

SUMMARY
Cancer heterogeneity impacts therapeutic response, driving efforts to discover over-arching rules that supersede variability. Here, we define pan-cancer binary classes based on distinct expression of YAP and YAP-responsive adhesion regulators. Combining informatics with in vivo and in vitro gain- and loss-of-function studies across multiple murine and human tumor types, we show that opposite pro- or anti-cancer YAP activity functionally defines binary YAPon or YAPoff cancer classes that express or silence YAP, respectively. YAPoff solid cancers are neural/neuroendocrine and frequently RB1–/–, such as retinoblastoma, small cell lung cancer, and neuroendocrine prostate cancer. YAP silencing is intrinsic to the cell of origin, or acquired with lineage switching and drug resistance. The binary cancer groups exhibit distinct YAP-dependent adhesive behavior and pharmaceutical vulnerabilities, underscoring clinical relevance. Mechanistically, distinct YAP/TEAD enhancers in YAPoff or YAPon cancers deploy anti-cancer integrin or pro-cancer proliferative programs, respectively. YAP is thus pivotal across cancer, but in opposite ways, with therapeutic implications.
Graphical Abstract
In brief
Pearson et al. demonstrate that YAP/TAZ, well-known oncogenes, are tumor suppressors in a large group of cancers. Pan-cancer analyses reveal that opposite YAP/TAZ expression, adhesive behavior, and oncogenic versus tumor suppressor YAP/TAZ activity functionally stratify binary cancer classes, which interchange to drive drug resistance. Contrasting YAPoff/YAPon classes exhibit unique vulnerabilities, facilitating therapeutic selection.
INTRODUCTION
Tumor heterogeneity and plasticity complicate cancer diagnosis and therapeutics, thus there is much interest in deducing over-arching rules that span distinct tumor types (Balanis et al., 2019; Chen et al., 2018, 2019; Hoadley et al., 2018; Priestley et al., 2019). These efforts can expose vulnerabilities with broad therapeutic relevance, such as strategies that exploit different classes of p53 mutation (Hientz et al., 2017). Here, starting with the goal of understanding unique properties of small cell cancers, we delineate a pan-cancer rule based on opposite expression and activity of a single transcriptional complex.
Neuroendocrine (NE) cancers arise from hormone/neurotransmitter-secreting cells, or evolve from adenocarcinomas through lineage conversion (Rindi and Inzani, 2020). Small cell neuroendocrine (SCN) cancers are a highly lethal subset, and often exhibit metastasis at presentation; e.g., small cell lung cancer (SCLC) has a dismal 5-year survival of <6% and originates from endogenous lung NE cells (Huang et al., 2018; Lázaro et al., 2019; Sutherland et al., 2011; Yang et al., 2018), or conversion of non-SCLC (NSCLC) to SCLC linked to drug resistance (reviewed in Rudin et al., 2019). SCN prostate cancer is rare at presentation, but up to 17% of castration-resistant cases develop pure or mixed NE histology, with reduced overall survival versus non-NE castration-resistant disease (Aggarwal et al., 2018). Such lineage switching will rise with increased use of targeted adenocarcinoma therapies, thus there is much interest in new SCN cancer vulnerabilities (e.g., Balanis et al., 2019; Oser et al., 2019; Zhang et al., 2020).
The RB1 tumor suppressor gene is inactivated in most SCLCs, and is lost during NE lineage transitions in both NSCLC and prostate cancer (reviewed in Beltran et al., 2019; Rudin et al., 2019). The RB1 gene was identified in the pediatric cancer, retinoblastoma, which is also a small cell cancer (Rajwanshi et al., 2009), but arises from neurons, as opposed to NE cells (Xu et al., 2014). RB1 loss approaches 100% in these cancers, yet is far rarer in most other solid tumors, but this context specificity is un-explained. SCN cancers grow as semi- or non-adherent cells in culture, akin to hematopoietic tumors, but most solid cancers adhere via interactions between adhesion molecules (e.g., integrins) and extracellular matrix (ECM) components. The driving force behind opposite pan-cancer adhesive behavior is unclear.
The paralogs YAP and TAZ (WWTR1) are oncogenic drivers in human solid cancers (Zanconato et al., 2016; Zheng and Pan, 2019). These transcriptional coactivators interact with DNA-binding transcription factors (TFs). TEAD1–4 recruit YAP to remote enhancers populated with AP1 complexes to induce cell-cycle genes and promote cancer (Liu et al., 2016; Stein et al., 2015; Verfaillie et al., 2015; Zanconato et al., 2015; Zhao et al., 2008). In contrast, ectopically expressing YAP in the liquid cancer, multiple myeloma, promotes p73-mediated apoptosis (Cottini et al., 2014). In line with an anti-cancer function, YAP is absent in hematopoietic cancers. Intriguingly, most cases of SCLC may be YAP deficient (Rudin et al., 2019), but whether that reflects a tumor suppressor role, whether TAZ behaves in this way, and/or the extent to which YAP/TAZ silencing is critical in other solid cancers is unknown. Finally, it is unclear whether contrasting YAP/TAZ levels influence therapeutic vulnerability.
Here, we connect contrasting levels and opposite pro- or anti-cancer activity of YAP/TAZ to context-specific transformation upon RB1 loss, opposite adhesive behavior, and distinct pan-cancer vulnerabilities, which tumors exploit to evade therapeutics. We tie these opposing activities to unique YAP/TEAD enhancer deployment in YAP-deficient or YAP-positive cancers. Divergent behavior of a single transcriptional complex thus defines binary pan-cancer classes, exposing broadly relevant therapeutic opportunities.
RESULTS
Obligate YAP and TAZ absence in the retinoblastoma and SCLC cells of origin
Removing Rb1 and its paralog Rbl1 (p107) causes murine retino-blastoma, but penetrance is partial (Chen et al., 2004). Tumors were Yap/Taz positive (Figure 1A), but despite their oncogenic role in most solid cancers (Zanconato et al., 2016; Zheng and Pan, 2019), penetrance rose when one, and more so both, genes were deleted (Figure 1B and S1A). These data prove Yap/Taz tumor suppressor activity in vivo, and explain low penetrance murine retinoblastoma. Moreover, while ectopic Yap initially promoted growth of Rb1 or Rb1/Rbl1-null clones, division petered out, generating benign Yap+ lesions, but never malignancy (Figures S1B–S1E). Thus, both loss-of-function (LoF) and gain-of-function (GoF) assays show that Yap suppresses cancer in Rb1–/– murine retina.
Figure 1. Obligate YAP/TAZ silencing in retinoblastoma.
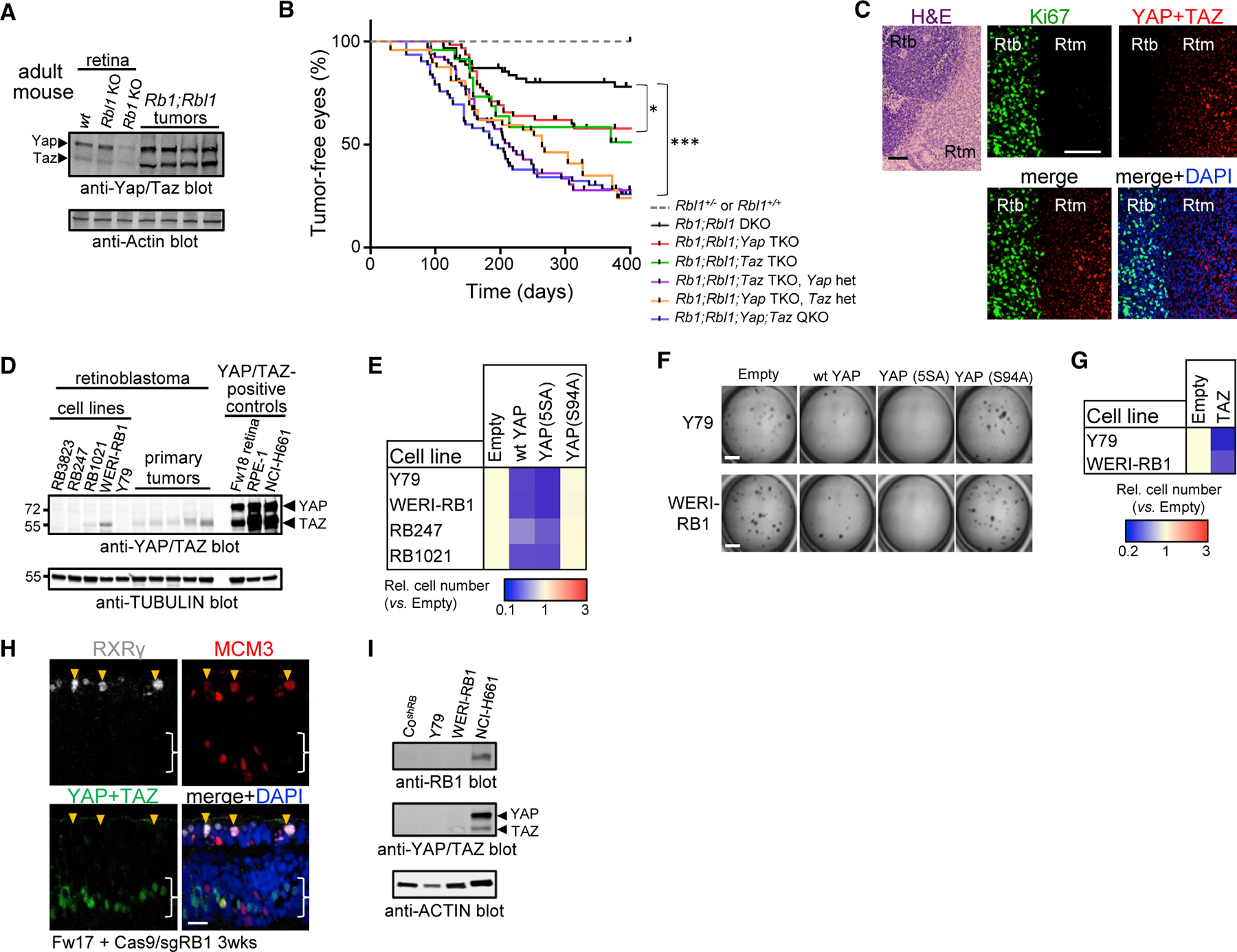
(A) Western blots of murine retinal or tumor lysates.
(B) Kaplan-Meyer from αCre mice of the indicated genotypes. *p < 0.01, ***p < 0.0001, log rank (Mantel-Cox) test. DKO/TKO/QKO, double/triple/quadruple knockout; het, heterozygous.
(C) H&E and immunofluorescence (IF) in human retinoblastoma (Rtb) and retinoma (Rtm). Scale bar, 100 mm.
(D) Western blots of Rtb cell lines and tumors.
(E) Ectopic YAP or hyperactive YAP5SA, but not YAPS94A, impedes growth of Rtb lines (n = 3, p < 0.01).
(F) Soft agar assays of Rtb lines expressing the indicated YAP constructs. Scale bars, 3 mm.
(G) Ectopic TAZ impedes growth of Rtb lines (n = 3, p < 0.001).
(H) IF in human Fw17 RB1–/– retinal explants. Orange arrowheads, YAP– dividing (MCM3+) cones (RXRγ+); white brackets, YAP+ progenitors/Müller glia. Scale bar, 20 mm.
(I) Western blots of human RB1-depleted cone line (CoshRB). NCI-H661, control for RB1, YAP/TAZ. See also Figure S1.
Strikingly, YAP/TAZ were absent in human retinoblastoma, but expressed in retinoma (Figure 1C), an RB1–/–, but benign, postmitotic ‘‘dead-end’’ or barrier for progression (Dimaras et al., 2008). Matching the immunofluorescence data, YAP/TAZ were undetectable/low in human retinoblastoma tumors and cell lines (Figure 1D). Expressing YAP or TAZ inhibited retinoblastoma 2D or 3D growth, which overactive YAP (YAP5SA) enhanced, and deleting ectopic YAP ablated (Figures 1E–1G and S1F–S1H). Thus, unlike other solid cancers, YAP/TAZ suppress retinoblastoma.
Human retinoblastoma arises from cones, which are normally post-mitotic, but divide without RB1 (Xu et al., 2014). In human fetal explants, dividing progenitors expressed YAP/TAZ, but ectopically dividing RB1–/– cones did not (Figure 1H and S1I). Moreover, RB1-deficient primary cones we derived from fetal retina also lacked YAP/TAZ (Figure 1I). Thus, YAP/TAZ absence is intrinsic to the retinoblastoma cell of origin.
Next, we assessed SCLC, a highly lethal RB1–/– cancer. Among seven cell lines, representing three subtypes (A: ASCL1; N: NEUROD1; P: POU2F3, Figure S2A) (Mollaoglu et al., 2017; Rudin et al., 2019), none were YAP+, and only one expressed TAZ, but weakly relative to NSCLC (Figure 2A). Immunohistochemistry (IHC) showed that primary SCLC lacks YAP/TAZ, unlike NSCLC (Figure 2B). Transcriptome analysis revealed low YAP/TAZ mRNA levels in NE versus other lung cancers, and in SCLC patient-derived xenografts (PDXs), YAP/TAZ proteins were well below NSCLC PDX levels (Figures 2C and 2D). Yap/ Taz mRNAs were low in murine SCLC (Figure 2E), and in both CgrpCreER;Rb1f/f;p53f/f (Song et al., 2012) and adenoviral (Ad)-Cre-Rb1f/f;p53f/f SCLC, Yap/Taz proteins were absent, contrasting normal lung and murine EGFR-driven NSCLC (Figures 2F and 2G). Thus, retinoblastoma and SCLC are predominantly YAP/TAZ– (‘‘YAPoff’’) cancers, contrasting YAP/TAZ+ (‘‘YAPon’’) cancers.
Figure 2. Obligate YAP/TAZ silencing in SCLC.
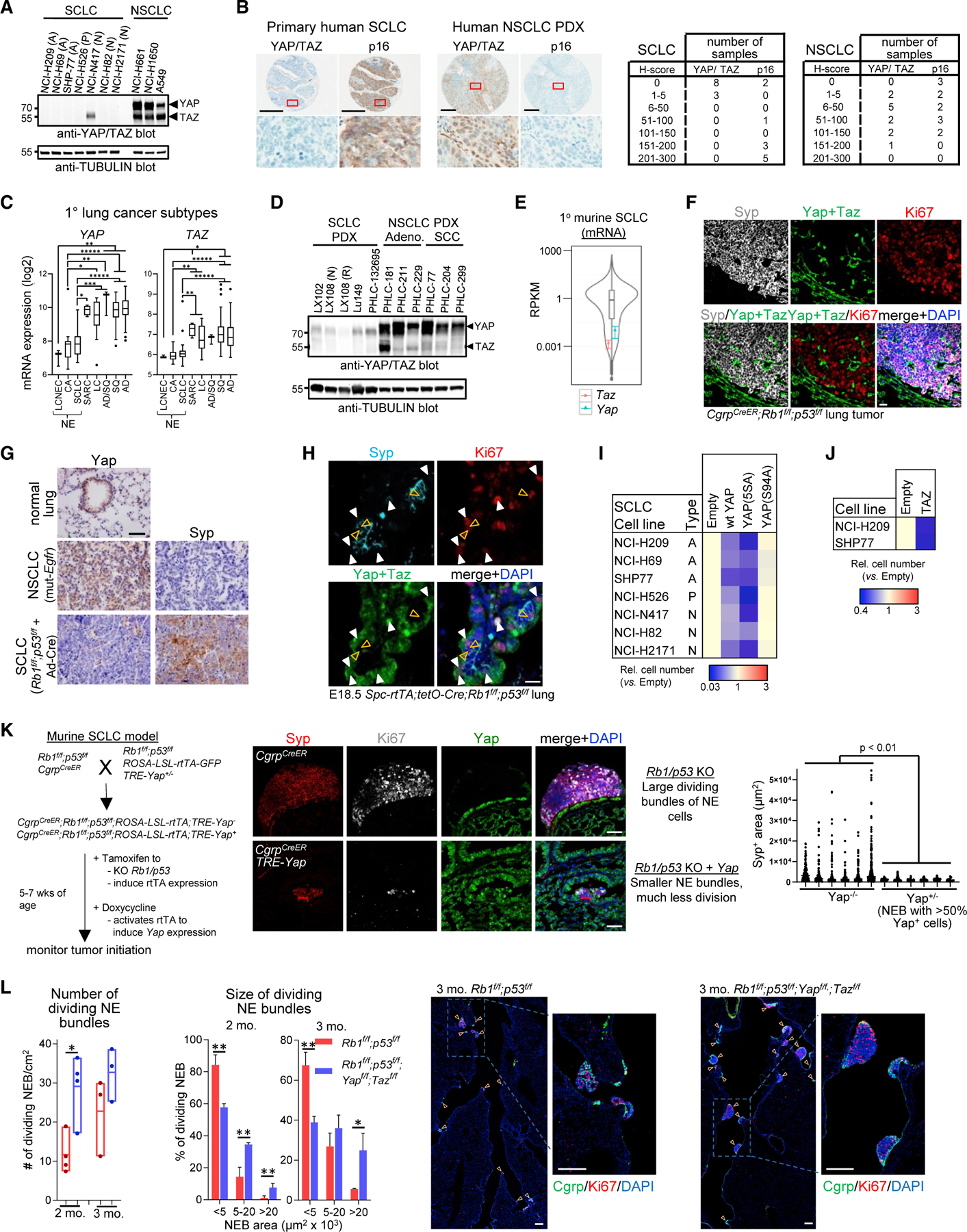
(A) Western blots of SCLC, NSCLC cell lines.
(B) YAP/TAZ and p16 (marker for RB1 loss) IHC in primary human SCLC and NSCLC PDXs (left) and scoring (right). Scale bars, 300 µm.
(C) YAP and TAZ mRNA in primary human lung tumors. *p < 0.05, **p < 0.01, ***p < 0.001, *****p < 1 × 10–5.
(D) Western blots of YAP/TAZ in SCLC and NSCLC PDXs. PDXs are from different donors, except the paired treatment naive (N) and cisplatin/etoposide-resistant (R) LX108 samples.
(E) Violin plot of low Yap and Taz mRNA in murine SCLC.
(F) Yap/Taz IF in CgrpCreER;Rb1f/f;p53f/f SCLC. Neighboring airway/alveolar cells express Yap/Taz but not dividing Syp+ NE cells. Note rare Syp–;Ki67–;Yap/Taz+ cells within tumor. Scale bar, 20 µm.
(G) IHC for Ccsp-rtTA;tetO-EGFRL858R NSCLC and Ad-CMV-Cre-Rb1f/f;p53f/f;Ptenf/+ SCLC. Scale bar, 100 µm.
(H) IF on embryonic day 18.5 (E18.5) Spc-rtTA;tetO-Cre;Rb1f/f;p53f/f lungs. Orange arrows, Yap– dividing (Ki67+) NE cells (Syp+); white arrows, Yap+ dividing non-NE cells (Syp–). Scale bar, 20 µm.
(I) Ectopic YAP or hyperactive YAP5SA (n = 3, p < 0.05) but not YAPS94A impede growth of SCLC lines.
(J) Ectopic TAZ suppresses growth of SCLC lines (n = 3, p < 0.0001).
(K) Outline for murine SCLC +/– Yap (left), images (middle), and area (right) of control and Yap+ SCLC 3 months after tam/dox. NEB, NE bundle. t test with Welch’s correction. Scale bars, 50 µm.
(L) Graphs show number (left) and size (middle) of dividing NEB in Rb1f/f;p53f/f or Rb1f/f;p53f/f;Yapf/f;Tazf/f mice 2 or 3 months after Ad-CMV-Cre administration. Mean ± SD, n = 3 (3 months) or 4 (2 months) mice/genotype; *p < 0.05, **p < 0.01. Representative images (right) are from 3 month mice. Scale bars, 200 µm. See also Figure S2.
Rb1/p53-deficient pulmonary NE cells (PNECs) generate SCLC (Semenova et al., 2015; Sutherland et al., 2011), so we asked whether, like human RB1–/– cones, they are naturally YAP/TAZ–. In SPC-rtTA;tetO-Cre;Rb1f/f;p53f/f mice, doxycycline (dox) treatment during gestation induces ectopic division in many Rb1/p53 null lung lineages (Akeno et al., 2017; Simpson et al., 2009). After birth, ectopic division ceases, except in PNECs. At embryonic day 18.5, abnormally dividing but cancer-resistant ATII and Club cells expressed Yap/Taz, but cancer-prone PNECs did not (Figures 2H and S2B). Lineage tracing in tamoxifen-treated CgrpCreER;Rb1f/f;p53f/f mice, which specifically targets NE cells (Song et al., 2012), confirmed that Rb1/p53-null PNECs lack Yap/Taz, as did adult wild-type PNECs (Figures S2C–S2D). Thus, Rb1/p53 loss triggers division in many lung cell types, but abundant Yap+ lineages are tumor-resistant while rare tumor-prone NE cells (<0.5% of the lung), are Yap–. Our lung and retina data tie YAP/TAZ status to context-dependent susceptibility to cancer after RB1 loss.
Next, we performed GoF studies in models of SCLC. Ectopic YAP/TAZ inhibited growth of human YAPoff SCLC lines, but not YAPon NSCLC lines (Figures 2I, 2J, and S2E–S2I). To assess whether YAP blocks SCLC initiation in vivo, we combined CgrpCreER;Rb1f/f;p53f/f mice with a Cre-inducible, tetracycline (tet)-activated transactivator (lox-STOP-lox-rtTA-GFP) and tetresponsive Yap transgene (TRE-Yap) (Gregorieff et al., 2015) (Figure 2K). Yap dramatically reduced expansion of Rb1/p53-null PNECs (Figure 2K). Rare, large, dividing NE clusters emerged, but expressed minimal Yap (Figure S2J). To test whether Yap also inhibits established SCLC, we treated Rb1f/f;p53f/f;lox-STOP-lox-rtTA-GFP;TRE-Yap mice with Ad-Cre, then dox-induced Yap 3 months after tumor initiation. Despite only mosaic induction, Yap reduced tumor load and prolonged survival (Figure S2K), and Yap+ regions retained an NE phenotype and NE marker expression, but exhibited large cell instead of small cell morphology and significantly reduced division (Figures S2L–S2M). Our extensive GoF data suggest that reactivating YAP expression is a therapeutic option for SCLC.
