation of bone marrow-derived macrophages from cryopreserved mouse bone marrow cells. PloS one 5, e15263. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Martin E, Nathan C, and Xie QW (1994). Role of interferon regulatory factor 1 in induction of nitric oxide synthase. J. Exp. Med 180, 977–984. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mata-Haro V, Cekic C, Martin M, Chilton PM, Casella CR, and Mitchell TC (2007). The Vaccine Adjuvant Monophosphoryl Lipid A as a TRIF-Biased Agonist of TLR4. Science 316, 1628. [DOI] [PubMed] [Google Scholar]
- Mehta AK, Gracias DT, and Croft M (2018). TNF activity and T cells. Cytokine 101, 14–18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Monie A, Hung C-F, Roden R, and Wu TC (2008). Cervarix: a vaccine for the prevention of HPV 16, 18-associated cervical cancer. Biologics 2, 97–105. [PMC free article] [PubMed] [Google Scholar]
- Nakasone ES, Askautrud HA, Kees T, Park JH, Plaks V, Ewald AJ, Fein M, Rasch MG, Tan YX, Qiu J, et al. (2012). Imaging tumor-stroma interactions during chemotherapy reveals contributions of the microenvironment to resistance. Cancer Cell 21, 488–503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Parker BS, Rautela J, and Hertzog PJ (2016). Antitumour actions of interferons: implications for cancer therapy. Nat. Rev. Cancer 16, 131–144. [DOI] [PubMed] [Google Scholar]
- Pathria P, Louis TL, and Varner JA (2019). Targeting Tumor-Associated Macrophages in Cancer. Trends Immunol. 40, 310–327. [DOI] [PubMed] [Google Scholar]
- Peranzoni E, Lemoine J, Vimeux L, Feuillet V, Barrin S, Kantari-Mimoun C, Bercovici N, Guerin M, Biton J, Ouakrim H, et al. (2018). Macrophages impede CD8 T cells from reaching tumor cells and limit the efficacy of anti-PD-1 treatment. Proc. Natl. Acad. Sci. U S A 115, E4041–E4050. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Pogge von Strandmann E, Reinartz S, Wager U, and Muller R (2017). Tumor-Host Cell Interactions in Ovarian Cancer: Pathways to Therapy Failure. Trends Cancer 3, 137–148. [DOI] [PubMed] [Google Scholar]
- Pozzi L-AM, Maciaszek JW, and Rock KL (2005). Both Dendritic Cells and Macrophages Can Stimulate Naive CD8 T Cells In Vivo to Proliferate, Develop Effector Function, and Differentiate into Memory Cells. J. Immunol 175, 2071–2081. [DOI] [PubMed] [Google Scholar]
- Rodell CB, Arlauckas SP, Cuccarese MF, Garris CS, Li R, Ahmed MS, Kohler RH, Pittet MJ, and Weissleder R (2018). TLR7/8-agonist-loaded nanoparticles promote the polarization of tumour-associated macrophages to enhance cancer immunotherapy. Nat. Biomed. Eng 2, 578–588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rodriguez GM, Galpin KJC, McCloskey CW, and Vanderhyden BC (2018). The Tumor Microenvironment of Epithelial Ovarian Cancer and Its Influence on Response to Immunotherapy. Cancers (Basel) 10, 242. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rozenblit M, Hendrickx W, Heguy A, Chiriboga L, Loomis C, Ray K, Darvishian F, Egeblad M, Demaria S, Marincola FM, et al. (2019). Transcriptomic profiles conducive to immune-mediated tumor rejection in human breast cancer skin metastases treated with Imiquimod. Sci. Rep 9, 8572. