

Abstract
Chromodomain Helicase DNA Binding Protein 1 Like (CHD1L) is an oncogene implicated in tumor progression, multidrug resistance, and metastasis in many types of cancer. In this article, we describe the optimization of the first lead CHD1L inhibitors (CHD1Li) through drug design and medicinal chemistry. More than thirty CHD1Li were synthesized and evaluated using a variety of colorectal cancer (CRC) tumor organoid models and functional assays. The results led to the prioritization of six lead CHD1Li analogs with improved potency and antitumor activity, and drug-like properties including metabolic stability and in vivo pharmacokinetics. Furthermore, lead CHD1Li 6.11 proved to be an oral bioavailable antitumor agent significantly reducing the tumor volume of CRC xenografts generated from isolated quasi mesenchymal cells (M-Phenotype), which possess enhanced tumorigenic properties. In conclusion, we report the optimization of first-in-class inhibitors of oncogenic CHD1L as a novel therapeutic strategy with potential for the treatment of cancer.
Graphical Abstract
INTRODUCTION
Chromodomain Helicase DNA Binding Protein 1 Like (CHD1L), also known as Amplified in Liver Cancer 1 (ALC1), is a chromatin remodeling enzyme that has emerged as an oncogene implicated in the pathology of many types of cancer.1–3 In the clinical setting, CHD1L amplification and overexpression are associated with metastatic cancer, poor prognosis, and low overall survival for patients with ovarian, breast, glioma, lung, and gastrointestinal (GI) cancers.3–10 In addition, we have determined CHD1L as a marker of poor prognosis, metastasis, and low overall survival for colorectal cancer (CRC).11
CHD1L is unique from other chromatin remodelers and has a diverse repertoire of cellular functions.12 CHD1L is essential for poly ADP-ribose (PAR) polymerase (PARP) mediated DNA repair and knockdown of CHD1L sensitizes tumor cells to DNA damaging agents.1 Two recent reports validate CHD1L as a significant factor promoting drug resistance to PARP inhibitors via CHD1L-mediated nucleosome sliding, alleviating PARP trapping by PARP inhibitors.13, 14 Knockout of CHD1L sensitizes BRCA1/2 mutant HR-deficient tumor cells to PARP inhibition causing cell death in vitro and loss of tumor growth with increased survival in vivo.13, 14 We have characterized CHD1L as a required component of the TCF/LEF-transcription factor complex (denoted henceforth as TCF-transcription)11, which is linked as a driver of GI cancers and many other cancers.15–20 We and others have determined this complex to be a master regulator of epithelial-mesenchymal transition (EMT) that promotes epithelial-mesenchymal plasticity (EMP).21–25 In particular, we demonstrated the TCF-transcription is upregulated in isolated quasi-mesenchymal cell phenotypes, compared to other EMT phenotypes, promoting increased cancer stem cell (CSC) stemness and invasiveness.21
Taken together, we hypothesize that targeted small molecule inhibitors of CHD1L may be an effective therapeutic strategy to treat CRC and other cancers. Recently, we reported the high-throughput screening (HTS) drug discovery and hit-to-lead validation of the first-in-class CHD1L inhibitors (CHD1Li).11 Herein, we describe the medicinal chemistry optimization and biological evaluation and structure activity relationship (SAR) of CHD1Li analogs based off lead 6.0. We demonstrate that lead analog 6.11 displays improved pharmacokinetics (PK) compared to 6.0, including oral bioavailability and in vivo antitumor efficacy against CRC tumor xenografts.
RESULTS AND DISCUSSION
Medicinal Chemistry.
The synthesis of lead CHD1Li 6.0 has not been reported in the literature. Thus, our first goal was to develop the synthesis of 6.0 and optimize the chemistry to efficiently prepare analogs for ligand-based drug design. To accomplish this goal, we began our synthesis from commercially available starting material p-phenylenediamine (1) and dichloropyrimidine analogs (2.1-2.3) (Scheme 1). Compounds 1 and 2 were reacted in the presence of triethylamine to obtain a selective nucleophilic aromatic substitution, providing intermediates 3.1-3.3 in good yield ranging from 70–83%. A second nucleophilic aromatic substitution with pyrrolidine afforded the core pyrimidine pharmacophore of 6.0. Using propanephosphonic acid anhydride (T3P), commercially available thiophene (5) was coupled to provide 6.0, 6.1, and 6.2 with yields ranging from 77–84%. Analog 6.3 was produced from 6.1 in the presence of iodomethane and sodium hydride. This chemistry provided us several analogs through modification of the pyrimidine ring. Initially, we also utilized this synthetic approach to modify the thiophene aromatic ring coupled as amides to the phenylenediamine ring.
Scheme 1.
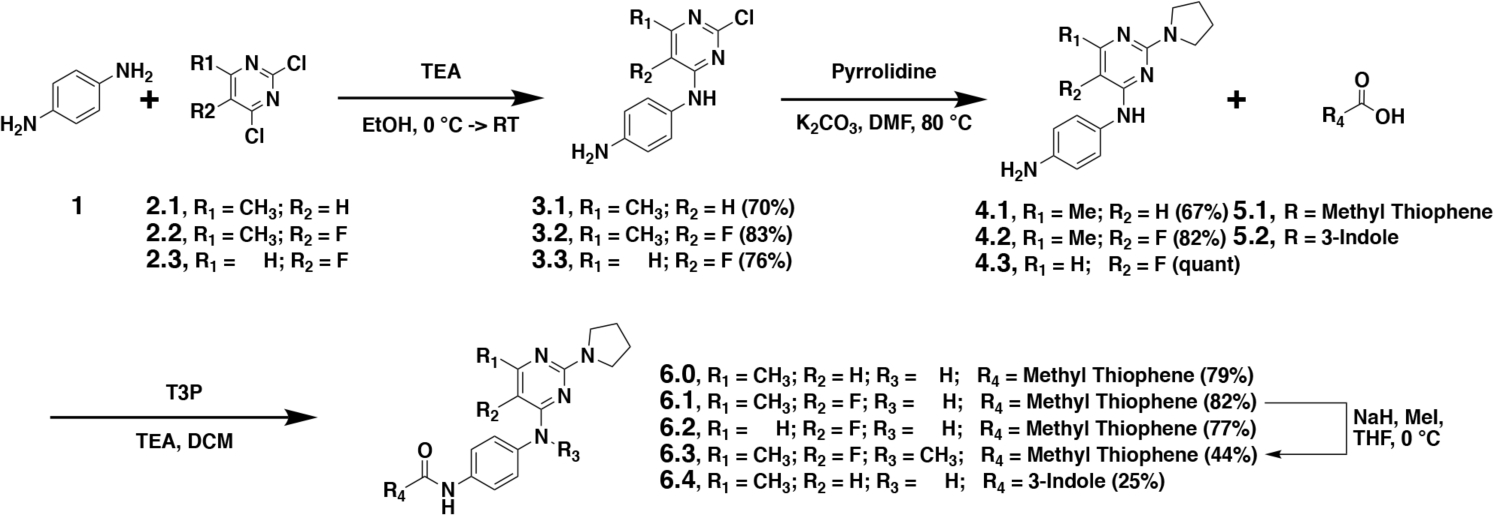
The synthesis of lead CHD1Li 6.0 and analogs 6.1–6.4. Percent yield for each compound is shown in parentheses.
We observed significant challenges with this synthetic approach to produce 6.4 (Scheme 1), encountering low yields and difficulty in purification. Therefore, we further optimized our synthesis starting with a tert-butoxycarbonyl (BOC) protected phenylenediamine, which resulted in a significant increase in purity of the desired substitution at the 4-position of the pyrimidine ring, facilitating the pyrrolidine substitution in the 2-position. Unfortunately, after BOC deprotection the challenges persisted with peptide coupling of the 3-indole carboxylic acid to produce 6.4. However, increasing the carbon spacer between the aromatic rings and the carboxylic acid functionality allowed for efficient peptide coupling, leading to our optimized syntheses of CHD1Li (Scheme 2A).
Scheme 2.
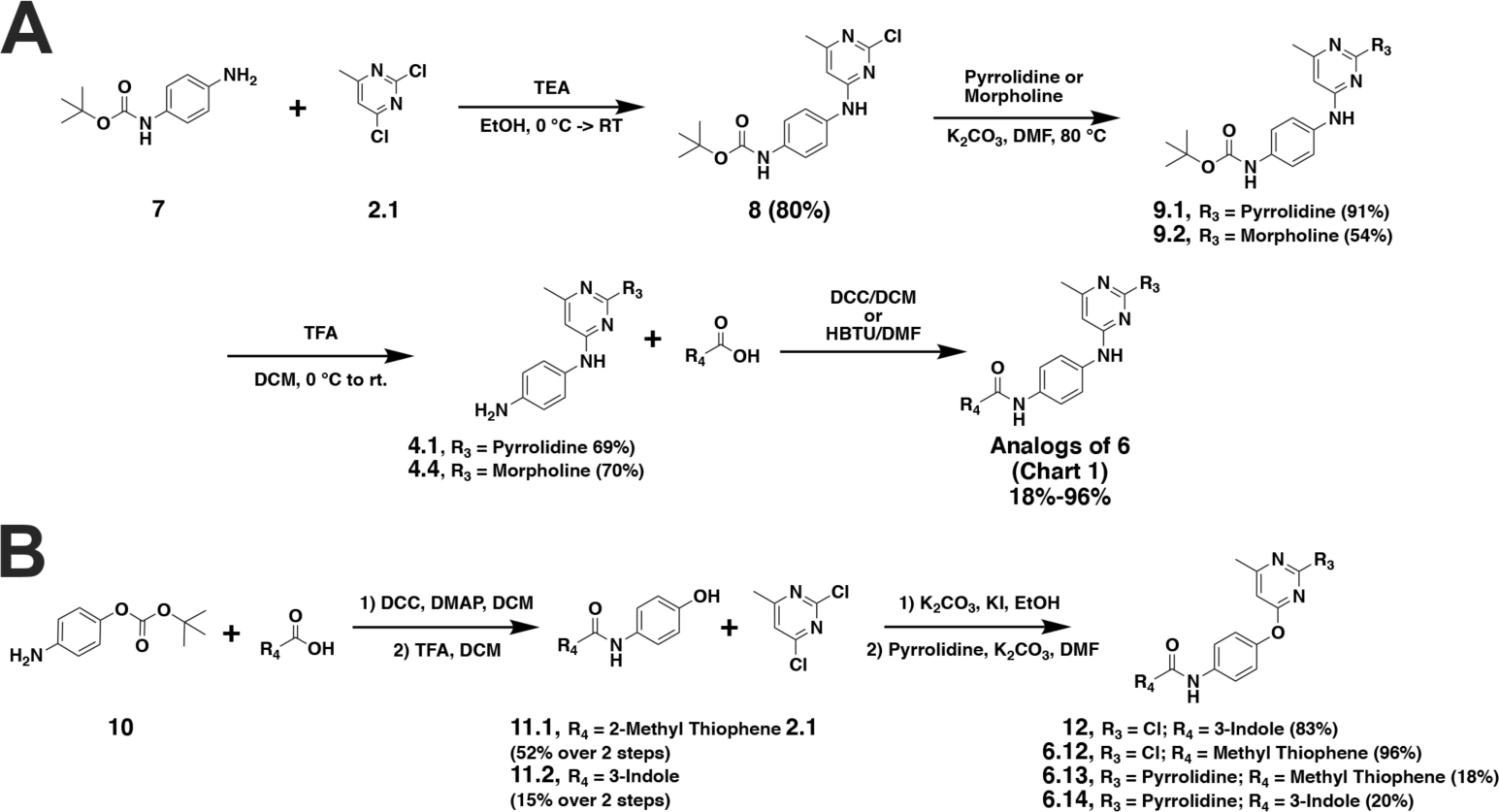
The optimized syntheses of CHD1Li 6.0 analogs. Percent yield for each compound is shown in parentheses or as a range for analogs of 6.0.
The cell-based antitumor activity of CHD1Li 6.4 (R4 = indole) maintained potency compared to 6.1, 6.2, and 6.3, which were modified at the pyrimidine ring (R1, R2, and R3) or the adjacent aniline (SI Table S1). Thus, we hypothesized that substitution of the thiophene at R4 with different aromatic rings would potentially improve the biological activity of CHD1Li 6.0 analogs. Thus, we focused our medicinal chemistry primarily at the R4 position. In Scheme 2, we utilized BOC protecting groups to facilitate derivatization of R4 with various aromatic groups, including thiophene, indole, azaindole, benzimidazole, and quinoline rings (Scheme 2A). We substituted pyrrolidine for morpholine at R3. Finally, to investigate the necessity of the aniline linkage of 6.0 we generated ether linked analogs (Scheme 2B). Taken together, the facile synthetic approaches in Scheme 1 and 2 produced >30 novel CHD1Li analogs including 6.0 (Chart 1).
Chart 1.
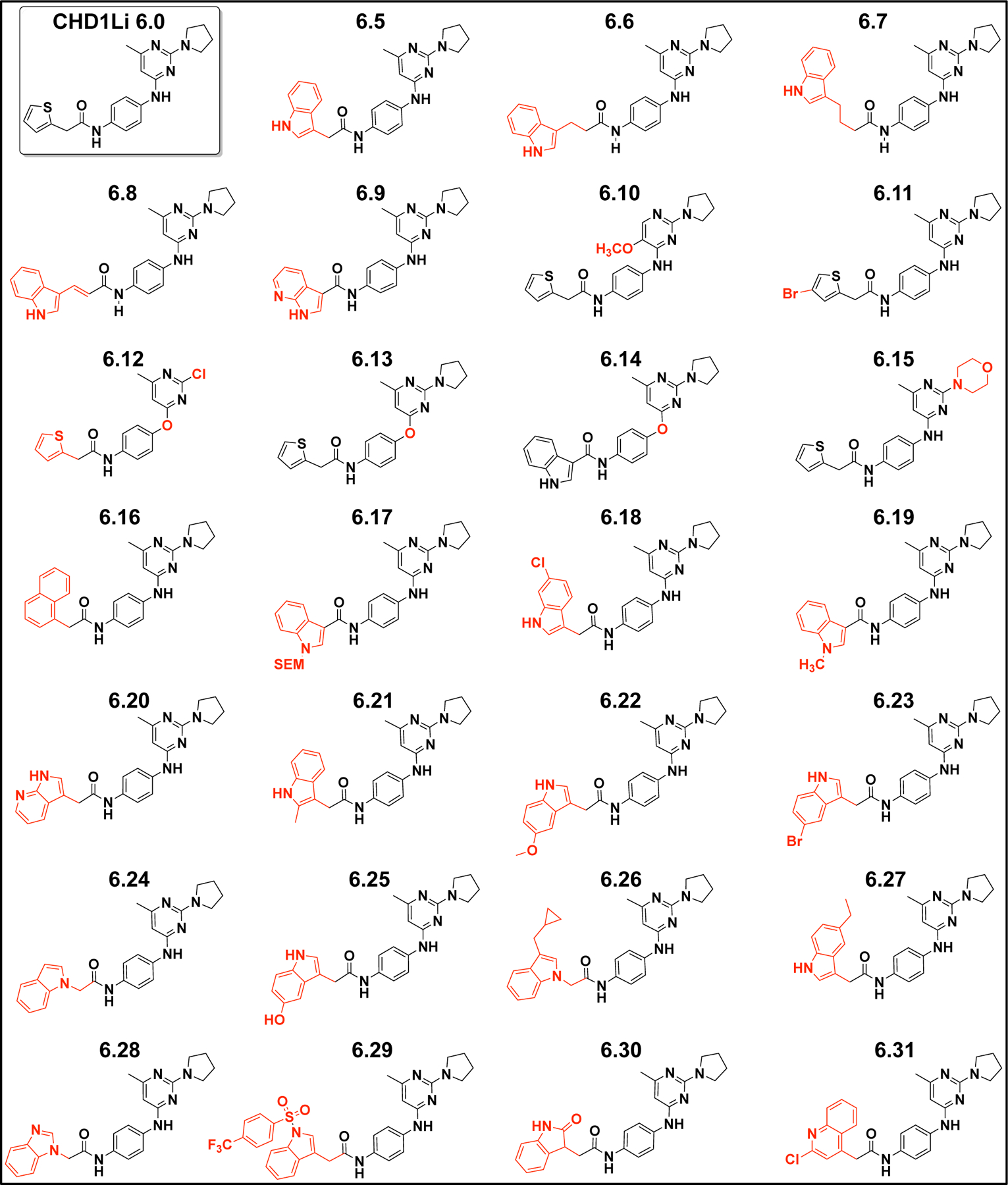
The structures of CHD1Li 6.0 analogs with structural differences highlighted in red.
In vitro Biological Evaluation of CHD1Li.
Previously, we demonstrated that CHD1Li 6.0 inhibits EMT and induces mesenchymal-epithelial transition (MET, i.e. reverse EMT), promoting apoptosis in CRC tumor organoid models.11 Thus, we first assessed CHD1Li for their ability to inhibit the viability of SW620 tumor organoids (SI Table S1). In parallel, we examined CHD1Li for their ability to inhibit the ATPase activity of recombinant catalytic CHD1L (cat-CHD1L) enzyme, using a single dose of 20 μM for each CHD1Li (SI Figure S1).11 Taken together, the cytotoxicity against SW620 tumor organoids and inhibitory activity of cat-CHD1L were used to prioritize lead CHD1Li. The most potent CHD1Li with low μM dose-dependent inhibition concentration 50% (IC50) activity against tumor organoids and cat-CHD1L were 6.5 (R4 = indole), 6.11 (R4 = 4-bromo-thiophene), 6.16 (R4 = naphthyl), 6.18 (R4 = 7-Chloro-indole), 6.21(2-methyl-indole), and 6.31 (R4 = 2-chloro-quinoline) (Figure 1), which were prioritized for further biological interrogation of antitumor activity. Furthermore, the diversity of the aromatic ring structures at R4 among these analogs would provide insight into the SAR of this novel class of CHD1Li.
Figure 1. Lead CHD1Li determined by tumor organoid cytotoxicity and enzymatic inhibition of cat-CHD1L.
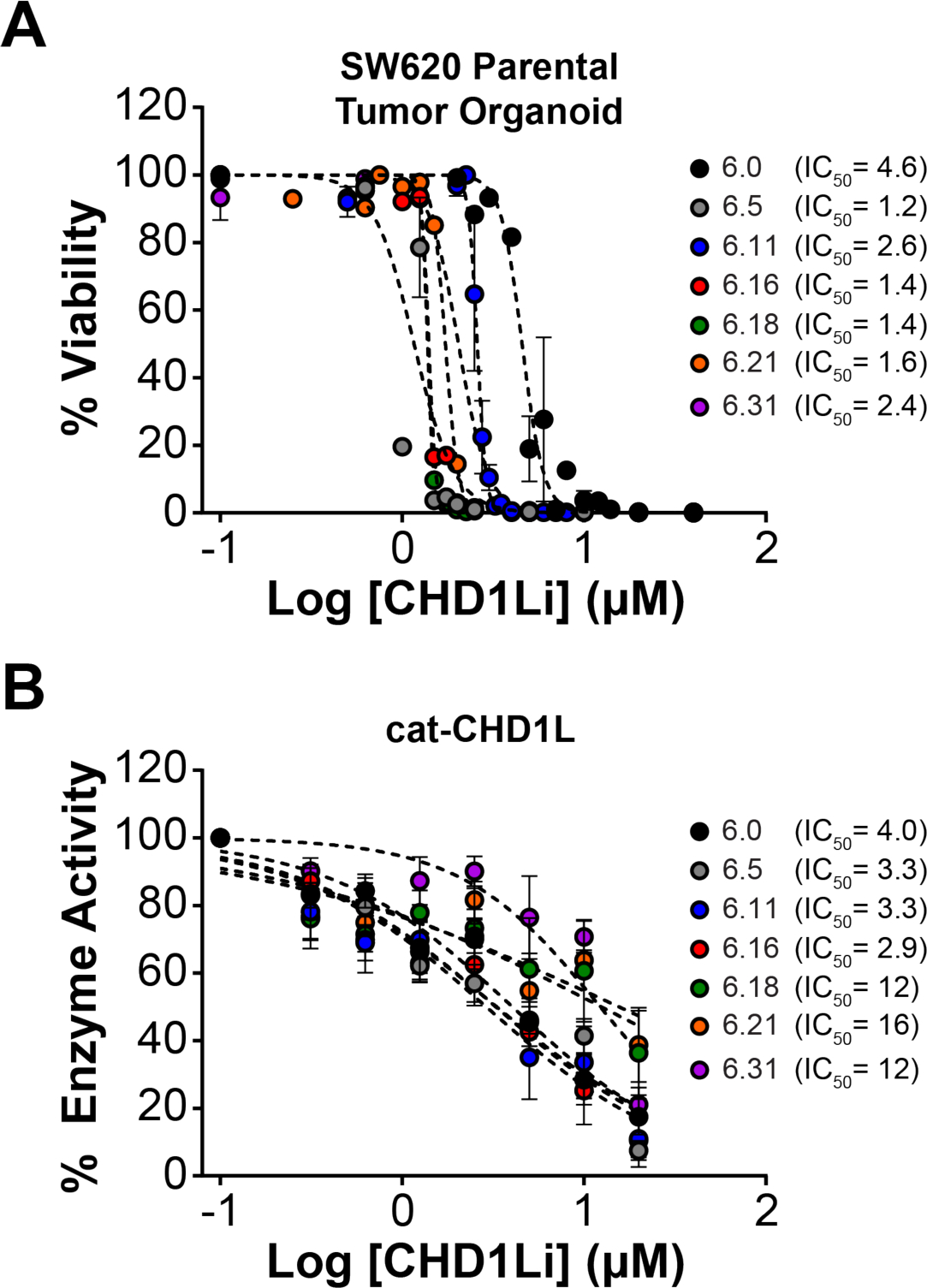
(A) Dose-response of lead CHD1L inhibitor compounds measuring SW620 tumor organoid viability. (B) Enzymatic inhibition of the cat-CHD1L recombinant protein by lead CHD1Li. Data is normalized to DMSO control and is shown as mean ± SEM of triplicate experiments.