The observation that YAP/TAZ are undetectable in the SCLC cell of origin, coupled with our GoF data, suggest tumor suppressor activity. Conceivably, however, trace levels could be essential for SCLC. To test this idea, we compared tumorigenesis in Rb1f/f;p53f/f and Rb1f/f;p53f/f; Yapf/f;Tazff mice treated with Ad-Cre. Yap/Taz loss did not inhibit tumorigenesis, and instead tumors were larger (Figure 2L). The increase may reflect tumor suppression by trace Yap/Taz in NE cells, more efficient conversion of non-NE cells (Ferone et al., 2020), and/or loss of indirect tumor suppression from neighboring cells (Moya et al., 2019), although these were Yap/Taz+ (Figure S2N). Irrespective, these in vivo data show, unequivocally, that Yap/Taz are not essential oncogenes in SCLC, and instead suppress this cancer.
YAP/TAZ and integrin/adhesion genes stratify binary pan-cancer classes
To define other YAP/TAZ-deficient cancers, we surveyed 1,036 lines in the Cancer Cell Line Encyclopedia (CCLE). YAP/TAZ exhibited a striking off/on pattern (Figure 3A), raising the hypothesis that binary expression and opposite functions of YAP/TAZ may provide a useful stratification for human cancers. As an unbiased test, we used principal-component analysis (PCA) to assess CCLE transcriptomes. YAP/TAZ were top scoring genes on the positive side of principal component 1 (PC1+), which defined binary YAPoff and YAPon classes (Figures 3B and S3A; Table S1). PC2 and, more effectively, PC3 distinguished two groups of YAPoff cancers: hematopoietic (cluster A), and solid neural/NE cancers (cluster B) from many tissues, including lung, prostate, thyroid, breast, gastrointestinal, and ganglia (Figures 3B, 3C, S3B, and S3C). PC3 also parsed YAPon cancers into two groups (clusters C/D). PCA of Sanger lines gave similar results (Figure S3D). Western blots confirmed no/very low YAP/TAZ in YAP-off cancers of the breast, pancreas, ganglia, and prostate, similar to SCLC (Figure 3D). RB1 mutation was enriched in cluster B, extending our retinoblastoma/SCLC data (Figure 3E). Gene set enrichment analysis (GSEA) showed that PC1+ genes are Hippo signaling (e.g., YAP, TAZ, CYR61, AMOTL2), adhesion, and ECM components (e.g., the integrin ITGB5; Figures 3C and 3F; Table S1). PC3+ and PC1–, highest in blood cancers (cluster A, YAPoff), were enriched for hematopoietic genes. PC3– was enriched for REST-repressed neural genes, consistent with high levels in solid YAPoff cancers, which have neural/NE features. Thus, YAP/TAZ and correlated integrin/ECM genes in PC1+ parse cancers into binary classes that are further defined by PC3 genes.
Figure 3. YAP, TAZ, and their integrin/ECM/adhesion targets stratify binary cancer classes.
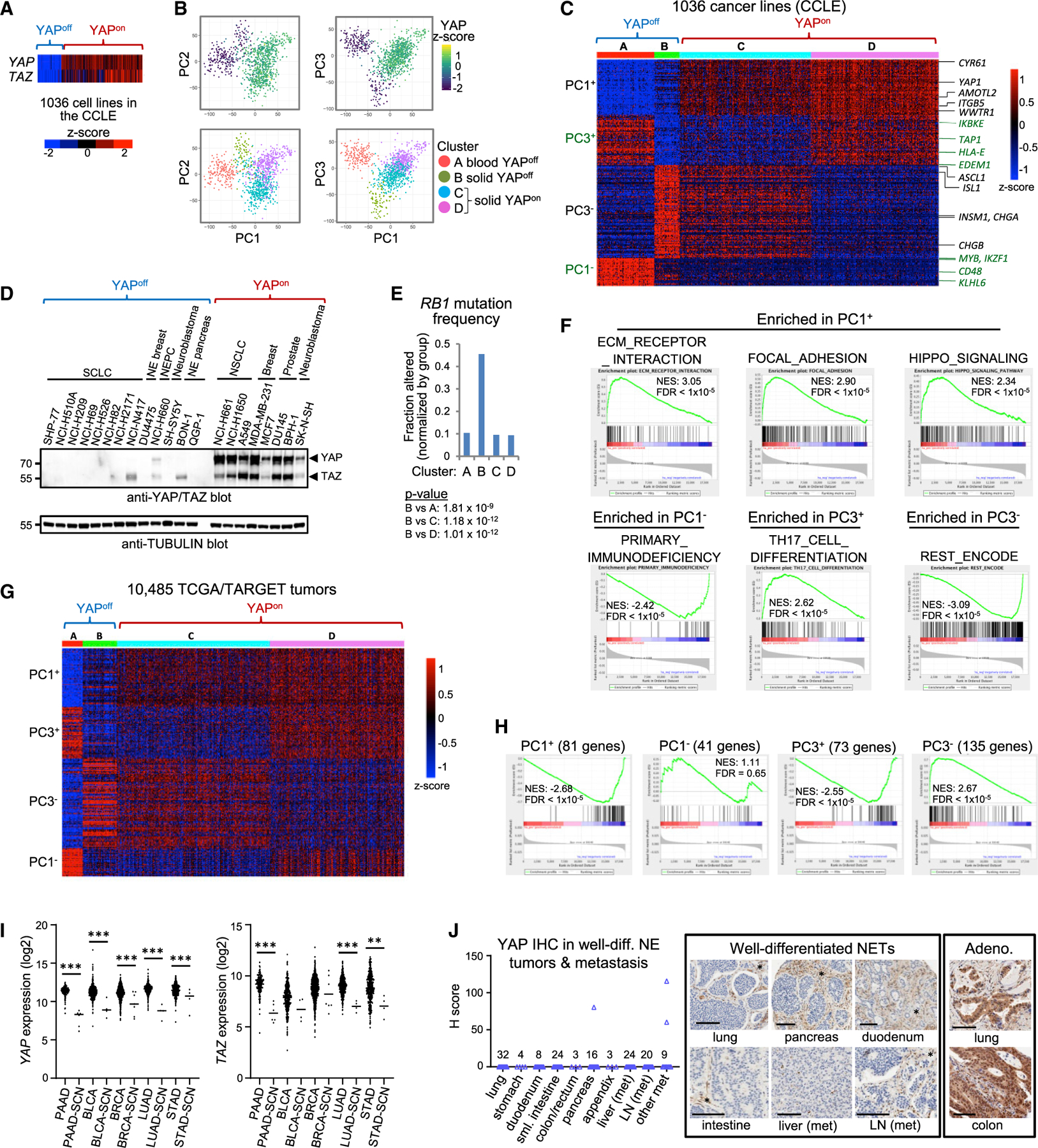
(A) YAP/TAZ mRNA across the CCLE.
(B) PCA on CCLE transcriptome data.
(C) k-Means clustering of the top (>1.8%) PC1/PC3 genes.
(D) Western blots of YAPoff and YAPon lines.
(E) RB1 mutation frequency (clusters from C); Fisher’s exact test.
(F) GSEA of KEGG pathways or ENCODE/ChEA TF binding to ranked PC1/PC3 gene lists.
(G) Expression of the top 330 PC1/3 genes in TCGA/TARGET cancers.
(H) GSEA of the top PC1/3 gene sets from (C) versus an SCN cancer signature (Balanis et al., 2019).
(I) YAP/TAZ mRNA expression in the indicated TCGA cancers.
(J) YAP IHC in lung and gastrointestinal well-differentiated NETs and metastases (met). Similar results with a YAP/TAZ antibody. LN, lymph node. *YAP+ stroma/ endothelia. Scale bars, 100 µm.
Next, we assessed primary cancers. Strikingly, the top 330 PC1/3 genes stratified 10,485 TCGA/TARGET cancers into YAPoff/YAPon classes (Figures 3G, S3B, and S3C). Like cell lines, YAPoff cancers were cluster A/blood and cluster B/solid cancers from neural/NE tissues, while YAPon cancers comprised clusters C/D similar to the CCLE (Figure S3C). PC1+/PC3+ genes were down-regulated and PC3- genes up-regulated in a signature of SCN cancers (Figure 3H) (Balanis et al., 2019). TCGA ‘‘adenocarcinomas’’ include SCN variants of pancreas, bladder, breast, lung, and stomach cancer (Balanis et al., 2019), and YAP/TAZ were down-regulated in these tumors (Figure 3I). Well-differentiated NE tumors (NETs) are histologically distinct from SCN cancers, and among 143 NETs from stomach, lung, duodenum, small and large intestine, pancreas, and appendix, as well as liver and lymph node metastases, we easily detected YAP/TAZ in mesenchyme and vasculature, but they were absent in 97.9% (140) of these tumors (Figure 3J; Table S2). While there may be exceptions, our data suggest that most cancers stratify into binary YAPoff and YAPon classes.
Low levels of YAP/TAZ-correlated adhesion genes could reflect transcriptional or post-transcriptional regulation. Hippo pathway kinases can destabilize YAP/TAZ, but depleting MST1 or overexpressing dominant-negative LATS1 did not induce YAP or TAZ (Figure S3E). Mining chromatin immunoprecipitation sequencing (ChIP-seq) data from YAPoff lines revealed YAP and YAP-correlated PC1+ gene promoters lack RNA polymerase II, unlike anti-correlated PC1– genes, and the opposite pattern applied in YAPon cells (Figure S3F). ChIP-qPCR of activating and repressive epigenetic marks at YAP, TAZ, AJUBA, and CYR61 promoters indicated opposite patterns in YAPoff versus YAPon cell lines (Figure S3G). Thus, epigenetic regulation defines the binary YAPoff or YAPon pan-cancer states, respectively.
Binary classes exhibit opposite YAP/TAZ function, contrasting vulnerabilities, and therapy-driven drug resistance
YAP/TAZ suppress solid YAPoff cancers (Figures 1 and 2), and YAP also inhibits multiple myeloma (Cottini et al., 2014), a liquid YAPoff cancer (Figures 3C and S3C). In contrast, many YAPon solid cancers require YAP/TAZ (Zanconato et al., 2016; Zheng and Pan, 2019). Thus, an intriguing hypothesis is that opposite YAP/TAZ function defines most cancers. Mining the Cancer Dependency Map (DepMap) (Meyers et al., 2017), a functional genomics effort to define essential genes in hundreds of cancer cell lines, revealed that deleting YAP or TAZ did not affect 92% of YAPoff cases, contrasting dependency on either gene in 65% of YAPon lines, and in 5/5 YAPon lines where either YAP or TAZ are dispensable, deleting both revealed redundancy (Figures 4A–4C, S4A, and S4B). One study claimed that RNAi-depletion of TAZ weakly impedes retinoblastoma cell line growth (Zhang et al., 2015), but dual YAP/TAZ-deletion had no effect in these or other YAPoff cell lines (Figures 4C and S4B). Moreover, as in 11 retinoblastoma/SCLC cell lines (Figures 1E and 2I), expressing YAP inhibited growth of breast, pancreas, and prostate YAPoff NE cancer lines (Figures 4D and S4C). Combined with our in vivo and in vitro GoF/LoF studies in YAPoff cancers, these pan-cancer data suggest that it is not only opposite expression, but also opposite YAP/TAZ function that defines pan-cancer binary classes.
Figure 4. Binary cancer classes exhibit contrasting YAP/TAZ function and vulnerabilities.
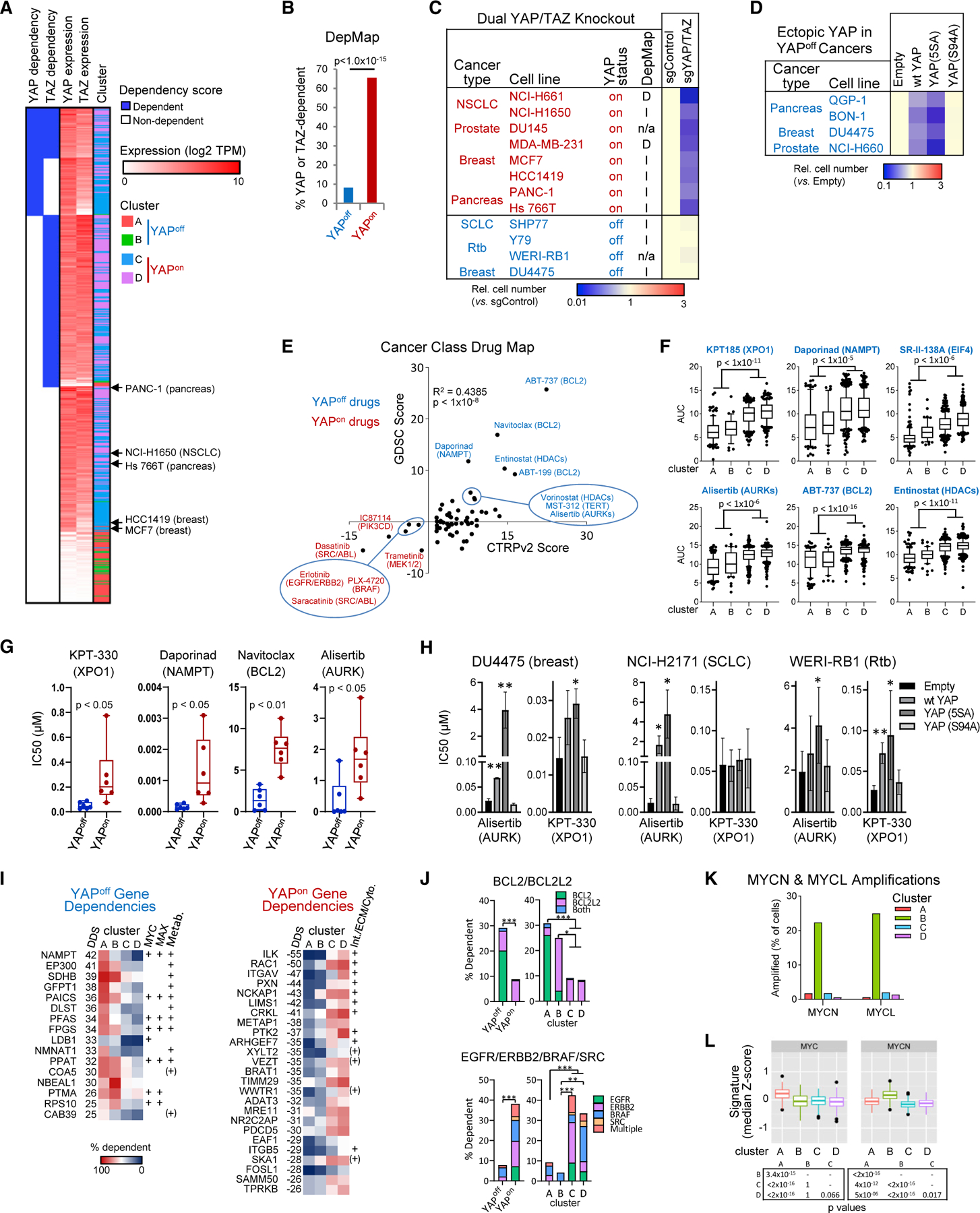
(A) YAP/TAZ dependency and mRNA levels in DepMap cell lines. The indicated YAPon lines were tested in dual KO assays (see panel C).
(B) YAP or TAZ dependency in YAPoff or YAPon lines; Fisher’s test.
(C) YAP/TAZ KO suppresses growth of YAPon (n = 3, p < 0.05) but not YAPoff cells. D, I, n/a, YAP/TAZ-dependent, -independent, or not available in DepMap.
(D) Ectopic YAP or YAP5SA, but not YAPS94A, impede growth of indicated YAPoff lines (n = 3, p < 0.05).
(E) Drug map of YAPoff solid cancer-selective (blue) or YAPon cancer-selective (red) drugs in CTRPv2 and GDSC databases.
(F) Area under the curve (AUC) of select top scoring drugs in CTRPv2 database.
(G) Validation of indicated YAPoff-selective drugs.
(H) Effect of the indicated expression vectors on the sensitivity of YAPoff lines to YAPoff-selective drugs. Mean ± SD, n ≥ 3; *p < 0.05, **p < 0.01.
(I) Top dependency genes selective for cluster A/B (YAPoff) or cluster C/D (YAPon) cell lines. MYC/MAX-bound targets and metabolic (Metab.) genes (YAPoff), or genes related to integrin signaling, ECM, or cytoskeletal regulation (Int./ECM/Cyto., YAPon) are indicated; +, from Enrichr; (+), from the literature.
(J) DepMap-defined dependency of different cancer classes on select gene combinations. Fisher’s test: *p < 0.05, **p < 0.01, ***p < 0.0001.
(K) MYCN or MYCL amplification in cancer classes.
(L) MYC and MYCN signature scores in cancer classes. Pairwise Wilcoxon test, Bonferroni correction.
Next, we searched for class-selective vulnerabilities, which could have broad therapeutic implications. Employing CTRP and GDSC drug databases, we identified several high-confidence cluster B-selective drugs. Inhibitors of the NAD synthesis enzyme NAMPT, aurora kinases, the anti-apoptotic BCL family, and histone deacetylases (HDACs), were hits in both databases (Figure 4E), and inhibitors of the nuclear transporter XPO1 and the elongation factor EIF4 had multiple hits in the CTRP database (Table S3). Moreover, these drugs also potently inhibited cluster A cancers, revealing general YAPoff selectivity (Figures 4F and S4D). Notably, aurora kinase inhibitors are synthetic lethal with RB1 loss (Gong et al., 2019; Oser et al., 2019), which is enriched in YAPoff solid cancers (Figure 3E), and XPO1 and BCL or HDAC inhibitors are potent against leukemia and SCN tumors, respectively (Balanis et al., 2019; Verbeke et al., 2020). We validated several YAPoff-selective drugs, and expressing YAP and/or constitutively active YAPS5A reduced sensitivity in several YAPoff cancer types (Figures 4G and 4H). Thus, the unique drug profile of YAPoff cancers depends on YAP silencing.
To further validate and expand therapeutic targets, we assessed genetic vulnerabilities in YAPoff cancers. Using DepMap we calculated differential gene dependency scores between YAPoff and YAPon cancers (Figure 4I; Table S4). Genes for drug targets that are indispensable in all cells (e.g., XPO1, EIF4) cannot be validated genetically, and single-gene deletion for redundant families (e.g., BCL/HDAC) may have little/no effect. Nevertheless, the fraction of lines that required BCL2 and/or BCL2L2 were significantly higher in YAPoff cancers (Figure 4J). In striking agreement with YAPoff sensitivity to NAMPT inhibitors (Figure 4E), top single-gene dependencies included both NAMPT and its partner, NAD synthesis enzyme, NMNAT (Figure 4I). YAPoff-dependency genes were enriched for metabolic enzymes, including three of the six-member purinosome that drives de novo purine synthesis (Figures 4I and S4E; Table S4). This gene set was also enriched for direct MYC/MAX binding (Figure 4I; Table S4), and MYC drives these metabolic pathways (Stine et al., 2015). Furthermore, MYCN or MYCL amplification was enriched in solid (cluster B) YAPoff cancers (Figure 4K), and both cluster A and cluster B YAPoff cancers had higher levels of MYC- or MYCN-induced gene signatures than YAPon cancers (Figure 4L). Contrasting the YAPoff drug map, YAPon cancers exhibited elevated sensitivity to tyrosine (e.g., SRC, ERBB, EGFR) or serine/threonine (e.g., BRAF) kinase inhibitors and, in agreement, the fraction of YAPon cancers dependent on the corresponding genes was higher in YAPon versus YAPoff cancers (Figures 4E and 4J). In addition, YAPon gene dependencies were enriched in integrin/ECM/cytoskeletal regulators (Figure 4I), mirroring high expression in YAPon cancers (Figures 3C and 3F; Table S1). Thus, YAPoff and YAPon cancers exhibit distinct drug vulnerabilities, genetic dependencies complement these contrasts, and class specificity mirrors divergent mutational spectra and transcriptional profiles that typify YAPoff and YAPon cancers.
The discovery of contrasting vulnerabilities in YAPoff versus YAPon classes suggested that cancer may switch binary classes to escape therapeutics. Indeed, conversion of adenocarcinoma to SCN cancer is a lethal, therapy-driven event associated with RB1 loss, an NE lineage switch, and altered drug sensitivity (Beltran et al., 2019), all features consistent with YAPon to YAPoff transformation. To explore this hypothesis, we assessed relevant murine cancer models. In lung cancer with mixed adenocarcinoma and NE histology, only the former was Yap+ (Figure 5A). Similarly, Yap protein was expressed in Pten–/– (so-called single KO [SKO]) prostate adeno-carcinoma (PrAd), but absent in two models of late-stage NE prostate cancer (NEPC) involving Rb1/Pten (DKO) or Rb1/ Pten/p53 loss (TKO) (Figure 5B). Mapping our class-defining PC1/PC3 gene set onto transcriptome data revealed that SKO PrAd are YAPon cancers, whereas advanced DKO or TKO NEPC are YAPoff cancers (cf. Figures 5C and 3C). DKO tumors are initially PrAd, and convert to NEPC starting at around weeks 20–25, mimicking anti-androgen-induced lineage conversion in human prostate cancer (Ku et al., 2017). Strikingly, early-stage 12-week DKO PrAd exhibited a YAPon transcriptome (Figure 5C). To quantitatively assess these differences, PCA was used to define a ranked gene list differentiating SKO and 12-week DKO PrAd from late-stage DKO and TKO NEPC (Table S5). GSEA validated the YAPon versus YAPoff classification of PrAd versus NEPC, respectively (Figure 5D). In the TKO model, PrAd to PNEC conversion starts earlier, at 11 weeks and, while we lacked transcriptome data from this stage, nascent NEPC regions were Yap–, starkly contrasting neighboring Yap+ adenocarcinoma (Figure 5E).
Figure 5. Epithelial-to-SCN cancer is a YAPon to YAPoff switch.