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CM, Pryer N, Daniel D, Hwang ES, Rugo HS, and Coussens LM (2014). Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell 26, 623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Salim T, Sershen CL, and May EE (2016). Investigating the Role of TNF-alpha and IFN-gamma Activation on the Dynamics of iNOS Gene Expression in LPS Stimulated Macrophages. PLoS One 11, e0153289. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Schroder K, Hertzog PJ, Ravasi T, and Hume DA (2004). Interferon-gamma: an overview of signals, mechanisms and functions. J. Leukoc. Biol 75, 163–189. [DOI] [PubMed] [Google Scholar]
- Stuehr DJ, and Nathan CF (1989). Nitric oxide. A macrophage product responsible for cytostasis and respiratory inhibition in tumor target cells. J. Exp. Med 169, 1543–1555. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tap WD, Wainberg ZA, Anthony SP, Ibrahim PN, Zhang C, Healey JH, Chmielowski B, Staddon AP, Cohn AL, Shapiro GI, et al. (2015). Structure-Guided Blockade of CSF1R Kinase in Tenosynovial Giant-Cell Tumor. N. Engl. J. Med 373, 428–437. [DOI] [PubMed] [Google Scholar]
- ten Brinke A, van Schijndel G, Visser R, de Gruijl TD, Zwaginga JJ, and van Ham SM (2010). Monophosphoryl lipid A plus IFNgamma maturation of dendritic cells induces antigen-specific CD8+ cytotoxic T cells with high cytolytic potential. Cancer Immunol. Immunother 59, 1185–1195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Todd PA, and Goa KL (1992). Interferon gamma-1b. A review of its pharmacology and therapeutic potential in chronic granulomatous disease. Drugs 43, 111–122. [DOI] [PubMed] [Google Scholar]
- Trinchieri G (2003). Interleukin-12 and the regulation of innate resistance and adaptive immunity. Nat. Rev. Immunol 3, 133–146. [DOI] [PubMed] [Google Scholar]
- Vacchelli E, Sistigu A, Yamazaki T, Vitale I, Zitvogel L, and Kroemer G (2015). Autocrine signaling of type 1 interferons in successful anticancer chemotherapy. Oncoimmunology 4, e988042. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wainberg ZA, Eisenberg PD, Sachdev JC, Weise AM, Kaufman DR, Hutchinson M, Tong S, Aromin I, Hu-Lieskovan S, and Patnaik A (2016). Phase 1/2a study of double immune suppression blockade by combining a CSF1R inhibitor (pexidartinib/PLX3397) with an anti PD-1 antibody (pembrolizumab) to treat advanced melanoma and other solid tumors. J. Clin. Oncol 34, TPS465. [Google Scholar]
- Walton J, Blagih J, Ennis D, Leung E, Dowson S, Farquharson M, Tookman LA, Orange C, Athineos D, Mason S, et al. (2016). CRISPR/Cas9-Mediated Trp53 and Brca2 Knockout to Generate Improved Murine Models of Ovarian High-Grade Serous Carcinoma. Cancer Res. 76, 6118–6129. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wiemann B, and Starnes CO (1994). Coley’s toxins, tumor necrosis factor and cancer research: A historical perspective. Pharmacol. Ther 64, 529–564. [DOI] [PubMed] [Google Scholar]
