

Abstract
The tumor immune microenvironment (TIME) is commonly infiltrated by diverse collections of myeloid cells. Yet, the complexity of myeloid-cell identity and plasticity has challenged efforts to define bona fide populations and determine their connections to T-cell function and their relationship to patient outcome. Here, we have leveraged single-cell RNA-sequencing analysis of several mouse and human tumors and found that monocyte–macrophage diversity is characterized by a combination of conserved lineage states as well as transcriptional programs accessed along the differentiation trajectory. We also found in mouse models that tumor monocyte-to-macrophage progression was profoundly tied to regulatory T cell (Treg) abundance. In human kidney cancer, heterogeneity in macrophage accumulation and myeloid composition corresponded to variance in, not only Treg density, but also the quality of infiltrating CD8+ T cells. In this way, holistic analysis of monocyte-to-macrophage differentiation creates a framework for critically different immune states.
Introduction
A key component of most immune responses, including those to cancers, are mononuclear phagocyte cell populations, which share common features of phagocytosis, tissue repair, and immunoregulation but diverge in functional specialization. Conventional dendritic cells (cDCs) are positioned in tissues to initiate and sustain adaptive T-cell responses (1), whereas macrophages engage in high rates of phagocytosis and tissue remodeling (2). Self-renewing tissue-resident macrophages are seeded during embryonic development (3), whereas inflammatory stimuli prompt infiltration of adult hematopoietic stem cell–derived monocytes that give rise to tumor macrophages (4–7). These monocyte-derived macrophages preferentially accumulate as tumors progress (8) and may predominate in regulating the ongoing antitumor T-cell response (9).
Macrophages consist of numerous subset populations that have been identified across tissues (10–13). Therapeutic blockade of key epigenetic and signaling pathways has demonstrated their amenability to transcriptional reprogramming (14), but how phenotypic diversity arises remains poorly understood. Recruited bloodborne monocytes exhibit plasticity in differentiation potential and can acquire features of macrophages and/or DCs depending on the inflammatory setting (5,6,10,15–17). In addition, early studies demonstrated that macrophage exposure to type 1- or type 2-associated cytokines induces “M1” or “M2” cellular programs, respectively, and a model was put forth in which myeloid cells are polarized to be pro- (“M1”) or anti- (“M2”) inflammatory (18–20). Although this nomenclature was thereafter understood to require nuance to account for additional plasticity (21), it remains undetermined if these binary programs are applicable to describe tumor macrophage differentiation in vivo.
Myeloid phenotypic diversity has challenged efforts to utilize myeloid populations as biomarkers for patient treatment options and outcome. cDCs are critical for coordinating antitumor T-cell immunity (22–25) and higher cDC abundance is broadly associated with improved cancer patient survival, although additional tumor immune microenvironment (TIME) features may inform functionality (23,24,26). In contrast, macrophages have largely been considered to be pro-tumorigenic (2,14) and monocytes have often been described as myeloid-derived suppressor cells (MDSCs) (27). Yet, several studies have indicated that macrophages are not consistently a negative predictor of patient prognosis (28–31), and increased levels of circulating monocytes were unexpectedly linked to patient responsiveness to immune checkpoint blockade (ICB) (32). These contrary findings speak to the need for improved resolution of myeloid-cell categorization and phenotype in order to dissect heterogenous responses amongst cancer patients.
Using single-cell RNA-sequencing (scRNA-seq), we uncovered transcriptional heterogeneity amongst tumor-infiltrating myeloid cells and distinguished monocyte and macrophage lineage- and activation-induced programs shared between multiple mouse tumor models and human kidney cancer samples. Monocyte differentiation is dynamically regulated, and we found that regulatory T cell (Treg) density was one immunoregulatory axis capable of modulating macrophage density. Further comprehensive analysis of key myeloid populations revealed distinct network connections between different myeloid-cell types and T-cell subsets, including Tregs and effector T cells. This is consistent with an archetypal organization of immune systems in tumors — collections of cell types that move together as modules (33) — and improved classification of patients such that we could identify those with effective antitumor T-cell responses.
Materials & Methods
Mice
The following mice were housed and/or bred under specific pathogen-free conditions at the University of California, San Francisco Animal Barrier Facility: C57BL/6J (The Jackson Laboratory), MMTV-PyMT-mCherry-OVA transgenic (34), and Foxp3-DTR (The Jackson Laboratory). All mice were handled in accordance with NIH and American Association of Laboratory Animal Care standards, and experiments were approved by the Institutional Animal Care and Use Committee of the University of California, San Francisco.
Human Tumor Samples
Renal cell carcinoma (RCC), melanoma, and head and neck tumor samples were transported from various cancer operating rooms or outpatient clinics. All patients provided informed written consent to the UCSF IPI clinical coordinator group for tissue collection under a UCSF IRB approved protocol (UCSF IRB# 20–31740) in accordance with the Declaration of Helsinki guidelines. Patients were selected without regard to prior treatment and 21 RCC (all primary tumors), 22 melanoma (20 primary and 2 metastasis tumors) and 4 head and neck tumors (all primary tumors) were collected. All samples were defined as primary tumor or metastasis by pathology assistants. Samples were obtained after surgical excision with biopsies taken to confirm the presence of tumor cells. Freshly resected samples were placed in ice-cold DPBS (Thermo Fisher Scientific, catalog no. 14190144) or Leibovitz’s L-15 medium (Thermo Fisher Scientific, catalog no. 11415064) in a 50 mL conical tube and immediately transported to the laboratory for sample labeling and processing. As described below (Human Tissue Processing and Flow Cytometry Analysis), the whole tissue underwent digestion and processing to generate a single-cell suspension. In the event that part of the tissue was sliced and preserved for imaging analysis, the remaining portion of the tissue sample was used for flow cytometry analysis.
Tumor Cell Lines
B16-F10 cells (ATCC, CRL-6475) were purchased in 2015 and stock vials were generated from an initial thaw. Cells in use were taken from early passages and maintained consistent and homogenous morphological characteristics, during which time they were tested for Mycoplasma. B16-F10-ZsGreen was previously generated in our laboratory as described (35) and tested for maintained expression of ZsGreen by flow cytometry. After thawing, tumor cells were cultured at 37°C in 5% CO2 in DMEM (Thermo Fisher Scientific, catalog no. A4192101), 10% FCS (Benchmark, catalog no. 100–106), Penicillin, Streptomycin, and L-Glutamine (Thermo Fisher Scientific, catalog no. 10378016). Cells were generally cultured for 2–5 days (0–1 passages) before use for subcutaneous injection.
Mouse Tumor Cell Injections and Growth
Prior to injection, adherent B16-F10 or B16-ZsGreen tumor cells were dissociated with 0.05% Trypsin-EDTA (Thermo Fisher Scientific, catalog no. 25300120) and washed 2–3X with DPBS (Thermo Fisher Scientific, catalog no. 14190144). 1.0×105 – 2.5×105 cells were resuspended in DPBS and mixed 1:1 with Matrigel GFR (Corning, catalog no. 356231). Mice were injected subcutaneously with a volume of 50 μl either unilaterally or bilaterally depending on the experimental setup. Tumor tissue was harvested approximately 12–16 days later.
MMTV-PyMT-mCherry-OVA transgenic mice were bred and genotyped for the transgene by PCR. Spontaneous tumor growth was monitored by measuring with electronic calipers. Tumors were harvested when the mice were approximately 20–30 weeks of age such that palpable tumors had developed but had not exceeded a size of 100mm2.
In Vivo Mouse Treatments
To deplete Treg cells, Foxp3-DTR and control mice were injected intraperitoneally with 500ng of unnicked diptheria toxin (List Biologics, catalog no. 150). Mice were typically injected 9, 10, and 12 days following initial inoculation with tumor cells (see Mouse Tumor Cell Injections and Growth).
For specified experiments, wild-type mice were injected intraperitoneally 7, 9, 10, 11, and 13 days following tumor injection with 250 μg of anti-mouse CTLA-4 IgG2c (modified clone 9D9, Bristol-Myers-Squibb), mouse IgG2C isotype, anti-mouse CTLA-4 IgG1 (modified clone 9D9, Bristol-Myers-Squibb), or mouse IgG1 isotype.
Mouse Tissue Processing and Flow Cytometry Analysis
Mouse tumor tissue was harvested and enzymatically digested with 0.2mg/ml DNase I (Sigma-Aldrich, catalog no. D5025), 100U/ml Collagenase I (Worthington Biochemical, catalog no. LS004197), and 500U/ml Collagenase Type IV (Worthington Biochemical, catalog no. LS004189) for 30 minutes at 37°C while under constant agitation. Blood was collected via cardiac puncture from mice that were euthanized by overdose with 2.5% Avertin. Blood samples were treated with 175 mM NH4Cl for 5 minutes at room temperature to lyse red blood cells.
Samples were filtered, washed with stain media (DPBS, 2% FCS), and resuspended in stain media. Cells from this single-cell suspension were washed with DPBS and stained with Zombie NIR fixable viability dye (BioLegend, catalog no. 423106) for 30 minutes at 4°C. Cells were washed and resuspended in stain media containing anti-CD16/32 (BioXCell, catalog no. BE0307), 2% rat serum (Thermo Fisher Scientific, catalog no. 10710C), 2% Armenian hamster serum (Innovative Research, catalog no. IGHMA-SER), and antibodies against surface proteins of interest. Cells were stained for 30 minutes at 4°C. In some experiments, cells were then washed and stained for intracellular protein levels, for which they were fixed, permeabilized, and stained according to BD Cytofix/Cytoperm Kit (BD Biosciences, catalog no. 554655) or the FoxP3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, catalog no. 00–5523-00).
The following antibodies were from Biolegend: anti-mouse CD45 (clone A20, catalog no. 110727), anti-mouse Ly-6C (clone HK1.4, catalog no. 128037), anti-mouse CD11b (clone M1/70, catalog no. 101257), anti-mouse CD11c (clone N418, catalog no. 117339), anti-mouse MHC-II (clone M5/114.15.2, catalog no. 107622), anti-mouse F4/80 (clone BM8, catalog no. 123135, 123107, or 123131), anti-mouse CD24 (clone M1/69, catalog no. 101822), anti-mouse Ly-6G (clone IA8, catalog no. 127645), anti-mouse NK1.1 (clone PK136, catalog no. 108749), anti-mouse CD90.2 (clone 30-H12, catalog no. 105331), anti-mouse/human CD45R/B220 (clone RA3–6B2, catalog no. 103246), anti-mouse CD301b (clone URA-1, catalog no. 146814 or 146803), anti-mouse CD64 (clone X54–5/7.1, catalog no. 139306), anti-mouse CD127 (clone A7R34, 135031). The following antibodies were from BD Biosciences: anti-mouse Siglec-F (clone E50–2440, catalog no. 740956), anti-mouse CD106 (clone 429, catalog no. 745672). The following antibodies were from R&D: anti-mouse/human ARG1 (polyclonal, catalog no. IC5868F), normal sheep IgG (polyclonal, catalog IC016F). The following antibodies were from Thermo Fisher Scientific: anti-mouse FoxP3 (clone FJK-16s, catalog no. 48–5773-82).
Following staining, cells were washed, resuspended in stain media, and analyzed on a BD Biosciences Fortessa or sorted with a BD Biosciences FACSAria Fusion. Flow cytometry data was analyzed using FlowJo software (BD Biosciences, version 9 or 10).
Human Tissue Processing and Flow Cytometry Analysis
Human tumor or metastatic tissue was thoroughly chopped with surgical scissors and transferred to gentleMACS C Tubes (Miltenyi Biotec) containing 20 uL/mL Liberase TL (5 mg/ml, Roche, catalog no. 5401020001) and 50 U/ml DNAse I (Roche, catalog no. 10104159001) in RPMI 1640 (Thermo Fisher Scientific catalog no. 11875093) per 0.3 g tissue. gentleMACS C Tubes were installed onto the gentleMACS Octo Dissociator (Miltenyi Biotec) and incubated for 45 minutes according to the manufacturer’s instructions. Samples were then quenched with 15 mL of sort buffer (DPBS, 2% FCS, 2mM EDTA), filtered through 100 μm filters, and spun down. Red blood cell lysis was performed with 175 mM NH4Cl if needed. Cells were incubated with Human FcX (Biolegend, catalog no. 422301) to prevent non-specific antibody binding. Cells were then washed in DPBS and incubated with Zombie Aqua Fixable Viability Dye (Thermo Fisher Scientific, catalog no. L34957). Following viability dye, cells were washed with sort buffer and incubated with cell surface antibodies that had been diluted in the BV stain buffer (BD Biosciences, catalog no. 563794) for 30 minutes on ice. Cells were subsequently fixed in either Fixation Buffer (BD Biosciences, catalog no. 554655) or in Foxp3/Transcription Factor Staining Buffer Set (Thermo Fisher Scientific, catalog no. 00–5523-00) if intracellular staining was required.