We have developed and validated a novel fluorescent EMT dual-reporter suitable for 2D and 3D high-content imaging that is an effective tool to measure EMP in real time while simultaneously tracking the spectrum of EMT cellular phenotypes.21 EMT phenotypes can be isolated by FACS based on dual-reporter fluorescence and interrogated as individual cell populations in long-term culture, including stable xE/xM (RFP−/GFP−) and quasi-EMT populations of E (RFP+), E/M (RFP+/GFP+), and M (GFP+).21 Isolated EMT phenotypes display unequivocal differences morphologically and metabolically, and in tumorigenic potential. Moreover, in CRC, these phenotypic differences are driven by TCF-transcription. The correlation between cytotoxicity and CHD1L inhibition are consistent with the inhibition of CHD1L-mediated TCF-transcription in CRC.11, 21 In particular, we demonstrate that the more tumorigenic CSC quasi-M-Phenotype has upregulated TCF-transcription compared to the other EMT phenotypes (Figure 2A). Thus, we treated M-Phenotype SW620 and HCT116 cells with lead CHD1Li and observed a dose-dependent decrease in TCF-transcription with all lead CHD1Li IC50 values in the low μM range (Figure 2B and C). Notably, this potency is consistent with the potency of inhibiting recombinant cat-CHD1L (Figure 1B).
Figure 2. CHD1Li downregulate CHD1L mediated TCF-transcription in M-Phenotype cells.
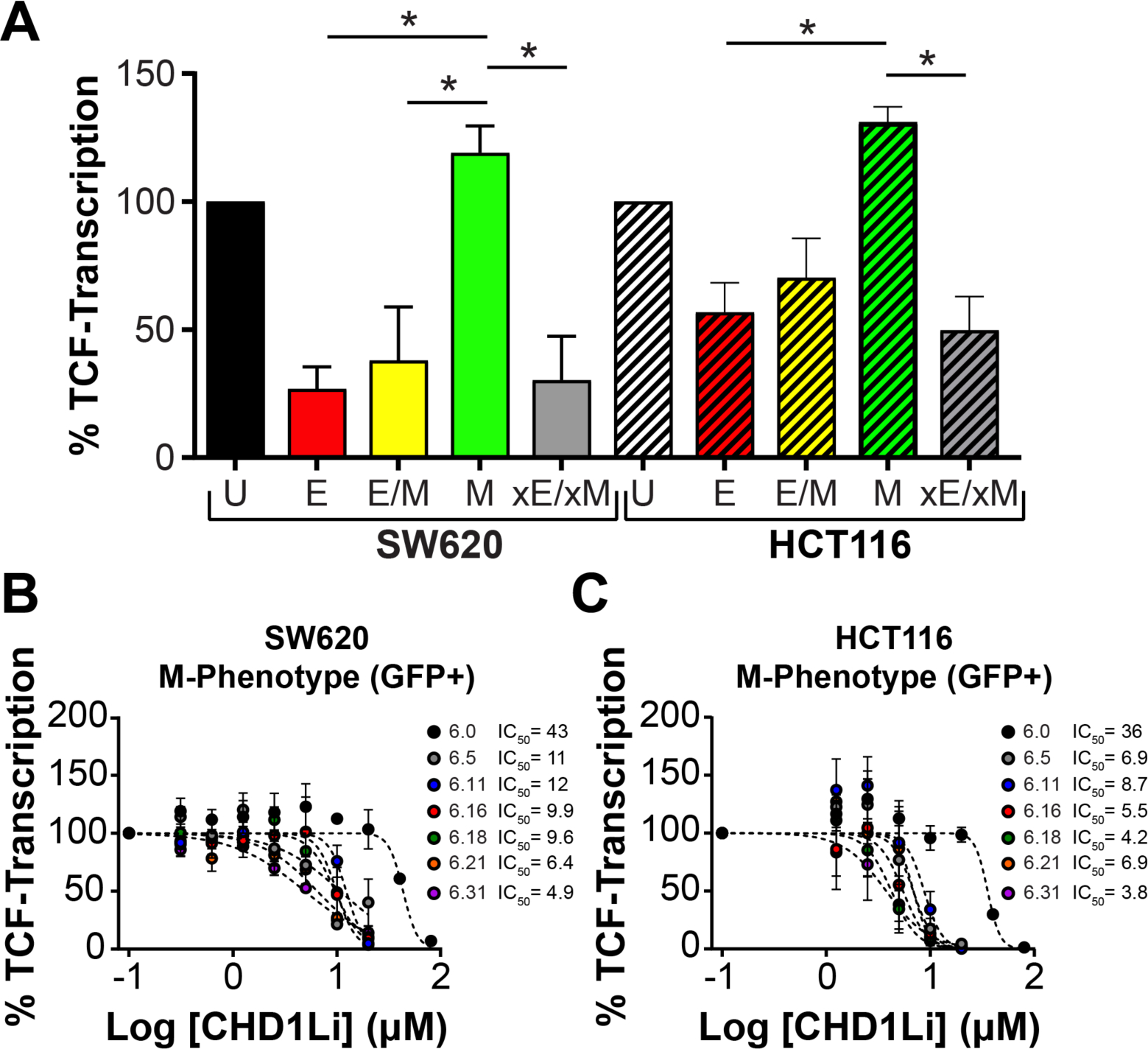
(A) TCF-transcriptional activity in isolated SW620 and HCT116 EMT phenotypes. P-values were calculated by one-way ANOVA where *P<0.05. Dose-response graphs of (B) SW620 and (C) HCT116 M-Phenotype monolayer cell culture treated with lead CHD1Li for 24 h, measuring TCF-transcription via the TOPflash luminescent reporter assay. Data is normalized to cell viability and is shown as mean ± SEM of duplicate experiments. Cell phenotypes are defined as U (unsorted), quasi-epithelial (E, RFP+), -hybrid (E/M, RFP+/GFP+); -mesenchymal (M, GFP+); and stable epithelial/mesenchymal population (xE/xM, RFP−/GFP−).
Next, we measured CHD1Li cytotoxicity in both cell lines (SW620 and HCT116) and patient tumor organoids (CRC042 and CRC102) (Figure 3). Lead CHD1Li displayed potent antitumor activity in SW620 and HCT116 isolated M-Phenotype tumor organoids, inhibiting cell viability with low μM IC50 values (Figure 3A and B). Likewise, CHD1Li had potent cytotoxicity against CRC042 and CRC102 patient tumor organoids (Figure 3C and D). These results underscore the potent antitumor activity of CHD1Li in a variety of CRC cell models, including CRC patient tumor organoids that are considered to be ideal for the study of cancer ex vivo.26
Figure 3. CHD1Li are potent cytotoxic agents in CRC cell line and patient tumor organoids.
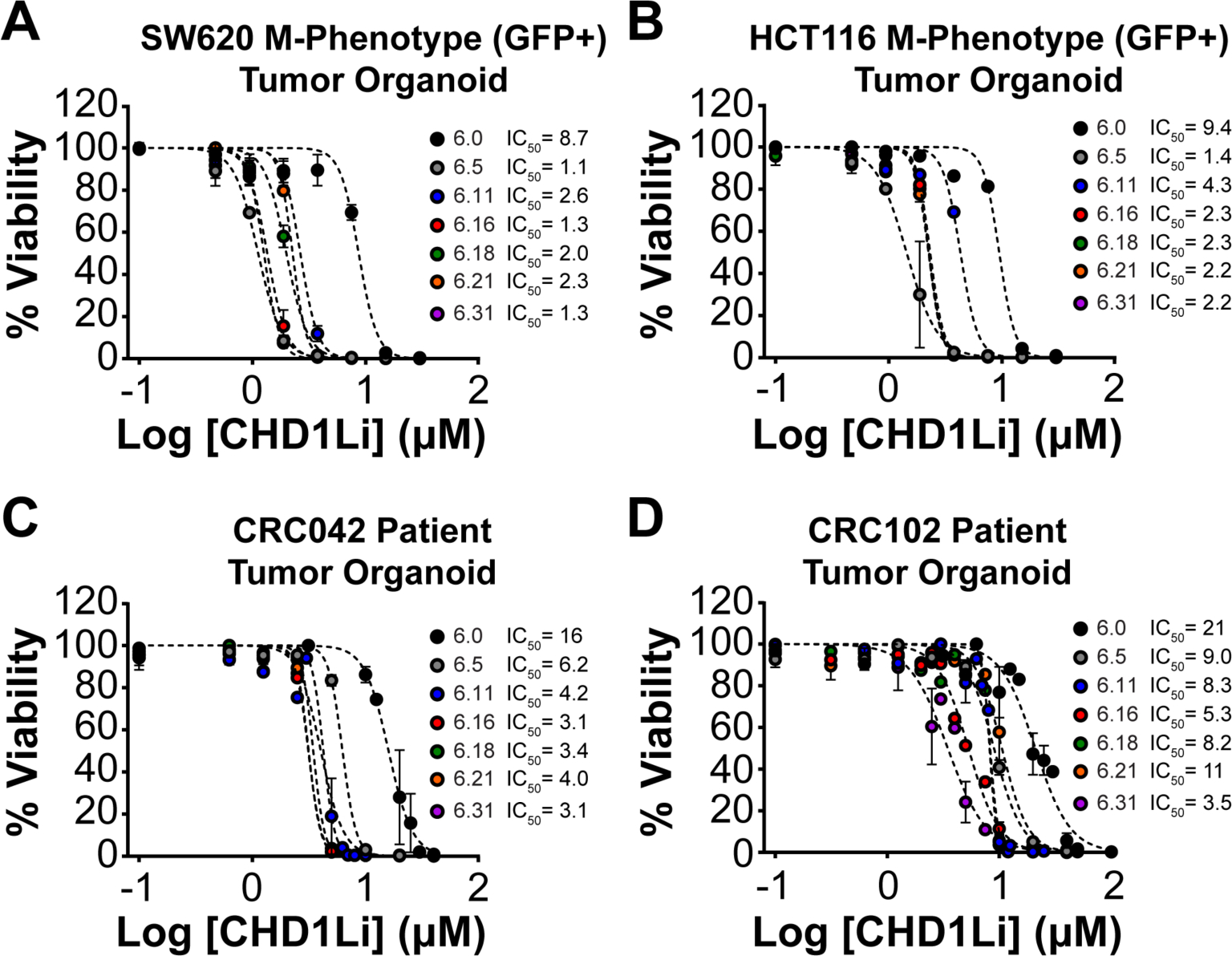
(A and B) Dose-response graphs of lead CHD1Li, measuring cell viability after 72 h of treatment of isolated M-Phenotype SW620 and HCT116 tumor organoids. (C and D) Dose-response graphs of lead CHD1Li, measuring cell viability after 72 h of treatment of CRC042 and CRC102 patient tumor organoids. The data was normalized to DMSO (vehicle) and is shown as the mean ± SEM of triplicate experiments with technical replicates (n = 3) for each experiment.
We then evaluated CHD1Li for their ability to inhibit EMT and/or induce MET by simultaneously measuring the fluorescent signals of the EMT dual-reporter (VimPro-GFP and EcadPro-RFP), using the high-content imaging methodology previously described.21 Indeed, CHD1Li induce MET in SW620 and HCT116 M-Phenotype tumor organoids characterized by dose-dependent downregulation of vimentin with concomitant upregulation of E-cadherin expression (Figure 4). To quantify E-cadherin upregulation we generated a non-linear regression model to determine the dose at which a 2-fold increase of RFP fluorescent signal occurs. Representative dual-reporter M-Phenotype HCT116 tumor organoid images treated with CHD1Li 6.5 are shown (Figure 4E). A marked downregulation of the VimPro-GFP is observed, while an upregulation of EcadPro-RFP is noted as the treatment dose increases, and this result is consistent with our prior results obtained for CHD1Li 6.0.11 Previously, we demonstrated that small molcule reversion of TCF-driven EMT in M-Phenotype tumor organoids promotes an epithelial state that is significantly less tumorigenic.21 We also demonstreated that small molecule inhibition of TCF-driven EMT significantly inhibited the tumorigenic properties regardless of the EMT phenotype.11, 21 Thus, we hypothesize that the induction of MET by CHD1Li promotes an altered epithelial state that is sensitive to the antitumor activity of CHD1Li.
Figure 4. CHD1Li induce MET in M-Phenotype SW620 and HCT116 tumor organoids.
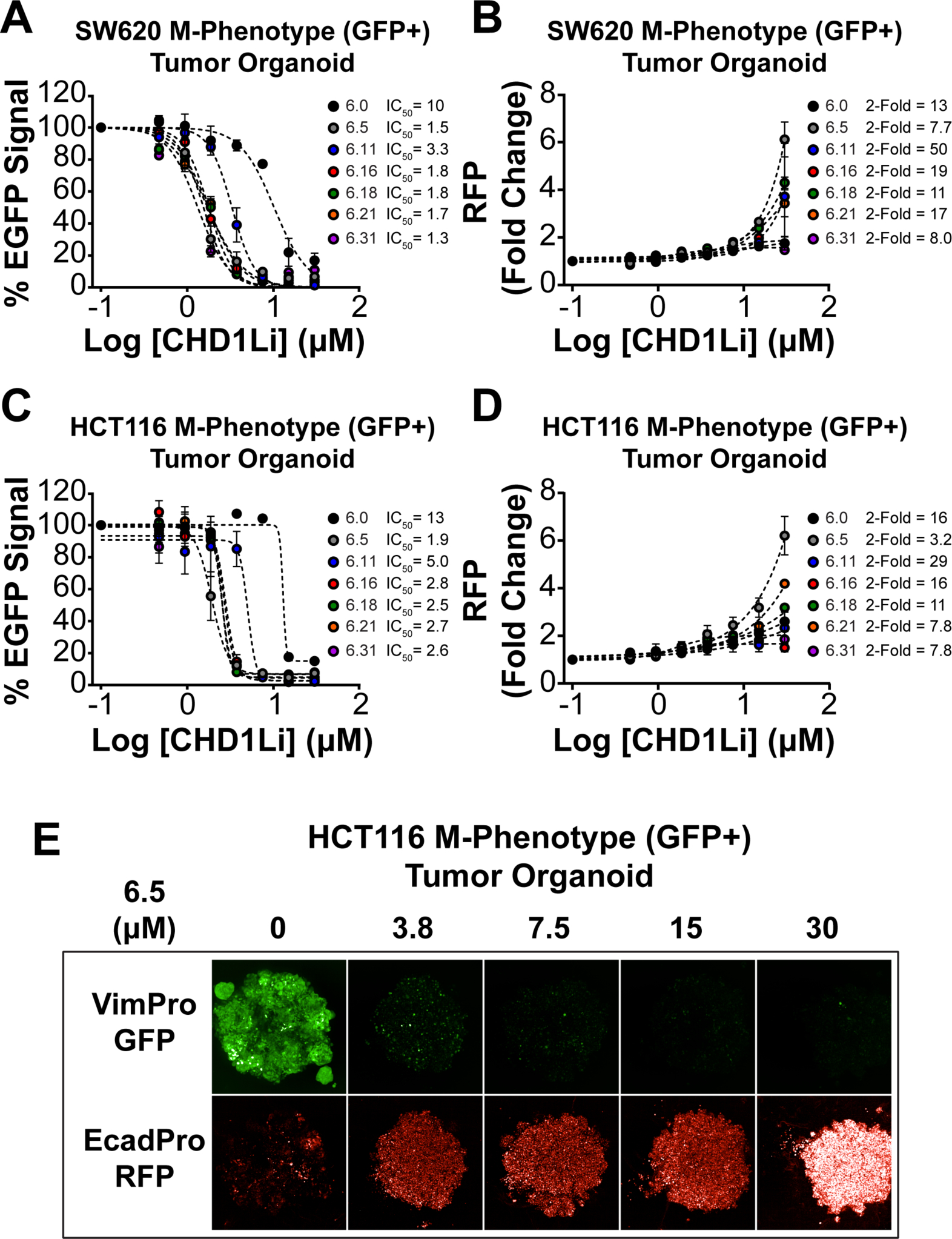
(A and C) Dose-response graphs of the downregulation of VimPro-GFP promoter activity measured by EGFP fluorescence of SW620 and HCT116 tumor organoids treated with lead CHD1Li. (B and D) Fold change upregulation of EcadPro-RFP promoter activity measured through RFP fluorescent signal in SW620 and HCT116 tumor organoids after treatment with lead CHD1Li. (E) Representative maximum projection confocal images of HCT116 tumor organoids after treatment with compound 6.5 for both VimPro-GFP and EcadPro-RFP promoter activity. Data is shown in mean ± SEM of duplicate experiments.
TCF-driven EMT is linked as a mechanism enabling mesenchymal cells with increased tumorigenic properties, including CSC stemness self-renewal, resistance to apoptosis, and metastatic potential.27, 28 This fact is consistent with our published results that isolated M-Phenotype tumor cells have significantly increased CSC stemness when compared to other isolated phenotypes.21 Furthermore, we have reported that CHD1Li 6.0 significantly inhibits CSC stemness,11 based on the clonogenic colony formation assay.29, 30 Thus, we evaluated CHD1Li for their ability to inhibit CSC stemness in SW620 and HCT116 M-Phenotype cells, using high-content imaging.29 CHD1Li effectively inhibited colony formation over a low μM to nM range (Figure 5A and B). CHD1Li 6.31 was the most potent with an IC50 value of 300 nM in SW620 cells and 200 nM in HCT116 cells. Taken together, CHD1Li prove to be effective antitumor agents that prevent CHD1L-mediated TCF-transcription, which in turn inhibits EMT and induces MET, resulting in loss of CSC stemness while promoting cytotoxicity to tumor cells.
Figure 5. Cancer cell stemness is greatly reduced by CHD1L inhibitors.
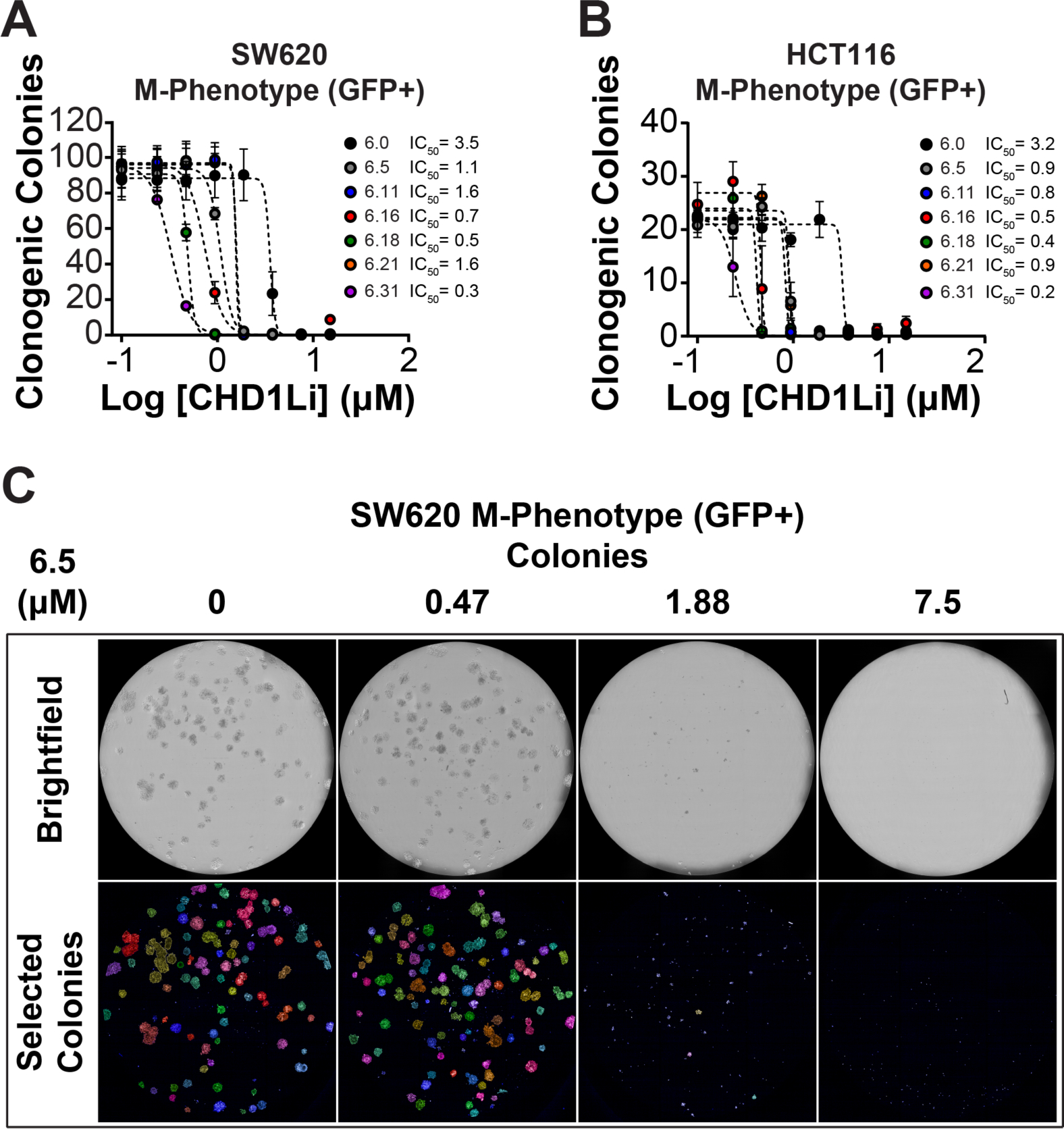
(A) Number of clonogenic colonies formed after continuous lead CHD1Li treatment in SW620 cells. (B) Number of clonogenic colonies formed after continuous treatment with lead CHD1L inhibitors in HCT116 cells. (C) Representative brightfield and pseudo-colored images of CSC colonies treated with CHD1Li 6.5. The data is represented as the mean ± SEM of duplicate experiments using triplicate technical replicates.