- Wynn TA, Chawla A, and Pollard JW (2013). Macrophage biology in development, homeostasis and disease. Nature 496, 445–455. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xie QW, Whisnant R, and Nathan C (1993). Promoter of the mouse gene encoding calcium-independent nitric oxide synthase confers inducibility by interferon gamma and bacterial lipopolysaccharide. J. Exp. Med 177, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang L, Liu Q, Zhang X, Liu X, Zhou B, Chen J, Huang D, Li J, Li H, Chen F, et al. (2020). DNA of neutrophil extracellular traps promotes cancer metastasis via CCDC25. Nature 583, 133–138. [DOI] [PubMed] [Google Scholar]
- Yigit R, Massuger LF, Figdor CG, and Torensma R (2010). Ovarian cancer creates a suppressive microenvironment to escape immune elimination. Gynecol. Oncol 117, 366–372. [DOI] [PubMed] [Google Scholar]
- Yin M, Li X, Tan S, Zhou HJ, Ji W, Bellone S, Xu X, Zhang H, Santin AD, Lou G, and Min W (2016). Tumor-associated macrophages drive spheroid formation during early transcoelomic metastasis of ovarian cancer. J. Clin. Invest 126, 4157–4173. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang W, Lim S-M, Hwang J, Ramalingam S, Kim M, and Jin J-O (2021). Monophosphoryl lipid A-induced activation of plasmacytoid dendritic cells enhances the anti-cancer effects of anti-PD-L1 antibodies. Cancer Immunol. Immunother 70, 689–700. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zitvogel L, Galluzzi L, Kepp O, Smyth MJ, and Kroemer G (2015). Type I interferons in anticancer immunity. Nat. Rev. Immunol 15, 405–414. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data file S1. List of upregulated and downregulated genes in the MPLA+IFNγ, MPLA, and IFNγ treatment groups, related to Figure 3. RNA-seq data were collected following the experimental design in Figure 3A. After the combined MPLA+INFγ treatment, 179 genes were upregulated and 36 genes were downregulated in PyMT tumors, compared to vehicle. After treatment with MPLA alone, 20 genes were upregulated and 5 genes were downregulated. After treatment with INFγ alone, 33 genes were upregulated and 15 genes were downregulated.
Movie S1. Un-activated breast tumor-derived macrophages do not kill cancer cells, related to Figure 1. Macrophages and PyMT tumor cells were isolated from the primary tumors of MMTV-PyMT mice, and macrophages were stained with CellTracker™ Deep Red Dye, while tumor cells were stained with CellTracker™ CMFDA Dye. Deep Red dye is falsely colored green for clarity. Time indicated is time after imaging was initiated.
Movie S2. MPLA+IFNγ activates breast tumor-derived macrophages to kill cancer cells, related to Figure 1. Macrophages and PyMT tumor cells were isolated from the primary tumors of MMTV-PyMT mice, and macrophages were stained with CellTracker™ Deep Red Dye, while tumor cells were stained with CellTracker™ CMFDA Dye. Deep Red dye is falsely colored green for clarity. MPLA with mouse IFNγ was added before imaging was initiated. Time indicated is time after imaging was initiated.
Supplemental Figures and Tables
Data Availability Statement
RNA-seq data have been deposited in the Sequence Read Archive (SRA) with the accession number GSE146211 (also listed in the Key Resources Table).