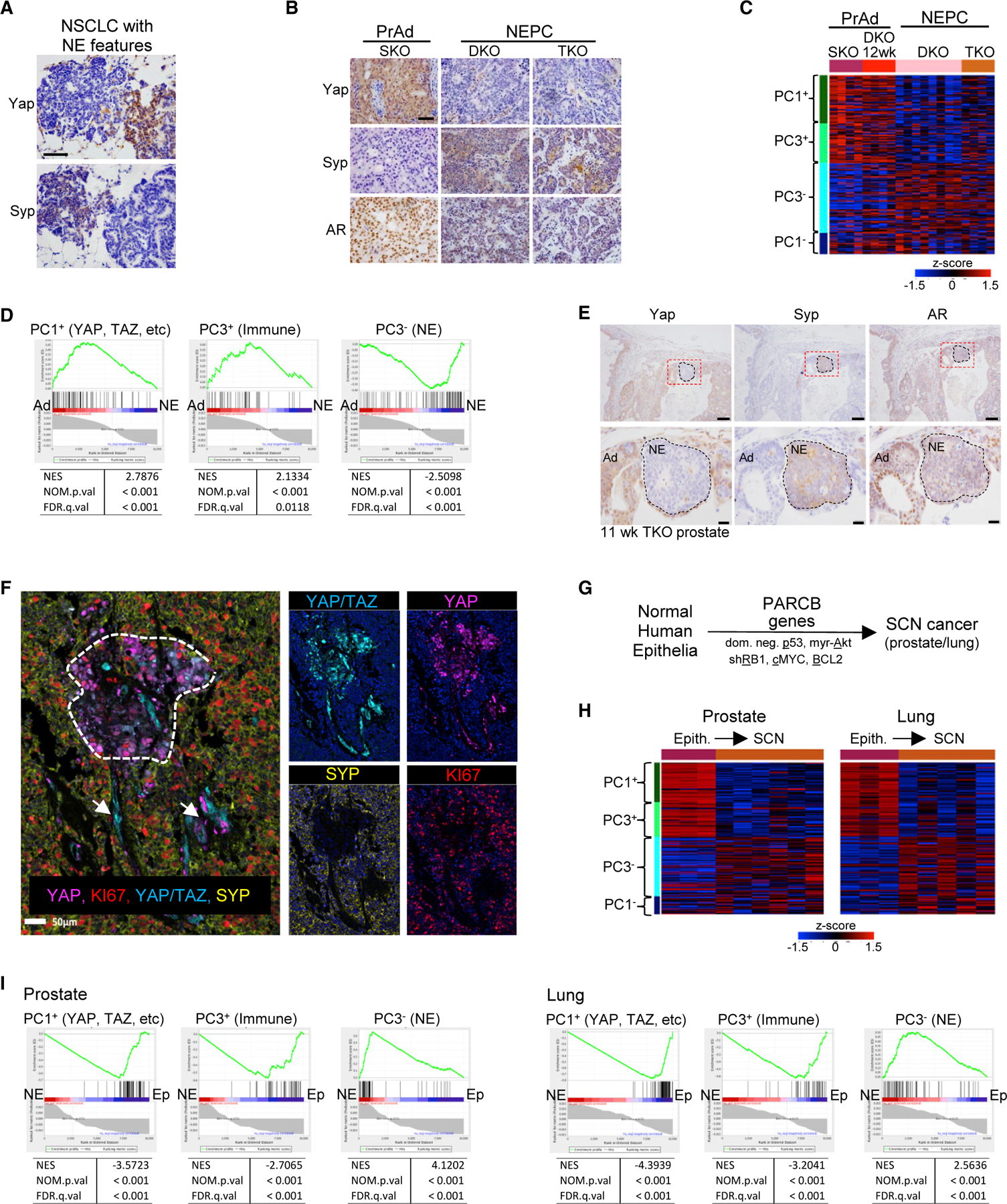
(A) IHC on a murine lung model with mixed NSCLC (Syp–/Yap+) and NE (Syp+/Yap–) histology. Scale bar, 100 µm.
(B) IHC in murine PrAd (SKO: Pten–/–) or NEPC (DKO: Pten–/–;Rb1–/– or TKO:Pten–/–;Rb1–/–;p53–/–). Scale bar, 100 µm.
(C) PC1/3 gene levels in murine prostate cancer (cf. Figure 3C).
(D) PC1/3 gene enrichment in a ranked list (Table S5) distinguishing murine PrAd and NEPC confirms these prostate tumors are YAPon and YAPoff cancers, respectively.
(E) IHC on emerging NE tumors in 11-week murine TKO prostate. Scale bars, 100 µm (top), 20 µm (bottom).
(F) Multispectral IHC of mixed human NEPC (SYP+) and PrAd (SYP–). Only PrAd (dotted area) and blood vessels (arrows) stain for YAP or YAP/TAZ.
(G) PARCB gene code to reprogram human primary epithelia to SCN cancer (Park et al., 2018).
(H) PC1/3 gene levels in primary human epithelia or SCN cancer derived as in (G) (cf. Figure 3C).
(I) PC1/3 gene enrichment in a ranked list (Table S5) distinguishing the two states in (G) confirms the PARCB genes induce YAPon to YAPoff reprogramming.
See also Table S5.
Next, we asked whether NE cancer conversion in humans is also a YAPon to YAPoff class switch. We used H&E staining plus multispectral IHC to analyze 17 pure PrAd, 11 mixed NEPC/PrAd, and 9 NEPC samples. Pure PrAd and NEPC tumors were YAP/TAZ positive and negative, respectively, and, in mixed tumors, YAP/TAZ staining was confined to PrAd regions or blood vessels, and absent from NEPC (Figure 5F). In a second approach, we took advantage of a model in which primary human epithelia are reprogrammed to aggressive SCN cancer through the ‘‘PARCB’’ code involving RB1/p53 inactivation plus cMYC/AKT/BCL2 activation (Figure 5G) (Park et al., 2018). Mining the associated transcriptome changes revealed a YAPon to YAPoff transformation, both in lung and prostate (Figures 5H and 5I). These murine and human data reveal that highly lethal NE lineage switches in YAPon lung or PrAd drive YAPoff cancer, highlighting the potential value of YAPoff-selective therapies (Figures 4E–4G, S4D, and S4E).
YAPon- and YAPoff-specific gene targets drive opposite adhesive behavior of binary cancer classes
To deduce why YAP has opposite pro- or anti-cancer effects, we performed RNA sequencing (RNA-seq) with five empty vector- or YAP-transduced YAPoff lines, and compared published YAP-dependent gene sets from five YAPon cell lines. YAP mainly induced genes in YAPoff lines (Figure S5A). Either rank-rank hypergeometric overlap or GSEA clustered YAPoff and YAPon datasets separately, implying distinct YAP targets underpin binary classes (Figures 6A and S5B). We confirmed that YAP induces proliferative genes (G1-S transition, DNA replication, etc.) in YAPon cells (Hiemer et al., 2015; Kim et al., 2016; Li et al., 2014; Park et al., 2016; Zanconato et al., 2015), but YAP did not affect these gene sets in YAPoff cells and, instead, activated the PC1+ integrin/ECM/adhesion genes silenced in this class (Figures 6B, S5C, and S5D; Table S6). Unsupervised clustering of the top YAP targets confirmed induction of integrins, collagens, laminins, and downstream cytoskeletal components (e.g., myosins, FLNA) in YAPoff cells, contrasting induction of cyclins, minichromosome maintenance (MCM), and other cell-cycle genes in YAPon cells, while well-known YAP targets, such as CTGF and CYR61, were induced in both classes (mixed) (Figure 6C; Table S6).
Figure 6. Distinct YAP targets drive opposite adhesive behavior of binary cancer classes.
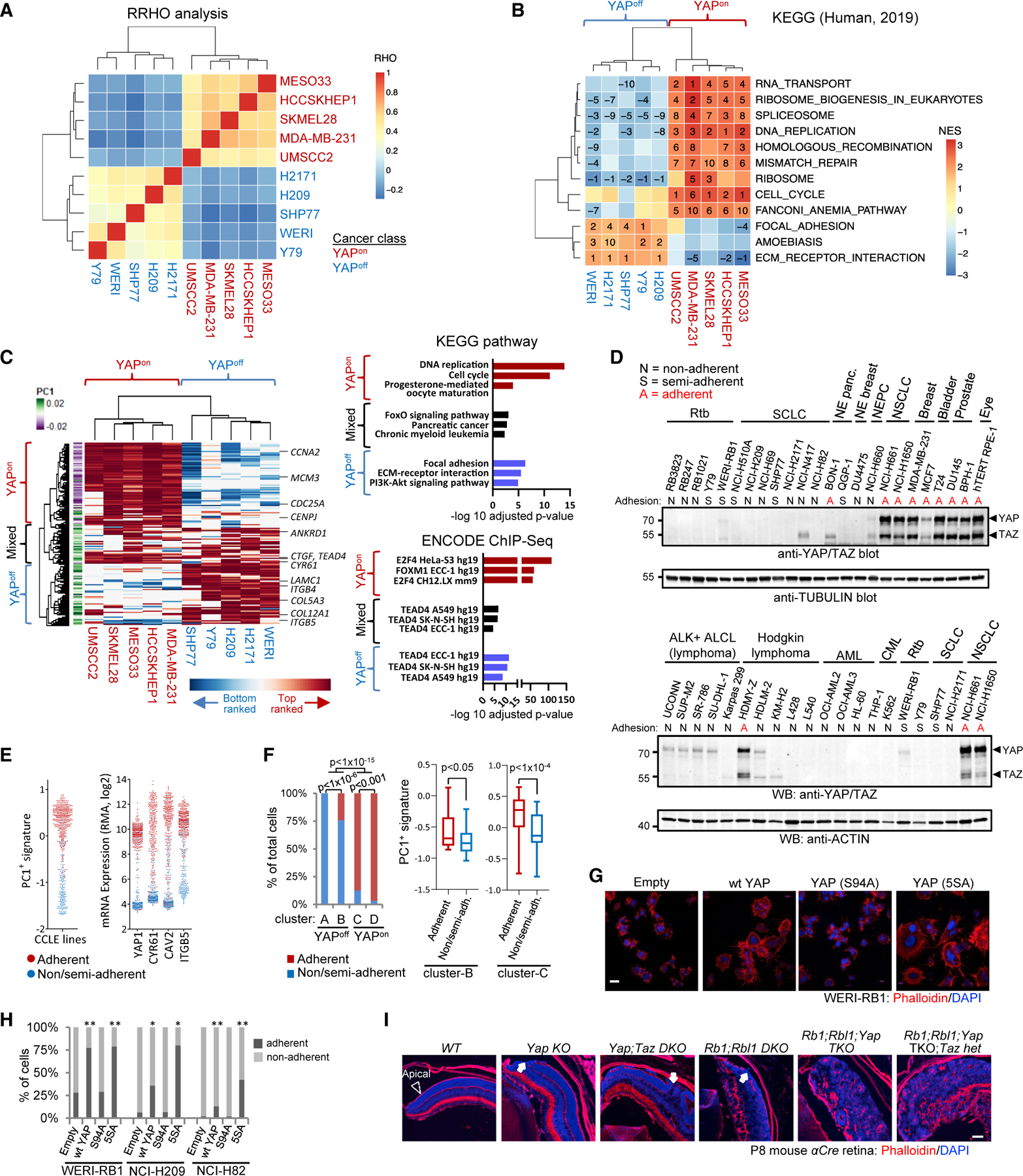
(A) Hierarchical clustering of RHO coefficients from rank-rank hypergeometric overlap (RRHO) to compare YAP targets in YAPon and YAPoff lines.
(B) Hierarchical clustering of KEGG pathways enriched in YAP targets. Numbers: rank (stats in Table S6).
(C) Unsupervised clustering of the top YAP-induced genes (left). Graphs of the top enriched KEGG pathways and ENCODE TFs (right).
(D) YAP/TAZ western blots and adhesion type for indicated cell lines.
(E) PC1+ signature scores (left) and levels of select genes (right) in CCLE lines with indicated adhesive behavior.
(F) Proportion of adherent and non/semi-adherent cell lines in cancer classes (left; Fisher’s test); PC1+ signature score in the adherent and non/semi-adherent cell lines from clusters B and C (right).
(G) Phalloidin (F-actin) staining of WERI-RB1 Rtb cells transduced with the indicated vectors.
(H) YAP increases adhesion in YAPoff cell lines. n R 3; Fisher’s test: *p < 0.05, **p < 0.01.
(I) Phalloidin staining of retina from aCre mice of the indicated genotypes (abbreviations, Figure 1B). Arrowhead, apical surface; arrows, apical breaks.
YAPon- or YAPoff -dependent adhesion/ECM genes suggested that binary classes may have distinct adhesive behavior. Indeed, YAPoff cancers from many different tissues were non- or semi-adherent in culture, whereas YAPon lines were adherent (Figure 6D). The Hodgkin lymphoma line, HDMY-Z, unlike other lymphomas is adherent and it expressed YAP/TAZ at levels seen in YAPon cancers (Figure 6D). Comparison of hundreds of cell lines showed that PC1+ genes exhibit opposite expression across non-adherent versus adherent cell lines (Figure 6E). Among YAPoff lines, all cluster A and most cluster B lines were non-adherent, while most YAPon lines were adherent, and within clusters B and C, adherence correlated with PC1+ gene levels (Figure 6F). Ectopic YAP induced adhesion of human YAPoff cancer lines (Figures 6G and 6H), prompting us to ask if YAP/TAZ influence adhesion in vivo. Basal integrin/ECM signaling engages cytoskeletal components to drive apical constriction required for tissue morphogenesis (Fernandes et al., 2014) and, intriguingly, Yap/Taz loss disrupted the apical surface in the Rb1/Rbl1 null retina at P8, commensurate with the subsequent increase in tumor penetrance in adults (cf. Figures 6I and 1B). Thus, adding to distinct transcriptional, pharmaceutical and genetic profiles, YAPoff and YAPon cancers also display opposite adhesive properties.
Differential enhancer deployment dictates pro- or anti-cancer YAP/TEAD functions
Next, we investigated the mechanism underlying contrasting YAP targets in binary cancers. In oncogenic contexts, YAP/ TAZ associate with TEADs and AP-1 to promote cancer (Liu et al., 2016; Stein et al., 2015; Verfaillie et al., 2015; Zanconato et al., 2015; Zhao et al., 2008), whereas it binds p73 to drive apoptosis in multiple myeloma (Cottini et al., 2014), a YAPoff liquid cancer. To test YAP-TEAD involvement in suppression of solid YAPoff cancers, we used a mutant (YAPS94A) that disrupts TEAD binding (Zhao et al., 2008). YAPS94A failed to induce cytostasis, drug resistance, adhesion, or gene induction in YAPoff cancer cells (Figures 1E, 1F, 2I, 4D, 4H, 6G, 6H, and S5A). In contrast, mutating YAP WW domains, required for binding to p73 and other factors (Piccolo et al., 2014), did not alter YAP-induced cytostasis (Figure S6A). YAP-mediated growth inhibition was linked to reduced S- and increased G2/M-phase cells with minimal apoptosis (Figures S6B and S6C), consistent with p73 independence. We confirmed that TEAD binds ectopic YAP but not YAPS94A in YAPoff cells (Figures 7A and S6D). Deleting TEAD1, the predominant TEAD in SHP77 SCLC cells blocked, whereas overexpressing TEAD3 or TEAD4 enhanced YAP-mediated cytostasis (Figures 7B and 7C). Moreover, a hybrid protein with TEAD4-DNA-binding domain fused to the VP64-activation domain recapitulated YAP-induced cytostasis (Figure 7D). Thus, YAP suppresses YAPoff cancers through TEAD binding and gene activation, identical to the oncogenic mechanism in YAPon cancers (He et al., 2019; Kapoor et al., 2014; Ota and Sasaki, 2008).
Figure 7. Enhancer usage dictates opposite YAP/TEAD effects.
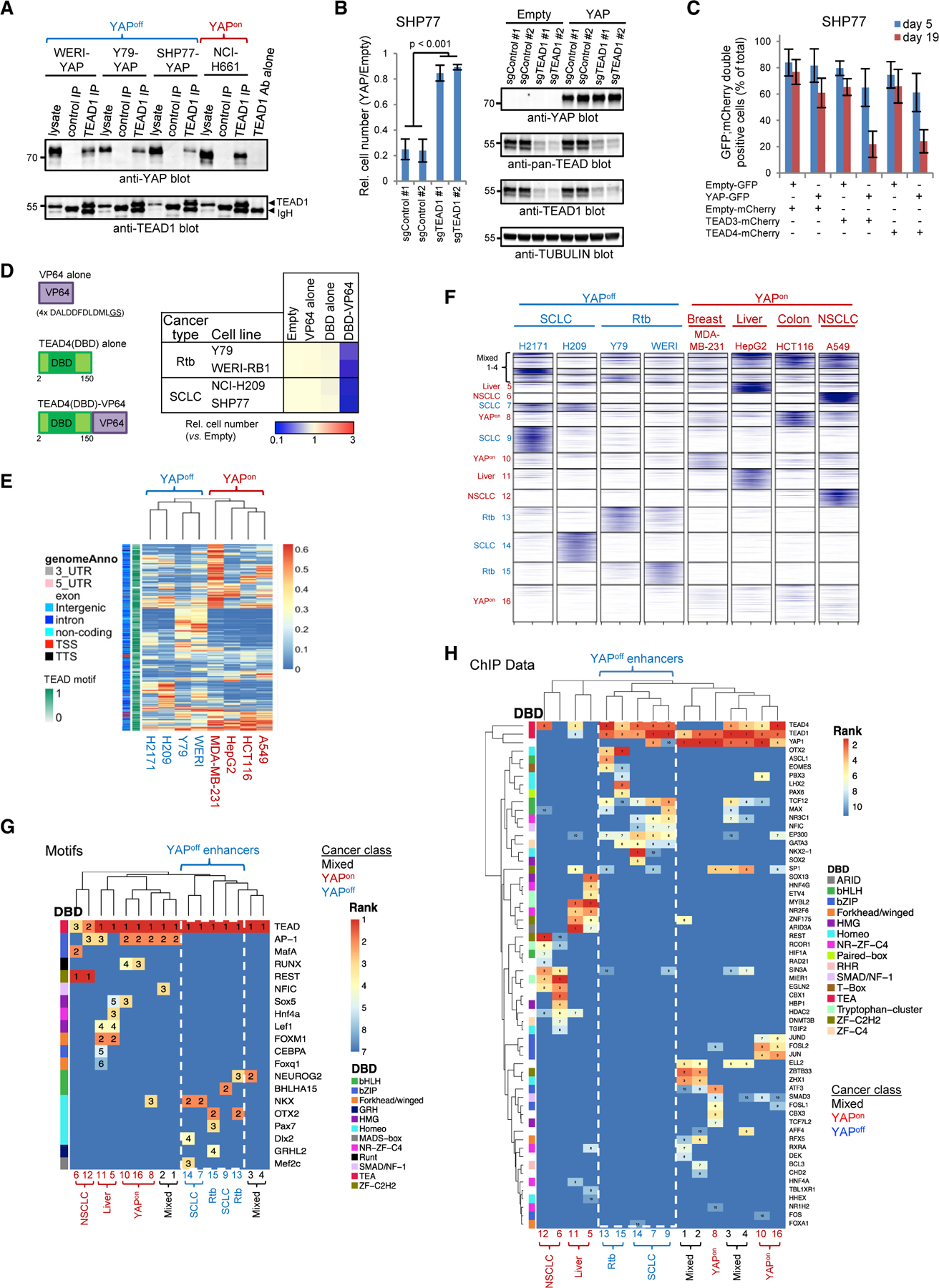
(A) TEAD1/YAP coimmunoprecipitation in YAPon and YAP-transduced YAPoff cells.
(B) TEAD1 deletion prevents YAP-induced cytostasis. Mean ± SD, n = 4.
(C) Ectopic TEAD3 or TEAD4 enhances YAP-meditated cytostasis. Mean ± SD, n = 3.
(D) TEAD4-VP64 impedes YAPoff cell line growth (n = 3, p < 0.001).
(E) Hierarchical clustering of the top 1,000 TEAD4 ChIP-seq peaks from each YAPon or YAP-transduced YAPoff line.
(F) k-Means clustering of TEAD4 peaks defines 16 enhancer groups.
(G) Unsupervised hierarchical clustering of enriched TF motifs in the 16 TEAD4-enhancer groups. Numbers: motif rank.
(H) Hierarchical clustering of co-bound TFs in the 16 TEAD enhancer groups.
See also Figures S6 and S7 and Table S7.
To understand contrasting YAP/TEAD function, we performed TEAD4 ChIP-seq in four YAPoff lines plus/minus YAP, comparing our results with published TEAD4 ChIP-seq data from four YAPon lines. YAP enhanced existing binding sites and greatly expanded the target repertoire of TEAD4 in YAPoff cancers, suggesting cooperativity, likely aided by increased TEAD levels (Figures S7A–S7C). We noted tissue-specific TEAD sub-clusters in YAPoff data but, more importantly, YAPoff and YAPon cancers formed separate superclusters (Figure 7E), indicating distinct targeting in each class.
TEADs cooperate with AP1 to induce cell-cycle genes in YAPon cancers (Liu et al., 2016; Stein et al., 2015; Verfaillie et al., 2015; Zanconato et al., 2015), thus we asked if they employ distinct factors in YAPoff cancers. For this, we first classified 16 distinct enhancer clusters using k-means clustering (Figures 7F and S7D; Table S7). Most YAPoff enhancers were remote (Figure S7E), similar to YAPon TEAD enhancers (Liu et al., 2016; Stein et al., 2015; Verfaillie et al., 2015; Zanconato et al., 2015). TEAD-bound clusters 1–4 were common to multiple YAPoff and YAPon lines, but others were specific to cell lines, cancer types, or cancer classes (Figure 7F). We then searched for features that distinguish YAP/TEAD enhancers in YAPoff and YAPon cancer classes. Motif analysis revealed TEAD sites at all enhancers, validating the dataset (Figure 7G). However, whereas YAPon enhancers contained AP1/MafA, FOXM1, or REST motifs, YAPoff enhancers lacked AP1 sites, and were enriched in motifs for lineage-determining basic-helix-loop-helix (bHLH) and homeobox TFs (Figure 7G). To validate these results, we mined 2,829 ChIP-seq datasets covering 485 TFs. Again, unsupervised clustering separated YAPon and YAPoff enhancers, sub-clustered retino-blastoma and SCLC-specific enhancers, and confirmed enrichment of homeobox (OTX2/PBX3/LHX2/NKX2–1) or bHLH (ASCL1/TCF12/MAX) factors at YAPoff enhancers, but AP1 (JUN/FOS/ATF family) or REST at YAPon enhancers (Figure 7H). Consistent with tissue of origin, SCLC and retinoblastoma elements bound TFs that influence lung (e.g., NKX) or retinal (e.g., OTX2) development, respectively (Figures 7G and 7H). Thus, while YAP/TEAD cooperate with AP1 in YAPon cancers, they collaborate with neurogenic bHLH/homeobox TFs in YAPoff cancers, exposing a high-level regulatory rule distinguishing binary cancer classes.