In Silico Studies and Structure Activity Relationships of CHD1Li.
During the course of our ligand-based drug design and in vitro biological evaluation, the 3D-structure of CHD1L had not been reported. However, just prior to this publication, a structure was reported (PDB:7EPU).31 Thus, we utilized this structure as a model to help elucidate the promising biological activity of CHD1Li. We conducted molecular modeling using the Schrödinger Molecular Modeling Suite (version 2021–2) to gain insight on the potential protein-CHD1Li interactions.
Binding site determination and molecular dynamics simulation.
We investigated potential binding sites on the CHD1L structure, considering the various domain organization (Figure 6A). CHD1L is comprised of a C-terminal macro domain that is connected to the N-terminal ATPase domain, consisting of two ATPase lobes (C-ATPase and N-ATPase) (Figure 6A).32, 33 The macro domain, in part, functions as a switch regulating CHD1L ATPase by autoinhibition through binding of the C-ATPase lobe. SiteMap calculations were performed on the reported CHD1L structure to elucidate the potential binding sites for CHD1Li (SI Table S2). Although the results suggest the PAR binding site of the macro domain is a feasible binding site, this site was ruled out since CHD1Li potently inhibit cat-CHD1L, which is missing the macro domain. The molecular modeling data indicated several other potential binding sites for CHD1Li, including a loop region of the C-ATPase lobe, an N-ATPase allosteric site, and a C-ATPase allosteric site. Overall, the most plausible binding site revealed from molecular modeling is the C-ATPase lobe allosteric site (Figure 6B). This allosteric pocket has a site score of 1 with the largest pocket size and volume and is adjacent to the N-ATPase lobe ATP binding site.
Figure 6.
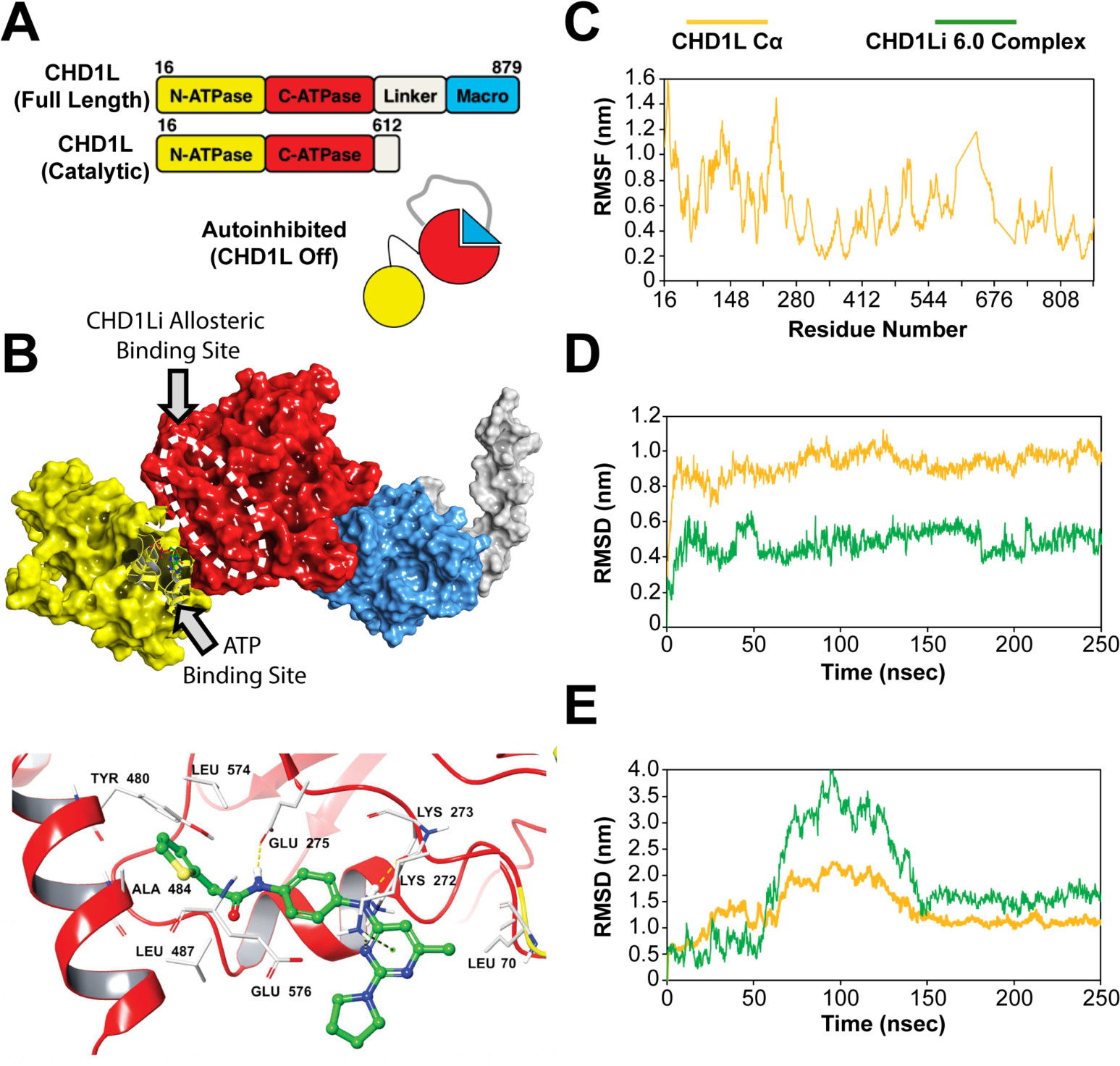
(A) CHD1L domain architecture showing the autoinhibited protein state. (B) Molecular surface representation of the protein (PDB ID: 7EPU) showing the most plausible SiteMap-predicted CHD1Li binding site (white dashed oval). The structure is color coded, including macrodomain (blue), linker region (grey), C-ATPase (red) domain, and N-ATPase domain (yellow). A bound ADP, part of the CHD1L crystal, is shown as a ball and stick model in the N-ATPase ATP binding site. Note, the PDB structure shown in panel B is missing part of the linker region, which normally extends to the C-ATPase domain and is depicted in the CHD1L cartoon in panel A. Below the surface representation is the lead CHD1Li 6.0 bound in the C-ATPase allosteric site. (C) RMSF plot of the apo CHD1L residues highlighting the reduced structural flexibility of C-ATPase compared to N-ATPase. (D) RMSD evolution of CHD1Li 6.0 at the C-ATPase and (E) N-ATPase allosteric sites, respectively over the 250 ns molecular dynamics trajectory
To further evaluate the C-ATPase allosteric binding site as a potential target for CHD1Li, molecular dynamics (MD) simulation was performed over 250 ns on apo CHD1L. A plot of the protein residues root-mean-square fluctuation (RMSF) showed the reduced flexibility of C-ATPase lobe compared to N-ATPase lobe (Figure 6C). High structural fluctuations (1.0–1.4 nm) occurred primarily at the N-ATPase lobe. Consistent with this result, similar structural fluctuations of this lobe during MD was also reported by Lehmann and colleagues.33 Additionally, the MD simulation of docked CHD1Li 6.0 (Figure 6B, lower panel) highlighted the stability of CHD1Li 6.0 bound at this site relative to the N-ATPase allosteric site. The root-mean-square deviations (RMSD) of CHD1Li 6.0 complex also showed that bound 6.0 stabilized the protein while equilibrating at an average RMSD of 0.5 nm after 50 ns (Figure 6D). In contrast, 6.0 experienced large deviations (>3.5 nm) at the N-ATPase allosteric site for 100 ns before converging at an average RMSD of 1.6 nm for the remaining 100 ns (Figure 6E), which indicates that 6.0 drifted from the binding site. The computed distances between the protein and ligand also highlight this phenomenon (SI Table S3 and SI Figure S2). Taken together, the results corroborate the selection of the C-ATPase allosteric site as the most plausible binding site for CHD1Li.
Molecular docking.
Having established a preferred binding site for ligand docking, models of the CHD1Li complexes were generated using glide and induced fit docking protocols were used to identify the nature of possible hydrophobic interactions (Figure 7). Analysis of the results suggests that CHD1Li target affinity is influenced by the number of hydrophobic contacts providing low-energy complexation (SI Table S4). For instance, CHD1Li with IC50 ≤4.0 μM (Figure 1B) have 30–39 hydrophobic contacts and strong affinity (docking score >6) for the C-ATPase allosteric site (Figure 7A–C and SI Table S4). The trend in binding free energy (MMGBSA ΔG bind) also aligns with the experimental inhibitory potency of CHD1Li.
Figure 7.
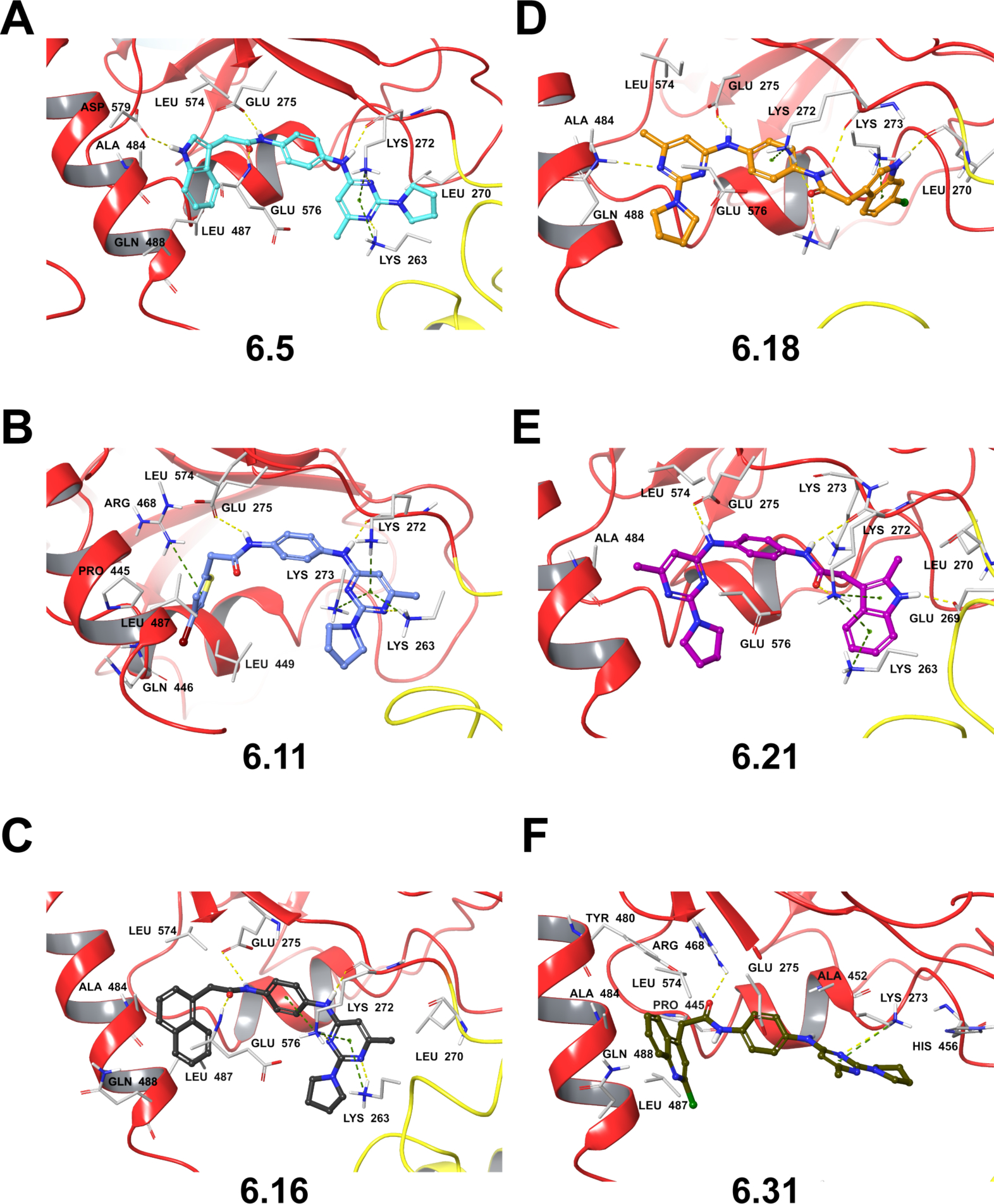
Molecular docking poses for lead CHD1Li in the C-ATPase allosteric site of CHD1L. Panels A-F depict the 3D-representation of the binding pose for lead CHD1Li as indicated. Depicted interactions include hydrogen bond (yellow dashed) and π-cation (green dashed).
Further analysis of the complexes reveals that the CHD1Li (except 6.18 and 6.21) similarly orient in the binding site with aryl rings extending towards α-H7 of C-ATPase allosteric site to form hydrophobic contacts with Ala 484, Leu 487, and Gln 488 (Figure 7A–C, and F). In contrast, 6.18 and 6.21 have an opposite configuration (Figure 7D–E). The CHD1Li (except 6.31) also consistently featured hydrogen bond interactions between the phenylenediamine ring aniline and amide groups with Lys 272 and Glu 275, respectively. This in silico result is consistent with the biological activity of CHD1Li, and loss of the aniline hydrogen bond doner due to an N-methyl (6.3), or alteration of the pyrimidine ring (6.1 and 6.2) leads to loss of inhibitor activity against cat-CHD1L and in cell-based models. Taken together, these hydrogen bonding interactions may be important to CHD1Li target binding and antitumor activity. Additionally, the π-cation interactions observed between the pyrimidine ring and lysine residues 263, 272, and 273 also constitute prominent interactions favoring CHD1Li binding. Of note, CHD1Li 6.11 is the only analog to display all three of these interactions with lysine residues 263, 272, and 273 (Figure 7B).
Molecular dynamics substantiates the improved biological activity of 6.11 over 6.0.
The molecular docking data suggests that the bromo-thiophene ring may influence the increased hydrophobic contacts observed with 6.11 and affinity to CHD1L relative to 6.0. To substantiate this, a MD simulation of the CHD1Li 6.11 complex was performed and compared to CHD1Li 6.0. The trajectory analysis showed that 6.11 maintained an average RMSD of 0.4 nm for the first 110 ns of the simulation and 0.6 nm for the remaining 140 ns (SI Figure S3). Moreover, 6.11 has lower values for the average CHD1Li complex energy and distance to protein, respectively, compared to 6.0 (SI Table S3). The thiophene ring in 6.11 also formed π-cation interaction with Arg 468 (Figure 7B). Additionally, during MD we observed π-π interactions with Tyr 480, which lasted for 25% (62.5 ns) of the simulation and this interaction is absent in CHD1Li 6.0 (SI Figure S3)
In vivo Biological Evaluation of CHD1Li.
We previously determined that lead CHD1Li 6.0 has a good in vivo disposition, including a plasma half-life of 3 h in mice.11 We postulated that the half-life of 6.0 may be affected due to liver metabolism of the thiophene ring. Thiophene rings may or may not form reactive metabolites (e.g. S-oxidation), and substituted thiophenes are more stable to liver metabolizing enzymes.34 Moreover, 6.0 does not display any liver toxicity when treating mice at a maximum tolerated dose of 50 mg/kg by intraperitoneal (IP) administration daily over five days.11 Thus, thiophene reactive metabolites leading to toxicity does not appear to be a limiting adverse effect. Prior to conducting in vivo studies with CHD1Li, we conducted in vitro mouse microsomal stability studies with select CHD1Li that represent a range of structural diversity, including 6.0, 6.4, 6.5, 6.11, and 6.31 (SI Figure S4). CHD1Li 6.11 proved to be the most metabolically stable when exposed to microsomes with a 2-fold longer half-life of 130.3 minutes compared to 67.2 minutes for CHD1Li 6.0. Therefore, 6.11 was prioritized for in vivo evaluation.
Using CD-1 mice, we administered 6.11 by IP injection at a dose of 50 mg/kg and assessed the pharmacokinetics (PK) of 6.11, including elimination half-life (t1/2λ) from plasma, liver, and fat tissues (Figure 8A). The t1/2λ of 6.11 in the plasma and tissues is 8 h, which is ~3-fold longer than 6.0. Using the same dose, we assessed the oral bioavailability of 6.11 after oral gavage (PO) and found that 6.11 is bioavailable with 44% uptake in the plasma and a t1/2λ of 8 h (Figure 8A). The in vivo half-life of 6.11 is consistent with its in vitro microsomal stability, indicating that the bromothiophene moiety of 6.11 significantly improves the in vivo PK by increasing its stability to liver enzymes compared to 6.0.
Figure 8. In vivo Pharmacokinetics and Efficacy of 6.11.
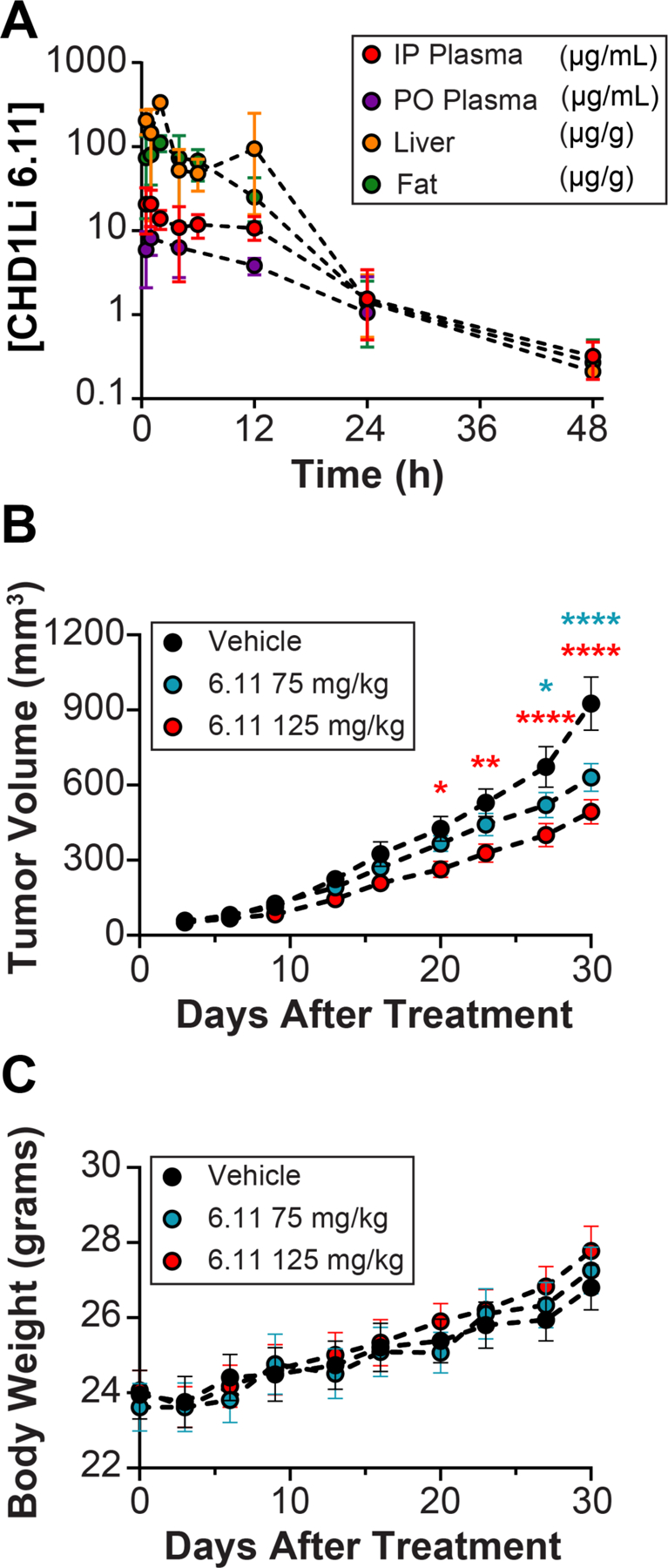
(A) Pharmacokinetics in CD-1 female mice. Concentration of CDH1Li 6.11 over 48 h in plasma, liver, and fat. For administration IP = intraperitoneal and PO = oral gavage. (B) SW620 GFP+ tumor flank model showing the anti-tumor activity of CHD1Li 6.11 in nude athymic mice after 28 days of PO treatment. (C) Measured body weight of nude athymic mice treated PO with 6.11 over 28 days.