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
| Mouse monoclonal anti-mouse iNOS | BD Biosciences | Cat #610431 |
| Goat polyclonal anti-mouse CD206 | R&D Systems | Cat #AF2535 |
| Mouse monoclonal anti-β-actin | Santa Cruz Biotechnology | Cat #sc-47778 |
| Rabbit monoclonal anti-NFκB p65 | Cell Signaling Technology | Cat #8242 |
| Rabbit monoclonal anti-Phospho-NFκB p65 (Ser536) (93H1) | Cell Signaling Technology | Cat #3033 |
| Rabbit monoclonal anti-STAT1 | Cell Signaling Technology | Cat #14994 |
| Rabbit monoclonal anti-Phospho-STAT1 (Tyr701) (58D6) | Cell Signaling Technology | Cat #9167 |
| Mouse monoclonal anti-GAPDH | Santa Cruz Biotechnology | Cat #sc-32233 |
| IRDye 800CW Goat anti-Mouse IgG Secondary Antibody | LI-COR | Cat #926-32210 |
| IRDye 800CW Donkey anti-Goat IgG Secondary Antibody | LI-COR | Cat #926-32214 |
| Goat polyclonal anti-Neutrophil Elastase | Santa Cruz Biotechnology | Cat #sc-9521 |
| BV785™ anti-mouse CD45 Antibody | BioLegend | Cat #103149 |
| Alexa Fluor® 647 anti-mouse CD3 Antibody | BioLegend | Cat #100209 |
| APC/Cy7 anti-mouse CD3 Antibody | BioLegend | Cat #100221 |
| APC anti-mouse/human CD11b Antibody | BioLegend | Cat #101212 |
| FITC anti-mouse CD4 Antibody | BioLegend | Cat #100510 |
| PE anti-mouse CD8a Antibody | BioLegend | Cat #100708 |
| APC/Cy7 anti-mouse CD8a Antibody | BioLegend | Cat #100714 |
| PE/Cy5 anti-mouse CD19 Antibody | BioLegend | Cat #115509 |
| PE/Cy7 anti-mouse CD11c Antibody | BioLegend | Cat #117318 |
| APC/Cy7 anti-mouse CD11c Antibody | BioLegend | Cat #117324 |
| APC/Cy7 anti-mouse Ly6C Antibody | BioLegend | Cat #128026 |
| FITC anti-mouse Ly6G Antibody | BioLegend | Cat #127605 |
| PerCP/Cy5.5 anti-mouse F4/80 Antibody | BioLegend | Cat #123127 |
| Alexa Fluor® 488 anti-mouse F4/80 Antibody | BioLegend | Cat #123120 |
| APC anti-mouse CD326 (EpCAM) Antibody | BioLegend | Cat #118214 |
| PE/Cy7 anti-mouse CD40 Antibody | BioLegend | Cat #124621 |
| FITC anti-mouse CD206 (MMR) Antibody | BioLegend | Cat #141704 |
| FITC anti-mouse CD69 Antibody | BioLegend | Cat #104505 |
| PE anti-mouse CD107a (LAMP-1) Antibody | BioLegend | Cat #121612 |
| FITC anti-mouse CD279 (PD-1) Antibody | BioLegend | Cat #135213 |
| PerCP/Cy5.5 anti-mouse/human CD44 Antibody | BioLegend | Cat #103032 |
| APC anti-mouse CD62L Antibody | BioLegend | Cat #104412 |
| APC anti-mouse H-2Kb bound to SIINFEKL Antibody | BioLegend | Cat #141605 |
| Alexa Fluor® 647 anti-human CD326 (EpCAM) Antibody | BioLegend | Cat #324212 |
| BV510™ anti-human CD45 Antibody | BioLegend | Cat #368525 |
| APC anti-human CD68 Antibody | BioLegend | Cat #333810 |
| PE Rat Anti-CD11b Clone M1/70 (RUO) | BD Biosciences | Cat #557397 |
| CD335 (NKp46) Monoclonal Antibody (29A1.4), eFluor 450 | Invitrogen | Cat #48335180 |
| iNOS Monoclonal Antibody (CXNFT), PE | Invitrogen | Cat #12592082 |
| CD103 (Integrin alpha E) Monoclonal Antibody (2E7), FITC | Invitrogen | Cat #11103182 |
| PE anti-mouse CD40 Antibody | BioLegend | Cat #124610 |