The following antibodies were from BD Biosciences: anti-human HLA-DR (clone G46–6, catalog no. 564040), anti-human CD56 (clone NCAM16.2, catalog no. 564448), anti-human CD127 (clone HIL-7R-M21, catalog no. 563225) anti-human CD25 (clone 2A3, catalog no. 340939), anti-human CD45RO (clone UCHL1, catalog no. 561889), anti-human PD-1 (clone EH12, catalog no.563789), anti-human CTLA-4 (clone BNI3, catalog no. 565931), and anti-human CD64 (clone 10.1, catalog no. 564425). The following antibodies were from Thermo Fisher Scientific: anti-human CD45 (clone HI30, catalog no. 47–0459-42), anti-human CD3ε (clone OKT3, catalog no. 46–0037-42), anti-human FoxP3 (clone 236A/E7, catalog no. 25–4777-41), anti-human Ki-67 (SolA15, catalog no. 11–5698-82), anti-human CD19 (clone H1B19, catalog no. 45–0199-42), anti-human CD20 (clone 2H7, catalog no. 45–0209-42), anti-human CD56 (clone CMSSB, catalog no. 46–0567-42), and anti-human CD11c (clone 3.9, catalog no. 56–0116-42). The following antibodies were from Biolegend: anti-human CD4 (clone S3.5, catalog no. 100455), anti-human CD8α (clone RPA-T8, catalog no. 301039), anti-human CD38 (clone HIT2, catalog no. 303523), anti-human CD16 (clone 3G8, catalog no. 302039), CD1C/BDCA-1 (clone L161, catalog no. 331515), anti-human CD14, (clone M5E2, catalog no. 301837) anti-human CD304 (clone 12C2, catalog no. 354503), and streptavidin. Anti-human BDCA-3 (clone AD5–14H12, catalog no. 130–098-843) was purchased from Miltenyi Biotec.
Stained cells were washed and analyzed on a BD Biosciences Fortessa or sorted with a BD Biosciences FACSAria Fusion. Flow cytometry data was analyzed using FlowJo software (BD Biosciences, version 10.6). Clustering and heatmap analyses were performed using Morpheus (Broad Institute).
Single-Cell RNA-Sequencing Data Generation
Mouse B16 samples were generated over 2 independent experiments. Samples were pooled from at least 5 mice per experiment to ensure representation across a cohort of tumor-bearing mice. In the first experiment, CD45+CD90–B220–NK1.1–Ly6G– cells that were Ly6C–MHC-II+ or Ly6C+CD11b+ were sorted as 1 bulk myeloid sample. Individual monocyte (CD45+CD90–B220–NK1.1–Ly6G–Ly6C+CD11b+) and macrophage (CD45+CD90–B220–NK1.1–Ly6G–Ly6C–MHC-II+F4/80+CD24loCD11clo/hi) populations from these tumors were also sorted. Each of these samples were processed separately, but in parallel, for scRNA-seq analysis. In the second experiment, B16 tumor myeloid cells (CD45+CD90–B220–NK1.1–Ly6G– cells that were CD11b+ and/or CD11c+) were sorted from control or Foxp3-DTR mice. Blood myeloid cells (CD45+CD90B220–NK1.1–Ly6G– cells that were CD11b+ and/or CD11c+) were also sorted from these B16 tumor-bearing wildtype mice. Each of these 3 samples were processed separately, but in parallel, for scRNA-seq analysis. In a subsequent experiment, myeloid cells (CD45+CD90–B220–NK1.1–Ly6G–CD11b+ and/or CD11c+) from a mouse PyMT tumor were sorted. In addition, myeloid cells (CD45+CD3ε–CD19–CD20–CD56–HLA-DRdim/hi) from a total of 1 RCC, 4 head and neck, and 6 melanoma biopsies were sorted and processed individually as they became available.
Once sorted, cells were resuspended at a concentration of 1×103 cells/μl in media (DPBS, 0.04% BSA) and loaded onto the Chromium Controller (10X Genomics). Samples underwent single-cell encapsulation and cDNA library preparation using the Chromium Single Cell 3’ v1 or v2 Reagent Kits (10X Genomics, catalog no. 120237). The cDNA library was sequenced on an Ilumina HiSeq 4000 or Illumina Novaseq.
Single-Cell RNA-Sequencing Data Processing
Sequencing data was processed using the 10X Genomics Cell Ranger V1.2 pipeline. Fastq files were generated from Ilumina bcl files with the Cell Ranger subroutine mkfastq. Fastq files were then processed with Cell Ranger’s count to align RNA reads against UCSC mm10 or GRCh38 genomics for mouse and human cells, respectively, using the aligner STAR (36). Redundant unique molecular identifiers (UMI) were filtered, and a gene–cell barcode matrix was generated with count. Mkfastq and count were run with default parameters.
For mouse B16 tumor samples, the gene–cell barcode matrix was passed to the R software package Seurat (v2.3.0) (37) for all downstream analyses. Genes that were expressed in at least 3 cells were included. Cells that did not express at least 200 genes were excluded, as were those that contained >5% reads associated with cell cycle genes (38,39). For mouse PyMT and human tumor samples, raw feature–barcode matrices were loaded into Seurat (v3.1.5) (40) and genes with fewer than 3 UMIs were dropped from the analyses. Matrices were further filtered to remove events with greater than 20% percent mitochondrial content, events with greater than 50% ribosomal content, or events with fewer than 200 total genes. The cell cycle state of each cell was assessed using a published set of genes associated with various stages of human mitosis (41).
Using Seurat’s ScaleData function, read counts were log2 transformed and scaled using each cell’s proportion of cell cycle genes as a nuisance factor. A set of highly variable genes was generated by Seurat’s FindVariableGenes function, which were used for principal component analysis (PCA). Genes associated with principal components (selected following visualization with scree plots) were used for graph-based cluster identification and dimensionality reduction using t-distributed stochastic neighbor embedding (t-SNE) or Uniform Manifold Approximation and Projection (UMAP) analysis. Seurat’s FindAllMarkers function was used for subsequent cluster-based analyses, including cluster marker identification and differentially expressed (DE) gene analyses.
Single-Cell RNA-Sequencing Signature Generation
To generate mouse monocyte- and macrophage-specific gene signatures, sorted monocyte, CD11clo macrophage, and CD11chi macrophage samples were aggregated, log2 transformed, and scaled using Seurat. DE gene analysis was performed using FindMarkers with the parameters log N fold change > 1.5 and a min.pct of 0.25. Genes were selected by ranked fold change and the criteria that min.pct1/min.pct2 > 1.5. Genes used for cell cycle regression analysis were excluded. The top 10 genes (or fewer if less remained) were median normalized and aggregated, scaled 0–1, and plotted on specific t-SNE plots.
Gene signatures for cellular programs such as metabolism (42), “M1” and “M2” polarization (43), and MHC-II–associated genes (GSEA, REACTOME_MHC_CLASS_II_ANTIGEN_PRESENTATION), previously published cell types (44,45), and cell populations identified here were generated by taking the mean of log-normalized expression for a particular set of genes related to the specific pathway or phenotype. To visualize the distribution of these scores across cells, we binarized the distribution of the score at the 70th percentile unless specified otherwise and overlaid on the calculated t-SNE coordinates.
For correlation analysis of “M1” and “M2” genes, the expression of each gene in the signatures was calculated for each B16 tumor Csf1r+Mafb+ cluster cell and binarized at the median. A cross-correlation gene–gene matrix was obtained using the R cor function with method=”pearson”.
Single-Cell RNA-Sequencing Sample Aggregation
Pairwise comparison analyses were performed between B16 tumor myeloid-cell clusters from wildtype and Treg-depleted mice. For this, 1 sample from wildtype mice and 1 sample from Foxp3-DTR mice were used, along with an additional wildtype sample that had been generated in a previous independent experiment. The 3 objects were first transformed from Seurat v2 to Seurat v3. The raw UMI counts were renormalized using person residuals from “regularized negative binomial regression,” with sequencing depth a covariate in a generalized linear model via the R sctransform package (46). Pairwise “anchor” cells were identified between the 3 objects using the original wild-type mouse sample as a reference via the Seurat FindIntegrationAnchors function. Briefly, each pair of samples was reduced to a lower dimensional space using diagonalized Canonical Correlation Analysis (CCA) using the top 3,000 genes with the highest dispersions. The canonical correlation vectors were normalized using L2-normalization. Multiple Nearest Neighbors (MNNs) across datasets were identified for each cell in each dataset and cell–cell similarities are used as anchors to integrate the datasets together using the Seurat IntegrateData function.
For the integration of all human samples, the individually processed samples were normalized, and variance stabilized using negative binomial regression via the scTransform method offered by Seurat (46). Counts matrices were merged into a single Seurat object and the batch (or library) of origin was stored in the metadata of the object. The log-normalized counts were reduced to a lower dimension using PCA and the individual libraries were aligned in the shared PCA space in a batch-aware manner (each individual library was considered a batch) using the Harmony algorithm (47). The resulting Harmony components were used to generate a batch-corrected UMAP, and to identify clusters of transcriptionally similar cells across each of the 11 samples.
Single-Cell RNA-Sequencing Pseudotime Analysis
Raw UMI counts from the cleaned and processed Seurat objects for the control and Treg-depleted mouse experiment were extracted and coerced into Monocle2 (48,49). CellDataSet objects were generated, normalizing the data using a negative binomial distribution with fixed variance (negbinom.size). Each object was independently processed to identify a pseudotime trajectory. Briefly, each object was processed to estimate per-cell coverage and sequencing depth (estimateSizeFactors) and gene dispersions (estimateDispersions). Cells were further filtered to retain high-quality cells with ≥500 genes and genes were filtered to retain only those in at least 10 cells. The dataset was reduced to 2 dimensions using the DDRTree algorithm and the marker genes that differentiated the Ly6c2+Hp+ monocytes and C1qa+ macrophage clusters from other cell types were used to guide the trajectory inference. Relative pseudotime was obtained through a linear transformation relative to the cells with the lowest and highest pseudotimes (1-min_pseudotime)/max_pseudotime. The “wave” plots were constructed using the Seurat LogNormalized counts and the relative pseudotime described above for the top 5 genes per cluster as identified by scRNA-seq.
Human samples were analyzed with Monocle3 (48,49), and the cell_data_set object along with the batch-corrected PCA and UMAP embeddings were imported directly from the Seurat object. Each cell-specific trajectory was inferred by reverse embedding the UMAP coordinates using the DDRTree method. Relative pseudotime was obtained as described for the mouse tumor samples.
TCGA Survival Analyses
Tumor RNAseq counts and transcripts-per-million (TPM) values for 985 kidney renal clear cell carcinoma (KIRC) samples from the Toil recompute data in the TCGA Pan-Cancer (PANCAN) cohort (50) were downloaded from the UCSC Xena browser (51). A gene signature score for each patient was calculated using the gene signature score method below. The feature gene signature scores were calculated using an m x n matrix where m represented the TPM normalized log2 counts per million (logCPM) expression of the feature signature genes and n represented the selected sample set (52). The expression of each gene was converted to percentile ranks across the samples using the SciPy Python module (53). The top and bottom 30 percentile were then used to define low and high patients for each respective signature unless otherwise noted.
Statistical analysis and data visualization
Comparisons between groups were analyzed using GraphPad Prism software. Experimental group allocation was determined by genotype or by random designation when all wild-type mice were used. Error bars represent mean ± SEM calculated with Prism unless otherwise noted. Comparisons between 2 groups were analyzed with Student’s t-test. For statistical measures between more than 2 groups, one-way ANOVA was performed unless otherwise noted. Nonsignificant comparisons are not shown. Investigators were not blinded to experiment group assignment during experimental data generation or analyses. The R packages Seurat and ggplot2 were used to generate figures.
Data Availability
The expression matrices for the scRNA-seq samples reported in this paper can be found with the following GEO accession numbers: GSE188548, GSE184096, GSE159913, and GSE184398. Code used to generate the scRNA-seq analyses are included on Github (https://github.com/UCSF-DSCOLAB/mujal_et_al_MonoMac_2021).
Results
Mouse B16 Tumors Harbor a Diversity of Myeloid States
Subcutaneous implantation of B16 melanoma cells is a well-established mouse tumor model with abundant infiltration of monocytes, macrophages, and cDCs (23). To study these cells along their differentiation trajectories, we used conventional markers to sort bulk myeloid populations, along with reference populations of Ly6C+ monocytes and two tumor-associated macrophage (TAM) populations, distinguished based on level of expression of CD11c and MHC-II (23) (Fig 1A, Supplementary Fig S1A). Each of these samples were then subjected to scRNA-seq analysis.
Figure 1. ScRNA-seq analysis of mouse B16 tumor myeloid cells maps transcriptional heterogeneity amongst monocytes and TAMs.