We performed dose finding and maximum tolerated dose (MTD) studies using athymic nude mice, which indicated that doses of 6.11 at or below 150 mg/kg administered by PO 5-days/week over 21 days were well tolerated, assessed grossly by weight and daily normal behavior. After determining the MTD, the antitumor efficacy of 6.11 was measured in athymic nude mouse xenografts baring SW620 isolated M-Phenotype flank tumors. CHD1Li 6.11 was administered by PO at doses of 75 mg/kg and 125 mg/kg 5-days/week over 30 days. Mice treated with 6.11 showed a significant decrease in tumor volume beginning from day 20, becoming highly significant after 28 days of treatment compared to the vehicle control (Figure 8B). As with the MTD studies, 6.11 was well tolerated by mice during this study with no adverse gross toxicity. In fact, we observed increased body weight for all groups during the treatment period along with no abnormal observations associated with gross toxicity (Figure 8C). These results highlight the antitumor potential of CHD1Li. Furthermore, CHD1L has numerous biological functions12 that could be exploited in combination with drugs that target other antitumor mechanisms, underlining CHD1L as an attractive anticancer drug target.
Structure-activity relationship Summary.
From the cat-CHD1L inhibition and cell-based assays, a valuable SAR emerged to describe the favorable antitumor activity of CHD1Li (Chart 2). When morpholine (6.15) is substituted for pyrrolidine, we observe a significant loss in biological activity. However, the pyrrolidine functionality appears to maintain a good balance of lipophilicity and hydrophilicity. In fact, the pyrrolidine group facilitates aqueous solubility of lead CHD1Li, an ideal drug-like property. Interestingly, when the pyrimidine ring is substituted with electron withdrawing groups such as fluorine (6.1) or methoxy (6.10) we observed a significant loss in activity. Perhaps electronegative groups in proximity of the adjacent aniline may inductively withdraw electron density decreasing the pKa of the aniline. As result, this may lead to a negatively charged molecule a physiological pH, which is likely stabilized by resonance. Our results also demonstrate that the aniline may be important for CHD1L inhibition as it is consistently observed as a Hydrogen bond donor with Lys 272 in molecular modeling. This is substantiated by the observed significant loss of activity of the ether analogs (6.12, 6.13, and 6.14), which would function as a hydrogen bond acceptor. Likewise, the aniline N-methyl analog (6.3) has diminished biological activity. Next, the carbon chain connecting the aromatic rings at position R (Chart 2) appears to be optimal at one carbon. The 2- and 3-carbon analogs (6.6 and 6.7) result in loss of activity. Interestingly, the 2-carbon trans-double bond (6.8) restores activity albeit less than the 1-carbon analogs. It was apparent early on in our drug design campaign that the thiophene group of the parent analog (6.0) could be modified with a variety of aromatic rings at position R (Chart 2), which generally display improved potency and biological activity. In addition, substitution of the thiophene parent analog (6.0) with indole (6.4 and 6.5), 2-chloro-quinoline (6.31), or 4-bromo-thiophene (6.11) increased the metabolic stability to mouse liver microsomes (SI Figure S2). CHD1Li 6.11 proved to consistently have the most promising in vitro biological activity and metabolic stability. Consistent with the in vitro metabolic stability, in vivo PK studies show that 6.11 has an ~3-fold longer plasma half-life compared to 6.0. In addition, the drug-like properties of 6.11 extended to its oral bioavailability with 44% penetrating the circulatory system after oral administration to mice (Figure 8).
Chart 2.
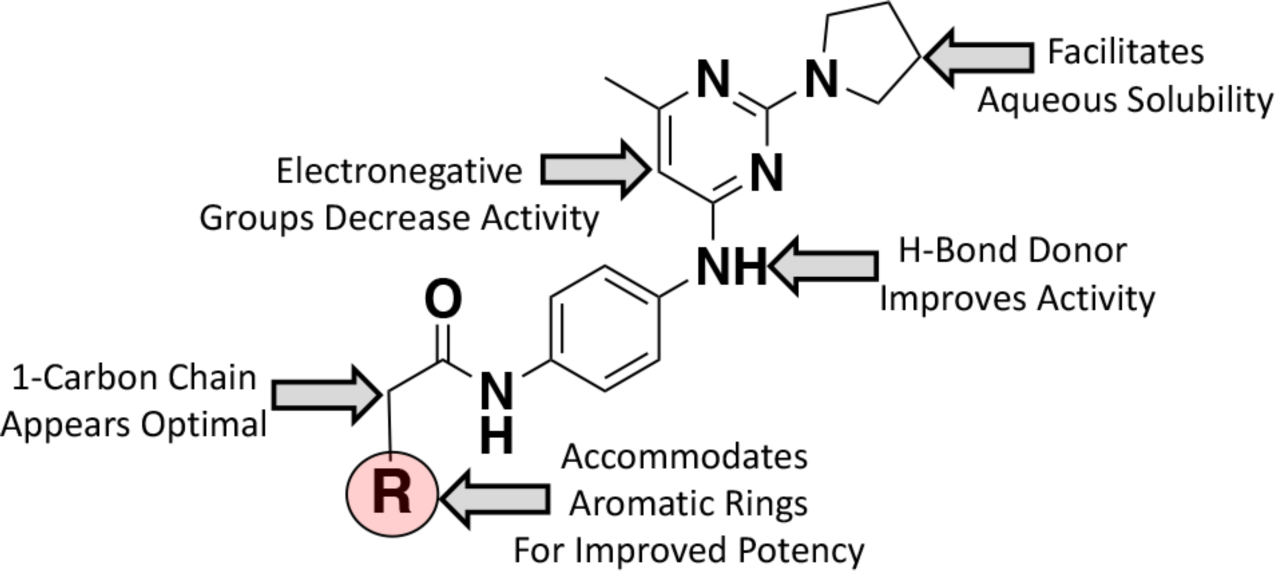
Schematic depiction of the SAR summary for CHD1Li.
CONCLUSION
CHD1L has recently surfaced as an oncogene that is involved in several types of cancers. However, there are no clinically available therapeutic agents that target CHD1L. Herein, we expanded on our previous HTS discovery of the first CHD1Li lead drug 6.0.11 Our medicinal chemistry approach has improved the potency, pharmacokinetics, bioavailability, and efficacy of this class of CHD1Li. We validated CHD1Li leads through enzymatic inhibition of cat-CHD1L, cytotoxicity in both parental and EMT dual-reporter isolated M-Phenotype CRC tumor organoids, CRC patient tumor organoids, TCF-transcriptional downregulation, modulation of EMP, reduction of CSC stemness, and mouse microsomal stability. All these findings pointed to 6.11 as a lead candidate to perform in vivo studies. The in vivo studies confirmed that CHD1Li 6.11 is a potent oral bioavailable antitumor agent that is well-tolerated by mice at the doses tested. In addition, due to CHD1L playing multiple biological roles, we expect that the anti-tumor activity of CHD1Li will synergize when combined with clinically used drugs and radiation therapy. Finally, we expect this work to lay the foundation for the clinical translation of CHD1Li lead drugs that may be effective alone and in combination with other clinical therapies to treat GI cancers and other cancers driven by CHD1L.
EXPERIMENTAL SECTION
General Experimental Methods.
All commercial chemicals were used as supplied unless otherwise stated. All solvents used were dried and distilled using standard procedures. Thin layer chromatography (TLC) was performed using Aluminum backed plates coated with 60Å Silica gel F254 (Sorbent Technologies, Norcross, GA, USA). Plates were visualized using a UV lamp (λmax = 254 nm). Column chromatography was carried out using 230–400 mesh 60Å silica gel or using a Teledyne Isco Combiflash next gen 300+ chromatography system with high performance gold columns. NMR spectra were recorded on a Bruker Avance III 400 (1H 400 MHz, 13C 100 MHz). All chemical shifts are recorded in parts per million (ppm), referenced to residual solvent frequencies (1H NMR: Me4Si = 0, CDCl3 = 7.26, D2O = 4.79, CD3OD = 4.87 or 3.31, DMSO-d6 = 2.50, Acetone-d6 = 2.05 and 13C NMR: CDCl3 = 77.16; CD3OD = 49.0, DMSO-d6 = 39.5, Acetone-d6 = 29.9 Coupling constants (J) values are expressed in hertz (Hz). The following splitting abbreviations were used: s = singlet, d = doublet, t = triplet, q = quartet, p = pentet, m = multiplet, br = broad, dd = doublet of doublets, dt = doublet of triplets, td = triplet of doublets. Melting points (m.p.) were determined using a Stuart melting point apparatus (SMP20). Infrared (IR) spectra were recorded on a Bruker ALPHA platinum ATR FTIR. Oils and solids were examined neat. High resolution mass spectrometry (HRMS) analyses were recorded using Q Exactive mass spectrometer (ThermoFisher, San Jose, CA) operated independently in positive or negative ion mode, scanning in full MS mode (2 μscans) from 150 to 1500 m/z at 140,000 resolution, with 4 kV spray voltage, 45 sheath gas, 15 auxiliary gas. Acquired data were then converted from raw to mzXML file format using Mass Matrix (Cleveland, OH).
Compound Purity:
All final compounds were determined to be ≥95% pure by HPLC analysis, using a Shimadzu prominence HPLC equipped with a photodiode array detector (PDA) and LunaR Omega Polar C18 column (5 μm, 100 Å, 250 mm × 4.6 mm). Compounds were eluted using a flow rate of 0.525 mL/min, with a gradient of water/ methanol, with 0.1%TFA in water / 0.1%TFA in methanol over 25 min. HPLC purity traces are shown in the SI for each final compound.
Chemistry.
N-(4-aminophenyl)-2-chloro-6-methyl-pyrimidin-4-amine (3.1).
To 0.5 g (3.06 mmol) of 2,4-dichloro-6-methylpyrmidine (2) dissolved in 10 mL of ethanol at 0 °C were added 513.7 μL (1.2 equivalents, 372.5 mg, 3.68 mmol) of triethyl amine, and 330.5 mg (3.06 mmol) of p-phenylenediamine (1). The reaction mixture was warm to room temperature and for 8h. The solvents were removed under reduced pressure and the resulting residue was chromatographed on silica gel using 40% hexane in ethyl acetate as the eluent to afford 500 mg (70% yield) of the 3.1. Rf = 0.40; m.p. 157–159 °C; 1H NMR (400 MHz, CDCl3) δ 7.507 (s, N-H), 7.014 (d, J=8.6 Hz, 2H), 6.681 (d, J=8.6 Hz, 2H), 6.162 (s, 1H), 3.777 (s, N-H, 2H), 2.239 (s, 3H); 13C-NMR (100 MHz, CDCl3); 168.384, 164.430, 160.118, 145.450, 127.560, 126.906, 115.827, 100.182, 23.936; IR (neat) υmax 33214.14, 1590.07, 1506.88, 1424.41, 1214.54, 1028.66, 970.00, 905.78, 826.69, 757.43, 547.51, 510.10; ESI-HRMS [M+H]+ calculated for C11H11ClN4, 234.6870, found 235.0735.
N-(2-chloro-5-fluoro-6-methylpyrimidin-4-yl)benzene-1,4-diamine (3.2).
2,4-dichloro-5-fluoro-6-methylpyrimidine (2.2) (250 mg, 1.381 mmol, 1.0 equiv) was dissolved in ethanol (10 mL) and cooled in an ice bath. Triethyl amine (231 μL, 1.657 mmol, 1.2 equiv) and p-phenylenediamine (1) (324.1 mg, 1.381 mmol, 1.0 equiv) were added and the reaction was allowed to warm to room temperature and stir for 15h. The solvent was removed under reduced pressure and the crude mixture was purified via column chromatography using 60% ethyl acetate in Hexanes to provide 3.2 (290 mg, 83% yield) as a dark yellow solid. TLC (60% ethyl acetate in hexanes), Rf = 0.40; m.p. 157–159 °C; 1H NMR (400 MHz, CDCl3) δ 7.324 (d, J=8.7 Hz, 2H), 6.746 (s, 1H), 6.685 (d, J=8.7 Hz, 2H), 3.667 (s, N-H, 2H), 2.368 (d, J=3.0 Hz, 3H); 13C-NMR (100 MHz, CDCl3); 153.422, 151.036, 150.891, 150.742, 144.413, 143.984, 141.895, 128.176, 123.111, 115.638, 17.025; IR (neat) υmax 3328.86, 1616.04, 1507.65, 1281.35, 829.94, 830.88, 624.12, 562.34, 511.93; ESI-HRMS [M+H]+ calculated for C11H13ClFN4, 252.6774, found 253.0640.
N-(2-chloro-5-fluoropyrimidin-4-yl)benzene-1,4-diamine (3.3).
2,4-dichloro-5-fluoropyrimidine (2.3) (500 mg, 2.995 mmol, 1.0 equiv) was dissolved in ethanol (20 mL) and cooled in an ice bath. Triethyl amine (501.57 μL, 3.593 mmol, 1.2 equiv) and p-phenylenediamine (1) (323.88 mg, 2.995 mmol, 1.0 equiv) were added and the reaction was allowed to warm to room temperature and stir for 8h. The solvent was removed under reduced pressure and the crude mixture was purified via column chromatography using 60% ethyl acetate in Hexanes to provide 3.3 (544 mg, 76% yield) as a tan solid. TLC (60% ethyl acetate in hexanes), Rf = 0.36; m.p. 155–157 °C; 1H NMR (400 MHz, CDCl3) δ 7.985(d, J=2.7 Hz, 1H), 7.351 (d, J=8.7 Hz, 2H), 6.801 (s, N-H), 6.703 (d, J=8.7 Hz, 2H), 3.691 (s, N-H, 2H); 13C-NMR (100 MHz, CDCl3)154.642, 151.535, 151.433, 146.62, 144.223, 143.900, 140.533, 140.331, 127.753, 123.174, 115.620; IR (neat) υmax 3014.43, 1627.78, 1580.69, 1506.69, 1323.90, 1235.19, 946.92, 816.77, 746.87, 690.44, 641.61, 592.67, 514.39, 430.10; ESI-HRMS [M+H]+ calculated for C10H8ClFN4, 238.6504, found 239.0484.
N-(6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)benzene-1,4-diamine (4.1).
3.1 (15.21g, 0.0412 mol) was dissolved in 60 mL of dichloromethane and treated with 10 mL of TFA at 0 °C, resulting in a red colored solution. The reaction was to warmed to room temperature and allowed to stir for 3h. The reaction was concentrated and redissolved in 10% methanol and dichloromethane, then washed with bicarb and water. The organic later was dried over sodium sulfate and concentrated, purified via column chromatography using 10% methanol in dichloromethane to produce 4.1 (7.6 g, 69% yield) as an orange solid. TLC (5% methanol/dichloromethane), Rf = 0.18; m.p. 190–192 °C; 1H NMR (400 MHz, CDCl3) δ 7.116 (d, J=8.6 Hz, 2H), 6.677 (d, J=8.6 Hz, 2H), 6.190 (s, N-H), 5.680 (s, 1H), 3.542–3.575 (m, 4H), 2.188 (s, 3H), 1.920–1.953 (m, 4H); 13C-NMR (100 MHz, CDCl3) 192.975, 162.718, 160.872, 143.621, 130.275, 125.357, 115.779, 91.608, 46.674, 25.689, 24.546; IR (neat) υmax 1755.53, 1658.71, 1612.71, 1548.12, 1504.15, 1403.49, 1251.05, 1138.16, 890.44, 696.38, 517.52; ESI-HRMS [M+H]+ calculated for C15H19N5, 269.3520, found 270.1700.
N-(2-chloro-5-fluoro-6-methylpyrimidin-4-yl)benzene-1,4-diamine (4.2).
3.2 (260 mg, 1.029 mmol, 1.0 equiv) was dissolved in DMF (29 mL) and treated with potassium carbonate (156.4 mg, 1.132 mmol, 1.1 equiv) and pyrrolidine (422.5 μL, 5.145 mmol, 5.0 equiv). The reaction was heated to 80 °C for 8h then diluted with ethyl acetate and washed with water and a 5% lithium chloride solution. The organic layer was dried over sodium sulfate, concentrated under reduced pressure, then purified via column chromatography using 10% methanol in ethyl acetate, to provide 4.2 (243 mg, 82% yield) as a brown solid. TLC (10% methanol/ethyl acetate), Rf = 0.57; m.p. 180–182 °C; 1H NMR (400 MHz, CDCl3) δ 7.467 (d, J=8.6 Hz, 2H), 6.674 (d, J=8.6 Hz, 2H), 6.436 (s, N-H), 3.497–3.530 (m, 4H), 2.271 (d, J=2.9 Hz, 3H), 1.914–1.947 (m, 4H); 13C-NMR (100 MHz, CDCl3)156.073, 149.566, 149.460, 149.193, 149.062, 142.081, 139.428, 139.056, 130.876, 121.623, 115.600, 47.019, 25.812, 17.361; IR (neat) υmax 3185.05, 1600.39, 1506.17, 1444.25, 1238.75, 826.91, 762.06, 509.83; ESI-HRMS [M+H]+ calculated for C15H18FN5, 287.3424, found 288.1606.
N-(5-fluoro-2-(pyrrolidin-1-yl)pyrimidin-4-yl)benzene-1,4-diamine (4.3).
3.3 (310 mg, 1.30 mmol, 1.0 equiv) was dissolved in DMF (36 mL) and treated with potassium carbonate (197.6 mg, 1.43 mmol, 1.1 equiv) and pyrrolidine (533.8 μL, 6.5 mmol, 5.0 equiv). The reaction was heated to 80 °C for 8h then diluted with ethyl acetate and washed with water and brine. The organic layer was dried over sodium sulfate and concentrated under reduced pressure to provide 4.3 as a dark yellow solid, which was carried on crude. TLC (10% methanol/dichloromethane), Rf = 0.59; m.p. 179–181 °C; 1H NMR (400 MHz, CDCl3) δ 7.851 (d, J=3.7 Hz, 1H), 7.469 (d, J=8.7 Hz, 2H), 6.682 (d, J=8.7 Hz, 2H), 6.494 (s, N-H), 3.497–3.530 (m, 4H), 1.934–1.967 (m, 4H); 13C-NMR (100 MHz, CDCl3)156.889, 149.984, 149.886, 142.406, 141.214, 139.910, 139.717, 138.808, 130.346, 122.189, 121.827, 115.572, 47.026, 37.755, 25.808; IR (neat) υmax 3388.87, 1598.56, 1568.68, 1500.63, 1447.20, 1227.27, 930.93, 831.66, 763.87, 496.87; ESI-HRMS [M+H]+ calculated for C14H16FN5, 273.3154, found 274.1450.
N-(4-((6-methyl-2-(pyrrolidine-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(thiophen-2-yl)acetamide (6.0)
4.1 (262.0 mg, 0.973 mmol, 1.0 equiv) was dissolved in dichloromethane (40 mL, anhydrous) then treated with 5.1 (145.3 mg, 1.02 mmol, 1.05 equiv), DMAP (118.9 mg, 0.973 mmol, 1.0 equiv), and then DCC (251 mg, 1.22 mmol, 1.25 equiv) under nitrogen. The reaction was allowed to stir for 8h then the material was concentrated onto silica gel and purified via column chromatography using 1:1 ethyl acetate:dichloromethane and 3 % methanol to provide compound 6.0 (302.8 mg, 79% yield) as a yellow solid. TLC (10% methanol/ethyl acetate), Rf = 0.49; m.p. 186–188 °C; 1H NMR (400 MHz, CDCl3) δ 7.611 (s, N-H), 7.389 (s, 4H), 7.273–7.289 (dd, 1H), 7.012–7.032 (m, 2H), 6.600 (s, N-H), 5.762 (s, 1H), 3.919 (s, 2H), 3.537–3.570 (m, 4H), 2.204 (s, 3H), 1.904–1.938 (s, 4H); 13C-NMR (100 MHz, CDCl3); 167.993, 166.584, 161.249, 160.656, 136.446, 135.883, 132.826, 127.768, 127.625, 126.049, 121.643, 120.996, 92.885, 46.727, 38.504, 25.626, 24.380; IR (neat) υmax 2862.14, 1572.75, 1500.37, 1398.29, 1330.32, 1226.59, 1169.02, 830.35, 782.81, 681.54, 513.32; ESI-HRMS [M+H]+ calculated for C21H23N5OS, 393.5090, found 394.1680.