| FITC anti-mouse CD8a Antibody | BioLegend | Cat #100706 |
| Rat monoclonal anti-PyMT antigen | Abcam | Cat #ab15085 |
| Alexa Fluor 488 goat anti-Rat IgG secondary antibody | Invitrogen | Cat #A11006 |
| Alexa Fluor® 647 anti-mouse CD4 Antibody | BioLegend | Cat #100424 |
| Rat monoclonal anti-Mouse F4/80 | Bio-Rad | Cat #MCA497R |
| Rabbit anti-Phospho-NFkB p65 (Ser276) Antibody | Invitrogen | Cat #PA5-37718 |
| Rabbit polyclonal anti-CFP | Bio-Rad | Cat #AHP2986 |
| Alexa Fluor 488 goat anti-rabbit IgG secondary antibody | Invitrogen | Cat #A11070 |
| Goat polyclonal anti-Mouse IL-12 | R&D Systems | Cat #AF-419-NA |
| Rat monoclonal anti-Mouse TNF-alpha | R&D Systems | Cat #MAB4101 |
| Goat polyclonal anti-Mouse CXCL9/MIG | R&D Systems | Cat #AF-492-NA |
| InVivoMAb anti-mouse CD8α | BioXCell | Cat #BE0004-1 |
| InVivoMAb anti-mouse CD4 | BioXCell | Cat #BE0003-1 |
| InVivoMAb anti-mouse CSF1R (CD115) | BioXCell | Cat #BE0213 |
| InVivoMAb anti-mouse CSF1 | BioXCell | Cat #BE0204 |
| InVivoPlus anti-mouse PD1 | BioXCell | Cat #BP0273 |
| Biological Samples | ||
| Metastatic pleural effusions from breast cancer patients | New York University | IRB #i17-01382 |
| Chemicals, Peptides, and Recombinant Proteins | ||
| Matrigel | Corning | Cat #356231 |
| Human M-CSF | ProSpec | Cat #CYT-308 |
| L-glutamine | Gibco | Cat #25030081 |
| Sodium pyruvate | Gibco | Cat #11360070 |
| 2-mercaptoethanol | Gibco | Cat #31350010 |
| B27 supplement | Gibco | Cat #17504044 |
| Human EGF | ProSpec | Cat #CYT-217 |
| Insulin | Gibco | Cat #12585014 |
| Collagenase/hyaluronidase | STEMCELL Technologies | Cat #07912 |
| DNase I | Roche | Cat #4716728001 |
| Dispase | STEMCELL Technologies | Cat #07913 |
| Collagenase D | Sigma | Cat #11088866001 |
| DNase I | Sigma | Cat #045362820 |
| Liberase DL | Sigma | Cat #05466202001 |
| TrypLE Express Enzyme | Gibco | Cat #12605010 |
| Mouse M-CSF | ProSpec | Cat #CYT-439 |
| Fibronectin | Sigma Aldrich | Cat #F1141 |
| CellTracker™ Deep Red Dye | Invitrogen | Cat #C34565 |
| CellTracker™ Green CMFDA Dye | Invitrogen | Cat #C7025 |
| LPS | Sigma Aldrich | Cat #L4391 |
| MPLA for in vitro experiments | InvivoGen | Cat #tlrl-mpls |
| MPLA for in vivo experiments | InvivoGen | Cat #vac-mpls |
| PolyI:C | InvivoGen | Cat #tlrl-picw |
| Mouse IFNγ | R&D Systems | Cat #485-MI/CF |
| Human IFNγ | R&D Systems | Cat #285-IF/CF |
| TMP195 | MedChemExpress | Cat #HY-18361 |
| IPI-549 | MedChemExpress | Cat #HY-100716 |
| PLX3397 (Pexidartinib) | MedChemExpress | Cat #HY-16749 |
| L-NIL (N6-(1-Iminoethyl)-L-lysine hydrochloride) | TOCRIS | Cat #1139 |
| Creatine | Selleckchem | Cat #S5588 |
| JSH-23 | Selleckchem | Cat #S7351 |
| SR11302 | TOCRIS | Cat #2476 |
| CellTiter 96® AQueous One Solution reagent | Promega | Cat #G3582 |
| PowerUp™ SYBR™ Green Master Mix | Applied Biosystems | Cat #A25780 |
| Taqman™ Universal Master Mix II, no UNG | Applied Biosystems | Cat #4440040 |
| RIPA lysis buffer | Thermo Fisher Scientific | Cat #89990 |
| Protease and phosphatase inhibitor | Thermo Fisher Scientific | Cat #78440 |
| Cisplatin | MBL | Cat #JM-1550-1000 |