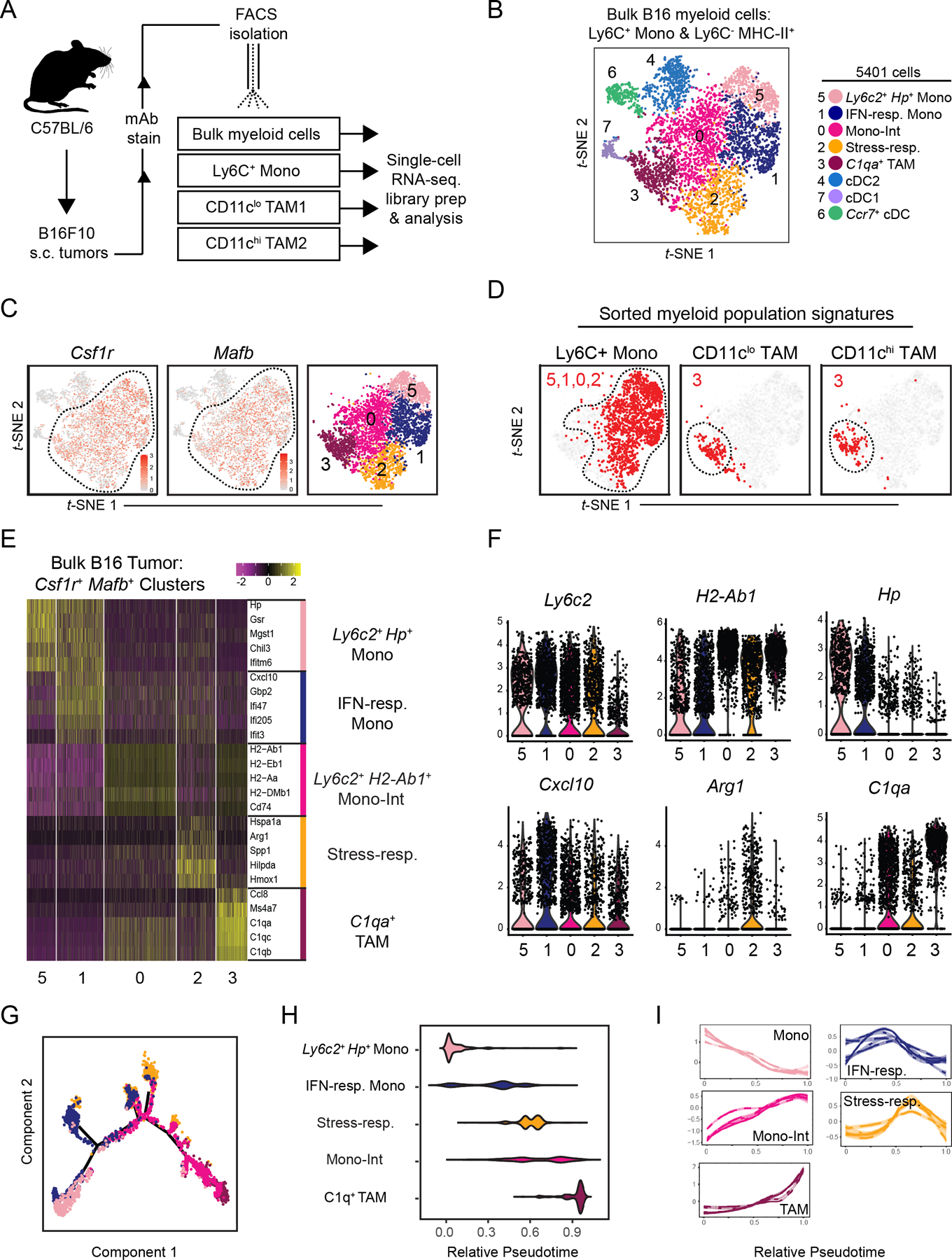
(A) Schematic illustration of workflow for isolation of specified myeloid cell populations from B16 tumors subcutaneously implanted in wild-type C57Bl/6 mice.
(B) t-SNE plot of graph-based clustering of Ly6C+CD11b+ monocytes and Ly6C–MHCII+ myeloid cells that were sorted and pooled from at least 5 B16 tumors, and underwent scRNA-seq (A). Each dot represents a single cell.
(C) Expression of Csf1r (left) and Mafb (middle) on t-SNE plot of bulk myeloid cells (B), and display of selected Csf1r+Mafb+ clusters (right).
(D) Expression of gene signatures specific to Ly6C+ monocyte, CD11clo TAM1, or CD11chi TAM2 populations (A, Supplementary Fig. S1F) displayed on t-SNE plot of Csf1r+Mafb+ myeloid cells (C). Cells with top median of signature expression level labeled in red.
(E) Heatmap displaying expression levels of top 5 DE genes between Csf1r+Mafb+ cell clusters (C). Genes ranked by fold change.
(F) Expression levels of selected genes amongst Csf1r+Mafb+ cell clusters (C).
(G) Differentiation trajectory model using Monocle analysis of cells from Csf1r+Mafb+ clusters (C). Color coding corresponds to previous labels (B).
(H) Graph of relative pseudotime values of Csf1r+Mafb+ cluster cells (C) from Monocle analysis (G).
(I) Expression levels of cluster-specific genes (E) over relative pseudotime (H). Each line corresponds to an individual gene.
Within the bulk myeloid population, t-SNE clustering yielded eight transcriptionally-distinct cell populations (Fig. 1B, Supplementary Fig S1B–C), including three Flt3+Kit+ cDC populations (Clusters 4, 6, 7), which were marked by signatures specific to cDC1s, cDC2s, and conserved cDC activation programs (Supplementary Fig. S1D, Supplementary Table S1) (23,24,26). The remaining myeloid cells (Clusters 0, 1, 2, 3, 5) broadly expressed Csf1r and Mafb (Fig. 1C), indicative of monocytes and macrophages. In addition, these cells broadly expressed Ccr2 and modest but appreciable levels of Cx3cr1 (Supplementary Fig. S1E). Having focused on the stimulatory capacity of cDCs in previous work (23,24,45), here we focused on the diversity of monocytes and macrophages as it related to the TIME.
To align transcriptional cell type categorization with flow cytometry analysis, we generated cell type–specific gene signatures from the scRNA-seq analysis of the FACS-sorted monocytes and TAMs (Fig. 1A, Supplementary Fig. S1F). When applied (Fig. 1D), these indicated that four Csf1r+Mafb+ populations (Clusters 0, 1, 2, 5) expressed monocyte-specific genes. The four monocyte populations expressed Ly6c2, but varied in levels of other monocyte-associated genes (e.g., Hp, Chil3) and, as found in Cluster 0, also expressed TAM-associated genes (e.g. H2-Ab1, C1qa, Ms4a7) (Fig. 1E–F). Monocyte-like clusters were differentiated from one another by cellular activation programs. For example, Cluster 1 (“IFN-responsive”) was specifically enriched for interferon (IFN)-inducible genes such as Cxcl10, Gbp2, and IFIT-family members. Cluster 2 (“stress-responsive”) cells expressed Arg1 and were enriched for cellular stress processes, including oxidative stress–responsive genes and heat-shock protein genes such as Hmox1, Hspa1a, Hilpda, Bnip3, Ero1l, and Ndrg1 (Fig. 1F, Supplementary Fig. S1G). In contrast to the heterogeneity observed amongst monocytes, signatures for both populations of TAMs localized within Cluster 3 (Fig. 1D).
We applied pseudotime analysis (48) to generate a model of tumor monocyte-to-macrophage differentiation (Fig. 1G–H, Supplementary Fig. S1H). This model placed Cluster 5 Ly6c2+Hp+ monocytes and Cluster 3 C1qa+ TAMs at opposite ends of a linear trajectory, consistent with our expectations. Cluster 0 monocytes occupied the continuum between them and expressed a combination of both monocyte- and TAM-associated gene signatures such that we designated these cells “Intermediate monocytes” (“Mono-Int”). Kinetic analysis of cluster-enriched genes confirmed gradual downregulation of Ly6c2+Hp+ monocyte-associated genes and up-regulation of “Mono-Int”- and TAM-associated genes along the pseudotime trajectory (Fig. 1I). This transcriptional model thus supported a framework of progressive monocyte-to-TAM differentiation, in which Ly6C down-regulation is paired with up-regulation of CD64, MHC-II, and F4/80 (15) (Fig. Supplementary S1I–J). In contrast, IFN- and stress-responsive cells occupied distinct branches that diverged from the dominant differentiation trajectory at intermediate timepoints (Fig. 1H–I, Supplementary Fig S1H).
Heterogeneous Acquisition of ‘Stress-’ and ‘IFN-Responsive’ Cellular Programs
To gain higher resolution on the differentiation trajectories within the monocyte/macrophage lineage, we performed cluster analysis on the sorted monocyte and TAM samples. Sorted monocytes expressed Ly6c2 and contained clusters similar to those identified within the bulk myeloid-cell sample (Fig. 2A, Supplementary Fig S2A), indicating that these cells may not consist purely of early-stage monocytes but also include some cells that have acquired macrophage attributes (Supplementary Fig. S1J). Cluster analysis of CD11clo and CD11chi TAMs, however, resolved diversity beyond the C1qa+ TAM signature (Fig. 2B, Supplementary Fig. S2B–C) including identifying clusters enriched for cell cycle–related genes, and an Mgl2+ TAM subset that expressed immune modulators such as Ccl6, Il1b, and Retnla as compared to the C1qa+ cluster, which more highly expressed genes such as Ms4a7. Although these cells had not formed a distinct population in our original analysis of bulk myeloid cells (Fig. 1), we did retrospectively detect Mgl2+ cells in that scRNA-seq dataset, as well as by flow cytometry (Supplementary Fig. S2D). TAM-subset clusters were also accompanied by an Arg1+ stress-responsive cluster akin to that found in the sorted monocytes (Fig. 2B, Supplementary Fig. S2B–C). Indeed, re-clustering of the entire stress-responsive cluster from the bulk tumor myeloid sample revealed that this program was acquired by monocytes, “Mono-Int” and TAMs (Fig. 2C, Supplementary Fig. S2E).
Figure 2. ScRNA-seq analysis highlights layering of microenvironment-induced programs during tumor monocyte-to-macrophage differentiation.
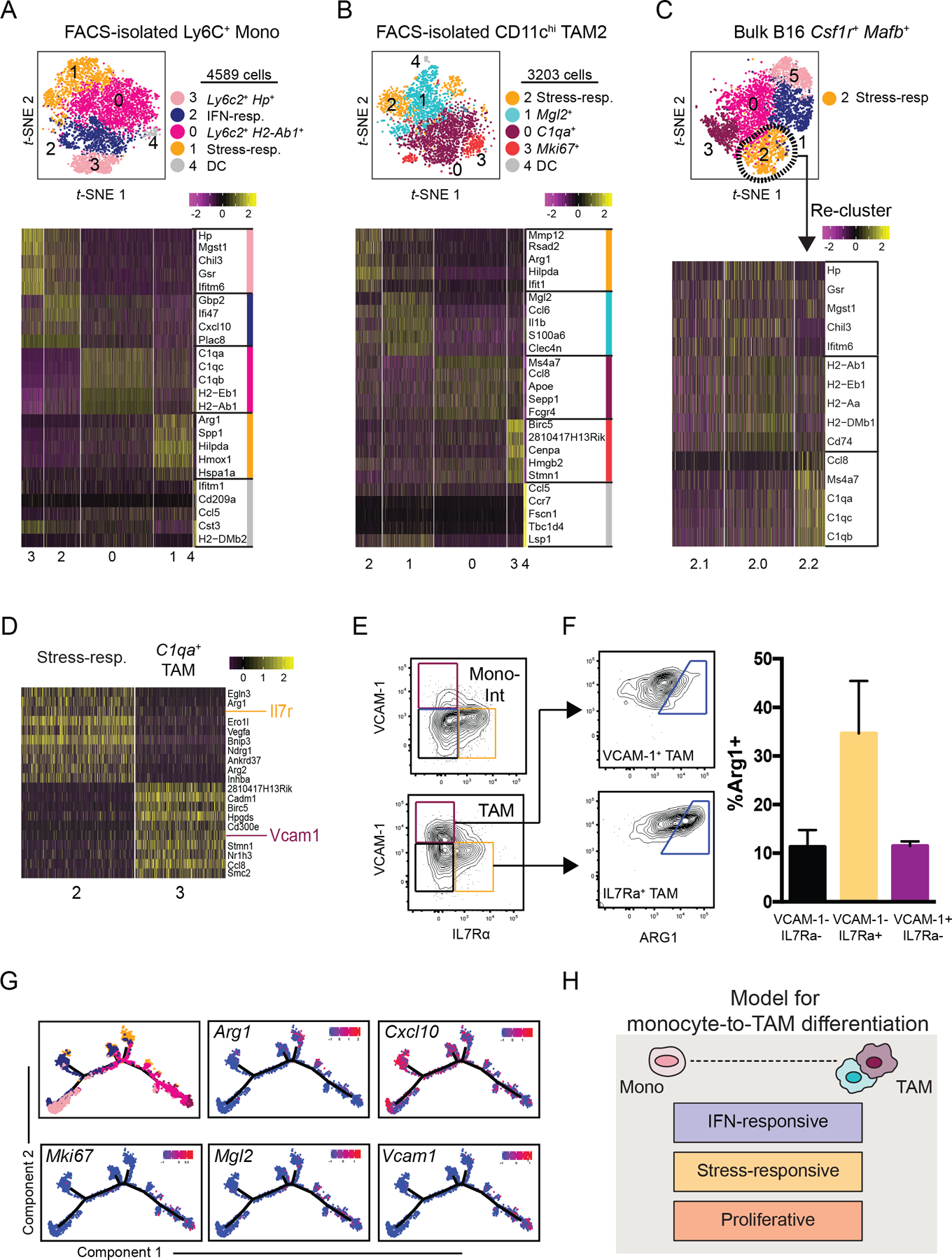
(A) t-SNE plot of graph-based clustering (top) of Ly6C+ monocytes sorted from B16 tumors and processed for scRNA-seq (Fig. 1A), and heatmap displaying expression levels of top 5 DE genes between clusters (bottom) with genes ranked by fold change.
(B) t-SNE plot and graph-based clustering (top) of CD11chi TAMs sorted from B16 tumors and processed for scRNA-seq (Fig 1A), and heatmap displaying expression levels of top 5 DE genes between clusters (bottom) with genes ranked by fold change.
(C) Stress-responsive cells (Cluster 2) from bulk B16 myeloid cells (Fig. 1B) were selected for further clustering analysis (top). Heatmap of expression levels of monocyte- and macrophage-specific genes (Fig. 1E) by Cluster 2 sub-cluster (bottom).
(D) Heatmap of DE gene expression levels between Cluster 2 and Cluster 3 of bulk tumor myeloid cell sample (Fig. 1B). Genes ranked by degree of exclusivity to a given cluster (min.pct1/min.pct2).
(E) Expression levels of IL7Rα and VCAM-1, as assessed by flow cytometry, of “Mono-Int” (Ly6C+CD64+) (top) and TAMs (Ly6C–F4/80+CD64+) (bottom) from B16 tumors.