N-(4-((5-fluoro-6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(thiophen-2-yl)acetamide (6.1).
4.2 (450 mg, 1.566 mmol, 1.0 equiv) was dissolved in DCM (15.0 mL, anhydrous) and treated with 2-thiopheneacetic acid (5.1) (244.9 mg, 1.723 mmol, 1.1 equiv), and triethylamine (546.4 μL, 3.915 mmol, 2.5 equiv). The reaction was allowed to stir for 5 min. then propanephosphonic acid anhydride (1.85 mL, 3.132 mmol, 2.0 equiv) was added and the reaction was allowed to stir for 15h. The reaction was then quenched with ice water and extracted with dichloromethane (x3). The organic layer was dried over sodium sulfate and concentrated under reduced pressure and purified via column chromatography using 10% methanol in ethyl acetate to provide 6.1 as a yellow solid (529 mg, 82% yield). TLC (10% methanol/ethyl acetate), Rf = 0.42; m.p. 245–247 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.093 (s, N-H), 8.950 (s, N-H), 7.763 (d, J=8.9 Hz, 2H), 7.493 (d, J=8.9 Hz, 2H), 7.375–7.391 (dd, 1H), 6.963–6.982 (m, 2H) 3.844 (s, 2H), 3.391–3.423 (m, 4H), 2.187 (d, J=2.9 Hz, 3H), 1.860–1.892 (m, 4H); 13C-NMR (100 MHz, DMSO-d6); 167.579, 155.240, 148.710, 149.054, 138.597, 137.281, 136.205, 135.432, 133.499, 126.622, 126.211, 124.986, 120.444, 119.297, 46.517, 37.493, 25.128, 17.109; IR (neat) υmax 3256.79, 1654.91, 1621.65, 1587.57, 1501.62, 1417.54, 1292.95, 1226.40, 826.57, 689.26, 548.39, 512.40; ESI-HRMS [M+H]+ calculated for C21H22FN5OS, 411.4994, found 412.1587.
N-(4-((5-fluoro-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(thiophen-2-yl)acetamide (6.2).
4.1 (50.0 mg, 0.183 mmol, 1.0 equiv) was dissolved in DCM (15.0 mL, anhydrous) and treated with 2-thiopheneacetic acid (5.1) (28.6 mg, 0.201 mmol, 1.1 equiv), and triethylamine (63.9 μL, 0.458 mmol, 2.5 equiv). The reaction was allowed to stir for 5 min. then propanephosphonic acid anhydride (0.194 mL, 0.366 mmol, 2.0 equiv) was added and the reaction was allowed to stir for 15h. The reaction was then quenched with ice water and extracted with dichloromethane (x3). The organic layer was dried over sodium sulfate and concentrated under reduced pressure and purified via column chromatography using 10% methanol in ethyl acetate to provide 6.1 as a yellow solid (56 mg, 77% yield). TLC (10% methanol/ethyl acetate), Rf = 0.61; m.p. 245–247 °C; 1H NMR (400 MHz, CD3OD) δ 10.112 (s, N-H), 9.110 (s, N-H), 7.955 (d, J=3.9 Hz, 1H), 7.778 (d, J=8.9 Hz, 2H), 7.511 (d, J=8.9 Hz, 2H), 7.377–7.393 (dd, 1H), 6.964–6.983 (m, 2H) 3.848 (s, 2H), 3.402–3.434 (m, 4H), 1.874–1.907 (m, 4H); 13C-NMR (100 MHz, CD3OD); 167.617, 156.244, 149.151, 149.046, 140.693, 140.541, 140.350, 138.276, 137.262, 135.08, 133.735, 126.627, 126.223, 124.994, 120.588, 119.306, 46.568, 37.498, 25.115; IR (neat) υmax 3256.79, 1654.91, 1621.65, 1587.57, 1501.62, 1417.54, 1292.95, 1226.40, 826.57, 689.26, 548.39, 512.40; ESI-HRMS [M+H]+ calculated for C20H20FN5OS, 397.4724, found 398.1431.
N-(4-((5-fluoro-6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)(methyl)amino)phenyl)-2-(thiophen-2-yl)acetamide (6.3).
6.1 (13 mg, 0.0316 mmol, 1.0 equiv) was dissolved in THF (1.0 mL, anhydrous) and treated with sodium hydride (1.52 mg, 0.0379 mmol, 1.2 equiv) at 0 °C under nitrogen. The reaction was allowed to stir for 10 min. then iodomethane (3.0 μL, 0.047 mmol, 1.5 equiv) was added and the reaction was allowed to stir for 15h. The reaction was then quenched with ice water and extracted with ethyl acetate (x3). The organic layer was dried over sodium sulfate and concentrated under reduced pressure and purified via 1000mm prep plate using 3% methanol in dichloromethane to provide 6.3 as a yellow oil (6.8 mg, 44% yield). TLC (5 % methanol/ dichloromethane), Rf = 0.62; m.p. 178–180 °C; 1H NMR (400 MHz, CDCl3) δ 7.795 (d, J=8.7 Hz, 2H), 7.154–7.166 (dd, 1H), 7.129 (d, J=8.7 Hz, 2H), 6.875–6.897 (m, 1H), 6.715–6.736 (m, 1H), 3.674 (s, 2H), 3.547–3.580 (m, 4H), 3.284 (s, 3H), 2.311 (s. 3H), 1.967–1.991 (m, 4H); 13C-NMR (100 MHz, CDCl3); 170.285, 155.868, 148.927, 139.213, 138.053, 137.039, 127.982, 126.533, 126.362, 126.270, 125.259, 124.801, 120.274, 47.142, 37.865, 35.280, 25.822, 17.541; IR (neat) υmax 1583.85, 1508.03, 1441.51, 1231.31, 910.69, 729.28; ESI-HRMS [M+H]+ calculated for C22H24FN5OS, 425.5264, found 426.1741.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-1H-indole-3-carboxamide (6.4).
Indole-3-carboxylic acid (11.6 mg, 0.0722 mmol, 1.2 equiv) was slurried with DIPEA (12.6 μL, 0.0722 mmol, 1.2 equiv) in DMF (anhydrous, 0.5 mL) and was treated with HBTU (27.4 mg, 0.0722 mmol, 1.2 equiv) in DMF (anhydrous, 0.2 mL). The reaction was allowed to stir for 15 min at RT then 4.1 (16.2 mg, 0.0602 mmol, 1.0 equiv) in DMF (anhydrous, 0.2 mL) was added dropwise and the reaction was allowed to continue stirring for another 8h at RT. The reaction was diluted with dichloromethane and washed with water and brine. The organic layer was dried over sodium sulfate and concentrated onto silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.4 as a white solid (7.5 mg, 25% yield). TLC (3 % methanol/ dichloromethane), Rf = 0.20; m.p. 182–184 °C; 1H NMR (400 MHz, CDCl3) δ 8.182 (d, J=7.7 Hz, 1H), 7.973 (s, 1H), 7.896 (s, N-H), 7.662 (s, 4H), 7.450 (d, J=7.7 Hz, 1H), 7.155–7.238 (m, 2H), 5.963 (s, 1H), 3.574–3.594 (m, 4H), 2.274 (s, 3H), 2.022–2.050 (m, 4H); 13C-NMR (100 MHz, CDCl3); 166.616, 164.856, 162.165, 138.153, 136.427, 135.951, 129.349, 127.596, 123.682, 122.418, 122.314, 122.137, 122.074, 112.812, 112.005, 97.046, 79.467, 36.944, 31.641, 26.260, 20.453; IR (neat) υmax 1505.51, 1232.99, 1176.73, 833.69, 743.65, 552.85; ESI- HRMS [M+H]+ calculated for C24H24N6O, 412.4970, found 413.2066.
tert-butyl (4-((2-chloro-6-methylpyrimidin-4-yl)amino)phenyl)carbamate (8).
2,4-dichloro-6-methylpyrimidine (2.1) (2.36g, 0.0145 mol, 1.05 equiv) was dissolved in 30 mL of absolute ethanol and cooled with an ice bath and triethyl amine (2.5 mL, 0.0179 mol, 1.3 equiv) was added. tert-butyl (4-aminophenyl)carbamate (2.87g, 0.0138 mol, 1.0 equiv) was dissolved in 15 mL of absolute ethanol and transferred to an additional funnel. The aniline was added dropwise and the reaction was allowed to warm to room temperature. After 24h, the reaction was heated to 40 °C until the reaction was complete. The solvent was removed under reduced pressure and purified via column chromatography using 5–50% ethyl acetate in hexanes to produce 8 (3.88g, 80% yield) as an orange solid. TLC (20% ethyl acetate/dichloromethane), Rf = 0.49; m.p. 109–111 °C; 1H NMR (400 MHz, CD3OD) δ 7.371–7.437 (m, 4H), 6.446 (s, 1H), 2.281 (s, 3H), 1.513 (s, 9H); 13C-NMR (100 MHz, CD3OD) 168.015, 163.907, 160.978, 155.334, 137.049, 134.613, 123.140, 120.421, 80.832, 54.787, 28.713, 23.136; IR (neat) υmax 1723.38, 1591.78, 1518.26, 1398.75, 1310.28, 1221.08, 1152.21, 1024.67, 970.18, 910.69, 835.64, 735.57, 515.54; ESI-HRMS [M+H]+ calculated for C16H19ClN4O2, 334.8040, found 335.1257.
tert-butyl (4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl) amino)phenyl)carbamate (9.1).
8 (15.16 g, 0.0454 mol, 1.0 equiv) was dissolved in 60 mL anhydrous DMF. Potassium Carbonate (6.59 g, 0.0477 mol, 1.1 equiv) was added followed pyrrolidine (15.15 mL, 0.181 mol, 4.0 equiv) and the reaction was heated to 80 °C for 8h. The reaction was then diluted with ethyl acetate and washed with water then brine. The organic layer was dried over sodium sulfate and concentrated under reduced vacuum, resulting in an orange solid. The crude material was run through a plug of silica gel with 5% methanol in dichloromethane, and concentrated to produce carbamate 9.1 (15.21 g, 91% yield) as a yellow solid. TLC (3% methanol/dichloromethane), Rf = 0.28; m.p. 118–120 °C; 1H NMR (400 MHz, CDCl3) δ 7.303–7.352 (m, 4H), 6.428 (s, N-H), 6.294 (s, N-H), 5.767 (s, 1H), 3.532–3.609 (m, 4H), 2.219 (s, 3H), 1.928–1.961 (m, 4H), 1.520 (s, 9H); 13C-NMR (100 MHz, CDCl3) 166.567, 161.606, 160.709, 153.112, 134.881, 134.083, 122.437, 119.567, 92.520, 80.474, 46.660, 28.447, 25.602, 24.318; IR (neat) υmax 2971.67, 1718.69, 1569.84, 1505.67, 1399.06, 1227.56, 1155.05, 1050.05, 750.41, 515.91; ESI-HRMS [M+H]+ calculated for C20H27H5O2, 369.4690, found 370.2225.
tert-butyl (4-((6-methyl-2-morpholinopyrimidin-4-yl)amino)phenyl)carbamate (9.2).
8 (199.0 mg, 0.594 mmol, 1.0 equiv) was dissolved in acetone (3.4 mL) and cooled with an ice bath. Sodium carbonate (69.3 mg, 0.653 mmol, 1.1 equiv) was added followed by morpholine (53.0 μL, 0.612 mmol, 1.03 equiv) in 1.0 mL of acetone, dropwise. The ice bath was removed and the reaction was heated to 80 °C for 8h. The reaction was diluted with ethyl acetate, then washed with water and brine. The organic layer was dried over sodium sulfate, concentrated under reduced pressure, then purified via column chromatography using 1% methanol in dichloromethane to produce 9.2 (122.88 mg, 54% yield) as a white solid. TLC (5% acetone in dichloromethane), Rf = 0.18; m.p. 226–228 °C; 1H NMR (400 MHz, CDCl3) δ 7.317–7.349 (m, 2H), 7.243–7.264 (m, 2H), 6.465 (s, N-H), 6.314 (s, N-H), 5.816 (s, 1H), 3.742–3.765 (m, 8H), 2.203 (s, 3H), 1.520 (s, 9H); 13C-NMR (100 MHz, CDCl3) 167.048, 162.072, 161.944, 153.257, 134.747, 134.264, 123.308, 119.610, 93.338, 80.766, 67.148, 44.511, 28.500, 24.474; IR (neat) υmax 2947.41, 1702.51, 1579.06, 1489.70, 1357.90, 1230.06, 1155.92, 1004.35, 811.14, 746.29, 516.15; ESI-HRMS [M+H]+ calculated for C20H27N5O3, 385.4680, found 386.2170.
2-(1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)acetamide (6.5).
4.1 (70.0 mg, 0.260 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated with DMAP (31.8 mg, 0.260 mmol, 1.0 equiv), DCC (67.1 mg, 0.325 mmol, 1.25 equiv), and 2-(1H-indol-3-yl)acetic acid (47.8 mg, 0.273 mmol, 1.05 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.5 (92.0 mg, 83 % yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.24; m.p. 197–199 °C; 1H NMR (400 MHz, CD3OD) δ 7.894 (s, N-H), 7.603–7.638 (m, 3H), 7.429 (d, J=8.9 Hz, 2H), 7.357 (d, J=8.2 Hz, 1H), 7.226 (s, 1H), 7.109 (t, 1H), 7.028 (t, 1H), 5.831 (s, 1H), 3.807 (s, 2H), 3.527–3.560 (m, 4H), 2.187 (s, 3H), 1.969–1.978 (m, 4H); 13C-NMR (100 MHz, CD3OD) 172.991, 162.612, 161.617, 138.384, 138.140, 134.053, 128.607, 124.802, 122.560, 121.985, 121.160, 119.975, 119.411, 112.328, 109.563, 95.337, 79.466, 47.804, 34.895, 26.455, 23.433; IR (neat) υmax 1503.05, 1401.52, 1202.57, 828.19, 738.45, 512.00; ESI-HRMS [M+H]+ calculated for C25H26N6O, 426.5240, found 427.2220.
3-(1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) propanamide (6.6).
4.1 (30.1 mg, 0.112 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated with DMAP (13.7 mg, 0.112 mmol, 1.0 equiv), DCC (28.9 mg, 0.14 mmol, 1.25 equiv), and 3-(1H-indol-3-yl)propanoic acid (23.3 mg, 0.123 mmol, 1.1 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.6 (38.3 mg, 78% yield) as a light orange solid. TLC (3% methanol/dichloromethane), Rf = 0.20; m.p. 89–90 °C; 1H NMR (400 MHz, CD3OD) δ 7.888 (s, N-H), 7.558–7.629 (m, 3H), 7.399 (d, J=8.9 Hz, 2H), 7.317 (d, J=8.0 Hz, 1H), 7.061–7.097 (m, 2H), 6.979–7.015 (t, 1H), 5.834 (s, 1H), 3.529–3.562 (m, 4H), 3.127–3.165 (m, 2H), 2.700–2.738 (t, 2H), 2.190 (s, 3H), 1.947–1.980 (m, 4H); 13C-NMR (100 MHz, CD3OD) 174.149, 166.029, 162.608, 161.578, 138.254, 138.174, 134.099, 128.543, 122.992, 122.286, 121.974, 121.142, 119.538, 119.330, 115.073, 112.169, 95.353, 79.460, 47.808, 39.114, 26.452, 23.426, 22.627; IR (neat) υmax 1570.80, 1503.13, 1399.39, 1227.17, 791.94, 738.86, 514.31; ESI-HRMS [M+H]+ calculated for C26H28N6O, 440.5510, found 441.2382.
4-(1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)butanamide (6.7).
4.1 (26.7 mg, 0.0992 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated with DMAP (12.1 mg, 0.0992 mmol, 1.0 equiv), DCC (25.6 mg, 0.124 mmol, 1.25 equiv), and 4-(1H-indol-3-yl)butanoic acid (20.2 mg, 0.109 mmol, 1.1 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.7 (43 mg, 95% yield) as a light orange solid. TLC (5% methanol/dichloromethane), Rf = 0.33; m.p. 157–159 °C; 1H NMR (400 MHz, CD3OD) δ 7.656 (s, N-H), 7.415 (d, J=8.9 Hz, 2H), 7.324 (d, J=7.9 Hz, 1H), 7.223 (d, J=8.9 Hz, 2H), 7.100 (d, J=8.0 Hz, 1H), 6.750–6.875 (m, 3H), 5.609 (s, 1H), 3.250–3.350 (m, 4H), 2.616 (t, 2H), 2.188 (t, 2H), 1.967 (s, 3H), 1.877 (m, 2H), 1.660–1.750 (m, 4H); 13C-NMR (100 MHz, CD3OD) 174.403, 165.762, 162.519, 161.371, 138.179, 138.136, 134.163, 128.769, 122.968, 122.176, 121.839, 121.154, 119.419, 119.384, 115.626, 112.132, 95.418, 79.435, 47.780, 37.599, 28.738, 27.761, 26.403, 25.708, 23.363; IR (neat) υmax 2861.61, 1570.45, 1503.18, 1398.69, 1229.00, 785.54, 738.42, 511.98; ESI-HRMS [M+H]+ calculated for C27H30N6O, 454.5780, found 455.2534.
(E)-3-(1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acrylamide (6.8).
4.1 (19.0 mg, 0.0706 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated wthi(E)-3-(1H-indol-3-yl)acrylic acid (13.2 mg, 0.0706 mmol, 1.0 equiv), HBTU (34.8 mg, 0.0918 mmol, 1.3 equiv), and DIPEA (25.0 mL, 0.141 mmol, 1.1 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was diluted with ethyl acetate and washed with water and brine. The organic layer was dried over sodium sulfate and concentrated, then purified via column chromatography using 0–10% methanol in dichloromethane to produce the desired product 6.8 (20 mg, 63 % yield) as a yellow oil. TLC (3% methanol/dichloromethane, run twice), Rf = 0.33; m.p. 188–190 °C; 1H NMR (400 MHz, CD3OD) δ 7.948 (d, J=7.9 Hz, 1H), 7.880 (d, J=15.6 Hz, 1H), 7.669 (s, 4H), 7.626 (s, 1H), 7.446 (d, J=7.9 Hz, 1H), 7.176–7.252 (m, 2H), 6.746 (d, J=15.6 Hz, 1H), 5.986 (s, 1H), 3.539–3.630 (bs, 4H), 2.296 (s, 3H), 2.072 (bs, 4H); 13C-NMR (100 MHz, CD3OD) 168.323, 162.306, 139.243, 137.042, 135.967, 135.664, 131.257, 126.624, 123.692, 121.838, 121.798, 121.453, 121.117, 116.081, 114.168, 113.084, 96.286, 79.465, 36.938, 31.638 26.347, 21.908; IR (neat) υmax 3107.31, 1581.55, 1508.82, 1403.71, 1343.98, 1241.42, 1180.48, 829.45, 743.93; ESI-HRMS [M+H]+ calculated for C26H26N6O, 438.5330, found 439.2223.
1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)- 1H-pyrrolo[2,3-b]pyridine-3-carboxamide (6.9).
4.1 (13.2 mg, 0.049 mmol, 1.0 equiv) was dissolved in anhydrous DFM (0.5 mL) then treated with 1H-pyrrolo[2,3-b]pyridine-3-carboxylic acid (8.0 mg, 0.049 mmol, 1.0 equiv), HBTU (21.4 mg, 0.056 mmol, 1.15 equiv), and DIPEA (9.8 mL, 0.056 mmol, 1.15 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was diluted with ethyl acetate and washed with water and brine. The organic layer was dried over sodium sulfate and concentrated, then purified via column chromatography using 0–10% methanol in dichloromethane to produce the desired product 6.9 (17.4 mg, 86 % yield) as a yellow solid. TLC (3% methanol/dichloromethane), Rf= 0.24 (3%methanol/dichloromethane); m.p. 295–297 °C; 1H NMR (400 MHz, CD3OD) δ 12.234 (s, N-H), 8.482–8.505 (m, 1H), 8.440 (s, 1H), 8.296–8.311 (m, 1H), 7.692 (s, 4H), 7–195-7.226 (dd, 1H), 5.926 (s, 1H), 3.509 (bs, 4H), 2.177 (s, 3H), 1.924 (bs, 4H); 13C-NMR (100 MHz, CDCl3) 162.263, 160.458, 148.461, 143.662, 129.324, 128.763, 128.564, 120.305, 120.199, 120.197, 119.935, 118.752, 117.094, 109.395, 99.522, 46.569, 24.964; IR (neat) υmax 3297.80, 1661.79, 1576.88, 1501.18, 1450.36, 1338.97, 1221.50, 1012.59, 825.14, 689.68, 596.71, 510.91; ESI-HRMS [M+H]+ calculated for C23H23N7O, 413.4850, found 414.2022.