A YAP-integrin axis drives cytostasis in YAPoff cancer cells
To identify genes required to mediate cytostasis in YAPoff cancers we ran CRISPR screens against ~1,000 YAP targets, PC1+ genes, and controls. A top hit was integrin-β5 (ITGB5), and validation assays confirmed its loss inhibits YAP-induced cytostasis, not YAP expression or activity (Figures 8A, 8B, S8A, and S8B). ITGB5 is a remarkable hit, because it is a PC1+ gene (off in YAPoff, on in YAPon cancers; Figure 3C), dimerizes with integrin-αV (ITGAV) (Raab-Westphal et al., 2017), a high-rank essential gene in YAPon cancers (Figure 4I), and was induced by YAP in 4/5 YAPoff cancer cell lines, but unaffected by YAP-loss in 5/5 published YAPon lines (Figure 6C). We confirmed differential regulation of ITGB5 protein by YAP in multiple YAPoff and YAPon lines (Figures 8C and S8C). ITGB5 regulation in YAPon cells may be complex, as it is not YAP dependent, but can be induced by supra-physiological YAP levels (Cordenonsi et al., 2011). Irrespective, our data highlight a class-specific cytostatic YAP-ITGB5 axis, explaining why these genes must be down-regulated in YAPoff cancers.
Figure 8. ITGAV/ITGB5 are effectors of YAP-induced cytostasis.
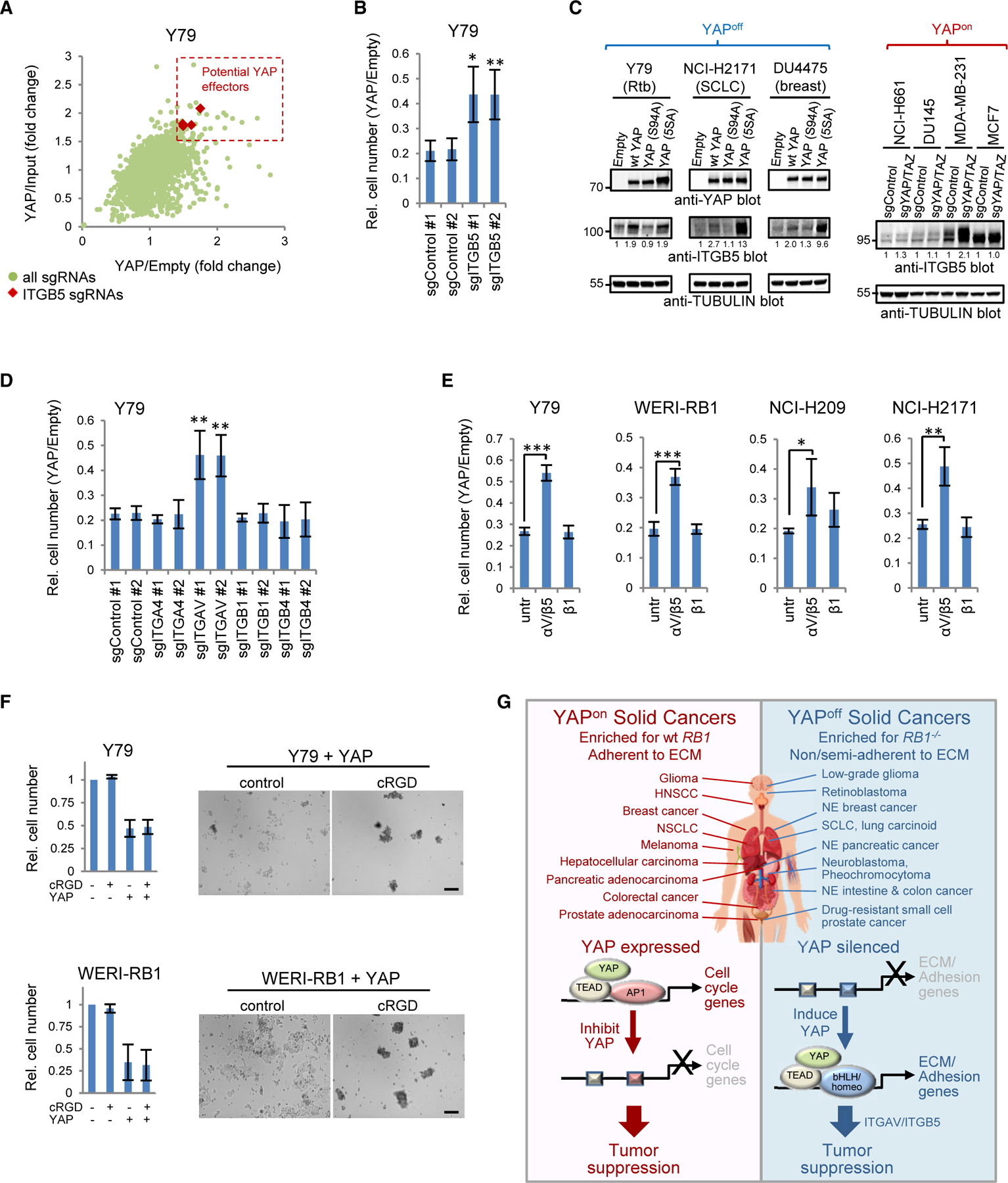
(A) Scatterplot of fold difference between YAP- and empty vector-transduced or input cells for each single-guide RNA in the CRISPR screen.
(B) ITGB5 knockout rescues YAP-induced cytostasis.
(C) Western blots in YAP-transduced YAPoff or YAP/TAZ-knockout YAPon lines.
(D) Knockout of ITGAV, but not other integrins, alleviates YAP-induced cytostasis.
(E) ITGAV/B5, but not ITGB1, blocking antibody alleviates YAP-induced cytostasis. All panels: mean ± SD, n ≥ 3. *p < 0.05, **p < 0.01, ***p < 0.001 versus singleguide control or untreated (untr) cells.
(F) Cyclic RGD peptide (cRGD) blocks YAP-induced adhesion (images), but not cytostasis (graphs). Scale bars, 200 µm.
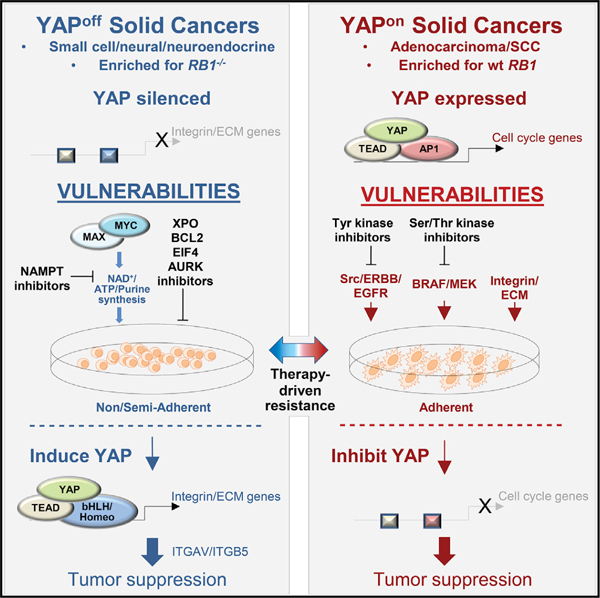
(G) Summary of contrasting YAP/TEAD mechanisms of action in solid YAPon and YAPoff cancers.
See also Figure S8.
Next, we asked whether the ITGB5 partner, ITGAV, influences growth suppression. Indeed, deleting ITGAV, but not other integrins, rescued cell growth (Figures 8D and S8D). Moreover, an ITGAV/B5, but not ITGB1, blocking antibody alleviated YAP-induced growth suppression in YAPoff lines (Figure 8E). The ITGAV/ITGB5 dimer is thus a key effector of YAP-induced cytostasis in YAPoff cancer cells.
Integrins mediate cell-matrix adhesion, but also have adhesion-independent functions (Ferraris et al., 2014; Seguin et al., 2014). A cyclic RGD peptide that disrupts integrin-ECM interactions prevented adhesion of YAP-transduced YAPoff cells, but did not rescue cytostasis (Figure 8F). These data contrast the genetic and antibody blocking assays above tying ITGAV/B5 to YAP-induced cytostasis. ITGAV/B5-blocking antibodies inhibited YAP-induced adhesion of some (e.g., Y79), but not all (e.g., WERI-RB1), YAPoff lines (Figure S8E). Thus, while YAP drives both adhesion and cytostasis in YAPoff cancer cells, they are uncoupled, focusing attention on adhesion-independent functions of ITGAV/ITGB5. Altogether, our transcriptome, chromatin binding, and GoF and LoF studies reveal that YAP silencing is vital in solid YAPoff cancers to avoid deployment of a cytostatic integrin program centered on ITGAV/B5, contrasting the opposite pro-cancer role in YAPon cancers (Figure 8G).
DISCUSSION
Over-arching cancer principles can expose therapies with broad relevance. Here, starting with the goal of understanding distinct genetic and adhesive features of small cell versus other solid cancers, we uncovered a binary pan-cancer classification scheme that pivots on contrasting YAP activity. Unlike YAP-dependent YAPon solid cancers, a large group of neural/NE solid cancers co-silence YAP/TAZ with integrins and other adhesion regulators. Opposite anti- or pro-cancer YAP activity in YAPoff versus YAPon cancers underlies these transcriptome differences and, mechanistically, a unique YAP/TEAD enhancer repertoire in YAPoff solid cancers deploys integrin-αV/β5 to drive cytostasis. This YAP-centric cancer classification scheme has interesting therapeutic implications, because the binary groups exhibit distinct vulnerabilities, and therapy-driven lineage switching exploits these differences to drive drug resistance.
Small molecules that disrupt YAP/TEAD activity show promise in YAPon cancers (Crawford et al., 2018; Gill et al., 2018; Kurppa et al., 2020). However, targeting YAP would, by definition, be futile in YAPoff cancers, and in YAPon prostate and lung adenocarcinomas may favor evolution of drug-resistant YAPoff SCN cancer (Beltran et al., 2016, 2019; Lee et al., 2017; Niederst et al., 2015; Tan et al., 2014). Our LoF and GoF data, in multiple cancer/tissue types, in vivo, in vitro, and in murine and human contexts, expose YAP derepression as a potential avenue to tackle solid YAPoff cancers, which our functional genomics data extends to include the YAP-regulated integrin pair, ITGAV/ITGB5. Methods to derepress these genes await discovery, but our studies also highlight approved drugs that selectively inhibit YAPoff cancers, several of which target metabolic dependencies (e.g., NAD/purine/ ATP synthesis) that correlate with high MYC family activity. High MYC activity provides a logical explanation as to how YAPoff cancers sustain division, contrasting reliance on YAP in YAPon cancers. We find that expressing YAP reduces sensitivity to YAPoff-selective therapies, predicting YAPoff-to-YAPon switching as a potential resistance mechanism. YAP expression alone seems insufficient to cause such a switch, because we find it hinders growth of murine retinoblastoma or SCLC in vivo, consistent with YAP presence in benign, cytostatic human retinoma. Conversion to a malignant YAPon state may require cooperation between YAP and other pathways, such as NOTCH signaling (Ireland et al., 2020; Tlemsani et al., 2020). Irrespective, our binary scheme opens a different perspective on cancer treatment, based on opposite activity of a single transcription complex.
Cancer resistance is the common response to ‘‘oncogenic’’ mutations. For example, RB1 loss predisposes lung neuroendocrine or retinal cone cells to cancer, but these lineages represent only ~0.4% or ~3% of these tissues. We find that the cell of origin in each case is intrinsically YAP deficient, and in cancers where RB1 loss is a late event, we link this genetic event to silencing of YAP and the adhesion gene targets that define binary cancers. Removing YAP/TAZ may sensitize cancer-resistant lineages to transformation upon RB1 loss, as we observed in murine retina, although there are likely additional requirements, such as the elevated MYC/AKT activity plus p53 loss that converts human RB1–/– epithelia to NE cancer (Park et al., 2018). Notably, cone photoreceptors provide naturally high levels of MYCN and the p53 inhibitor MDM2 (Xu et al., 2009), MYC family amplification and p53 loss is common in SCLC (Rudin et al., 2019), and deleting the AKT inhibitor PTEN promotes murine retinoblastoma and SCLC (Cui et al., 2014; Song et al., 2012; Xie et al., 2015). Why certain RB1–/– lineages are far more susceptible to transformation than others puzzled the field for decades. Our data expose YAP/TAZ silencing as a key component of the underlying code.
Why TEAD complexes target different enhancers in YAPoff versus YAPon cancers is an intriguing issue for future studies. The co-bound bHLH/homeobox proteins we identified (e.g., ASCL1, NEUROD1, OTX2) specify YAP/TAZ-deficient lineages that are sensitive to RB1 loss in lung and retina (Borges et al., 1997; Neptune et al., 2008; Nishida et al., 2003). Lineage, therefore, seems to determine opposite YAP/TAZ activity. Potentially, lineage-determining bHLH/homeobox proteins may compete with AP1, consistent with YAP/TEAD/homeobox protein interactions in embryonic stem cells (Beyer et al., 2013). bHLH neurogenic proteins are indispensable in small cell cancers (Rudin et al., 2019), providing a logical explanation as to why YAP/ TAZ must be silenced in these cancers. Extensive proteomics in YAPoff cancers will be required to test this model.
In summary, our results show that YAP and TAZ profoundly influence context-specific responses to RB1 loss, expose a large YAPoff cancer class with pharmaceutical, genetic, metabolic, and adhesive profiles that contrast YAPon cancers, and pinpoint alternate enhancer engagement and a YAP-integrin axis as a key component of opposite molecular behavior in YAPoff cancers. The data highlight multiple therapeutic avenues for YAPoff cancers, including untreatable SCN cancers of the prostate and lung.
STAR☆METHODS
RESOURCE AVAILABILITY
Lead contact
Further information and requests for resources and reagents should be directed to and will be fulfilled by the lead contact, Rod Bremner (bremner@lunenfeld.ca).
Materials availability
Vectors generated in this study can be obtained from the lead contact.
Data and code availability
RNA-seq and ChIP-seq data have been deposited at GEO under accession codes GSE144972 and GSE144973. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.
EXPERIMENTAL MODEL AND SUBJECT DETAILS
Cell culture, PDXs and virus production
The retinoblastoma cell lines RB1021, RB3823 and RB247 (kindly provided by Brenda Gallie) were grown in Iscove’s media supplemented with 10% FBS, 0.0004% (v/v) β-mercaptoethanol, and 0.010 mg/mL insulin (Thermo Fisher) (To et al., 2012). Y79 and WERI-RB1 retinoblastoma cells, SH-SY5Y and SK-N-SH neuroblastoma lines (kindly provided by Meredith Irwin), along with the DU4475 breast cancer (kindly provided by Eldad Zacksenhaus), BPH-1 (benign prostate), HCC1419 (breast cancer), and NCI-H526 (SCLC) cell lines were cultured in RPMI 1640 media supplemented with 10% FBS. SHP77, NCI-H510A and NCI-H82 SCLC lines, as well as NCI-H661 and NCI-H1650 NSCLC cells were maintained in RPMI 1640 media containing 10% FBS and 10mM HEPES. NCI-H209, NCI-H69 and NCI-N417 SCLC cells were grown in RPMI 1640 supplemented with 7.5% FBS. NCI-H2171 SCLC cells were cultured in DMEM:F12 media with 5% FBS, 0.005 mg/ml insulin, 0.01 mg/ml bovine transferrin (Thermo Fisher), 30 nM sodium selenite (Sigma), 10 nM hydrocortisone (Sigma), 10 nM β-estradiol (Sigma) and extra 2mM L-glutamine (for final conc. of 4.5 mM). NCI-H660 NEPC cells were maintained in RPMI 1640 supplemented the same as NCI-H2171 culture media. The NE pancreatic cell lines, QGP-1 and BON-1 (kindly provided by Dr. Todd McMullen), were grown in DMEM media supplemented with 10% FBS, 0.01 mg/mL insulin, 10 nM hydrocortisone, 0.005 mg/mL transferrin, 10 ng/ml Somatostatin (Sigma) and 10 ng/ml Glycyl-L-histidyl-L-lysine (Sigma). A549 (NSCLC), SW-1573 (NSCLC), MCF7 (breast cancer), MDA-MB-231 (breast cancer), DU145 (prostate cancer), PANC-1 (pancreatic adenocarcinoma), Hs 766T (pancreatic adenocarcinoma) and Lenti-X 293T cells were cultured in DMEM media with 10% FBS. T24 bladder cancer cells were cultured in McCoy’s 5a media with 10% FBS. ALK+ Anaplastic Large Cell Lymphoma (Karpas 299, SUP-M2, SU-DHL-1, SR-786 and UCONN) and the HDMY-Z Hodgkin lymphoma cell lines were cultured in RPMI-1640 containing 10% FBS and 50 µM 2-mercaptoethanol, while Hodgkin lines KM-H2, L428, L540 and HDLM-2 were cultured in similar media with 20% FBS. The leukemia lines OCI-AML2 and OCI-AML3 were cultured in AMEM supplemented with 10% FBS and THP-1, HL-60 and K562 in IMDM with 10% FBS. All cells were maintained at 37◦C and 5% CO2, and were routinely screened for mycoplasma contamination by PCR.
SCLC PDXs (kindly provided by Charles Rudin) have been described previously (Gardner et al., 2017; Lok et al., 2017). Lentiviruses were generated using Lenti-X 293T cells and the psPax2 and pMD2.G packaging vectors. Following collection, virus was concentrated by ultracentrifugation. Retroviruses were produced using the Phoenix-eco packaging line and concentrated by ultracentrifugation.
Human retinal explants and generation of a human cone cell line
Human fetal retinas were obtained from the Morgentaler Clinic in Toronto with approval from the Research Ethics Board (REB #13–0132-E) of Mount Sinai Hospital in Toronto, Canada. All donors read the consent form approved by the REB before surgical procedures, and voluntarily donated fetal eye samples. The gestational age was estimated by a combination of clinic intakes, ultrasound, crown-rump, and fetal foot length measurements where possible (FitzSimmons et al., 1994; Shepard, 1975). Eye samples collected were held on ice for up to 6 hr in retina culture medium (IMDM with 10% FBS and 1X Antibiotic-Antimycotic, Life Technologies).
FW17 retina were dissected in sterile IMDM and cut radially. Tissue fragments were transferred onto cell-culture inserts (Millipore) with photoreceptor side down (Jin and Xiang, 2012). Inserts with retinal fragments were quickly put in 6-well plates with 1300 µl of prewarmed retina culture medium, and incubated at 37◦C with 5% CO2. Explants were transduced the next day with CRISPR/Cas9 lentivirus expressing a control, non-targeting or RB1-targeting sgRNA. Three weeks later, explants were harvested and processed for immunostaining.
To generate a human cone cell line, a Fw17 eye was sterilized in 70% ethanol for 3 seconds, rinsed twice in cold phosphate-buffered saline (PBS), and transferred to IMDM for retinal dissection. Retina was dissociated with Papain Dissociation System (Worthington Biochemical, Cat# LK003150). Briefly, retina was incubated in papain solution for about 15 min at 37◦C and 5% CO2, with gentle pipetting every 5 min. After dissociation to ~20-cell clusters, cells were suspended in 10 volumes of sterile phosphate buffered saline (PBS), pelleted by centrifugation at 300g for 10 min, and washed with PBS, followed by digestion in 0.05% trypsin/EDTA with gentle pipetting to produce single cell suspension. Cells were washed in 10 volumes of retina culture medium, resuspended in culture medium, then transduced with shRB1 lentivirus and replicating cells selected. Lineage was confirmed using the expression of the cone markers RXRγ, and L/M Opsin.
Mouse lines and experiments
All animal experiments were conducted with ethical approval from the respective animal care committees of each research institute, and all experiments conformed to the relevant regulatory standards. All mice were maintained on mixed backgrounds, and siblings utilized in comparisons.
Retina
α-Cre;Rb1f/f;Rbl1−/− (Chen et al., 2004) and Yapf/f;Tazf/f (Reginensi et al., 2013) mice were interbred to generate α-Cre;Rb1f/ f;Rbl1−/−;Yapf/f;Tazf/f experimental mice and controls used for survival analysis (Figures 1A, 1B, and S1A) and apical polarity studies (Figure 6I). Mice were a mixed 129Sv x C57BL/6J genetic background. Littermates from multiple breeding pairs were assessed, e.g. To compare Rb1f/f;p107−/− and Rb1f/f;p107−/−;Yapf/f mice we used litters from several aCre;Rb1f/f;p107−/−;Yapf/+x Rb1f/f;p107−/−;Yapf/+breeding pairs. Subretinal injections (Livne-Bar et al., 2006) of Cre-GFP or YAP-Cre-GFP retroviruses were performed on newborn P0 Rb1f/f or Rb1f/f;Rbl1−/− pups, which were then harvested for analysis at the indicated time points (Figures S1B–S1E).
Lung
We used six models to study cell-of-origin and/or cancer in lung. The CgrpCreER;Rb1f/f;p53f/f SCLC model (Figures 2F and S2C) is described in (Song et al., 2012), and was crossed with Rb1f/f;p53f/f;ROSA-rtTA+/+;TRE-Yap+/− mice (Gregorieff et al., 2015) to generate Rb1f/f;p53f/f;CgrpCreER+/−;ROSA-rtTA+/−;TRE-Yap+/− or TRE-Yap−/− experimental mice (Figures 2K and S2J). 5–7 wk old mice were injected IP with 0.225 mg/g tamoxifen (Sigma) daily for four consecutive days, then placed on a continuous doxycycline-containing diet. Mice were harvested 3 months after tamoxifen treatment and lungs processed for frozen sections. For Ad-Cre induction of Rb1/p53 null SCLC, combined with Yap/Taz loss (Figures 2L and S2N) or Yap transgene induction (Figures S2K–S2M), Ad5CMVCre (University of Iowa Viral Vector Core Facility, Iowa City, IA) was delivered to the lungs of P2–3 mice of the indicated genotypes by intranasal inhalation (10 µL at 1×108 pfu). For EGFR-driven NSCLC (Figure 2G) we used the previously described CcsprtTA;tetO-EGFRL858R NSCLC model (Politi et al., 2006; Tichelaar et al., 2000). At weaning, mice were fed chow containing doxycycline, activating rtTA and inducing expression of EGFRL858R in lung respiratory epithelial cells. Moribund mice were euthanized and tissues harvested. To compare Yap expression in the latter NSCLC model with that in a SCLC model (Figure 2G), we delivered Ad5CMVCre to the lungs of 6–8 week old Ptenf/+;Rb1f/f;p53f/f mice by intratracheal injection (50 µL at 1×108 pfu), as described previously (Cui et al., 2014; DuPage et al., 2009). To obtain a mixed model of NSCLC and SCLC (Figure 5A), we used the same intratracheal injection technique, but in Ptenf/f;Rb1f/f;p53f/f mice. Finally, we studied Yap/Taz expression in Rb1/p53 null E18.5 embryonic lung using SPC-rtTA;tetO-Cre;Rb1f/f;p53f/f mice (Figures 2H and S2B), treated as described previously (Akeno et al., 2015; Perl et al., 2002a; 2002b; Simpson et al., 2009). Briefly, timed pregnant females were fed doxycycline-containing diet throughout gestation to induced Cre and Rb1;p53 gene deletion. E18.5 embryos were then harvested and lungs processed for immunostaining.