(F) Example (left) and quantification (right) of intracellular ARG1 expression by VCAM-1+ (top) or IL7rα+ (bottom) TAMs from B16 tumors using flow cytometry. ARG1+ gating determined by isotype control. Data are representative of 2 independent experiments with 3–5 mice per experiment (mean ± SEM).
(G) Expression levels of selected genes along differentiated trajectory generated by Monocle (Fig. 1G).
(H) Schematic model of tumor monocyte-to-macrophage differentiation that integrates lineage-associated and microenvironmentally-induced transcriptional programs.
Segregated expression of stress-responsive genes and canonical TAM-associated genes suggested divergent transcriptional programs and we sought to determine if these populations could also be distinguished by flow cytometry. Differential gene expression analysis of the stress-responsive and C1qa+ TAM clusters from our bulk myeloid-cell sample revealed cluster-specific expression of cell surface genes Il7r and Vcam1, respectively (Fig. 2D). Using the same gating as in Supplementary Fig. S1A, we confirmed this split in both “Mono-Int” and TAMs (Fig. 2E) and we found enriched arginase 1 (ARG1) expression in both IL-7Rα+ populations (Fig. 2E–F, Supplementary Fig. S2F). As expected from the single-cell transcriptional analysis, VCAM1+ cells were more abundantly found within TAMs (Fig. 2E–F, Supplementary Fig. S2F).
Together, this dissection of sorted cell populations lent support to a model in which monocytes and TAMs exist in a differentiation trajectory, along which cells can adopt specialized cellular programs (Fig. 2G–H). Some programs, such as those associated with Mgl2+ or Vcam1+ TAMs, selectively emerged later, in mature TAMs. Others, such as IFN-induced signaling or stress-responsiveness may be more universally accessible across differentiation stages. In addition, we detected populations of IFN-responsive monocytes in the peripheral blood of B16 tumor-bearing mice (Supplementary Fig. S2G–H), perhaps suggesting that systemic IFN signaling, or other induction of this program, may define monocytes prior to tumor entry. In contrast, stress-responsive populations were not detected in the blood, suggesting that microenvironmental cues in the TIME likely induce this activation program locally. Further studies are warranted to explore if these programs directly influence monocyte differentiation processes or act as ‘layers’ that accessorize a canonical differentiation trajectory.
Mouse Tumor Macrophage Subset Heterogeneity Does Not Reflect “M1/M2” Polarization
Macrophage exposure to type-1 or type-2 cytokines in vitro results in “M1” and “M2” transcriptional signatures that are often used to describe ‘pro-inflammatory,’ or ‘anti-inflammatory’ and wound-healing processes, respectively (18–20). To address whether “M1/M2” polarization was a useful construct to define tumor macrophage diversity in vivo, we tested how “M1” and “M2” gene signatures (43) corresponded to the tumor myeloid-cell subsets profiled here. Using correlation and clustering analyses (Fig. 3A, Supplementary Fig. S3A), we found that, contrary to in vitro findings, tumor myeloid cells were marked by broad expression of both “M1”- and “M2”-associated genes, and we did not observe substantial correlation of gene expression within “M1” or “M2” gene groups across single cells. These data suggest that although tumor myeloid cells can express individual “M1” and “M2” genes, they rarely do so in any distinguishably consistent way during unperturbed tumor growth. We next processed tumor myeloid cells from MMTV-PyMT spontaneous mammary carcinomas for scRNA-seq analysis, sorting on Lin–CD11c+ and/or CD11b+ cells to capture the full cadre of myeloid populations including MHCII+/– cells (Supplementary Fig. S1A). We found that MMTV-PyMT tumors shared populations with the identical signatures as those defined for B16 tumors in Figure 1, albeit in different proportions, and also showed a lack of co-association between “M1” and “M2” signatures amongst the clusters (Fig 3B–C, Supplementary Fig. S3B–E).
Figure 3. B16 and PyMT tumor monocyte–macrophage heterogeneity can be attributed to diversity in transcriptional and metabolic programs, but not “M1/M2” polarization.
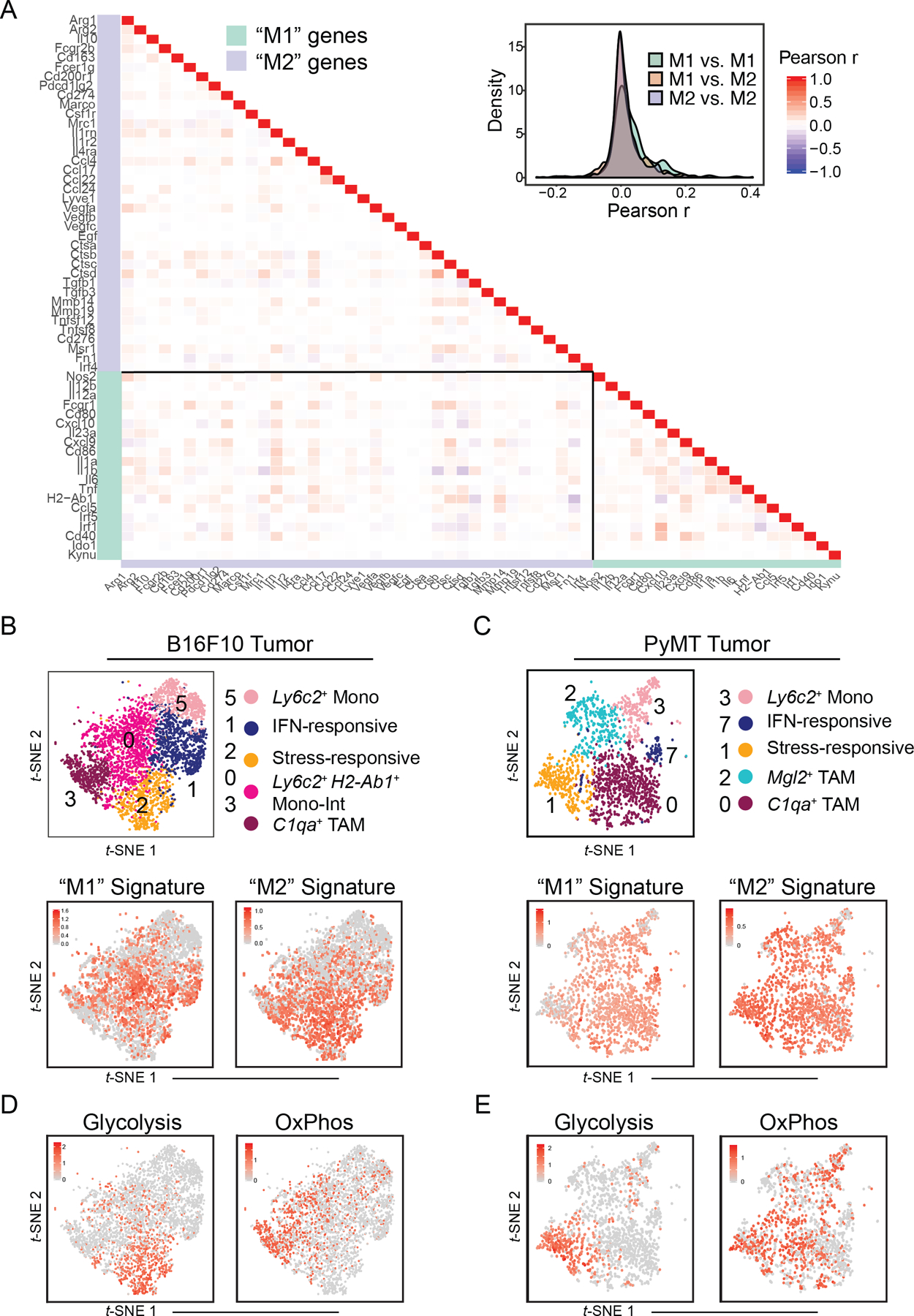
(A) Heatmap (left) and density plot (right) of Pearson r coefficient scores between “M1”- and “M2”-associated gene expression levels within Csf1r+Mafb+ cells from B16 tumors (Fig. 1C).
(B) t-SNE plot of Csf1r+Mafb+ clusters from B16 tumors (top; Fig. 1C) with expression levels of “M1” (bottom, left) and “M2” (bottom, right) gene signatures (A) displayed. Cells with top median of signature expression level labeled in red.
(C) t-SNE plot and graph-based clustering of Csf1r+Mafb+ clusters of myeloid cells that were sorted from 1 PyMT tumor and processed for scRNA-seq in an independent experiment (top; Supplementary Fig. 3B). Expression levels of “M1” (bottom, left) and “M2” (bottom, right) gene signatures (A) displayed. Cells with top 70 percentile of signature expression level labeled in red.
(D) Expression levels of glycolysis (left) and oxidative phosphorylation (“OxPhos”) (right) gene signatures (Supplementary Fig. 3F) displayed on t-SNE plot of Csf1r+Mafb+ clusters from B16 tumors (Fig. 1C). Cells with top 70 percentile of signature expression level labeled in red.
(E) Expression levels of glycolysis (left) and oxidative phosphorylation (“OxPhos”) (right) gene signatures (Supplementary Fig. 3F) displayed on t-SNE plot of Csf1r+Mafb+ clusters from PyMT tumors (C). Cells with top 70 percentile of signature expression level labeled in red.
While myeloid-cell populations appeared to be largely defined by differentiation stage and activation programs, we considered whether other core cellular features could help to further distinguish subsets across diverse microenvironments. It is now increasingly appreciated that metabolic reprogramming accompanies differentiation of immune cells, including macrophages (54). Indeed, assessment of metabolism-related genes (42) demonstrated that glycolysis-associated genes were specifically enriched in the stress-responsive cell cluster, whereas genes pertaining to oxidative phosphorylation were specifically enriched in C1qa+ TAMs in two distinct mouse models (Fig. 3D–E, Supplementary Fig. S3F). This suggests that these populations have additional important biological features in common—namely those coupled to distinct bioenergetic processes and demands.
Together, our data provides compelling evidence that “M1” and “M2” pathways have limited use in defining in vivo tumor myeloid-cell differentiation and subset plasticity during normal tumor development. Rather, common microenvironmentally-induced programs and associated metabolic programs may yield greater insight in efforts to transcriptionally define and selectively target monocyte/TAM subsets.
Human RCC-infiltrating monocytes and macrophages mirror murine populations
We next assessed how the mouse monocyte/macrophage transcriptional programs we identified might compare to those from human cancers. We performed scRNA-seq analysis on HLA-DRdim/+Lin– myeloid cells sorted from an RCC sample, which are described to have substantial myeloid-cell infiltration (55), as well as 6 melanoma and 4 head and neck cancer samples (Fig. 4A–B, Supplementary Fig. S4A). Signatures derived from previously described blood myeloid-cell populations (11,44) guided cluster identification and exclusion of cDCs (Supplementary Fig. S4B). Analysis of the CSF1R+MAFB+ clusters revealed a heterogenous collection of monocytes and macrophages with varying levels of CD14 and CD16 (Fig 4C–E).
Figure 4. Human RCC and mouse tumor myeloid cell compartments exhibit shared transcriptional features.
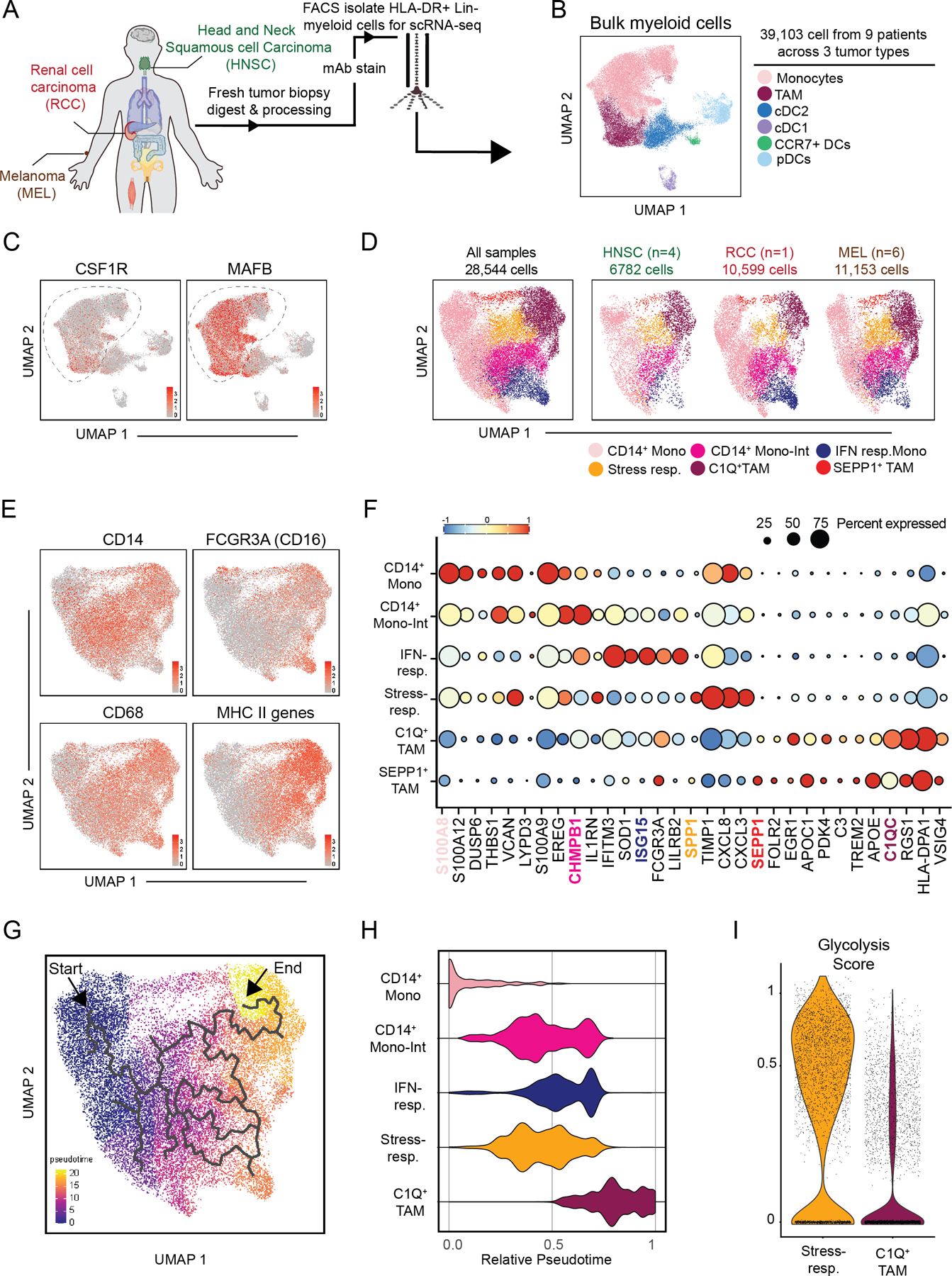
(A) Schematic of the 1 human RCC, 6 melanomas, and 4 head and neck biopsy samples processed for scRNAseq analysis.