N-(4-((5-methoxy-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(thiophen-2-yl)acetamide (6.10).
The synthesis of 6.10 utilized scheme 1 methodology and the following starting materials (SI Scheme S3). 4.4 (31.0 mg, 0.109 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated with DMAP (13.3 mg, 0.109 mmol, 1.0 equiv), DCC (35.5 mg, 0.136 mmol, 1.25 equiv), and 2-(thiophen-2-yl)acetic acid (15.5 mg, 0.109 mmol, 1.0 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.10 (30.8 mg, 70% yield) as a light orange solid. TLC (8% methanol/dichloromethane), Rf = 0.4; m.p. 226–228 °C; 1H NMR (400 MHz, CDCl3) δ 7.716 (d, J=8.9 Hz, 2H), 7.665 (s, 1H), 7.410 (d, J=8.9 Hz, 2H), 7.345 (s, 1H), 7.298–7.315 (dd, 1H), 7.083 (s, 1H), 7.039–7.058 (m, 2H), 3.948 (s, 2H), 3.819 (s, 3H), 3.519–3.551 (m, 4H), 1.945–1.977 (m, 4H); 13C-NMR (100 MHz, CDCl3) 167.786, 155.389, 151.795, 136.178, 135.906, 132.876, 132.313, 127.966, 127.763, 126.215, 120.652, 119.885, 57.190, 47.047, 38.617, 31.086, 25.796; IR (neat) υmax 3297.80, 1661.79, 1576.88, 1501.18, 1450.36, 1338.97, 1221.50, 1012.59, 825.14, 689.68, 596.71, 510.91; ESI-HRMS [M+H]+ calculated for C21H23N5O2S, 409.5080, found 410.1631.
2-(4-bromothiophen-2-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.11).
4.1 (2.33 g, 0.00866 mol, 1.0 equiv) was dissolved in anhydrous dichloromethane (100 mL) then treated with DMAP (1.06 g, 0.00866 mol, 1.0 equiv), DCC (2.23 g, 0.0108 mol, 1.25 equiv), and 2-(4-bromothiophen-2-yl)acetic acid (2.11 g, 0.00952 mol, 1.1 equiv). The reaction was allowed to stir for 24h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.11 (3.76 mg, 92% yield) as a brown solid. TLC (8% methanol/dichloromethane), Rf = 0.42; m.p. 195–197 °C; 1H NMR (400 MHz, CDCl3) δ 7.386–7.434 (m, 4H), 7.325 (s, N-H), 7.197 (s, 1H), 6.957 (s, 1H), 6.370 (s, N-H), 5.777 (s, 1H), 3.885 (s, 2H), 3.547–3.580 (m, 4H), 2.224 (s, 3H), 1.925–1.959 (m, 4H); 13C-NMR (100 MHz, CDCl3) 166.809, 161.075, 137.195, 136.470, 132.471, 130,052, 123.159, 121.889, 121.607, 120.978, 109.899, 99.980, 92.628, 46.618, 38.423, 25.531, 24.357; IR (neat) υmax 2862.68, 1568.07, 1501.64, 1400.13, 1333.38, 1231.08, 785.83, 563.89; ESI-HRMS [M+H]+ calculated for C21H22BrN5OS, 471.0728, found 472.0787.
N-(4-((2-chloro-6-methylpyrimidin-4-yl)oxy)phenyl)-2-(thiophen-2-yl)acetamide (6.12).
11.1 (63.0 mg, 0.27 mool, 1.0 equiv) was dissolved in ethanol (absolute, 3 mL) then treated with 2.1 (44.1 mg, 0.270 mmol, 1.0 equiv), potassium carbonate (44.8 mg, 0.324 mmol, 1.2 equiv) then a crystal of KI. The reaction was allowed to stir at room temperature for 8h, which was then concentrated onto silica gel and purified via column chromatography using 0–3% methanol in dichloromethane to produce 6.12 (92.8 mg, 96% yield) was found as an off-white solid. TLC (3% methanol/dichloromethane), Rf = 0.45; m.p. 176–177 °C; 1H NMR (400 MHz, CDCl3) δ 7.566 (s, N-H), 7.494 (d, J=8.9 Hz, 2H), 7.287–7.304 (dd, 1H), 7.018–7.063 (m, 4H), 6.564 (s, 1H), 3.937 (s, 2H), 2.453 (s, 3H); 13C-NMR (100 MHz, CDCl3) 171.760, 170.920, 168.118, 160.102, 148.396, 135.564, 135.517, 127.912, 127.728, 126.211, 122.103, 121.936, 121.570, 121.238, 115.629, 105.169, 38.549, 24.044; IR (neat) υmax 1655.62, 1578.47, 1507.89, 1323.18, 1207.23, 846.34, 793.77, 690.66, 481.83; ESI-HRMS [M+H]+ calculated for C17H14ClN3O2S, 359.8280, found 360.0552.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)oxy)phenyl)-2-(thiophen-2-yl)acetamide (6.13).
6.12 (44.3 mg, 0.123 mmol, 1.0 equiv) was dissolved in DMF (anhydrous, 1 mL) and then treated with potassium carbonate (20.3 mg, 0.148 mmol, 1.2 equiv) and pyrrolidine (20.6 mL, 0.247 mmol, 2.0 equiv). The mixture was heated to 80 °C for 8h, then diluted in dichloromethane and washed with water then brine. The organic layer was dried over sodium sulfate, concentrated then purified via column chromatography using 30–60% ethyl acetate in hexanes to provide 6.13 (8.5 mg, 18% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.43; m.p. 187–189 °C; 1H NMR (400 MHz, CDCl3) δ 7.449 (d, J= 8.9 Hz, 2H), 7.310–7.350 (m, 1H), 7.049–7.102 (m, 3H), 5.727 (s, 1H), 3.960 (s, 2H), 3.484 (bs, 4H), 2.258 (s, 3H), 1.882–1.936 (m, 4H); 13C-NMR (100 MHz, CDCl3) 170.292, 169.595, 167.758, 160.509, 149.532, 135.530, 134.331, 127.895, 127.678, 126.174, 122.203, 120.974, 93.152, 46.590, 38.484, 25.418, 24.404; IR (neat) υmax 1655.18, 1575.53, 1503.62, 1330.12, 1204.97, 961.26, 844.36, 792.73, 688.69, 567.34, 519.75, 480.84; ESI-HRMS [M+H]+ calculated for C17H14ClN3O2S, 394.4930, found 395.1522.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)oxy)phenyl)-1H-indole-3-carboxamide (6.14).
11.2 (9.0 mg, 0.024 mmol, 1.0 equiv) was dissolved in 1.0 mL anhydrous DMF. Potassium Carbonate (4.3 mg, 0.0312 mol, 1.3 equiv) was added followed pyrrolidine (10 mL, 0.119 mmol, 5.0 equiv) and the reaction was heated to 80 °C for 8h. The reaction was then diluted with ethyl acetate and washed with water then brine. The organic layer was dried over sodium sulfate and concentrated under reduced vacuum, resulting in an orange solid. The crude material was run through a plug of silica gel with 5% methanol in dichloromethane and concentrated to produce 6.14 (4.1 mg, 40% yield) as a yellow solid. TLC (3% methanol/dichloromethane), Rf = 0.20; m.p. 172–174 °C; 1H NMR (400 MHz, CDCl3) δ 8.710 (s, N-H), 8.054–8.094 (m, 1H), 7.886 (d, J=4.0 Hz, 1H), 7.733 (s, N-H), 7.678 (d, J=8.9 Hz, 2H), 7.457–7.500 (m, 1H), 7.300–7.343 (m, 2H), 7.173 (d, J= 8.9 Hz, 2H), 5.778 (s, 1H), 3.522 (bs, 4H), 2.281 (s, 3H), 1.909–1.943 (m, 4H); 13C-NMR (100 MHz, CDCl3) 170.631, 169.798, 163.563, 149.229, 136.539, 135.440, 128.389, 124.854, 123.429, 122.408, 122.143, 121.465, 120.113, 112.729, 112.252, 93.360, 60.557, 46.804, 25.581, 24.551, 21.214, 14.348; IR (neat) υmax 3325.82, 1575.09, 1506.16, 1425.63, 1248.26, 1094.00, 949.73, 808.19, 775.56, 487.07; ESI-HRMS [M+H]+ calculated for C24H23N5O2, 413.4810, found 414.1908.
N-(4-((6-methyl-2-morpholinopyrimidin-4-yl)amino)phenyl)-2-(thiophen-2-yl)acetamide (6.15).
4.1 (17.5 mg, 0.061 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated with DMAP (7.5 mg, 0.061 mmol, 1.0 equiv), DCC (19.9 mg, 0.0763 mmol, 1.25 equiv), and 5.1 (9.1 mg, 0.064 mmol, 1.05 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.15 (14 mg, 56% yield) as a dark solid. TLC (5% methanol/dichloromethane), Rf = 0.60; m.p. 196–198 °C; 1H NMR (400 MHz, CD3OD) δ 7.454–7.522 (q, 4H), 7.270 (d, J=5.7 Hz, 1H), 6.995 (bs, 1H), 6.952–6.974 (t, 1H), 5.907 (s, 1H), 3.870 (s, 2H), 3.723 (s, 8H), 2.190 (s, 3H); 13C-NMR (100 MHz, CD3OD) 170.716, 166.644, 163.165, 162.779, 137.897, 134.192, 127.716, 127.482, 125.742, 121.808, 121.653, 96.230, 67.886, 45.856, 38.672, 23.833; IR (neat) υmax 2842.93 1654.21, 1579.80, 1545.32, 1486.92, 1439.58, 1398.31, 1354.51, 1232.28, 1104.29, 993.25, 830.14, 786.59, 696.36, 483.49; ESI-HRMS [M+H]+ calculated for C21H23N5O2S, 409.5080, found 410.1630.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(naphthalen-1-yl)acetamide (6.16).
4.1 (33.6 mg, 0.125 mmol, 1.0 equiv) was dissolved in anhydrous dichloromethane (1.5 mL) then treated with DMAP (15.3 mg, 0.125 mmol, 1.0 equiv), DCC (32.2 mg, 0.156 mmol, 1.25 equiv), and 2-(naphthalen-1-yl) acetic acid (25.6 mg, 0.137 mmol, 1.1 equiv). The reaction was allowed to stir for 12h at room temperature. Upon completion, the reaction was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol produced the desired product 6.16 (22 mg, 40% yield) as an off white solid. TLC (3% methanol/dichloromethane, run twice), Rf = 0.5; m.p. 275–278 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.168 (s, N-H, 1H), 8.946 (s, N-H, 1H), 8.144 (d, J=8.1 Hz, 1H), 7.937 (d, J=7.9 Hz, 1H), 7.830–7.855 (dd, 1H), 7.620 (d, J=8.9 Hz 1H), 7.457–7.582 (m, 6H), 5.824 (s, 1H), 4.121 (s, 2H), 3.435–3.467 (m, 4H), 2.113 (s, 3H), 1.864–1.896 (m, 4H); 13C NMR (100 MHz, DMSO-d6) 168.533, 164.598, 160.633, 159.945, 136.439, 133.358, 132.978, 132.628, 131.997, 128.411, 127.776, 127.161, 126.065, 125.671, 125.539, 124.213, 119.633, 119.483, 93.799, 46.185, 40.620, 25.019, 23.938; IR (neat) υmax 1660.02, 1575.10, 1503.95, 1401.14, 1227.15, 787.35, 558.68; ESI-HRMS [M+H]+ calculated for C27H27N5O, 437.5470, found 438.2273.
2-(6-chloro-1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.18).
4.1 (25.8 mg, 0.096 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (12.8 mg, 0.105 mmol, 1.1 equiv), DCC (24.8 mg, 0.12 mmol, 1.25 equiv) and 2-(6-chloro-1H-indol-3-yl)acetic acid (22.1 mg, 0.105 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at RT. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.18 (25.4 mg, 57% yield) as an off white solid. TLC (2% Methanol, 20% acetone, 78% dichloromethane), Rf = 0.57; m.p. 146–148 °C; 1H NMR (400 MHz, CD3OD) δ 7.632 (d, J=8.9 Hz, 2H), 7.568 (d, J=8.5 Hz, 1H), 7.438 (d, J=8.9 Hz, 2H), 7.354 (d, J=2.0 Hz, 1H), 7.242 (s, 1H), 6.994–7.019 (dd, 1H), 5.825 (s, 1H), 3.784 (s, 2H), 3.529–3.563 (m, 4H), 2.189 (s, 3H), 1.949–1.983 (m, 4H); 13C-NMR (100 MHz, CD3OD) 172.615, 162.616, 159.239, 138.467, 138.412, 134.068, 128.481, 127.362, 125.759, 121.970, 121.184, 120.604, 120.468, 112.127, 110.057, 95.356, 76.019, 47.820, 34.666, 26.458, 23.418; IR (neat) υmax 1660.51, 1571.67, 1505.47, 1399.30, 795.55; ESI-HRMS [M+H]+ calculated for C25H25ClN6O, 460.9660, found 461.1834.
1-methyl-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-1H-indole-3-carboxamide (6.19).
4.1 (60 mg, 0.223 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (30 mg, 0.245 mmol, 1.1 equiv), DCC (57.5 mg, 0.279 mmol, 1.25 equiv) and 1-methyl-1H-indole-3-carboxylic acid (39 mg, 0.223 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at RT. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.19 (23.6 mg, 25% yield) as a light yellow solid. TLC (5% methanol/dichloromethane, run twice), Rf = 0.58; m.p. 262–264 °C; 1H NMR (400 MHz, CD3OD) δ 8.186 (d, J=8.1 Hz, 1H), 7.945 (s, 1H), 7.679 (d, J=8.9 Hz, 2H), 7.571 (d, J=8.9 Hz, 2H), 7.435 (d, J=8.1 Hz, 1H), 7.250–7.291 (t, 1H), 7.187–7.227 (t, 1H), 5.857 (s, 1H), 3.855 (s, 3H), 3.544–3.577 (m, 4H), 2.201 (s, 3H), 1.958–1.991 (m, 4H); 13C-NMR (100 MHz, CD3OD) 166.950, 165.212, 162.641, 161.568, 138.775, 138.012, 134.530, 133.132, 128.188, 123.729, 122.537, 122.328, 121.277, 111.161, 110.925, 95.376, 79.464, 47.819, 36.937, 33.480, 26.463, 23.422; IR (neat) υmax 3307.96, 1571.45, 1502.60, 1399.70, 1227.73, 1100.50, 741.44; ESI-HRMS [M+H]+ calculated for C25H26N6O, 426.5240, found 427.2227.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(1H-pyrrolo[2,3-b]pyridine-3-yl)acetamide (6.20)
4.1 (25.2 mg, 0.0936 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (12.6 mg, 0.103 mmol, 1.1 equiv), DCC (24.1 mg, 0.117 mmol, 1.25 equiv) and 2-(1H-pyrrolo[2,3-b]pyridin-3-yl)acetic acid (39 mg, 0.223 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.20 (36.0 mg, 90% yield) as a white solid. TLC (20% acetone, 2% methanol, 78% dichloromethane, run twice), Rf = 0.35; m.p. 236–238 °C; 1H NMR (400 MHz, CD3OD) δ 8.183 (d, J=5.1 Hz, 1H), 8.092 (d, J=7.9 Hz, 1H), 7.831 (s, 1H), 7.629 (d, J=9.0 Hz, 2H), 7.460 (d, J=9.0 Hz, 2H), 7.092–7.124 (dd, 1H), 5.849 (s, 1H), 3.818 (s, 2H), 3.545–3.578 (m, 4H), 2.204 (s, 3H), 1.965–1.999 (m, 4H); 13C-NMR (100 MHz, CD3OD) 172.217, 162.578, 149.416, 143.291, 138.215, 134.262, 129.094, 125.774, 121.924, 121.851, 121.317, 116.466, 109.195, 101.393, 95.543, 47.878, 34.712, 26.440, 23.082; IR (neat) υmax 2864.93, 1572.18, 1502.48, 1398.08, 1229.57, 753.09, 515.55; ESI-HRMS [M+H]+ calculated for C24H25N7O, 427.5120, found 428.2178.
2-(2-methyl-1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.21).
4.1 (20.8 mg, 0.0773 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (9.44 mg, 0.0773 mmol, 1.1 equiv), DCC (19.9 mg, 0.0966 mmol, 1.25 equiv) and 2-(2-methyl-1H-indol-3-yl)acetic acid (15.4 mg, 0.0773 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.21 (24.1 mg, 73% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.42; m.p. 142–144 °C; 1H NMR (400 MHz, CD3OD) δ 7.603 (d, J=8.9 Hz, 2H), 7.481 (d, J=7.6 Hz, 1H), 7.394 (d, J=8.9 Hz, 2H), 7.249 (d, J=7.6 Hz, 1H), 6.947–7.038 (m, 2H), 5.802 (s, 1H), 3.739 (s, 2H), 3.492–3.525 (m, 4H), 2.417 (s, 3H), 2.168 (s, 3H), 1.908–1.941 (m, 4H); 13C-NMR (100 MHz, CD3OD). 173.092, 165.696, 162.506, 161.309, 138.322, 137.089, 134.671, 133.965, 129.856, 122.078, 121.634, 121.134, 119.853, 118.485, 111.417, 105.226, 95.441, 47.791, 33.567, 26.409, 23.316, 11.539; IR (neat) υmax 1571.52, 1505.98, 1399.07, 741.35; ESI-HRMS [M+H]+ calculated for C26H28N6O, 440.5510, found 441.2381.
2-(5-methoxy-1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.22).
4.1 (21.6 mg, 0.0802 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (9.80 mg, 0.0802 mmol, 1.1 equiv), DCC (20.7 mg, 0.100 mmol, 1.25 equiv) and 2-(5-methoxy-1H-indol-3-yl)acetic acid (17.3 mg, 0.0843 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.22 (15.5 mg, 44% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.55; m.p. 167–169 °C; 1H NMR (400 MHz, CD3OD) δ 7.632 (d, J=8.9 Hz, 2H), 7.440 (d, J=8.9 Hz, 2H), 7.248 (d, J=8.7 Hz, 1H), 7.199 (s, 1H), 7.121 (d, J=2.0 Hz 1H), 6.760–6.788 (dd, 1H), 5.844 (s, 1H), 3.802 (s, 3H), 3.775 (s, 2H), 3.552 (bm, 4H), 2.195 (s, 3H), 1.974 (bm, 4H); 13C-NMR (100 MHz, CD3OD); 173.054, 162.612, 155.223, 137.780, 134.158, 133.356, 128.899, 125.518, 121.988, 121.257, 113.029, 112.906, 109.361, 101.367, 95.418, 79.464, 56.255, 47.828, 35.031, 26.454, 23.724, 20.049; IR (neat) υmax 2864.55, 1573.52, 1504.15, 1398.88, 1224.02, 790.75, 513.02; ESI-HRMS [M+H]+ calculated for C26H28N6O2, 456.5500, found 457.2329.
2-(5-bromo-1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.23).
4.1 (17.0 mg, 0.0632 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (7.72 mg, 0.0632 mmol, 1.1 equiv), DCC (16.3 mg, 0.079 mmol, 1.25 equiv) and 2-(5-bromo-1H-indol-3-yl)acetic acid (17.7 mg, 0.0695 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.23 (30.7 mg, 99% yield) as a white solid. TLC (5% methanol/dichloromethane), Rf = 0.47; m.p. 151–153 °C; 1H NMR (400 MHz, CD3OD) δ 7.899 (s, 1H), 7.786 (d, J=1.9 Hz, 1H), 7.641 (d, J=8.9 Hz, 2H), 7.448 (d, J=8.9 Hz 2H), 7.264–7.272 (m, 2H), 7.183–7.210 (dd, 1H), 5.843 (s, 1H), 3.769 (s, 2H), 3.518–3.598 (m, 4H), 2.193 (s, 3H), 1.948–2.003 (m, 4H); 13C-NMR (100 MHz, CD3OD) 172.565, 162.617, 161.530, 155.127, 138.382, 136.716, 134.086, 130.445, 126.290, 125.261, 122.170, 122.017, 121.233, 113.973, 113.149, 109.537, 95.377, 47.823, 34.618, 26.455, 23.383; IR (neat) υmax 2862.30, 1570.94, 1504.59, 1399.41, 1225.39, 789.49, 513.22; ESI-HRMS [M+H]+ calculated for C25H25BrN6O, 504.4200, found 505.1327.
2-(1H-indol-1-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)acetamide (6.24).