Prostate
We used three models of prostate cancer (Figures 5B–5E). Male PB-Cre4; Ptenf/f (SKO) mice were used to model prostate adenocarcinoma, and male PB-Cre4;Ptenf/f;Rb1f/f (DKO) or PB-Cre4;Ptenf/f;Rb1f/f;p53f/f (TKO) mice were used to model NEPC. These models are described in (Ku et al., 2017; Lesche et al., 2002; Sun et al., 2006; Wang et al., 2003; Wu et al., 2001; Zhou et al., 2006). Briefly, recombination of floxed alleles was induced by the Cre4 transgene under control of the prostate epithelial cell-specific probasin promoter (PB-Cre4).
Confirmation of recombined floxed alleles
Cre-mediated recombination of floxed Rb1, p53, Yap and Taz alleles was confirmed in retina and lung models by PCR (data not shown, primers and diagnostic fragment sizes are in Table S8). Tissue was scraped from 14 µm-thick OCT-embedded frozen tissue sections on glass slides using a 30 G x 1/2” hypodermic needle and extracted by alkaline lysis (25 mM NaOH, 0.2 mM EDTA, pH 12) with heating at 95oC for ~1 hr followed by neutralization (40 mM Tris-HCl, pH 5). Rb1 and p53 primers have been described (Meuwissen et al., 2003), and all primer sequences are listed in Table S8. PCR was performed with 500 nM each primer using REDExtract-N-Amp PCR ReadyMix (Sigma) and the following cycling conditions: 94oC for 3 min; 35 cycles of 94oC for 0:40 min, 60oC (Rb1, p53, Yap and Taz F1-R1) or 63oC (Taz F2-R2) for 0:40 min, 72oC for 1 min; 72oC for 10 min. PCR products were resolved on 2% agarose gels.
METHOD DETAILS
Plasmid cloning
Lentiviral vectors to express YAP, TAZ and TEADs were generated using a modified pLKO.1 backbone (Stewart et al., 2003) from which the U6 promoter and shRNA cassette were removed. For YAP expression in retinoblastoma cells and TAZ in all lines the original hPGK promoter was used. For all other constructs the hPGK was replaced with an EFS promoter. The puromycin (puro) resistance gene in pLKO was replaced with IRES-GFP-T2A-puro or IRES-mCherry sequence. YAP, TAZ or YAP mutants were cloned upstream of the IRES-GFP while TEAD and TEAD4-VP64 constructs were cloned into the mCherry vector. Retroviruses for subretinal injections utilized the pMXIE backbone expressing Yap-IRES-CreGFP or CreGFP alone. CRISPR sgRNA sequences are listed in Table S8 and were cloned into the LentiCRISPR v2 vector (Addgene #52961) (Sanjana et al., 2014). The RB1 shRNA (sequence: CCACATTATTTCT AGTCCAAA, TRCN0000040163) was cloned into the pLKO.1 lentiviral vector modified to carry a GFP-puromycin selection cassette.
Growth curves and soft agar assays
For YAP/TAZ knockouts, cells were transduced with control or a YAP/TAZ sgRNA virus and selected in puromycin. Knockout was confirmed in bulk populations by Western blotting and/or genomic DNA sequencing followed by TIDE analysis (Brinkman et al., 2014) 8–10 days after selection, and cells plated for growth curves. Final cell counts were conducted 9 days after plating. New knockout populations were generated for each biological replicate.
For ectopic expression of YAP, TAZ or TEAD4-VP64, cells were transduced so that >90% expressed the indicated gene as determined by YAP, GFP and/or mCherry staining and flow cytometry and/or fluorescent microscopy. For growth curves, cells were seeded five days (seven days for NCI-H660) after viral transduction, and then counted 10 days later for all lines except slower-growing H660 NEPC cells which were counted 5 wks later. For sgYAP (rescue), sgTEAD1, or sgITG experiments in SHP77 and/or Y79 cells, stable bulk knockout populations were generated, then transduced with control or YAP virus as above. Final cell counts were conducted 15 days after YAP virus transduction. At least three independent knockout populations were generated for each sgRNA. For experiments with co-expression of YAP and TEAD3 or TEAD4, SHP77 cells were transduced with control, YAP-IRES-GFP or TEAD-IRES-mCherry viruses and the percentage of GFP/mCherry double-positive cells was assessed by flow cytometry after 5 days (plateau of YAP/TEAD expression) and 19 days on a Beckman Coulter Gallios flow cytometer and Kaluza analysis software.
For Integrin blocking experiments, cells were transduced with control (Empty) or YAP lentiviruses, then the following day left un-treated or treated with ITGAV/B5 (sc-81632) or ITGB1 (sc-13590) blocking antibodies at 2.5 µg/ml. Fresh antibody was added to the cells every 3–4 days and final cell counts conducted 15 days after YAP transduction.
For soft agar assays, 1×103 Y79, SHP77 or WERI-RB1 cells were seeded in 0.4% 2-hydroxyethyl agarose (Sigma), which was overlaid on 0.6% bottom agar in a 24-well plate. After 3–4 weeks, cells were fixed in MeOH and stained with crystal violet.
Cell cycle profiling by flow cytometry
SCLC and retinoblastoma cell lines transduced with YAP-GFP or control GFP viruses were pulsed with EdU (10 µm) for 20 min (Y79/ SHP77) or 30 min (WERI/H2171), harvested and fixed in 4% PFA for 10 min. After permeabilization, cells were stained for goat anti-GFP (1 hr), washed and stained using a donkey anti-goat Alexa488 secondary antibody (30 min). EdU was then labelled for 30 min using standard Click chemistry with an Alexa647-conjugated azide (Thermo Fisher). After washing cells were counterstained using the FxCycle violet DNA dye (Thermo Fisher) and analyzed on a Beckman Coulter Gallios flow cytometer and Kaluza analysis software.
Drug studies
For drug studies, six YAPoff (WERI-RB1, NCI-H82, NCI-H69, NCI-H2171, DU4475 and SH-SY5Y) and six YAPon (NCI-H661, NCI-H1650, SW-1573, PANC-1, DU145 and MDA-MB-231) lines were used. Cells were seeded in 96-well plates ~24 hrs prior to treatment, then left untreated or treated in triplicate with dose curves of the indicated inhibitors (all from Selleck Chemicals). After 72 hrs, viability was measured using Alamar blue. To test the role of YAP in drug sensitivity, cells were transduced with the indicated YAP vectors as above, then seeded five days after transduction and treated as above.
CRISPR/Cas9 screen
A custom pooled sgRNA library of four sgRNAs per gene and 50 non-targeting controls (Table S8) was cloned into the LentiCRISPR v2 lentiviral backbone. sgRNA sequences were from the human Brunello library (Doench et al., 2016) and non-targeting controls from GeCKOv2 (Sanjana et al., 2014). Y79 cells were transduced with the pooled library (MOI = 0.3), then four days later selected in puromycin (2 mg/ml). Six days later, an input gDNA sample was collected, then cells transduced with control (empty vector) or YAP lentiviruses. After 15 days gDNA was collected and was extracted using the Qiagen DNEasy Blood and Tissue Kit and sgRNA sequences amplified with primers containing Illumina i5 and i7 adaptor barcodes. Gel-purified PCR products were quantified by qPCR using SsoAdvanced Universal SYBR Green Supermix (BioRad) on a BioRad CFX96 Touch Real-Time PCR Detection System, and loaded onto an Illumina NextSeq 500 running at 22 dark cycles and 26 light cycles with Illumina NextSeq 500/550 Hi Output Kit v2.5 (75 Cycles). Real-time base call (.bcl) files were converted to FASTQ files using Illumina bcl2fastq2 conversion software v2.17 (on CentOS 6.0 data storage and computation linux servers).
Western blotting and immunoprecipitations
Cells and tissues were lysed in RIPA lysis buffer containing protease/phosphatase inhibitor cocktail (Cell Signaling Technologies) and PMSF, followed by brief sonication then centrifugation to remove insoluble material. Equal protein amounts were resolved by SDS-PAGE, transferred to nitrocellulose membrane, and then probed with the indicated antibodies (see key resources table and Table S8). Blots were imaged using a Li-Cor Odyssey Infrared imaging system. Immunoprecipitations were performed (Lee et al., 2013) with 2 mg of control or anti-TEAD1 antibody and Protein G agarose beads.
Immunostaining
For immunofluorescence on human retinal explants, murine retina and murine embryonic lung samples were harvested and fixed in 4% paraformaldehyde for 1hr (murine retina) or overnight (human retina and mouse lung) at 4◦C, dehydrated in 30% sucrose over-night and then processed for frozen sections. For adult mice, lungs were inflated with 4% PFA and fixed in 4% PFA overnight, then washed and dehydrated in 30% sucrose overnight prior to processing for frozen sections. After heat-assisted antigen retrieval in pH 6.0 citrate buffer (except human retinal explant samples, which were stained without antigen retrieval), sections were stained with the indicated primary antibodies overnight (see key resources table and Table S8), followed by detection with fluorescently-conjugated secondary antibodies. For Yap or Yap/Taz staining in the mouse lung, the Tyramide SuperBoost amplification kit (Thermo Fisher) was used for detection as per the manufacturer’s protocol. FFPE human retinoblastoma/retinoma samples were deparaffinized and rehydrated, then stained as above.
Immunohistochemistry (IHC) was performed on FFPE tissue microarrays or PFA-fixed mouse tissues following standard procedures. Analysis of murine SCLC +/− Yap induction and scoring of human SCLC, NSCLC PDXs and NET TMAs was performed by expert pathologists (TW, AKW). De-identified prostate tumor tissues were provided under an IRB approved protocol at Roswell Park Cancer Center. FFPE tissues were sectioned at 4 mm for immunohistochemical and multispectral immunostaining with indicated antibodies (see key resources table and Table S8). All stains were validated using recommended tissue controls and evaluated for correct compartment specific staining by pathologists. Staining was performed with a BondRX (Leica Biosystems) automated stainer. Multispectral staining was performed after antibodies were optimized for standard immunohistochemistry and uniplex immunofluorescence staining. Slides were imaged using the Vectra Polaris spectral imaging system (PerkinElmer). Each fluorophore from Opal Polaris reagent kit was visualized using MOTiF™ technology, which generates unmixed whole slide scans at 40x magnification using a separate filter cube corresponding to its emission wavelength. The images were unmixed using a spectral library and individual fluorophores were separated by the InFormTM software. The cell populations were visualized using cell segmentation and phenotype cell tool from Inform 1.1 (PerkinElmer). Threshold for positive staining and accuracy of phenotypes were confirmed by pathologist supervision (AKW).
RNA extraction, qPCR and RNA-seq
Five days after transduction with Empty vector, wild type or S94A YAP (SHP77 and Y79), or Empty vector or 5SA YAP (H209, H2171 and WERI), cells were harvested and RNA extracted using the RNeasy mini kit (Qiagen). For qPCR, cDNA was generated using Superscript II Reverse Transcriptase and qPCR performed using Power SYBR Green PCR master mix (Thermo Fisher) on a Bio-Rad C1000 Touch Real Time PCR system. The primers used are listed in Table S8.
For RNA-seq, RNA was assessed using a Fragment Analyzer, and high quality total RNA was subjected to library preparation using the Illumina TruSeq Stranded mRNA Library Preparation Kit according to the manufacturer’s instructions. Library fragment size was checked using an Agilent Fragment Analyzer, and then quantified with qPCR using SsoAdvanced Universal SYBR Green Supermix (BioRad) on a BioRad CFX96 Touch Real-Time PCR Detection System (WERI, H209 and H2171 lines) or using KAPA SYBR FAST Universal 2X qPCR Master Mix (Kapa Biosystems) running in 7900HT Fast Real Time PCR System (Applied Biosystems) (Y79 and SHP77 cells). Quality checked libraries from WERI, H209 and H2171 cells were loaded onto an Illumina NextSeq 500 run with Illumina NextSeq 500/550 Hi Output Kit v2.5 (75 Cycles). Real-time base call (.bcl) files were converted to FASTQ files using Illumina bcl2fastq2 conversion software v2.17 (on CentOS 6.0 data storage and computation linux servers). Libraries from Y79 and SHP77 cells were loaded on a flowcell for cluster generation using c-Bot and TruSeq PE Cluster Kit v3 (Illumina), and sequencing performed using a HiSeq2000 system and the TruSeq SBS Kit v3 (pair-ended 200 cycles, Illumina). The real-time base call (.bcl) files were converted to fastq files using CASAVA 1.8.2 (Illumina, on CentOS 6.0 data storage and computation linux servers).
For analysis of RNA-seq data, FASTQ files were assessed by fastQC (https://www.bioinformatics.babraham.ac.uk/projects/fastqc) for sequence quality before further processing and analysis. RSEM (Li and Dewey, 2011) was used for transcript quantification, with reference genome GRCh37.p13, and the resulting gene expression, as a matrix of raw counts, was applied to DESeq2 (Love et al., 2014) for differential gene expression analysis in each cell line. Gene expression data of YAPon cancer cell lines was obtained from GEO datasets (GSE54617, GSE66949, GSE66082, GSE68599, GSE56157) and differential expression analysis was obtained using the GEO online tool, GEO2R, for each microarray dataset.
To compare YAP regulated genes in different cells across studies on distinct platforms, we used ranked gene lists. We first defined a set of common genes for this study by overlapping official gene symbols annotated from different platforms. Then we ranked genes by signed log2 p-values of differential expression. Rank Rank Hypergeometric Overlap (RRHO) was performed to compare similarity of the ranked gene lists using the R package RRHO and RRHO2 (Cahill et al., 2018; Plaisier et al., 2010), and the Spearman’s rank correlation coefficient (Spearman’s rho) between each pair of ranked gene lists were clustered and displayed as a heatmap using the R package ‘‘NMF’’ (Gaujoux and Seoighe, 2010). Gene Set Enrichment Analysis (GSEA) (Subramanian et al., 2005) was conducted on the pre-ranked gene lists to identify biological processes and pathways regulated by YAP. The gene sets used in this analysis were obtained from Enrichr (Chen et al., 2013; Kuleshov et al., 2016). Terms with frequencies <10 or >2000 were excluded. The top 10 enriched terms from each of the 10 cell lines were merged and clustered by their Normalized Enrichment Score (NES) values. We also compared our RNAseq data in YAPoff lines with an additional four published transcriptome sets from YAPon lines, generated using RNAseq data from A549 & H1299 NSCLC lung cancer (siRNA depletion of YAP, GSE151200), CAOV2 ovarian cancer (siRNA depletion of YAP, GSE146353), and mel537 melanoma (siRNA depletion of both YAP & TAZ, GSE146918), and obtained the same conclusions using either RRHO or GSEA.
For analysis of top YAP-induced genes we used a ranked list consisting of the top and bottom 2000 ranked genes from the above ranked list. We then selected and merged the top 100 genes from each cell line. To identify targets induced in multiple contexts, we removed genes that were not ranked in the top 500 from at least three different cell lines, which resulted in a final list of 339 genes; the median number of genes induced in YAPoff or YAPon datasets was > 550, thus 500 was chosen as a conservative cutoff for target selection. The rank of those 339 genes from each cell line was then clustered using the aheatmap function in the R package ‘‘NMF’’. Gene lists from each of the 3 major clusters (class-specific or mixed) were then subjected to Enrichr analysis to obtain top KEGG and ENCODE TF binding terms.
ChIP, MeDIP and ChIP-seq
For TEAD4 ChIPs, control or YAP-expressing SCLC and retinoblastoma lines were harvested 5 days after viral transduction and processed for ChIP (Ni and Bremner, 2007; Ni et al., 2008) with 2 µg of antibody and 20 µl of Dynabeads (sheep anti-mouse or anti-rabbit, Invitrogen). ChIP for histone modifications or RNA POLII were similarly performed using parental cells.
For methylated DNA IPs (MeDIP), RNase-free genomic DNA was sonicated to generate 100–400bp fragments, PCR purified/ concentrated, then heat denatured (95◦C for 1 min) and cooled on ice. Approximately 5 µg of sheared DNA was added to 5mC-antibody (10 µg) pre-bound to Dynabeads in IP buffer (10 mM Na3PO4 pH 7.0, 140 mM NaCl, 0.05% Triton X-100). IPs were incubated 4 hrs at 4◦C, washed 3 × 10 min in IP buffer and then eluted. Samples were then digested with Proteinase K (2 hrs at 50◦C) and DNA purified.
For ChIP or MeDIP-qPCR, samples were analyzed by absolute quantification using Power SYBR Green PCR master mix (Thermo Fisher) on a Bio-Rad C1000 Touch Real Time PCR system. The primers used are listed in Table S8. ChIPs were corrected for control IgG samples and calculated as a percent of the input chromatin.
For TEAD4 ChIP-seq, sequencing libraries were prepared using the Illumina TruSeq ChIP Sample Prep Kit (cat#: IP-202–101) according to the manufacturers protocol with 18 cycles for final PCR enrichment. Samples were size selected using PippinHT (Sage Science) with 2% gel, targeting 300 bp fragments (start at 239 bp, end at 361 bp, and tight on Range Flag). Sizes were confirmed using an Agilent Fragment Analyzer and quantified by qPCR using SsoAdvanced Universal SYBR Green Supermix (BioRad) on a BioRad CFX96 Touch Real-Time PCR Detection System. Quality checked libraries were loaded onto an Illumina NextSeq 500, run with Illumina NextSeq 500/550 Hi Output Kit v2.5 (75 Cycles). Real-time base call (.bcl) files were converted to fastq files using Illumina bcl2fastq2 conversion software v2.17 (on CentOS 6.0 data storage and computation linux servers).
The sequence quality of ChIP-seq data were evaluated using fastQC and then aligned to the human reference genome build hg19 by using Bowtie2 (Langmead and Salzberg, 2012) with default parameters. Redundant reads were removed and only uniquely mapping reads were kept for further analysis using SAMtools (Li et al., 2009). To assess the consistency of replicate experiments and identify possible external and/or contamination across different cell lines, the association between duplicate samples within each line and across all four cell lines was examined using the Spearman Correlation of read counts in 10 kb bins over the entire genome after excluding the genomic regions on the ‘‘Black List’’ (Amemiya et al., 2019) using DeepTools (Ramírez et al., 2016).
The Homer package (Heinz et al., 2010) version 4.10 was used for peak calling and de novo motif discovery of the ChIP-seq data. We first called peaks in individual replicates using input as background control in order to assess the consistency between replicates, the resolution of the called peak regions, the IP efficiency and specificity before further analysis. Next, we generated a high-confidence unified peak set with two replicates of each condition in H209, WERI and Y79 cells by calling the getDifferentialPeaksReplicates function. For H2171 cells, only the peaks called in replicate1 were used. The peak regions on the ‘‘Black List’’ were excluded from further analysis. Normalized signal coverage (fragment pileup per million reads, SPMR) was calculated using the MACS2 (Zhang et al., 2008) callpeak function and background signals were subtracted from IP with the bdgcmp function. The result bedGraph files were converted to bigwig files with the UCSC tool bedGraphToBigWig (Kent et al., 2010). The bigwig files were used to display TEAD4 signal coverage with the Integrative Genomics Viewer (IGV) (Robinson et al., 2011) and for further clustering analysis and heatmap.
To compare TEAD4 genomic binding events between two conditions (empty vector control and YAP-expressing cells), we overlapped unified peaks from the two conditions in each cell line using the Homer’s mergePeaks function to obtain a list of merged unique peak regions. We then mapped the normalized signals in bigwig files from each replicate into 10 bp bins of a 300 bp window centered at each of the merged regions for each replicate in each condition correspondingly to get a set of signal coverage matrices. For each line, the matrices were then plotted as heatmaps of signals in: 1) regions sorted by average signal values per region in YAP-expressing cells, and 2) regions clustered by K-means algorithm. Examples of the clustered regions were displayed in the IGV.
To compare TEAD4 binding in YAP-expressing YAPoff cells to YAPon cells, we compared our data with published TEAD4 ChIP-seq data in four YAPon cells: A549, HCT116, HepG2 and MDA-MB-231. The aligned reads for A549, HCT116 and HepG2 cells were downloaded from the ENCODE project (http://genome.ucsc.edu/ENCODE/downloads.html) and data for MDA-MB-231 was obtained from GEO dataset GSE66081. A set of unified peaks was called with replicates in each cell using the same parameters as described above. We selected the top 1000 peaks ranked by peak score for each cell line and overlapped them into a list of merged unique peak regions as described above. The normalized genomic signal coverage in bigwig file was obtained by using pooled replicates for each cell, except in H2171 where only replicate1 was used. We evaluated the similarity of genomic TEAD4 binding in each pair of cells by their co-localized binding intensity. To achieve this, we first subset the merged peak set by filtering out singletons, which did not overlap with peaks from other cells, and then generated a matrix of binding intensity scores with overlapped peak regions as rows and cells as columns. Each score was calculated as the average signal coverage in a 200 bp window centered at peak center and scaled into range 0 to 1 in each column by using the rescale function of the R package ‘‘scale’’ (https://scales.r-lib.org). The score matrix was ordered in both columns and rows by hierarchical clustering using the “euclidean” distance and displayed as heatmap with a dendrogram to show the relationship of the cells by their TEAD4 bindings. The aheatmap function of R package ‘‘NMF’’ (Gaujoux and Seoighe, 2010) was used for the clustering and heatmap plotting.