(B) UMAP plot of graph-based clustering of bulk myeloid (Lin–HLA-DR+) cells sorted from human biopsy samples (A).
(C) Gene expression levels of CSF1R (left) and MAFB (right) displayed on UMAP plot of human tumor-infiltrating myeloid cells (B).
(D) UMAP plot of graph-based clustering of CSF1R+MAFB+ cells (C) with cells from all human biopsy samples (left) or specified cancer type (right) displayed.
(E) Expression levels of selected genes (CD14, FCGR3A, CD68) or gene signature (MHC-II-associated genes) displayed on t-SNE plot of CSF1R+MAFB+ clusters (C).
(F) Expression of selected genes expressed by CSF1R+MAFB+ clusters (C).
(G) Differentiation trajectory model generated by Monocle analysis of CSF1R+MAFB+ clusters (C).
(H) Relative pseudotime values of early-stage CD14+ monocytes, CD14+ “Mono-Int”, C1Q+ TAMs, IFN-responsive cells, and stress-response TAM clusters (C) from Monocle analysis (G).
(I) Expression levels of glycolysis-associated gene signature by cells in stress-responsive and C1Q+ TAM cells (B).
As in mouse models, we detected early-stage CD14+S100A8+ classical monocytes along with terminally-differentiated C1QC+ TAMs (Fig. 4 E–F, Supplementary Table S2). Another population was CD14+ and differentially expressed LYPD3 and MHC-II genes, consistent with intermediate differentiation of monocytes towards TAM (“Mono-Int”; Fig. 4E–F). A population of FCGR3A+ non-classical monocytes also expressed IFN-stimulated genes and thus appears to functionally represent ‘IFN-responsive’ cells (Fig. 4E–F, Supplementary Fig. S4C).
Finally, we found that there were a mix of cells on the monocyte–macrophage trajectory that expressed the stress-responsive program identified in mice, including the gene SPP1 (Fig. 4F, Supplementary Fig. S4C). When compared further to C1Q+ TAMs, this SPP1+ cluster was less mature based on higher expression of monocytic markers (i.e. S100A genes) and lower expression of MHC-II–related genes (Fig. Supplementary S4D). This analysis revealed also a population marked by expression of the antioxidant gene SEPP1 (Fig. 4F). These cells largely resembled C1Q+ TAMs but were enriched for FOLR2 (Fig. 4F, Supplementary Fig. S4E). FOLR2 is a marker previously associated with tissue-resident macrophages in breast cancer (56). Pseudotime analysis recapitulated a monocyte-to-macrophage differentiation trajectory (Fig. 4G–H) but did not connect the SEPP1+ cluster to the other monocyte–macrophage subsets, potentially due to the distinct ontogeny of tissue-resident macrophages, and thus this cluster was not considered for further trajectory analysis (Fig. 4G–H).
As in the mouse samples, the stress- and IFN-responsive programs aligned over the monocyte-to-macrophage trajectory, although in these samples, IFN-responsive monocytes appeared more advanced in differentiation stage. Again, there was broad co-expression of “M1”- and “M2”-associated genes across the populations (Supplementary Fig. S4G). Also, as in mice, there was a striking enrichment of a glycolytic signature (42) within the stress-responsive (SPP1+) cluster as compared to the C1Q+ TAMs, supporting the notion that these cells were functional orthologs in the two species (Fig 4I). Altogether, these data confirm the limitation of “M1” and “M2” applicability in human tumors and illustrate the ability of other pathways to better define myeloid-cell subsets in vivo.
Treg depletion elicits reprogramming of the tumor myeloid-cell compartment
Myeloid-cell density can vary across patients (55), but how myeloid-cell infiltration and differentiation is collectively regulated in human cancer is still not well understood. When we quantified myeloid-cell populations in 20 RCC patient biopsies using flow cytometry we found that the proportion of myeloid cells amongst live immune cells was increased in tumors of greater size and later stages (Supplementary Fig. S5A). Closer examination revealed that the ratio of macrophages-to-monocytes was also specifically increased in more advanced tumors (Fig. 5A, top). This suggested that the balance between monocytes and macrophages is dynamically regulated and that tumor growth is tied to higher macrophage density.
Figure 5. Immunosuppressive Treg cells promote tumor monocyte-to-macrophage differentiation.
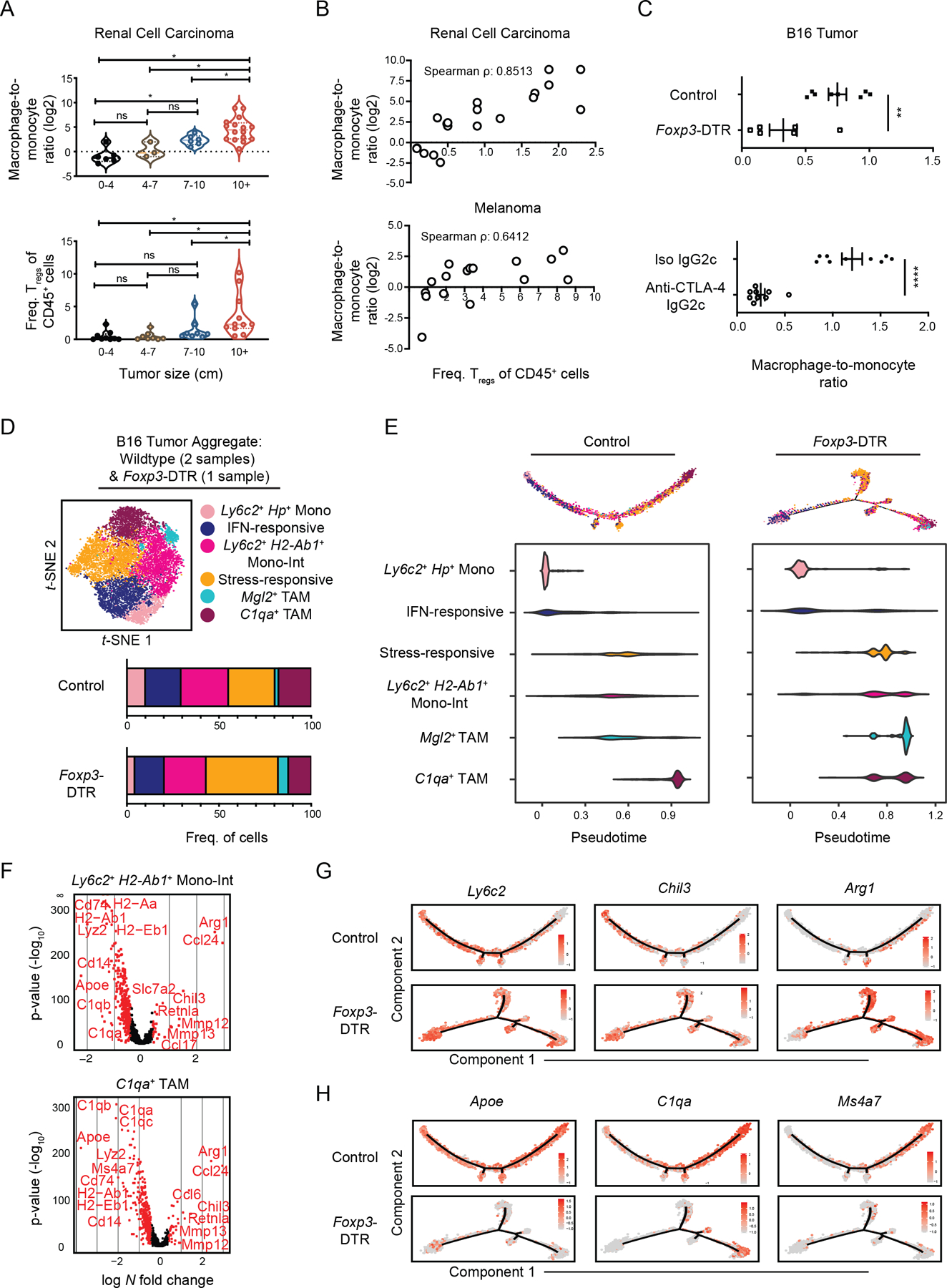
(A) 20 human RCC biopsies were measured and processed for flow cytometric analysis. The ratio of macrophage-to-monocyte (log2) cell numbers (top) and Treg frequency amongst CD45+ cells (bottom) were quantified. Samples were acquired and pooled for analysis. *p<0.05, Kruskal Wallis rank test. Dashed lines represent the median and dotted lines represent 25th percentile and 75th percentile.
(B) Dot plot and Spearman’s correlation coefficient of macrophage-to-monocyte cell number ratio (log2) and Treg frequency within CD45+ cells in 20 human RCC (top) and 16 melanoma (bottom) biopsies that were analyzed by flow cytometry. Samples were acquired and pooled for analysis.
(C) Quantification of the ratio between macrophages (Ly6C–F4/80+CD64+) and monocytes (Ly6C+CD11b+) cell number ratio in B16 tumors of DT-treated control and Foxp3-DTR mice (top), or of wild-type mice treated with depleting anti-CTLA-4 (IgG2c clone) or isotype antibody (bottom). Data is representative of at least 2 independent experiments with 3–9 mice per group per experiment (mean ± SEM). **p <0.01, ****p<0.0001, unpaired t-test.
(D) t-SNE plot of graph-based clustering (top) of B16-infiltrating Csf1r+Mafb+ cells from wildtype mice (Fig. 1) which were aggregated with DT-treated control and Foxp3-DTR mice (Supplementary Fig. 5D) from a second independent experiment in which tumors from at least 5 mice were pooled. Cell numbers in specified clusters were quantified (bottom).
(E) Differentiation trajectory model generated from Monocle analysis (top) and relative pseudotime values (bottom) of Csf1r+Mafb+ cluster cells from B16 tumors from DT-treated control (left) and FoxP3-DTR mice (right).
(F) Volcano plot displaying DE genes between B16 tumor “Mono-Int” (top) and C1qa+ TAM (bottom) cluster cells from DT-treated control and FoxP3-DTR mice (D). Genes with > 0.4 log-fold changes and an adjusted p value of 0.05 (based on Bonferroni correction) are highlighted in red. Genes of interest labeled.
(G) Expression of selected monocyte-associated genes displayed on the differentiation trajectory (E) of control (top) or Foxp3-DTR (bottom) B16 tumor-infiltrating Csf1r+Mafb+ cells.
(H) Expression of selected macrophage-associated genes displayed on the differentiation trajectory (E) of control (top) or Foxp3-DTR (bottom) B16 tumor-infiltrating Csf1r+Mafb+ cells.
We thus sought other immunosuppressive parameters that might work in concert with increased macrophage accumulation. Tregs are a potent immunosuppressive force in the TIME and ablation can result in tumor clearance (24,57). We found that Tregs accumulated as kidney tumor size increased (Fig. 5A, bottom), and that Treg abundance correlated well with macrophage-to-monocyte ratios in RCC as well as melanoma (Fig. 5B). The positive correlation between Treg and macrophage density spurred us to ask whether one caused the other. Using Foxp3-DTR mice, we found that depletion of Tregs dramatically reduced the macrophage-to-monocyte ratio in mouse B16 tumors (Fig. 5C, top) as assessed by flow cytometry. These results were phenocopied by treatment of mice with an anti–CTLA-4 that specifically depletes tumor Tregs (24,58) (Fig. 5C, bottom, Supplementary Fig. S5B–C). The shift in macrophage-to-monocyte ratio observed following both methods of Treg loss preceded subsequent tumor growth control (24,58).
To further examine how Tregs may be influencing monocyte and macrophage proportions, we performed scRNA-seq analysis on mouse tumor Lin–CD11b+ and/or CD11c+ myeloid cells from B16 tumor-bearing control and FoxP3-DTR mice. Csf1r+Mafb+ clusters from this experiment were aggregated with those from the original wild-type B16 tumor sample in Figure 1 and we observed similar cell populations across both experiments and treatment conditions (Fig. 5D, Supplementary Fig. S5D–E). Cluster proportions were modestly shifted with Treg loss (Fig. 5D), but cells from control and Treg-depleted tumors shared similar differentiation trajectories (Fig. 5E). However, Monocle analysis revealed differences in the accumulation of cells along the trajectory. Namely, whereas tumor monocytes, “Mono-Int”, and TAMs from the control sample acquired progressively increased pseudotime scores, “Mono-Int”, and TAM populations in the Foxp3-DTR sample did not exhibit sequential increases in pseudotime scores (Fig. 5E). In effect, TAM progression appeared stunted following depletion of Tregs.