4.1 (16.6 mg, 0.0617 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (7.5 mg, 0.0617 mmol, 1.1 equiv), DCC (16.0 mg, 0.077 mmol, 1.25 equiv) and 2-(1H-indol-1-yl)acetic acid (11.3 mg, 0.0648 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.24 (12.0 mg, 47% yield) as an off white solid. TLC (3% methanol/dichloromethane), Rf = 0.32; m.p. 275–277 °C; 1H NMR (400 MHz, CD3OD) δ 7.121 (d, J=7.8 Hz, 1H), 6.887 (d, J=8.9 Hz, 2H), 6.756–6.825 (m, 4H), 6.708 (t, 1H), 6.599–6.650 (m, 2H), 6.087 (d, J=3.2 Hz, 1H), 5.230 (s, 1H), 4.381 (s, 2H), 2.960–2.993 (m, 3H), 1.642 (s, 3H), 1.377–1.410 (m, 4H); 13C-NMR (100 MHz, CD3OD) 173.133, 162.616, 138.337, 136.690, 136.060, 134.116, 128.780, 124.921, 123.207, 122.022, 121.234, 117.835, 112.158, 109.187, 95.39434.965, 30.173, 26.451, 23.333, 17.153; IR (neat) υmax 1668.49, 1576.69, 1502.37, 1400.50, 1330.94, 1227.86, 794.97, 735.87, 568.62, 509.47; ESI-HRMS [M+H]+ calculated for C25H26N6O, 426.5240, found 427.2224.
2-(5-hydroxy-1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.25).
4.1 (44.2 mg, 0.164 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (20.0 mg, 0.164 mmol, 1.0 equiv), DCC (42.3 mg, 0.205 mmol, 1.25 equiv) and 2-(5-hydroxy-1H-indol-3-yl)acetic acid (33.0 mg, 0.173 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.25 (15.5 mg, 21% yield) as a white solid. TLC (5% methanol/dichloromethane), Rf = 0.28; m.p. 196–198 °C; 1H NMR (400 MHz, CD3OD) δ 7.625 (d, J=8.9 Hz, 2H), 7.456 (d, J=8.9 Hz, 2H), 7.173–7.203 (m, 2H), 6.986 (d, J=3.2 Hz, 1H), 6.675–6.703 (dd, 1H), 5.877 (s, 1H), 3.746 (s, 2H), 3.547–3.581 (m, 4H), 2.221 (s, 3H), 1.961–2.025 (m, 4H); 13C-NMR (100 MHz, CD3OD) 173.106, 162.529, 151.451, 151.370, 134.650, 133.095, 129.301, 125.657, 125.397, 121.978, 121.555, 112.809, 112.696, 108.727, 103.674, 103.594, 95.786, 52.325, 35.009, 31.977, 26.407; IR (neat) υmax 3258.66, 1578.16, 1506.67, 1397.36, 1228.60, 793.25; ESI-HRMS [M+H]+ calculated for C25H26N6O2, 442.5230, found 443.2172.
2-(3-(cyclopropylmethyl)-1H-indol-1-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)acetamide (6.26).
4.1 (17.0 mg, 0.0632 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (7.72 mg, 0.0632 mmol, 1.1 equiv), DCC (16.3 mg, 0.079 mmol, 1.25 equiv) and 2-(3-(cyclopropylmethyl)-1H-indol-1-yl)acetic acid (16.1 mg, 0.0663 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.26 (23.7 mg, 78% yield) as an off white solid. TLC (3% methanol/dichloromethane), Rf = 0.27; m.p. 275–277 °C; 1H NMR (400 MHz, CD3OD) δ 8.311 (s, 1H), 8.280 (d, J=7.8 Hz, 1H), 7.780 (s, 1H), 7.636 (d, J= 8.9 Hz, 2H), 7.528 (d, J=8.9 Hz, 2H), 7.435 (d, J= 7.8 Hz, 1H), 7.218–7.305 (m, 2H), 5.918 (s, 1H), 5.109 (s, 2H), 3.554–3.586 (m, 4H), 2.605–2.667 (m, 1H), 2.250 (s, 3H), 1.991–2.024 (m, 4H), 1.956 (s, 2H), 1.121–1.158 (m, 2H), 0.949–0.995 (m, 2H); 13C-NMR (100 MHz, CD3OD) 197.917, 166.991, 162.055, 139.009, 138.658, 138.587, 137.137, 134.355, 127.271, 124.469, 123.493, 123.177, 121.899, 121.535, 118.579, 110.702, 96.389, 49.285, 50.561, 26.188, 22.943, 21.585, 18.662, 10.523; IR (neat) υmax 2009.39, 1508.43, 1386.73, 1167.26, 1065.42, 946.58, 744.69; ESI-HRMS [M+H]+ calculated for C29H32N6O2, 494.5990, found 495.2484.
2-(5-ethyl-1H-indol-3-yl)-N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl) acetamide (6.27).
4.1 (15.0 mg, 0.056 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (6.84 mg, 0.056 mmol, 1.1 equiv), DCC (14.4 mg, 0.070 mmol, 1.25 equiv) and 2-(5-ethyl-1H-indol-3-yl)acetic acid (11.9 mg, 0.0585 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.27 (24.8 mg, 98% yield) as an off white solid. TLC (3% methanol/dichloromethane), Rf = 0.30; m.p. 177–179 °C; 1H NMR (400 MHz, CD3OD) δ 7.628 (d, J=8.9 Hz, 2H), 7.417–7.439 (m, 3H), 7.268 (d, J=8.9 Hz, 1H), 7.189 (s, 1H), 6.984 (d, J=8.4 Hz 1H), 5.838 (s, 1H), 3.789 (s, 2H), 3.530–3.563 (m, 4H), 2.685–2.742 (q, 2H), 2.191 (s, 3H), 1.950–1.984 (m, 4H), 1.253 (t, 3H); 13C-NMR (100 MHz, CD3OD) 173.133, 162.616, 138.337, 138.332, 136.690, 136.060, 134.116, 128.780, 124.921, 123.207, 122.022, 121.234, 117.835, 112.158, 109.187, 95.394, 47.829, 34.965, 30.173, 26.452, 23.333, 17.153; IR (neat) υmax 2957.96, 1504.91, 1399.77, 1253.22, 803.62; ESI-HRMS [M+H]+ calculated for C27H30N6O, 454.5780, found 455.2535.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(1-((4-(trifluoromethyl) phenyl)sulfonyl)-1H-indol-3-yl)acetamide (6.28).
4.1 (15.6 mg, 0.0580 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (7.1 mg, 0.0580 mmol, 1.1 equiv), DCC (15.0 mg, 0.0725 mmol, 1.25 equiv) and 2-(1H-benzo[d]imidazol-1-yl)acetic acid (10.7 mg, 0.0609 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.28 (16.9 mg, 68% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.27; m.p. 175–177 °C; 1H NMR (400 MHz, DMSO-d6) δ 10.368 (s, 1H), 8.229 (s, 1H), 7.641–7.678 (m, 3H), 7.486–7.542 (m, 3H), 7.190–7.270 (m, 2H), 5.857 (s, 1H), 5.148 (s, 2H), 3.446–3.505 (m, 4H), 2.135 (s, 3H), 1.885–1.916 (m, 4H); 13C-NMR (100 MHz, DMSO-d6) 179.572, 164.927, 144.989, 143.167, 134.379, 122.336, 121.465, 119.673, 119.323, 110.280, 72.465, 63.055, 48.568, 47.275, 46.295, 24.955; IR (neat) υmax 3093.18, 1581.23, 1502.31, 1403.46, 1298.11, 1230.31, 1175.88, 835.05, 738.59; ESI- HRMS [M+H]+ calculated for C24H25N7O, 427.5120, found 428.2177.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(1-((4-(trifluoromethyl) phenyl)sulfonyl)-1H-indol-3-yl)acetamide (6.29).
4.1 (15.6 mg, 0.0580 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with DMAP (7.1 mg, 0.0580 mmol, 1.1 equiv), DCC (15.0 mg, 0.0725 mmol, 1.25 equiv) and 2-(1H-benzo[d]imidazol-1-yl)acetic acid (10.7 mg, 0.0609 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.29 (16.9 mg, 68% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.47; m.p. 230–232 °C; 1H NMR (400 MHz, CD3OD) δ 8.113 (d, J=8.4 Hz, 2H), 8.000 (d, J=8.4 Hz, 1H), 7.804 (d, J=8.4 Hz, 1H), 7.612–7.676 (m, 4H), 7.460 (d, J=8.9 Hz, 2H), 7.344–7.383 (t, 1H), 7.264–7.302 (t, 1H), 5.878 (s, 1H), 3.787 (s, 2H), 3.554–3.586 (m, 4H), 2.219 (s, 3H), 1.978–2.010 (m, 4H); 13C-NMR (100 MHz, CD3OD) 170.699, 162.550, 142.731, 136.537, 136.446, 136.116, 132.265, 128.837, 127.658, 126.295, 126.000, 124.954, 121.830, 121.473, 121.061, 119.402, 114.738, 101.399, 79.474, 47.965, 33.994, 26.429; IR (neat) υmax 1649.02, 1577.42, 1505.48, 1400.80, 1319.87, 1169.71, 1126.77, 1059.29, 830.21, 784.74, 713.73, 607.93, 558.38, 427.28; ESI-HRMS [M+H]+ calculated for C32H29F3N6O3S, 634.6782, found 635.2022.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(2-oxoindolin-3-yl) acetamide (6.30).
4.1 (22.0 mg, 0.0817 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (1.5 mL) and treated with HBTU (40.3 mg, 0.106 mmol, 1. equiv), 2-(2-oxoindolin-3-yl)acetic acid (17.2 mg, 0.090 mmol, 1.1 equiv) and then DIPEA (28.3 mL, 0.163 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was concentrated on to silica gel and purified via column chromatography using 5–20% methanol in dichloromethane to provide 6.30 (16.0 mg, 45% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.27; m.p. 170–172 °C; 1H NMR (400 MHz, CD3OD) δ 7.637 (d, J=8.9 Hz, 2H), 7.495 (d, J=8.9 Hz, 2H), 7.192–7.252 (q, 2H), 6.961–6.999 (t, 1H), 6.917 (d, J=7.8 Hz, 1H), 5.952 (s, 1H), 3.882–3.915 (m, 1H), 3.581–3.654 (m, 4H), 3.060–3.111 (dd, 1H), 2.746–2.818 (m, 1H), 2.277 (s, 3H), 2.036 (m, 4H); 13C-NMR (100 MHz, CD3OD) 181.633, 170.864, 162.145, 143.659, 136.567, 135.491, 130.594, 129.266, 125.084, 123.288, 122.212, 121.651, 110.921, 96.940, 55.830, 38.068, 36.945, 31.639, 26.245, 20.701, 13.165; IR (neat) υmax 3323.06, 1571.06, 1502.57, 1335.88, 1231.30, 816.45, 663.70, 520.32; ESI-HRMS [M+H]+ calculated for C25H26N6O2, 442.5230, found 443.2175.
N-(4-((6-methyl-2-(pyrrolidin-1-yl)pyrimidin-4-yl)amino)phenyl)-2-(1-((4-(trifluoromethyl) phenyl)sulfonyl)-1H-indol-3-yl)acetamide (6.31).
4.1 (62.0 mg, 0.230 mmol, 1.0 equiv) was dissolved in dichloromethane, anhydrous (6.0 mL) and treated with DMAP (28.1 mg, 0.230 mmol, 1.1 equiv), DCC (59.3 mg, 0.288 mmol, 1.25 equiv) and 2-(2-chloroquinolin-4-yl)acetic acid (56.0 mg, 0.253 mmol, 1.0 equiv). The vial was purged with nitrogen and allowed to stir for 24h at room temperature. The solution was filtered through a pad of celite and concentrated on to silica gel. Purification via column chromatography using 1:1 ethyl acetate/dichloromethane and 3% methanol provided 6.31 (13.5 mg, 57% yield) as a white solid. TLC (3% methanol/dichloromethane), Rf = 0.58; m.p. 260–262 °C; 1H NMR (400 MHz, CDCl3) δ 8.199 (d, J=8.5 Hz, 1H), 7.987 (d, J=8.5 Hz, 1H), 7.798–7.839 (t, 1H), 7.658–7.710 (m, 3H), 7.553 (s, 1H), 7.486 (d, J=8.9 Hz, 2H), 5.859 (s, 1H), 4.243 (s, 2H), 3.558–3.591 (m, 4H), 2.214 (s, 3H), 1.976–2.009 (m, 4H); 13C-NMR (100 MHz, CDCl3) 167.354, 165.894, 161.314, 160.216, 150.517, 147.781, 145.224, 136.193, 133.217, 130.757, 128.675, 127.402, 126.491, 124.017, 123.331, 121.492, 120.797, 116.000, 93.281, 46.653, 40.236, 25.483, 23.457; IR (neat) υmax 1755.53, 1658.71, 1612.71, 1548.21, 1504.15, 1403.49, 1251.05, 1138.16, 890.44, 696.38, 517.52 ESI-HRMS [M+H]+ calculated for C26H25ClN6O, 472.9770, found 473.1834.
Cell Lines.
Cell lines were purchased directly from ATCC and used as indicated. Engineered cell lines previously reported were STR profiled for authenticity. All cell lines were tested for bacterial and mycoplasma contamination before use. Deidentified patient sample cells were obtained from the CU Cancer Center GI tissue bank.
Cell Culture.
SW620 and HCT116 cell lines were obtained from American Type Culture Collection (ATCC) (Manassas, VA) and grown in RPMI-1640 media supplemented with 5% fetal bovine serum (FBS) in a humidified incubator at 37 °C and 5% CO2. Cells were expanded in 10 cm2 tissue culture-treated dishes (ThermoFisher, Waltham, MA) following ATCC protocols. Epithelial-Mesenchymal transition (EMT) dual reporter cell lines (SW620 and HCT116 E, E/M, and M) were generated, characterized, and maintained as previously outlined.21 Both wildtype and dual reporter cell lines were harvested and prepared for experiments by aspirating media, washing with 10 mL PBS, detaching with 1 mL of Trypsin-EDTA at 0.25% (ThermoFisher), and neutralizing with 4 mL of complete growth medium. Cells were counted using a Bio-Rad TC20 automated cell counter (Bio-Rad, Hercules, CA) by Trypan Blue (1:1) live/dead cell exclusion.
Cell Line Tumor Organoid Culture.
Tumor organoids were prepared by plating at 2,000 cells/well into CellCarrier Spheroid Ultra-Low Attachment (ULA) 96-well plates (Cat. No. 6055330, PerkinElmer, Waltham, MA) or Clear Round Bottom ULA 96-well plates (Cat. No. 7007, Corning). Briefly, the plated cells were centrifuged at 1,000 RPM to promote cellular aggregation, afterwards a final concentration of 2% Matrigel (Corning) was added, and the plates were placed in a 37 °C and 5% CO2 humidified incubator for 72 h to reach proper tumor organoid structure. Tumor organoids were then treated with CHD1Li compounds for an additional 72 h in dose response assays and analyzed for EMT reversal and cytotoxicity.
cat-CHD1L Enzyme ATPase Assay.
CHD1L enzyme inhibition assay was performed as described previously.11 All reactions were carried out using low volume non-binding surface 384-well plates (Corning Inc., Corning, NY). 800 nM cat-CHD1L, 200 nM mononucleosome (Active Motif, Carlsbad, CA), and various concentrations of inhibitors were preincubated at 37 °C for 10 min in 1x buffer containing 50 mmol/L Tris pH 7.5, 50 mmol/L NaCl, 5 mmol/L MgCl2, 2 mmol/L DTT, and 5% glycerol. The reaction was initiated by the addition of 10 μmol/L ATP (New England Biolabs, Ipswich, MA) to a total volume of 10 μL and incubated at 37 °C for 1 hour. ATPase activity was measured by adding 500 nmol/L of Phosphate Sensor (ThermoFisher) measuring excitation (430 nm) and emission (450 nm) immediately on an Envision plate reader (PerkinElmer). Background signal was determined by using all assay components excluding the enzyme.
Patient Tumor Organoid Culture.
CRC042 and CRC102 patient cell samples were expanded and cultured as PDTOs using reported methodologies and reagents.35, 36 Briefly, patient cells were washed with PBS, digested with TrypLE, and filtered through a 100 μm cell strainer before being used in 3D cell culture. Patient cells were seeded at 5,000 cells per well in 96-well plates, coated with 2% Matrigel, and allowed to self-assemble as PDTOs over 72 h.
Tumor Organoid Cytotoxicity.
CHD1Li cytotoxicity was assessed using CellTiter Glo 3D (Cat. No. G9681, Promega, Madison, WI). Tumor organoids were manually transferred from Clear Round Bottom ULA 96-well plates to Corning 96-or 384-well solid bottom white plates (Cat. No. 655083, Greiner Bio-One, Monroe, NC). CellTiter Glo 3D was added at a 1:1 ratio, incubated for 45 min at room temperature on an orbital shaker at 450 RPM. Finally, luminescence was quantified using an Envision Plate Reader (PerkinElmer). Cell cytotoxicity was normalized to 0.5% DMSO as vehicle control.
3D High-Content Imaging and Analysis of EMT Dual-Reporter Activity.
Tumor organoids were imaged using the Opera Phenix high-content screening system (PerkinElmer) in confocal mode utilizing a 10x air objective (NA 0.3). The following excitation and emission (Ex/Em) wavelengths were employed: RFP (561/570-630) and GFP (488/500-550). Organoids were also imaged in Brightfield to segment and perform high-content analysis of dual reporter specific fluorescence intensity. RFP fluorescent signal correlates with E-cadherin promoter activity, while GFP fluorescent signal is correlated with vimentin promoter activity. Both fluorescent signals were quantified and normalized as previously described.21
TCF-transcriptional Reporter Assay.
Assay has been adapted and modified from.21, 22, 37 SW620 cells were plated into duplicate 96-well plates at 20,000 cells/well (HCT116 at 10,000 cells/well), one white solid bottom plate was used to assess TCF-transcriptional activity and one clear plate was used to measure total protein by BCA assay for normalization purpose. Cell lines were allowed to attach overnight and then were transiently transfected with TOPflash plasmid (Millipore, Billerica, MA) using TransIT-LT1 transfection reagent (Mirus Bio, Madison, WI) for 72 h. Afterwards, cells from the white solid bottom plate were carefully washed with PBS and a 1:1 ratio of PBS: One-Glo Luciferase Assay System (Promega) was added, incubated for 10 min, and luminescence was quantified on Envision plate reader (PerkinElmer) for TCF-activity. As a control, cells from the clear plate were lysed with Mammalian Cell Lysis Buffer (Promega) and the total protein from each well was quantified with BCA assay (ThermoFisher). Experiments were replicated three times (n = 4 for each replicate)
Clonogenic Assay.
Cancer stem cell colony formation after CHD1Li treatment was assessed as previously described.29 In brief, HCT116 cells were plated at an optimal cell concentration to avoid colonies merging over a growth period of 7–10 days. SW620 and HCT116 M-Phenotype cells were plated at 200 and 75 cells/well, respectively, into 96-well μClear CellStar black plates (Greiner Bio-One). Fresh media and drug treatments were replenished every three days. Images were acquired using the Opera Phenix HCS system and colony number, area (μm2), and confluence (μm2) were quantified and analyzed using the Harmony software. Experiments were replicated three times (n = 3 for each condition).
Molecular modeling.
In silico studies were conducted using the Schrodinger (version 2021–2) and OPLS4 force field.
Ligand and protein preparation.
The structures of lead CHD1Li were drawn by inputting their SMILES strings into the 2D sketcher. The minimized 3-D structures were then generated with LigPrep while assigning their ionization and protonation states at pH 7.2 ± 0.2 with Epik. The minimized CHD1L structure, on the other hand, was obtained by processing the reported crystal structure (PDB ID: 7EPU) with the Protein Preparation Wizard. The preprocessing included assigning bond orders (using the Chemical Components Dictionary [CCD] database), adding hydrogen atoms and missing side chains, and generating heteroatomic states with EpiK at pH 7.2 ± 0.2. The bound ADP and co-crystallized antibody were removed, followed by hydrogen bond network optimization with PROPKA at pH 7.2. The protein structure was subjected to a restrained minimization of RMSD 0.30 Å.
Binding site determination and molecular docking.
SiteMap calculations were performed on the minimized CHD1L structure using the default settings. The returned sites’ coordinates were defined for receptor grid generation while setting the receptor vdW scaling as 1.0 and the inner and outer grid box sizes at 10 and 30 Å, respectively, on all dimensions. Subsequently, glide rigid docking (ligand vdW scaling = 0.8) was performed with the parent lead 6.0 to generate a CHD1L complex for each site. The best model, selected using the calculated MMGBSA binding free energy, was utilized as input structure for induced fit docking (IFD) to account for protein’s structural flexibility and ligand-induced conformational changes. The employed standard protocol included a brief constrained refinement of the protein to RMSD 0.18 Å and auto trimming of side chains, as well as an extra-precision mode for the glide redocking stage. The best poses were selected using the parameters in Table S3. This “integrated molecular docking” approach gave a more accurate reflection of the experimental CHD1L inhibition than using each glide or IFD alone.
Molecular dynamics simulation.