To further explore putative co-bound transcription factors at TEAD4 sites, we first classified the entire merged peak regions into 16 clusters by K-means clustering of TEAD4 binding signals mapped into 10 bp bins of a 300 bp window centered at peak center. We use the ReMap online database and annotation tool (Chè neby et al., 2018) to define overlap between TEAD4 sites and 485 TFs from ENCODE and published studies in each of the 16 clusters. The statistical significance of the enrichment was computed by comparing the number of overlapped regions with the number of overlaps obtained using random regions sampled with the same size and number as the regions in each TEAD4 binding group. We ranked the co-bound TFs by their enrichment p-values and selected the top 10 enriched TFs in each group to form a matrix of ranks with the TFs as rows and clusters as columns. We then used the previously described hierarchical clustering and heatmap to show association of the co-bound TFs with TEAD4-bound clusters. We also conducted de novo motif discovery in each of the 16 TEAD4-bound clusters.
Pan-cancer transcriptomics analysis
Microarray gene expressions data of cancer cell lines, ‘‘CCLE_Expression_Entrez_2012–09-29.gct’’ (18988 probes and 1037 samples) and ‘‘CCLE_sample_info_file_2012–10-18.txt’’ were downloaded from the Cancer Cell Line Encyclopedia (CCLE; https://portals.broadinstitute.org/ccle) (Barretina et al., 2012). Outliers and samples with missing values were removed. Gene expression values were obtained from the mean expression values of probes collapsing by corresponding genes. Genes with zero expression in all samples were removed, and the remaining 1036 samples with expression data for 18900 genes were used for further analysis. Principle component analysis (PCA) was applied on standard scaled data of genes using the ‘‘prcomp’’ function of R. YAP and TAZ expression in each sample, as z-scores, was projected onto the PC1 and PC2/PC3 dimensions to display the pattern of YAP expression across the cell lines distributed in the 2D space. We found that PC3 performed better than PC2 to separate two distinct YAPoff groups. Genes with rotation values >0.018 and <−0.018 in PC1 and PC3 were selected as features for further clustering analysis. Using the scaled expression values of the feature genes, samples were classified into four clusters with the ‘‘k-means’’ function of R and displayed as a heatmap using the ‘‘aheatmap’’ function of the R package NMF (Gaujoux and Seoighe, 2010). The frequency of the ‘‘Site Primary’’ and the ‘‘cancer type’’ of the samples in each super-cluster were displayed as bar charts. The ‘‘cancer type’’ was defined based on the ‘‘Hist Subtype1’’ and combined with the ‘‘Site Primary’’ and ‘‘Histology’’ information. The distribution of YAP and TAZ expression in each cluster (A-D) was displayed as a box plot. The GSEA analysis was applied to pre-ranked gene lists by using PC1 and PC3 rotation values. Mutational data for the four clusters (A-D) were obtained from cBioportal (Cerami1 et al., 2012; Gao et al., 2013). The MYC signature was based on ‘‘validated targets of c-Myc transcriptional activation’’ from the NCI-Nature Pathway Interaction Database, and the MYCN signature was calculated from shMYCN knockdown in IMR32 neuroblastoma cells (Valentijn et al., 2012). Scores were applied to CCLE cell line data as per (McCurdy et al., 2017). Groups were compared using a pair-wise Wilcoxon Test with Bonferroni correction for multiple testing (Figure 2L).
RNA-seq gene expression data from primary tumor samples from TCGA (Cancer Genome Atlas Research Network et al., 2013) and TARGET (Ma et al., 2018) data sets, ‘‘TcgaTargetGtex_RSEM_Hugo_norm_count.txt’’ and ‘‘TcgaTargetGTEX_phenotype.txt’’ were downloaded from UCSC Xena (https://xena.ucsc.edu/). Only cancer samples were extracted for this study. Outliers and samples with missing values were removed. The remaining 10,485 samples were applied for further analysis. The top PC1/PC3 genes mapped above were extracted and scaled for k-means clustering into four clusters. The clustered gene expression matrix with the features was displayed as a heatmap as before. Bar charts for frequencies of the ‘‘Site Primary’’ and the ‘‘cancer type’’ of the cancer samples in each super-clusters were similarly plotted as before. The ‘‘cancer type’’ was defined by the “detailed category” in the phenotype. All plotting except the heatmaps was conducted by using the R package ‘‘ggplot2’’ (Wickham, 2016).
Tissue-specific transcriptomics analysis
Analysis of YAP/TAZ mRNA in human lung cancer subtypes (Figure 2C) was extracted from (Clinical Lung Cancer Genome Project (CLCGP) and Network Genomic Medicine (NGM), 2013) and subtypes with <3 samples were excluded. Statistical analysis was performed using a Kruskal-Wallis H-test (YAP H-value: 99.8, p-value: 5.97 × 10−17; TAZ H-value: 82.7, p-value: 1.48 × 10−13) followed by Conover’s post-hoc test with a Holm p-value adjustment. Yap/Taz mRNA levels in murine lung cancers (Figure 2E) were extracted from (Semenova et al., 2016). For analysis of the SCN subset in the TCGA (Figure 3I), previously defined SCN samples (Balanis et al., 2019) were used. Cancer types with >2 SCN samples with a SCN score of >50 were included and YAP/TAZ expression was compared to the remaining non-SCN samples for each cancer type.
For the analysis of transcriptome changes during lineage switching (Figure 5) we used murine and human datasets. DESeq2 normalized transcriptome data from murine prostate cancer (Ku et al., 2017) was Log2 transformed and the top 10,000 highly variable genes (hvgs) were used for PCA, which separated adenocarcinoma (SKO and 12 wk DKO) from NEPC (DKO and TKO end stage tumors) in a single dimension (Table S5). This prostate cancer rotation vector was used as a ranked list for GSEA analysis together with the PC1/3 gene-sets from Figure 3C that define YAPoff and YAPon cancers. For this we identified 74 PC1+ (which include Yap & Taz), 33 PC1-, 61 PC3+, and 108 PC3- murine orthologs of the human PC1/3 gene-sets. We also generated 96 random control gene sets of comparable sizes from background (total genes in the data set after removing low-counts genes with maximum counts < 5). A similar approach was used to analyze transcriptome changes during PARCB induced reprogramming of human primary epithelia to SCN cancer (Figures 5G–5I), except that the transcriptome data (Park et al., 2018) was normalized with UQ-FPKM and Log2 transformed prior to PCA/GSEA analyses.
Cancer class genetic and pharmaceutical dependencies
Data for gene dependencies from genome-wide CRISPR/Cas9 screens were obtained from the Cancer DepMap (DepMap Public 20Q2, https://depmap.org/portal/download/) (Meyers et al., 2017). First, we determined the percentage of cell lines in each cluster (A-D) that are dependent on a particular gene; as per DepMap, genes were considered essential if they had a dependency score > 0.5. Next, we averaged the percentages for cluster-A and -B (YAPoff lines), and subtracted the average of the percentages for clusters-C and -D (YAPon lines) to obtain a YAPoff vs. YAPon Differential Dependency Score (DDS), and focused on genes with a DDS of ≥ 25 or ≤ −25. Genes which were essential in over 50% of lines in all four clusters were excluded. In addition, to avoid biases caused by a single cluster, we excluded genes for which the percentage of dependent lines in either A or B was less than 10 percentage points different from the percentage of dependent lines in either C or D. These criteria yielded 16 YAPoff and 31 YAPon gene dependencies, and we performed Enrichr analysis with all YAPoff and the top 25 YAPon hits to obtain enriched TF binding features (ENCODE/ChEA), pathways (KEGG, BioPlanet), and GO Biological Processes.
Drug sensitivity data (AUC values) were obtained from the Cancer Therapeutics Response Portal (CTRPv2) (Seashore-Ludlow et al., 2015) or Genomics of Drug Sensitivity in Cancer (GDSC1 & GDSC2) (Iorio et al., 2016). For each compound, p-values comparing the four clusters (A-D) and median AUC values for each cluster were calculated. The mean p-value comparing cluster-B to clusters-C or -D was -log10-transformed and multiplied by the mean fold difference in AUC between the groups to calculate a weighted sensitivity score. Compounds scoring >1.5 or <−1.5 in CTRPv2 were compared to values obtained for the GDSC1/2 databases to obtain high-confidence hits.
QUANTIFICATION AND STATISTICAL ANALYSIS
Unless stated otherwise in the above Method Details or figure legends, unpaired 2-tailed t-tests determined using Prism software (GraphPad) were used for sample comparisons. Statistical details of individual experiments can be found in the related figure legends, where n indicates the number of mice (in vivo experiments) or independent biological replicates (in vitro cell line experiments).
Supplementary Material
KEY RESOURCES TABLE
| REAGENT or RESOURCE | SOURCE | IDENTIFIER |
|---|---|---|
| Antibodies | ||
|
| ||
| Mouse monoclonal anti-YAP/TAZ (clone 63.7) | Santa Cruz Biotechnology | Cat# sc-101199; RRID: AB_1131430 |
| Rabbit monoclonal anti-YAP/TAZ (clone D24E4) | Cell Signaling Technology | Cat# 8418; RRID: AB_10950494 |
| Rabbit monoclonal anti-YAP (clone D8H1X) | Cell Signaling Technology | Cat# 14074; RRID: AB_2650491 |
| Mouse monoclonal anti-YAP (clone 1A12) | Cell Signaling Technology | Cat# 12395; RRID: AB_2797897 |
| Mouse monoclonal anti-GFP (clone B-2) | Santa Cruz Biotechnology | Cat# sc-9996; RRID: AB_627695 |
| Goat polyclonal anti-GFP | Abcam | Cat# ab6673; RRID: AB_305643 |
| Chicken polyclonal anti-GFP | Abcam | Cat# ab13970; RRID: AB_300798 |
| Mouse monoclonal anti-Ki67 (clone B56) | BD Biosciences | Cat# 550609; RRID: AB_393778 |
| Rat monoclonal anti-Ki67 (clone SolA15) | ThermoFisher | Cat# 14–5698-82; RRID: AB_10854564 |
| Rabbit monoclonal anti-Human Ki67 (clone SP6) | ThermoFisher | Cat# RM-9106-S1; RRID: AB_149792 |
| Guinea pig polyclonal anti-Synaptophysin | Synaptic Systems | Cat# 101 004; RRID: AB_1210382 |
| Rabbit monoclonal anti-Synaptophysin (clone MRQ-40) | Origene | Cat# TA327740 |
| Rabbit monoclonal anti-Synaptophysin (clone D8F6H) | Cell Signaling Technology | Cat# 36406; RRID: AB_2799098 |
| Rabbit polyclonal anti-CGRP | Sigma-Aldrich | Cat# C8198; RRID: AB_259091 |
| Rabbit polyclonal anti-Androgen Receptor (AR) | Santa Cruz Biotechnology | Cat# sc-816; RRID: AB_1563391 |
| Rabbit polyclonal anti-CCSP (CC10) | Seven Hills Bioreagents | WRAB-3950 |
| Rabbit polyclonal anti-pro SPC | Abcam | Cat# ab40879; RRID: AB_777473 |
| Mouse monoclonal anti-PARP-1 (clone C2–10) | Santa Cruz Biotechnology | Cat# sc-53643; RRID: AB_785086 |
| Mouse monoclonal anti-a TUBULIN (clone DM1A) | Santa Cruz Biotechnology | Cat# sc-32293; RRID: AB_628412 |
| Mouse monoclonal anti-TEAD1 (clone 31/TEF-1) | BD Biosciences | Cat# 610923; RRID: AB_398238 |
| Rabbit monoclonal anti-pan TEAD (clone D3F7L) | Cell Signaling Technology | Cat# 13295; RRID: AB_2687902 |
| Mouse monoclonal anti-TEAD4 (clone N-G2) | Santa Cruz Biotechnology | Cat# sc-101184; RRID: AB_2203086 |
| Mouse monoclonal anti-ASCL1 (clone D-7) | Santa Cruz Biotechnology | Cat# sc-374104; RRID: AB_10918561 |
| Mouse monoclonal anti-NEUROD (clone A-10) | Santa Cruz Biotechnology | Cat# sc-46684; RRID: AB_671759 |
| Mouse monoclonal anti-MYCN (clone B8.4.B) | Santa Cruz Biotechnology | Cat# sc-53993, RRID:AB_831602 |
| Goat polyclonal anti-MYCL | Novus Biologicals | Cat# AF4050 |
| Mouse monoclonal anti-RB1 (clone G99–549) | BD Biosciences | Cat# 554164; RRID: AB_395279 |
| Mouse monoclonal anti-b-ACTIN (clone AC-15) | Sigma-Aldrich | Cat# A5441; RRID: AB_476744 |
| Mouse monoclonal anti-RXRg (clone A-2) | Santa Cruz Biotechnology | Cat# sc-365252; RRID: AB_10850062 |
| Goat polyclonal anti-MCM3 (N-19) | Santa Cruz Biotechnology | Cat# sc-9850; RRID: AB_2142269 |
| Mouse monoclonal anti-ITGB5 (clone F-5) | Santa Cruz Biotechnology | Cat# sc-398214 |
| Rabbit monoclonal anti-ITGB1 (clone D2E5) | Cell Signaling Technology | Cat# 9699; RRID: AB_11178800 |
| Rabbit monoclonal anti-ITGB4 (clone D8P6C) | Cell Signaling Technology | Cat# 14803; RRID: AB_2798620 |
| Rabbit monoclonal anti-ITGA4 (clone D2E1) | Cell Signaling Technology | Cat# 8440; RRID: AB_2797643 |
| Rabbit polyclonal anti-ITGAV (clone D2E5) | Cell Signaling Technology | Cat# 4711; RRID: AB_2128178 |
| Rabbit polyclonal anti-Histone H3 (trimethyl Lys4) | Abcam | Cat# ab8580; RRID: AB_306649 |
| Rabbit polyclonal anti-Histone H3, (trimethyl Lys27) | Millipore | Cat# 07–449; RRID: AB_310624 |
| Rabbit polyclonal anti-Histone H3 (trimethyl Lys9) | Abcam | Cat# ab8898; RRID: AB_306848 |
| Rabbit polyclonal anti-Histone H3 (acetyl Lys27) | Abcam | Cat# ab4729; RRID: AB_2118291 |
| Mouse monoclonal anti-RNA polymerase II CTD repeat YSPTSPS antibody [clone 4H8] | Abcam | Cat# ab5408; RRID: AB_304868 |
| Mouse monoclonal anti-Methylcytidine | Eurogentec | Cat# BI-MECY-0100; RRID: AB_2616058 |
| Mouse monoclonal anti-ITGAV/B5 (clone P1F6) | Santa Cruz Biotechnology | Cat# sc-81632; RRID: AB_1123634 |
| Mouse monoclonal anti-ITGB1 (clone P5D2) | Santa Cruz Biotechnology | Cat# sc-13590, RRID:AB_627008 |
|
| ||
| Bacterial and virus strains | ||
|
| ||
| Ad5CMVCre | University of Iowa Viral Vector Core Facility | VVC-U of Iowa-5 |
|
| ||
| Biological samples | ||
|
| ||
| Human fetal retinas | Morgentaler Clinic | N/A |
| SCLC patient derived xenografts | Dr. Charles Rudin | Gardner et al., 2017; Lok et al., 2017 |
| Paraffin-embedded SCLC TMA | Princess Margaret Cancer Centre | N/A |
| Paraffin-embedded NSCLC PDX TMA | Princess Margaret Cancer Centre | N/A |
| Paraffin-embedded NET TMA | Roswell Park Cancer Institute | N/A |
| Paraffin-embedded primary prostate cancer tissue samples | Roswell Park Cancer Institute | N/A |
|
| ||
| Chemicals, peptides, and recombinant proteins | ||
|
| ||
| Insulin, human recombinant, zinc solution | ThermoFisher | Cat# 12585014 |
| Transferrin, Bovine (Holo form) | ThermoFisher | Cat# 11107047 |
| Sodium selenite | Sigma-Aldrich | Cat# S5261 |
| Hydrocortisone | Sigma-Aldrich | Cat# H0888 |
| b-Estradiol | Sigma-Aldrich | Cat# E2758 |
| Somatostatin | Sigma-Aldrich | Cat# S1763 |
| Glycyl-L-histidyl-L-lysine | Sigma-Aldrich | Cat# G7387 |
| REDExtract-N-Amp PCR ReadyMix | Sigma-Aldrich | Cat# R4775 |
| 2-hydroxyethyl agarose | Sigma-Aldrich | Cat# A4018 |
| Crystal Violet | Sigma-Aldrich | Cat# C6158 |
| EdU (5-ethynyl-2’-deoxyuridine) | ThermoFisher | Cat# E10187 |
| Paraformaldehyde | Electron Microscopy Sciences | Cat# 15710 |
| Alexa Fluor 647 Azide, Triethylammonium Salt | ThermoFisher | Cat# A10277 |
| FxCycle Violet Stain | ThermoFisher | Cat# F10347 |
| AlamarBlue Cell Viability Reagent | ThermoFisher | Cat# DAL1100 |
| Alisertib | Selleck Chemicals | Cat# S1133 |
| KPT-330 (Selinexor) | Selleck Chemicals | Cat# S7252 |
| Navitoclax | Selleck Chemicals | Cat# S1001 |
| Daporinad | Selleck Chemicals | Cat# S2799 |
| SsoAdvanced Universal SYBR Green Supermix | Bio-Rad | Cat# 1725270 |
| Protease/Phosphatase Inhibitor Cocktail | Cell Signaling Technology | Cat# 5872S |
| Phenylmethanesulfonyl fluoride (PMSF) solution | Sigma-Aldrich | Cat# 93482 |
| Protein G Agarose Beads | Cell Signaling Technology | Cat# 37478S |
| Cyclic RGD (cRGD) peptide | Sigma-Aldrich | Cat# SCP0111 |
| Superscript II Reverse Transcriptase | ThermoFisher | Cat# 18064014 |
| Power SYBR Green PCR Master Mix | ThermoFisher | Cat# 4368708 |
| Dynabeads M-280 Sheep Anti-Mouse IgG | ThermoFisher | Cat# 11202D |
| Dynabeads M-280 Sheep Anti-Rabbit IgG | ThermoFisher | Cat# 11204D |
| Proteinase K, recombinant, PCR grade | ThermoFisher | Cat# EO0491 |
|
| ||
| Critical commercial assays | ||
|
| ||
| Papain Dissociation System | Worthington Biochemical | Cat# LK003150 |
| Alexa Fluor 488 Tyramide SuperBoost Kit, goat anti-rabbit IgG | ThermoFisher | Cat# B40943 |
| Alexa Fluor 594 Tyramide SuperBoost Kit, goat anti-mouse IgG | ThermoFisher | Cat# B40915 |
| TruSeq Stranded mRNA Library Prep Kit | Illumina | Cat# 20020594; Cat# RS-122–2101 |
| TruSeq ChIP Sample Prep Kit | Illumina | Cat# IP-202–101 |
|
| ||
| Deposited data | ||
|
| ||
| RNA-seq from YAP-transduced SCLC and retinoblastoma cell lines | This paper | GSE144972 |
| TEAD4 ChIP-seq in SCLC and retinoblastoma cell lines | This paper | GSE144973 |
| Microarray from YAP/TAZ knockdown SK-Hep1 cells | Park et al., 2016 | GSE54617 |
| Microarray from YAP/TAZ knockdown UM-SCC-2 cells | Hiemer et al., 2015 | GSE66949 |
| Microarray from YAP/TAZ knockdown MDA-MB-231 cells | Zanconato et al., 2015 | GSE66082 |
| Microarray from YAP/TAZ knockdown SKMEL28 cells | Kim et al., 2016 | GSE68599 |
| Microarray from YAP/TAZ knockdown Meso-33 cells | Li et al., 2014 | GSE56157 |
| TEAD4 ChIP-seq from A549, HCT116 and HepG2 cells | ENCODE project | http://genome.ucsc.edu/ENCODE/downloads.html |
| TEAD4 ChIP-seq from MDA-MB-231 cells | Zanconato et al., 2015 | GSE66081 |
| Cancer Cell Line Encyclopedia microarray gene expression data | Barretina et al., 2012 | https://portals.broadinstitute.org/ccle |
| TCGA and TARGET RNA-seq gene expression data from primary tumor samples | Cancer Genome Atlas Research Network et al., 2013; Ma et al., 2018 | https://xena.ucsc.edu/ |
| Gene expression data from primary human lung tumors | Clinical Lung Cancer Genome Project (CLCGP) and Network Genomic Medicine (NGM), 2013 | http://www.uni-koeln.de/med-fak/clcgp/ |
| RNA-seq from murine prostate cancer models | Ku et al., 2017 | GSE90891 |
| RNA-seq from normal epithelia and PARCB-transformed SCN tumors | Park et al., 2018 | GSE118206 |
| Cancer DepMap Public 20Q2 | Meyers et al., 2017 | https://depmap.org/portal/download/ |
| Cancer Therapeutics Response Portal (CTRPv2) drug sensitivity data | Seashore-Ludlow et al., 2015 | https://portals.broadinstitute.org/ctrp/ |
| Genomics of Drug Sensitivity in Cancer (GDSC1 & GDSC2) drug sensitivity data | Iorio et al., 2016 | https://www.cancerrxgene.org/ |
|
| ||
| Experimental models: Cell lines | ||
|
| ||
| RB1021 | Dr. Brenda Gallie (To et al., 2012) | RRID:CVCL_S624 |
| RB3823 | Dr. Brenda Gallie (To et al., 2012) | RRID:CVCL_ZF07 |
| RB247 | Dr. Brenda Gallie (To et al., 2012) | RRID:CVCL_2704 |
| Y79 | Rod Bremner lab | RRID: CVCL_1893 |
| WERI-RB1 | Rod Bremner lab | RRID: CVCL_1792 |
| NCI-H526 | ATCC | Cat# CRL-5811; RRID: CVCL_1569 |
| SHP77 | ATCC | Cat# CRL-2195; RRID: CVCL_1693 |
| NCI-H510A | ATCC | Cat# HTB-184; RRID: CVCL_1565 |
| NCI-H82 | ATCC | Cat# HTB-175; RRID: CVCL_1591 |
| NCI-H2171 | ATCC | Cat# CRL-5929; RRID: CVCL_1536 |
| NCI-H209 | Susan P.C. Cole lab | RRID: CVCL_1525 |
| NCI-H69 | Susan P.C. Cole lab | RRID: CVCL_1579 |
| NCI-N417 | Susan P.C. Cole lab | RRID: CVCL_1602 |
| NCI-H660 | ATCC | Cat# CRL-5813; RRID: CVCL_1576 |
| DU4475 | Dr. Eldad Zacksenhaus | RRID: CVCL_1183 |
| QGP-1 | Dr. Todd McMullen | RRID: CVCL_3143 |
| BON-1 | Dr. Todd McMullen | RRID:CVCL_3985 |
| SH-SY5Y | Dr. Meredith Irwin | RRID: CVCL_0019 |
| SK-N-SH | Dr. Meredith Irwin | RRID: CVCL_0531 |
| NCI-H661 | ATCC | Cat# HTB-183; RRID: CVCL_1577 |
| NCI-H1650 | ATCC | Cat# CRL-5883; RRID: CVCL_1483 |
| A549 | Rod Bremner lab | RRID: CVCL_0023 |
| SW-1573 | Susan P.C. Cole lab | RRID: CVCL_1720 |
| MCF7 | Rod Bremner lab | RRID: CVCL_0031 |
| MDA-MB-231 | Rod Bremner lab | RRID: CVCL_0062 |
| HCC1419 | Dr. Benjamin Neel | RRID: CVCL_1251 |
| DU145 | Dr. Jan Jongstra | RRID: CVCL_0105 |
| BPH-1 | Rod Bremner lab | RRID: CVCL_1091 |
| PANC-1 | Rod Bremner lab | RRID: CVCL_0480 |
| Hs 766T | Rod Bremner lab | RRID: CVCL_0334 |
| T24 | Jeff L. Wrana lab | RRID: CVCL_0554 |
| Karpas 299 | Dr. Rob Ingham | RRID: CVCL_1324 |
| SUP-M2 | Dr. Rob Ingham | RRID: CVCL_2209 |
| SU-DHL-1 | Dr. Rob Ingham | RRID: CVCL_0538 |