In addition to increased expression of inflammatory and immunomodulatory genes (e.g., Ccl24, Arg1, Retnla, Mmp12, Mmp13, Nos2), expression of monocyte-associated genes was sustained in TAMs from Treg-depleted tumors (Fig. 5F–G, Supplementary Table S3). Moreover, expression of genes tied to macrophage differentiation (e.g., C1qa, H2-Ab1, Apoe, Ms4a7) were decreased across stages of differentiation (Fig. 5H, Supplementary Fig. S5F), further indicating these TAMs were more immature. Our analysis suggests that Treg-depletion may impair implementation of TAM transcriptional programs, a remodeling detected early during tumor monocyte differentiation. Altogether these findings support a model in which Treg abundance promotes an accumulation of terminally-differentiated TAMs in both mouse and human tumors.
Multiparametric immune-cell analysis improves classification of kidney cancer patients
Given this association between T-cell subset density and TAM maturation, we sought to further explore how features of tumor macrophage infiltration could be harnessed to reliably inform features of patient outcome, such as survival. Analysis of TCGA KIRC samples using a myeloid gene signature from CiberSort (59) demonstrated that patients with varying levels of overall myeloid-cell density did not significantly differ in their survival (Fig. 6A, left). We next stratified TCGA patient data based on levels of the monocyte–macrophage lineage genes CSF1R and MAFB, finding that patients with higher levels of these had modest improvements in outcome (Fig. 6A, middle). As these genes are not strictly macrophage-specific, we leveraged our scRNA-seq analyses of human RCC samples to generate signature scores based on the ratio between macrophage and monocyte (Fig 4E). However, no significant differences in survival were revealed using this metric (Fig. 6A, right).
Figure 6. Multiparametric analysis of tumor myeloid composition identifies kidney cancer patients with effector CD8+ T-cell responses and improved survival rates.
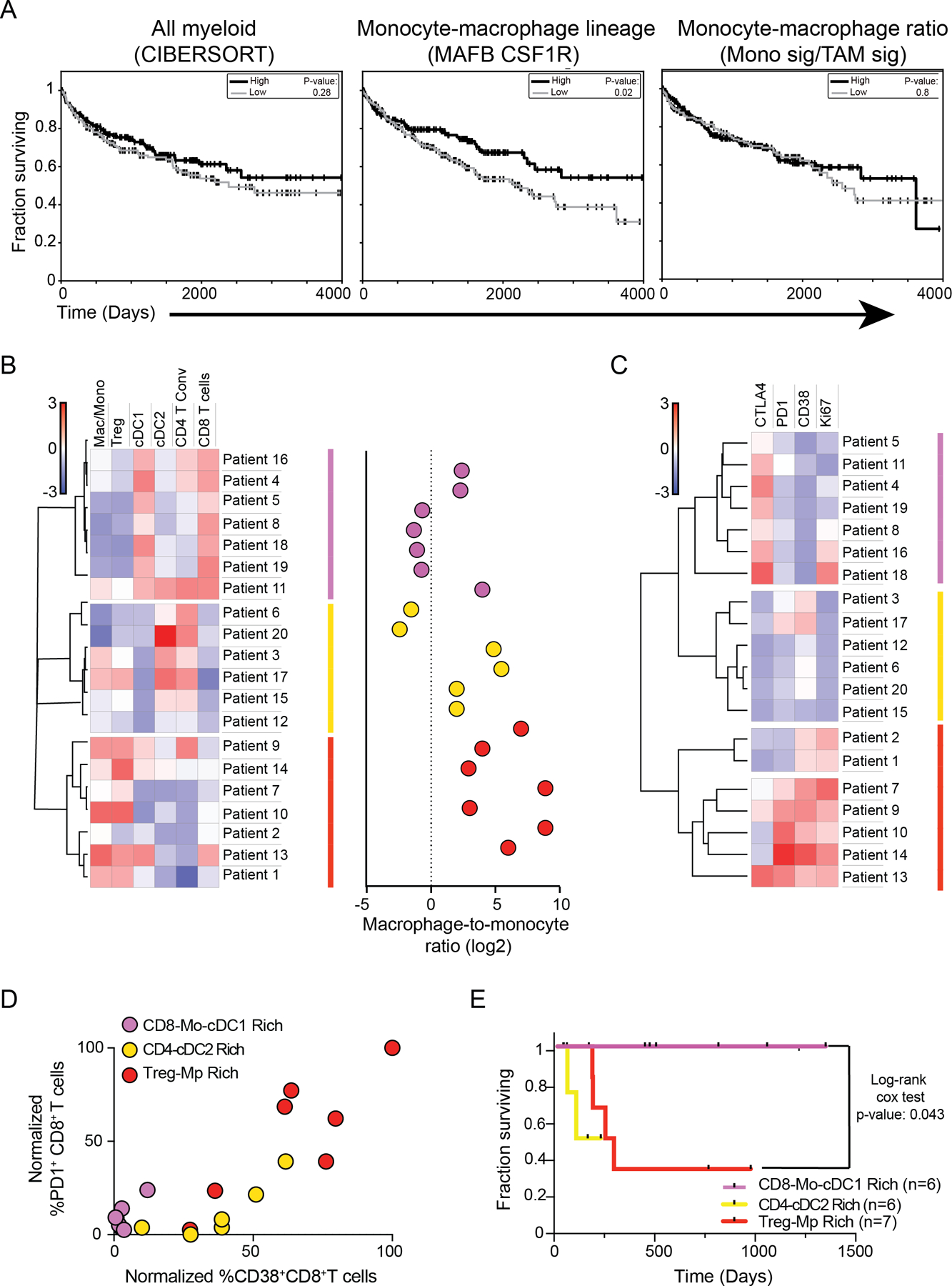
(A) Survival curves of kidney tumor patients whose TCGA tumor samples exhibited high (33%) or low (33%) levels of expression levels of pan-myeloid cell gene signatures derived from CIBERSORT (left), MAFB and CSF1R (middle), or ratio of monocyte-to-TAM gene signatures (Fig. 4) (right), analyzed with log-rank test.
(B) Heatmap of specified immune cell population frequencies (left) and the ratio of macrophage-to-monocytes (right) detected in 20 human kidney tumor samples by flow cytometry.
(C) Heatmap of specified surface receptor or Ki-67 expression frequencies amongst CD8+ T cells from 20 human kidney tumor samples that were analyzed with flow cytometry.
(D) Quantification of the frequency of CD8+ T cells from 20 human tumor kidney samples that are PD1+ or CD38+. Labeling of dots corresponds to patient groups (B,C).
(E) Survival curves of kidney cancer patients in cohort analyzed with log-rank test (B-D).
As TAM density did not appear to robustly inform patient outcome, we sought to test how TAM abundance corresponds with other immune parameters and may stratify kidney cancer patients, using flow cytometry analysis of their biopsies. We thus performed unbiased clustering analysis using measurements of myeloid-cell, Treg and conventional T cell (Tconv)-frequencies. This revealed three groups of patients that were characterized by nearly binary (all high or all low) levels of CD8+ T cells, CD4+ Tconv, cDC2s, and cDC1s (Fig. 6B). These groups could be further parsed by varied macrophage–monocyte density: low (pink), moderate (yellow) and high (red) (Fig. 6B, right). Consistent with our previous finding (Fig. 5), the group with the highest degree of macrophage differentiation was associated with the consistent presence of Treg infiltration (Treg–Mp). Of two groups with lowest macrophage differentiation, one (CD8–Mo–cDC1; pink) was distinguished by notable infiltration of cDC1s, which are critical for CD8+ T-cell responses (22,23), and that group presented with uniformly high CD8+ T-cell infiltration. A second group had higher frequencies of both CD4+ T cells and cDC2s along with a variable amount of Tregs, consistent with the demonstrated role of cDC2s in supporting both CD4+ Tconv and Treg responses (24). Further, the same patients clustered together again based on phenotypic analysis of the CD8+ T-cell compartment (Fig. 6C). Notably, CD8+ T cells from the CD8–Mo–cDC1 group (pink) expressed low levels of the exhaustion markers PD-1 and CD38 (Fig. 6C–D) and were also distinguished by higher expression of the checkpoint regulator CTLA-4, which may indicate ongoing activation (60) (Fig. 6C–D). In contrast, the Treg–Mp (red) group showed the highest levels of both exhaustion markers and proliferative capacity (i.e., Ki-67).
In testament to the heightened antitumor CD8+ T-cell profile associated with low macrophage and Treg abundance but high cDC1 density, the subset of patients with these attributes (pink) had dramatically improved survival, showing no mortality for over three years (Fig. 6E). This multiparametric clustering parsed patients with the highest survival rates more profoundly from our data set than the sole metric of cDC1 infiltration (Fig. Supplementary Fig. S6A). Similarly, although sole measurement of a cDC1 signature alone corresponded to higher survival rates amongst TCGA patients (Fig. Supplementary Fig. S6B) in support of previous studies (23,45), a combined measurement of the ratio of cDC1s to macrophages through combined gene signatures using the C1Q+ macrophage gene signature (Fig. Supplementary Fig. S6C–D) allowed for identification of kidney cancer patients with better survival. Thus, fine-tuned stratification of the kidney cancer TIME provided the resolution critical for identifying three biologically-distinct patient classes including this CD8–Mo–cDC1 group, which defines patients with the best CD8+ T-cell infiltration and outcome.
Discussion
Here we undertook scRNA-seq analysis of tumor monocytes and macrophages to determine the key hallmarks of their transcriptional diversity. We found two types of differentiation manifest during tumor development. On the one hand we found a classical lineage differentiation trajectory that progresses from monocytes-to-macrophages in a way that has been long appreciated (61) with a discernable ‘intermediate’ monocyte (“Mono-Int”) cell population. A “Mono-Int” population is, for reference, well-described in other settings. For example, Randolph and colleagues detect ‘intermediate’ monocytes in lymphoid and non-lymphoid tissue in steady-state conditions (62), and fluorescent real-time lineage tracing identifies cells undergoing that transition during allergic challenge (63).
On the other hand, we found two differentiation layers – ‘stress-responsive’ and ‘IFN-responsive’ – that co-exist along that trajectory and that were shared across multiple mouse models as well as a profiled human RCC biopsy (Fig. 1, 4). These programs were also present in other recently published studies (11,13,64,65). For example, in a pan-cancer study, Cheng et al. discern myeloid populations whose primary distinction is their expression of IFN-induced genes (e.g., ISG15+ TAMs) (11). Similarly, we noted that the stress-responsive population shares characteristics with cells historically contained within MDSCs (i.e. Arg1 expression and glycolytic programming) (27). A notable difference in our interpretation compared to these previous reports lies in our incorporation of these layers within the monocyte–macrophage differentiation axis, rather than proposing them as a unique trajectory. Through independent profiling of purified monocytes and macrophages in our study and pseudotime analysis (Fig. 1–2), we find the stress-responsive signatures evident in both cell populations and indeed across them. In additional support of such a view, we found that an IFN-responsive signature was enriched amongst monocytes in one mouse model and macrophages in another (Fig. 1, 3). We believe that this indicates that macrophages can differentiate in two dimensions – progression through the classical lineage as well as acquisition of specialized states characterized by examples of IFN or stress exposure. For these reasons, we prefer employing a nomenclature system that integrates the degree of monocyte-to-macrophage differentiation first, followed by additional transcriptional and functional qualities. Intuitively, this is similar to CD4+ T cells that can differentiate along a naïve–effector–memory axis while also being able to layer on Th1/Th2/Th17 programs.
Despite the comparison with CD4+ T cells, we do not find any populations, nor indeed any cells, that have an exclusively “M1” or “M2” signature (Fig. 3). Individual genes such as Arg1 are associated with certain clusters, as some have observed (11), but both correlation and signature analyses fail to identify any of the described ‘M1’ or ‘M2’ genes as either being selectively linked with one another in single cells, or as key classifiers of cell clusters. To this extent, the ‘M1/M2’ nomenclature has provided direction in the fruitful study of myeloid-cell signaling and differentiation but does not appear to be accurately categorize distinct differentiation states, at least for tumors in vivo. We note the absence of data to the contrary of this conclusion in other recent reports (43,55,65), although of course individual nomenclature (e.g., “M2-like”) is clearly a matter of choice and needs only discussion as to which part of the in vitro signature might be biologically relevant.
One important aspect of myeloid cell biology that requires further elaboration is how to identify IFN- and stress-responsive phenotypes. For example, Gubin et al. use iNOS as a marker by flow cytometry to define the IFN-stimulated population induced by ICB (12), whereas Cheng et al. utilize ISG15 (11). Particularly in the former study, which studied macrophage identity following ICB therapies, varied levels of type I and II IFNs may also modulate properties of this differentiation layer. In the case of ‘stress-responsive’ populations, our data also point to IL-7Rα expression, which may indicate involvement of TSLP signaling through heterodimeric pairing with TSLPR (66). An important set of conserved genes for ‘stress-responsive’ macrophages, taken from our manuscript, is their consistent and significant enrichment for glycolytic genes, particularly in comparison to conventional C1q ‘mature’ TAMs. Given that HIF-1 is known to induce glycolytic genes under inflammatory and/or hypoxic conditions (54), this finding raises the question of whether these cells are selected for hypoxic environments where oxidative phosphorylation may not proceed, as well as their specific function. Going forward, the use of multiplexed imaging technologies such as ion beam imaging (MIBI) and single-cell spatial transcriptomics will enable this question to be addressed.