| Sodium citrate solution | Sigma | Cat #C3821 |
| ABTS Substrate Solution | Thermo Fisher Scientific | Cat #37615 |
| Fc receptor blocker | Innovex | Cat #NB309 |
| Goat serum | Dako | Cat #X0907 |
| Mounting media | Electron Microscopy Sciences | Cat #17985-16 |
| OVA-Q4H7 peptide | AnaSpec | Cat #AS-64405 |
| Critical Commercial Assays | ||
| RNeasy Mini kit | Qiagen | Cat #74104 |
| TaqMan™ Reverse Transcription Reagents | Invitrogen | Cat #N8080234 |
| Cell Death Detection ELISA Kit | Roche | Cat #11774425001 |
| Proteome Profiler Mouse XL Cytokine Array Kit | R&D Systems | Cat #ARY028 |
| Mouse CXCL9/MIG DuoSet ELISA | R&D Systems | Cat #DY492 |
| Mouse CCL5/RANTES DuoSet ELISA | R&D Systems | Cat #DY478 |
| DuoSet ELISA Ancillary Reagent Kit 2 | R&D Systems | Cat #DY008 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat #20020594 |
| Fixation/Permeabilization Solution Kit | BD Biosciences | Cat #554714 |
| Comprehensive Chemistry test of serum | IDEXX BioAnalytics | Cat #6006 |
| CD8a+ T Cell Isolation Kit, mouse | Miltenyi Biotec | Cat #130-104-075 |
| Deposited Data | ||
| RNA-seq data | This paper | GEO: GSE146211 |
| Experimental Models: Cell Lines | ||
| Mouse: 4T1 cells | ATCC | Cat #CRL-2539 |
| Mouse: ID8-p53−/− cells | Walton et al., 2016 | N/A |
| Experimental Models: Organisms/strains | ||
| MMTV-PyMT mice (on C57BL/6 background) | Jackson Laboratory | Cat #022974 |
| MMTV-PyMT mice (on FVB/N background) | Jackson Laboratory | Cat #002374 |
| MMTV-PyMT-chOVA mice (on C57BL/6 background) | Engelhardt et al., 2012 | N/A |
| ACTB-ECFP mice | Jackson Laboratory | Cat #004218 |
| OT1 mice | Jackson Laboratory | Cat #003831 |
| Tlr4 KO mice (on C57BL/6 background) | Jackson Laboratory | Cat #029015 |
| MMTV-PyMT; ACTB-ECFP mice (on C57BL/6 background) | This paper | N/A |
| Oligonucleotides | ||
| Primers for human CD40, ACTB, NOS2, TNF, MRC1, IL12B, IL10, mouse Stat1, Stat2, Ifit1, Irgm2, Gbp2, Cxcl9, Igfbp3, Col4a1, Actb, Tnf, Il12b, Il10, Nos2, Mrc1, Arg1, see Table S2 | This paper | N/A |
| Taqman Probe for mouse Tbp | Thermo Fisher Scientific | Mm00446973-m1 |
| Taqman Probe for mouse Cd86 | Thermo Fisher Scientific | Mm00444543-m1 |
| Taqman Probe for mouse H2-Q2 | Thermo Fisher Scientific | Mm04213368-g1 |
| Software and Algorithms | ||
| STAR | Dobin et al., 2012 | N/A |
| FeatureCounts | Liao et al., 2013 | N/A |
| DESeq2 | Love et al., 2014 | N/A |
| Other | ||
| CD326 (EpCAM) microbeads, human | Miltenyi Biotec | Cat #130-061-101 |
| CD11b MicroBeads, human and mouse | Miltenyi Biotec | Cat #130-049-601 |
| 24-well glass bottom plate | MatTek | Cat #P24G-1.5-13-F |
| PVDF membrane | Bio-Rad | Cat #1620177 |
| Relfex 7 mm wound clips | CellPoint Scientific | Cat #203-1000 |
| 96-well Enzyme ImmunoAssay/Radio Immuno-Assay (EIA/RIA) plates | Costar | Cat #3590 |
| Not TC-treated culture dishes | Corning | Cat #430597 |
| Cell culture insert for 24-well Plate with 1.0 μm Transparent PET Membrane | Corning | Cat #353104 |
| Rodent Chow 5053 With PLX3397 Diet | Research Diets, Inc. | N/A |
| Rodent Chow 5053 With IPI-549 Diet | Research Diets, Inc. | N/A |