| SR-786 | Dr. Rob Ingham | RRID: CVCL_1711 |
| UCONN | Dr. Rob Ingham | RRID: CVCL_A693 |
| HDMY-Z | Dr. Rob Ingham | RRID: CVCL_1273 |
| KM-H2 | Dr. Rob Ingham | RRID: CVCL_1330 |
| L428 | Dr. Rob Ingham | RRID: CVCL_1361 |
| L540 | Dr. Rob Ingham | RRID: CVCL_1362 |
| HDLM-2 | Dr. Rob Ingham | RRID: CVCL_0009 |
| OCI-AML2 | Dr. Mark Minden | RRID: CVCL_1619 |
| OCI-AML3 | Dr. Mark Minden | RRID: CVCL_1844 |
| THP-1 | Dr. Aaron Schimmer | RRID: CVCL_0006 |
| HL-60 | Dr. Aaron Schimmer | RRID: CVCL_0002 |
| K562 | Dr. Aaron Schimmer | RRID: CVCL_0004 |
| Lenti-X 293T | Takara/Clontech | Cat# 632180 |
|
| ||
| Experimental models: Organisms/strains | ||
|
| ||
| Mouse: αCre: Tg(Pax6-cre,GFP)2Pgr | Dr. Peter Gruss | MGI:3052661 |
| Mouse: Rb1f/f: Rb1tm2Brn | Dr. Anton Berns | MGI:1931018 |
| Mouse: Rbl1−/−: Rbl1tm1Mru | Dr. Michael Rudnicki | MGI:1857798 |
| Mouse: Yapf/f: Yap1tm1Hmc | Reginensi et al., 2013 | MGI:5486298 |
| Mouse: Tazf/f: Wwtr1tm1Hmc | Reginensi et al., 2013 | MGI:5486299 |
| Mouse: p53f/f: Trp53tm1Brn | NCI Mouse Repository | Strain# 01XC2; MGI:1931011 |
| Mouse: CgrpCreER: Calcatm1.1(cre/ERT2)Ptch | Dr. Pao-Tien Chuang (Song et al., 2012) | MGI:5460801 |
| Mouse: TRE-Yap: TRE-HA-YapTg | Gregorieff et al., 2015 | N/A |
| Mouse: ROSA-rtTA: Gt(ROSA)26Sortm1(rtTA,EGFP)Nagy | Dr. Andras Nagy | MGI:3583817 |
| Mouse: Ccsp-rtTA: Tg(Scgb1a1-rtTA)1Jaw | Tichelaar et al., 2000 | MGI:2445707 |
| Mouse: tetO-EGFRL858R: Tg(tetO-EGFR*L858R)56Hev | Politi et al., 2006 | MGI:3690078 |
| Mouse: Ptenf/f: Ptentm1Hwu | Lesche et al., 2002 | MGI:2156086 |
| Mouse: SPC-rtTA: SFTPC-rtTA | Perl et al., 2002a | N/A |
| Mouse: tetO-Cre: Tg(tetO-cre)1Jaw | Perl et al., 2002b | MGI:2679524 |
| Mouse: PB-Cre4: Tg(Pbsn-cre)4Prb | Wu et al., 2001 | MGI:2385927 |
|
| ||
| Oligonucleotides | ||
|
| ||
| See Table S8 for oligonucleotide sequences used | ||
| for CRISPR sgRNA vectors, the CRIPSR library, | ||
| RT-qPCR, ChIP-qPCR and mouse genotyping. | ||
|
| ||
| Recombinant DNA | ||
|
| ||
| psPax2 | Didier Trono | Addgene plasmid #12260; RRID: Addgene_12260 |
| pMD2.G | Didier Trono | Addgene plasmid #12259; RRID: Addgene_12259 |
| LentiCRISPR v2 | Sanjana et al., 2014 | Addgene plasmid #52961; RRID: Addgene_52961 |
| pLKO.1-puro | Stewart et al., 2003 | Addgene plasmid #8453; RRID: Addgene_8453 |
|
| ||
| Software and algorithms | ||
|
| ||
| Kaluza, Flow Cytometry analysis software | Beckman Coulter | https://www.mybeckman.ca/flow-cytometry/software/kaluza |
| Prism | GraphPad | https://www.graphpad.com/scientific-software/prism/ |
| RSEM | Li and Dewey, 2011 | https://deweylab.github.io/RSEM/ |
| DESeq2 | Love et al., 2014 | https://bioconductor.org/packages/release/bioc/html/DESeq2.html |
| RRHO | Plaisier et al., 2010 | https://bioconductor.org/packages/release/bioc/html/RRHO.html |
| RRHO2 | Cahill et al., 2018 | https://github.com/RRHO2/RRHO2 |
| DeepTools | Ramírez et al., 2016 | https://github.com/deeptools/deeptools |
| Homer package | Heinz et al., 2010 | http://homer.ucsd.edu/homer/ |
| NMF | Gaujoux and Seoighe, 2010 | https://cran.r-project.org/web/packages/NMF/index.html |
| fastQC | N/A | https://www.bioinformatics.babraham.ac.uk/projects/fastqc |
| Bowtie2 | Langmead and Salzberg, 2012 | http://bowtie-bio.sourceforge.net/bowtie2 |
| SAMtools | Li et al., 2009 | http://samtools.sourceforge.net |
| MACS2 | Zhang et al., 2008 | https://github.com/macs3-project/MACS |
| Integrative Genomics Viewer (IGV) | Robinson et al., 2011 | https://software.broadinstitute.org/software/igv/ |
| UCSC tool bedGraphToBigWig | Kent WJ., 2010 | https://anaconda.org/bioconda/ucsc-bedgraphtobigwig |
| ReMap | Chèneby et al., 2018 | http://remap.univ-amu.fr/ |
| GSEA | Subramanian, et al., 2005 | https://www.gsea-msigdb.org/gsea/index.jsp |
| R package ‘‘ggplot2’’ | Wickham, 2016 | https://cran.r-project.org/web/packages/ggplot2 |
| R package ‘‘scales’’ | N/A | https://scales.r-lib.org |
Highlights.
YAP/TAZ silencing in multiple cancers reflects their tumor suppressor activity
Cancers can be parsed into binary YAPon and YAPoff classes
Binary transcriptomes drive distinct adhesive, metabolic, genetic, and drug profiles
Alternate YAP enhancers explain anti-cancer integrin-aVb5 program in YAPoff cancers
ACKNOWLEDGMENTS
We thank Pao-Tien Chuang for CgrpCreER mice, Brenda Gallie for retinoblastoma samples, Meredith Irwin for neuroblastoma lines, Eldad Zacksenhaus for breast cancer lines, Todd McMullen for NE pancreatic lines, Rob Ingham for lymphoma cells lines, Mark Minden and Aaron Schimmer for leukemia cell lines, Charles M Rudin for SCLC PDXs, Ayla Pearson for assistance with data analysis, Hanna Rosenheck for help with multispectral imaging, Michael Parsons at the LTRI Flow Cytometry Core for help with flow cytometry, Louise Brown of the LTRI Imaging Core for assistance with imaging, Kin Chan at the LTRI Network Biology Collaborative Centre (nbcc.lunenfeld.ca) for RNA-seq and ChIP-seq services, and Rene Bernards, Dan Durocher, Helen McNeill, Jeremy Sivak and Valerie Wallace for helpful comments. The NBCC is supported by the Canada Foundation for Innovation, the Ontario Genomics, and Genome Canada and Ontario Genomics (OGI-139). J.D.P. is a recipient of a post-doctoral fellowship from the Canadian Institutes of Health Research (CIHR). The project was supported by funding from the CIHR (grant no. 153128), The Cancer Research Society, and the Krembil Foundation (to R.B.); a CIHR Foundation Program Grant (FDN 143252) and a Terry Fox Research Institute Program Project Grant (TFRI Project no. 1107) (to J.L.W.); and research in the lab of B.H.L. is supported by funding from the Conquer Cancer Foundation of ASCO, International Association for the Study of Lung Cancer (IASLC), Ontario Molecular Pathology Research Network of the Ontario Institute for Cancer Research, Lung Cancer Research Foundation, Canada Foundation for Innovation, Cancer Research Society, CIHR, National Institute of Health/National Cancer Institute (U01CA253383), Clinical and Translational Science Center at Weill Cornell Medical Center and MSKCC (UL1TR00457).
Footnotes
SUPPLEMENTAL INFORMATION
Supplemental information can be found online at https://doi.org/10.1016/j.ccell.2021.06.016.
DECLARATION OF INTERESTS
B.H.L. reports honoraria and non-financial support from AstraZeneca, and has received research funding from Pfizer and AstraZeneca not related to this work.
REFERENCES
- Aggarwal R, Huang J, Alumkal JJ, Zhang L, Feng FY, Thomas GV, Weinstein AS, Friedl V, Zhang C, Witte ON, et al. (2018). Clinical and genomic characterization of treatment-emergent small-cell neuroendocrine prostate cancer: a multi-institutional prospective study. J. Clin. Oncol 36, 2492–2503. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeno N, Miller AL, Ma X, and Wikenheiser-Brokamp KA (2015). p53 suppresses carcinoma progression by inhibiting mTOR pathway activation. Oncogene 34, 589–599. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Akeno N, Reece AL, Callahan M, Miller AL, Kim RG, He D, Lane A, Moulton JS, and Wikenheiser-Brokamp KA (2017). TRP53 mutants drive neuroendocrine lung cancer through loss-of-function mechanisms with gain-of-function effects on chemotherapy response. Mol. Cancer Ther 16, 2913–2926. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Amemiya HM, Kundaje A, and Boyle AP (2019). The ENCODE blacklist: identification of problematic regions of the genome. Sci. Rep 9, 9354. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Balanis NG, Sheu KM, Esedebe FN, Patel SJ, Smith BA, Park JW, Alhani S, Gomperts BN, Huang J, Witte ON, et al. (2019). Pan-cancer convergence to a small-cell neuroendocrine phenotype that shares susceptibilities with hematological malignancies. Cancer Cell 36, 17–34.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Barretina J, Caponigro G, Stransky N, Venkatesan K, Margolin AA, Kim S, Wilson CJ, Lehár J, Kryukov GV, Sonkin D, et al. (2012). The Cancer Cell Line Encyclopedia enables predictive modelling of anticancer drug sensitivity. Nature 483, 603–607. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Prandi D, Mosquera JM, Benelli M, Puca L, Cyrta J, Marotz C, Giannopoulou E, Chakravarthi BVSK, Varambally S, et al. (2016). Divergent clonal evolution of castration-resistant neuroendocrine prostate cancer. Nat. Med 22, 298–305. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beltran H, Hruszkewycz A, Scher HI, Hildesheim J, Isaacs J, Yu EY, Kelly K, Lin D, Dicker A, Arnold J, et al. (2019). The role of lineage plasticity in prostate cancer therapy resistance. Clin. Cancer Res [DOI] [PMC free article] [PubMed] [Google Scholar]
- Beyer TA, Weiss A, Khomchuk Y, Huang K, Ogunjimi AA, Varelas X, and Wrana JL (2013). Switch enhancers interpret TGF-β and Hippo signaling to control cell fate in human embryonic stem cells. Cell Rep 5, 1611–1624. [DOI] [PubMed] [Google Scholar]
- Borges M, Linnoila RI, van de Velde HJ, Chen H, Nelkin BD, Mabry M, Baylin SB, and Ball DW (1997). An achaete-scute homologue essential for neuroendocrine differentiation in the lung. Nature 386, 852–855. [DOI] [PubMed] [Google Scholar]
- Brinkman EK, Chen T, Amendola M, and van Steensel B (2014). Easy quantitative assessment of genome editing by sequence trace decomposition. Nucleic Acids Res 42, e168. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cahill KM, Huo Z, Tseng GC, Logan RW, and Seney ML (2018). Improved identification of concordant and discordant gene expression signatures using an updated rank-rank hypergeometric overlap approach. Sci. Rep 8, 9588. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cancer Genome Atlas Research Network, Weinstein JN, Collisson EA, Mills GB, Shaw KRM, Ozenberger BA, Ellrott K, Shmulevich I, Sander C, and Stuart JM (2013). The Cancer Genome Atlas pan-cancer analysis project. Nat. Genet 45, 1113–1120. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cerami1 E, Gao J, Dogrusoz U, Gross BE, Sumer SO, Aksoy BA, Jacobsen A, Byrne CJ, Heuer ML, Larsson E, et al. (2012). The cBio cancer genomics portal: an open platform for exploring multidimensional cancer genomics data. Cancer Discov 2, 401–404. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen D, Livne-Bar I, Vanderluit JL, Slack RS, Agochiya M, and Bremner R (2004). Cell-specific effects of RB or RB/p107 loss on retinal development implicate an intrinsically death-resistant cell-of-origin in retino-blastoma. Cancer Cell 5, 539–551. [DOI] [PubMed] [Google Scholar]
- Chen EY, Tan CM, Kou Y, Duan Q, Wang Z, Meirelles GV, Clark NR, and Ma’ayan A (2013). Enrichr: interactive and collaborative HTML5 gene list enrichment analysis tool. BMC Bioinformatics 14, 128. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Zhang Y, Gibbons DL, Deneen B, Kwiatkowski DJ, Ittmann M, and Creighton CJ (2018). Pan-cancer molecular classes transcending tumor lineage across 32 cancer types, multiple data platforms, and over 10,000 cases. Clin. Cancer Res 24, 2182–2193. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chen F, Chandrashekar DS, Varambally S, and Creighton CJ (2019). Pan-cancer molecular subtypes revealed by mass-spectrometry-based proteomic characterization of more than 500 human cancers. Nat. Commun 10, 5679. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Chèneby J, Gheorghe M, Artufel M, Mathelier A, and Ballester B (2018). ReMap 2018: an updated atlas of regulatory regions from an integrative analysis of DNA-binding ChIP-seq experiments. Nucleic Acids Res 46, D267–D275. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Clinical Lung Cancer Genome Project (CLCGP), and Network Genomic Medicine (NGM) (2013). A genomics-based classification of human lung tumors. Sci. Transl Med 5, 209ra153. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Cordenonsi M, Zanconato F, Azzolin L, Forcato M, Rosato A, Frasson C, Inui M, Montagner M, Parenti AR, Poletti A, et al. (2011). The Hippo transducer TAZ confers cancer stem cell-related traits on breast cancer cells. Cell 147, 759–772. [DOI] [PubMed] [Google Scholar]
- Cottini F, Hideshima T, Xu C, Sattler M, Dori M, Agnelli L, ten Hacken E, Bertilaccio MT, Antonini E, Neri A, et al. (2014). Rescue of Hippo coactivator YAP1 triggers DNA damage-induced apoptosis in hematological cancers. Nat. Med 20, 599–606. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Crawford JJ, Bronner SM, and Zbieg JR (2018). Hippo pathway inhibition by blocking the YAP/TAZ-TEAD interface: a patent review. Expert Opin. Ther. Pat 28, 867–873. [DOI] [PubMed] [Google Scholar]
- Cui M, Augert A, Rongione M, Conkrite K, Parazzoli S, Nikitin AY, Ingolia N, and MacPherson D (2014). PTEN is a potent suppressor of small cell lung cancer. Mol. Cancer Res 12, 654–659. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Dimaras H, Khetan V, Halliday W, Orlic M, Prigoda NL, Piovesan B, Marrano P, Corson TW, Eagle RC, Squire JA, et al. (2008). Loss of RB1 induces non-proliferative retinoma: increasing genomic instability correlates with progression to retinoblastoma. Hum. Mol. Genet 17, 1363–1372. [DOI] [PubMed] [Google Scholar]
- Doench JG, Fusi N, Sullender M, Hegde M, Vaimberg EW, Donovan KF, Smith I, Tothova Z, Wilen C, Orchard R, et al. (2016). Optimized sgRNA design to maximize activity and minimize off-target effects of CRISPR-Cas9. Nat. Biotechnol 34, 184–191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- DuPage M, Dooley AL, and Jacks T (2009). Conditional mouse lung cancer models using adenoviral or lentiviral delivery of Cre recombinase. Nat. Protoc 4, 1064–1072. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Fernandes VM, McCormack K, Lewellyn L, and Verheyen EM (2014). Integrins regulate apical constriction via microtubule stabilization in the Drosophila eye disc epithelium. Cell Rep 9, 2043–2055. [DOI] [PubMed] [Google Scholar]
- Ferone G, Lee MC, Sage J, and Berns A (2020). Cells of origin of lung cancers: lessons from mouse studies. Genes Dev 34, 1017–1032. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ferraris GMS, Schulte C, Buttiglione V, De Lorenzi V, Piontini A, Galluzzi M, Podestà A, Madsen CD, and Sidenius N (2014). The interaction between uPAR and vitronectin triggers ligand-independent adhesion signalling by integrins. EMBO J 33, 2458–2472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- FitzSimmons J, Fantel A, and Shepard TH (1994). Growth parameters in mid-trimester fetal Turner syndrome. Early Hum. Dev 38, 121–129. [DOI] [PubMed] [Google Scholar]
- Gao J, Aksoy BA, Dogrusoz U, Dresdner G, Gross B, Sumer SO, Sun Y, Jacobsen A, Sinha R, Larsson E, et al. (2013). Integrative analysis of complex cancer genomics and clinical profiles using the cBioPortal. Sci. Signal 6, pl1. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gardner EE, Lok BH, Schneeberger VE, Desmeules P, Miles LA, Arnold PK, Ni A, Khodos I, de Stanchina E, Nguyen T, et al. (2017). Chemosensitive relapse in small cell lung cancer proceeds through an EZH2-SLFN11 axis. Cancer Cell 31, 286–299. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gaujoux R, and Seoighe C (2010). A flexible R package for nonnegative matrix factorization. BMC Bioinformatics 11, 367. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gill MK, Christova T, Zhang YY, Gregorieff A, Zhang L, Narimatsu M, Song S, Xiong S, Couzens AL, Tong J, et al. (2018). A feed forward loop enforces YAP/TAZ signaling during tumorigenesis. Nat. Commun 9, 3510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Gong X, Du J, Parsons SH, Merzoug FF, Webster Y, Iversen PW, Chio L-C, Van Horn RD, Lin X, Blosser W, et al. (2019). Aurora A kinase inhibition is synthetic lethal with loss of the RB1 tumor suppressor gene. Cancer Discov 9, 248–263. [DOI] [PubMed] [Google Scholar]
- Gregorieff A, Liu Y, Inanlou MR, Khomchuk Y, and Wrana JL (2015). Yap-dependent reprogramming of Lgr5(+) stem cells drives intestinal regeneration and cancer. Nature 526, 715–718. [DOI] [PubMed] [Google Scholar]
- He L, Yuan L, Sun Y, Wang P, Zhang H, Feng X, Wang Z, Zhang W, Yang C, Zeng YA, et al. (2019). Glucocorticoid receptor signaling activates TEAD4 to promote breast cancer progression. Cancer Res 79, 4399–4411. [DOI] [PubMed] [Google Scholar]
- Heinz S, Benner C, Spann N, Bertolino E, Lin YC, Laslo P, Cheng JX, Murre C, Singh H, and Glass CK (2010). Simple combinations of lineage-determining transcription factors prime cis-regulatory elements required for macrophage and B cell identities. Mol. Cell 38, 576–589. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hiemer SE, Zhang L, Kartha VK, Packer TS, Almershed M, Noonan V, Kukuruzinska M, Bais MV, Monti S, and Varelas X (2015). A YAP/TAZ-regulated molecular signature is associated with oral squamous cell carcinoma. Mol. Cancer Res 13, 957–968. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hientz K, Mohr A, Bhakta-Guha D, and Efferth T (2017). The role of p53 in cancer drug resistance and targeted chemotherapy. Oncotarget 8, 8921–8946. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Hoadley KA, Yau C, Hinoue T, Wolf DM, Lazar AJ, Drill E, Shen R, Taylor AM, Cherniack AD, Thorsson V, et al. (2018). Cell-of-origin patterns dominate the molecular classification of 10,000 tumors from 33 types of cancer. Cell 173, 291–304.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Huang Y-H, Klingbeil O, He X-Y, Wu XS, Arun G, Lu B, Somerville TDD, Milazzo JP, Wilkinson JE, Demerdash OE, et al. (2018). POU2F3 is a master regulator of a tuft cell-like variant of small cell lung cancer. Genes Dev 32, 915–928. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Iorio F, Knijnenburg TA, Vis DJ, Bignell GR, Menden MP, Schubert M, Aben N, Gonçalves E, Barthorpe S, Lightfoot H, et al. (2016). A landscape of pharmacogenomic interactions in cancer. Cell 166, 740–754. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ireland AS, Micinski AM, Kastner DW, Guo B, Wait SJ, Spainhower KB, Conley CC, Chen OS, Guthrie MR, Soltero D, et al. (2020). MYC drives temporal evolution of small cell lung cancer subtypes by reprogramming neuroendocrine fate. Cancer Cell 38, 60–78. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Jin K, and Xiang M (2012). In vitro explant culture and related protocols for the study of mouse retinal development. Methods Mol. Biol 884, 155–165. [DOI] [PubMed] [Google Scholar]
- Kapoor A, Yao W, Ying H, Hua S, Liewen A, Wang Q, Zhong Y, Wu C-J, Sadanandam A, Hu B, et al. (2014). Yap1 activation enables bypass of oncogenic Kras addiction in pancreatic cancer. Cell 158, 185–197. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kent WJ, Zweig AS, Barber G, Hinrichs AS, and Karolchik D (2010). BigWig and BigBed: enabling browsing of large distributed datasets. Bioinformatics 26, 2204–2207. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kim MH, Kim J, Hong H, Lee S-H, Lee J-K, Jung E, and Kim J (2016). Actin remodeling confers BRAF inhibitor resistance to melanoma cells through YAP/TAZ activation. EMBO J 35, 462–478. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ku SY, Rosario S, Wang Y, Mu P, Seshadri M, Goodrich ZW, Goodrich MM, Labbé DP, Gomez EC, Wang J, et al. (2017). Rb1 and Trp53 cooperate to suppress prostate cancer lineage plasticity, metastasis, and antiandrogen resistance. Science 355, 78–83. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kuleshov MV, Jones MR, Rouillard AD, Fernandez NF, Duan Q, Wang Z, Koplev S, Jenkins SL, Jagodnik KM, Lachmann A, et al. (2016). Enrichr: a comprehensive gene set enrichment analysis web server 2016 update. Nucleic Acids Res 44, W90–W97. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Kurppa KJ, Liu Y, To C, Zhang T, Fan M, Vajdi A, Knelson EH, Xie Y, Lim K, Cejas P, et al. (2020). Treatment-induced tumor dormancy through YAP-mediated transcriptional reprogramming of the apoptotic pathway. Cancer Cell 37, 104–122.e12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Langmead B, and Salzberg SL (2012). Fast gapped-read alignment with Bowtie 2. Nat. Methods 9, 357–359. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lázaro S, Pérez-Crespo M, Lorz C, Bernardini A, Oteo M, Enguita AB, Romero E, Hernández P, Tomás L, Morcillo MÁ, et al. (2019). Differential development of large-cell neuroendocrine or small-cell lung carcinoma upon inactivation of 4 tumor suppressor genes. Proc. Natl. Acad. Sci. U.S.A 116, 22300–22306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lee J-K, Lee J, Kim S, Kim S, Youk J, Park S, An Y, Keam B, Kim D-W, Heo DS, et al. (2017). Clonal history and genetic predictors of transformation into small-cell carcinomas from lung adenocarcinomas. J. Clin. Oncol 35, 3065–3074. [DOI] [PubMed] [Google Scholar]