Our investigation of monocyte–macrophage differentiation led us to explore how its regulation could inform our understanding of antitumor immunity. Analysis of RCC and melanoma patient cohorts revealed an increase in macrophage-to-monocyte ratios with tumor grade, a rise that coincided with Treg density and was Treg-dependent. Tregs exert potent immunosuppression and are thought to restrain T-cell activity and antitumor responses through modulation of DC stimulatory capacity, production of immunosuppressive cytokines and substrates, and competitive usage of growth factors and metabolic byproducts (24,67,68). It is becoming clear now that tumor Tregs also strongly influence the monocyte–macrophage lineage, likely through multiple mechanisms. In a recent study, tumor Tregs promoted tumor macrophage numbers by supporting their mitochondrial capacity and viability (69). Here, our scRNA-seq data demonstrates that early-stage monocytes and “Mono-Int” cells are already unable to properly implement TAM-associated transcriptional programs in the absence of Tregs, indicating that Tregs also fuel macrophage differentiation processes. This liaison between Tregs and macrophages mirrors one identified in the adipose fat of lean mice, where Tregs are thought to actively maintain homeostasis and hold inflammatory macrophages at bay (70,71). Similarly, during the resolution of injury and inflammation in skeletal muscle and heart tissue, a transition from pro- to anti-inflammatory macrophages occurs in a manner that appears to rely on Treg accumulation (72,73). That Tregs may act on tumor macrophages in a similar fashion offers another example of how the TIME can exploit immune programs of “accommodation” that are otherwise in place to achieve tissue homeostasis in the face of perturbations (74).
Accumulation of a broad swath of macrophages in the TIME has previously been implicated with poor outcome (75). Consistent with this but at higher resolution, we detected a group of kidney cancer patients for whom high macrophage-to-monocyte abundance was associated with diminished T-cell infiltration and exhaustion of those cells detected, concurring with other reports (55,64). Our manuscript thus points to an emerging trio of Tregs, macrophages, and exhausted T cells, whereby effector T cells may be corrupted through direct cellular interactions with TAMs, as has been suggested by observations of TAM–CD8+ T-cell co-localization in clear cell RCC (ccRCC) (64), or indirectly through macrophage-induced Treg expansion and activity (8,76) or DC suppression (24,77).
Yet, high myeloid cell infiltration or skewed macrophage-to-monocyte ratios alone were not prognostic for KIRC patient survival. Indeed, although macrophages have often been found to be negatively associated with patient outcome, macrophage abundance as a sole biomarker has not been universally useful with prior studies similarly reporting instances in which macrophage abundance is not informative for patient cohorts with specific cancer sub-types, treatment regimens, or tumor stage (78–81). Clustering analysis of kidney TIME composition using comprehensive immune parameters, however, uncovered an archetype characterized by low macrophage-to-monocyte differentiation in conjunction with high cDC1 infiltration. These patients (CD8–Mo–cDC1) had elevated infiltration of CD8+ T cells with low surface expression of proteins associated with exhaustion and highly enhanced survival rates (Fig. 6, pink). Notably, recent work focused on ascertaining the different immune archetypes across solid tumors suggests that these patient groups, though most frequent in kidney cancer, span cancer types including frequent representation in colorectal and bladder tumors (33).
Identification of a CD8–Mo–cDC1archetype emphasizes the value of integrating multiparametric biomarkers as a means to better parse patient outcome and to establish principles of TIME organization. Given that T-cell activity appears to be collectively influenced by multiple immune cell populations with distinct partnering patterns, our analysis suggests that dual targeting of TIME axes may elicit the best CD8+ T-cell responses. For example, reprogramming and/or depletion of macrophages may relieve active suppression (2,14) and strategies that boost cDC1 recruitment and survival (1) may further benefit even those with favorable macrophage-to-monocyte density. It is also notable that this protective archetype is specifically enriched for monocytes. Indeed, monocyte differentiation into macrophages may not be inevitable and accumulation of “Mono-Int” cells have been detected in multiple forms of inflammation (10,16,82,83). Additionally, the potential importance of monocytes is indicated by their increased numbers in the blood of ICB responsive as compared to non-responsive melanoma patients (32). In ccRCC patients, IFN-responsive TAMs exhibited lower levels of HLA-DR, reminiscent of the “Mono-Int” cells described here, and higher levels of these ISGhi TAMs were predictive of survival after tyrosine kinase inhibitor treatment (79). Such a relationship opens questions across cancer type; namely, whether “Mono-Int” are distinct in their antitumor function, and how might monocytes be additive or synergistic with cDC1s to drive antitumor CD8+ T cells?
Altogether these findings contribute to the endeavor of clarifying useful distinctions in myeloid cell gene expression and highlight settings in which multiparametric analysis of tumor myeloid cell composition can inform patient immune archetype and guide development of relevant therapies.
Supplementary Material
Synopsis.
Single-cell RNA-sequencing analyses of mouse and human tumor-infiltrating myeloid cells show that monocyte-to-macrophage differentiation is marked by conventional lineage-associated and microenvironmental-induced cellular programs. Multiparametric analysis of myeloid composition, including monocyte-to-macrophage differentiation, helps to identify patients with improved survival.
Acknowledgments
We would like to thank E. Wan and the Institute for Human Genetics at UCSF for helping prepare samples for scRNA-seq as well as the UCSF Flow Cytometry Core for maintenance of flow cytometers and sorters. We would like to also thank J.J. Engelhardt and Bristol-Myers-Squibb for Fc-modified anti-CTLA-4 antibodies.
Funding Information
This work was supported by the US National Institutes of Health (R01CA197363 and U01CA217864 to M.F.K). A.M.M. is supported by the Cancer Research Institute as a Cancer Research Institute/Amgen Fellow. Acquisition and analysis of certain human samples was partially funded by contributions from Abbvie, Amgen, Bristol-Myers Squibb, and Pfizer as part of the UCSF Immunoprofiler Initiative.
Footnotes
Declaration of Interests
M.F.K. is a founder and shareholder in Pionyr Immunotherapeutics and Foundery Innovations, which develop novel immunotherapeutics that target diseases including cancers. M.B and J.L.P. are shareholders in Pionyr Immunotherapeutics.
References
- 1.Wculek SK, Cueto FJ, Mujal AM, Melero I, Krummel MF, Sancho D. Dendritic cells in cancer immunology and immunotherapy. Nat Rev Immunol. 2020;20:7–24. [DOI] [PubMed] [Google Scholar]
- 2.DeNardo DG, Ruffell B. Macrophages as regulators of tumour immunity and immunotherapy. Nat Rev Immunol. 2019;19:369–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 3.Ginhoux F, Guilliams M. Tissue-Resident Macrophage Ontogeny and Homeostasis. Immunity. 2016;44:439–49. [DOI] [PubMed] [Google Scholar]
- 4.Franklin RA, Liao W, Sarkar A, Kim MV, Bivona MR, Liu K, et al. The cellular and molecular origin of tumor-associated macrophages. Science. 2014;344:921–925. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 5.Arwert EN, Harney AS, Entenberg D, Wang Y, Sahai E, Pollard JW, et al. A Unidirectional Transition from Migratory to Perivascular Macrophage Is Required for Tumor Cell Intravasation. Cell Reports. 2018;23:1239–48. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 6.Qian B-Z, Li J, Zhang H, Kitamura T, Zhang J, Campion LR, et al. CCL2 recruits inflammatory monocytes to facilitate breast-tumour metastasis. Nature. 2011;475:222–5. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 7.Loyher P-L, Hamon P, Laviron M, Meghraoui-Kheddar A, Goncalves E, Deng Z, et al. Macrophages of distinct origins contribute to tumor development in the lung. J Exp Med. 2018;215:2536–53. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 8.Casanova-Acebes M, Dalla E, Leader AM, LeBerichel J, Nikolic J, Morales BM, et al. Tissue-resident macrophages provide a pro-tumorigenic niche to early NSCLC cells. Nature. 2021;1–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 9.Zhu Y, Herndon JM, Sojka DK, Kim K-W, Knolhoff BL, Zuo C, et al. Tissue-Resident Macrophages in Pancreatic Ductal Adenocarcinoma Originate from Embryonic Hematopoiesis and Promote Tumor Progression. Immunity. 2017;47:323–338.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 10.Etzerodt A, Tsalkitzi K, Maniecki M, Damsky W, Delfini M, Baudoin E, et al. Specific targeting of CD163+ TAMs mobilizes inflammatory monocytes and promotes T cell–mediated tumor regression. J Exp Med. 2019;216:2394–411. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 11.Cheng S, Li Z, Gao R, Xing B, Gao Y, Yang Y, et al. A pan-cancer single-cell transcriptional atlas of tumor infiltrating myeloid cells. Cell. 2021;184:792–809.e23. [DOI] [PubMed] [Google Scholar]
- 12.Gubin MM, Esaulova E, Ward JP, Malkova ON, Runci D, Wong P, et al. High-Dimensional Analysis Delineates Myeloid and Lymphoid Compartment Remodeling during Successful Immune-Checkpoint Cancer Therapy. Cell. 2018;175:1014–1030.e19. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 13.Zhang L, Li Z, Skrzypczynska KM, Fang Q, Zhang W, O’Brien SA, et al. Single-Cell Analyses Inform Mechanisms of Myeloid-Targeted Therapies in Colon Cancer. Cell. 2020;181:442–459.e29. [DOI] [PubMed] [Google Scholar]
- 14.Cassetta L, Pollard JW. Targeting macrophages: therapeutic approaches in cancer. Nat Rev Drug Discov. 2018;17:887–904. [DOI] [PubMed] [Google Scholar]
- 15.Jakubzick CV, Randolph GJ, Henson PM. Monocyte differentiation and antigen-presenting functions. Nat Rev Immunol. 2017;17:349–362. [DOI] [PubMed] [Google Scholar]
- 16.Iijima N, Mattei LM, Iwasaki A. Recruited inflammatory monocytes stimulate antiviral Th1 immunity in infected tissue. Proc National Acad Sci. 2011;108:284–9. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 17.Plantinga M, Guilliams M, Vanheerswynghels M, Deswarte K, Branco-Madeira F, Toussaint W, et al. Conventional and Monocyte-Derived CD11b+ Dendritic Cells Initiate and Maintain T Helper 2 Cell-Mediated Immunity to House Dust Mite Allergen. Immunity. 2013;38:322–35. [DOI] [PubMed] [Google Scholar]
- 18.Nathan CF, Murray HW, Wiebe ME, Rubin BY. Identification of interferon-gamma as the lymphokine that activates human macrophage oxidative metabolism and antimicrobial activity. J Exp Med. 1983;158:670–89. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 19.Stein M, Keshav S, Harris N, Gordon S. Interleukin 4 potently enhances murine macrophage mannose receptor activity: a marker of alternative immunologic macrophage activation. J Exp Med. 1992;176:287–92. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 20.Mills CD, Kincaid K, Alt JM, Heilman MJ, Hill AM. M-1/M-2 Macrophages and the Th1/Th2 Paradigm. J Immunol. 2000;164:6166–73. [DOI] [PubMed] [Google Scholar]
- 21.Murray PJ, Allen JE, Biswas SK, Fisher EA, Gilroy DW, Goerdt S, et al. Macrophage Activation and Polarization: Nomenclature and Experimental Guidelines. Immunity. 2014;41:14–20. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 22.Roberts EW, Broz ML, Binnewies M, Headley MB, Nelson AE, Wolf DM, et al. Critical Role for CD103+/CD141+ Dendritic Cells Bearing CCR7 for Tumor Antigen Trafficking and Priming of T Cell Immunity in Melanoma. Cancer Cell. 2016;30:324–36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 23.Broz ML, Binnewies M, Boldajipour B, Nelson AE, Pollack JL, Erle DJ, et al. Dissecting the Tumor Myeloid Compartment Reveals Rare Activating Antigen-Presenting Cells Critical for T Cell Immunity. Cancer Cell. 2014;26:638–52. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 24.Binnewies M, Mujal AM, Pollack JL, Combes AJ, Hardison EA, Barry KC, et al. Unleashing Type-2 Dendritic Cells to Drive Protective Antitumor CD4+ T Cell Immunity. Cell. 2019;177:556–571.e16. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 25.Hildner K, Edelson BT, Purtha WE, Diamond M, Matsushita H, Kohyama M, et al. Batf3 Deficiency Reveals a Critical Role for CD8a+ Dendritic Cells in Cytotoxic T Cell Immunity. Science. 2008;322:1097–100. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 26.Maier B, Leader AM, Chen ST, Tung N, Chang C, LeBerichel J, et al. A conserved dendritic-cell regulatory program limits antitumour immunity. Nature. 2020;580:257–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 27.Veglia F, Sanseviero E, Gabrilovich DI. Myeloid-derived suppressor cells in the era of increasing myeloid cell diversity. Nat Rev Immunol. 2021;1–14. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 28.Ruffell B, Coussens LM. Macrophages and therapeutic resistance in cancer. Cancer Cell. 2015;27:462–472. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 29.Roxburgh CSD, McMillan DC. The role of the in situ local inflammatory response in predicting recurrence and survival in patients with primary operable colorectal cancer. Cancer Treat Rev. 2012;38:451–66. [DOI] [PubMed] [Google Scholar]