Model systems of 6.0 and 6.11 for MD simulation were generated from the system builder panel using SPC solvent models in an orthorhombic box with 1.0 nm periodic boundary conditions on all sides. The systems were neutralized with chloride ions while keeping NaCl salt concentration at 50 mM. MD simulations were achieved with the Desmond module. The model systems were first relaxed in short MD simulations using the NVT and NPT ensembles. The production MD simulation was thereafter performed with the NPT ensemble using the Nose-Hoover chain and Martyna-Tobias-Klein as the thermostat and barostat methods, respectively, at a constant temperature and pressure of 300 K and 1.01 bar, respectively. The simulation lasted for 250 ns while recording the trajectory at 250 ps intervals. Trajectory analysis was subsequently achieved using relevant Python scripts.
In Vitro Microsomal Stability Drug Preparation.
Microsomal stability of CHD1Li was assessed using male CD-1 mouse microsomes (ThermoFisher, Cat. MSMCPL). The reactions were prepared at the following reagent concentrations: mouse microsomes (0.5 mg/mL), phosphate buffer at pH 7.4 (44 mM KH2PO4, 56 mM K2HPO4), UDPGA (3 mM, Sigma-Aldrich Cat. U6751), alamethicin (25 μg, Sigma-Aldrich, A4665), MgCl2 (1 mM), NADPH (1 mM, Sigma-Aldrich Cat. 481973), and CHD1Li or testosterone (10 μM, Sigma-Aldrich T1500) at a final concentration of 0.1% of DMSO. Reactions were pre-incubated for 5 min at 37 °C. Afterwards, each microsome stability reaction was initiated by the addition of CHD1Li or testosterone as control and incubated in a water bath at 37 °C. The timepoints assessed were: 0, 5, 15, 30, 45, and 60 min. To quench each reaction at their respective timepoints, 100 μL was removed from each tube and mixed with 100 μL of acetonitrile (Sigma-Aldrich, Cat. 34998-4X4L). Finally, samples were centrifuged at 3,000 RPM for 5 min and 100 μL of the supernatant was extracted to be analyzed by mass spectrometry.
MS Analysis of Drug Microsomal Stability.
A Prominence HPLC (Shimadzu, Kyoto, Japan) coupled to a QTrap 4500 mass spectrometer (AB Sciex, Farmingham, MA) was used for this analysis. The mass spectrometer was operated in the ESI+ mode and all settings were optimized by manually tuning to infused standard solutions. Global mass spectrometer parameter settings were selected to give the highest average sensitivity for all compounds of interest. Global mass spectrometer settings are as follows: curtain gas, 40 psi; collision gas, high; ion spray voltage, 4500 V; source temperature, 700 °C; ion source gas 1 and 2, 50 psi; declustering potential, 85 V; entrance potential, 10 V; and collision cell exit potential, 14 V. The collision energy was optimized separately for each compound. Data was collected using the scheduled multiple reaction mode. Each compound was analyzed in a separate method (LC-MS/MS transitions below). Quantitation was performed using an external calibration curve for each compound. Analytical separation was achieved with a Phenomenex kinetex C18 column [2.1 × 100 mm, 2.6 μm]. The column was held at 40 °C and eluted at 0.6 mL/min with a gradient of 0.1% formic acid (A) and 0.1% formic acid in 9:1 acetonitrile:water (B) with a total runtime of 10 min. Chromatographic separation was achieved with a linear gradient (time, % of solvent B): 0–0.5 min, 5% B; 0.5–4.5 min, 5–55% B; 4.5–6.5 min, 55–94% B; 6–7 min, 94–5% B; and then isocratic for 3 minutes at 5% B to re-equilibrate the column. The first three minutes of eluent was diverted to waste to reduce contamination of the front end of the mass spectrometer. Calibration curves were prepared from stock standard solutions, which were prepared in DMSO. Calibration standards were prepared in 20% DMSO in water. Calibration curves typically contained 25 fmols – 2 pmols on column for testosterone, and 25 fmols – 1 pmol on column for compounds 6, 6.4, 6.5, 6.11, and 6.31. Data were fit to a mono-exponential decay graph to obtain first-order rate constant and the microsomal half-life was determined using the expression, 0.693/rate constant.38
In vivo PK studies.
Animal dosing, sample collection, and analysis was carried out in the Pharmacology Laboratory of the Drug Development & Discovery Shared Resource of the University of Colorado Cancer Center. The following animal studies were approved by the Institutional Animal Care and Use Committee at Colorado State University. 18 Female CD-1 mice were purchased from Charles River Laboratories. Animals were allowed to acclimate and were approximately 25 g when treated with 6.11. Mice were dosed by oral gavage (PO) with 6.11 prepared in DMSO (20%), PEG 400 (40%), and PBS (pH 7.4, 40%). The drug solution was made at 12.5 mg/mL so that a 25 g mouse would receive 0.10 mL for a final dose of 50 mg/kg. Following dosing, mice were sacrificed by cardiac exsanguination under isoflurane anesthesia and blood collected in sodium heparin treated syringes. Blood samples were then centrifuged at 8,000 × g for 10 min and the resulting plasma collected and frozen at 80 °C until analysis. Samples were analyzed using a Shimadzu Nexera X2 LC30AD pumps equipped with a DGU-20A5R DeGasser (Shimadzu, Japan). Mass spectrometry was performed using an AB Sciex 6500 Qtrap Triple Quadruple (Sciex, Framingham, MA). Blank pooled plasma samples from CD-1 mice were used for the preparation of standards and QC samples. Samples were prepared by adding 100 μL of plasma to 1.5 mL polypropylene microcentrifuge tubes. For the standard and QC samples, 10 μL of 10X 6.11 was added and for unknown samples 10 μL of acetonitrile:MilliQ H2O (2:1) was added. As an internal standard, 10 μL of 6.1 (100 ng/mL) was added. In addition, 200 μL of acetonitrile was added to each sample as solvent. Samples were then vortexed for 10 min and centrifuged at 13,100 × g for 10 min. Finally, 200 μL of supernatant was collected and transferred to autosampler vials with glass low volume inserts and submitted for LCMS analysis.
In Vivo CHD1Li 6.11 Efficacy Study.
Animal studies were conducted in accordance with the protocol procedures approved by the Institutional Animal Care and Use Committee at CU AMC. Female athymic nude mice, 8 weeks of age, were purchased from Envigo (Hsd:Athymic Nude-Foxn1nu (069)). A dose range finding study with 6.11 was conducted ranging from 50 to 200 mg/kg (n =1 animal per dose). Maximum tolerated dose studies were conducted using 125- and 150-mg/kg (n = 5 per dose) administered by PO 5x/week over 21 days. The MTD studies indicated that at doses at or below 150 mg/kg were well tolerated, assessed grossly by animal weight with no observed deterioration in regular behavior. SW620 EMT dual-reporter M-Phenotype cells were grown in RPMI 1640 (Gibco, Cat. No. 11875) supplemented with 5% FBS, then harvested and suspended at 2 × 106 cells/100 μL in 1:1 Matrigel (Corning, Cat. No. 356231) and RPMI 1640. Mice were anesthetized with 1–5% inhaled isoflurane then injected subcutaneously, 100 μL into each flank. At 5 days, mice were randomized into 3 groups, 1) vehicle (10% DMSO, 90% PEG 400); 2) treatment with 6.11 at 75 mg/kg; 3) treatment with 6.11 at 125 mg/kg. All treatments were given once a day, 5 days a week, via oral gavage (PO) for 28–31 days. Mice were weighed and tumor volumes were determined twice per week throughout the entire study. Tumor volumes were measured by caliper and calculated using the formula 4/3*4.13*(L/2)*(W/2)*(H/2).
Statistical Analysis
All statistical analyses were performed using Prism v9.0 (GraphPad Software Inc., La Jolla, CA). Data were collected using three experimental replicates (N=3) with three technical replicates (n=3) for each experiment, unless otherwise noted. For each figure presented including SI figures, error bars represent the SEM from three experimental means. IC50 values were calculated from the best fit curves within the 95% confidence interval. P-value significance is represented as *P <0.05; **P <0.01; ***P <0.001; ****P <0.0001.
Supplementary Material
Acknowledgement
This research was supported by grants from the NIH NCI 1R01CA251361-01 and SPARK/REACH awarded to D.V.L., and a grant from the University of Colorado Cancer Center (UCCC) Developmental Therapeutics Program awarded to D.V.L. and D.L.G. We also thank CU AMC shared resource (SR) laboratories, including the Drug Discovery and Development (D3SR), Mass Spectrometry (MSSR), and Flow Cytometry (FCSR). These shared resources are supported in part by the UCCC, an NIH NCI designated center (P30CA046934). The D3SR is also supported by the CU AMC Center for Drug Discovery, which was established with generous grant funding from The ALSAM Foundation and through CU AMC institutional support.
Abbreviations
- CSC
cancer stem cell
- CHD1L
chromodomain helicase DNA binding protein 1 like
- ALC1
also known as amplified in liver cancer 1
- CRC
colorectal cancer
- DCM
dichloromethane
- DCC
dicyclohexylcarbodiimide
- DIPEA
diisopropyl ethyl amine
- DMAP
dimethylamino pyridine
- DMF
dimethylformamide
- EMT
epithelial-mesenchymal transition
- MET
and mesenchymal-epithelial transition
- EMP
epithelial-mesenchymal plasticity
- TCF/LEF
T cell factor/lymphoid enhancer factor
- GI
gastrointestinal
- HBTU
N, N’- 2-(1H-benzotriazole-1-yl)-1,1,3,3-tetramethyluronium hexafluorophosphate
- TEA
triethyl amine
- TLC
Thin layer chromatography
- TFA
trifluoroacetic acid
Footnotes
CONFLICT OF INTEREST STATEMENT
The authors declare no conflicts of interest.
Supporting Information
The supporting information contains SI data figures and tables; the synthesis and methods to prepare key starting materials and intermediates; compound structural characterization data for all novel compounds: NMR, HRMS, and IR spectra; HPLC purity traces for all final compounds demonstrating ≥95% purity.
We declare that no authors have been removed from the author list.
REFERENCES
- (1).Ahel D; Horejsi Z; Wiechens N; Polo SE; Garcia-Wilson E; Ahel I; Flynn H; Skehel M; West SC; Jackson SP; Owen-Hughes T; Boulton SJ Poly(ADP-ribose)-dependent regulation of DNA repair by the chromatin remodeling enzyme ALC1. Science 2009, 325, 1240–1243. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (2).Ma NF; Hu L; Fung JM; Xie D; Zheng BJ; Chen L; Tang DJ; Fu L; Wu Z; Chen M; Fang Y; Guan XY Isolation and characterization of a novel oncogene, amplified in liver cancer 1, within a commonly amplified region at 1q21 in hepatocellular carcinoma. Hepatology 2008, 47, 503–510. [DOI] [PubMed] [Google Scholar]
- (3).Cheng W; Su Y; Xu F CHD1L: a novel oncogene. Molecular cancer 2013, 12, 170. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (4).Sun J; Zhang L; Zhao H; Qiu X; Chen W; Wang D; Ban N; Fan S; Shen C; Xia X; Ji B; Wang Y CHD1L regulates cell cycle, apoptosis, and migration in glioma. Cell. Mol. Neurobiol 2016, 36, 565–576. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (5).He LR; Ma NF; Chen JW; Li BK; Guan XY; Liu MZ; Xie D Overexpression of CHD1L is positively associated with metastasis of lung adenocarcinoma and predicts patients poor survival. Oncotarget 2015, 6, 31181–31190. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (6).Wu J; Zong Y; Fei X; Chen X; Huang O; He J; Chen W; Li Y; Shen K; Zhu L Presence of CHD1L over-expression is associated with aggressive tumor biology and is a novel prognostic biomarker for patient survival in human breast cancer. PLoS One 2014, 9, e98673. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (7).Su Z; Zhao J; Xian G; Geng W; Rong Z; Wu Y; Qin C CHD1L is a novel independent prognostic factor for gastric cancer. Clinical & translational oncology : official publication of the Federation of Spanish Oncology Societies and of the National Cancer Institute of Mexico 2014, 16, 702–707. [DOI] [PubMed] [Google Scholar]
- (8).Hyeon J; Ahn S; Park CK CHD1L Is a marker for poor prognosis of hepatocellular carcinoma after surgical resection. Korean J Pathol 2013, 47, 9–15. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (9).He WP; Zhou J; Cai MY; Xiao XS; Liao YJ; Kung HF; Guan XY; Xie D; Yang GF CHD1L protein is overexpressed in human ovarian carcinomas and is a novel predictive biomarker for patients survival. BMC cancer 2012, 12. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (10).Chen L; Chan TH; Yuan YF; Hu L; Huang J; Ma S; Wang J; Dong SS; Tang KH; Xie D; Li Y; Guan XY CHD1L promotes hepatocellular carcinoma progression and metastasis in mice and is associated with these processes in human patients. J Clin Invest 2010, 120, 1178–1191. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (11).Abbott JM; Zhou Q; Esquer H; Pike L; Broneske TP; Rinaldetti S; Abraham AD; Ramirez DA; Lunghofer PJ; Pitts TM; Regan DP; Tan AC; Gustafson DL; Messersmith WA; LaBarbera DV First-in-class inhibitors of oncogenic CHD1L with preclinical activity against colorectal cancer. Mol Cancer Ther 2020, 19, 1598–1612. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (12).Xiong X; Lai X; Li A; Liu Z; Ma N Diversity roles of CHD1L in normal cell function and tumorigenesis. Biomark Res 2021, 9:16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (13).Verma P; Zhou Y; Cao Z; Deraska PV; Deb M; Arai E; Li W; Shao Y; Puentes L; Li Y; Patankar S; Mach RH; Faryabi RB; Shi J; Greenberg RA ALC1 links chromatin accessibility to PARP inhibitor response in homologous recombination-deficient cells. Nat Cell Biol 2021, 23, 160–171. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (14).Juhasz S; Smith R; Schauer T; Spekhardt D; Mamar H; Zentout S; Chapuis C; Huet S; Timinszky G The chromatin remodeler ALC1 underlies resistance to PARP inhibitor treatment. Sci Adv 2020, 6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (15).van de Wetering M; Sancho E; Verweij C; de Lau W; Oving I; Hurlstone A; van der Horn K; Batlle E; Coudreuse D; Haramis AP; Tjon-Pon-Fong M; Moerer P; van den Born M; Soete G; Pals S; Eilers M; Medema R; Clevers H The β-catenin/TCF-4 complex imposes a crypt progenitor phenotype on colorectal cancer cells. Cell 2002, 111, 241–250. [DOI] [PubMed] [Google Scholar]
- (16).Ram Makena M; Gatla H; Verlekar D; Sukhavasi S; M KP; K CP Wnt/beta-Catenin Signaling: The Culprit in Pancreatic Carcinogenesis and Therapeutic Resistance. Int J Mol Sci 2019, 20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (17).Batlle E; Henderson JT; Beghtel H; van den Born MM; Sancho E; Huls G; Meeldijk J; Robertson J; van de Wetering M; Pawson T; Clevers H β-catenin and TCF mediate cell positioning in the intestinal epithelium by controlling the expression of EphB/EphrinB. Cell 2002, 111, 251–263. [DOI] [PubMed] [Google Scholar]
- (18).He S; Tang S WNT/beta-catenin signaling in the development of liver cancers. Biomed. Pharmacother 2020, 132, 110851. [DOI] [PubMed] [Google Scholar]
- (19).Polakis P Wnt signaling in cancer. Cold Spring Harb Perspect Biol 2012, 4. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (20).Clevers H Wnt/β-catenin signaling in development and disease. Cell 2006, 127, 469–480. [DOI] [PubMed] [Google Scholar]
- (21).Esquer H; Zhou Q; Nemkov T; Abraham AD; Rinaldetti S; Chen YC; Zhang X; Orman MV; D’Alessandro A; Ferrer M; Messersmith WA; LaBarbera DV Isolating and targeting the real-time plasticity and malignant properties of epithelial-mesenchymal transition in cancer. Oncogene 2021, 40, 2884–2897. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (22).Zhou Q; Abraham AD; Li L; Babalmorad A; Bagby S; Arcaroli JJ; Hansen RJ; Valeriote FA; Gustafson DL; Schaack J; Messersmith WA; LaBarbera DV Topoisomerase IIα mediates TCF-dependent epithelial-mesenchymal transition in colon cancer. Oncogene 2016, 35, 4990–4999. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (23).Yang J; Antin P; Berx G; Blanpain C; Brabletz T; Bronner M; Campbell K; Cano A; Casanova J; Christofori G; Dedhar S; Derynck R; Ford HL; Fuxe J; Garcia de Herreros A; Goodall GJ; Hadjantonakis AK; Huang RYJ; Kalcheim C; Kalluri R; Kang Y; Khew-Goodall Y; Levine H; Liu J; Longmore GD; Mani SA; Massague J; Mayor R; McClay D; Mostov KE; Newgreen DF; Nieto MA; Puisieux A; Runyan R; Savagner P; Stanger B; Stemmler MP; Takahashi Y; Takeichi M; Theveneau E; Thiery JP; Thompson EW; Weinberg RA; Williams ED; Xing J; Zhou BP; Sheng G; Association EMTI Guidelines and definitions for research on epithelial-mesenchymal transition. Nat Rev Mol Cell Biol 2020, 21, 341–352. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (24).Sánchez-Tilló E; de Barrios O; Siles L; Cuatrecasas M; Castells A; Postigo A β-catenin/TCF4 complex induces the epithelial-to-mesenchymal transition (EMT)-activator ZEB1 to regulate tumor invasiveness. Proc. Natl. Acad. Sci. U.S.A 2011, 108, 19204–19209. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (25).Kroger C; Afeyan A; Mraz J; Eaton EN; Reinhardt F; Khodor YL; Thiru P; Bierie B; Ye X; Burge CB; Weinberg RA Acquisition of a hybrid E/M state is essential for tumorigenicity of basal breast cancer cells. Proc Natl Acad Sci U S A 2019, 116, 7353–7362. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (26).Drost J; Clevers H Organoids in cancer research. Nat Rev Cancer 2018, 18, 407–418. [DOI] [PubMed] [Google Scholar]
- (27).Scheel C; Weinberg RA Cancer stem cells and epithelial–mesenchymal transition: Concepts and molecular links. Semin. Cancer Biol 2012, 22, 396–403. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (28).Chaffer CL; San Juan BP; Lim E; Weinberg RA EMT, cell plasticity and metastasis. Cancer Metast. Rev 2016, 35, 645–654. [DOI] [PubMed] [Google Scholar]
- (29).Esquer H; Zhou Q; Abraham AD; LaBarbera DV Advanced High-Content-Screening Applications of Clonogenicity in Cancer. SLAS Discov 2020, 25, 734–743. [DOI] [PubMed] [Google Scholar]
- (30).Franken NA; Rodermond HM; Stap J; Haveman J; van Bree C Clonogenic assay of cells in vitro. Nat. Protoc 2006, 1, 2315–2319. [DOI] [PubMed] [Google Scholar]
- (31).Wang L; Chen K; Chen Z Structural basis of ALC1/CHD1L autoinhibition and the mechanism of activation by the nucleosome. Nat Commun 2021, 12, 4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (32).Wang L; Chen K; Chen Z Structural basis of ALC1/CHD1L autoinhibition and the mechanism of activation by the nucleosome. Nature Communications 2021, 12, 4057. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (33).Lehmann LC; Hewitt G; Aibara S; Leitner A; Marklund E; Maslen SL; Maturi V; Chen Y; van der Spoel D; Skehel JM; Moustakas A; Boulton SJ; Deindl S Mechanistic Insights into Autoinhibition of the Oncogenic Chromatin Remodeler ALC1. Molecular Cell 2017, 68, 847–859.e847. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (34).Gramec D; Peterlin Masic L; Sollner Dolenc M Bioactivation potential of thiophene-containing drugs. Chem. Res. Toxicol 2014, 27, 1344–1358. [DOI] [PubMed] [Google Scholar]
- (35).Drost J; Karthaus WR; Gao D; Driehuis E; Sawyers CL; Chen Y; Clevers H Organoid culture systems for prostate epithelial and cancer tissue. Nat Protoc 2016, 11, 347–358. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (36).Sato T; Stange DE; Ferrante M; Vries RG; Van Es JH; Van den Brink S; Van Houdt WJ; Pronk A; Van Gorp J; Siersema PD; Clevers H Long-term expansion of epithelial organoids from human colon, adenoma, adenocarcinoma, and Barrett’s epithelium. Gastroenterology 2011, 141, 1762–1772. [DOI] [PubMed] [Google Scholar]
- (37).Abraham AD; Esquer H; Zhou Q; Tomlinson N; Hamill BD; Abbott JM; Li L; Pike LA; Rinaldetti S; Ramirez DA; Lunghofer PJ; Gomez JD; Schaack J; Nemkov T; D’Alessandro A; Hansen KC; Gustafson DL; Messersmith WA; LaBarbera DV Drug Design Targeting T-Cell Factor-Driven Epithelial-Mesenchymal Transition as a Therapeutic Strategy for Colorectal Cancer. J. Med. Chem 2019, 62, 10182–10203. [DOI] [PMC free article] [PubMed] [Google Scholar]
- (38).Shargel L; Andrew B; Wu-Pong S Applied biopharmaceutics & pharmacokinetics. Appleton & Lange Stamford: 1999; Vol. 264. [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.