- Lee JKH, Pearson JD, Maser BE, and Ingham RJ (2013). Cleavage of the JunB transcription factor by caspases generates a carboxyl-terminal fragment that inhibits activator protein-1 transcriptional activity. J. Biol. Chem 288, 21482–21495. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lesche R, Groszer M, Gao J, Wang Y, Messing A, Sun H, Liu X, and Wu H (2002). Cre/loxP-mediated inactivation of the murine Pten tumor suppressor gene. Genesis 32, 148–149. [DOI] [PubMed] [Google Scholar]
- Li B, and Dewey CN (2011). RSEM: accurate transcript quantification from RNA-seq data with or without a reference genome. BMC Bioinformatics 12, 323. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li H, Handsaker B, Wysoker A, Fennell T, Ruan J, Homer N, Marth G, Abecasis G, and Durbin R; 1000 Genome Project Data Processing Subgroup (2009). The sequence alignment/map format and SAMtools. Bioinformatics 25, 2078–2079. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Li W, Cooper J, Zhou L, Yang C, Erdjument-Bromage H, Zagzag D, Snuderl M, Ladanyi M, Hanemann CO, Zhou P, et al. (2014). Merlin/ NF2 loss-driven tumorigenesis linked to CRL4(DCAF1)-mediated inhibition of the hippo pathway kinases Lats1 and 2 in the nucleus. Cancer Cell 26, 48–60. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Liu X, Li H, Rajurkar M, Li Q, Cotton JL, Ou J, Zhu LJ, Goel HL, Mercurio AM, Park J-S, et al. (2016). Tead and AP1 coordinate transcription and motility. Cell Rep 14, 1169–1180. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Livne-Bar I, Pacal M, Cheung MC, Hankin M, Trogadis J, Chen D, Dorval KM, and Bremner R (2006). Chx10 is required to block photoreceptor differentiation but is dispensable for progenitor proliferation in the post-natal retina. Proc. Natl. Acad. Sci. U S A 103, 4988–4993. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Lok BH, Gardner EE, Schneeberger VE, Ni A, Desmeules P, Rekhtman N, de Stanchina E, Teicher BA, Riaz N, Powell SN, et al. (2017). PARP inhibitor activity correlates with SLFN11 expression and demonstrates synergy with temozolomide in small cell lung cancer. Clin. Cancer Res 23, 523–535. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Love MI, Huber W, and Anders S (2014). Moderated estimation of fold change and dispersion for RNA-seq data with DESeq2. Genome Biol 15, 550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ma X, Liu Y, Liu Y, Alexandrov LB, Edmonson MN, Gawad C, Zhou X, Li Y, Rusch MC, Easton J, et al. (2018). Pan-cancer genome and transcriptome analyses of 1,699 paediatric leukaemias and solid tumours. Nature 555, 371–376. [DOI] [PMC free article] [PubMed] [Google Scholar]
- McCurdy SR, Pacal M, Ahmad M, and Bremner R (2017). A CDK2 activity signature predicts outcome in CDK2-low cancers. Oncogene 36, 2491–2502. [DOI] [PubMed] [Google Scholar]
- Meuwissen R, Linn SC, Linnoila RI, Zevenhoven J, Mooi WJ, and Berns A (2003). Induction of small cell lung cancer by somatic inactivation of both Trp53 and Rb1 in a conditional mouse model. Cancer Cell 4, 181–189. [DOI] [PubMed] [Google Scholar]
- Meyers RM, Bryan JG, McFarland JM, Weir BA, Sizemore AE, Xu H, Dharia NV, Montgomery PG, Cowley GS, Pantel S, et al. (2017). Computational correction of copy number effect improves specificity of CRISPR-Cas9 essentiality screens in cancer cells. Nat. Genet 49, 1779–1784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Mollaoglu G, Guthrie MR, Böhm S, Brägelmann J, Can I, Ballieu PM, Marx A, George J, Heinen C, Chalishazar MD, et al. (2017). MYC drives progression of small cell lung cancer to a variant neuroendocrine subtype with vulnerability to aurora kinase inhibition. Cancer Cell 31, 270–285. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Moya IM, Castaldo SA, Van den Mooter L, Soheily S, Sansores-Garcia L, Jacobs J, Mannaerts I, Xie J, Verboven E, Hillen H, et al. (2019). Peritumoral activation of the Hippo pathway effectors YAP and TAZ suppresses liver cancer in mice. Science 366, 1029–1034. [DOI] [PubMed] [Google Scholar]
- Neptune ER, Podowski M, Calvi C, Cho J-H, Garcia JGN, Tuder R, Linnoila RI, Tsai M-J, and Dietz HC (2008). Targeted disruption of NeuroD, a proneural basic helix-loop-helix factor, impairs distal lung formation and neuroendocrine morphology in the neonatal lung. J. Biol. Chem 283, 21160–21169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ni Z, and Bremner R (2007). Brahma-related gene 1-dependent STAT3 recruitment at IL-6-inducible genes. J. Immunol 178, 345–351. [DOI] [PubMed] [Google Scholar]
- Ni Z, Abou El Hassan M, Xu Z, Yu T, and Bremner R (2008). The chromatin-remodeling enzyme BRG1 coordinates CIITA induction through many interdependent distal enhancers. Nat. Immunol 9, 785–793. [DOI] [PubMed] [Google Scholar]
- Niederst MJ, Sequist LV, Poirier JT, Mermel CH, Lockerman EL, Garcia AR, Katayama R, Costa C, Ross KN, Moran T, et al. (2015). RB loss in resistant EGFR mutant lung adenocarcinomas that transform to small-cell lung cancer. Nat. Commun 6, 6377. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Nishida A, Furukawa A, Koike C, Tano Y, Aizawa S, Matsuo I, and Furukawa T (2003). Otx2 homeobox gene controls retinal photoreceptor cell fate and pineal gland development. Nat. Neurosci 6, 1255–1263. [DOI] [PubMed] [Google Scholar]
- Oser MG, Fonseca R, Chakraborty AA, Brough R, Spektor A, Jennings RB, Flaifel A, Novak JS, Gulati A, Buss E, et al. (2019). Cells lacking the RB1 tumor suppressor gene are hyperdependent on aurora B kinase for survival. Cancer Discov 9, 230–247. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ota M, and Sasaki H (2008). Mammalian Tead proteins regulate cell proliferation and contact inhibition as transcriptional mediators of Hippo signaling. Development 135, 4059–4069. [DOI] [PubMed] [Google Scholar]
- Park JW, Lee JK, Sheu KM, Wang L, Balanis NG, Nguyen K, Smith BA, Cheng C, Tsai BL, Cheng D, et al. (2018). Reprogramming normal human epithelial tissues to a common, lethal neuroendocrine cancer lineage. Science 362, 91–95. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Park Y-Y, Sohn BH, Johnson RL, Kang M-H, Kim SB, Shim J-J, Mangala LS, Kim JH, Yoo JE, Rodriguez-Aguayo C, et al. (2016). Yes-associated protein 1 and transcriptional coactivator with PDZ-binding motif activate the mammalian target of rapamycin complex 1 pathway by regulating amino acid transporters in hepatocellular carcinoma. Hepatology 63, 159–172. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Perl A-KT, Tichelaar JW, and Whitsett JA (2002a). Conditional gene expression in the respiratory epithelium of the mouse. Transgenic Res 11, 21–29. [DOI] [PubMed] [Google Scholar]
- Perl A-KT, Wert SE, Nagy A, Lobe CG, and Whitsett JA (2002b). Early restriction of peripheral and proximal cell lineages during formation of the lung. Proc. Natl. Acad. Sci. U S A 99, 10482–10487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Piccolo S, Dupont S, and Cordenonsi M (2014). The biology of YAP/TAZ: hippo signaling and beyond. Physiol. Rev 94, 1287–1312. [DOI] [PubMed] [Google Scholar]
- Plaisier SB, Taschereau R, Wong JA, and Graeber TG (2010). Rank-rank hypergeometric overlap: identification of statistically significant overlap between gene-expression signatures. Nucleic Acids Res 38, e169. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Politi K, Zakowski MF, Fan P-D, Schonfeld EA, Pao W, and Varmus HE (2006). Lung adenocarcinomas induced in mice by mutant EGF receptors found in human lung cancers respond to a tyrosine kinase inhibitor or to down-regulation of the receptors. Genes Dev 20, 1496–1510. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Priestley P, Baber J, Lolkema MP, Steeghs N, de Bruijn E, Shale C, Duyvesteyn K, Haidari S, van Hoeck A, Onstenk W, et al. (2019). Pan-cancer whole-genome analyses of metastatic solid tumours. Nature 575, 210–216. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Raab-Westphal S, Marshall JF, and Goodman SL (2017). Integrins as therapeutic targets: successes and cancers. Cancers (Basel) 9, 110. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rajwanshi A, Srinivas R, and Upasana G (2009). Malignant small round cell tumors. J. Cytol 26, 1–10. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Ramírez F, Ryan DP, Grüning B, Bhardwaj V, Kilpert F, Richter AS, Heyne S, Dündar F, and Manke T (2016). deepTools2: a next generation web server for deep-sequencing data analysis. Nucleic Acids Res 44, W160–W165. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Reginensi A, Scott RP, Gregorieff A, Bagherie-Lachidan M, Chung C, Lim D-S, Pawson T, Wrana J, and McNeill H (2013). Yap- and Cdc42-dependent nephrogenesis and morphogenesis during mouse kidney development. Plos Genet 9, e1003380. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rindi G, and Inzani F (2020). Neuroendocrine neoplasm update: toward universal nomenclature. Endocr. Relat. Cancer 27, R211–R218. [DOI] [PubMed] [Google Scholar]
- Robinson JT, Thorvaldsdóttir H, Winckler W, Guttman M, Lander ES, Getz G, and Mesirov JP (2011). Integrative genomics viewer. Nat. Biotechnol 29, 24–26. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Rudin CM, Poirier JT, Byers LA, Dive C, Dowlati A, George J, Heymach JV, Johnson JE, Lehman JM, MacPherson D, et al. (2019). Molecular subtypes of small cell lung cancer: a synthesis of human and mouse model data. Nat. Rev. Cancer 19, 289–297. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sanjana NE, Shalem O, and Zhang F (2014). Improved vectors and genome-wide libraries for CRISPR screening. Nat. Methods 11, 783–784. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seashore-Ludlow B, Rees MG, Cheah JH, Cokol M, Price EV, Coletti ME, Jones V, Bodycombe NE, Soule CK, Gould J, et al. (2015). Harnessing connectivity in a large-scale small-molecule sensitivity dataset. Cancer Discov 5, 1210–1223. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Seguin L, Kato S, Franovic A, Camargo MF, Lesperance J, Elliott KC, Yebra M, Mielgo A, Lowy AM, Husain H, et al. (2014). An integrin β3-KRAS-RalB complex drives tumour stemness and resistance to EGFR inhibition. Nat. Cell Biol 16, 457–468. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova EA, Nagel R, and Berns A (2015). Origins, genetic landscape, and emerging therapies of small cell lung cancer. Genes Dev 29, 1447–1462. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Semenova EA, Kwon M-C, Monkhorst K, Song J-Y, Bhaskaran R, Krijgsman O, Kuilman T, Peters D, Buikhuisen WA, Smit EF, et al. (2016). Transcription factor NFIB is a driver of small cell lung cancer progression in mice and marks metastatic disease in patients. Cell Rep 16, 631–643. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Shepard TH (1975). Growth and Development of Human Embryo and Fetus (W.B. Saunders; ). [Google Scholar]
- Simpson DS, Mason-Richie NA, Gettler CA, and Wikenheiser-Brokamp KA (2009). Retinoblastoma family proteins have distinct functions in pulmonary epithelial cells in vivo critical for suppressing cell growth and tumorigenesis. Cancer Res 69, 8733–8741. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Song H, Yao E, Lin C, Gacayan R, Chen M-H, and Chuang P-T (2012). Functional characterization of pulmonary neuroendocrine cells in lung development, injury, and tumorigenesis. Proc. Natl. Acad. Sci. U.S.A 109, 17531–17536. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stein C, Bardet AF, Roma G, Bergling S, Clay I, Ruchti A, Agarinis C, Schmelzle T, Bouwmeester T, Schübeler D, et al. (2015). YAP1 exerts its transcriptional control via TEAD-mediated activation of enhancers. Plos Genet 11, e1005465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stewart SA, Dykxhoorn DM, Palliser D, Mizuno H, Yu EY, An DS, Sabatini DM, Chen ISY, Hahn WC, Sharp PA, et al. (2003). Lentivirus-delivered stable gene silencing by RNAi in primary cells. RNA 9, 493–501. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Stine ZE, Walton ZE, Altman BJ, Hsieh AL, and Dang CV (2015). MYC, metabolism, and cancer. Cancer Discov 5, 1024–1039. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Subramanian A, Tamayo P, Mootha VK, Mukherjee S, Ebert BL, Gillette MA, Paulovich A, Pomeroy SL, Golub TR, Lander ES, et al. (2005). Gene set enrichment analysis: a knowledge-based approach for interpreting genome-wide expression profiles. Proc. Natl. Acad. Sci. U.S.A 102, 15545–15550. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sun H, Chang Y, Schweers B, Dyer MA, Zhang X, Hayward SW, and Goodrich DW (2006). An E2F binding-deficient Rb1 protein partially rescues developmental defects associated with Rb1 nullizygosity. Mol. Cell. Biol 26, 1527–1537. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Sutherland KD, Proost N, Brouns I, Adriaensen D, Song J-Y, and Berns A (2011). Cell of origin of small cell lung cancer: inactivation of Trp53 and Rb1 in distinct cell types of adult mouse lung. Cancer Cell 19, 754–764. [DOI] [PubMed] [Google Scholar]
- Tan H-L, Sood A, Rahimi HA, Wang W, Gupta N, Hicks J, Mosier S, Gocke CD, Epstein JI, Netto GJ, et al. (2014). Rb loss is characteristic of prostatic small cell neuroendocrine carcinoma. Clin. Cancer Res 20, 890–903. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Tichelaar JW, Lu W, and Whitsett JA (2000). Conditional expression of fibroblast growth factor-7 in the developing and mature lung. J. Biol. Chem 275, 11858–11864. [DOI] [PubMed] [Google Scholar]
- Tlemsani C, Pongor L, Elloumi F, Girard L, Huffman KE, Roper N, Varma S, Luna A, Rajapakse VN, Sebastian R, et al. (2020). SCLC-CellMiner: a resource for small cell lung cancer cell line genomics and pharmacology based on genomic signatures. Cell Rep 33, 108296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- To K-H, Pajovic S, Gallie BL, and Thériault BL (2012). Regulation of p14ARF expression by miR-24: a potential mechanism compromising the p53 response during retinoblastoma development. BMC Cancer 12, 69. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Valentijn LJ, Koster J, Haneveld F, Aissa RA, van Sluis P, Broekmans MEC, Molenaar JJ, van Nes J, and Versteeg R (2012). Functional MYCN signature predicts outcome of neuroblastoma irrespective of MYCN amplification. Proc. Natl. Acad. Sci. U.S.A 109, 19190–19195. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Verbeke D, Demeyer S, Prieto C, de Bock CE, De Bie J, Gielen O, Jacobs K, Mentens N, Verhoeven BM, Uyttebroeck A, et al. (2020). The XPO1 inhibitor KPT-8602 synergizes with dexamethasone in acute lymphoblastic leukemia. Clin. Cancer Res [DOI] [PubMed] [Google Scholar]
- Verfaillie A, Imrichova H, Atak ZK, Dewaele M, Rambow F, Hulselmans G, Christiaens V, Svetlichnyy D, Luciani F, Van den Mooter L, et al. (2015). Decoding the regulatory landscape of melanoma reveals TEADS as regulators of the invasive cell state. Nat. Commun 6, 6683. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Wang S, Gao J, Lei Q, Rozengurt N, Pritchard C, Jiao J, Thomas GV, Li G, Roy-Burman P, Nelson PS, et al. (2003). Prostate-specific deletion of the murine Pten tumor suppressor gene leads to metastatic prostate cancer. Cancer Cell 4, 209–221. [DOI] [PubMed] [Google Scholar]
- Wickham H (2016). ggplot2: Elegant Graphics for Data Analysis (Springer-Verlag; ). [Google Scholar]
- Wu X, Wu J, Huang J, Powell WC, Zhang J, Matusik RJ, Sangiorgi FO, Maxson RE, Sucov HM, and Roy-Burman P (2001). Generation of a prostate epithelial cell-specific Cre transgenic mouse model for tissue-specific gene ablation. Mech. Development 101, 61–69. [DOI] [PubMed] [Google Scholar]
- Xie C, Lu H, Nomura A, Hanse EA, Forster CL, Parker JB, Linden MA, Karasch C, and Hallstrom TC (2015). Co-deleting Pten with Rb in retinal progenitor cells in mice results in fully penetrant bilateral retinoblastomas. Mol. Cancer 14, 93. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XL, Fang Y, Lee TC, Forrest D, Gregory-Evans C, Almeida D, Liu A, Jhanwar SC, Abramson DH, and Cobrinik D (2009). Retinoblastoma has properties of a cone precursor tumor and depends upon cone-specific MDM2 signaling. Cell 137, 1018–1031. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Xu XL, Singh HP, Wang L, Qi D-L, Poulos BK, Abramson DH, Jhanwar SC, and Cobrinik D (2014). Rb suppresses human cone-precursor-derived retinoblastoma tumours. Nature 514, 385–388. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Yang D, Denny SK, Greenside PG, Chaikovsky AC, Brady JJ, Ouadah Y, Granja JM, Jahchan NS, Lim JS, Kwok S, et al. (2018). Intertumoral heterogeneity in SCLC is influenced by the cell type of origin. Cancer Discov 8, 1316–1331. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Forcato M, Battilana G, Azzolin L, Quaranta E, Bodega B, Rosato A, Bicciato S, Cordenonsi M, and Piccolo S (2015). Genome-wide association between YAP/TAZ/TEAD and AP-1 at enhancers drives oncogenic growth. Nat. Cell Biol [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zanconato F, Cordenonsi M, and Piccolo S (2016). YAP/TAZ at the roots of cancer. Cancer Cell 29, 783–803. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang H, Christensen CL, Dries R, Oser MG, Deng J, Diskin B, Li F, Pan Y, Zhang X, Yin Y, et al. (2020). CDK7 inhibition potentiates genome instability triggering anti-tumor immunity in small cell lung cancer. Cancer Cell 37, 37–54.e9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Liu T, Meyer CA, Eeckhoute J, Johnson DS, Bernstein BE, Nusbaum C, Myers RM, Brown M, Li W, et al. (2008). Model-based analysis of ChIP-seq (MACS). Genome Biol 9, R137. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhang Y, Xue C, Cui H, and Huang Z (2015). High expression of TAZ indicates a poor prognosis in retinoblastoma. Diagn. Pathol 10, 187. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhao B, Ye X, Yu J, Li L, Li W, Li S, Yu J, Lin JD, Wang C-Y, Chinnaiyan AM, et al. (2008). TEAD mediates YAP-dependent gene induction and growth control. Genes Dev 22, 1962–1971. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zheng Y, and Pan D (2019). The hippo signaling pathway in development and disease. Dev. Cell 50, 264–282. [DOI] [PMC free article] [PubMed] [Google Scholar]
- Zhou Z, Flesken-Nikitin A, Corney DC, Wang W, Goodrich DW, Roy-Burman P, and Nikitin AY (2006). Synergy of p53 and Rb deficiency in a conditional mouse model for metastatic prostate cancer. Cancer Res 66, 7889–7898. [DOI] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
RNA-seq and ChIP-seq data have been deposited at GEO under accession codes GSE144972 and GSE144973. This paper analyzes existing, publicly available data. These accession numbers for the datasets are listed in the key resources table.
This paper does not report original code.
Any additional information required to reanalyze the data reported in this paper is available from the lead contact upon request.