- 30.Quatromoni JG, Eruslanov E. Tumor-associated macrophages: function, phenotype, and link to prognosis in human lung cancer. Am J Transl Res. 2012;4:376–89. [PMC free article] [PubMed] [Google Scholar]
- 31.Edin S, Wikberg ML, Dahlin AM, Rutegård J, Öberg Å, Oldenborg P-A, et al. The Distribution of Macrophages with a M1 or M2 Phenotype in Relation to Prognosis and the Molecular Characteristics of Colorectal Cancer. Plos One. 2012;7:e47045. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 32.Krieg C, Nowicka M, Guglietta S, Schindler S, Hartmann FJ, Weber LM, et al. High-dimensional single-cell analysis predicts response to anti-PD-1 immunotherapy. Nat Med. 2018;24:144–53. [DOI] [PubMed] [Google Scholar]
- 33.Combes AJ, Samad B, Tsui J, Chew NW, Yan P, Reeder GC, et al. A Pan-Cancer Census of Dominant Tumor Immune Archetypes. Biorxiv. 2021;2021.04.26.441344. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 34.Engelhardt JJ, Boldajipour B, Beemiller P, Pandurangi P, Sorensen C, Werb Z, et al. Marginating Dendritic Cells of the Tumor Microenvironment Cross-Present Tumor Antigens and Stably Engage Tumor-Specific T Cells. Cancer Cell. 2012;21:402–417. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 35.Headley MB, Bins A, Nip A, Roberts EW, Looney MR, Gérard A, et al. Visualization of immediate immune responses to pioneer metastatic cells in the lung. Nature. 2016;531:513–517. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 36.Dobin A, Davis CA, Schlesinger F, Drenkow J, Zaleski C, Jha S, et al. STAR: ultrafast universal RNA-seq aligner. Bioinformatics. 2013;29:15–21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 37.Satija R, Farrell JA, Gennert D, Schier AF, Regev A. Spatial reconstruction of single-cell gene expression data. Nat Biotechnol. 2015;33:495–502. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 38.Kowalczyk MS, Tirosh I, Heckl D, Rao TN, Dixit A, Haas BJ, et al. Single-cell RNA-seq reveals changes in cell cycle and differentiation programs upon aging of hematopoietic stem cells. Genome Res. 2015;25:1860–1872. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 39.Macosko EZ, Basu A, Satija R, Nemesh J, Shekhar K, Goldman M, et al. Highly Parallel Genome-wide Expression Profiling of Individual Cells Using Nanoliter Droplets. Cell. 2015;161:1202–1214. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 40.Stuart T, Butler A, Hoffman P, Hafemeister C, Papalexi E, Mauck WM, et al. Comprehensive Integration of Single-Cell Data. Cell. 2019;177:1888–1902.e21. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 41.Dominguez D, Tsai Y-H, Gomez N, Jha DK, Davis I, Wang Z. A high-resolution transcriptome map of cell cycle reveals novel connections between periodic genes and cancer. Cell Res. 2016;26:946–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 42.Argüello RJ, Combes AJ, Char R, Gigan J-P, Baaziz AI, Bousiquot E, et al. SCENITH: A Flow Cytometry-Based Method to Functionally Profile Energy Metabolism with Single-Cell Resolution. Cell Metab. 2020;32:1063–1075.e7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 43.Azizi E, Carr AJ, Plitas G, Cornish AE, Konopacki C, Prabhakaran S, et al. Single-Cell Map of Diverse Immune Phenotypes in the Breast Tumor Microenvironment. Cell. 2018;174:1293–1308.e36. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 44.Villani A-C, Satija R, Reynolds G, Sarkizova S, Shekhar K, Fletcher J, et al. Single-cell RNA-seq reveals new types of human blood dendritic cells, monocytes, and progenitors. Science [Internet]. 2017;356:eaah4573. Available from: http://science.sciencemag.org/content/sci/356/6335/eaah4573.full.pdf [DOI] [PMC free article] [PubMed] [Google Scholar]
- 45.Barry KC, Hsu J, Broz ML, Cueto FJ, Binnewies M, Combes AJ, et al. A natural killer–dendritic cell axis defines checkpoint therapy–responsive tumor microenvironments. Nat Med. 2018;24:1178–91. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 46.Hafemeister C, Satija R. Normalization and variance stabilization of single-cell RNA-seq data using regularized negative binomial regression. Genome Biol. 2019;20:296. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 47.Korsunsky I, Millard N, Fan J, Slowikowski K, Zhang F, Wei K, et al. Fast, sensitive and accurate integration of single-cell data with Harmony. Nat Methods. 2019;16:1289–96. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 48.Trapnell C, Cacchiarelli D, Grimsby J, Pokharel P, Li S, Morse M, et al. The dynamics and regulators of cell fate decisions are revealed by pseudotemporal ordering of single cells. Nat Biotechnol. 2014;32:381–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 49.Qiu X, Mao Q, Tang Y, Wang L, Chawla R, Pliner HA, et al. Reversed graph embedding resolves complex single-cell trajectories. Nat Methods. 2017;14:979–82. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 50.Vivian J, Rao AA, Nothaft FA, Ketchum C, Armstrong J, Novak A, et al. Toil enables reproducible, open source, big biomedical data analyses. Nat Biotechnol. 2017;35:314–6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 51.Goldman MJ, Craft B, Hastie M, Repečka K, McDade F, Kamath A, et al. Visualizing and interpreting cancer genomics data via the Xena platform. Nat Biotechnol. 2020;38:675–8. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 52.Robinson MD, Oshlack A. A scaling normalization method for differential expression analysis of RNA-seq data. Genome Biol. 2010;11:R25. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 53.Virtanen P, Gommers R, Oliphant TE, Haberland M, Reddy T, Cournapeau D, et al. SciPy 1.0: fundamental algorithms for scientific computing in Python. Nat Methods. 2020;17:261–72. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 54.O’Neill LAJ, Kishton RJ, Rathmell J. A guide to immunometabolism for immunologists. Nat Rev Immunol. 2016;16:553–65. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 55.Chevrier S, Levine JH, Zanotelli VRT, Silina K, Schulz D, Bacac M, et al. An Immune Atlas of Clear Cell Renal Cell Carcinoma. Cell. 2017;169:736–749.e18. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 56.Ramos RN, Missolo-Koussou Y, Gerber-Ferder Y, Bromley C, Bugatti M, Núñez NG, et al. Tissue-resident FOLR2+ macrophages associate with tumor-infiltrating CD8+ T cells and with increased survival of breast cancer patients. Biorxiv. 2021;2021.04.12.439412. [Google Scholar]
- 57.Bos PD, Plitas G, Rudra D, Lee SY, Rudensky AY. Transient regulatory T cell ablation deters oncogene-driven breast cancer and enhances radiotherapy. J Exp Med. 2013;210:2435–66. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 58.Selby MJ, Engelhardt JJ, Quigley M, Henning KA, Chen T, Srinivasan M, et al. Anti-CTLA-4 Antibodies of IgG2a Isotype Enhance Antitumor Activity through Reduction of Intratumoral Regulatory T Cells. Cancer Immunol Res. 2013;1:32–42. [DOI] [PubMed] [Google Scholar]
- 59.Newman AM, Liu CL, Green MR, Gentles AJ, Feng W, Xu Y, et al. Robust enumeration of cell subsets from tissue expression profiles. Nat Methods. 2015;12:453–7. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 60.Krummel MF, Allison JP. CD28 and CTLA-4 have opposing effects on the response of T cells to stimulation. J Exp Medicine. 1995;182:459–465. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 61.van Furth R, Cohn ZA. The Origin and Kinetics of Mononuclear Phagocytes. J Exp Medicine. 1968;128:415–35. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 62.Jakubzick C, Gautier EL, Gibbings SL, Sojka DK, Schlitzer A, Johnson TE, et al. Minimal Differentiation of Classical Monocytes as They Survey Steady-State Tissues and Transport Antigen to Lymph Nodes. Immunity. 2013;39:599–610. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 63.Sen D, Jones SM, Oswald EM, Pinkard H, Corbin K, Krummel MF. Tracking the Spatial and Functional Gradient of Monocyte-To-Macrophage Differentiation in Inflamed Lung. Plos One. 2016;11:e0165064. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 64.Braun DA, Street K, Burke KP, Cookmeyer DL, Denize T, Pedersen CB, et al. Progressive immune dysfunction with advancing disease stage in renal cell carcinoma. Cancer Cell. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 65.Bi K, He MX, Bakouny Z, Kanodia A, Napolitano S, Wu J, et al. Tumor and immune reprogramming during immunotherapy in advanced renal cell carcinoma. Cancer Cell. 2021; [DOI] [PMC free article] [PubMed] [Google Scholar]
- 66.Pandey A, Ozaki K, Baumann H, Levin SD, Puel A, Farr AG, et al. Cloning of a receptor subunit required for signaling by thymic stromal lymphopoietin. Nat Immunol. 2000;1:59–64. [DOI] [PubMed] [Google Scholar]
- 67.Plitas G, Rudensky AY. Regulatory T Cells in Cancer. Annu Rev Cancer Biol. 2020;4:459–77. [Google Scholar]
- 68.Vignali DAA, Collison LW, Workman CJ. How regulatory T cells work. Nat Rev Immunol. 2008;8:523–32. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 69.Liu C, Chikina M, Deshpande R, Menk AV, Wang T, Tabib T, et al. Treg Cells Promote the SREBP1-Dependent Metabolic Fitness of Tumor-Promoting Macrophages via Repression of CD8+ T Cell-Derived Interferon-γ. Immunity. 2019;51:381–397.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 70.Cipolletta D, Cohen P, Spiegelman BM, Benoist C, Mathis D. Appearance and disappearance of the mRNA signature characteristic of Treg cells in visceral adipose tissue: age, diet, and PPARγ effects. Proc National Acad Sci. 2015;112:482–487. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 71.Lumeng CN, Bodzin JL, Saltiel AR. Obesity induces a phenotypic switch in adipose tissue macrophage polarization. J Clin Invest. 2007;117:175–84. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 72.Weirather J, Hofmann UDW, Beyersdorf N, Ramos GC, Vogel B, Frey A, et al. Foxp3+ CD4+ T Cells Improve Healing After Myocardial Infarction by Modulating Monocyte/Macrophage Differentiation. Circ Res. 2014;115:55–67. [DOI] [PubMed] [Google Scholar]
- 73.Panduro M, Benoist C, Mathis D. Treg cells limit IFN-γ production to control macrophage accrual and phenotype during skeletal muscle regeneration. Proc National Acad Sci. 2018;115:201800618. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 74.Mujal AM, Krummel MF. Immunity as a continuum of archetypes. Science. 2019;364:28–9. [DOI] [PubMed] [Google Scholar]
- 75.DeNardo DG, Brennan DJ, Rexhepaj E, Ruffell B, Shiao SL, Madden SF, et al. Leukocyte Complexity Predicts Breast Cancer Survival and Functionally Regulates Response to Chemotherapy. Cancer Discov. 2011;1:54–67. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 76.Mondini M, Loyher P-L, Hamon P, Thoré MGD, Laviron M, Berthelot K, et al. CCR2-dependent recruitment of Tregs and monocytes following radiotherapy is associated with TNFα-mediated resistance. Cancer Immunol Res. 2019;7:canimm.0633.2018. [DOI] [PubMed] [Google Scholar]
- 77.Ruffell B, Chang-Strachan D, Chan V, Rosenbusch A, Ho CMT, Pryer N, et al. Macrophage IL-10 blocks CD8+ T cell-dependent responses to chemotherapy by suppressing IL-12 expression in intratumoral dendritic cells. Cancer Cell. 2014;26:623–637. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 78.Medrek C, Pontén F, Jirström K, Leandersson K. The presence of tumor associated macrophages in tumor stroma as a prognostic marker for breast cancer patients. Bmc Cancer. 2012;12:306. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 79.Krishna C, DiNatale RG, Kuo F, Srivastava RM, Vuong L, Chowell D, et al. Single-cell sequencing links multiregional immune landscapes and tissue-resident T cells in ccRCC to tumor topology and therapy efficacy. Cancer Cell. 2021;39:662–677.e6. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 80.Hakimi AA, Voss MH, Kuo F, Sanchez A, Liu M, Nixon BG, et al. Transcriptomic Profiling of the Tumor Microenvironment Reveals Distinct Subgroups of Clear cell Renal Cell Cancer - Data from a Randomized Phase III Trial. Cancer Discov. 2019;9:CD-18–0957. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 81.McDermott DF, Huseni MA, Atkins MB, Motzer RJ, Rini BI, Escudier B, et al. Clinical activity and molecular correlates of response to atezolizumab alone or in combination with bevacizumab versus sunitinib in renal cell carcinoma. Nat Med. 2018;24:749–57. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 82.Soudja SM, Ruiz AL, Marie JC, Lauvau G. Inflammatory Monocytes Activate Memory CD8+ T and Innate NK Lymphocytes Independent of Cognate Antigen during Microbial Pathogen Invasion. Immunity. 2012;37:549–62. [DOI] [PMC free article] [PubMed] [Google Scholar]
- 83.Bain CC, Scott CL, Uronen-Hansson H, Gudjonsson S, Jansson O, Grip O, et al. Resident and pro-inflammatory macrophages in the colon represent alternative context-dependent fates of the same Ly6Chi monocyte precursors. Mucosal Immunol. 2013;6:498–510. [DOI] [PMC free article] [PubMed] [Google Scholar]
Associated Data
This section collects any data citations, data availability statements, or supplementary materials included in this article.
Supplementary Materials
Data Availability Statement
The expression matrices for the scRNA-seq samples reported in this paper can be found with the following GEO accession numbers: GSE188548, GSE184096, GSE159913, and GSE184398. Code used to generate the scRNA-seq analyses are included on Github (https://github.com/UCSF-DSCOLAB/mujal_et_al_MonoMac_2021).